Chapter 8 Haematological disease
Introduction and general aspects




The formation of blood cells (haemopoiesis)
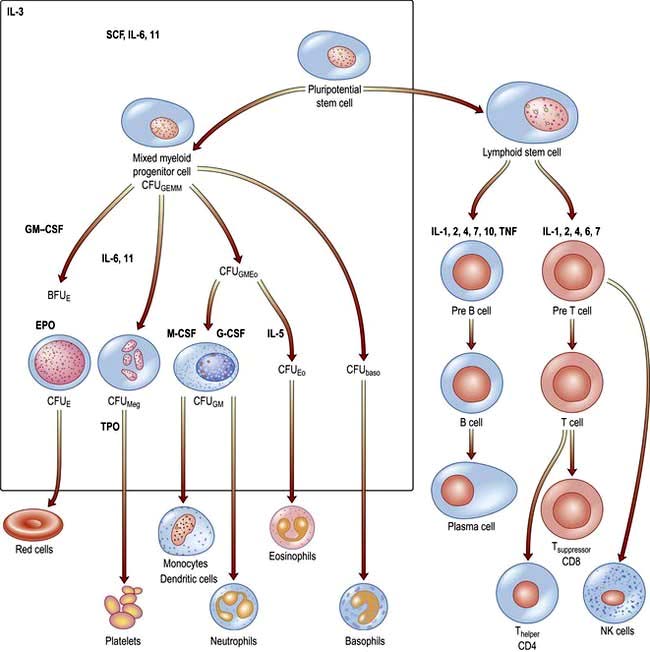
There are two major ancestral cell lines derived from the pluripotential stem cell: lymphocytic and myeloid (non-lymphocytic) cells (Fig. 8.1). The former gives rise to T and B cells. The myeloid stem cell gives rise to the progenitor CFU-GEMM (colony-forming unit, granulocyte–erythrocyte–monocyte–megakaryocyte). The CFU-GEMM can go on to form CFU-GM, CFU-Eo, and CFU-Meg, each of which can produce a particular cell type (i.e. neutrophils, eosinophils and platelets) under appropriate growth conditions. The progenitor cells such as CFU-GEMM cannot be recognized in bone marrow biopsies but are recognized by their ability to form colonies when haemopoietic cells are immobilized in a soft gel matrix. Haemopoiesis is under the control of growth factors and inhibitors, and the microenvironment of the bone marrow also plays a role in its regulation.
Haemopoietic growth factors
Haemopoietic growth factors are glycoproteins, which regulate the differentiation and proliferation of haemopoietic progenitor cells and the function of mature blood cells. They act on the cytokine-receptor superfamily expressed on haemopoietic cells at various stages of development to maintain the haemopoietic progenitor cells and to stimulate increased production of one or more cell lines in response to stresses such as blood loss and infection (Fig. 8.1).
These haemopoietic growth factors including erythropoietin, interleukin 3 (IL-3), IL-6, -7, -11, -12, β-catenin, stem cell factor (SCF, Steel factor or C-kit ligand) and Fms-tyrosine kinase 3 (Flt3) act via their specific receptor on cell surfaces to stimulate the cytoplasmic janus kinase (JAK) (see p. 25). This major signal transducer activates tyrosine kinase causing gene activation in the cell nucleus. Colony-stimulating factors (CSFs, the prefix indicating the cell type, see Fig. 8.1), as well as interleukins and erythropoietin (EPO) regulate the lineage committed progenitor cells.
Stem cell diseases
The clonal proliferation of bone marrow stem cells leads to diseases including leukaemia (see p. 451), polycythaemia vera (see p. 402), myelofibrosis (see p. 404), paroxysmal nocturnal haemoglobinuria (see p. 401). Failure of stem cell growth leads to aplastic anaemia (see p. 385).
Peripheral blood
Automated cell counters are used to measure the haemoglobin concentration (Hb) and the number and size of red cells, white cells and platelets (Table 8.1). Other indices can be derived from these values. A carefully evaluated blood film is still an essential adjunct to the above, as definitive abnormalities of cells can be seen.
 The mean corpuscular volume (MCV) of red cells is a useful index and is used to classify anaemia (see p. 376).
The mean corpuscular volume (MCV) of red cells is a useful index and is used to classify anaemia (see p. 376).





 C-reactive protein (CRP) is a pentraxin, one of the proteins produced in the acute-phase response. It is synthesized exclusively in the liver and rises within 6 hours of an acute event. The CRP level rises with fever (possibly triggered by IL-1, IL-6 and TNF-α and other cytokines), in inflammatory conditions and after trauma. It follows the clinical state of the patient much more rapidly than the ESR and is unaffected by the level of Hb, but it is less helpful than the ESR or plasma viscosity in monitoring chronic inflammatory diseases. The measurement of CRP is easy and quick to perform using an immunoassay that can be automated. High-sensitivity assays have shown that increased levels may predict future cardiovascular disease (see p. 728).
C-reactive protein (CRP) is a pentraxin, one of the proteins produced in the acute-phase response. It is synthesized exclusively in the liver and rises within 6 hours of an acute event. The CRP level rises with fever (possibly triggered by IL-1, IL-6 and TNF-α and other cytokines), in inflammatory conditions and after trauma. It follows the clinical state of the patient much more rapidly than the ESR and is unaffected by the level of Hb, but it is less helpful than the ESR or plasma viscosity in monitoring chronic inflammatory diseases. The measurement of CRP is easy and quick to perform using an immunoassay that can be automated. High-sensitivity assays have shown that increased levels may predict future cardiovascular disease (see p. 728).
| Male | Female | |
|---|---|---|
|
Hb (g/L) |
135–175 |
115–160 |
|
PCV (haematocrit; L/L) |
0.4–0.54 |
0.37–0.47 |
|
RCC (1012/L) |
4.5–6.0 |
3.9–5.0 |
|
MCV (fL) |
80–96 |
|
|
MCH (pg) |
27–32 |
|
|
MCHC (g/L) |
320–360 |
|
|
RDW (%) |
11–15 |
|
|
WBC (109/L) |
4.0–11.0 |
|
|
Platelets (109/L) |
150–400 |
|
|
ESR (mm/h) |
<20 |
|
|
Reticulocytes |
0.5–2.5% (50–100 × 109/L) |
|
ESR, erythrocyte sedimentation rate; Hb, haemoglobin; MCH, mean corpuscular haemoglobin; MCHC, mean corpuscular haemoglobin concentration; MCV, mean corpuscular volume of red cells; PCV, packed cell volume; RCC, red cell count; RDW, red blood cell distribution width; WBC, white blood count.
The red cell
Erythropoiesis
 Reticulocytes contain residual ribosomal RNA and are still able to synthesize Hb. They remain in the marrow for about 1–2 days and are released into the circulation, where they lose their RNA and become mature red cells (erythrocytes) after another 1–2 days. Mature red cells are non-nucleated biconcave discs.
Reticulocytes contain residual ribosomal RNA and are still able to synthesize Hb. They remain in the marrow for about 1–2 days and are released into the circulation, where they lose their RNA and become mature red cells (erythrocytes) after another 1–2 days. Mature red cells are non-nucleated biconcave discs.
 Nucleated red cells (normoblasts) are not normally present in peripheral blood, but are present if there is extramedullary haemopoiesis and in some marrow disorders (see leucoeryothroblastic anaemia, p. 413).
Nucleated red cells (normoblasts) are not normally present in peripheral blood, but are present if there is extramedullary haemopoiesis and in some marrow disorders (see leucoeryothroblastic anaemia, p. 413).

 Erythropoietin is a hormone which controls erythropoiesis. The gene for erythropoietin is on chromosome 7 and codes for a heavily glycosylated polypeptide of 165 amino acids. Erythropoietin has a molecular weight of 30 400 and is produced in the peritubular cells in the kidneys (90%) and in the liver (10%). Its production is regulated mainly by tissue oxygen tension. Production is increased if there is hypoxia from whatever cause, e.g. anaemia or cardiac or pulmonary disease. The erythropoietin gene is one of a number of genes that is regulated by the hypoxic sensor pathway. The 3′-flanking region of the erythropoietin gene has a hypoxic response element, which is necessary for the induction of transcription of the gene in hypoxic cells. Hypoxia-inducible factor 1 (HIF-1) is a transcription factor, which binds to the hypoxia response element and acts as a master regulator of several genes that are responsive to hypoxia. Erythropoietin stimulates an increase in the proportion of bone marrow precursor cells committed to erythropoiesis, and CFU-E are stimulated to proliferate and differentiate. Increased ‘inappropriate’ production of erythropoietin occurs in certain tumours such as renal cell carcinoma and other causes (see Table 8.15).
Erythropoietin is a hormone which controls erythropoiesis. The gene for erythropoietin is on chromosome 7 and codes for a heavily glycosylated polypeptide of 165 amino acids. Erythropoietin has a molecular weight of 30 400 and is produced in the peritubular cells in the kidneys (90%) and in the liver (10%). Its production is regulated mainly by tissue oxygen tension. Production is increased if there is hypoxia from whatever cause, e.g. anaemia or cardiac or pulmonary disease. The erythropoietin gene is one of a number of genes that is regulated by the hypoxic sensor pathway. The 3′-flanking region of the erythropoietin gene has a hypoxic response element, which is necessary for the induction of transcription of the gene in hypoxic cells. Hypoxia-inducible factor 1 (HIF-1) is a transcription factor, which binds to the hypoxia response element and acts as a master regulator of several genes that are responsive to hypoxia. Erythropoietin stimulates an increase in the proportion of bone marrow precursor cells committed to erythropoiesis, and CFU-E are stimulated to proliferate and differentiate. Increased ‘inappropriate’ production of erythropoietin occurs in certain tumours such as renal cell carcinoma and other causes (see Table 8.15).
Haemoglobin synthesis
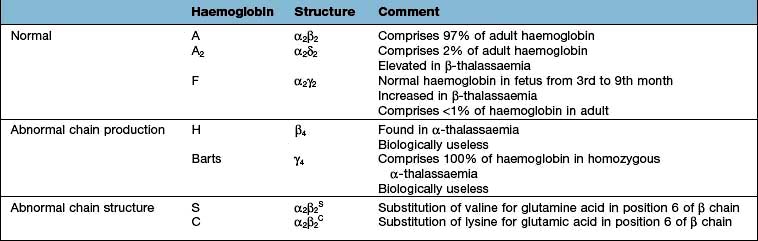
Haemoglobin performs the main functions of red cells – carrying O2 to the tissues and returning CO2 from the tissues to the lungs. Each normal adult Hb molecule (HbA) has a molecular weight of 68 000 and consists of two α and two β globin polypeptide chains (α2β2). HbA comprises about 97% of the Hb in adults. Two other haemoglobin types, HbA2 (α2δ2) and HbF (α2γ2), are found in adults in small amounts (1.5–3.2% and <1%, respectively) (see p. 390).
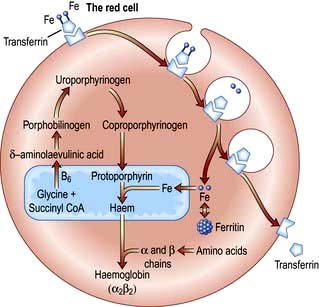
Haemoglobin synthesis occurs in the mitochondria of the developing red cell (Fig. 8.2). The major rate-limiting step is the conversion of glycine and succinic acid to δ-aminolaevulinic acid (ALA) by ALA synthase. Vitamin B6 is a coenzyme for this reaction, which is inhibited by haem and stimulated by erythropoietin. Two molecules of δ-ALA condense to form a pyrrole ring (porphobilinogen). These rings are then grouped in fours to produce protoporphyrins and with the addition of iron haem is formed. Haem is then inserted into the globin chains to form a haemoglobin molecule. The structure of Hb is shown in Figure 8.3.
Haemoglobin function
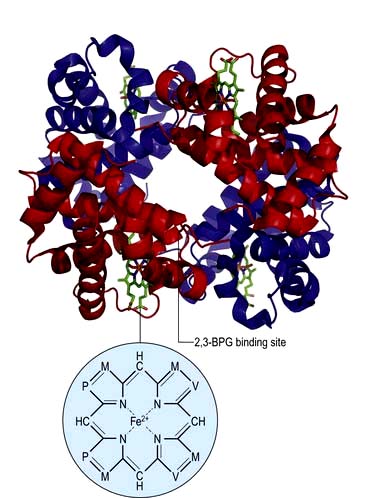
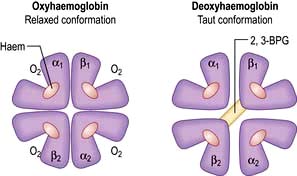
In adult haemoglobin (Hb), a haem group is bound to each of the four globin chains; the haem group has a porphyrin ring with a ferrous atom which can reversibly bind one oxygen molecule. The haemoglobin molecule exists in two conformations, R and T. The T (taut) conformation of deoxyhaemoglobin is characterized by the globin units being held tightly together by electrostatic bonds (Fig. 8.4). These bonds are broken when oxygen binds to haemoglobin, resulting in the R (relaxed) conformation in which the remaining oxygen binding sites are more exposed and have a much higher affinity for oxygen than in the T conformation. The binding of one oxygen molecule to deoxyhaemoglobin increases the oxygen affinity of the remaining binding sites – this property is known as ‘cooperativity’ and is the reason for the sigmoid shape of the oxygen dissociation curve. Haemoglobin is, therefore, an example of an allosteric protein. The binding of oxygen can be influenced by secondary effectors – hydrogen ions, carbon dioxide and red-cell 2,3-bisphosphoglycerate (2,3-BPG). Hydrogen ions and carbon dioxide added to blood cause a reduction in the oxygen-binding affinity of haemoglobin (the Bohr effect). Oxygenation of haemoglobin reduces its affinity for carbon dioxide (the Haldane effect). These effects help the exchange of carbon dioxide and oxygen in the tissues.
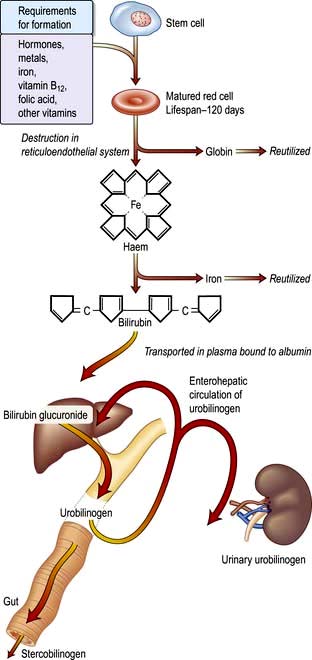
A summary of normal red cell production and destruction is given in Figure 8.5.
Anaemia
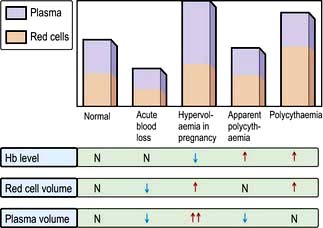
Anaemia is present when there is a decrease in Hb in the blood below the reference level for the age and sex of the individual (Table 8.1). Alterations in the Hb may occur as a result of changes in the plasma volume, as shown in Figure 8.6. A reduction in the plasma volume will lead to a spuriously high Hb – this is seen with dehydration and in the clinical condition of apparent polycythaemia (see p. 404). A raised plasma volume produces a spurious anaemia, even when combined with a small increase in red cell volume as occurs in pregnancy.
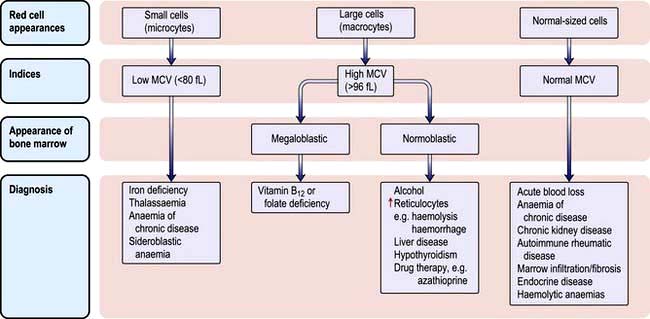
The various types of anaemia, classified by MCV, are shown in Figure 8.7. There are three major types:



Clinical features













Investigations
Peripheral blood
A low Hb should always be evaluated with:

 The white blood cell (WBC) count
The white blood cell (WBC) count

 The reticulocyte count (as this indicates marrow activity)
The reticulocyte count (as this indicates marrow activity)
 The blood film, as abnormal red cell morphology (see Fig. 8.9) may indicate the diagnosis. Where two populations of red cells are seen, the blood film is said to be dimorphic. This may, for example, be seen in patients with ‘double deficiencies’ (e.g. combined iron and folate deficiency in coeliac disease, or following treatment of anaemic patients with the appropriate haematinic).
The blood film, as abnormal red cell morphology (see Fig. 8.9) may indicate the diagnosis. Where two populations of red cells are seen, the blood film is said to be dimorphic. This may, for example, be seen in patients with ‘double deficiencies’ (e.g. combined iron and folate deficiency in coeliac disease, or following treatment of anaemic patients with the appropriate haematinic).
Bone marrow
Techniques for obtaining bone marrow are shown in Practical Box 8.1.
 Practical Box 8.1
Practical Box 8.1
Techniques for obtaining bone marrow
The technique should be explained to the patient and consent obtained.















Examination of the bone marrow is performed to further investigate abnormalities found in the peripheral blood (Practical Box 8.1). Aspiration provides a film which can be examined by microscopy for the morphology of the developing haemopoietic cells. The trephine provides a core of bone which is processed as a histological specimen and allows an overall view of the bone marrow architecture, cellularity and presence/absence of abnormal infiltrates.





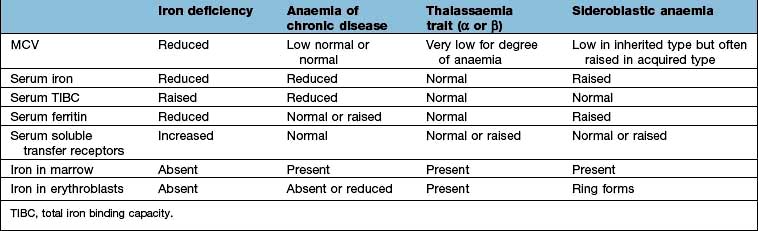
Microcytic anaemia
The other causes of a microcytic hypochromic anaemia are anaemia of chronic disease, sideroblastic anaemia and thalassaemia. In thalassaemia (see p. 390), there is a defect in globin synthesis, in contrast to the other three causes of microcytic anaemia where the defect is in the synthesis of haem.
Iron
Absorption
Factors influencing iron and haem iron absorption (Fig. 8.8) are shown in Table 8.2.
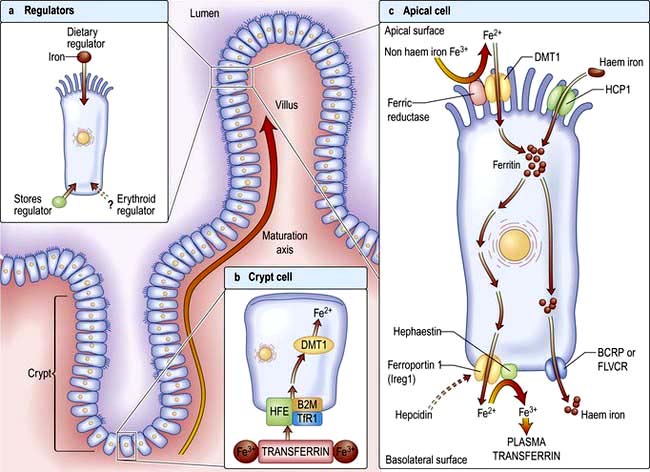
Dietary haem iron is more rapidly absorbed than non-haem iron derived from vegetables and grain. Most haem is absorbed in the proximal intestine, with absorptive capacity decreasing distally. The intestinal haem transporter HCP1 (haem carrier protein 1) has been identified and found to be highly expressed in the duodenum. It is upregulated by hypoxia and iron deficiency. Some haem iron may be reabsorbed intact into circulation via the cell by two exporter proteins – BCRP (breast cancer resistant protein) and FLVCR (feline leukaemia virus subgroup C) (Fig. 8.8).
Once inside the mucosal cell, iron may be transferred across the cell to reach the plasma, or be stored as ferritin; the body’s iron status at the time the absorptive cell developed from the crypt cell is probably the crucial deciding factor. Iron stored as ferritin will be lost into the gut lumen when the mucosal cells are shed; this regulates iron balance. The mechanism of transport of iron across the basolateral surface of mucosal cells involves a transporter protein, ferroportin 1 (FPN 1) through its iron-responsive element (IRE). This transporter protein requires an accessory, multicopper protein, hephaestin (Fig. 8.8).
Transport in the blood
The normal serum iron level is about 13–32 µmol/L; there is a diurnal rhythm with higher levels in the morning. Iron is transported in the plasma bound to transferrin, a β-globulin that is synthesized in the liver. Each transferrin molecule binds two atoms of ferric iron and is normally one-third saturated. Most of the iron bound to transferrin comes from macrophages in the reticuloendothelial system and not from iron absorbed by the intestine. Transferrin-bound iron becomes attached by specific receptors to erythroblasts and reticulocytes in the marrow and the iron is removed (Fig. 8.2).


Iron deficiency




Clinical features
The symptoms of anaemia are described on page 375. The well known clinical features of iron deficiency listed below are generally only seen in cases of very longstanding iron deficiency:






Investigations
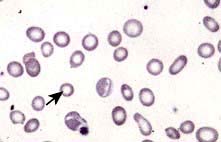
 Blood count and film. A characteristic blood film is shown in Figure 8.9. The red cells are microcytic (MCV <80 fL) and hypochromic (MCH (mean corpuscular haemoglobin) <27 pg). There is poikilocytosis (variation in shape) and anisocytosis (variation in size). Target cells are seen.
Blood count and film. A characteristic blood film is shown in Figure 8.9. The red cells are microcytic (MCV <80 fL) and hypochromic (MCH (mean corpuscular haemoglobin) <27 pg). There is poikilocytosis (variation in shape) and anisocytosis (variation in size). Target cells are seen.
 Serum iron and iron-binding capacity. The serum iron falls and the total iron-binding capacity (TIBC) rises in iron deficiency compared with normal. Iron deficiency is regularly present when the transferrin saturation (i.e. serum iron divided by TIBC) falls below 19% (Table 8.3).
Serum iron and iron-binding capacity. The serum iron falls and the total iron-binding capacity (TIBC) rises in iron deficiency compared with normal. Iron deficiency is regularly present when the transferrin saturation (i.e. serum iron divided by TIBC) falls below 19% (Table 8.3).
 Serum ferritin. The level of serum ferritin reflects the amount of stored iron. The normal values for serum ferritin are 30–300 µg/L (11.6–144 nmol/L) in males and 15–200 µg/L (5.8–96 nmol/L) in females. In simple iron deficiency, a low serum ferritin confirms the diagnosis. However, ferritin is an acute-phase reactant, and levels increase in the presence of inflammatory or malignant diseases. Very high levels of ferritin may be observed in hepatitis and in a rare disease, haemophagocytic lymphohistiocytosis (p. 80).
Serum ferritin. The level of serum ferritin reflects the amount of stored iron. The normal values for serum ferritin are 30–300 µg/L (11.6–144 nmol/L) in males and 15–200 µg/L (5.8–96 nmol/L) in females. In simple iron deficiency, a low serum ferritin confirms the diagnosis. However, ferritin is an acute-phase reactant, and levels increase in the presence of inflammatory or malignant diseases. Very high levels of ferritin may be observed in hepatitis and in a rare disease, haemophagocytic lymphohistiocytosis (p. 80).
 Serum soluble transferrin receptors. The number of transferrin receptors increases in iron deficiency. The results of this immunoassay compare well with results from bone marrow aspiration at estimating iron stores. This assay can help to distinguish between iron deficiency and anaemia of chronic disease (Table 8.3), and may avoid the need for bone marrow examination even in complex cases. It may sometimes be helpful in the investigation of complicated causes of anaemia.
Serum soluble transferrin receptors. The number of transferrin receptors increases in iron deficiency. The results of this immunoassay compare well with results from bone marrow aspiration at estimating iron stores. This assay can help to distinguish between iron deficiency and anaemia of chronic disease (Table 8.3), and may avoid the need for bone marrow examination even in complex cases. It may sometimes be helpful in the investigation of complicated causes of anaemia.
 Other investigations. These will be indicated by the clinical history and examination. Investigations of the gastrointestinal tract are often required to determine the cause of the iron deficiency (see p. 257).
Other investigations. These will be indicated by the clinical history and examination. Investigations of the gastrointestinal tract are often required to determine the cause of the iron deficiency (see p. 257).
Differential diagnosis
The presence of anaemia with microcytosis and hypochromia does not necessarily indicate iron deficiency. The most common other causes are thalassaemia, sideroblastic anaemia and anaemia of chronic disease, and in these disorders the iron stores are normal or increased. The differential diagnosis of microcytic anaemia is shown in Table 8.3.



Anaemia of chronic disease
The serum iron and the TIBC are low, and the serum ferritin is normal or raised because of the inflammatory process. The serum soluble transferrin receptor level is normal (Table 8.3). Stainable iron is present in the bone marrow, but iron is not seen in the developing erythroblasts. Patients do not respond to iron therapy, and treatment is, in general, that of the underlying disorder. Recombinant erythropoietin therapy is used in the anaemia of renal disease (see p. 623), and occasionally in inflammatory disease (rheumatoid arthritis, inflammatory bowel disease).
Sideroblastic anaemia
Sideroblastic anaemias are inherited or acquired disorders characterized by a refractory anaemia, a variable number of hypochromic cells in the peripheral blood, and excess iron and ring sideroblasts in the bone marrow. The presence of ring sideroblasts is the diagnostic feature of sideroblastic anaemia. There is accumulation of iron in the mitochondria of erythroblasts owing to disordered haem synthesis forming a ring of iron granules around the nucleus that can be seen with Perls’ reaction. The blood film is often dimorphic; ineffective haem synthesis is responsible for the microcytic hypochromic cells. Sideroblastic anaemias can be inherited as an X-linked disease transmitted by females. Acquired causes include myelodysplasia, myeloproliferative disorders, myeloid leukaemia, drugs (e.g. isoniazid), alcohol misuse and lead toxicity. It can also occur in other disorders such as rheumatoid arthritis, carcinomas, megaloblastic and haemolytic anaemias. A structural defect in δ-aminolaevulinic acid (ALA) synthase, the pyridoxine-dependent enzyme responsible for the first step in haem synthesis (Fig. 8.2), has been identified in one form of inherited sideroblastic anaemia. Primary acquired sideroblastic anaemia is one of the myelodysplastic syndromes (see p. 405) and this is the cause of the vast majority of cases of sideroblastic anaemia in adults. Lead toxicity is described in Chapter 17.
Normocytic anaemia
Normocytic, normochromic anaemia is seen in anaemia of chronic disease, in some endocrine disorders (e.g. hypopituitarism, hypothyroidism and hypoadrenalism) and in some haematological disorders (e.g. aplastic anaemia and some haemolytic anaemias) (Fig. 8.7). In addition, this type of anaemia is seen acutely following blood loss.
Macrocytic anaemias
Megaloblastic anaemia
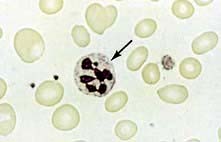
Megaloblastic anaemia is characterized by the presence in the bone marrow of erythroblasts with delayed nuclear maturation because of defective DNA synthesis (megaloblasts). Megaloblasts are large and have large immature nuclei. The nuclear chromatin is more finely dispersed than normal and has an open stippled appearance (Fig. 8.10). In addition, giant metamyelocytes are frequently seen in megaloblastic anaemia. These cells are about twice the size of normal cells and often have twisted nuclei. Megaloblastic changes occur in:
 Vitamin B12 deficiency or abnormal vitamin B12 metabolism
Vitamin B12 deficiency or abnormal vitamin B12 metabolism



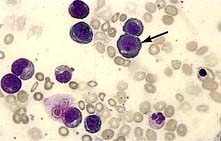
Haematological findings
 Anaemia may be present. The MCV is characteristically >96 fL unless there is a co-existing cause of microcytosis when there may be a dimorphic picture with a normal/low average MCV.
Anaemia may be present. The MCV is characteristically >96 fL unless there is a co-existing cause of microcytosis when there may be a dimorphic picture with a normal/low average MCV.
 The peripheral blood film shows oval macrocytes with hypersegmented polymorphs with six or more lobes in the nucleus (Fig. 8.11).
The peripheral blood film shows oval macrocytes with hypersegmented polymorphs with six or more lobes in the nucleus (Fig. 8.11).

Biochemical basis of megaloblastic anaemia
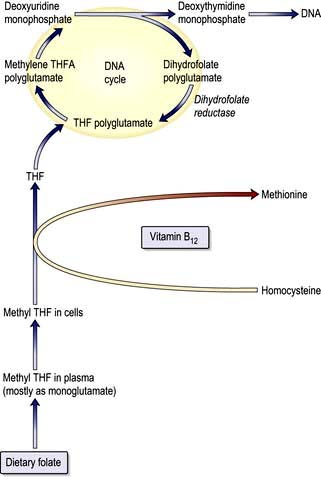
The key biochemical problem common to both vitamin B12 and folate deficiency is a block in DNA synthesis owing to an inability to methylate deoxyuridine monophosphate to deoxythymidine monophosphate, which is then used to build DNA (Fig. 8.12). The methyl group is supplied by the folate coenzyme, methylene tetrahydrofolate.
Vitamin B12
Structure and function
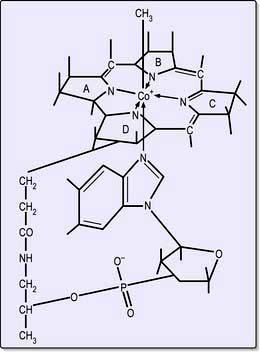
Cobalamins consist of a planar group with a central cobalt atom (corrin ring) and a nucleotide set at right-angles (Fig. 8.13). Vitamin B12 was first crystallized as cyanocobalamin, but the main natural cobalamins have deoxyadenosyl-, methyl- and hydroxocobalamin groups attached to the cobalt atom.
Vitamin B12 deficiency
There are a number of causes of B12 deficiency and abnormal B12 metabolism (Table 8.4). The most common cause of vitamin B12 deficiency in adults is pernicious anaemia. Malabsorption of vitamin B12 because of pancreatitis, coeliac disease or treatment with metformin is mild and does not usually result in significant vitamin B12 deficiency.
Table 8.4 Vitamin B12 deficiency and abnormal B12 utilization: further causes (see text)
| Low dietary intake | Abnormal utilization |
|---|---|
|
Vegans |
Congenital transcobalamin II deficiency |
|
|
Nitrous oxide (inactivates B12) |
|
Impaired absorption |
|
|
Stomach |
|
|
Pernicious anaemia |
|
|
Gastrectomy |
|
|
Congenital deficiency of intrinsic factor |
|
|
Small bowel |
|
|
Ileal disease or resection |
|
|
Bacterial overgrowth |
|
|
Tropical sprue |
|
|
Fish tapeworm (Diphyllobothrium latum) |
|
Pernicious anaemia
Pathology
Autoimmune gastritis (see p. 247) affecting the fundus is present with plasma cell and lymphoid infiltration. The parietal and chief cells are replaced by mucin-secreting cells. There is achlorhydria and absent secretion of intrinsic factor. The histological abnormality can be improved by corticosteroid therapy, which supports an autoimmune basis for the disease.
Clinical features
The neurological changes, if left untreated for a long time, can be irreversible. These neurological abnormalities occur only with very low levels of serum B12 (<60 ng/L or 50 pmol/L) and occasionally occur in patients who are not clinically anaemic. The classical neurological features are those of a polyneuropathy progressively involving the peripheral nerves and the posterior and eventually the lateral columns of the spinal cord (subacute combined degeneration, see p. 1147). Patients present with symmetrical paraesthesiae in the fingers and toes, early loss of vibration sense and proprioception, and progressive weakness and ataxia. Paraplegia may result. Dementia, psychiatric problems, hallucinations, delusions, and optic atrophy may occur from vitamin B12 deficiency.
Investigations
 Haematological findings show the features of a megaloblastic anaemia as described on page 381.
Haematological findings show the features of a megaloblastic anaemia as described on page 381.
 Bone marrow shows the typical features of megaloblastic erythropoiesis (Fig. 8.10), although it is frequently not performed in cases of straightforward macrocytic anaemia and a low serum vitamin B12.
Bone marrow shows the typical features of megaloblastic erythropoiesis (Fig. 8.10), although it is frequently not performed in cases of straightforward macrocytic anaemia and a low serum vitamin B12.





Differential diagnosis
Pernicious anaemia should be distinguished from other causes of vitamin B12 deficiency (Table 8.4). Any disease involving the terminal ileum or bacterial overgrowth in the small bowel can produce vitamin B12 deficiency (see p. 268). Gastrectomy can lead, in the long term, to vitamin B12 deficiency.
Folic acid
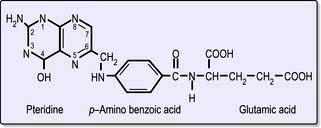
Folic acid monoglutamate is not itself present in nature but occurs as polyglutamates. Folates are present in food as polyglutamates in the reduced dihydrofolate or tetrahydrofolate (THF) forms (Fig. 8.14), with methyl (CH3), formyl (CHO) or methylene (CH2) groups attached to the pteridine part of the molecule. Polyglutamates are broken down to monoglutamates in the upper gastrointestinal tract, and during the absorptive process these are converted to methyl THF monoglutamate, which is the main form in the serum. The methylation of homocysteine to methionine requires both methylcobalamin and methyl THF as coenzymes. This reaction is the first step in which methyl THF entering cells from the plasma is converted into folate polyglutamates. Intracellular polyglutamates are the active forms of folate and act as coenzymes in the transfer of single carbon units in amino acid metabolism and DNA synthesis (Fig. 8.12).
Folate deficiency
The causes of folate deficiency are shown in Table 8.5. The main cause is poor intake, which may occur alone or in combination with excessive utilization or malabsorption. The body’s reserves of folate, unlike vitamin B12, are low (about 10 mg). On a deficient diet, folate deficiency develops over the course of about 4 months, but folate deficiency may develop rapidly in patients who have both a poor intake and excess utilization of folate (e.g. patients in intensive care units).
| Nutritional (major cause) | Excess utilization |
|---|---|
|
Poor intake |
Physiological |
|
Old age |
Pregnancy |
|
Poor social conditions |
Lactation |
|
Starvation |
Prematurity |
|
Alcohol excess (also causes impaired utilization) |
Pathological |
|
Haematological disease with excess red cell production, e.g. haemolysis |
|
|
Poor intake due to anorexia |
|
|
Gastrointestinal disease, e.g. partial gastrectomy, coeliac disease, Crohn’s disease |
Malignant disease with increased cell turnover |
|
Inflammatory disease |
|
|
Cancer |
Metabolic disease, e.g. homocystinuria |
|
Antifolate drugs |
|
|
Anticonvulsants: |
Haemodialysis or peritoneal dialysis |
|
Phenytoin |
|
|
Primidone |
Malabsorption |
|
Methotrexate |
Occurs in small bowel disease, but the effect is minor compared with that of anorexia |
|
Pyrimethamine |
|
|
Trimethoprim |
|
|
|
Macrocytosis without megaloblastic changes




 Some haematological disorders (e.g. aplastic anaemia, sideroblastic anaemia, pure red cell aplasia)
Some haematological disorders (e.g. aplastic anaemia, sideroblastic anaemia, pure red cell aplasia)
 Drugs (e.g. hydroxycarbamide, azathioprine)
Drugs (e.g. hydroxycarbamide, azathioprine)
 Cold agglutinins due to autoagglutination of red cells (see p. 398) (the MCV decreases to normal with warming of the sample to 37°C).
Cold agglutinins due to autoagglutination of red cells (see p. 398) (the MCV decreases to normal with warming of the sample to 37°C).
An increased number of reticulocytes also leads to a raised MCV because they are large cells.
Anaemia due to marrow failure (aplastic anaemia)
Aplastic anaemia is due to a reduction in the number of pluripotential stem cells (Fig. 8.1) together with a fault in those remaining or an immune reaction against them so that they are unable to repopulate the bone marrow. Failure of only one cell line may also occur, resulting in isolated deficiencies such as the absence of red cell precursors in pure red cell aplasia. Evolution to myelodysplasia, paroxysmal nocturnal haemoglobinuria (PNH) or acute myeloblastic leukaemia occurs in some cases, probably owing to the emergence of an abnormal clone of haemopoietic cells.
Causes
A list of causes of aplasia is given in Table 8.6. Immune mechanisms are probably responsible for most cases of idiopathic acquired aplastic anaemia and play a part in at least the persistence of many secondary cases. Activated cytotoxic T cells in blood and bone marrow are responsible for the bone marrow failure.
Investigations



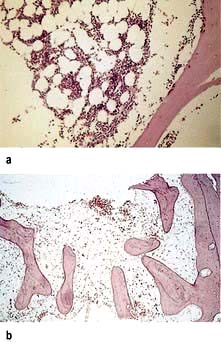
Differential diagnosis
This is from other causes of pancytopenia (Table 8.7). A bone marrow trephine is essential for assessment of the bone marrow cellularity.
Treatment and prognosis
The main danger is infection and stringent measures should be undertaken to avoid this (see also p. 448). Any suspicion of infection in a severely neutropenic patient should lead to immediate institution of broad-spectrum parenteral antibiotics. Supportive care including transfusions of red cells and platelets should be given as necessary. The cause of the aplastic anaemia must be eliminated if possible.



Immunosuppressive therapy is recommended for:
 Patients with severe disease over the age of 40
Patients with severe disease over the age of 40
 Younger patients with severe disease without an HLA-identical sibling donor
Younger patients with severe disease without an HLA-identical sibling donor
 Patients who do not have severe disease but who are transfusion-dependent.
Patients who do not have severe disease but who are transfusion-dependent.
Levels of haemopoietic growth factors (Fig. 8.1) are normal or increased in most patients with aplastic anaemia, and are ineffective as primary treatment.
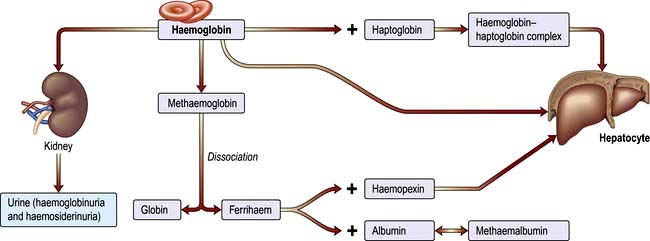
Haemolytic anaemias: an introduction
Breakdown of normal red cells occurs in the macrophages of the bone marrow, liver and spleen (Fig. 8.5).
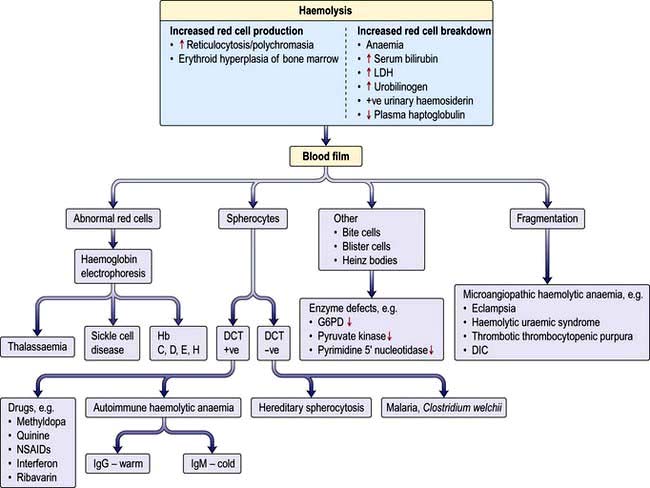
Evidence for haemolysis
Increased red cell breakdown is accompanied by increased red cell production. This is shown in Figure 8.16.
Inherited haemolytic anaemia
Red cell membrane defects
The normal red cell membrane consists of a lipid bilayer crossed by integral proteins with an underlying lattice of proteins (or cytoskeleton), including spectrin, actin, ankyrin and protein 4.1, attached to the integral proteins (Fig. 8.18).
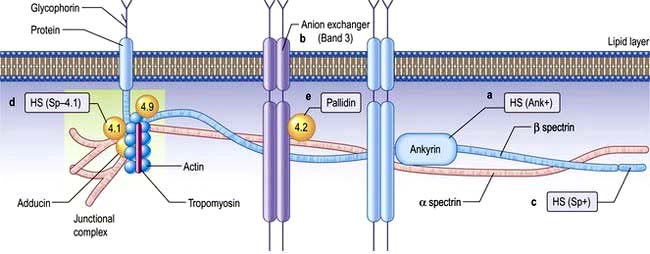
Hereditary spherocytosis (HS)
HS is the most common inherited haemolytic anaemia in northern Europeans, affecting 1 in 5000. It is inherited in an autosomal dominant manner, but in 25% of patients, neither parent is affected and it is presumed that HS has occurred by spontaneous mutation or is truly recessive. HS is due to defects in the red cell membrane, resulting in the cells losing part of the cell membrane as they pass through the spleen, possibly because the lipid bilayer is inadequately supported by the membrane skeleton. The best-characterized defect is a deficiency in the structural protein spectrin, but quantitative defects in other membrane proteins have been identified (Fig. 8.18), with ankyrin defects being the most common. The abnormal red cell membrane in HS is associated functionally with an increased permeability to sodium, and this requires an increased rate of active transport of sodium out of the cells which is dependent on ATP produced by glycolysis. The surface-to-volume ratio decreases, and the cells become spherocytic. Spherocytes are more rigid and less deformable than normal red cells. They are unable to pass through the splenic microcirculation so they have a shortened lifespan.
Clinical features
The condition may present with jaundice at birth. However, the onset of jaundice can be delayed for many years and some patients may go through life with no symptoms and are detected only during family studies. The patient may eventually develop anaemia, splenomegaly and ulcers on the leg. As in many haemolytic anaemias, the course of the disease may be interrupted by aplastic, haemolytic and megaloblastic crises. Aplastic anaemia usually occurs after infections, particularly with erythro (parvo) virus, whereas megaloblastic anaemia is the result of folate depletion owing to the hyperactivity of the bone marrow. Chronic haemolysis leads to the formation of pigment gallstones (see p. 351).
Investigations





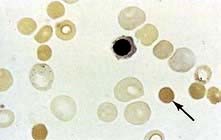
Treatment
Splenectomy is indicated in hereditary spherocytosis to relieve symptoms due to anaemia or splenomegaly, reverse growth failure and prevent recurrent gallstones. It is best to postpone splenectomy until after childhood, as sudden overwhelming fatal infections, usually due to encapsulated organisms such as pneumococci, may occur (see p. 406). Splenectomy should be preceded by appropriate immunization and followed by lifelong penicillin prophylaxis (see Box 8.3). In addition to the well known risk of bacterial infection, there is also some evidence that there is a significant risk of adverse arterial and venous thromboembolic events after splenectomy performed for hereditary spherocytosis.
Hereditary elliptocytosis
This disorder of the red cell membrane is inherited in an autosomal dominant manner and has a prevalence of 1 in 2500 in Caucasians. The red cells are elliptical due to deficiencies of protein 4.1 or the spectrin/actin/4.1 complex which leads to weakness of the horizontal protein interaction and to the membrane defect (Fig. 8.18). Clinically it is a similar condition to HS but milder. Only a minority of patients have anaemia and only occasional patients require splenectomy.
Haemoglobin abnormalities
In early embryonic life, haemoglobins Gower 1, Gower 2 and Portland predominate. Later, fetal haemoglobin (HbF), which has two α and two γ chains, is produced (Fig. 8.20). There is increasing synthesis of β chains from 13 weeks’ gestation and at term there is 80% HbF and 20% HbA. The haemoglobin switch from HbF to HbA occurs after birth when the genes for γ chain production are further suppressed and there is rapid increase in the synthesis of β chains. BCL IIA, a zinc finger protein, is one of a number of proteins that suppress γ gene expression. There is little HbF produced (normally <1%) from 6 months after birth. The δ chain is synthesized just before birth and HbA2 (α2δ2) remains at a level of about 2% throughout adult life (Table 8.9).
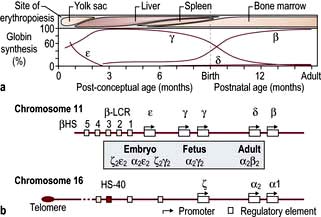
Figure 8.20 Normal development switches in globin expression.
(From Higgs DR, Engel JD, Stamatoyannopoulos G. Thalassaemia. Lancet 2012; 379:373–383, with permission.)
Globin chains are synthesized in the same way as any protein (see p. 42). A normal individual has four α-globin chain genes (Fig. 8.20) with two α-globin genes on each haploid genome (genes derived from one parent). These are situated close together on chromosome 16. The genes controlling the production of ε, γ, δ and β chains are close together on chromosome 11. The globin genes are arranged on chromosomes 16 and 11 in the order in which they are expressed and combine to give different haemoglobins. Normal haemoglobin synthesis is discussed on page 374.



The thalassaemias
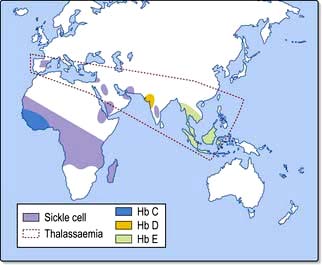
The thalassaemias affect people throughout the world (Fig. 8.21). Normally, there is balanced (1:1) production of α and β chains. The defective synthesis of globin chains in thalassaemia leads to ‘imbalanced’ globin chain production, leading to precipitation of globin chains within the red cell precursors and resulting in ineffective erythropoiesis. Precipitation of globin chains in mature red cells leads to haemolysis.
β-Thalassaemia
In homozygous β-thalassaemia, either no normal β chains are produced (β0) or β-chain production is very reduced (β+). There is an excess of α chains, which precipitate in erythroblasts and red cells causing ineffective erythropoiesis and haemolysis. The excess α chains combine with whatever β, δ and γ chains are produced, resulting in increased quantities of HbA2 and HbF and, at best, small amounts of HbA. In heterozygous β-thalassaemia there is usually symptomless microcytosis with or without mild anaemia. Table 8.10 shows the findings in the homozygote and heterozygote for the common types of β-thalassaemia.
| Type of thalassaemia | Findings in homozygote | Findings in heterozygote |
|---|---|---|
|
β+ |
Thalassaemia major HbA + F + A2 |
Thalassaemia minor HbA2 raised |
|
β0 |
Thalassaemia major HbF + A2 |
Thalassaemia minor HbA2 raised |
|
δβ |
Thalassaemia intermedia |
Thalassaemia minor HbF 5–15% |
|
HbF only |
HbA2 normal |
|
|
δβ+ (Lepore) |
Thalassaemia major or intermedia |
Thalassaemia minor |
|
HbF and Lepore |
Hb Lepore 5–15% |
|
|
HbA2 normal |
Hb Lepore is a cross-fusion product of δ and β globin genes.
Adapted with permission from Weatherall DJ. Disorders of the synthesis of function of haemoglobin. In: Weatherall DJ, Warrell DA, Cox TM, Firth JD (eds) Oxford Textbook of Medicine, 5th edn. Oxford: Oxford University Press; 2010.
Molecular genetics
Clinical syndromes
Clinically, β-thalassaemia can be divided into the following:



Thalassaemia minor (trait)
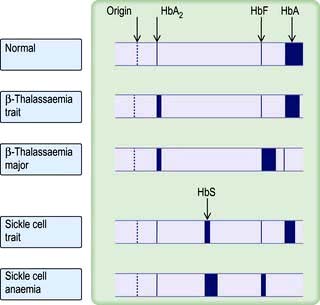
This common carrier state (heterozygous β-thalassaemia) is asymptomatic. Anaemia is mild or absent. The red cells are hypochromic and microcytic with a low MCV and MCH, and it may be confused with iron deficiency. However, the two are easily distinguished, as in thalassaemia trait the serum ferritin and the iron stores are normal (Table 8.2). The RDW is usually normal (see p. 373). Hb electrophoresis usually shows a raised HbA2 and often a raised HbF (Fig. 8.22). Iron should not be given to these patients unless they also have proven coincidental iron deficiency.
Thalassaemia intermedia
Patients may have splenomegaly and bone deformities. Recurrent leg ulcers, gallstones and infections are also seen. It should be noted that these patients may be iron overloaded despite a lack of regular blood transfusions. This is caused by excessive iron absorption which results from the underlying dyserythropoiesis (see iron absorption, p. 377).
Thalassaemia major (Cooley’s anaemia)
Most children affected by homozygous β-thalassaemia present during the first year of life with:
 Failure to thrive and recurrent bacterial infections
Failure to thrive and recurrent bacterial infections

 Extramedullary haemopoiesis that soon leads to hepatosplenomegaly and bone expansion, giving rise to the classical thalassaemic facies (Fig. 8.23a).
Extramedullary haemopoiesis that soon leads to hepatosplenomegaly and bone expansion, giving rise to the classical thalassaemic facies (Fig. 8.23a).
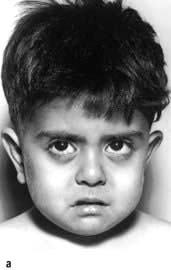
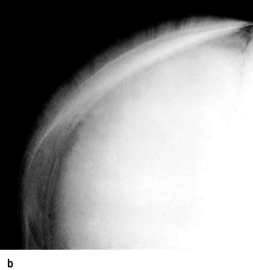
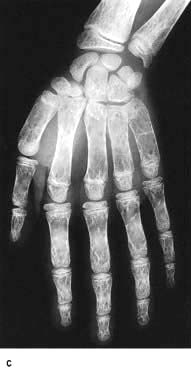
Skull X-rays in these children show the characteristic ‘hair on end’ appearance of bony trabeculation as a result of expansion of the bone marrow into cortical bone (Fig. 8.23b). The expansion of the bone marrow is also shown in an X-ray of the hand (Fig. 8.23c).
Management
 Long-term folic acid supplements are required.
Long-term folic acid supplements are required.

 If transfusion requirements increase, splenectomy may help, although this is usually delayed until after the age of 6 years because of the risk of infection. Prophylaxis against infection is required for patients undergoing splenectomy (see p. 406).
If transfusion requirements increase, splenectomy may help, although this is usually delayed until after the age of 6 years because of the risk of infection. Prophylaxis against infection is required for patients undergoing splenectomy (see p. 406).

 Intensive treatment with desferrioxamine has been reported to reverse damage to the heart in patients with severe iron overload, but excessive doses of desferrioxamine may cause cataracts, retinal damage and nerve deafness. Infection with Yersinia enterocolitica occurs in iron-loaded patients treated with desferrioxamine. Iron overload should be periodically assessed by measuring the serum ferritin and by assessment of hepatic iron stores by MRI.
Intensive treatment with desferrioxamine has been reported to reverse damage to the heart in patients with severe iron overload, but excessive doses of desferrioxamine may cause cataracts, retinal damage and nerve deafness. Infection with Yersinia enterocolitica occurs in iron-loaded patients treated with desferrioxamine. Iron overload should be periodically assessed by measuring the serum ferritin and by assessment of hepatic iron stores by MRI.

 Prenatal diagnosis and gene therapy are discussed on page 43.
Prenatal diagnosis and gene therapy are discussed on page 43.
 Patients’ partners should be tested. If both partners have β-thalassaemia trait, there is a one in four chance of such pregnancy resulting in a child having β-thalassaemia major. Therefore, couples in this situation must be offered prenatal diagnosis (see p. 43).
Patients’ partners should be tested. If both partners have β-thalassaemia trait, there is a one in four chance of such pregnancy resulting in a child having β-thalassaemia major. Therefore, couples in this situation must be offered prenatal diagnosis (see p. 43).
α-Thalassaemia
Molecular genetics
In contrast to β-thalassaemia, α-thalassaemia is often caused by gene deletions, although mutations of the α-globin genes may also occur. The gene for α-globin chains is duplicated on both chromosomes 16, i.e. a normal person has a total of four α-globin genes. Deletion of one α-chain gene (α+) or both α-chain genes (α0) on each chromosome 16 may occur (Table 8.11). The former is the most common of these abnormalities.
 Four-gene deletion (deletion of both genes on both chromosomes); there is no α-chain synthesis and only Hb Barts (γ4) is present. Hb Barts cannot carry oxygen and is incompatible with life (Table 8.9 and Table 8.11). Infants are either stillborn at 28–40 weeks or die very shortly after birth. They are pale, oedematous and have enormous livers and spleens – a condition called hydrops fetalis.
Four-gene deletion (deletion of both genes on both chromosomes); there is no α-chain synthesis and only Hb Barts (γ4) is present. Hb Barts cannot carry oxygen and is incompatible with life (Table 8.9 and Table 8.11). Infants are either stillborn at 28–40 weeks or die very shortly after birth. They are pale, oedematous and have enormous livers and spleens – a condition called hydrops fetalis.
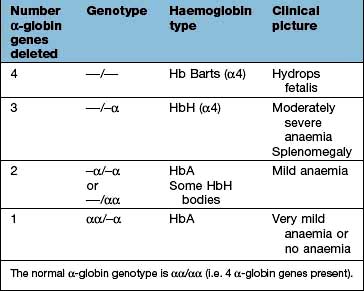
 Three-gene deletion; HbH disease, which is common in parts of Asia, has four β chains with low levels of HbA and Hb Barts. HbA2 is normal or reduced. HbH does not transport oxygen and precipitates in erythroblasts and erythrocytes. There is moderate anaemia (Hb 70–100 g/L) and splenomegaly (thalassaemia intermedia). The patients are not usually transfusion-dependent.
Three-gene deletion; HbH disease, which is common in parts of Asia, has four β chains with low levels of HbA and Hb Barts. HbA2 is normal or reduced. HbH does not transport oxygen and precipitates in erythroblasts and erythrocytes. There is moderate anaemia (Hb 70–100 g/L) and splenomegaly (thalassaemia intermedia). The patients are not usually transfusion-dependent.


Sickle syndromes
Pathogenesis
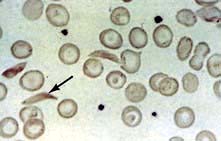
Deoxygenated HbS molecules are insoluble and polymerize. The flexibility of the cells is decreased and they become rigid and take up their characteristic sickle appearance (Fig. 8.24). This process is initially reversible but, with repeated sickling, the cells eventually lose their membrane flexibility and become irreversibly sickled. This is due to dehydration, partly caused by potassium leaving the red cells via calcium activated potassium channels called the Gados channel. These irreversibly sickled cells are dehydrated and dense and will not return to normal when oxygenated. Sickling can produce:


Depending on the type of haemoglobin chain combinations, three clinical syndromes occur:



Sickle cell anaemia
Clinical features



Long-term problems
Bones are a common site for vaso-occlusive episodes, leading to chronic infarcts. Avascular necrosis of hips, shoulders, compression of vertebrae and shortening of bones in the hands and feet occur. These episodes are the common cause for the painful crisis. Osteomyelitis is commoner in sickle cell disease and is caused by Staphylococcus aureus, Staph. pneumoniae and salmonella (see p. 534). Occasionally, hip joint replacement may be required.
Infections are common in tissues susceptible to vasoocclusion, e.g. bones, lungs, kidneys.
Cholelithiasis. Pigment stones occur as a result of chronic haemolysis.
Liver. Chronic hepatomegaly and liver dysfunction are caused by trapping of sickle cells.
Renal. Chronic tubulointerstitial nephritis occurs (see p. 596).
Investigations
 Blood count. The level of Hb is in the range 60–80 g/L with a high reticulocyte count (10–20%).
Blood count. The level of Hb is in the range 60–80 g/L with a high reticulocyte count (10–20%).
 Blood films can show features of hyposplenism (see Fig. 8.29) and sickling (Fig. 8.24).
Blood films can show features of hyposplenism (see Fig. 8.29) and sickling (Fig. 8.24).

 Hb electrophoresis (Fig. 8.22) is always needed to confirm the diagnosis. There is no HbA, 80–95% HbSS and 2–20% HbF.
Hb electrophoresis (Fig. 8.22) is always needed to confirm the diagnosis. There is no HbA, 80–95% HbSS and 2–20% HbF.
 The parents of the affected child will show features of sickle cell trait.
The parents of the affected child will show features of sickle cell trait.
Management
Precipitating factors (see above) should be avoided or treated quickly. The complications requiring inpatient management are shown in Table 8.12.
Acute painful attacks require supportive therapy with intravenous fluids, and adequate analgesia. Oxygen and antibiotics are only given if specifically indicated. Crises can be extremely painful and require strong, usually narcotic, analgesia. Morphine is the drug of choice. Milder pain can sometimes be relieved by codeine, paracetamol and NSAIDs (Box 8.1).
 Box 8.1
Box 8.1
Management of acute painful crisis in opioid naive adults with sickle cell disease


















Prophylaxis is with penicillin 500 mg daily and vaccination with polyvalent pneumococcal and Haemophilus influenzae type b vaccine (see p. 406). Folic acid is given to all patients with haemolysis.
Sickle cell trait
These individuals have no symptoms unless extreme circumstances cause anoxia, such as flying in non-pressurized aircraft. Sickle cell trait gives some protection against Plasmodium falciparum malaria (see p. 144), and consequently the sickle gene has been seen as an example of a balanced polymorphism (where the advantage of the malaria protection in the heterozygote is balanced by the mortality of the homozygous condition). Typically there is 60% HbA and 40% HbS. It should be emphasized that unlike thalassaemia trait, the blood count and film of a person with sickle cell trait are normal. The diagnosis is made by a positive sickle test or by Hb electrophoresis (Fig. 8.22).
Prenatal screening and diagnosis of severe haemoglobin abnormalities
Of the offspring of parents who both have either β-thalassaemia or sickle cell trait, 25% will have β-thalassaemia major or sickle cell anaemia, respectively. Recognition of these heterozygous states in parents and family counselling provide a basis for antenatal screening and diagnosis (p. 44).
Metabolic disorders of the red cell
Red cell metabolism
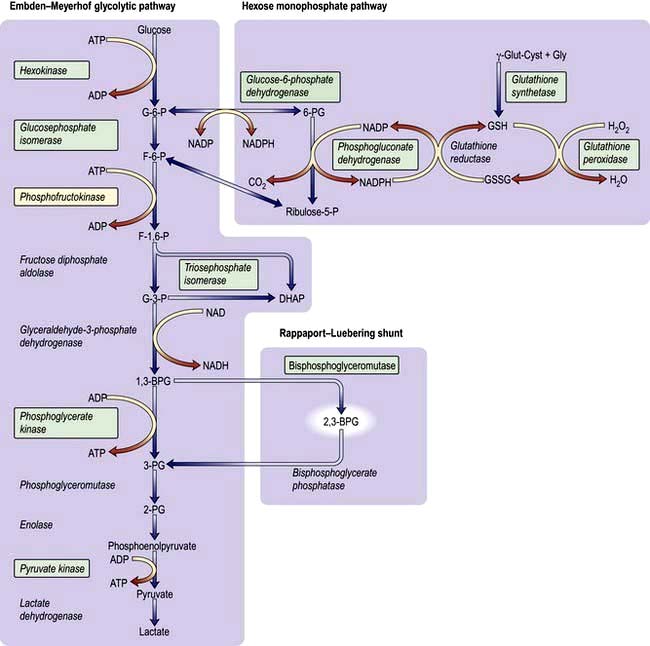
The enzyme systems responsible for producing energy and reducing power are (Fig. 8.25):
 The glycolytic (Embden–Meyerhof) pathway, in which glucose is metabolized to pyruvate and lactic acid with production of ATP
The glycolytic (Embden–Meyerhof) pathway, in which glucose is metabolized to pyruvate and lactic acid with production of ATP

 Rigidity due to cross-linking of spectrin, which decreases membrane flexibility (see Fig. 8.18) and causes ‘leakiness’ of the red cell membrane
Rigidity due to cross-linking of spectrin, which decreases membrane flexibility (see Fig. 8.18) and causes ‘leakiness’ of the red cell membrane

2,3-BPG is formed from a side-arm of the glycolytic pathway (Fig. 8.25). It binds to the central part of the Hb tetramer, fixing it in the low-affinity state (Fig. 8.4). A decreased affinity with a shift in the oxygen dissociation curve to the right enables more oxygen to be delivered to the tissues.
Glucose-6-phosphate dehydrogenase (G6PD) deficiency
The enzyme G6PD holds a vital position in the hexose monophosphate shunt (Fig. 8.25), oxidizing glucose-6-phosphate to 6-phosphoglycerate with the reduction of NADP to NADPH. The reaction is necessary in red cells where it is the only source of NADPH, which is used via glutathione to protect the red cell from oxidative damage. G6PD deficiency is a common condition that presents with a haemolytic anaemia and affects millions of people throughout the world, particularly in Africa, around the Mediterranean, the Middle East (around 20%) and South-east Asia (up to 40% in some regions).
Clinical syndromes
 Acute drug-induced haemolysis (Table 8.13) – usually dose-related
Acute drug-induced haemolysis (Table 8.13) – usually dose-related
 Favism (ingestion of fava beans)
Favism (ingestion of fava beans)


 Infections and acute illnesses will also precipitate haemolysis in patients with G6PD deficiency.
Infections and acute illnesses will also precipitate haemolysis in patients with G6PD deficiency.
Table 8.13 Drugs causing haemolysis in glucose-6-phosphate deficiency
|
|
Mothballs containing naphthalene can also cause haemolysis.
FURTHER READING
Abboud MR. Hematopoietic stem-cell transplantation for adults with sickle cell disease. N Engl J Med 2009; 361:2380–2381.
Cappellini MD, Fiorelli G. Glucose-6-phosphate dehydrogenase deficiency. Lancet 2008; 371:64–72.
Gladwin MT, Vichinsey E. Pulmonary complications of sickle cell disease. N Engl J Med 2008; 357:2254–2265.
Shander A, Sazama K. Clinical consequences of iron overload from chronic red blood cell transfusions, its diagnosis, and its management by chelation therapy. Transfusion 2010; 50:1144–1155.
Taher AT, Musallam KM, Cappellini MD et al. Optimal management of β thalassaemia intermedia. Br J Haematol 2011; 152:512–523.
Investigations
 Blood count is normal between attacks.
Blood count is normal between attacks.
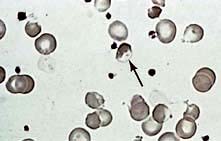
 During an attack the blood film may show irregularly contracted cells, bite cells (cells with an indentation of the membrane), blister cells (cells in which the Hb appears to have become partially detached from the cell membrane; Fig. 8.26), Heinz bodies (best seen on films stained with methyl violet) and reticulocytosis.
During an attack the blood film may show irregularly contracted cells, bite cells (cells with an indentation of the membrane), blister cells (cells in which the Hb appears to have become partially detached from the cell membrane; Fig. 8.26), Heinz bodies (best seen on films stained with methyl violet) and reticulocytosis.






Pyruvate kinase deficiency
This is the most common defect of red cell metabolism after G6PD deficiency; it affects thousands rather than millions of people. The site of the defect is shown in Figure 8.25. There is reduced production of ATP, causing rigid red cells. Homozygotes have haemolytic anaemia and splenomegaly. It is inherited as an autosomal recessive.
Investigations
 Anaemia of variable severity is present (Hb 50–100 g/L). The oxygen dissociation curve is shifted to the right as a result of the rise in intracellular 2,3-BPG, and this reduces the severity of symptoms due to anaemia.
Anaemia of variable severity is present (Hb 50–100 g/L). The oxygen dissociation curve is shifted to the right as a result of the rise in intracellular 2,3-BPG, and this reduces the severity of symptoms due to anaemia.
 Blood film shows distorted (‘prickle’) cells and a reticulocytosis.
Blood film shows distorted (‘prickle’) cells and a reticulocytosis.
 Pyruvate kinase activity is low (affected homozygotes have levels of 5–20%).
Pyruvate kinase activity is low (affected homozygotes have levels of 5–20%).
Pyrimidine 5′ nucleotidase deficiency
This autosomal disorder produces a haemolytic anaemia with basophilic stippling of the red cells. The enzyme degrades pyrimidine nucleotides to cytidine and uridine (pentose phosphate shunt), which in turn leads to the degradation of RNA in the reticulocytes. Lack of the enzyme results in accumulation of partially degraded RNA, which shows as basophilic stippling in mature red cells. The enzyme is also inhibited by lead (see p. 922) and thus basophilic stippling is seen in lead poisoning. The hereditary form can be diagnosed by measuring the enzyme in erythrocytes. A screening test using the ultraviolet absorption spectrum of red cells is available.
Acquired haemolytic anaemia
These anaemias may be divided into those due to immune, non-immune, or other causes (Table 8.8).






Miscellaneous causes
 Various toxic substances can disrupt the red cell membrane and cause haemolysis (e.g. arsenic, and products of Clostridium welchii).
Various toxic substances can disrupt the red cell membrane and cause haemolysis (e.g. arsenic, and products of Clostridium welchii).

 Hypersplenism (see p. 406) results in a reduced red cell survival, which may also contribute to the anaemia seen in malaria.
Hypersplenism (see p. 406) results in a reduced red cell survival, which may also contribute to the anaemia seen in malaria.
 Extensive burns result in denaturation of red cell membrane proteins and reduced red cell survival.
Extensive burns result in denaturation of red cell membrane proteins and reduced red cell survival.
 Some drugs (e.g. dapsone, sulfasalazine) cause oxidative haemolysis with Heinz bodies.
Some drugs (e.g. dapsone, sulfasalazine) cause oxidative haemolysis with Heinz bodies.

Autoimmune haemolytic anaemias
Autoimmune haemolytic anaemias (AIHA) are acquired disorders resulting from increased red cell destruction due to red cell autoantibodies. These anaemias are characterized by the presence of a positive direct antiglobulin (Coombs’) test, which detects the autoantibody on the surface of the patient’s red cells (Fig. 8.27).
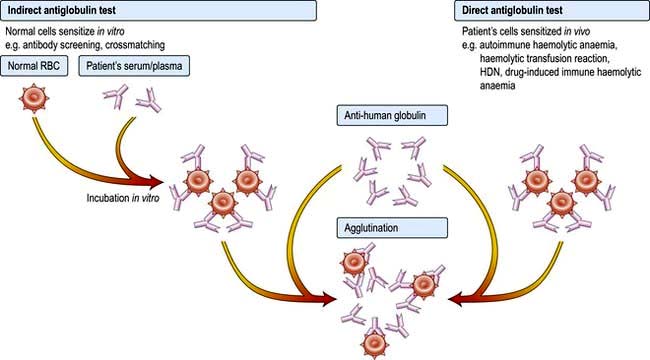
AIHA is divided into ‘warm’ and ‘cold’ types, depending on whether the antibody attaches better to the red cells at body temperature (37°C) or at lower temperatures. The major features and the causes of these two forms of AIHA are shown in Table 8.14. In warm AIHA, IgG antibodies predominate and the direct antiglobulin test is positive with IgG alone, IgG and complement or complement only. In cold AIHA, the antibodies are usually IgM. They easily elute off red cells, leaving complement, which is detected as C3d.
Table 8.14 Causes and major features of autoimmune haemolytic anaemias
| Warm | Cold | |
|---|---|---|
|
Temperature at which antibody attaches best to red cells |
37°C |
Lower than 37°C |
|
Type of antibody |
IgG |
IgM |
|
Direct Coombs’ test |
Strongly positive |
Positive |
|
Causes of primary conditions |
Idiopathic |
Idiopathic |
|
Causes of secondary condition |
Autoimmune rheumatic disorders, e.g. SLE |
Infections, e.g. infectious mononucleosis, Mycoplasma pneumoniae, other viral infections (rare) |
|
Chronic lymphocytic leukaemia |
||
|
Lymphomas |
||
|
Hodgkin’s lymphoma |
||
|
Carcinomas |
||
|
Drugs, many including methyldopa, penicillins, cephalosporins, NSAIDs, quinine, interferon |
Immune destruction of red cells
IgG antibodies frequently do not activate complement and the coated red cells undergo extravascular haemolysis (Fig. 8.28). They are either completely phagocytosed in the spleen through an interaction with Fc receptors on macrophages, or they lose part of the cell membrane through partial phagocytosis and circulate as spherocytes until they become sequestered in the spleen. Some IgG antibodies partially activate complement, leading to deposition of C3b on the red cell surface, and this may enhance phagocytosis as macrophages also have receptors for C3b.
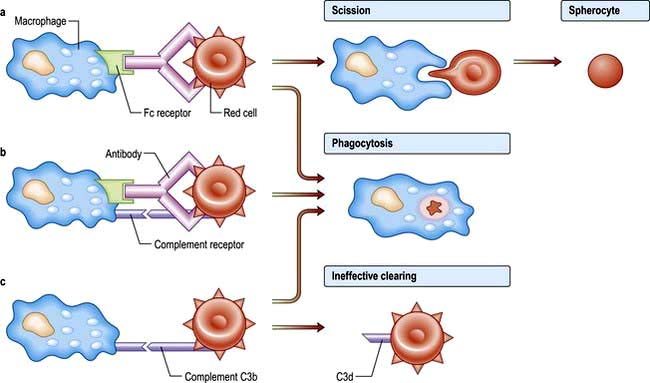
‘Warm’ autoimmune haemolytic anaemias
Clinical features
These anaemias may occur at all ages and in both sexes, although they are most frequent in middle-aged females. They can present as a short episode of anaemia and jaundice but they often remit and relapse and may progress to an intermittent chronic pattern. The spleen is often palpable. Infections or folate deficiency may provoke a profound fall in the haemoglobin level. Autoimmune haemolytic anaemias are primary or secondary. A history of blood transfusions and infections, exposure to drugs or vaccination and the general clinical condition make the secondary form likely. If there is a suspicion of a drug related anaemia then stopping the drug is the obvious measure. The clinical examination will reveal any lymphadenopathy or splenomegaly. The commonest underlying cause is a lymphoproliferative disorder (Table 8.14).
Investigations
 Haemolytic anaemia is evident (see p. 387).
Haemolytic anaemia is evident (see p. 387).
 Spherocytosis is present as a result of red cell damage.
Spherocytosis is present as a result of red cell damage.

 Autoantibodies may have specificity for the Rh blood group system (e.g. for the e-antigen).
Autoantibodies may have specificity for the Rh blood group system (e.g. for the e-antigen).
 Autoimmune thrombocytopenia and/or neutropenia may also be present (Evans’ syndrome).
Autoimmune thrombocytopenia and/or neutropenia may also be present (Evans’ syndrome).
 Abdominal CT scan for the detection of splenomegaly or abdominal lymphoma.
Abdominal CT scan for the detection of splenomegaly or abdominal lymphoma.
‘Cold’ autoimmune haemolytic anaemias
Chronic cold haemagglutinin disease (CHAD)
Investigations
 Red cells agglutinate in the cold or at room temperature. Agglutination is sometimes seen in the sample tube after cooling but is more easily seen on the peripheral blood film made at room temperature. The agglutination is reversible after warming the sample. The agglutination may cause a spurious increase in the MCV (see p. 376).
Red cells agglutinate in the cold or at room temperature. Agglutination is sometimes seen in the sample tube after cooling but is more easily seen on the peripheral blood film made at room temperature. The agglutination is reversible after warming the sample. The agglutination may cause a spurious increase in the MCV (see p. 376).
 Cold agglutinin test (the titre is markedly elevated in CHAD to >1:512 )
Cold agglutinin test (the titre is markedly elevated in CHAD to >1:512 )
 Direct antiglobulin test is positive with complement (C3d) alone.
Direct antiglobulin test is positive with complement (C3d) alone.

Drug-induced immune haemolytic anaemia
 Antibodies to the drug only, e.g. quinidine, rifampicin. Immune complexes attach to red cells, and may cause acute and severe intravascular haemolysis, sometimes associated with kidney injury. The haemolysis usually resolves quickly once the drug is withdrawn.
Antibodies to the drug only, e.g. quinidine, rifampicin. Immune complexes attach to red cells, and may cause acute and severe intravascular haemolysis, sometimes associated with kidney injury. The haemolysis usually resolves quickly once the drug is withdrawn.


Confirmation of the diagnosis of drug-induced immune haemolytic anaemia requires:




Alloimmune haemolytic anaemia
Antibodies produced in one individual react with the red cells of another. This situation occurs in haemolytic disease of the newborn, haemolytic transfusion reactions (see p. 400) and after allogeneic bone marrow, renal, liver, cardiac or intestinal transplantation when donor lymphocytes transferred in the allograft (‘passenger lymphocytes’) may produce red cell antibodies against the recipient and cause haemolytic anaemia.
Haemolytic disease of the newborn (HDN)
HDN due to ABO incompatibility is usually mild and exchange transfusion is rarely needed. HDN due to RhD incompatibility has become much less common in developed countries following the introduction of anti-D prophylaxis (see below). HDN may be caused by antibodies against antigens in many blood group systems (e.g. other Rh antigens such as c and E, and Kell, Duffy and Kidd; see p. 407).



Postnatal management
 a cord Hb of <120 g/L (normal cord Hb is 136–196 g/L)
a cord Hb of <120 g/L (normal cord Hb is 136–196 g/L)
 a cord bilirubin of >60 µmol/L
a cord bilirubin of >60 µmol/L


Further exchange transfusions may be necessary to remove the unconjugated bilirubin.