[level-membership-for-cardiovascular-category]
34 Guidelines for Managing Pacemaker and Implantable Defibrillator Advisories
Pacemakers and implantable cardiac-defibrillators (ICDs), two of the most remarkable technologic advances in medical history, have undergone significant product modifications since the first human implants.1 For example, although pacemakers became a clinically useful tool in the 1950s, initial models were large, electrically powered, and impractical for long-term patient use. Similarly, early ICDs, initially implanted in humans in 1980 and FDA approved in 1985, were cumbersome, required a thoracotomy for implantation, and had limited reprogramming and data storage capabilities.1,2 Since then, remarkable advances in pacemaker and ICD technology have resulted in substantial reductions in generator size, an exponential increase in computer memory, and advances in lead technology that have reduced diameter, improved chronic pacing thresholds, and with the advent of transvenous leads, greatly facilitated safe lead implantation.3,4 The resultant marked reduction in morbidity and mortality associated with device implantation has led to the widespread adoption of implantable arrhythmia device therapy to the benefit of many patients.
 Pacemaker and ICD System Performance
Pacemaker and ICD System Performance
Definitions and Assessment of Device Performance
Pacemaker and ICD systems, consisting of a generator and lead, must be able to reliably monitor cardiac electrical activity and deliver life-sustaining therapy to the heart when needed. Device reliability measures the freedom of an ICD or pacemaker generator or lead from specific structural and electrical failures.5,6 Reliability is usually reported as the percentage of devices still implanted and functioning appropriately at a given point in time (prevalence) or a failure rate per unit of time, such as failure rate per month (incidence). Device performance is a more comprehensive assessment of device quality, clinical usability, freedom from failure (malfunction), and conformance to applicable regulatory labeling. Device performance depends on a number of factors, including device design; materials and manufacturing methods; implanting physician skill and technique; patient characteristics (e.g., age, anatomy, activity level); and the expertise of the caregivers providing postimplant care. Box 34-1 highlights common terms used to describe device performance.5,6
Box 34-1
Definitions of Device Performance Terms
Pacemaker and ICD generators and leads may be removed from service because they are malfunctioning or for reasons unrelated to device performance (e.g., patient death, device infection, device upgrade). A malfunction occurs when an implanted device fails to meet its performance specifications or otherwise perform as intended. Although ideally the mechanism of a device malfunction should be confirmed by direct bench laboratory analysis, in many cases this is not possible. For example, malfunctioning ICD leads often are not explanted because of the hazards associated with lead extraction, or leads are damaged by the extraction process itself, making analysis difficult. In addition, devices may fail to perform as expected in ways not identifiable by bench analysis; for example, a design flaw resulting in an increased risk of perforation or dislodgment may be difficult to distinguish from a complication caused by a physician’s implant technique.6 Accurate and useful assessment of overall device performance and malfunctions demands monitoring, analysis, and reporting not only of device failures, but also of adverse clinical events when such events may be caused by device performance issues.
Generator Performance and Mechanisms of Malfunction
The importance of monitoring pacemaker and ICD performance has long been recognized. For example, the Bilitch Registry was started in 1974 and evaluated 22786 pacemakers from 6 sites over a 19.5-year period and 3520 ICDs from 13 sites over almost 12 years, ending in December 1993.7,8 Failures involving 50 different generator models were reported.7,8 Other long-term device failure registries reported 700 cases of pacemaker and ICD generator malfunction between 1988 and 1996, including 370 “dangerous” cases involving loss of output or decrease in pulse amplitude.7,9
More modern device performance is reflected by data compiled from manufacturer annual reports, submitted to the U.S Food And Drug Administration (FDA) from 1990 and 2002.10 Because pacemakers and ICDs represent “life-sustaining” therapies for many patients, the FDA requires manufacturers to submit annual reports detailing the number of device implants by model number and information concerning device malfunctions. From 1990 to 2002, 17,323 devices (8834 pacemakers and 8489 ICDs) were explanted because of confirmed malfunction.10 The annual number of malfunctions ranged from 366 to 1152 for pacemakers and 134 to 2228 for ICDs (Fig. 34-1). The device malfunction replacement rate (the number of devices removed in a given year because of malfunction indexed to the number of device implants in that same year) during the study period was 20.7 per 1000 implants for ICDs and 4.6 per 1000 implants for pacemakers.10
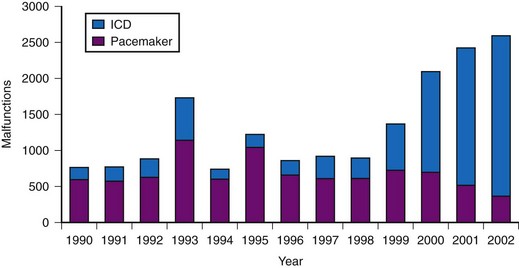
Figure 34-1 Annual number of malfunctions for pacemakers and ICDs (1990-2002).
(Adapted from Maisel WH et al: Pacemaker and ICD generator malfunctions: analysis of Food and Drug Administration annual reports. JAMA 295:1901-1906, 2006.)
A meta-analysis of active device registries (including the Bilitch Registry, the Danish Pacemaker and ICD Register, and the United Kingdom [UK] Pacemaker and ICD Registry) provides additional insights into overall pacemaker and ICD generator performance as well as trends in device reliability.11 During 2.1 million pacemaker person-years of observation over 22 years, pacemaker malfunctions were observed in 2981 devices. Pacemaker reliability improved greatly during the study period, from an annual pacemaker malfunction rate of 12.4 malfunctions per 1000 person-years in 1983 to less than 1.0 malfunction per 1000 person-years in the latter study years (Fig. 34-2). Malfunctions in ICDs were observed during the 17-year study period in 384 devices during 14,821 person-years of observation. Annual ICD malfunction rates ranged from a high of 52.5 malfunctions per 1000 person-years in 1989 to a low of 5.6 malfunctions per 1000 person-years in 1998. A significant improvement in ICD reliability occurred during the decade beginning in 1988 until a transient increase in the malfunction rate was observed in the late 1990s and early 2000s (Fig. 34-3). Device reliability again improved greatly beginning in 2003. Overall, the annual ICD malfunction rate was about 20-fold higher than the pacemaker malfunction rate (26.5 vs. 1.3 malfunctions per 1000 person-years).11
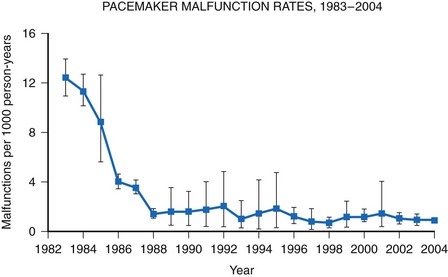
Figure 34-2 Pacemaker malfunction rates: 1983-2004.
(Adapted from Maisel WH: Pacemaker and ICD generator reliability: meta-analysis of device registries. JAMA 295:1929-1934, 2006.)
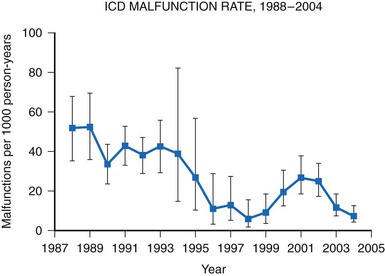
Figure 34-3 Annual ICD malfunction rates: 1988-2004.
(Adapted from Maisel WH: Pacemaker and ICD generator reliability: meta-analysis of device registries. JAMA 295:1929-1934, 2006.)
Analysis of FDA annual reports that include manufacturer-confirmed analysis of device malfunctions demonstrates that hardware abnormalities are by far the most common type of device malfunction requiring device replacement for both pacemakers and ICDs.10 Overall, hardware problems represent 79.8% of observed malfunctions. Of these, battery/capacitor abnormalities (23.6%) and electrical issues (27.1%) accounted for half the total device failures.10 Firmware (integral device software) abnormalities and miscellaneous problems (e.g., physical damage, foreign material contamination, manufacturing errors) were much less frequent causes of device malfunctions. In only 4.7% of cases could the manufacturer confirm device malfunction but not determine the cause. Battery/capacitor abnormalities account for a higher percentage of device malfunctions in ICDs than in pacemakers (31.7% vs. 15.8%).10 Miscellaneous electrical problems, such as broken wires, current leakage, and electrical short circuits, represented similar percentages of malfunctions in pacemakers and in ICDs.
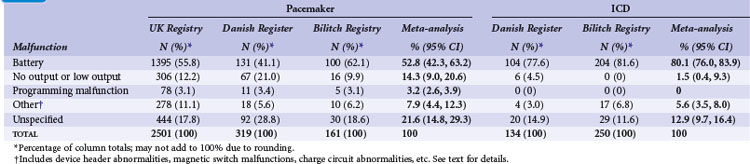
Meta-analysis of active device registries has yielded similar findings.11 Battery malfunctions, most often premature battery failure, were the most common cause of device failure in the Bilitch Registry, the Danish Pacemaker and ICD Register, and the UK Pacemaker and ICD Registry11 (Table 34-1). For pacemakers, an additional 14.3% of device malfunctions were caused by low-output or no-output states. A number of root causes, however, can masquerade as a battery abnormality, including rapid premature battery depletion, electrical short circuits, inappropriate high current drains, or hermetic seal abnormalities. Miscellaneous other types of device failures, such as programming malfunctions, device header abnormalities, magnetic switch malfunctions, and charge circuit abnormalities (for ICDs), accounted for the majority of the other reported device malfunctions.11
Lead Performance and Mechanisms of Malfunction
Pacemaker and ICD lead performance is more difficult to assess than generator performance because malfunctioning leads are often not explanted, and even when they are, leads are often damaged during the explant process.6,12 In addition, reported lead performance varies widely depending on study design, performance definitions, physician and patient characteristics, implant methodology, duration and method of follow-up, and lead models studied. These factors explain some of the observed variability in lead performance.12
Manufacturer product reports describe the performance of atrial, right ventricular, left ventricular, and high-voltage leads and demonstrate lead survival probabilities for most leads of 92% to 99% at 5 years after initial implant.13–17 However, most of these lead survival estimates are significantly limited due to potential underreporting of device malfunctions, insufficient patient follow-up, lead surveillance based on voluntary reporting, and lack of uniform definitions of lead performance and malfunction.6,12 Some manufacturers have utilized prospective, multicenter lead studies in an effort to overcome these limitations. However, small sample sizes or slow enrollment can undermine the ability of these studies to accurately identify underperforming leads in a timely manner.18 Also, they may fail to identify important differences in lead performance between models.
Other data sources also provide estimates of lead reliability. In Denmark, all lead implantations are entered into a longitudinal registry.19 Ten-year lead survival for unipolar and bipolar pacemaker leads implanted since 1993 was reported as 96.5% and 97.8%, respectively, and reliability has improved over time.19
Eckstein et al.20 conducted a retrospective analysis of 1317 consecutive patients who received ICD systems (including 38 different ICD lead models) at three centers in Germany between 1993 and 2004. Follow-up after implantation included noninvasive routine lead evaluation every 3 to 6 months. For the study, “lead failure” was defined as a lead-related problem requiring surgical revision performed at the discretion of the treating physician. Abnormalities were classified as either structural (insulation defects or lead fracture) or functional (far-field sensing; T-wave or physiologic oversensing, noise from contact with another lead, unstable impedance measurements, R-wave reduction, or loss of capture). During a median follow-up of 6.4 years, 38 ICD leads required surgical revision, resulting in a reported cumulative ICD lead survival rate of 97.5% at 5 years.20 A number of prior published studies report on the reliability and durability of ICD leads.21 Reported ICD lead “survival” varies from 91% to 99% at 2 years, 85% to 98% at 5 years, and 60% to 72% at 8 years12,21–33 (Fig. 34-4).
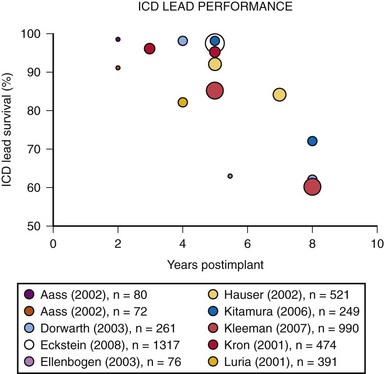
Figure 34-4 Results of selected studies of ICD lead performance.
(From Maisel WH, Kramer DB: ICD lead performance. Circulation 117:2721-2723, 2008.)
Importantly, the definition of ICD lead “survival” or lead “performance” varies among published studies, making it difficult to compare manufacturers or lead models.12 Typically, lead malfunction is defined as electrical abnormalities on lead testing, a chest radiograph consistent with a fracture, or evidence of oversensing unrelated to cardiac signals. Other studies, however, rely on physician clinical judgment and require replacement of the ICD lead in order to consider the lead to have malfunctioned.20 In most published studies, thresholds for action are poorly defined and ambiguous.
Although conceptually simple, pacemaker and ICD leads are complicated devices with lead designs that vary from model to model. These design differences may include variations in insulation, cable/conductor, length, diameter, and fixation mechanism.34 Knowledge about lead abnormalities mainly comes from manufacturer analysis of returned products, which is a rich source of information that manufacturers utilize to support product improvements and enhancements.4 Mechanical or electrical abnormalities may develop in components such as insulation, conductors, the connector, the terminal pin, or the stimulation electrode. In addition to failure mechanisms that are intrinsic to the lead, extrinsic factors (lead damage from trauma, mishandling, lead dislodgment, etc.) may cause pacemaker and ICD leads to provide insufficient therapy.6
The clinical implications of a lead malfunction vary depending on the type of malfunction and the individual patient’s clinical condition. Notably, some structurally normal leads may provide insufficient therapy (e.g., lack of clinical improvement with cardiac resynchronization therapy), inappropriate therapy (e.g., shock delivery for rapid atrial fibrillation), or may need to be removed from service as a result of issues unrelated to the lead, such as the patient’s underlying illness, physiology, implant technique, or device upgrade (e.g., from pacemaker to ICD).6 Importantly, patient and physician characteristics—in addition to lead design—affect ICD lead performance.4 For example, patients with an ICD lead failure are eight times more likely to experience a second failure than a patient without a failure.20
Unfortunately, the tools available to detect impending ICD lead failure are limited. Fluoroscopy, radiography, electrical testing, or direct visualization may be used to detect lead abnormalities, but in many respects these methods are quite rudimentary, imprecise, and insensitive. Long-term, remote monitoring has provided insights into the sometimes intermittent nature of lead abnormalities and has allowed earlier detection of lead performance issues.12
The Heart Rhythm Society (HRS) Task Force on Lead Performance Policies and Guidelines proposed a classification scheme for pacemaker and ICD leads removed from service6 (Fig. 34-5).

Surveillance of Device Performance
The goal of post-market surveillance is to “enhance the public health by reducing the incidence of medical device adverse experiences.”35 The current surveillance system relies on regulators, medical device manufacturers, health care providers, hospitals and other medical care facilities, and patients to report device malfunctions.
Regulatory authorities utilize several different methods to conduct post-market surveillance. For example, the FDA uses spontaneous reporting systems, analysis of large health care databases, scientific studies, registries, and field inspection of facilities5,6,35 (Table 34-2). Historically, the FDA has depended primarily on a passive, “adverse event” reporting system, relying on patients and the health care industry to identify and report adverse events, including rare but serious occurrences. Manufacturers are required to report to the FDA any medical device–related event or malfunction that may have caused or could cause a serious injury or death. Hospitals, nursing homes, and other medical facilities are required to report serious device-related injuries to the manufacturer and device-related deaths to both the manufacturer and the FDA.
The FDA annually receives more than 220,000 adverse event reports regarding medical devices of all types, including some that involve pacemakers, ICDs, or leads.36 The vast majority of reports are provided by manufacturers; fewer than 10,000 come directly from medical facilities. Postmortem device interrogation is rarely performed.37 Health care professionals and patients are encouraged, but not required, to report suspected device-related adverse events, but the FDA receives only several thousand reports from health care providers annually, and physicians, in particular, rarely report events.4–6,35,36
The Manufacturer and User Facility Device Experience (MAUDE) database was established to assist with adverse event reporting and information dissemination for medical devices of all types.38 It contains more than 1 million adverse event reports, including voluntary reports since June 1993 and manufacturer reports since August 1996.35,38 Selected information from this database is publicly searchable via the Internet, and the database has been successfully used to identify concerning safety signals.39,40 However, because submitted adverse event reports are sometimes cryptic or incomplete, it is often difficult to determine if a true device malfunction or patient injury has occurred. Furthermore, significant underreporting of device malfunctions and the absence of denominator data make it difficult to determine the true rate of an observed abnormality41 (Fig. 34-6). For example, all product experience reports from Italy related to one ICD lead were reviewed and demonstrated that only 126 of 454 physician product experience reports were accompanied by a returned lead—and 80% of the returns were related to implant procedures, not chronic use.42
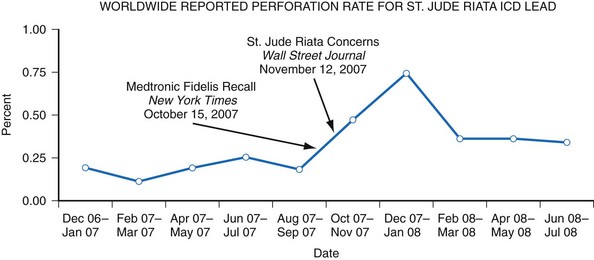
Figure 34-6 Worldwide St. Jude Riata ICD lead reported perforation rate.
(From Maisel WH: Implantable cardioverter-defibrillator lead complication: when is an outbreak out-of-bounds? Heart Rhythm 5:1673-1674, 2008.)
Recognizing this shortcoming, the FDA established the Medical Product Safety Network (MedSun) and HeartNet.43 This active surveillance system utilizes health care facilities and individuals specially trained in device adverse event reporting to identify problems in both device function and user error in the clinical setting. HeartNet in particular focuses on identifying, understanding, and solving problems with medical devices used in electrophysiology laboratories.43 In addition, many manufacturers utilize post-market studies to track the performance of their products. The Centers for Medicare and Medicaid Services (CMS) mandated National Cardiovascular Data Registry (NCDR) collects data on ICD generators and leads and is being better adapted to track longitudinal device performance.44,45 Remote monitoring of device performance has revolutionized pacemaker and ICD patient care by providing automated checks of device functionality and wireless notification of device performance to manufacturers and health care providers46 (see Chapter 32).
Device Recalls and Advisory Notices
Manufacturers and regulators occasionally become aware of devices that are not performing as expected. The terminology used to describe regulatory actions related to abnormal device behavior can be confusing to health care providers and patients because they vary by jurisdiction and are often used by the media and public inappropriately and interchangeably. The FDA defines a recall as “an action taken to address a problem with a medical device that violates FDA law.”5 Recalls occur when a medical device is defective and/or when it could constitute a risk to health. The FDA classifies recalls and safety alerts (collectively referred to as “advisories”) into four categories: class I, class II, class III, and safety alerts.7 Class I recalls are situations in which there is a reasonable probability that the use of or exposure to a product will cause serious adverse health consequences or death.7,47 Class II recalls are issued when use of product may cause temporary or medically reversible adverse health consequences or the probability of serious adverse health consequences is remote.7,47 Class III recalls mean that the use of the product is not likely to cause adverse health consequences.7,47 Product notifications or safety alerts are communications issued by a manufacturer to inform of an unreasonable risk of substantial harm from a medical device.7,47 In some cases, these situations are also considered recalls. Importantly, the term recall does not mean that an implanted device must be removed from a patient. Clinical management of advisory notices must be personalized and should reflect numerous factors related to the potential device malfunction, the patient risk profile, and physician and hospital expertise5,6 (see later discussion).
Threshold for Activation
The threshold for activation of an advisory notice may vary depending on the frequency of the device performance problem and the clinical implications of the malfunction.5,6,3,48 Even a single event, if it is systematic and associated with a significant risk for death or serious injury, may warrant early review by a manufacturer’s physician advisory committee. HRS has previously taken the position that it is inadvisable to determine a fixed percentage of device malfunctions or attempt to classify all of the particular types of malfunction that would automatically trigger a notification or advisory.5,6 Instead, HRS recommends regular review of data to determine when a pattern of inadequate device performance exists. In general, when a pattern of malfunction is recognized and associated with significant risk for patient harm, the manufacturer will retrieve affected, unimplanted devices from their sales force and hospital inventories. The timing and method of public notification as well as the clinical recommendations to health care providers and patients with advisory devices are made with involvement of industry, regulators, and arrhythmia experts.
Number, Rate, and Reasons for Advisory
More than 250,000 pulse generators, leads, and pacemaker/ICD programmers were subject to FDA recalls and safety alerts between 1974 and 1987. Furthermore, all five major manufacturers had an increase in the number of models of pulse generators, leads, and pacemaker/ICD programmers subject to FDA recalls and safety alerts between 1976 and 1995.7,49
From 1990 to 2000, there were 52 advisories affecting pacemaker and ICD generators. While recalls and safety alerts more often involved pacemakers than ICDs (67% vs. 35%; P = .002), the annual percentage of advisories involving ICDs increased between 1990 and 2000. Between January 1990 and December 2000, 408,500 pacemakers and 114,645 ICDs (523,145 total devices) were subject to recalls or safety alerts7 (Fig. 34-7). The annual number of affected devices varied from a low of 0 in 1990 to a high of 182,647 in 1999. Of the devices recalled from 1990 to 2000, 65% were recalled in 1999 and 2000, including 71% of the recalled pacemakers and 42% of the recalled ICDs. In total, since 1990, more than 60 advisories have affected over 1 million arrhythmia devices.3
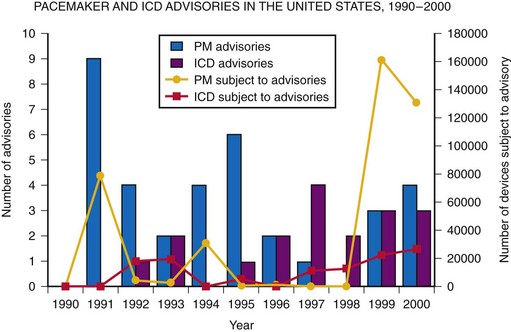
Figure 34-7 Annual number of pacemaker (PM) and ICD advisories: 1990-2000.
(Adapted from Maisel WH, et al: Recalls and safety alerts involving pacemakers and implantable cardioverter-defibrillator generators. JAMA 286:793-799, 2001.)
Of the advisories issued from 1990 to 2000, three-quarters were classified by the FDA as class I or II recalls.7 The majority (35 of 52 advisories, 67%) were issued because of hardware malfunctions such as electrical/circuitry malfunction, battery/capacitor malfunction, inadequate hermetic seal, defective crystals, or device header abnormalities. Firmware (integral device computer programming) errors accounted for an additional 10 (19%) advisories.7 Overall, hardware (54%) and firmware (41%) malfunctions accounted for 95% of all advisory devices. The remaining recalls and alerts resulted from either environment-device interactions (e.g., ICD interactions with magnetic fields) or to non-device-related problems (mislabeling of product, inappropriate product distribution).
The annual advisory rate for devices ranged from 0 to 27 per 100 person-years for pacemakers, 0 to 71 per 100 person-years for ICDs, and 0 to 25 per 100 person-years for total devices.7 The overall pacemaker and ICD generator advisory rate for 1990-2000 was 7.7 advisories per 100 person-years.7 The ICD advisory rate was significantly greater than the pacemaker advisory rate (16.4 vs. 6.7 advisories per 100 person-years). Notably, between 1995 and 2000 the advisory rate increased for pacemaker and ICD generators. Recent studies suggest as many as 25% of ICD patients are affected by an advisory.50 In addition, a changing pattern of causes of device advisories has been observed, with a threefold increase in the number of devices affected per advisory and a threefold increase in the number of devices affected by hardware advisories, (primarily from a 700-fold increase in electrical/circuitry abnormalities and a 20-fold increase in potential battery/capacitor malfunctions).51 Other hardware abnormalities (defects in the device header, hermetic seal, etc.) became less common. The number of devices recalled because of firmware abnormalities also more than doubled. Many of these observations are consistent with the changing design and manufacturing of pacemaker and ICD generators and the exponential growth in their computer memory and reliance on transistors and microprocessors.1,3,52
Communication After Device Malfunction Identified
Although transparency and timely disclosure are paramount to ensure patient safety, premature notification of generator or lead performance concerns can trigger unnecessary physician and patient anxiety and promote clinical overreaction.5,6 Premature or poorly communicated notifications about abnormal pacemaker or ICD system performance may result in unnecessary operations, lead extractions, and device replacements. The ultimate goal of communication is to promote patient safety and well-being. To achieve that goal, communication must be timely, succinct, and unambiguous so that patients and physicians are not needlessly alarmed or driven to hasty, unsafe, and unwise reactions. Communication from industry and health care providers to patients should not only identify the problem but also offer guidance, when possible, as to the clinical management and mitigation of patient risk.
The 2006 HRS Device Performance Policies and Guidelines provide a standardized Physician Device Advisory Notification form (Fig. 34-8) and Patient Device Advisory Notification letter to ensure that all critical information regarding device performance is communicated in an easily understood manner.5 In some cases, national heart rhythm societies have effectively established communication networks to disseminate important information rapidly to health care providers.53 Manufacturers typically attempt to contact patients affected by advisory using the patient registration information obtained at the initial device implantation. Health care providers should contact their patients as soon as possible after advisory notification to minimize patient anxiety and promote evidence-based clinical management.54

 Clinical Management of Device Advisory Notices
Clinical Management of Device Advisory Notices
Overview and Clinician Responsibilities
The setting of realistic expectations for patients during the informed-consent process is among the most important clinician tasks.5,6 Informed consent for a permanently implanted, potentially life-sustaining device should include a discussion of the clinical benefits and risks of device implantation as well as anticipated device and lead longevity, reliability expectations, and the potential for generator and lead performance abnormalities. In addition, routine clinical monitoring of device performance is recommended, with the frequency and method of follow-up determined by such factors as the patient’s medical status and the type and age of the implanted device.55 Remote device interrogation increasingly offers the ability to monitor device performance more closely and to detect device abnormalities earlier, potentially averting adverse clinical consequences of device failure.6,12 In many cases, close, routine, conservative monitoring of an advisory device is the safest clinical management option.5,6
Balancing Clinical Risks and Benefits
The appropriate clinical management of a pacemaker or ICD generator or lead advisory depends on a critical assessment of the risk/benefit balance in individual patients. Overall, clinician management of pacemaker and ICD advisories has been widely disparate, with some physicians recommending the replacement of most or all advisory devices, whereas others replace none or very few.56 Importantly, although generator or lead malfunctions can have important clinical consequences, so can the complications of prophylactic device replacement. For example, the Telectronics Accufix atrial J lead was susceptible to fracture; however, more patients died from lead removal than from the lead fracture itself.57 Even generator replacement for devices under advisory has been associated with a major complication rate of up to 5.8% and may be riskier than initial implantation.58,59 These cautionary tales emphasize the need for great discretion in management of advisories. Physicians must carefully consider patient- and device-specific factors that influence the balance of risks and benefits, while also considering the clinical experiences of their own institution with device replacement in different patient populations.60–62
Striking the proper clinical balance can be difficult. A low threshold to remove a device may unnecessarily expose patients to increased risks. However, too restrictive an approach to device replacement may leave patients vulnerable to potentially life-threatening device malfunction. Patient and device factors should be carefully considered, although no single factor should dictate the treatment decision6 (Box 34-2). Importantly, the expertise of the physician and the hospital where the patient receives care are also important factors in the decision-making process; patient outcomes generally improve with operator and medical center experience.63,64 Several analyses have attempted to develop treatment algorithms and define the patient populations who are best served by conservative or aggressive advisory management strategies.61,62,65 However, improvements in remote monitoring, lead extraction technologies, and procedural quality control can alter this calculus.
Clinical Management Options
The development of an individualized patient advisory clinical management strategy depends on numerous factors. In general, noninvasive management may be considered when the risk of advisory device malfunction is low, particularly for patients who are not pacemaker dependent, and for primary prevention ICD patients with a low probability of future therapy. In contrast, device revision or replacement should be considered if the risk of malfunction is substantial, if the malfunction is likely to lead to patient death or serious harm, and if the risk of device revision or replacement is less than the risk of patient harm from device malfunction. Because the risk of invasive management can be significant, and because remote monitoring offers the potential for close clinical follow-up, noninvasive management sometimes offers the lowest risk management strategy.6,12 Health care providers should communicate to the patient the risks and potential benefits of available clinical management strategies, including device revision or replacement, reprogramming the device to mitigate the risk of an adverse event from device malfunction, and routine or intensified follow-up. Patient anxiety caused by device performance concerns should also be carefully considered when developing a management strategy. In addition, patients should be provided updated device performance information whenever such information may impact the clinical management strategy.
Risks of Generator Replacement and Lead Placement
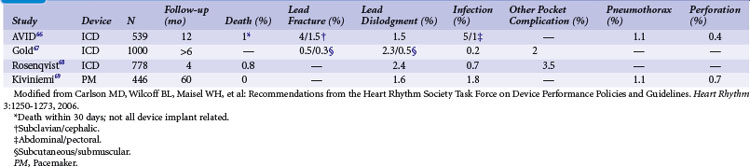
The risks of pacemaker and ICD implantation have been well described66–69 (Table 34-3). Infection rates (0.2%-1.8%) and lead dislodgment rates (1.5%-2.4%) are low but significant and may be higher for device replacement compared to initial implantation.58 The complication rate related to advisory generator replacement has been evaluated in a large, multicenter cohort of 2635 patients with advisory devices, 451 of which were replaced.70 The acute complication rate was 5.8%.58 Over almost 1 year of follow-up, 9.1% of patients undergoing replacement experienced complications, including 27 requiring reoperation and two procedure-related deaths.70 Notably, generator replacement was riskier in patients with multiple previous pocket interventions.70 Similarly, a single-center experience of 237 advisory devices resulted in a 5.5% major complication rate, including a 2.1% rate of complications requiring reoperation but no deaths.71 A major complication rate of 4.1% with no deaths was also observed among 222 patients undergoing advisory generator replacements.72 Importantly, not all medical centers demonstrate similar complication rates.73
Adjunctive medical therapy with anticoagulants and antiplatelet agents has been shown to increase the likelihood of bleeding complications from generator changes.74–77 Depending on the specific indications (e.g., recently implanted drug-eluting coronary stents), the timing of a device replacement may need to be carefully considered.77
Risks of Lead Extraction
Although lead extraction has historically been associated with a small but significant mortality risk, improvements in lead extraction technology and increased operator experience challenge the reliability of earlier estimates of morbidity and mortality associated with the procedure. For example, among a retrospective cohort of 348 patients undergoing 349 ICD lead extractions (mean implant duration of 27.5 months) at five high-volume extraction centers between May 2005 and August 2009, complete removal of the index lead was accomplished in every case with no major complications, and minor complications were observed in only 0.57% of patients. Simple traction was used in 49.7% of cases, with additional specialized extraction tools required in the remainder.78
In contrast, registry data from both American and European experiences with 7823 extraction procedures involving 12,833 pacemaker and ICD leads demonstrated a major complication rate of 1.6%.79 These studies identified implant duration of the oldest lead, ICD leads (compared to pacemaker leads), female gender, low-volume extraction hospitals, and the need for laser assistance as risk factors for adverse outcomes.79 Similarly, among 2561 pacemaker and ICD leads in 1684 patients undergoing laser lead extraction in the PLEXES Trial, 1.9% experienced a major complication, with 0.8% in-hospital mortality.80 A study of 2405 laser-assisted lead extractions in 1449 patients resulted in a 97.7% clinical success rate, 1.4% major adverse event rate, 0.28% procedure-related mortality, and 1.86% in-hospital mortality.81 Contemporary registries involving high-volume centers similarly report high procedural success rates and very low rates of serious complications (0.4%-0.9%).82,83
Several studies have demonstrated a significant learning curve associated with lead extraction, particularly for operators with fewer than 20 to 30 cases. Even experienced lead extractors continued to improve with time and volume.84–87 HRS guidelines particularly emphasize the importance of operator experience for procedural success and clinical safety.79
Notably, the favorable complication rates associated with lead extraction, which typically involves both lead removal and device system reimplantation, actually appear better than generator replacement alone. This apparent paradox relates to the terminology used to define major and minor complications; even those complications defined for study purposes as “minor” may be clinically important. The HRS Transvenous Lead Extraction Consensus Document defines minor complications to include: hematoma at implant site requiring reoperation, venous thrombosis of implant veins requiring medical intervention, vascular repair at extraction site, blood loss requiring transfusion, pulmonary embolism not requiring surgical intervention, and hemodynamically significant air embolism.79 Clearly, some of the “minor” events are also clinically relevant.
Review of device-assisted lead extraction adverse events reported to FDA revealed 57 deaths and 48 serious cardiovascular injuries between 1995 and 2008, with almost 40% occurring in the final 2 study years.88 Most alarming was the observation that some extraction injuries cannot be mitigated—even by emergency cardiac surgery. These uncommon but catastrophic events should serve as a reminder to consider carefully other advisory device clinical management options before embarking on an invasive and potentially harmful strategy. Indeed, not all advisory or malfunction leads need be removed. Lead abandonment rather than extraction may be an option for some patients with suitable anatomy. Abandoned leads do not appear to have a clinically important interaction with defibrillation threshold or sensing, and did not lead to increased thromboembolic complications, in a small study of 78 patients.89 This approach has also be proven effective in pediatric patients.90
Conservative Management and Device Reprogramming
Health care providers and patients may choose to monitor a device under advisory with a strategy of routine or enhanced surveillance, although this may not be applicable to all advisories and may not prevent serious outcomes in cases where manifestations of malfunction may be sudden and drastic.91 The availability of patient-managed, remote, and home monitoring devices may be particularly important in the early detection of problems in a patient known to be at risk for malfunction. In some cases, automated remote alerts have identified performance concerns before an important clinical adverse event.12 Patients, however, may not be aware of audible device alerts or may not interpret them correctly;92 as such, this approach must be individualized for appropriate patients. In some cases, device programming changes or software upgrades may help mitigate the clinical impact of an advisory or device performance issue. For example, algorithms can reduce the incidence of inappropriate shocks in patients with lead fracture.93,94
Device and Patient Factors Impacting Clinical Decision Making
Similarly, each patient’s clinical characteristics and individual risk profile influences the best clinical management strategy for their problematic device (see Box 34-2). For example, pacemaker dependence or past burden of tachyarrhythmia may influence the potential consequences of device malfunction for that patient. Duration since implant may also influence decision making; more recently implanted leads are generally easier to extract than older leads.79 Patient comorbidities influence clinical decision making as well. A patient’s overall prognosis from either their underlying cardiac pathology, as with advanced heart failure, or other conditions, such as malignancies, chronic obstructive pulmonary disease, or end-stage renal disease, may render device replacement excessively risky or imprudent because of the poor prognosis.
In general, noninvasive management of generator or lead advisories is preferable when the risk of malfunction is deemed to be low, particularly for patients who are not pacemaker dependent or who have not manifested clinical arrhythmia. However, device replacement or system revision merits consideration if the risk of malfunction is likely to lead to patient death or serious harm, and if the risk of revision or replacement is believed to be less than the risk of patient harm from generator or lead malfunction (Box 34-3).6
Box 34-3
Recommendations for Clinicians Managing Pacemaker and ICD Advisories
Data from Maisel WH, Hauser RG, Hammill SC, et al: Recommendations from the Heart Rhythm Society Task Force on Lead Performance Policies and Guidelines. Heart Rhythm 6:869-885, 2009.
Psychological Impact of Recalls and Advisories
Anxiety, depression, and posttraumatic stress disorder are common diagnoses in cardiac implantable electronic device (CIED) patients in the absence of device advisories.95,96 Clinicians should therefore remain alert to the potential emotional and psychological impact on patients of learning that their permanent device has become subject to an advisory.
Investigations of device advisory impact on patients’ psychological well-being have yielded mixed results. One study examining associations between ICD recalls and anxiety, depression, and quality of life (QOL) found that recalls reduced patient trust in the health care system and that more severe recalls (FDA class I vs. class II) decreased QOL.97 However, not all studies have demonstrated an untoward psychological impact. For example, in 86 patients with advisory Medtronic Marquis ICDs, there was no evidence of long-term psychological morbidity.98 Other investigations also demonstrate that QOL and psychological features are similar in patients with advisory ICDs and those with nonadvisory ICDs.99,100 However, some studies suggest that younger patients and those who previously received shocks may be more likely to experience advisory-related anxiety.101 Timely, quality physician communication is critical to maintaining patient trust and reducing patient anxiety.54 Patients who exhibit persistent, significant anxiety benefit from formal counseling.102
Although most patients with advisory devices do not experience significant, long-term anxiety, some patients may experience ongoing psychological impact. In some the clinical management strategy may need to be adjusted to account for and potentially alleviate significant symptoms.5,6
 Conclusion
Conclusion1 Maisel WH. Cardiovascular device development: lessons learned from pacemaker and implantable cardioverter-defibrillator therapy. Am J Ther. 2005;12:183-185.
2 Mirowski M, Reid PR, Mower MM, et al. Termination of malignant ventricular arrhythmias with an implanted automatic defibrillator in human beings. N Engl J Med. 1980;303:322-324.
3 Maisel WH. Safety issues involving medical devices: implications of recent implantable cardioverter-defibrillator malfunctions. JAMA. 2005;294:955-958.
4 Maisel WH, Hauser R. Proceedings of the ICD Lead Performance Conference. Heart Rhythm. 2008;5:1331-1338.
5 Carlson MD, Wilcoff BL, Maisel WH, et al. Recommendations from the Heart Rhythm Society Task Force on Device Performance Policies and Guidelines. Heart Rhythm. 2006;3:1250-1273.
6 Maisel WH, Hauser RG, Hammill SC, et al. Recommendations from the Heart Rhythm Society Task Force on Lead Performance Policies and Guidelines. Heart Rhythm. 2009;6:869-885.
7 Maisel WH, Sweeney MO, Stevenson WG, et al. Recalls and safety alerts involving pacemakers and implantable cardioverter-defibrillator generators. JAMA. 2001;286:793-799.
8 Song SL. Bilitch Report: performance of implantable cardiac rhythm management devices. Pacing Clin Electrophysiol. 1994;17:692-708.
9 Kawanishi DT, Song S, Furman S, et al. Failure rates of leads, pulse generators, and programmers have not diminished over the last 20 years: formal monitoring of performance is still needed. Bilitch Registry and STIMAREC. Pacing Clin Electrophysiol. 1996;19:1819-1823.
10 Maisel WH, Moynahan M, Zuckerman BD, et al. Pacemaker and ICD generator malfunctions: analysis of Food and Drug Administration annual reports. JAMA. 2006;295:1901-1906.
11 Maisel WH. Pacemaker and ICD generator reliability: meta-analysis of device registries. JAMA. 2006;295:1929-1934.
12 Maisel WH, Kramer DB. ICD lead performance. Circulation. 2008;117:2721-2723.
13 Biotronik Cardiac Rhythm Management Product Performance Report, January 2010. http://www.biotronik.com/sixcms/media.php/162/BIOTRONIK%20Product%20Performance%20Report%20January%202010.pdf.
14 . Boston Scientific CRM Product Performance Report 2010. Q2 Summary Edition http://www.bostonscientific.com/templatedata/imports/HTML/CRM/Product_Performance_Resource_Center/report_archives/q2_10_ppr.pdf
15 . Medtronic Cardiac Rhythm Disease Management Product Performance Report 2010. ed 1, issue 62 http://www.medtronic.com/crm/performance/downloads/mdt-prod-performance-2010-1-en.pdf
16 Sorin Group Cardiac Rhythm Management Product Performance Report. November 2009. http://www.sorin-crm.com/uploads/Media/SorinCRM_PPR-worldwide.pdf.
17 St. Jude Medical Cardiac Rhythm Management Product Performance Report. May 2010. http://www.sjmprofessional.com/Resources/reference-guides/~/media/Cardiac%20Pro/Reference%20and%20Resources/Reference%20Guides/Product_Performance_Report_CRM_May2010.ashx.
18 Maisel WH. Semper Fidelis: consumer protection for patients with implanted medical devices. N Engl J Med. 2008;358:985-987.
19 Danish Pacemaker and ICD Register 2007 Annual Report. Odense, Denmark. http://www.pacemaker.dk.
20 Eckstein J, Koller MT, Zabel M, et al. Necessity for surgical revision of defibrillator leads implanted long-term: causes and management. Circulation. 2008;117:2727-2733.
21 Aass H, Ilvento J. Short and medium time experience with a tined, multilumen steroid eluting defibrillation lead. J Interv Card Electrophysiol. 2002;6:81-86.
22 Kron J, Herre J, Renfroe EG, et al. Lead- and device-related complications in the Antiarrhythmics versus Implantable Defibrillators Trial. Am Heart J. 2001;141:92-98.
23 Dorwarth U, Frey B, Gugas M, et al. Transvenous defibrillation leads: high incidence of failure during long-term follow-up. J Cardiovasc Electrophysiol. 2003;14:38-43.
24 Ellenbogen KA, Wood MA, Shepard RK, et al. Detection and management of an implantable cardioverter defibrillator lead failure: incidence and clinical implications. J Am Coll Cardiol. 2003;41:73-80.
25 Luria D, Glikson M, Brady PA, et al. Predictors and mode of detection of transvenous lead malfunction in implantable defibrillators. Am J Cardiol. 2001;87:901-904.
26 Kleemann T, Becker T, Doenges K, et al. Annual rate of transvenous defibrillation lead defects in implantable cardioverter-defibrillators over a period of >10 years. Circulation. 2007;115:2474-2480.
27 Kitamura S, Satomi K, Kurita T, et al. Long-term follow-up of transvenous defibrillation leads: high incidence of fracture in coaxial polyurethane lead. Circ J. 2006;70:273-277.
28 Hauser RG, Cannom D, Hayes DL, et al. Long-term structural failure of coaxial polyurethane implantable cardioverter defibrillator leads. Pacing Clin Electrophysiol. 2002;25:879-882.
29 Borleffs CJW, Van Erven L, Van Bommel RJ, et al. Risk of failure of transvenous implantable cardioverter-defibrillator leads. Circ Arrhythm Electrophysiol. 2009;2:411-416.
30 Corbisiero R, Armbruster R. Does size really matter? A comparison of the Riata lead family based on size and its relation to performance. Pacing Clin Electrophysiol. 2008;31:722-726.
31 Koplan BA, Weiner S, Gilligan D, et al. Clinical and electrical performance of expanded polytetrafluoroethylene-covered defibrillator leads in comparison to traditional leads. Pacing Clin Electrophysiol. 2008;31:47-55.
32 Kupper B, Yee R, O’Hara G, et al. Do small (6.6 Fr.) active and passive fixation defibrillation leads perform as well as larger sized leads? A multi-centre analysis. Europace. 2007;9:657-661.
33 Luria D, Bar-Lev D, Gurevitz O, et al. Long-term performance of screw-in atrial pacing leads: a randomized comparison of J-shaped and straight leads. Pacing Clin Electrophysiol. 2005;28:898-902.
34 Maisel WH. Transvenous implantable cardioverter leads—the weakest link. Circulation. 2007;115:2461-2463.
35 Maisel WH. Medical device regulation: an introduction for the practicing physician. Ann Intern Med. 2004:296-302.
36 US Food and Drug Administration Center for Devices and Radiological Health Annual Reports. http://www.fda.gov/AboutFDA/CentersOffices/CDRH/CDRHReports/ucm109733.htm.
37 Kirkpatrick JN, Ghani SN, Burke MC, Knight BP. Postmortem interrogation and retrieval of implantable pacemakers and defibrillators: a survey of morticians and patients. J Cardiovasc Electrophysiol. 2007;18:478-482.
38 US Food and Drug Administration. MAUDE: Manufacturer and User Facility Device Experience. http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfmaude/search.cfm.
39 Shah JS, Maisel WH. Recalls and safety alerts affecting automated external defibrillators. JAMA. 2006;296:655-660.
40 Hauser RG, Kallinen LM, Almquist AK, et al. Early failure of a small-diameter high-voltage implantable cardioverter-defibrillator lead. Heart Rhythm. 2007;4:892-896.
41 Maisel WH. Implantable cardioverter-defibrillator lead complication: when is an outbreak out-of-bounds? Heart Rhythm. 2008;5:1673-1674.
42 Padeletti L, Pappone C, Curnis A, et al. Product-experience reporting on endocardial defibrillation leads: a 4-year national perspective. Expert Rev Med Devices. 2009;6:383-388.
43 US Food and Drug Administration. MedSun: Medical Product Safety. www.fda.gov/MedicalDevices/Safety/MedSunMedicalProductSafetyNetwork/default.htm.
44 National Cardiovascular Data Registry. ICD Registry. https://www.ncdr.com/webncdr/ICD/Default_ssl.aspx.
45 Hammill SC, Kremers MS, Kadish AH, et al. Review of the ICD Registry’s third year, expansion to include lead data and pediatric ICD procedures, and role for measuring performance. Heart Rhythm. 2009;6:1397-1401.
46 Duru F, Luechinger R, Scharf C, Brunckhorst C. Automatic impedance monitoring and patient alert feature in implantable cardioverter defibrillators: being alert for the unexpected!. J Cardiovasc Electrophysiol. 2005;16:444-448.
47 US Food and Drug Administration. Background and definitions. http://www.fda.gov/Safety/Recalls/ucm165546.htm.
48 Basile EM, Lorell BH. The Food and Drug Administration’s regulation of risk disclosure for implantable cardioverter-defibrillators: has technology outpaced the agency’s regulatory framework? Food Drug Law J. 2006;61:251-272.
49 Tyers GF. FDA recalls: how do pacemaker manufacturers compare? Ann Thorac Surg. 1989;48:390-396.
50 Mahajan T, Dubin AM, Atkins DL, et al. Impact of manufacturer advisories and FDA recalls of implantable cardioverter-defibrillator generators in pediatric and congenital heart disease patients. J Cardiovasc Electrophysiol. 2008;19:1270-1274.
51 Maisel WH, Stevenson WG, Epstein LM. Changing trends in pacemaker and implantable-cardioverter-defibrillator generator advisories. Pacing Clin Electrophysiol. 2002;25:1670-1678.
52 Levine PA, Stanton MS, Sims JJ. High quality performance of pacemakers and implantable defibrillators. Pacing Clin Electrophsyiol. 2002;25:1667-1669.
53 Krahn AD, Simpson CS, Parkash R, et al. Formation of a national network for rapid response to device and lead advisories: the Canadian Heart Rhythm Society Device Advisory Committee. Can J Cardiol. 2009;25:403-405.
54 Stutts LA, Conti JB, Aranda JMJr, et al. Patient evaluation of ICD recall communication strategies: a vignette study. Pacing Clin Electrophysiol. 2007;30:1105-1111.
55 Wilkoff BL, Auricchio A, Brugada J, et al. HRS/EHRA Expert Consensus on the monitoring of cardiovascular implantable electronic devices (CIED): description of the techniques, indications, personnel, frequency and ethical considerations. Heart Rhythm. 2008;5:907-925.
56 Maisel WH. Physician management of pacemaker and implantable cardioverter-defibrillator advisories. Pacing Clin Electrophysiol. 2004;27:437-442.
57 Kay GN, Brinker JA, Kawanishi DT, et al. Risks of spontaneous injury and extraction of an active fixation pacemaker lead: report of the Accufix Multicenter Clinical Study and Worldwide Registry. Circulation. 1999;100:2344-2352.
58 Gould PA, Krahn AD. Complications associated with implantable cardioverter-defibrillator replacement in response to device advisories. JAMA. 2006;295:1907-1911.
59 Klug D, Balde M, Pavin D, et al. Risk factors related to infections of implanted pacemakers and cardioverter-defibrillators: results of a large prospective study. Circulation. 2007;116:1349-1355.
60 Priori SG, Auricchio A, Nisam S, Yong P. To replace or not to replace: a systematic approach to respond to device advisories. J Cardiovasc Electrophysiol. 2009;20:164-170.
61 Amin MS, Ellenbogen KA. Should recent defibrillator and lead advisories affect decisions to refer patients for implantable cardioverter-defibrillator therapy? Curr Opin Cardiol. 2010;25:23-28.
62 Amin MS, Wood MA, Shepard RK, et al. Clinical judgment versus decision analysis for managing device advisories. Pacing Clin Electrophysiol. 2008;31:1236-1240.
63 Al-Khatib SM, Lucas FL, Jollis JG, et al. The relation between patients’ outcomes and the volume of cardioverter-defibrillator implantation procedures performed by physicians treating Medicare beneficiaries. J Am Coll Cardiol. 2005;46:1536-1540.
64 Curtis JP, Luebbert JJ, Wang Y, et al. Association of physician certification and outcomes among patients receiving an implantable cardioverter-defibrillator. JAMA. 2009;301:1661-1670.
65 Amin MS, Matchar DB, Wood MA, Ellenbogen KA. Management of recalled pacemakers and implantable cardioverter-defibrillators: a decision analysis model. JAMA. 2006;296:412-420.
66 Antiarrhythmics versus Implantable Defibrillators (AVID) Investigators. A comparison of antiarrhythmic-drug therapy with implantable defibrillators in patients resuscitated from near-fatal ventricular arrhythmias. N Engl J Med. 1997;337:1576-1583.
67 Gold MR, Peters RW, Johnson JW, Shorofsky SR. Complications associated with pectoral cardioverter-defibrillator implantation: comparison of subcutaneous and submuscular approaches. Worldwide Jewel Investigators. J Am Coll Cardiol. 1996;28:1278-1282.
68 Rosenqvist M, Beyer T, Block M, et al. Adverse events with transvenous implantable cardioverter-defibrillators: a prospective multicenter study. European 7219 Jewel ICD Investigators. Circulation. 1998;98:663-670.
69 Kiviniemi MS, Pirnes MA, Eranen HJ, et al. Complications related to permanent pacemaker therapy. Pacing Clin Electrophysiol. 1999;22:711-720.
70 Gould PA, Gula LJ, Champagne J, et al. Outcome of advisory implantable cardioverter-defibrillator replacement: one-year follow-up. Heart Rhythm. 2008;5:1675-1681.
71 Moore JWIII, Barrington W, Bazaz R, et al. Complications of replacing implantable devices in response to advisories: a single center experience. Int J Cardiol. 2009;134:42-46.
72 Costea A, Rardon DP, Padanilam BJ, et al. Complications associated with generator replacement in response to device advisories. J Cardiovasc Electrophysiol. 2008;19:266-269.
73 Anderson KP. Estimates of implantable cardioverter-defibrillator complications: caveat emptor. Circulation. 2009;119:1069-1071.
74 Tompkins C, Cheng A, Dalal D, et al. Dual antiplatelet therapy and heparin “bridging” significantly increase the risk of bleeding complications after pacemaker or implantable cardioverter-defibrillator device implantation. J Am Coll Cardiol. 2010;55:2376-2382.
75 Thal S, Moukabary T, Boyella R, et al. The relationship between warfarin, aspirin, and clopidogrel continuation in the peri-procedural period and the incidence of hematoma formation after device implantation. Pacing Clin Electrophysiol. 2010;33:385-388.
76 Jamula E, Douketis JD, Schulman S. Perioperative anticoagulation in patients having implantation of a cardiac pacemaker or defibrillator: a systematic review and practical management guide. J Thromb Haemost. 2008;6:1615-1621.
77 Kaluza GL, Joseph J, Lee JR, et al. Catastrophic outcomes of noncardiac surgery soon after coronary stenting. J Am Coll Cardiol. 2000;35:1288-1294.
78 Maytin M, Love CJ, Fischer A, et al. Multicenter experience with extraction of the Sprint Fidelis ICD lead. J Am Coll Cardiol. 56, 2010.
79 Wilkoff BL, Love CJ, Byrd CL, et al. Transvenous lead extraction: Heart Rhythm Society expert consensus on facilities, training, indications, and patient management. Heart Rhythm. 2009;6:1085-1104.
80 Byrd CL, Wilkoff BL, Love CJ, et al. Clinical study of the laser sheath for lead extraction: the total experience in the United States. Pacing Clin Electrophysiol. 2002;25:804-808.
81 Wazni O, Epstein LM, Carrillo RG, et al. Lead extraction in the contemporary setting: the LExICon study: an observational retrospective study of consecutive laser lead extractions. J Am Coll Cardiol. 2010;55:579-586.
82 Jones SOIV, Eckart RE, Albert CM, Epstein LM. Large, single-center, single-operator experience with transvenous lead extraction: outcomes and changing indications. Heart Rhythm. 2008;5:520-525.
83 Kennergren C, Bjurman C, Wiklund R, Gabel J. A single-centre experience of over one thousand lead extractions. Europace. 2009;11:612-617.
84 Wilkoff BL, Byrd CL, Love CJ, et al. Trends in intravascular lead extraction: analysis of data from 5339 procedures in 10 years. XIth World Symposium on Cardiac Pacing and Electrophysiology, Berlin. Pacing Clin Electrophysiol. 1999;22(6 pt II):A207.
85 Bracke FA, Meijer A, Van Gelder B. Learning curve characteristics of pacing lead extraction with a laser sheath. Pacing Clin Electrophysiol. 1998;21:2309-2313.
86 Smith HJ, Fearnot NE, Byrd CL, et al. Five-years experience with intravascular lead extraction. U.S. Lead Extraction Database. Pacing Clin Electrophysiol. 1994;17:2016-2020.
87 Ghosh N, Yee R, Klein GJ, et al. Laser lead extraction: is there a learning curve? Pacing Clin Electrophysiol. 2005;28:180-184.
88 Hauser RG, Katsiyiannis WT, Gornick CC, et al. Deaths and cardiovascular injuries due to device-assisted implantable cardioverter-defibrillator and pacemaker lead extraction. Europace. 2010;12:395-401.
89 Glikson M, Suleiman M, Luria DM, et al. Do abandoned leads pose risk to implantable cardioverter-defibrillator patients? Heart Rhythm. 2009;6:65-68.
90 Silvetti MS, Drago F. Outcome of young patients with abandoned, nonfunctional endocardial leads. Pacing Clin Electrophysiol. 2008;31:473-479.
91 Kallinen LM, Hauser RG, Lee KW, et al. Failure of impedance monitoring to prevent adverse clinical events caused by fracture of a recalled high-voltage implantable cardioverter-defibrillator lead. Heart Rhythm. 2008;5:775-779.
92 Simons EC, Feigenblum DY, Nemirovsky D, Simons GR. Alert tones are frequently inaudible among patients with implantable cardioverter-defibrillators. Pacing Clin Electrophysiol. 2009;32:1272-1275.
93 Kallinen LM, Hauser RG, Tang C, et al. Lead integrity alert algorithm decreases inappropriate shocks in patients who have Sprint Fidelis pace-sense conductor fractures. Heart Rhythm. 2010;7:1048-1055.
94 Swerdlow CD, Gunderson BD, Ousdigian KT, et al. Downloadable algorithm to reduce inappropriate shocks caused by fractures of implantable cardioverter-defibrillator leads. Circulation. 2008;118:2122-2129.
95 Sears SF, Todaro JF, Lewis TS, et al. Examining the psychosocial impact of implantable cardioverter-defibrillators: a literature review. Clin Cardiol. 1999;22:481-489.
96 Sears SF, Matchett M, Conti JB. Effective management of ICD patient psychosocial issues and patient critical events: clinical reviews. J Cardiovasc Electrophysiol. 2009;20:1297-1304.
97 Undavia M, Goldstein NE, Cohen P, et al. Impact of implantable cardioverter-defibrillator recalls on patients’ anxiety, depression, and quality of life. Pacing Clin Electrophysiol. 2008;31:1411-1418.
98 Birnie DH, Sears SF, Green MS, et al. No long-term psychological morbidity living with an implantable cardioverter-defibrillator under advisory: the Medtronic Marquis experience. Europace. 2009;11:26-30.
99 Gibson DP, Kuntz KK, Levenson JL, Ellenbogen KA. Decision-making, emotional distress, and quality of life in patients affected by the recall of their implantable cardioverter-defibrillator. Europace. 2008;10:540-544.
100 Cuculi F, Herzig W, Kobza R, Erne P. Psychological distress in patients with ICD recall. Pacing Clin Electrophysiol. 2006;29:1261-1265.
101 Sears SFJr, Conti JB. Psychological aspects of cardiac devices and recalls in patients with implantable cardioverter defibrillators. Am J Cardiol. 2006;98:565-567.
102 Fisher JD, Koulogiannis KP, Lewallen L, et al. The psychological impact of implantable cardioverter-defibrillator recalls and the durable positive effects of counseling. Pacing Clin Electrophysiol. 2009;32(8):1012-1016.
[/level-membership-for-cardiovascular-category][not-level-membership-for-cardiovascular-category]
34 Guidelines for Managing Pacemaker and Implantable Defibrillator Advisories
Pacemakers and implantable cardiac-defibrillators (ICDs), two of the most remarkable technologic advances in medical history, have undergone significant product modifications since the first human implants.1 For example, although pacemakers became a clinically useful tool in the 1950s, initial models were large, electrically powered, and impractical for long-term patient use. Similarly, early ICDs, initially implanted in humans in 1980 and FDA approved in 1985, were cumbersome, required a thoracotomy for implantation, and had limited reprogramming and data storage capabilities.1,2 Since then, remarkable advances in pacemaker and ICD technology have resulted in substantial reductions in generator size, an exponential increase in computer memory, and advances in lead technology that have reduced diameter, improved chronic pacing thresholds, and with the advent of transvenous leads, greatly facilitated safe lead implantation.3,4 The resultant marked reduction in morbidity and mortality associated with device implantation has led to the widespread adoption of implantable arrhythmia device therapy to the benefit of many patients.
Pacemaker and ICD System Performance
Definitions and Assessment of Device Performance
Pacemaker and ICD systems, consisting of a generator and lead, must be able to reliably monitor cardiac electrical activity and deliver life-sustaining therapy to the heart when needed. Device reliability measures the freedom of an ICD or pacemaker generator or lead from specific structural and electrical failures.5,6 Reliability is usually reported as the percentage of devices still implanted and functioning appropriately at a given point in time (prevalence) or a failure rate per unit of time, such as failure rate per month (incidence). Device performance is a more comprehensive assessment of device quality, clinical usability, freedom from failure (malfunction), and conformance to applicable regulatory labeling. Device performance depends on a number of factors, including device design; materials and manufacturing methods; implanting physician skill and technique; patient characteristics (e.g., age, anatomy, activity level); and the expertise of the caregivers providing postimplant care. Box 34-1 highlights common terms used to describe device performance.5,6
Box 34-1
Definitions of Device Performance Terms
Pacemaker and ICD generators and leads may be removed from service because they are malfunctioning or for reasons unrelated to device performance (e.g., patient death, device infection, device upgrade). A malfunction occurs when an implanted device fails to meet its performance specifications or otherwise perform as intended. Although ideally the mechanism of a device malfunction should be confirmed by direct bench laboratory analysis, in many cases this is not possible. For example, malfunctioning ICD leads often are not explanted because of the hazards associated with lead extraction, or leads are damaged by the extraction process itself, making analysis difficult. In addition, devices may fail to perform as expected in ways not identifiable by bench analysis; for example, a design flaw resulting in an increased risk of perforation or dislodgment may be difficult to distinguish from a complication caused by a physician’s implant technique.6 Accurate and useful assessment of overall device performance and malfunctions demands monitoring, analysis, and reporting not only of device failures, but also of adverse clinical events when such events may be caused by device performance issues.
Generator Performance and Mechanisms of Malfunction
The importance of monitoring pacemaker and ICD performance has long been recognized. For example, the Bilitch Registry was started in 1974 and evaluated 22786 pacemakers from 6 sites over a 19.5-year period and 3520 ICDs from 13 sites over almost 12 years, ending in December 1993.7,8 Failures involving 50 different generator models were reported.7,8 Other long-term device failure registries reported 700 cases of pacemaker and ICD generator malfunction between 1988 and 1996, including 370 “dangerous” cases involving loss of output or decrease in pulse amplitude.7,9
More modern device performance is reflected by data compiled from manufacturer annual reports, submitted to the U.S Food And Drug Administration (FDA) from 1990 and 2002.10 Because pacemakers and ICDs represent “life-sustaining” therapies for many patients, the FDA requires manufacturers to submit annual reports detailing the number of device implants by model number and information concerning device malfunctions. From 1990 to 2002, 17,323 devices (8834 pacemakers and 8489 ICDs) were explanted because of confirmed malfunction.10 The annual number of malfunctions ranged from 366 to 1152 for pacemakers and 134 to 2228 for ICDs (Fig. 34-1). The device malfunction replacement rate (the number of devices removed in a given year because of malfunction indexed to the number of device implants in that same year) during the study period was 20.7 per 1000 implants for ICDs and 4.6 per 1000 implants for pacemakers.10
Figure 34-1 Annual number of malfunctions for pacemakers and ICDs (1990-2002).
(Adapted from Maisel WH et al: Pacemaker and ICD generator malfunctions: analysis of Food and Drug Administration annual reports. JAMA 295:1901-1906, 2006.)
A meta-analysis of active device registries (including the Bilitch Registry, the Danish Pacemaker and ICD Register, and the United Kingdom [UK] Pacemaker and ICD Registry) provides additional insights into overall pacemaker and ICD generator performance as well as trends in device reliability.11 During 2.1 million pacemaker person-years of observation over 22 years, pacemaker malfunctions were observed in 2981 devices. Pacemaker reliability improved greatly during the study period, from an annual pacemaker malfunction rate of 12.4 malfunctions per 1000 person-years in 1983 to less than 1.0 malfunction per 1000 person-years in the latter study years (Fig. 34-2). Malfunctions in ICDs were observed during the 17-year study period in 384 devices during 14,821 person-years of observation. Annual ICD malfunction rates ranged from a high of 52.5 malfunctions per 1000 person-years in 1989 to a low of 5.6 malfunctions per 1000 person-years in 1998. A significant improvement in ICD reliability occurred during the decade beginning in 1988 until a transient increase in the malfunction rate was observed in the late 1990s and early 2000s (Fig. 34-3). Device reliability again improved greatly beginning in 2003. Overall, the annual ICD malfunction rate was about 20-fold higher than the pacemaker malfunction rate (26.5 vs. 1.3 malfunctions per 1000 person-years).11
Figure 34-2 Pacemaker malfunction rates: 1983-2004.
(Adapted from Maisel WH: Pacemaker and ICD generator reliability: meta-analysis of device registries. JAMA 295:1929-1934, 2006.)
Figure 34-3 Annual ICD malfunction rates: 1988-2004.
(Adapted from Maisel WH: Pacemaker and ICD generator reliability: meta-analysis of device registries. JAMA 295:1929-1934, 2006.)
Analysis of FDA annual reports that include manufacturer-confirmed analysis of device malfunctions demonstrates that hardware abnormalities are by far the most common type of device malfunction requiring device replacement for both pacemakers and ICDs.10 Overall, hardware problems represent 79.8% of observed malfunctions. Of these, battery/capacitor abnormalities (23.6%) and electrical issues (27.1%) accounted for half the total device failures.10 Firmware (integral device software) abnormalities and miscellaneous problems (e.g., physical damage, foreign material contamination, manufacturing errors) were much less frequent causes of device malfunctions. In only 4.7% of cases could the manufacturer confirm device malfunction but not determine the cause. Battery/capacitor abnormalities account for a higher percentage of device malfunctions in ICDs than in pacemakers (31.7% vs. 15.8%).10 Miscellaneous electrical problems, such as broken wires, current leakage, and electrical short circuits, represented similar percentages of malfunctions in pacemakers and in ICDs.
Meta-analysis of active device registries has yielded similar findings.11 Battery malfunctions, most often premature battery failure, were the most common cause of device failure in the Bilitch Registry, the Danish Pacemaker and ICD Register, and the UK Pacemaker and ICD Registry11 (Table 34-1). For pacemakers, an additional 14.3% of device malfunctions were caused by low-output or no-output states. A number of root causes, however, can masquerade as a battery abnormality, including rapid premature battery depletion, electrical short circuits, inappropriate high current drains, or hermetic seal abnormalities. Miscellaneous other types of device failures, such as programming malfunctions, device header abnormalities, magnetic switch malfunctions, and charge circuit abnormalities (for ICDs), accounted for the majority of the other reported device malfunctions.11
Lead Performance and Mechanisms of Malfunction
Pacemaker and ICD lead performance is more difficult to assess than generator performance because malfunctioning leads are often not explanted, and even when they are, leads are often damaged during the explant process.6,12 In addition, reported lead performance varies widely depending on study design, performance definitions, physician and patient characteristics, implant methodology, duration and method of follow-up, and lead models studied. These factors explain some of the observed variability in lead performance.12
Manufacturer product reports describe the performance of atrial, right ventricular, left ventricular, and high-voltage leads and demonstrate lead survival probabilities for most leads of 92% to 99% at 5 years after initial implant.13–17 However, most of these lead survival estimates are significantly limited due to potential underreporting of device malfunctions, insufficient patient follow-up, lead surveillance based on voluntary reporting, and lack of uniform definitions of lead performance and malfunction.6,12 Some manufacturers have utilized prospective, multicenter lead studies in an effort to overcome these limitations. However, small sample sizes or slow enrollment can undermine the ability of these studies to accurately identify underperforming leads in a timely manner.18 Also, they may fail to identify important differences in lead performance between models.
Other data sources also provide estimates of lead reliability. In Denmark, all lead implantations are entered into a longitudinal registry.19 Ten-year lead survival for unipolar and bipolar pacemaker leads implanted since 1993 was reported as 96.5% and 97.8%, respectively, and reliability has improved over time.19
Eckstein et al.20 conducted a retrospective analysis of 1317 consecutive patients who received ICD systems (including 38 different ICD lead models) at three centers in Germany between 1993 and 2004. Follow-up after implantation included noninvasive routine lead evaluation every 3 to 6 months. For the study, “lead failure” was defined as a lead-related problem requiring surgical revision performed at the discretion of the treating physician. Abnormalities were classified as either structural (insulation defects or lead fracture) or functional (far-field sensing; T-wave or physiologic oversensing, noise from contact with another lead, unstable impedance measurements, R-wave reduction, or loss of capture). During a median follow-up of 6.4 years, 38 ICD leads required surgical revision, resulting in a reported cumulative ICD lead survival rate of 97.5% at 5 years.20 A number of prior published studies report on the reliability and durability of ICD leads.21 Reported ICD lead “survival” varies from 91% to 99% at 2 years, 85% to 98% at 5 years, and 60% to 72% at 8 years12,21–33 (Fig. 34-4).
Figure 34-4 Results of selected studies of ICD lead performance.
(From Maisel WH, Kramer DB: ICD lead performance. Circulation 117:2721-2723, 2008.)
Importantly, the definition of ICD lead “survival” or lead “performance” varies among published studies, making it difficult to compare manufacturers or lead models.12 Typically, lead malfunction is defined as electrical abnormalities on lead testing, a chest radiograph consistent with a fracture, or evidence of oversensing unrelated to cardiac signals. Other studies, however, rely on physician clinical judgment and require replacement of the ICD lead in order to consider the lead to have malfunctioned.20 In most published studies, thresholds for action are poorly defined and ambiguous.
Although conceptually simple, pacemaker and ICD leads are complicated devices with lead designs that vary from model to model. These design differences may include variations in insulation, cable/conductor, length, diameter, and fixation mechanism.34 Knowledge about lead abnormalities mainly comes from manufacturer analysis of returned products, which is a rich source of information that manufacturers utilize to support product improvements and enhancements.4 Mechanical or electrical abnormalities may develop in components such as insulation, conductors, the connector, the terminal pin, or the stimulation electrode. In addition to failure mechanisms that are intrinsic to the lead, extrinsic factors (lead damage from trauma, mishandling, lead dislodgment, etc.) may cause pacemaker and ICD leads to provide insufficient therapy.6
The clinical implications of a lead malfunction vary depending on the type of malfunction and the individual patient’s clinical condition. Notably, some structurally normal leads may provide insufficient therapy (e.g., lack of clinical improvement with cardiac resynchronization therapy), inappropriate therapy (e.g., shock delivery for rapid atrial fibrillation), or may need to be removed from service as a result of issues unrelated to the lead, such as the patient’s underlying illness, physiology, implant technique, or device upgrade (e.g., from pacemaker to ICD).6 Importantly, patient and physician characteristics—in addition to lead design—affect ICD lead performance.4 For example, patients with an ICD lead failure are eight times more likely to experience a second failure than a patient without a failure.20
Unfortunately, the tools available to detect impending ICD lead failure are limited. Fluoroscopy, radiography, electrical testing, or direct visualization may be used to detect lead abnormalities, but in many respects these methods are quite rudimentary, imprecise, and insensitive. Long-term, remote monitoring has provided insights into the sometimes intermittent nature of lead abnormalities and has allowed earlier detection of lead performance issues.12
The Heart Rhythm Society (HRS) Task Force on Lead Performance Policies and Guidelines proposed a classification scheme for pacemaker and ICD leads removed from service6 (Fig. 34-5).
Surveillance of Device Performance
The goal of post-market surveillance is to “enhance the public health by reducing the incidence of medical device adverse experiences.”35 The current surveillance system relies on regulators, medical device manufacturers, health care providers, hospitals and other medical care facilities, and patients to report device malfunctions.
Regulatory authorities utilize several different methods to conduct post-market surveillance. For example, the FDA uses spontaneous reporting systems, analysis of large health care databases, scientific studies, registries, and field inspection of facilities5,6,35 (Table 34-2). Historically, the FDA has depended primarily on a passive, “adverse event” reporting system, relying on patients and the health care industry to identify and report adverse events, including rare but serious occurrences. Manufacturers are required to report to the FDA any medical device–related event or malfunction that may have caused or could cause a serious injury or death. Hospitals, nursing homes, and other medical facilities are required to report serious device-related injuries to the manufacturer and device-related deaths to both the manufacturer and the FDA.
The FDA annually receives more than 220,000 adverse event reports regarding medical devices of all types, including some that involve pacemakers, ICDs, or leads.36 The vast majority of reports are provided by manufacturers; fewer than 10,000 come directly from medical facilities. Postmortem device interrogation is rarely performed.37 Health care professionals and patients are encouraged, but not required, to report suspected device-related adverse events, but the FDA receives only several thousand reports from health care providers annually, and physicians, in particular, rarely report events.4–6,35,36
The Manufacturer and User Facility Device Experience (MAUDE) database was established to assist with adverse event reporting and information dissemination for medical devices of all types.38 It contains more than 1 million adverse event reports, including voluntary reports since June 1993 and manufacturer reports since August 1996.35,38 Selected information from this database is publicly searchable via the Internet, and the database has been successfully used to identify concerning safety signals.39,40 However, because submitted adverse event reports are sometimes cryptic or incomplete, it is often difficult to determine if a true device malfunction or patient injury has occurred. Furthermore, significant underreporting of device malfunctions and the absence of denominator data make it difficult to determine the true rate of an observed abnormality41 (Fig. 34-6). For example, all product experience reports from Italy related to one ICD lead were reviewed and demonstrated that only 126 of 454 physician product experience reports were accompanied by a returned lead—and 80% of the returns were related to implant procedures, not chronic use.42
Figure 34-6 Worldwide St. Jude Riata ICD lead reported perforation rate.
(From Maisel WH: Implantable cardioverter-defibrillator lead complication: when is an outbreak out-of-bounds? Heart Rhythm 5:1673-1674, 2008.)
Recognizing this shortcoming, the FDA established the Medical Product Safety Network (MedSun) and HeartNet.43 This active surveillance system utilizes health care facilities and individuals specially trained in device adverse event reporting to identify problems in both device function and user error in the clinical setting. HeartNet in particular focuses on identifying, understanding, and solving problems with medical devices used in electrophysiology laboratories.43 In addition, many manufacturers utilize post-market studies to track the performance of their products. The Centers for Medicare and Medicaid Services (CMS) mandated National Cardiovascular Data Registry (NCDR) collects data on ICD generators and leads and is being better adapted to track longitudinal device performance.44,45 Remote monitoring of device performance has revolutionized pacemaker and ICD patient care by providing automated checks of device functionality and wireless notification of device performance to manufacturers and health care providers46 (see Chapter 32).
[/not-level-membership-for-cardiovascular-category]
