Growth disorders and acromegaly
Normal growth
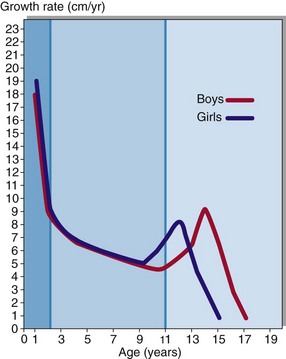
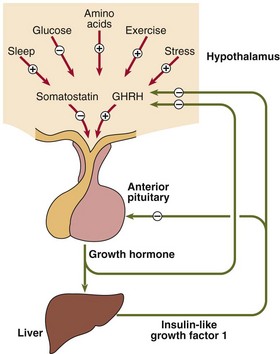
Growth in children can be divided into three stages (Fig 43.1). Rapid growth occurs during the first 2 years of life; the rate is influenced by conditions in utero, as well as the adequacy of nutrition in the postnatal period. The next stage is relatively steady growth for around 9 years and is controlled mainly by growth hormone (GH). If the pituitary does not produce sufficient growth hormone, the yearly growth rate during this period may be halved and the child will be of short stature. The growth spurt at puberty is caused by the effect of the sex hormones in addition to continuing GH secretion. The regulation of GH secretion is outlined in Figure 43.2.
Growth hormone insufficiency
 having parents who are both short
having parents who are both short
 inherited diseases such as achondroplasia, the commonest cause of severe dwarfism
inherited diseases such as achondroplasia, the commonest cause of severe dwarfism

 systemic chronic illness, such as renal disease, gastrointestinal disorders or respiratory disease
systemic chronic illness, such as renal disease, gastrointestinal disorders or respiratory disease


 Clinical note
Clinical noteExcessive growth



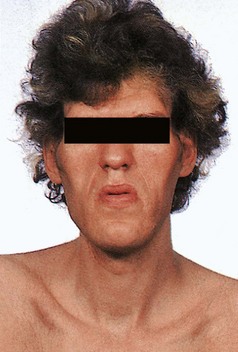
Acromegaly
Increased GH secretion later in life, after fusion of bony epiphyses, causes acromegaly (Fig 43.3). The most likely cause is a pituitary adenoma. Clinical features include:






Diagnosis
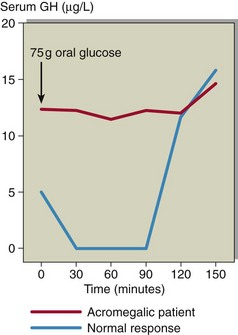
Formal diagnosis of acromegaly requires an oral glucose tolerance test with GH measurement (see pp. 82–83). Acromegalic patients do not suppress fully in response to hyperglycaemia (Fig 43.4), and indeed in some patients a paradoxical rise in GH may be observed.
Treatment
 Surgery. Trans-sphenoidal hypophysectomy is the first-line treatment for most acromegalic patients. Its success depends on the size of the tumour.
Surgery. Trans-sphenoidal hypophysectomy is the first-line treatment for most acromegalic patients. Its success depends on the size of the tumour.







