[level-membership-for-internal-medicine-category]
7 Haematological disease
Approach to the patient
Clinical features
Investigations
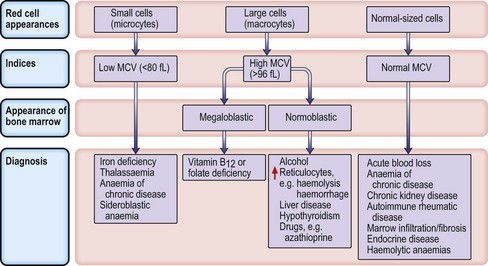
Microcytic anaemias
Iron deficiency anaemia
Investigations
Management
The thalassaemias
α-Thalassaemia
α-Thalassaemia is caused by deletions or mutations in one or more of the four α-globin genes (two genes on each copy of chromosome 16), leading to an excess of β-globin chains. Deletion of one gene results in silent α-thalassaemia, which is of no clinical significance and the blood picture is usually normal. Deletion of two genes results in α-thalassaemia trait (microcytosis with or without mild anaemia but with normal serum iron and ferritin). Haemoglobin H (HbH) disease is caused by a deletion of three α-globin genes, and is characterized by a moderate haemolytic anaemia (Hb 7–10 g/dL) and splenomegaly. Regular transfusion is rarely required, but increased haemolysis may occur with oxidant drugs, as in G6PD deficiency (p. 216), and folic acid should be administered throughout pregnancy. If all four α-globin genes are deleted (Hb Barts), there is a complete absence of normal Hb production, which is incompatible with life outside the uterus (hydrops fetalis).
β-Thalassaemia
• Management of thalassaemia major
Anaemia of chronic disease
A normochromic normocytic anaemia commonly occurs in association with chronic inflammatory and infective conditions (e.g. rheumatoid arthritis, malignancy, TB). The exact pathogenesis is complicated and contributing factors include a cytokine-mediated failure of iron utilization during erythropoiesis and high levels of hepcidin, which destroy ferroportin, limiting iron absorption in the intestinal cell. Red cell survival is decreased and there is an inappropriately low erythropoietin response for the level of anaemia. The MCV can be normal, or low in longstanding disease (resembling iron deficiency). The serum ferritin is usually normal or raised because of the inflammatory process. Iron is present in the bone marrow but not in developing erythroblasts.
Treatment
Address the underlying disorder, e.g. treat infection with antibiotics. Red cell transfusions may be necessary for symptomatic anaemia. Patients do not respond to oral iron, but coexisting iron deficiency should be identified and treated with parenteral iron (p. 202). Recombinant erythropoietin therapy at relatively high doses, e.g. 150–300 U/kg SC three times per week, is used, e.g. in rheumatoid arthritis, or intravenously in chronic kidney disease. Pegylated (PEG) erythropoietin is an alternative, administered once every 2 weeks.
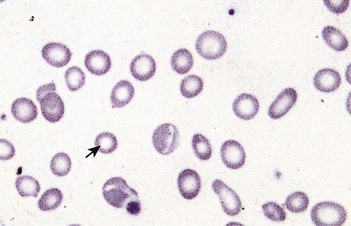
Macrocytic anaemias
Megaloblastic anaemia
Impaired DNA synthesis results in morphological abnormalities of blood cell precursors, characterized by delayed nuclear maturation (nuclear-cytoplasmic asynchrony). All haematopoietic cell lines (and other rapidly dividing cells) are affected, and in severe cases there may be pancytopenia (i.e. anaemia, leucopenia and thrombocytopenia). Causes are predominantly those leading to B12 and folate deficiencies, although drugs (azathioprine, hydroxycarbamide, zidovudine), myelodysplasia and rare enzyme deficiencies affecting DNA synthesis (e.g. orotic aciduria) can also cause macrocytosis and anaemia.
Investigations
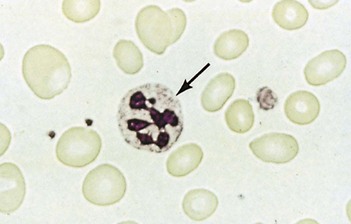
• Other investigations
Treatment
Aplastic anaemia
Treatment
Myelodysplastic syndromes
Investigations
Treatment
Haemolytic anaemias
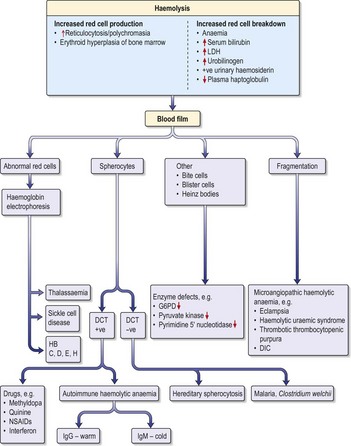
Inherited haemolytic anaemias
Hereditary spherocytosis
Hereditary spherocytosis (HS) is the commonest inherited cause of chronic haemolytic anaemia in Northern Europeans (1 in 5000 births). Inheritance is usually autosomal dominant but 25% of cases are due to spontaneous mutations. The red cells are poorly deformable due to abnormalities in the red cell cytoskeleton, so they become trapped in splenic sinusoids where they are destroyed. HS can present at any age (neonatal jaundice is common) and the clinical features are highly variable. Many patients are asymptomatic but features of chronic compensated haemolysis may be present, e.g. pigment gallstones, mild jaundice and splenomegaly. Aplastic crises sometimes occur after viral infections, e.g. erythrovirus B19. Investigations show anaemia and spherocytes on blood film, with evidence of haemolysis (see above).
• Treatment
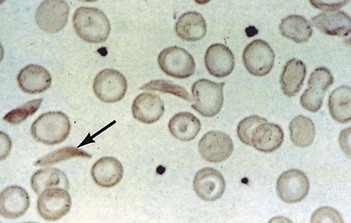
Sickle cell anaemia (HbSS)
Investigations
(See also demonstration of haemolysis, Fig. 7.4).
Clinical syndromes and management
• Analgesia
• Acute chest syndrome (Box 7.2)
Box 7.2
(Adapted from Rees DC et al. 2003.)
Management of acute painful crisis in opioid-naïve adults with sickle cell disease. Higher doses may be required for patients who have previously received opioids.
• Management of acute chest crises
Glucose-6-phosphate dehydrogenase (G6PD) deficiency
Clinical and laboratory features
Rapid intravascular haemolysis with symptomatic anaemia, jaundice and haemoglobinuria occurs 1–3 days after ingestion of certain drugs (Box 7.3). Infection can also precipitate haemolysis but the anaemia is usually mild. Favism (severe haemolysis after ingestion of fava beans) occurs with the Mediterranean variant and can be rapidly fatal. Chronic non-spherocytic haemolytic anaemia (in the absence of a precipitating cause) and neonatal jaundice are also seen in some types of G6PD deficiency.
Acquired haemolytic anaemias
Autoimmune haemolytic anaemias
Warm AIHA
Cold AIHA
Autoantibodies agglutinate red cells at temperatures < 32°C, causing cyanotic discoloration of the extremities in cold conditions. Complement activation and intravascular haemolysis occur when coated red cells return to 37°C on re-circulation.
Drug-induced immune haemolytic anaemia
Interaction between certain drugs and the red cell membrane can sometimes stimulate antibody formation, which may lead to extravascular or intravascular haemolysis (Box 7.3). Treatment is by withdrawal of the responsible drug.
Non-immune haemolytic anaemia
Paroxysmal nocturnal haemoglobinuria (PNH)
This is a rare form of chronic haemolytic anaemia, characterized by episodic intravascular haemolysis and haemoglobinuria. It is due to mutations in the X-linked gene PIG-A, which result in defective binding of CD55 and CD59 proteins to the cell surface, and failure to inactivate complement. Venous thromboses in unusual sites (e.g. Budd–Chiari syndrome) are a common feature. Transformation to aplastic anaemia (p. 208) or AML (p. 284) occurs in a proportion of patients
Myeloproliferative neoplasms
Polycythaemia vera (primary proliferative polycythaemia)
Management
• Radioactive 32P
Essential thrombocythaemia (ET)
This disorder is closely related to PV, but is usually characterized by an isolated thrombocytosis (platelets > 600–1000 × 109/L) with a normal Hb and white cell count. It is diagnosed by exclusion of other causes of thrombocytosis, e.g. iron deficiency or infection. A mutation in the JAK2 V617F gene is present in only 50% of patients.
Idiopathic myelofibrosis
Clinical features
Symptoms include anaemia, weakness, weight loss, night sweats, bruising, bleeding and infections. Most patients will have symptomatic (often massive) splenomegaly and hepatomegaly. The blood count and film show features of a leucoerythroblastic anaemia and characteristic ‘tear-drop’ poikilocytes. A bone marrow trephine is often diagnostic, by showing patchy cellularity with increased reticulin fibrosis. Cytogenetic and molecular analyses are required to differentiate myelofibrosis from CML (p. 285).
Treatment
• Symptomatic anaemia
Blood transfusion
Blood components
Blood components are prepared from single donations by simple separation methods. Component therapy (rather than use of whole blood) reduces the risks associated with transfusion of unnecessary blood constituents and makes the most economical use of each individual donation. Indications for blood components and suggested thresholds for platelet transfusion are listed in Box 7.4.
Procedure for red cell transfusion
 involves a mandatory meticulous final bedside check before blood or blood component transfusion is administered.
involves a mandatory meticulous final bedside check before blood or blood component transfusion is administered.
Complications and management of blood transfusion
Immunological complications
• Management
Non-immunological complications
Alternatives to blood transfusion
Haemostasis and thrombosis
Normal haemostasis
Vascular injury leads to endothelial damage and exposure of collagen within the vessel wall.
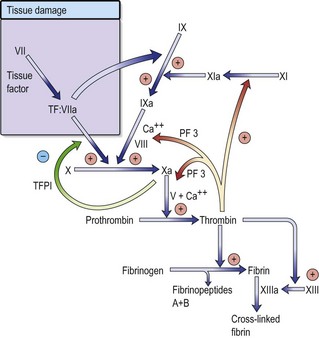
Regulation of coagulation is mediated by naturally occurring anticoagulants such as antithrombin, protein C and its cofactor, protein S. These inhibitors target various proteins within the coagulation cascade, in order to control thrombin generation and to help localize the haemostatic response to the site of vascular injury. Activation of the fibrinolytic system in response to vascular injury provides an additional level of control. Circulating plasminogen is converted to plasmin, which breaks down excess fibrin and fibrinogen into fibrin/fibrinogen degradation products (FDPs). One purpose of this mechanism is to recanalize blood vessels occluded by thrombus.
Approach to patients with bleeding disorders
Investigations
• Coagulation screen
Vascular disorders
Senile purpura and purpura due to steroids are both caused by atrophy of the vascular supporting tissue. Henoch–Schönlein purpura is described on p. 343. Episodes of inexplicable bleeding or bruising may represent abuse.
Platelet disorders
Thrombocytopenia
Peripheral destruction of platelets
Immune thrombocytopenic purpura (ITP)
• First-line therapy
Other immune thrombocytopenias
Drug-induced thrombocytopenia
This is most commonly due to immune mechanisms (when drugs stimulate antibody production); it is often dose-dependent but may be idiosyncratic. Immune mechanisms include antibodies directly binding to platelet membrane proteins or hapten-dependent antibodies, which are antibodies against drugs covalently bound to platelet membrane glycoproteins. The mechanism is similar to that of drug-induced immune haemolytic anaemia (p. 219).
Thrombotic thrombocytopenic purpura (TTP)
• Management
Platelet function disorders
Thrombocytosis
Inherited coagulation disorders
Haemophilia A
Clinical and laboratory features
Clinical features depend on the level of FVIII:C.
General management
• Severe haemophilia A
Other treatments
Haemophilia B (Christmas disease)
This is also an X-linked recessive condition but is caused by a deficiency of factor IX. The clinical features are identical to those of haemophilia A, but it is much rarer (1 in 30 000 males). The general management is similar to that for haemophilia A but desmopressin is not effective. Bleeding episodes should be treated with recombinant factor IX concentrates or pooled plasma products. 1 U of FIX per kg body weight raises the FIX level by 1 U/dL with a half-life of approximately 18 hours, so treatment and prophylactic doses are required less frequently than for haemophilia A. Inhibitor formation is rare, but where this does occur, bleeding episodes are managed with rVIIa as for haemophilia A.
Von Willebrand disease (VWD)
• General management
Acquired coagulation disorders
Vitamin K deficiency
Treatment
Disseminated intravascular coagulation (DIC)
Thrombosis
Arterial thrombosis
This is usually associated with atheromatous plaque rupture and vessel occlusion, and is discussed further in the relevant sections (see coronary artery disease, p. 249; cerebral vascular disease, p. 628; and peripheral vascular disease). Drug therapy includes the use of antiplatelet drugs and thrombolytic therapy (Box 7.5). Anticoagulants are used occasionally.
Antiplatelet drugs
The indications for and results of antiplatelet therapy are discussed in the appropriate sections (pp. 435 and 632).
Thrombolytic therapy
Venous thromboembolism (VTE)
Common risk factors include surgery (particularly in the elderly), malignant disease, immobility, a previous history of thrombosis, obesity, oral contraceptive use, HRT and pregnancy. Venous thrombosis is also increased in some blood disorders, including polycythaemia, essential thrombocythaemia and inherited or acquired thrombophilia (p. 242).
Clinical features
Diagnosis
Investigation of inherited and acquired thrombophilia
• General screening tests
Investigation of acquired thrombophilia
Prevention and treatment of VTE
Heparin
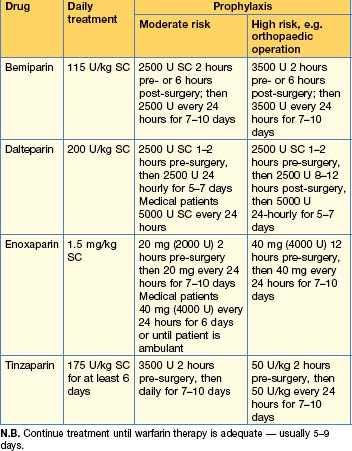
Alternatives to heparin
These and other Xa inhibitors e.g. apixaban are being more widely used, e.g. in atrial fibrillation.
Vitamin K antagonists
Warfarin (a coumarin) is the treatment of choice for the secondary management of VTE in most patients, because it has few side-effects apart from bleeding. Vitamin K antagonists interfere with the synthesis of coagulation factors, II, VII, IX and X (and natural anticoagulants protein C and protein S). The anticoagulant activity is monitored using a PT-based assay. However, the reference range for the PT assay varies according to the method used, so in order to simplify the management of oral anticoagulation, calculation of the International Normalized Ratio (INR) allows comparison of PT results between laboratories.
An example nomogram for starting warfarin therapy is shown in Table 7.2.
| Day | INR | Dose (mg) |
|---|---|---|
| 1 | < 1.4 | 10 |
| 2 | < 1.8 | 10 (or 5*) |
| > 1.8 | Refer to haematology dept for advice | |
| 3 | < 2 | 10 |
| 2.0–2.3 | 5 | |
| 2.4–2.7 | 4 | |
| 2.8–3.1 | 3 | |
| 3.2–3.4 | 2 | |
| 3.5–4.0 | 1 | |
| > 4.0 | Refer to haematology dept for advice | |
| 4 onwards (maintenance) | 1.6–1.7 | 7 |
| 1.8 | 6 & 7 alternate days | |
| 1.9 | 6 | |
| 2.0–2.1 | 5 & 7 alternate days | |
| 2.2–2.3 | 5 | |
| 2.4–2.6 | 4 & 5 alternate days | |
| 2.7–3.0 | 4 | |
| 3.1–3.5 | 3 & 4 alternate days | |
| 3.6–4.0 | 3 | |
| 4.2–4.5 | Miss 1 day, then 2 mg | |
| > 4.5 | Refer to haematology dept for advice |
* A lower starting dose may be required in patients with liver disease, excess alcohol users, body weight < 50 kg or congestive cardiac failure, and in the elderly.
(Adapted from Winter M, et al 2005.)
Table 7.3 Management of bleeding and excessive oral anticoagulation
| INR/severity of bleeding | Management |
|---|---|
(Adapted from British Committee for Standards in Haematology 1998)
Prevention of VTE
Treatment of established VTE
The aim of treatment is to prevent propagation and embolization of an existing venous thrombus until it resolves or organizes as a result of natural fibrinolytic activity. Initial therapy for the majority of patients is with LMWH, which has been shown to be at least as effective as UFH for the treatment of VTE, allows patients with DVT to be treated on an outpatient basis, and has a number of other advantages (p. 243). Differences in efficacy between LMWHs appear to be insignificant, so the choice of anticoagulant is usually guided by local policy. The recommended dose depends on the product used and patient weight, e.g. enoxaparin 1.5 mg/kg per day.
Where therapeutic anticoagulation with UFH is necessary, it is administered intravenously as a 75 U/kg bolus loading dose, followed by a continuous infusion of 18 U/kg/hour. The anticoagulant effect should be monitored by the APTT ratio 2–4 hours after the start of treatment or after a dose adjustment, and every 24 hours thereafter if no dose adjustment is required. The recommended APTT ratio is 1.5–2.5 times the control APTT (the sensitivity of reagents used between laboratories varies, so the APTT needs to be locally calibrated against an anti-Xa assay to ensure accuracy and reproducibility of the results).
• Peri-operative management of anticoagulation
Allford SL, Hunt BJ, et al. Guidelines on the diagnosis and management of the thrombotic microangiopathic haemolytic anaemias. Br J Haematol. 2003;120(4):556-573.
Baglin T, et al. Guidelines on the use and monitoring of heparin. Br J Haematol. 2006;133(1):19-34.
Blood Transfusion Task Force. Guidelines for the use of platelet transfusions. Br J Haematol. 2003;122(1):10-23. Jul
Bolton-Maggs PH, et al. Guidelines for the diagnosis and management of hereditary spherocytosis. Br J Haematol. 2004;126(4):455-474.
British Committee for Standards in Haematology (BCSH). Guidelines on oral anticoagulation, ed 3. Br J Haematol. 1998;101:374-387.
Campbell PJ, Green AL. Mechanisms of disease: the myeloproliferative disorders. N Engl J Med. 2006;355:2452.
Cazzola M, Malcovati L. Myelodysplastic syndromes — coping with ineffective hematopoiesis. N Engl J Med. 2005;352:536-538.
General Haematology Task Force. Guidelines for the investigation and management of idiopathic thrombocytopenic purpura in adults, children and in pregnancy. Br J Haematol. 2003;120(4):574-596.
George JW. N Engl J Med. 2006;354:1927-1935.
Hillmen P, et al. The complement inhibitor eculizumab in paroxysmal nocturnal hemoglobinuria. N Engl J Med. 2006;355:1233-1243.
Keel SB, Abkowitz JL. The microcytic red cell and the anaemia of inflammation. N Engl J Med. 2009;361:1904-1906.
O’Shaughnessy DF, et al. Guidelines for the use of fresh-frozen plasma, cryoprecipitate and cryosupernatant. Br J Haematol. 2004;126(1):11-28.
Rees DC, Olujohungbe AD, Parker NE, et al. Guidelines for the management of the acute painful crisis in sickle cell disease. Br J Haematol. 2003;120:744-752.
Rees DC, Williams TN, Gladwin MT. Sickle cell disease. Lancet. 2010;376:2018-2031.
Winter M, Keeling D, Sharpen F, et al. Haemostasis and Thrombosis Task Force of the British Committee for Standards in Haematology. Procedures for the outpatient management of patients with deep venous thrombosis. Clin Lab Haematol. 2005;27:61-66.
[/level-membership-for-internal-medicine-category][not-level-membership-for-internal-medicine-category]
7 Haematological disease
Approach to the patient
Clinical features
Investigations
Microcytic anaemias
Iron deficiency anaemia
Investigations
Management
The thalassaemias
α-Thalassaemia
α-Thalassaemia is caused by deletions or mutations in one or more of the four α-globin genes (two genes on each copy of chromosome 16), leading to an excess of β-globin chains. Deletion of one gene results in silent α-thalassaemia, which is of no clinical significance and the blood picture is usually normal. Deletion of two genes results in α-thalassaemia trait (microcytosis with or without mild anaemia but with normal serum iron and ferritin). Haemoglobin H (HbH) disease is caused by a deletion of three α-globin genes, and is characterized by a moderate haemolytic anaemia (Hb 7–10 g/dL) and splenomegaly. Regular transfusion is rarely required, but increased haemolysis may occur with oxidant drugs, as in G6PD deficiency (p. 216), and folic acid should be administered throughout pregnancy. If all four α-globin genes are deleted (Hb Barts), there is a complete absence of normal Hb production, which is incompatible with life outside the uterus (hydrops fetalis).
β-Thalassaemia
• Management of thalassaemia major
Anaemia of chronic disease
A normochromic normocytic anaemia commonly occurs in association with chronic inflammatory and infective conditions (e.g. rheumatoid arthritis, malignancy, TB). The exact pathogenesis is complicated and contributing factors include a cytokine-mediated failure of iron utilization during erythropoiesis and high levels of hepcidin, which destroy ferroportin, limiting iron absorption in the intestinal cell. Red cell survival is decreased and there is an inappropriately low erythropoietin response for the level of anaemia. The MCV can be normal, or low in longstanding disease (resembling iron deficiency). The serum ferritin is usually normal or raised because of the inflammatory process. Iron is present in the bone marrow but not in developing erythroblasts.
Treatment
Address the underlying disorder, e.g. treat infection with antibiotics. Red cell transfusions may be necessary for symptomatic anaemia. Patients do not respond to oral iron, but coexisting iron deficiency should be identified and treated with parenteral iron (p. 202). Recombinant erythropoietin therapy at relatively high doses, e.g. 150–300 U/kg SC three times per week, is used, e.g. in rheumatoid arthritis, or intravenously in chronic kidney disease. Pegylated (PEG) erythropoietin is an alternative, administered once every 2 weeks.
Macrocytic anaemias
Megaloblastic anaemia
Impaired DNA synthesis results in morphological abnormalities of blood cell precursors, characterized by delayed nuclear maturation (nuclear-cytoplasmic asynchrony). All haematopoietic cell lines (and other rapidly dividing cells) are affected, and in severe cases there may be pancytopenia (i.e. anaemia, leucopenia and thrombocytopenia). Causes are predominantly those leading to B12 and folate deficiencies, although drugs (azathioprine, hydroxycarbamide, zidovudine), myelodysplasia and rare enzyme deficiencies affecting DNA synthesis (e.g. orotic aciduria) can also cause macrocytosis and anaemia.
Investigations
• Other investigations
Treatment
Aplastic anaemia
Treatment
Myelodysplastic syndromes
Investigations
Treatment
Haemolytic anaemias
Inherited haemolytic anaemias
Hereditary spherocytosis
Hereditary spherocytosis (HS) is the commonest inherited cause of chronic haemolytic anaemia in Northern Europeans (1 in 5000 births). Inheritance is usually autosomal dominant but 25% of cases are due to spontaneous mutations. The red cells are poorly deformable due to abnormalities in the red cell cytoskeleton, so they become trapped in splenic sinusoids where they are destroyed. HS can present at any age (neonatal jaundice is common) and the clinical features are highly variable. Many patients are asymptomatic but features of chronic compensated haemolysis may be present, e.g. pigment gallstones, mild jaundice and splenomegaly. Aplastic crises sometimes occur after viral infections, e.g. erythrovirus B19. Investigations show anaemia and spherocytes on blood film, with evidence of haemolysis (see above).
• Treatment
Sickle cell anaemia (HbSS)
Investigations
(See also demonstration of haemolysis, Fig. 7.4).
Clinical syndromes and management
• Analgesia
• Acute chest syndrome (Box 7.2)
Box 7.2
(Adapted from Rees DC et al. 2003.)
Management of acute painful crisis in opioid-naïve adults with sickle cell disease. Higher doses may be required for patients who have previously received opioids.
• Management of acute chest crises
Glucose-6-phosphate dehydrogenase (G6PD) deficiency
Clinical and laboratory features
Rapid intravascular haemolysis with symptomatic anaemia, jaundice and haemoglobinuria occurs 1–3 days after ingestion of certain drugs (Box 7.3). Infection can also precipitate haemolysis but the anaemia is usually mild. Favism (severe haemolysis after ingestion of fava beans) occurs with the Mediterranean variant and can be rapidly fatal. Chronic non-spherocytic haemolytic anaemia (in the absence of a precipitating cause) and neonatal jaundice are also seen in some types of G6PD deficiency.
