Molecular Defects More than 90% of patients clinically classified as having MFS by the “Ghent criteria” have a mutation in the gene for fibrillin-1 (FBN1). Mutations in the same gene are found in a few patients who do not meet the Ghent criteria. Also, a few MFS patients without mutations in the FBN1 gene have mutations in the gene for TGF-β receptor 2 (TGFBR2). In addition, mutations in either TGFBR2 or TGFBR1 are found in the related Loeys-Dietz syndrome, which is characterized by aortic aneurysms, cleft palate, and hypertelorism. Mutations in the FBN2 gene, which is structurally similar to the FBN1 gene, are found in patients with MFS-like syndrome of congenital contractual arachnodactyly.
FBN1 gene mutations are scattered throughout its 65 coding exons. Most are private mutations, but about 10% are recurrent new mutations that are largely located in CpG sequences known to be “hot spots.” Most severe mutations are located in the central codons (24–32). About one-third of the mutations introduce premature termination codons, and about two-thirds are missense mutations that alter calcium-binding domains in the repetitive epidermal growth factor–like domains of the protein. Rarer mutations alter the processing of the protein. As in many genetic diseases, the severity of the phenotype cannot be predicted from the nature of the mutation.
The discovery that syndromes similar to MFS are caused by mutations in TGFBR1 and TGFBR2 refocused attention on structural similarity between fibrillin-1 and TGF-β binding proteins that sequester TGF-β in the extracellular matrix. As a result, some of the manifestations of MFS have been shown to arise from alterations in binding sites that modulate TGF-β bioavailability during development of the skeleton and other tissues. Likewise, TGFBR1 and TGFBR2 mutations in Loeys-Dietz syndrome alter TGF-β signaling. In both MFS and Loeys-Dietz syndrome, the pathogenic mechanisms involve increased TGF-β signaling, which contributes to aneurysm formation.
Diagnosis All patients with a suspected diagnosis of MFS should have a slit-lamp examination and an echocardiogram. Also, homocystinuria should be ruled out by amino acid analysis of plasma (Chap. 434e). The diagnosis of MFS according to the international Ghent standards places emphasis on major criteria that include presence of at least four skeletal abnormalities: ectopia lentis; dilation of the ascending aorta with or without dissection; dural ectasia; and a blood relative who meets the same criteria, with or without a DNA diagnosis. A final diagnosis is based on a balanced assessment of the major criteria together with several minor criteria. The absence of ocular changes suggests the Loeys-Dietz syndrome, and the presence of contractures with some of the signs of OI suggests congenital contractual arachnodactyly.
Diagnostic tests based on gene sequencing or detection of protein defects are available. These results are unlikely to alter the treatment or prognosis but are helpful to inform the patients and families and to rapidly exclude the diagnosis in unaffected family members.
|
TREATMENT |
MARFAN’S SYNDROME |
Propranolol or other β-adrenergic blocking agents are used to lower blood pressure and thereby delay or prevent aortic dilation. Surgical correction of the aorta, aortic valve, and mitral valve has been successful in many patients, but tissues are frequently friable. Patients should be advised that the risks are increased by severe physical exertion, emotional stress, and pregnancy.
The scoliosis tends to be progressive and should be treated by mechanical bracing and physical therapy if >20° or by surgery if it progresses to >45°. Dislocated lenses rarely require surgical removal, but patients should be followed closely for retinal detachment. The finding that MFS pathophysiology involves alterations in TGF-β signaling has raised the possibility of new therapeutic strategies. Attenuation of TGF-β signaling with agents such as angiotensin II receptor blockers (e.g., losartan) was effective in animal studies and has been very promising in small observational studies on MFS patients, significantly reducing progressive aortic enlargement. Based on these results, large randomized clinical trials of angiotensin receptor blockers in MFS are under way.
ELASTIN-RELATED DISEASES
Mutations in the elastin gene (ELN) have been found in patients with supravalvular aortic stenosis and skin that hangs in loose and redundant folds (cutis laxa). As indicated in Table 427-3, patients with several forms of EDS have similar changes in skin that were initially thought to reflect changes in elastin.
EPIDERMOLYSIS BULLOSA (EB)
EB has been defined as the category of heritable disorders involving skin that is specifically characterized by blistering as a result of friction. Using this criterion, it was possible to define subtypes by the ultrastructural layer of skin in which the cleavage and blistering occurred. These functional and anatomical criteria made it possible to establish that most patients with a specific subtype have mutations in genes coding for a structural protein, or a cell adherence protein, expressed in the corresponding layer of skin.
Classification and Incidence The four major types of EB are: (1) EB simplex in which cleavage occurs within the epidermis, (2) junctional EB in which cleavage occurs within the lamina lucida, (3) dystrophic EB in which cleavage occurs within the sublamina densa, and (4) Kindler’s syndrome with a mixed level of cleavage in different layers. Patients are then separated into major and minor subtypes based on clinical features and analysis of mutations.
The incidence of EB in the United States is about 1 in 50,000.
![]() Molecular Defects The distinctive anatomic locations in skin have made it possible to relate the clinical subtypes of EB to mutations for specific components. In EB simplex, mutations are found primarily in the genes for the major keratins of basal epithelial cells (keratins 5 and 14) and the cell adhesion proteins plectin, α6β4 integrin, plakophilin-1, and desmoplakin. Patients with the related syndrome, epidermolytic ichthyosis, have mutations in keratin 1 and keratin 10. In junctional EB, mutations occur in type XVII collagen, a laminin (laminin-332), and α6β4 integrin. In the severe syndrome of dystrophic EB, mutations are found in the gene that codes for type VII collagen, which forms long loops anchoring the epidermis to the dermis. Patients with more complex features of what is classified as Kindler’s syndrome have mutations in kindlin-1, a focal adhesion protein involved in integrin activation.
Molecular Defects The distinctive anatomic locations in skin have made it possible to relate the clinical subtypes of EB to mutations for specific components. In EB simplex, mutations are found primarily in the genes for the major keratins of basal epithelial cells (keratins 5 and 14) and the cell adhesion proteins plectin, α6β4 integrin, plakophilin-1, and desmoplakin. Patients with the related syndrome, epidermolytic ichthyosis, have mutations in keratin 1 and keratin 10. In junctional EB, mutations occur in type XVII collagen, a laminin (laminin-332), and α6β4 integrin. In the severe syndrome of dystrophic EB, mutations are found in the gene that codes for type VII collagen, which forms long loops anchoring the epidermis to the dermis. Patients with more complex features of what is classified as Kindler’s syndrome have mutations in kindlin-1, a focal adhesion protein involved in integrin activation.
Diagnosis and Treatment The diagnosis is based on skin that readily breaks and forms blisters from minor trauma. EB simplex is generally milder than junctional EB or dystrophic EB. Dystrophic EB variants usually have large and prominent scars. Precise classification within subtypes usually requires immunofluorescent mapping. DNA diagnostic tests have been developed as research tools but are not widely available. The treatment is symptomatic. Novel therapeutic approaches such as gene therapy, protein replacement therapy, and cell therapy are being explored.
ALPORT’S SYNDROME (AS)
AS is an inherited disorder characterized by hematuria and several associated features. It was not initially considered as a disorder of connective tissue. However, the search for mutations in the genes coding for the collagen found that most patients had mutations in collagen found in basement membranes (type IV). Four forms of AS are now recognized: (1) classic AS, which is inherited as an X-linked disorder with hematuria, sensorineural deafness, and conical deformation of the anterior surface of the lens (lenticonus); (2) an X-linked form associated with diffuse leiomyomatosis; (3) an autosomal recessive form; and (4) an autosomal dominant form. Both autosomal recessive and dominant forms can cause renal disease without deafness or lenticonus.
Incidence The incidence of AS is about 1 in 10,000 births in the general population and as high as 1 in 5000 in some ethnic groups. About 80% of AS patients have the classical X-linked variant.
![]() Molecular Defects Most patients have mutations in four of the six genes for the chains of type IV collagen (COL4A3, COL4A4, COL4A5, and COL4A6). The genes for the proteins are arranged in tandem pairs on different chromosomes in an unusual head-to-head orientation and with overlapping promoters; i.e., the COL4A1 and COL4A2 genes are head-to-head on chromosome 13q34, the COL4A3 and COL4A4 genes are on chromosome 2q35–37, and the COL4A5 and COL4A6 genes are on chromosome Xq22. The X-linked variants are caused by either mutations in the COL4A5 gene or by partial deletions of both of the adjacent COL4A4 and COL4A5 genes. The autosomal recessive variants are caused by mutations in either the COL4A3 or COL4A4 gene. The mutations responsible for the autosomal dominant variants are still unknown, but they have been mapped to the same locus as the COL4A3 and COL4A4 genes.
Molecular Defects Most patients have mutations in four of the six genes for the chains of type IV collagen (COL4A3, COL4A4, COL4A5, and COL4A6). The genes for the proteins are arranged in tandem pairs on different chromosomes in an unusual head-to-head orientation and with overlapping promoters; i.e., the COL4A1 and COL4A2 genes are head-to-head on chromosome 13q34, the COL4A3 and COL4A4 genes are on chromosome 2q35–37, and the COL4A5 and COL4A6 genes are on chromosome Xq22. The X-linked variants are caused by either mutations in the COL4A5 gene or by partial deletions of both of the adjacent COL4A4 and COL4A5 genes. The autosomal recessive variants are caused by mutations in either the COL4A3 or COL4A4 gene. The mutations responsible for the autosomal dominant variants are still unknown, but they have been mapped to the same locus as the COL4A3 and COL4A4 genes.
Diagnosis The diagnosis of classic AS is based on X-linked inheritance of hematuria, sensorineural deafness, and lenticonus. The lenticonus together with hematuria is pathognomonic of classic AS. The sensorineural deafness is primarily in the high-tone range. It can frequently be detected only by an audiogram and is usually not progressive. Because of the X-linked transmission, women are generally underdiagnosed and are usually less severely affected than men. The hematuria usually progresses to nephritis and may cause renal failure in late adolescence in affected males and at older ages in some women. Renal transplantation is usually successful.
ACKNOWLEDGMENTS
The authors acknowledge the contributions of Helena Kuivaniemi, Gerard Tromp, Leena Ala-Kokko, and Malwina Czarny-Ratajcak to this chapter in previous editions of Harrison’s. The authors also wish to thank David Sillence for his expert advice on the classifications of types of OI.
428 |
Hemochromatosis |
DEFINITION
Hemochromatosis is a common inherited disorder of iron metabolism in which dysregulation of intestinal iron absorption results in deposition of excessive amounts of iron in parenchymal cells with eventual tissue damage and impaired function in a wide range of organs. The iron-storage pigment in tissues is called hemosiderin because it was believed to be derived from the blood. The term hemosiderosis is used to describe the presence of stainable iron in tissues, but tissue iron must be quantified to assess body-iron status accurately (see below and Chap. 126). Hemochromatosis refers to a group of genetic diseases that predispose to iron overload, potentially leading to fibrosis and organ failure. Cirrhosis of the liver, diabetes mellitus, arthritis, cardiomyopathy, and hypogonadotropic hypogonadism are the major clinical manifestations.
Although there is debate about definitions, the following terminology is widely accepted.
1. Hereditary hemochromatosis is most often caused by a mutant gene, termed HFE, which is tightly linked to the HLA-A locus on chromosome 6p (see “Genetic Basis,” below). Persons who are homozygous for the mutation are at increased risk of iron overload and account for 80–90% of clinical hereditary hemochromatosis in persons of northern European descent. In such subjects, the presence of hepatic fibrosis, cirrhosis, arthropathy, or hepatocellular carcinoma constitutes iron overload–related disease. Rarer forms of non-HFE hemochromatosis are caused by mutations in other genes involved in iron metabolism (Table 428-1). The disease can be recognized during its early stages when iron overload and organ damage are minimal. At this stage, the disease is best referred to as early hemochromatosis or precirrhotic hemochromatosis.
|
CLASSIFICATION OF IRON OVERLOAD STATES |

2. Secondary iron overload occurs as a result of an iron-loading anemia, such as thalassemia or sideroblastic anemia, in which erythropoiesis is increased but ineffective. In the acquired iron-loading disorders, massive iron deposits in parenchymal tissues can lead to the same clinical and pathologic features as in hemochromatosis.
PREVALENCE
HFE-associated hemochromatosis mutations are among the most common inherited disease alleles, although the prevalence varies in different ethnic groups. It is most common in populations of northern European extraction in whom approximately 1 in 10 persons are heterozygous carriers and 0.3–0.5% are homozygotes. However, expression of the disease is variable and modified by several factors, especially alcohol consumption and dietary iron intake, blood loss associated with menstruation and pregnancy, and blood donation. Recent population studies indicate that approximately 30% of homozygous men develop iron overload–related disease and about 6% develop hepatic cirrhosis; for women, the figure is closer to 1%. Presumably there are as yet unidentified modifying genes responsible for expression and there is some early evidence to support this. Nearly 70% of untreated patients develop the first symptoms between ages 40 and 60. The disease is rarely evident before age 20, although with family screening (see “Screening for Hemochromatosis,” below) and periodic health examinations, asymptomatic subjects with iron overload can be identified, including young menstruating women.
In contrast to HFE-associated hemochromatosis, the non-HFE-associated forms of hemochromatosis (Table 428-1) are rare, but they affect all races and young people (juvenile hemochromatosis).
GENETIC BASIS
![]() A homozygous G to A mutation in the HFE gene resulting in a cysteine to tyrosine substitution at position 282 (C282Y) is the most common mutation. It is identified in 85–90% of patients with hereditary hemochromatosis in populations of northern European descent but is found in only 60% of cases from Mediterranean populations (e.g., southern Italy). A second, relatively common HFE mutation (H63D) results in a substitution of histidine to aspartic acid at codon 63. Homozygosity for H63D is not associated with clinically significant iron overload. Some compound heterozygotes (e.g., one copy each of C282Y and H63D) have mild to moderately increased body-iron stores but develop clinical disease only in association with cofactors such as heavy alcohol intake or hepatic steatosis. Thus, HFE-associated hemochromatosis is inherited as an autosomal recessive trait; heterozygotes have no, or minimal, increase in iron stores. However, this slight increase in hepatic iron can act as a cofactor that may modify the expression of other diseases such as porphyria cutanea tarda (PCT) or nonalcoholic steatohepatitis.
A homozygous G to A mutation in the HFE gene resulting in a cysteine to tyrosine substitution at position 282 (C282Y) is the most common mutation. It is identified in 85–90% of patients with hereditary hemochromatosis in populations of northern European descent but is found in only 60% of cases from Mediterranean populations (e.g., southern Italy). A second, relatively common HFE mutation (H63D) results in a substitution of histidine to aspartic acid at codon 63. Homozygosity for H63D is not associated with clinically significant iron overload. Some compound heterozygotes (e.g., one copy each of C282Y and H63D) have mild to moderately increased body-iron stores but develop clinical disease only in association with cofactors such as heavy alcohol intake or hepatic steatosis. Thus, HFE-associated hemochromatosis is inherited as an autosomal recessive trait; heterozygotes have no, or minimal, increase in iron stores. However, this slight increase in hepatic iron can act as a cofactor that may modify the expression of other diseases such as porphyria cutanea tarda (PCT) or nonalcoholic steatohepatitis.
Mutations in other genes involved in iron metabolism are responsible for non-HFE-associated hemochromatosis, including juvenile hemochromatosis, which affects persons in the second and third decades of life (Table 428-1). Mutations in the genes encoding hepcidin, transferrin receptor 2 (TfR2), and hemojuvelin (Fig. 428-1) result in clinicopathologic features that are indistinguishable from HFE-associated hemochromatosis. However, mutations in ferroportin, responsible for the efflux of iron from enterocytes and most other cell types, result in iron loading of reticuloendothelial cells and macrophages as well as parenchymal cells.
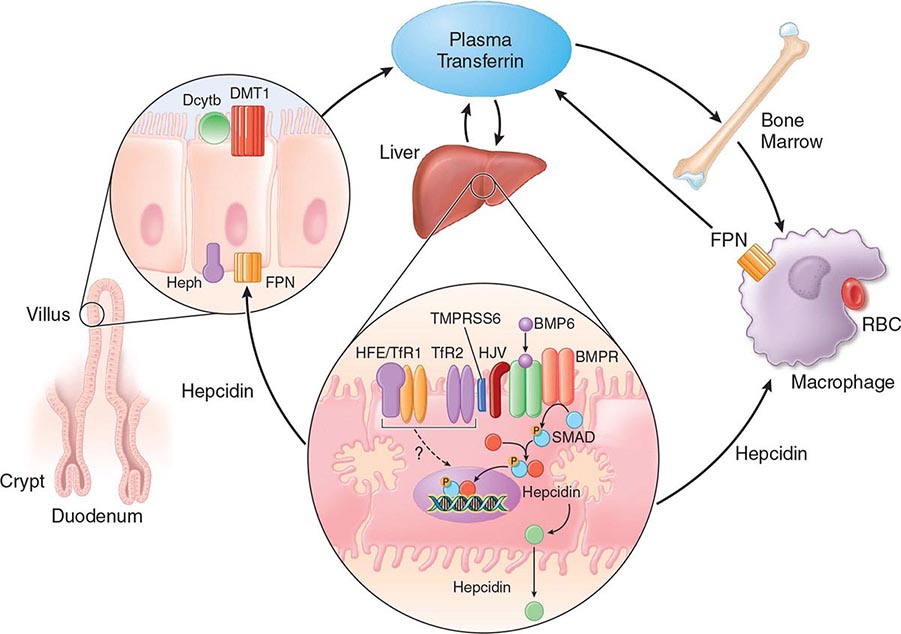
FIGURE 428-1 Pathways of normal iron homeostasis. Dietary inorganic iron traverses the brush border membrane of duodenal enterocytes via the divalent metal-ion transporter 1 (DMT1) after reduction of ferric (Fe3+) iron to the ferrous (Fe2+) state by duodenal cytochrome B (DcytB). Iron then moves from the enterocyte to the circulation via a process requiring the basolateral iron exporter ferroportin (FPN) and the iron oxidase hephaestin (Heph). In the circulation, iron binds to plasma transferrin and is thereby distributed to sites of iron utilization and storage. Much of the diferric transferrin supplies iron to immature erythrocyte cells in the bone marrow for hemoglobin synthesis. At the end of their life, senescent red blood cells (RBCs) are phagocytosed by macrophages, and iron is returned to the circulation after export through ferroportin. The liver-derived peptide hepcidin represses basolateral iron transport in the gut as well as iron released from macrophages and other cells and serves as a central regulator of body-iron traffic. Hepcidin responds to changes in body-iron requirements by signals mediated by diferric transferrin through two mechanisms. One involves HFE and TfR2, whereas the other involves hemojuvelin (HJV) and the bone morphogenetic protein (BMP)/SMAD pathway. TMPRSS6 is a protease that modulates HJV activity. Heme is metabolized by heme oxygenase within the enterocytes, and the released iron then follows the same pathway. Mutations in the genes encoding HFE, TfR2, hemojuvelin, and hepcidin all lead to decreased hepcidin release and increased iron absorption, resulting in hemochromatosis (Table 428-1).
PATHOPHYSIOLOGY
Normally, the body-iron content of 3–4 g is maintained such that intestinal mucosal absorption of iron is equal to iron loss. This amount is approximately 1 mg/d in men and 1.5 mg/d in menstruating women. In hemochromatosis, mucosal absorption is greater than body requirements and amounts to 4 mg/d or more. The progressive accumulation of iron increases plasma iron and saturation of transferrin and results in a progressive increase of plasma ferritin (Fig. 428-2). A liver-derived peptide, hepcidin, represses basolateral iron transport in the intestine and iron release from macrophages and other cells by binding to ferroportin. Hepcidin, in turn, responds to signals in the liver mediated by HFE, TfR2, and hemojuvelin (Fig. 428-1). Thus, hepcidin is a crucial molecule in iron metabolism, linking body stores with intestinal iron absorption.
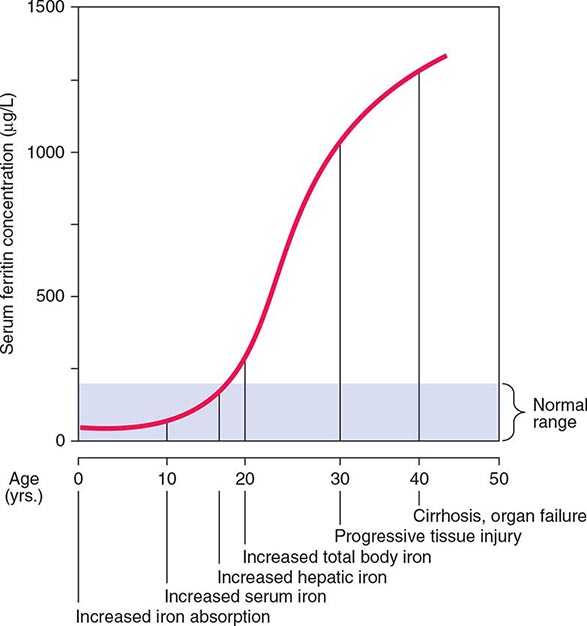
FIGURE 428-2 Sequence of events in genetic hemochromatosis and their correlation with the serum ferritin concentration. Increased iron absorption is present throughout life. Overt, symptomatic disease usually develops between ages 40 and 60, but latent disease can be detected long before this.
The HFE gene encodes a 343-amino-acid protein that is structurally related to MHC class I proteins (HFE). The basic defect in HFE-associated hemochromatosis is a lack of cell surface expression of HFE (due to the C282Y mutation). The normal (wild-type) HFE protein forms a complex with β2-microglobulin and transferrin receptor 1 (TfR1). The C282Y mutation completely abrogates this interaction. As a result, the mutant HFE protein remains trapped intracellularly, reducing TfR1-mediated iron uptake by the intestinal crypt cell. This impaired TfR1-mediated iron uptake leads to upregulation of the divalent metal transporter (DMT1) on the brush border of the villus cells, causing inappropriately increased intestinal iron absorption (Fig. 428-1). In advanced disease, the body may contain 20 g or more of iron that is deposited mainly in parenchymal cells of the liver, pancreas, and heart. Iron may be increased 50- to 100-fold in the liver and pancreas and 5- to 25-fold in the heart. Iron deposition in the pituitary causes hypogonadotropic hypogonadism in both men and women. Tissue injury may result from disruption of iron-laden lysosomes, from lipid peroxidation of subcellular organelles by excess iron, or from stimulation of collagen synthesis by activated stellate cells.
Secondary iron overload with deposition in parenchymal cells occurs in chronic disorders of erythropoiesis, particularly in those due to defects in hemoglobin synthesis or ineffective erythropoiesis such as sideroblastic anemia and thalassemia (Chap. 127). In these disorders, iron absorption is increased. Moreover, these patients require blood transfusions and are frequently treated inappropriately with iron. PCT, a disorder characterized by a defect in porphyrin biosynthesis (Chap. 430), can also be associated with excessive parenchymal iron deposits. The magnitude of the iron load in PCT is usually insufficient to produce tissue damage. However, some patients with PCT also have mutations in the HFE gene, and some have associated hepatitis C virus (HCV) infection. Although the relationship between these disorders remains to be clarified, iron overload accentuates the inherited enzyme deficiency in PCT and should be avoided along with other agents (alcohol, estrogens, haloaromatic compounds) that may exacerbate PCT. Another cause of hepatic parenchymal iron overload is hereditary aceruloplasminemia. In this disorder, impairment of iron mobilization due to deficiency of ceruloplasmin (a ferroxidase) causes iron overload in hepatocytes.
Excessive iron ingestion over many years rarely results in hemochromatosis. An important exception has been reported in South Africa among groups who brew fermented beverages in vessels made of iron. Hemochromatosis has been described in apparently normal persons who have taken medicinal iron over many years, but such individuals probably had genetic disorders.
The common denominator in all patients with hemochromatosis is excessive amounts of iron in parenchymal tissues. Parenteral administration of iron in the form of blood transfusions or iron preparations results predominantly in reticuloendothelial cell iron overload. This appears to lead to less tissue damage than iron loading of parenchymal cells.
In the liver, parenchymal iron is in the form of ferritin and hemosiderin. In the early stages, these deposits are seen in the periportal parenchymal cells, especially within lysosomes in the pericanalicular cytoplasm of the hepatocytes. This stage progresses to perilobular fibrosis and eventually to deposition of iron in bile-duct epithelium, Kupffer cells, and fibrous septa due to activation of stellate cells. In the advanced stage, a macronodular or mixed macro- and micronodular cirrhosis develops. Hepatic fibrosis and cirrhosis correlate significantly with hepatic iron concentration.
At autopsy, the enlarged nodular liver and pancreas are rusty in color. Histologically, iron is increased in many organs, particularly in the liver, heart, and pancreas, and, to a lesser extent, in the endocrine glands. The epidermis of the skin is thin, and melanin is increased in the cells of the basal layer and dermis. Deposits of iron are present around the synovial lining cells of the joints.
CLINICAL MANIFESTATIONS
C282Y homozygotes can be characterized by the stage of progression as follows: (1) a genetic predisposition without abnormalities; (2) iron overload without symptoms; (3) iron overload with symptoms (e.g., arthritis and fatigue); and (4) iron overload with organ damage—in particular, cirrhosis. Thus, many subjects with significant iron overload are asymptomatic. For example, in a study of 672 asymptomatic C282Y homozygous subjects—identified by either family screening or routine health examinations—there was hepatic iron overload (grades 2–4) in 56% and 34.5% of male and female subjects, respectively; hepatic fibrosis (stages 2–4) in 18.4% and 5.4%, respectively; and cirrhosis in 5.6% and 1.9%, respectively.
Initial symptoms are often nonspecific and include lethargy, arthralgia, change in skin color, loss of libido, and features of diabetes mellitus. Hepatomegaly, increased pigmentation, spider angiomas, splenomegaly, arthropathy, ascites, cardiac arrhythmias, congestive heart failure, loss of body hair, testicular atrophy, and jaundice are prominent in advanced disease.
The liver is usually the first organ to be affected, and hepatomegaly is present in more than 95% of symptomatic patients. Hepatic enlargement may exist in the absence of symptoms or of abnormal liver-function tests. Manifestations of portal hypertension and esophageal varices occur less commonly than in cirrhosis from other causes. Hepatocellular carcinoma develops in about 30% of patients with cirrhosis, and it is the most common cause of death in treated patients—hence the importance of early diagnosis and therapy. The incidence increases with age, it is more common in men, and it occurs almost exclusively in cirrhotic patients.
Excessive skin pigmentation is present in patients with advanced disease. The characteristic metallic or slate-gray hue is sometimes referred to as bronzing and results from increased melanin and iron in the dermis. Pigmentation usually is diffuse and generalized, but it may be more pronounced on the face, neck, extensor aspects of the lower forearms, dorsa of the hands, lower legs, and genital regions, as well as in scars.
Diabetes mellitus occurs in about 65% of patients with advanced disease and is more likely to develop in those with a family history of diabetes, suggesting that direct damage to the pancreatic islets by iron deposition occurs in combination with other risk factors. The management is similar to that of other forms of diabetes, although insulin resistance is more common in association with hemochromatosis. Late complications are the same as seen in other causes of diabetes mellitus.
Arthropathy develops in 25–50% of symptomatic patients. It usually occurs after age 50 but may occur as a first manifestation or long after therapy. The joints of the hands, especially the second and third metacarpophalangeal joints, are usually the first joints involved, a feature that helps to distinguish the chondrocalcinosis associated with hemochromatosis from the idiopathic form (Chap. 395). A progressive polyarthritis involving wrists, hips, ankles, and knees may also ensue. Acute brief attacks of synovitis may be associated with deposition of calcium pyrophosphate (chondrocalcinosis or pseudogout), mainly in the knees. Radiologic manifestations include cystic changes of the subchondral bones, loss of articular cartilage with narrowing of the joint space, diffuse demineralization, hypertrophic bone proliferation, and calcification of the synovium. The arthropathy tends to progress despite removal of iron by phlebotomy. Although the relation of these abnormalities to iron metabolism is not known, the fact that similar changes occur in other forms of iron overload suggests that iron is directly involved.
Cardiac involvement is the presenting manifestation in about 15% of symptomatic patients. The most common manifestation is congestive heart failure, which occurs in about 10% of young adults with the disease, especially those with juvenile hemochromatosis. Symptoms of congestive heart failure may develop suddenly, with rapid progression to death if untreated. The heart is diffusely enlarged; this may be misdiagnosed as idiopathic cardiomyopathy if other overt manifestations are absent. Cardiac arrhythmias include premature supraventricular beats, paroxysmal tachyarrhythmias, atrial flutter, atrial fibrillation, and varying degrees of atrioventricular block.
Hypogonadism occurs in both sexes and may antedate other clinical features. Manifestations include loss of libido, impotence, amenorrhea, testicular atrophy, gynecomastia, and sparse body hair. These changes are primarily the result of decreased production of gonadotropins due to impairment of hypothalamic-pituitary function by iron deposition. Adrenal insufficiency, hypothyroidism, and hypoparathyroidism are rare manifestations.
DIAGNOSIS
The association of (1) hepatomegaly, (2) skin pigmentation, (3) diabetes mellitus, (4) heart disease, (5) arthritis, and (6) hypogonadism should suggest the diagnosis. However, as stated above, significant iron overload may exist with none or only some of these manifestations. Therefore, a high index of suspicion is needed to make the diagnosis early. Treatment before permanent organ damage occurs can reverse the iron toxicity and restore life expectancy to normal.
The history should be particularly detailed in regard to disease in other family members; alcohol ingestion; iron intake; and ingestion of large doses of ascorbic acid, which promotes iron absorption (Chap. 96e). Appropriate tests should be performed to exclude iron deposition due to hematologic disease. The presence of liver, pancreatic, cardiac, and joint disease should be confirmed by physical examination, radiography, and standard function tests of these organs.
The degree of increase in total body iron stores can be assessed by (1) measurement of serum iron and the percent saturation of transferrin (or the unsaturated iron-binding capacity), (2) measurement of serum ferritin concentration, (3) liver biopsy with measurement of the iron concentration and calculation of the hepatic iron index (Table 428-2), and (4) magnetic resonance imaging (MRI) of the liver. In addition, a retrospective assessment of body-iron storage is also provided by performing weekly phlebotomy and calculating the amount of iron removed before iron stores are exhausted (1 mL blood = approximately 0.5 mg iron).
|
REPRESENTATIVE IRON VALUES IN NORMAL SUBJECTS, PATIENTS WITH HEMOCHROMATOSIS, AND PATIENTS WITH ALCOHOLIC LIVER DISEASE |
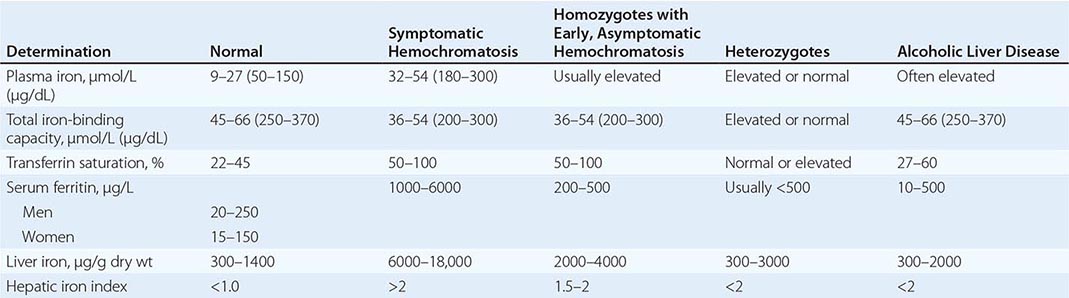
Each of these methods for assessing iron stores has advantages and limitations. The serum iron level and percent saturation of transferrin are elevated early in the course, but their specificity is reduced by significant false-positive and false-negative rates. For example, serum iron concentration may be increased in patients with alcoholic liver disease without iron overload; in this situation, however, the hepatic iron index is usually not increased as in hemochromatosis (Table 428-1). In otherwise healthy persons, a fasting serum transferrin saturation greater than 45% is abnormal and suggests homozygosity for hemochromatosis.
The serum ferritin concentration is usually a good index of body-iron stores, whether decreased or increased. In fact, an increase of 1 μg/L in serum ferritin level reflects an increase of about 5 mg in body stores. In most untreated patients with hemochromatosis, the serum ferritin level is significantly increased (Fig. 428-2 and Table 428-1), and a serum ferritin level >1000 μg/L is the strongest predictor of disease expression among individuals homozygous for the C282Y mutation. However, in patients with inflammation and hepatocellular necrosis, serum ferritin levels may be elevated out of proportion to body-iron stores due to increased release from tissues. Therefore, a repeat determination of serum ferritin should be carried out after acute hepatocellular damage has subsided (e.g., in alcoholic liver disease). Ordinarily, the combined measurements of the percent transferrin saturation and serum ferritin level provide a simple and reliable screening test for hemochromatosis, including the precirrhotic phase of the disease. If either of these tests is abnormal, genetic testing for hemochromatosis should be performed (Fig. 428-3).
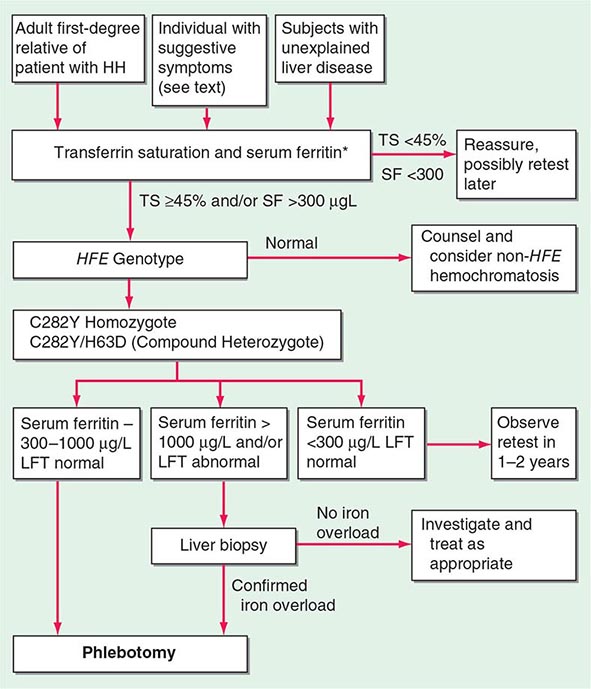
*For convenience both genotype and phenotype (iron tests) can be performed together at a single visit in first-degree relatives.
FIGURE 428-3 Algorithm for screening for HFE-associated hemochromatosis. HH, hereditary hemochromatosis, homozygous subject (C282Y +/+); LFT, liver function tests; SF, serum ferritin concentration; TS, transferrin saturation.
The role of liver biopsy in the diagnosis and management of hemochromatosis has been reassessed as a result of the widespread availability of genetic testing for the C282Y mutation. The absence of severe fibrosis can be accurately predicted in most patients using clinical and biochemical variables. Thus, there is virtually no risk of severe fibrosis in a C282Y homozygous subject with (1) serum ferritin level less than 1000 μg/L, (2) normal serum alanine aminotransferase values, (3) no hepatomegaly, and (4) no excess alcohol intake. However, it should be emphasized that liver biopsy is the only reliable method for establishing or excluding the presence of hepatic cirrhosis, which is the critical factor determining prognosis and the risk of developing hepatocellular carcinoma. Biopsy also permits histochemical estimation of tissue iron and measurement of hepatic iron concentration. Increased density of the liver due to iron deposition can be demonstrated by computed tomography (CT) or MRI, and with improved technology, MRI has become more accurate in determining hepatic iron concentration.
SCREENING FOR HEMOCHROMATOSIS
When the diagnosis of hemochromatosis is established, it is important to counsel and screen other family members (Chap. 84). Asymptomatic and symptomatic family members with the disease usually have an increased saturation of transferrin and an increased serum ferritin concentration. These changes occur even before the iron stores are greatly increased (Fig. 428-2). All adult first-degree relatives of patients with hemochromatosis should be tested for the C282Y and H63D mutations and counseled appropriately (Fig. 428-3). In affected individuals, it is important to confirm or exclude the presence of cirrhosis and begin therapy as early as possible. For children of an identified proband, testing for HFE of the other parent is helpful because if normal, the child is merely an obligate heterozygote and at no risk. Otherwise, for practical purposes, children need not be checked before they are 18 years old.
The role of population screening for hemochromatosis is controversial. Recent studies indicate that it is highly effective for primary care physicians to screen subjects using transferrin saturation and serum ferritin levels. Such screening also detects iron deficiency. Genetic screening of the normal population is feasible but is probably not cost effective.
|
TREATMENT |
HEMOCHROMATOSIS |
The therapy of hemochromatosis involves removal of the excess body iron and supportive treatment of damaged organs. Iron removal is best accomplished by weekly or twice-weekly phlebotomy of 500 mL. Although there is an initial modest decline in the volume of packed red blood cells to about 35 mL/dL, the level stabilizes after several weeks. The plasma transferrin saturation remains increased until the available iron stores are depleted. In contrast, the plasma ferritin concentration falls progressively, reflecting the gradual decrease in body-iron stores. One 500-mL unit of blood contains 200–250 mg of iron, and up to 25 g of iron or more may have to be removed. Therefore, in patients with advanced disease, weekly phlebotomy may be required for 1–2 years, and it should be continued until the serum ferritin level is <50 μg/L. Thereafter, phlebotomies are performed at appropriate intervals to maintain ferritin levels between 50 and 100 μg/L. Usually one phlebotomy every 3 months will suffice.
Chelating agents such as deferoxamine, when given parenterally, remove 10–20 mg of iron per day, which is much less than that mobilized by once-weekly phlebotomy. Phlebotomy is also less expensive, more convenient, and safer for most patients. However, chelating agents are indicated when anemia or hypoproteinemia is severe enough to preclude phlebotomy. Subcutaneous infusion of deferoxamine using a portable pump is the most effective means of its administration.
An effective oral iron chelating agent, deferasirox (Exjade), has recently become available but is still in clinical trials. This agent is effective in thalassemia and secondary iron overload, but its role in primary iron overload has yet to be established.
Alcohol consumption should be severely curtailed or eliminated because it increases the risk of cirrhosis in hereditary hemochromatosis nearly tenfold. Dietary adjustments are unnecessary, although vitamin C and iron supplements should be avoided. The management of hepatic failure, cardiac failure, and diabetes mellitus is similar to conventional therapy for these conditions. Loss of libido and change in secondary sex characteristics are managed with testosterone replacement or gonadotropin therapy (Chap. 411).
End-stage liver disease may be an indication for liver transplantation, although results are improved if the excess iron can be removed beforehand. The available evidence indicates that the fundamental metabolic abnormality in hemochromatosis is reversed by successful liver transplantation.
PROGNOSIS
The principal causes of death are cardiac failure, hepatocellular failure or portal hypertension, and hepatocellular carcinoma.
Life expectancy is improved by removal of the excessive stores of iron and maintenance of these stores at near-normal levels. The 5-year survival rate with therapy increases from 33 to 89%. With repeated phlebotomy, the liver decreases in size, liver function improves, pigmentation of skin decreases, and cardiac failure may be reversed. Diabetes improves in about 40% of patients, but removal of excess iron has little effect on hypogonadism or arthropathy. Hepatic fibrosis may decrease, but established cirrhosis is irreversible. Hepatocellular carcinoma occurs as a late sequela in patients who are cirrhotic at presentation. The apparent increase in its incidence in treated patients is probably related to their increased life span. Hepatocellular carcinoma rarely develops if the disease is treated in the precirrhotic stage. Indeed, the life expectancy of homozygotes treated before the development of cirrhosis is normal.
The importance of family screening and early diagnosis and treatment cannot be overemphasized. Asymptomatic individuals detected by family studies should have phlebotomy therapy if iron stores are moderately to severely increased. Assessment of iron stores at appropriate intervals is also important. With this management approach, most manifestations of the disease can be prevented.
ROLE OF HFE MUTATIONS IN OTHER LIVER DISEASES
![]() There is considerable interest in the role of HFE mutations and hepatic iron in several other liver diseases. Several studies have shown an increased prevalence of HFE mutations in PCT patients. Iron accentuates the inherited enzyme deficiency in PCT and clinical manifestations of PCT. The situation in nonalcoholic steatohepatitis (NASH) is less clear, but some studies have shown an increased prevalence of HFE mutations in NASH patients. The role of phlebotomy therapy, however, is unproven. In chronic HCV infection, HFE mutations are not more common, but some subjects have increased hepatic iron. Before initiating antiviral therapy in these patients, it is reasonable to perform phlebotomy therapy to remove excess iron stores, because this reduces liver enzyme levels.
There is considerable interest in the role of HFE mutations and hepatic iron in several other liver diseases. Several studies have shown an increased prevalence of HFE mutations in PCT patients. Iron accentuates the inherited enzyme deficiency in PCT and clinical manifestations of PCT. The situation in nonalcoholic steatohepatitis (NASH) is less clear, but some studies have shown an increased prevalence of HFE mutations in NASH patients. The role of phlebotomy therapy, however, is unproven. In chronic HCV infection, HFE mutations are not more common, but some subjects have increased hepatic iron. Before initiating antiviral therapy in these patients, it is reasonable to perform phlebotomy therapy to remove excess iron stores, because this reduces liver enzyme levels.
HFE mutations are not increased in frequency in alcoholic liver disease. Hemochromatosis in a heavy drinker can be distinguished from alcoholic liver disease by the presence of the C282Y mutation.
End-stage liver disease may also be associated with iron overload of the degree seen in hemochromatosis. The mechanism is uncertain, although studies have shown that alcohol suppresses hepatic hepcidin secretion. Hemolysis also plays a role. HFE mutations are uncommon.
A recent large population study has suggested that subjects homozygous for C282Y are at increased risk of breast and colorectal cancer.
GLOBAL CONSIDERATIONS
 The HFE mutation is of northern European origin (Celtic or Nordic) with a heterozygous carrier rate of approximately 1 in 10 (1 in 8 in Ireland). Thus, HFE-associated hemochromatosis is quite rare in non-European populations, e.g., Asia. However, non-HFE-associated hemochromatosis resulting from mutations in other genes involved in iron metabolism (Fig. 428-1) is ubiquitous and should be considered when one encounters iron overload.
The HFE mutation is of northern European origin (Celtic or Nordic) with a heterozygous carrier rate of approximately 1 in 10 (1 in 8 in Ireland). Thus, HFE-associated hemochromatosis is quite rare in non-European populations, e.g., Asia. However, non-HFE-associated hemochromatosis resulting from mutations in other genes involved in iron metabolism (Fig. 428-1) is ubiquitous and should be considered when one encounters iron overload.
429 |
Wilson’s Disease |
Wilson’s disease is an autosomal recessive disorder caused by mutations in the ATP7B gene, which encodes a membrane-bound, copper-transporting ATPase. Clinical manifestations are caused by copper toxicity and primarily involve the liver and the brain. Because effective treatment is available, it is important to make this diagnosis early.
The frequency of Wilson’s disease in most populations is about 1 in 30,000–40,000, and the frequency of carriers of ATP7B mutations is ∼1%. Siblings of a diagnosed patient have a 1 in 4 risk of Wilson’s disease, whereas children of an affected patient have about a 1 in 200 risk. Because a large number of inactivating mutations have been reported in the ATP7B gene, mutation screening for diagnosis is not routine, although this approach may be practical in the future. DNA haplotype analysis can be used to genotype siblings of an affected patient. A rare multisystem disorder of copper metabolism with features of both Menkes and Wilson’s diseases has been reported. It is termed the MEDNIK syndrome (mental retardation, enteropathy, deafness, neuropathy, ichthyosis, keratodermia) and is caused by mutations in the AP1S1 gene, which encodes an adaptor protein necessary for intracellular trafficking of copper pump proteins ATP7A (Menkes disease) and ATP7B (Wilson’s disease).
PATHOGENESIS
ATP7B protein deficiency impairs biliary copper excretion, resulting in positive copper balance, hepatic copper accumulation, and copper toxicity from oxidant damage. Excess hepatic copper is initially bound to metallothionein; liver damage begins as this storage capacity is exceeded, sometimes by 3 years of age. Defective copper incorporation into apoceruloplasmin leads to excess catabolism and low blood levels of ceruloplasmin. Serum copper levels are usually lower than normal because of low blood levels of ceruloplasmin, which normally binds >90% of serum copper. As the disease progresses, nonceruloplasmin serum copper (“free” copper) levels increase, resulting in copper buildup in other parts of the body (e.g., in the brain, with consequent neurologic and psychiatric disease).
CLINICAL PRESENTATION
Hepatic Features Wilson’s disease may present as hepatitis, cirrhosis, or hepatic decompensation. Patients typically present in the mid- to late teenage years in Western countries, although the age of presentation is quite broad and extends into the fifth decade of life. An episode of hepatitis may occur—with elevated serum aminotransferase levels, with or without jaundice—and then spontaneously regress. Hepatitis often recurs, and most of these patients eventually develop cirrhosis. Hepatic decompensation is associated with elevated serum bilirubin, reduced serum albumin and coagulation factors, ascites, peripheral edema, and hepatic encephalopathy. In severe hepatic failure, hemolytic anemia may develop because large amounts of copper derived from hepatocellular necrosis are released into the bloodstream. The association of hemolysis and liver disease makes Wilson’s disease a likely diagnosis.
Neurologic Features The neurologic manifestations of Wilson’s disease typically occur in patients in their early twenties, although the age of onset extends into the sixth decade of life. MRI and CT scans reveal damage in the basal ganglia and occasionally in the pons, medulla, thalamus, cerebellum, and subcortical areas. The three main movement disorders include dystonia, incoordination, and tremor. Dysarthria and dysphagia are common. In some patients, the clinical picture closely resembles that of Parkinson’s disease. Dystonia can involve any part of the body and eventually leads to grotesque positions of the limbs, neck, and trunk. Autonomic disturbances may include orthostatic hypotension and sweating abnormalities as well as bowel, bladder, and sexual dysfunction. Memory loss, migraine-type headaches, and seizures may occur. Patients have difficulty focusing on tasks, but cognition usually is not grossly impaired. Sensory abnormalities and muscular weakness are not features of the disease.
Psychiatric Features Half of patients with neurologic disease have a history of behavioral disturbances with onset in the 5 years before diagnosis. The features are diverse and may include loss of emotional control (temper tantrums, crying bouts), depression, hyperactivity, or loss of sexual inhibition.
Other Manifestations Some female patients have repeated spontaneous abortions, and most become amenorrheic prior to diagnosis. Cholelithiasis and nephrolithiasis occur with increased frequency. Some patients have osteoarthritis, particularly of the knee. Microscopic hematuria is common, and levels of urinary excretion of phosphates, amino acids, glucose, or urates may increase; however, a full-blown Fanconi syndrome is rare. Sunflower cataracts and Kayser-Fleischer rings (copper deposits in the outer rim of the cornea) may be seen. Electrocardiographic and other cardiac abnormalities have been reported but are not common.
DIAGNOSIS
Diagnostic tests for Wilson’s disease are listed in Table 429-1. Serum ceruloplasmin levels should not be used for definitive diagnosis, because they are normal in up to 10% of affected patients and are reduced in 20% of carriers. Kayser-Fleischer rings (Fig. 429-1) can be definitively diagnosed only by an ophthalmologist using a slit lamp. They are present in >99% of patients with neurologic/psychiatric forms of the disease and have been described very rarely in the absence of Wilson’s disease. Kayser-Fleischer rings are present in only ∼30–50% of patients diagnosed in the hepatic or presymptomatic state; thus, the absence of rings does not exclude the diagnosis.
|
USEFUL TESTS FOR WILSON’S DISEASE |
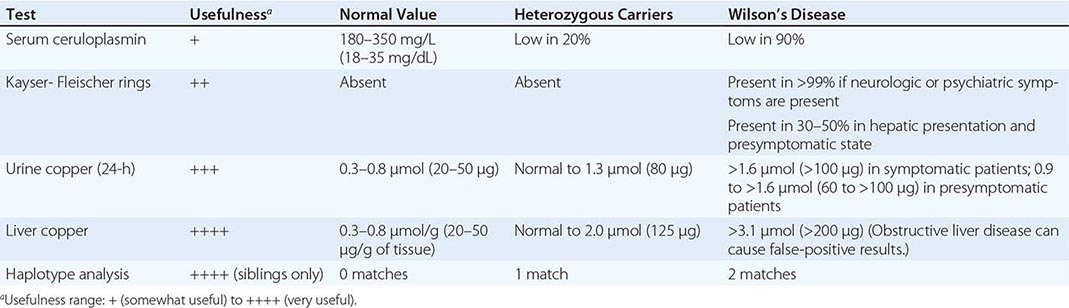
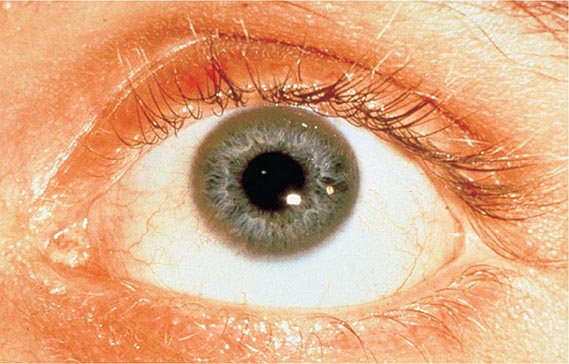
FIGURE 429-1 A Kayser-Fleischer ring. Although in this case, the brownish ring rimming the cornea is clearly visible to the naked eye, confirmation is usually made by slit-lamp examination.
Urine copper measurement is an important diagnostic tool, but urine must be collected carefully to avoid contamination. Symptomatic patients invariably have urine copper levels >1.6 μmol (>100 μg) per 24 h. Heterozygotes have values <1.3 μmol (<80 μg) per 24 h. About half of presymptomatic patients who are ultimately affected have diagnostically elevated urine copper values, but the other half have levels that are in an intermediate range between 0.9 and 1.6 μmol (60–100 μg) per 24 h. Because heterozygotes may have values up to 1.3 μmol (80 μg) per 24 h, patients in this range may require a liver biopsy for definitive diagnosis.
The gold standard for diagnosis remains liver biopsy with quantitative copper assays. Affected patients have values >3.1 μmol/g (>200 μg/g [dry weight] of liver). Copper stains are not reliable. False-positive results can occur with long-standing obstructive liver disease, which can elevate hepatic and urine copper concentrations and rarely causes Kayser-Fleischer rings.
|
TREATMENT |
WILSON’S DISEASE |
Recommended anticopper treatments are listed in Table 429-2. Penicillamine was previously the primary anticopper treatment but now plays only a minor role because of its toxicity and because it often worsens existing neurologic disease if used as initial therapy. If penicillamine is given, it should always be accompanied by pyridoxine (25 mg/d). Trientine is a less toxic chelator and is supplanting penicillamine when a chelator is indicated.
|
RECOMMENDED ANTICOPPER DRUGS FOR WILSON’S DISEASE |
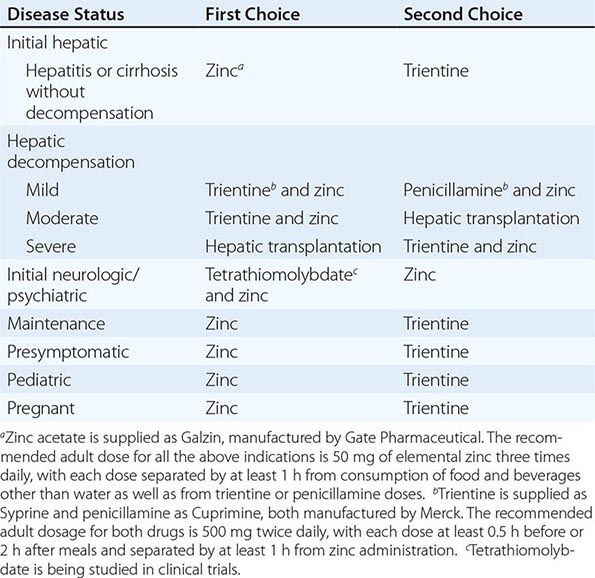
For patients with hepatitis or cirrhosis but without evidence of hepatic decompensation or neurologic/psychiatric symptoms, zinc is the therapy of choice although some experts advocate therapy with trientine. Zinc has proven efficacy in Wilson’s disease and is essentially nontoxic. It produces a negative copper balance by blocking intestinal absorption of copper, and it induces hepatic metallothionein synthesis, thereby sequestering additional toxic copper. All presymptomatic patients should be treated prophylactically because the disease is close to 100% penetrant.
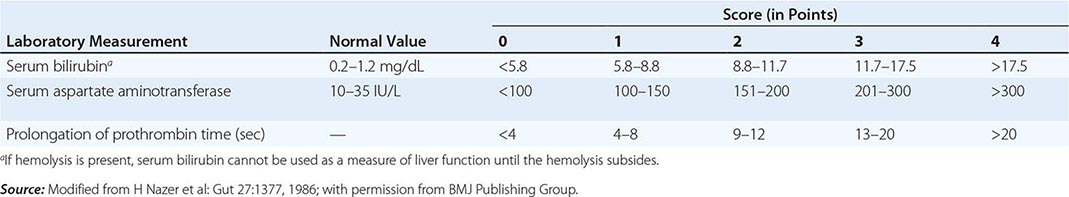
The first step in evaluating patients presenting with hepatic decompensation is to establish disease severity, which can be estimated with the Nazer prognostic index (Table 429-3). Patients with scores <7 can usually be managed with medical therapy. Patients with scores >9 should be considered immediately for liver transplantation. For patients with scores between 7 and 9, clinical judgment is required in deciding whether to recommend transplantation or medical therapy. A combination of trientine and zinc has been used to treat patients with Nazer scores as high as 9, but such patients should be watched carefully for indications of hepatic deterioration, which mandates transplantation.
|
PROGNOSTIC INDEX OF NAZER |
For initial medical treatment of patients with hepatic decompensation, the recommended regimen is a chelator (preferably trientine) plus zinc (Table 429-2). Zinc should not, however, be ingested simultaneously with trientine, which chelates zinc and forms therapeutically ineffective complexes. Administration of the two drugs should be separated by at least 1 h.
For initial neurologic therapy, tetrathiomolybdate is emerging as the drug of choice because of its rapid control of free copper, preservation of neurologic function, and low toxicity. Penicillamine and trientine should be avoided because both have a high risk of worsening the neurologic condition. Until tetrathiomolybdate is commercially available, zinc therapy is recommended. Although it is relatively slow-acting, zinc itself does not exacerbate neurologic abnormalities. Although hepatic transplantation may alleviate neurologic symptoms, it does so only by copper removal, which can be done more safely and inexpensively with anticopper drugs. Pregnant patients should be treated with zinc or trientine throughout pregnancy but without tight copper control because copper deficiency can be teratogenic.
Anticopper therapy must be lifelong. With treatment, liver function usually recovers after about a year although residual liver damage is usually present. Neurologic and psychiatric symptoms usually improve after 6–24 months of treatment.
MONITORING ANTICOPPER THERAPY
When trientine or penicillamine is first used, it is necessary to monitor for drug toxicity, particularly bone marrow suppression and proteinuria. Complete blood counts, standard biochemical profiles, and a urinalysis should be performed at weekly intervals for 1 month, then at twice-weekly intervals for 2 or 3 months, then at monthly intervals for 3 or 4 months, and at 4- to 6-month intervals thereafter.
The anticopper effects of trientine and penicillamine can be monitored by following 24-h “free” serum copper levels. Changes in urine copper levels are more difficult to interpret because excretion reflects the effect of the drug as well as body loading with copper. Free serum copper is calculated by subtracting the ceruloplasmin copper from the total serum copper. Each 10 mg/L (1 mg/dL) of ceruloplasmin contributes 0.5 μmol/L (3 μg/dL) of serum copper. The normal serum free copper value is 1.6–2.4 μmol/L (10–15 μg/dL); the level is often as high as 7.9 μmol/L (50 μg/dL) in untreated Wilson’s disease. With treatment, the serum free copper should be <3.9 μmol/L (<25 μg/dL).
Zinc treatment does not require monitoring of blood or urine for toxicity. Its only significant side effect is gastric burning or nausea in ∼10% of patients, usually with the first morning dose. This effect can be mitigated if the first dose is taken an hour after breakfast or if zinc is taken with a small amount of protein. Because zinc mainly affects stool copper, 24-h urine copper can be used to reflect body loading. The typical value in untreated symptomatic patients is >3.1 μmol (>200 μg) per 24 h. This level should decrease during the first 1–2 years of therapy to <2.0 μmol (<125 μg) per 24 h. A normal value (0.3–0.8 μmol [20–50 μg]) is rarely reached during the first decade of therapy and should raise concern about overtreatment (copper deficiency), the first sign of which is anemia and/or leukopenia.
GLOBAL CONSIDERATIONS
 The age of onset of clinical disease may be considerably younger in India and the Far East; in these regions, onset often occurs in children at only 5 or 6 years of age. The incidence of the disease may be increased in certain populations as a result of founder effects. For example, in Sardinia, the incidence may be 1 in 3000. In countries where penicillamine, trientine, and zinc acetate (as Galzin) are not available or are unaffordable, zinc salts such as gluconate or sulfate provide an alternative treatment option.
The age of onset of clinical disease may be considerably younger in India and the Far East; in these regions, onset often occurs in children at only 5 or 6 years of age. The incidence of the disease may be increased in certain populations as a result of founder effects. For example, in Sardinia, the incidence may be 1 in 3000. In countries where penicillamine, trientine, and zinc acetate (as Galzin) are not available or are unaffordable, zinc salts such as gluconate or sulfate provide an alternative treatment option.
430 |
The Porphyrias |
The porphyrias are metabolic disorders, each resulting from the deficiency of a specific enzyme in the heme biosynthetic pathway (Fig. 430-1 and Table 430-1). These enzyme deficiencies are inherited as autosomal dominant, autosomal recessive, or X-linked traits, with the exception of porphyria cutanea tarda (PCT), which usually is sporadic (Table 430-1). The porphyrias are classified as either hepatic or erythropoietic, depending on the primary site of overproduction and accumulation of their respective porphyrin precursors or porphyrins (Tables 430-1 and 430-2), although some have overlapping features. For example, PCT, the most common porphyria, is hepatic and presents with blistering cutaneous photosensitivity, which is typically characteristic of the erythropoietic porphyrias. The major manifestations of the acute hepatic porphyrias are neurologic, including neuropathic abdominal pain, peripheral motor neuropathy, and mental disturbances, with attacks often precipitated by dieting, certain drugs, and hormonal changes. While hepatic porphyrias are symptomatic primarily in adults, rare homozygous variants of the autosomal dominant hepatic porphyrias usually manifest clinically prior to puberty.
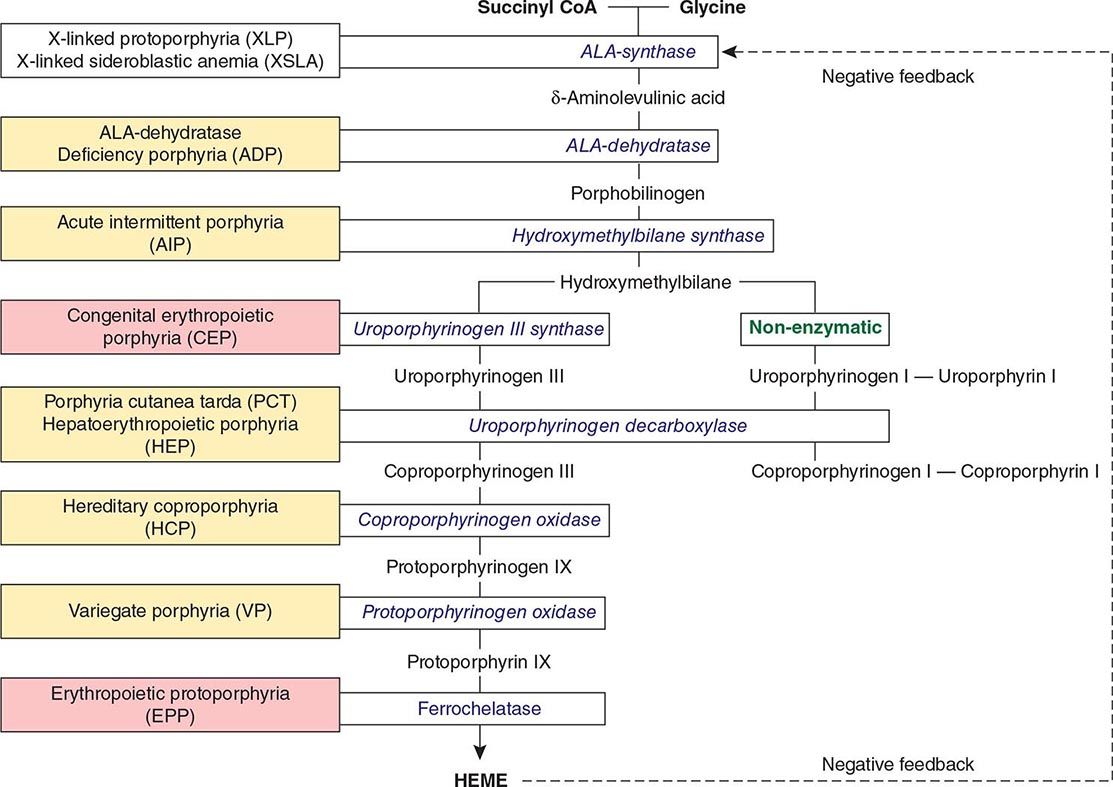
FIGURE 430-1 The human heme biosynthetic pathway indicating in linked boxes the enzyme that, when deficient, causes the respective porphyria. Hepatic porphyrias are shown in yellow boxes and erythropoietic porphyrias in pink boxes.
|
HUMAN PORPHYRIAS: MAJOR CLINICAL AND LABORATORY FEATURES |
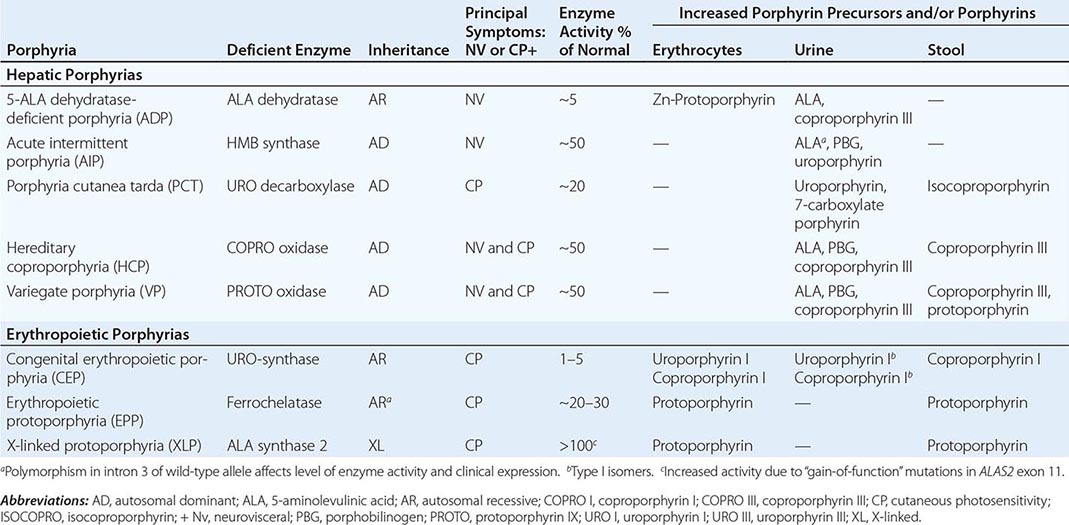
|
HUMAN HEME BIOSYNTHETIC ENZYMES AND GENES |
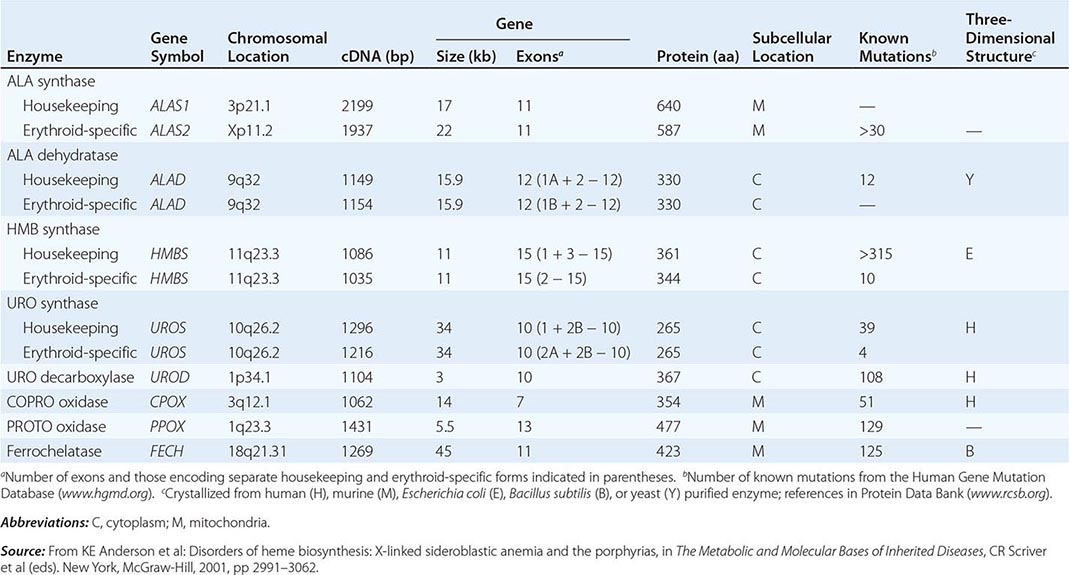
In contrast, the erythropoietic porphyrias usually present at birth or in early childhood with cutaneous photosensitivity, or in the case of congenital erythropoietic porphyria (CEP), even in utero as nonimmune hydrops fetalis. Cutaneous sensitivity to sunlight results from excitation of excess porphyrins in the skin by long-wave ultraviolet light, leading to cell damage, scarring, and disfigurement. Thus, the porphyrias are metabolic disorders in which environmental, physiologic, and genetic factors interact to cause disease.
Because many symptoms of the porphyrias are nonspecific, diagnosis is often delayed. Laboratory measurement of porphyrin precursors (5′-aminolevulinic acid [ALA] and porphobilinogen [PBG]) or porphyrins in urine, plasma, erythrocytes, or feces is required to confirm or exclude the various types of porphyria (see below). However, a definite diagnosis requires demonstration of the specific gene defect (Table 430-3). The genes encoding all the heme biosynthetic enzymes have been characterized, permitting identification of the mutations causing each porphyria (Table 430-2). Molecular genetic analyses now make it possible to provide precise heterozygote or homozygote identification and prenatal diagnoses in families with known mutations.
|
DIAGNOSIS OF ACUTE AND CUTANEOUS PORPHYRIAS |
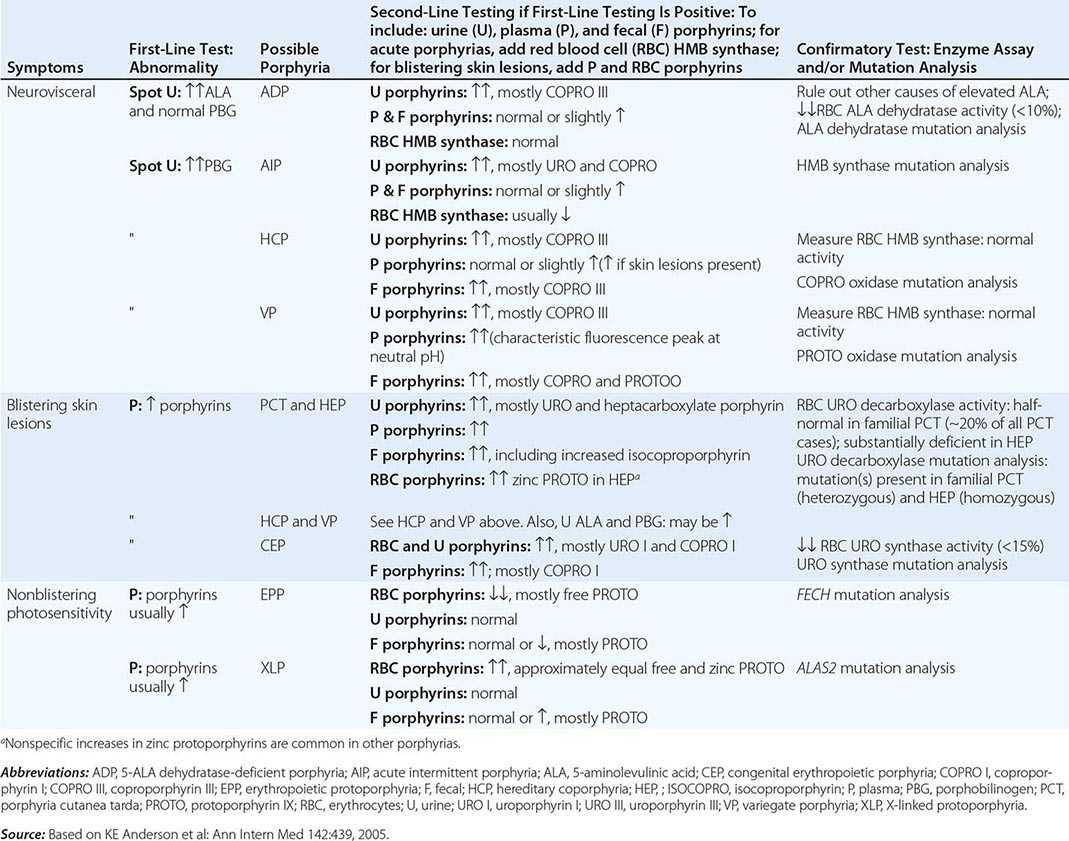
In addition to recent reviews of the porphyrias, informative and up-to-date websites are sponsored by the American Porphyria Foundation (www.porphyriafoundation.com) and the European Porphyria Initiative (www.porphyria-europe.org). An extensive list of unsafe and safe drugs for individuals with acute porphyrias is provided at the Drug Database for Acute Porphyrias (www.drugs-porphyria.com).
GLOBAL CONSIDERATIONS
 The porphyrias are panethnic metabolic diseases that affect individuals around the globe. The acute hepatic porphyrias—acute intermittent porphyria (AIP), hereditary coproporphyria (HCP), and variegate porphyria (VP)—are autosomal dominant disorders. The frequency of AIP, the most common acute hepatic porphyria, is ~1 in 20,000 among Caucasian individuals of Western European ancestry, and it is particularly frequent in Scandinavians, with a frequency of ~1 in 10,000 in Sweden. VP is particularly frequent in South Africa, where its high prevalence (>10,000 affected patients) is in part due to a genetic “founder effect.” The autosomal recessive acute hepatic porphyria, ALA dehydratase-deficient porphyria (ADP), is very rare, and less than 20 patients have been identified worldwide.
The porphyrias are panethnic metabolic diseases that affect individuals around the globe. The acute hepatic porphyrias—acute intermittent porphyria (AIP), hereditary coproporphyria (HCP), and variegate porphyria (VP)—are autosomal dominant disorders. The frequency of AIP, the most common acute hepatic porphyria, is ~1 in 20,000 among Caucasian individuals of Western European ancestry, and it is particularly frequent in Scandinavians, with a frequency of ~1 in 10,000 in Sweden. VP is particularly frequent in South Africa, where its high prevalence (>10,000 affected patients) is in part due to a genetic “founder effect.” The autosomal recessive acute hepatic porphyria, ALA dehydratase-deficient porphyria (ADP), is very rare, and less than 20 patients have been identified worldwide.
The erythropoietic protoporphyrias—CEP, erythropoietic protoporphyria (EPP), and X-linked protoporphyria (XLP)—also are panethnic. EPP is the most common porphyria in children, whereas CEP is very rare, with about 200 reported cases worldwide. The frequency of EPP varies globally because most patients have the common low expression FECH mutation that varies in frequency in different populations. It rarely occurs in Africans, is present in about 10% of whites, and is frequent (~30%) in the Japanese.
The autosomal recessive porphyrias—ADP, CEP, EPP, and hepatoerythropoietic porphyria (HEP)—are more frequent in regions with high rates of consanguineous unions. PCT, which is typically sporadic, occurs more frequently in countries in which its predisposing risk factors such as hepatitis C and HIV are more prevalent.
HEME BIOSYNTHESIS
Heme biosynthesis involves eight enzymatic steps in the conversion of glycine and succinyl-CoA to heme (Fig. 430-2 and Table 430-2). These eight enzymes are encoded by nine genes, as the first enzyme in the pathway, 5′-aminolevulinate synthase (ALA synthase), has two genes that encode unique housekeeping (ALAS1) and erythroid-specific (ALAS2) isozymes. The first and last three enzymes in the pathway are located in the mitochondrion, whereas the other four are in the cytosol. Heme is required for a variety of hemoproteins such as hemoglobin, myoglobin, respiratory cytochromes, and the cytochrome P450 enzymes (CYPs). Hemoglobin synthesis in erythroid precursor cells accounts for approximately 85% of daily heme synthesis in humans. Hepatocytes account for most of the rest, primarily for the synthesis of CYPs, which are especially abundant in the liver endoplasmic reticulum, and turn over more rapidly than many other hemoproteins, such as the mitochondrial respiratory cytochromes. As shown in Fig. 430-2, pathway intermediates are the porphyrin precursors, ALA and PBG, and porphyrins (mostly in their reduced forms, known as porphyrinogens). At least in humans, these intermediates do not accumulate in significant amounts under normal conditions or have important physiologic functions.
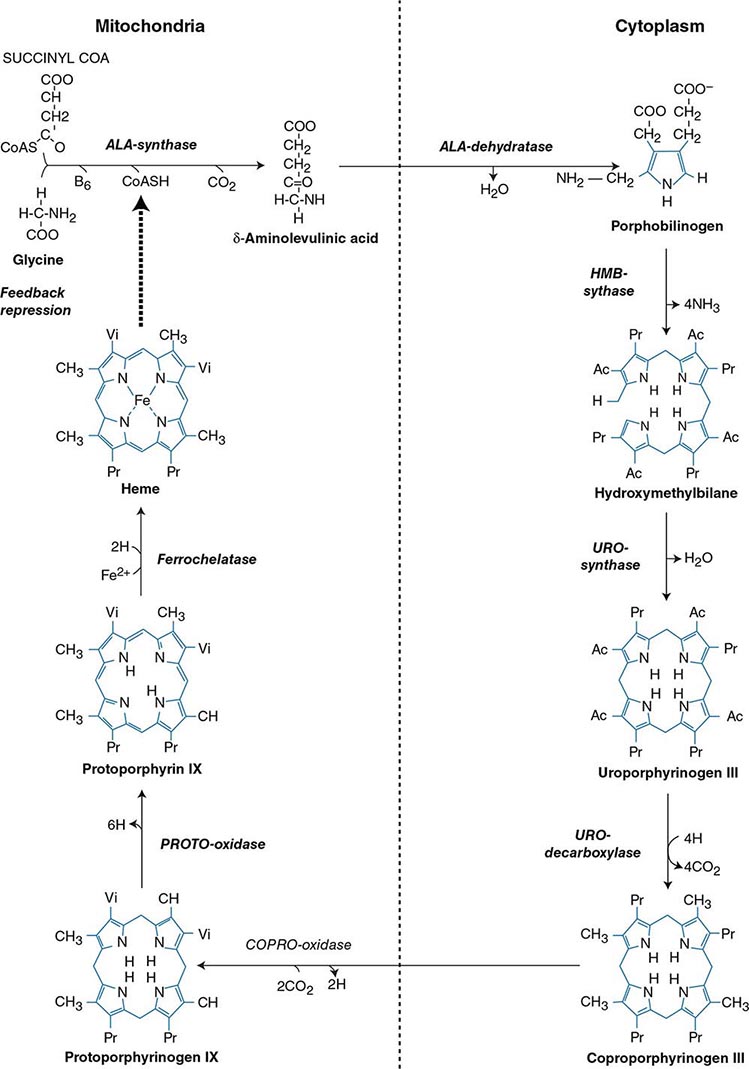
FIGURE 430-2 The heme biosynthetic pathway showing the eight enzymes and their substrates and products. Four of the enzymes are localized in the mitochondria and four in the cytosol.
The first enzyme, ALA synthase, catalyzes the condensation of glycine, activated by pyridoxal phosphate and succinyl coenzyme A, to form ALA. In the liver, this rate-limiting enzyme can be induced by a variety of drugs, steroids, and other chemicals. Distinct nonerythroid (e.g., housekeeping) and erythroid-specific forms of ALA synthase are encoded by separate genes located on chromosome 3p21.1 (ALAS1) and Xp11.2 (ALAS2), respectively. Defects in the erythroid gene ALAS2 that decrease its activity cause X-linked sideroblastic anemia (XLSA). Recently, gain-of-function mutations in the last exon (11) of ALAS2 that increase its activity have been shown to cause an X-linked form of EPP, known as X-linked protoporphyria (XLP).
The second enzyme, ALA dehydratase, catalyzes the condensation of two molecules of ALA to form PBG. Hydroxymethylbilane synthase (HMB synthase; also known as PBG deaminase) catalyzes the head-to-tail condensation of four PBG molecules by a series of deaminations to form the linear tetrapyrrole, HMB. Uroporphyrinogen III synthase (URO synthase) catalyzes the rearrangement and rapid cyclization of HMB to form the asymmetric, physiologic, octacarboxylate porphyrinogen, uroporphyrinogen (URO’gen) III.
The fifth enzyme in the pathway, uroporphyrinogen decarboxylase (URO decarboxylase), catalyzes the sequential removal of the four carboxyl groups from the acetic acid side chains of URO’gen III to form coproporphyrinogen (COPRO’gen) III, a tetracarboxylate porphyrinogen. This compound then enters the mitochondrion via a specific transporter, ABCB6, where COPRO oxidase, the sixth enzyme, catalyzes the decarboxylation of two of the four propionic acid groups to form the two vinyl groups of protoporphyrinogen (PROTO’gen) IX, a decarboxylate porphyrinogen. Next, PROTO oxidase oxidizes PROTO gen to protoporphyrin IX by the removal of six hydrogen atoms. The product of the reaction is a porphyrin (oxidized form), in contrast to the preceding tetrapyrrole intermediates, which are porphyrinogens (reduced forms). Finally, ferrous iron is inserted into protoporphyrin to form heme, a reaction catalyzed by the eighth enzyme in the pathway, ferrochelatase (also known as heme synthetase or protoheme ferrolyase).
REGULATION OF HEME BIOSYNTHESIS
Regulation of heme synthesis differs in the two major heme-forming tissues, the liver and erythron. In the liver, the concentration of “free” heme regulates the synthesis and mitochondrial translocation of the housekeeping form of ALA synthase 1. Heme represses the synthesis of the ALA synthase 1 mRNA and interferes with the transport of the enzyme from the cytosol into mitochondria. Hepatic ALA synthase 1 is increased by many of the same chemicals that induce the cytochrome P450 enzymes in the endoplasmic reticulum of the liver. Because most of the heme in the liver is used for the synthesis of cytochrome P450 enzymes, hepatic ALA synthase 1 and the cytochrome P450s are regulated in a coordinated fashion, and many drugs that induce hepatic ALA synthase 1 also induce the CYP genes. The other hepatic heme biosynthetic enzymes are presumably expressed at constant levels, although their relative activities and kinetic properties differ. For example, normal individuals have high activities of ALA dehydratase, but low activities of HMB synthase, the latter being the second rate-limiting step in the pathway.
In the erythron, novel regulatory mechanisms allow for the production of the very large amounts of heme needed for hemoglobin synthesis. The response to stimuli for hemoglobin synthesis occurs during cell differentiation, leading to an increase in cell number. In contrast, the erythroid-specific ALA synthase 2 is expressed at higher levels than the housekeeping enzyme, and erythroid-specific control mechanisms regulate other pathway enzymes as well as iron transport into erythroid cells. Separate erythroid-specific and nonerythroid or “housekeeping” transcripts are known for the first four enzymes in the pathway. As noted above, housekeeping and erythroid-specific ALA synthases are encoded by genes on different chromosomes, but for each of the next three genes in the pathway, both erythroid and nonerythroid transcripts are transcribed by alternative promoters from their single respective genes (Table 430-2).
CLASSIFICATION OF THE PORPHYRIAS
As mentioned above, the porphyrias can be classified as either hepatic or erythropoietic, depending on whether the heme biosynthetic intermediates that accumulate arise initially from the liver or developing erythrocytes, or as acute or cutaneous, based on their clinical manifestations. Table 430-1 lists the porphyrias, their principal symptoms, and major biochemical abnormalities. Four of the five hepatic porphyrias—AIP, HCP, VP, and ADP—present during adult life with acute attacks of neurologic manifestations and elevated levels of one or both of the porphyrin precursors, ALA and PBG, and are thus classified as acute porphyrias. Patients with ADP have presented in infancy and adolescence. The fifth hepatic disorder, PCT, presents with blistering skin lesions. HCP and VP also may have cutaneous manifestations similar to PCT.
The erythropoietic porphyrias—CEP, EPP, and the recently described XLP—are characterized by elevations of porphyrins in bone marrow and erythrocytes and present with cutaneous photosensitivity. The skin lesions in CEP resemble PCT but are usually much more severe, whereas EPP and XLP cause a more immediate, painful, and nonblistering type of photosensitivity. EPP is the most common porphyria to cause symptoms before puberty. Around 20% of EPP patients develop minor abnormalities of liver function, with up to about 5% developing hepatic complications that can become life-threatening. XLP has a clinical presentation similar to EPP causing photosensitivity and liver disease.
DIAGNOSIS OF PORPHYRIA
A few specific and sensitive first-line laboratory tests should be used whenever symptoms or signs suggest the diagnosis of porphyria (Table 430-3). If a first-line test is significantly abnormal, more comprehensive testing should follow to establish the type of porphyria, including the specific causative gene mutation.
Acute Porphyrias An acute porphyria should be suspected in patients with neurovisceral symptoms after puberty, such as abdominal pain, and when the initial clinical evaluation does not suggest another cause. The urinary porphyrin precursors (ALA and PBG) should be measured (Fig. 430-2). Urinary PBG is virtually always increased during acute attacks of AIP, HCP, and VP and is not substantially increased in any other medical condition. Therefore, this measurement is both sensitive and specific. A method for rapid, in-house testing for urinary PBG, such as the Trace PBG kit (Thermo Scientific), can be used. Results from spot (single-void) urine specimens are highly informative because very substantial increases in PBG are expected during acute attacks of porphyria. A 24-h collection can unnecessarily delay diagnosis. The same spot urine specimen should be saved for quantitative determination of ALA, PBG, and creatinine, in order to confirm the qualitative PBG result and also to detect patients with ADP. Urinary porphyrins may remain increased longer than porphyrin precursors in HCP and VP. Therefore, it is useful to measure total urinary porphyrins in the same sample, keeping in mind that urinary porphyrin increases are often nonspecific. Measurement of urinary porphyrins alone should be avoided for screening, because these may be increased in disorders other than porphyrias, such as chronic liver disease, and misdiagnoses of porphyria can result from minimal increases in urinary porphyrins that have no diagnostic significance. Measurement of erythrocyte HMB synthase is not useful as a first-line test. Moreover, the enzyme activity is not decreased in all AIP patients, a borderline low normal value is not diagnostic, and the enzyme is not deficient in other acute porphyrias.
Cutaneous Porphyrias Blistering skin lesions due to porphyria are virtually always accompanied by increases in total plasma porphyrins. A fluorometric method is preferred, because the porphyrins in plasma in VP are mostly covalently linked to plasma proteins and may be less readily detected by high-performance liquid chromatography (HPLC). The normal range for plasma porphyrins is somewhat increased in patients with end-stage renal disease.
Although a total plasma porphyrin determination will usually detect EPP and XLP, an erythrocyte protoporphyrin determination is more sensitive. Increases in erythrocyte protoporphyrin occur in many other conditions. Therefore, the diagnosis of EPP must be confirmed by showing a predominant increase in free protoporphyrin rather than zinc protoporphyrin. In XLP, both free and zinc protoporphyrin are markedly increased in approximately equal proportions. Interpretation of laboratory reports can be difficult, because the term free erythrocyte protoporphyrin sometimes actually represents zinc protoporphyrin.
More extensive testing is justified when an initial test is positive. A substantial increase in PBG may be due to AIP, HCP, or VP. These acute porphyrias can be distinguished by measuring urinary porphyrins (using the same spot urine sample), fecal porphyrins, and plasma porphyrins. Assays for COPRO oxidase or PROTO oxidase are not widely available. More specifically, mutation analysis by sequencing the genes encoding HMB synthase, COPRO oxidase, and PROTO oxidase will detect almost all disease-causing mutations, and will be diagnostic even when the levels of urinary ALA and PBG have returned to normal or near normal. The various porphyrias that cause blistering skin lesions are differentiated by measuring porphyrins in urine, feces, and plasma. These porphyrias also should be confirmed at the DNA level by the demonstration of the causative gene mutation(s). It is often difficult to diagnose or “rule out” porphyria in patients who have had suggestive symptoms months or years in the past, and in relatives of patients with acute porphyrias, because porphyrin precursors and porphyrins may be normal. In those situations, detection of the specific gene mutation in the index case can make the diagnosis and facilitate the diagnosis and genetic counseling of at-risk relatives. Consultation with a specialist laboratory and physician will assist in selecting the heme biosynthetic gene or genes to be sequenced.
THE HEPATIC PORPHYRIAS
Markedly elevated plasma and urinary concentrations of the porphyrin precursors, ALA and/or PBG, which originate from the liver, are especially evident during attacks of neurologic manifestations of the four acute porphyrias—ADP, AIP, HCP, and VP. In PCT, excess porphyrins also accumulate initially in the liver and cause chronic blistering of sun-exposed areas of the skin.
ALA DEHYDRATASE-DEFICIENT PORPHYRIA (ADP)
ADP is a rare autosomal recessive acute hepatic porphyria caused by a severe deficiency of ALA dehydratase activity. To date, there are only a few documented cases, some in children or young adults, in which specific gene mutations have been identified. These affected homozygotes had <10% of normal ALA dehydratase activity in erythrocytes, but their clinically asymptomatic parents and heterozygous relatives had about half-normal levels of activity and did not excrete increased levels of ALA. The frequency of ADP is unknown, but the frequency of heterozygous individuals with <50% normal ALA dehydratase activity was ~2% in a screening study in Sweden. Because there are multiple causes for deficient ALA dehydratase activity, it is important to confirm the diagnosis of ADP by mutation analysis.
Clinical Features The clinical presentation depends on the amount of residual ALA dehydratase activity. Four of the documented patients were male adolescents with symptoms resembling those of AIP, including abdominal pain and neuropathy. One patient was an infant with more severe disease, including failure to thrive beginning at birth. The earlier age of onset and more severe manifestations in this patient reflect a more significant deficiency of ALA dehydratase activity. Another patient developed an acute motor polyneuropathy at age 63 that was associated with a myeloproliferative disorder. He was heterozygous for an ALAD mutation that presumably was present in erythroblasts that underwent clonal expansion due to the bone marrow malignancy.
Diagnosis All patients had significantly elevated levels of plasma and urinary ALA and urinary coproporphyrin (COPRO) III; ALAD activities in erythrocytes were <10% of normal. Hereditary tyrosinemia type 1 (fumarylacetoacetase deficiency) and lead intoxication should be considered in the differential diagnosis because either succinylacetone (which accumulates in hereditary tyrosinemia and is structurally similar to ALA) or lead can inhibit ALA dehydratase, increase urinary excretion of ALA and COPRO III, and cause manifestations that resemble those of the acute porphyrias. Heterozygotes are clinically asymptomatic and do not excrete increased levels of ALA but can be detected by demonstration of intermediate levels of erythrocyte ALA dehydratase activity or a specific mutation in the ALAD gene. To date, molecular studies of ADP patients have identified nine point mutations, two splice-site mutations, and a two-base deletion in the ALAD gene (Human Gene Mutation Database; www.hgmd.org). The parents in each case were not consanguineous, and the index cases had inherited a different ALAD mutation from each parent. Prenatal diagnosis of this disorder is possible by determination of ALA dehydratase activity and/or gene mutations in cultured chorionic villi or amniocytes.
|
TREATMENT |
ALA-DEHYDRATASE DEFICIENT PORPHYRIA |
The treatment of ADP acute attacks is similar to that of AIP (see below). The severely affected infant referred to above was supported by hyperalimentation and periodic blood transfusions but did not respond to intravenous hemin and died after liver transplantation.
ACUTE INTERMITTENT PORPHYRIA (AIP)
This hepatic porphyria is an autosomal dominant condition resulting from the half-normal level of HMB synthase activity. The disease is widespread but is especially common in Scandinavia and Great Britain. Clinical expression is highly variable, and activation of the disease is often related to environmental or hormonal factors, such as drugs, diet, and steroid hormones. Attacks can be prevented by avoiding known precipitating factors. Rare homozygous dominant AIP also has been described in children (see below).
Clinical Features Induction of the rate-limiting hepatic enzyme ALA synthase in heterozygotes who have half-normal HMB synthase activity is thought to underlie the acute attacks in AIP. The disorder remains latent (or asymptomatic) in the great majority of those who are heterozygous for HMBS mutations, and this is almost always the case prior to puberty. In patients with no history of acute symptoms, porphyrin precursor excretion is usually normal, suggesting that half-normal hepatic HMB synthase activity is sufficient and that hepatic ALA synthase activity is not increased. However, under conditions where heme synthesis is increased in the liver, half-normal HMB synthase activity may become limiting, and ALA, PBG, and other heme pathway intermediates may accumulate and be excreted in the urine. Common precipitating factors include endogenous and exogenous steroids, porphyrinogenic drugs, alcohol ingestion, and low-calorie diets, usually instituted for weight loss.
The fact that AIP is almost always latent before puberty suggests that adult levels of steroid hormones are important for clinical expression. Symptoms are more common in women, suggesting a role for estrogens or progestins. Premenstrual attacks are probably due to endogenous progesterone. Acute porphyrias are sometimes exacerbated by exogenous steroids, including oral contraceptive preparations containing progestins. Surprisingly, pregnancy is usually well tolerated, suggesting that beneficial metabolic changes may ameliorate the effects of high levels of progesterone. Table 430-4 provides a partial list of the major drugs that are harmful in AIP (and also in HCP and VP). Extensive lists of unsafe and safe drugs are available on websites sponsored by the American Porphyria Foundation (www.porphyriafoundation.com) and the European Porphyria Initiative (www.porphyria-europe.org), and at the Drug Database for Acute Porphyrias website (www.drugs-porphyria.com). Reduced intake of calories and carbohydrate, as may occur with illness or attempts to lose weight, can also increase porphyrin precursor excretion and induce attacks of porphyria. Increased carbohydrate intake may ameliorate attacks. Studies in a knockout AIP mouse model indicate that the hepatic ALAS1 gene is regulated by the peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α). Hepatic PGC-1α is induced by fasting, which in turn activates ALAS1 transcription, resulting in increased heme biosynthesis. This finding suggests an important link between nutritional status and the attacks in acute porphyrias. Attacks also can be provoked by infections, surgery, and ethanol.
|
UNSAFE DRUGS IN PORPHYRIA |
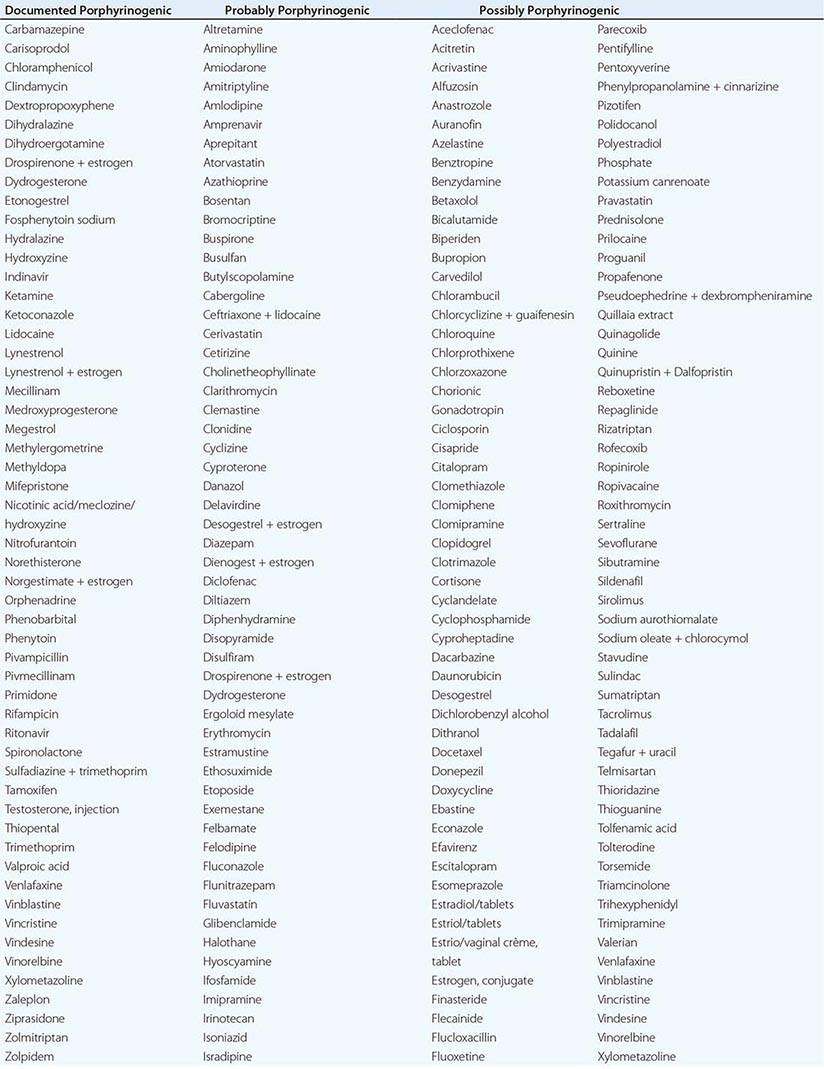

Because the neurovisceral symptoms rarely occur before puberty and are often nonspecific, a high index of suspicion is required to make the diagnosis. The disease can be disabling but is rarely fatal. Abdominal pain, the most common symptom, is usually steady and poorly localized but may be cramping. Ileus, abdominal distention, and decreased bowel sounds are common. However, increased bowel sounds and diarrhea may occur. Abdominal tenderness, fever, and leukocytosis are usually absent or mild because the symptoms are neurologic rather than inflammatory. Nausea; vomiting; constipation; tachycardia; hypertension; mental symptoms; pain in the limbs, head, neck, or chest; muscle weakness; sensory loss; dysuria; and urinary retention are characteristic. Tachycardia, hypertension, restlessness, tremors, and excess sweating are due to sympathetic overactivity.
The peripheral neuropathy is due to axonal degeneration (rather than demyelinization) and primarily affects motor neurons. Significant neuropathy does not occur with all acute attacks; abdominal symptoms are usually more prominent. Motor neuropathy affects the proximal muscles initially, more often in the shoulders and arms. The course and degree of involvement are variable and sometimes may be focal and involve cranial nerves. Deep tendon reflexes initially may be normal or hyperactive but become decreased or absent as the neuropathy advances. Sensory changes such as paresthesia and loss of sensation are less prominent. Progression to respiratory and bulbar paralysis and death occurs especially when the diagnosis and treatment are delayed. Sudden death may result from sympathetic overactivity and cardiac arrhythmia.
Mental symptoms such as anxiety, insomnia, depression, disorientation, hallucinations, and paranoia can occur in acute attacks. Seizures can be due to neurologic effects or to hyponatremia. Treatment of seizures is difficult because most antiseizure drugs can exacerbate AIP (clonazepam may be safer than phenytoin or barbiturates). Hyponatremia results from hypothalamic involvement and inappropriate vasopressin secretion or from electrolyte depletion due to vomiting, diarrhea, poor intake, or excess renal sodium loss. Persistent hypertension and impaired renal function may occur. When an attack resolves, abdominal pain may disappear within hours, and paresis begins to improve within days and may continue to improve over several years.
Homozygous dominant AIP is a rare form of AIP in which patients inherit HMBS mutations from each of their heterozygous parents and, therefore, have very low (<2%) enzyme activity. The disease has been described in a Dutch girl, two young British siblings, and a Spanish boy. In these homozygous affected patients, the disease presented in infancy with failure to thrive, developmental delay, bilateral cataracts, and/or hepatosplenomegaly. Urinary ALA and PBG concentrations were markedly elevated. All of these patients’ HMBS mutations (R167W, R167Q, and R172Q) were in exon 10 within five bases of each other. Studies of the brain magnetic resonance images (MRIs) of children with homozygous AIP have suggested damage primarily in white matter that was myelinated postnatally, while tracks that myelinated prenatally were normal. Most children with homozygous AIP die at an early age.
Diagnosis ALA and PBG levels are substantially increased in plasma and urine, especially during acute attacks, and become normal only after prolonged latency. For example, urinary PBG excretion during an attack is usually 50–200 mg/24 h (220–880 μmol/24 h) (normal, 0–4 mg/24 h, [0–18 μmol/24 h]), and urinary ALA excretion is 20–100 mg/24 h (150–760 μmol/24 h) (normal, 1–7 mg/24 h [8–53 μmol/24 h]). Because levels often remain high after symptoms resolve, the diagnosis of an acute attack in a patient with biochemically proven AIP is based primarily on clinical features. Excretion of ALA and PBG decreases over a few days after intravenous hemin administration. A normal urinary PBG level before hemin effectively excludes AIP as a cause for current symptoms. Fecal porphyrins are usually normal or minimally increased in AIP, in contrast to HCP and VP. Most AIP heterozygotes with no history of symptoms have normal urinary excretion of ALA and PBG. Therefore, the detection of the family’s HMBS mutation will diagnose asymptomatic family members.
Patients with HMBS mutations in the initiation of translation codon in exon 1 and in the intron 15′-splice donor site have normal enzyme levels in erythrocytes and deficient activity only in nonerythroid tissues. This occurs because the erythroid and housekeeping forms of HMB synthase are encoded by a single gene, which has two promoters. Thus, the enzyme assay may not be diagnostic, and genetic testing should be used to confirm the diagnosis.
More than 390 HMBS mutations have been identified in AIP, including missense, nonsense, and splicing mutations and insertions and deletions, with most mutations found in only one or a few families (Human Gene Mutation Database, www.hgmd.org). The prenatal diagnosis of a fetus at risk can be made with cultured amniotic cells or chorionic villi. However, this is seldom done, because the prognosis of individuals with HMBS mutations is generally favorable.
|
TREATMENT |
ACUTE INTERMITTENT PORPHYRIA |
During acute attacks, narcotic analgesics may be required for abdominal pain, and phenothiazines are useful for nausea, vomiting, anxiety, and restlessness. Chloral hydrate can be given for insomnia, and benzodiazepines are probably safe in low doses if a minor tranquilizer is required. Carbohydrate loading, usually with intravenous glucose (at least 300 g daily), may be effective in milder acute attacks of porphyria (without paresis, hyponatremia, etc.) if hemin is not available. Intravenous hemin is more effective and should be used as first-line therapy for all acute attacks. The standard regimen is 3–4 mg/kg of heme, in the form of lyophilized hematin (Recordati Pharmaceuticals), heme albumin (hematin reconstituted with human albumin), or heme arginate (Orphan Europe), infused daily for 4 days. Heme arginate and heme albumin are chemically stable and are less likely than hematin to produce phlebitis or an anticoagulant effect. Recovery depends on the degree of neuronal damage and usually is rapid if therapy is started early. Recovery from severe motor neuropathy may require months or years. Identification and avoidance of inciting factors can hasten recovery from an attack and prevent future attacks. Inciting factors are usually multiple, and removal of one or more hastens recovery and helps prevent future attacks. Frequent attacks that occur during the luteal phase of the menstrual cycle may be prevented with a gonadotropin-releasing hormone analogue, which prevents ovulation and progesterone production, or by prophylactic hematin administration.
The long-term risk of hypertension and chronic renal disease is increased in AIP; a number of patients have undergone successful renal transplantation. Chronic, low-grade abnormalities in liver function tests are common, and the risk of hepatocellular carcinoma is increased. Hepatic imaging is recommended at least yearly for early detection of these tumors.
An allogeneic liver transplant was performed on a 19-year-old female AIP heterozygote who had 37 acute attacks in the 29 months prior to transplantation. After transplantation, her elevated urinary ALA and BPG levels returned to normal in 24 h, and she did not experience acute neurologic attacks for more than 3 years after transplant. Two AIP patients had combined liver and kidney transplants secondary to uncontrolled acute porphyria attacks, chronic peripheral neuropathy, and renal failure requiring dialysis. Both patients had a marked improvement with no attacks and normal urinary PBG levels after transplantation, as well as improvement of their neuropathic manifestations. More recently, a group from the United Kingdom reported their experience with liver transplantation in 10 AIP patients with recurrent attacks that were refractory to medical management and impaired quality of life. Patients had a complete biochemical and symptomatic resolution after transplant. The investigators reported a high rate of hepatic artery thrombosis in their series. Clearly, liver transplantation is a high-risk procedure and should be considered as a last resort in patients with severe recurrent attacks. Recently, liver-directed gene therapy has proven successful in the prevention of drug-induced biochemical attacks in a murine model of human AIP, and clinical trials of AAV-HMBS gene transfer have been initiated. In addition, preclinical studies of a hepatic-targeted RNA interference (RNAi) therapy directed to inhibit the markedly elevated hepatic ALAS1 mRNA in the AIP mouse model prevented induced biochemical attacks and rapidly reduced the ALAS1 mRNA during an ongoing attack.
PORPHYRIA CUTANEA TARDA (PCT)
PCT, the most common of the porphyrias, can be either sporadic (type 1) or familial (type 2) and can also develop after exposure to halogenated aromatic hydrocarbons. Hepatic URO decarboxylase is deficient in all types of PCT, and for clinical symptoms to manifest, this enzyme deficiency must be substantial (~20% of normal activity or less); it is currently attributed to generation of an URO decarboxylase inhibitor in the liver, which forms uroporphomethene in the presence of iron and under conditions of oxidative stress. The majority of PCT patients (~80%) have no UROD mutations and are said to have sporadic (type 1) disease. PCT patients heterozygous for UROD mutations have familial (type 2) PCT. In these patients, inheritance of a UROD mutation from one parent results in half-normal enzyme activity in liver and all other tissues, which is a significant predisposing factor, but is insufficient by itself to cause symptomatic PCT. As discussed below, other genetic and environmental factors contribute to susceptibility for both types of PCT. Because penetrance of the genetic trait is low, many patients with familial (type 2) PCT have no family history of the disease. HEP is an autosomal recessive form of porphyria that results from the marked systemic deficiency of URO decarboxylase activity with clinical symptoms in childhood.
Clinical Features Blistering skin lesions that appear most commonly on the backs of the hands are the major clinical feature (Fig. 430-3). These rupture and crust over, leaving areas of atrophy and scarring. Lesions may also occur on the forearms, face, legs, and feet. Skin friability and small white papules termed milia are common, especially on the backs of the hands and fingers. Hypertrichosis and hyperpigmentation, especially of the face, are especially troublesome in women. Occasionally, the skin over sun-exposed areas becomes severely thickened, with scarring and calcification that resembles systemic sclerosis. Neurologic features are absent.
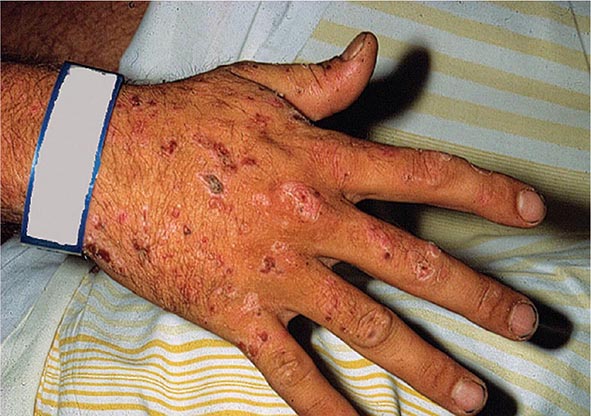
FIGURE 430-3 Typical cutaneous lesions in a patient with porphyria cutanea tarda. Chronic, crusted lesions resulting from blistering due to photosensitivity on the dorsum of the hand of a patient with porphyria cutanea tarda. (Courtesy of Dr. Karl E. Anderson; with permission.)
A number of susceptibility factors, in addition to inherited UROD mutations in type 2 PCT, can be recognized clinically and can affect management. These include hepatitis C, HIV, excess alcohol, elevated iron levels, and estrogens. The importance of excess hepatic iron as a precipitating factor is underscored by the finding that the incidence of the common hemochromatosis-causing mutations, hemochromatosis gene (HFE) mutations C282Y and H63D, are increased in patients with types 1 and 2 PCT (Chap. 428). Excess alcohol is a long-recognized contributor, as is estrogen use in women. HIV is probably an independent but less common risk factor that, like hepatitis C, does not cause PCT in isolation. Multiple susceptibility factors that appear to act synergistically can be identified in the individual PCT patient. Patients with PCT characteristically have chronic liver disease and sometimes cirrhosis and are at risk for hepatocellular carcinoma. Various chemicals can also induce PCT; an epidemic of PCT occurred in eastern Turkey in the 1950s as a consequence of wheat contaminated with the fungicide hexachlorobenzene. PCT also occurs after exposure to other chemicals, including di- and trichlorophenols and 2,3,7,8-tetrachlorodibenzo-(p)-dioxin (TCDD, dioxin).
Diagnosis Porphyrins are increased in the liver, plasma, urine, and stool. The urinary ALA level may be slightly increased, but the PBG level is normal. Urinary porphyrins consist mostly of uroporphyins and heptacarboxylate porphyrin, with lesser amounts of coproporphyrin and hexa- and pentacarboxylate porphyrins. Plasma porphyrins are also increased, and fluorometric scanning of diluted plasma at neutral pH can rapidly distinguish VP and PCT (Table 430-3). Isocoproporphyrins, which are increased in feces and sometimes in plasma and urine, are diagnostic for hepatic URO decarboxylase deficiency.
Type 2 PCT and HEP can be distinguished from type 1 by finding decreased URO decarboxylase in erythrocytes. URO decarboxylase activity in liver, erythrocytes, and cultured skin fibroblasts in type 2 PCT is approximately 50% of normal in affected individuals and in family members with latent disease. In HEP, the URO decarboxylase activity is markedly deficient, with typical levels of 3–10% of normal. Over 121 mutations have been identified in the UROD gene (Human Gene Mutation Database; www.hgmd.org). Of the mutations listed in the database, ~65% are missense or nonsense and ~10% are splice-site mutations. Most UROD mutations have been identified in only one or two families.
|
TREATMENT |
PORPHYRIA CUTANEA TARDA |
Alcohol, estrogens, iron supplements, and, if possible, any drugs that may exacerbate the disease should be discontinued, but this step does not always lead to improvement. A complete response can almost always be achieved by the standard therapy, repeated phlebotomy, to reduce hepatic iron. A unit (450 mL) of blood can be removed every 1–2 weeks. The aim is to gradually reduce excess hepatic iron until the serum ferritin level reaches the lower limits of normal. Because iron overload is not marked in most cases, remission may occur after only five or six phlebotomies; however, PCT patients with hemochromatosis may require more treatments to bring their iron levels down to the normal range. To document improvement in PCT, it is most convenient to follow the total plasma porphyrin concentration, which becomes normal some time after the target ferritin level is reached. Hemoglobin levels or hematocrits and serum ferritin should be followed closely to prevent development of iron deficiency and anemia. After remission, continued phlebotomy may not be needed. Plasma porphyrin levels are followed at 6- to 12-month intervals for early detection of recurrences, which are treated by additional phlebotomy.
An alternative when phlebotomy is contraindicated or poorly tolerated is a low-dose regimen of chloroquine or hydroxychloroquine, both of which complex with the excess porphyrins and promote their excretion. Small doses (e.g., 125 mg chloroquine phosphate twice weekly) should be given, because standard doses can induce transient, sometimes marked increases in photosensitivity and hepatocellular damage. Recent studies indicate that low-dose hydroxychloroquine is as safe and effective as phlebotomy in PCT. Hepatic imaging can diagnose or exclude complicating hepatocellular carcinoma. Treatment of PCT in patients with end-stage renal disease is facilitated by administration of erythropoietin.
HEREDITARY COPROPORPHYRIA (HCP)
HCP is an autosomal dominant hepatic porphyria that results from the half-normal activity of COPRO oxidase. The disease presents with acute attacks, as in AIP. Cutaneous photosensitivity also may occur, but much less commonly than in VP. HCP patients may have acute attacks and cutaneous photosensitivity together or separately. HCP is less common than AIP and VP. Homozygous dominant HCP and harderoporphyria, a biochemically distinguishable variant of HCP, present with clinical symptoms in children (see below).
Clinical Features HCP is influenced by the same factors that cause attacks in AIP. The disease is latent before puberty, and symptoms, which are virtually identical to those of AIP, are more common in women. HCP is generally less severe than AIP. Blistering skin lesions are identical to PCT and VP and begin in childhood in rare homozygous cases.
Diagnosis COPRO III is markedly increased in the urine and feces in symptomatic patients, and often persists, especially in feces, when there are no symptoms. Urinary ALA and PBG levels are increased (but less than in AIP) during acute attacks, but may revert to normal more quickly than in AIP when symptoms resolve. Plasma porphyrins are usually normal or only slightly increased, but they may be higher in cases with skin lesions. The diagnosis of HCP is readily confirmed by increased fecal porphyrins consisting almost entirely of COPRO III, which distinguishes it from other porphyrias.
Although the diagnosis can be confirmed by measuring COPRO oxidase activity, the assays for this mitochondrial enzyme are not widely available and require cells other than erythrocytes. To date, over 64 mutations have been identified in the CPOX gene, 67% of which are missense or nonsense (Human Gene Mutation Database; www.hgmd.org). Detection of a CPOX mutation in a symptomatic individual permits the identification of asymptomatic family members.
|
TREATMENT |
HEREDITARY COPROPORPHYRIA |
Neurologic symptoms are treated as in AIP (see above). Phlebotomy and chloroquine are not effective for the cutaneous lesions.
VARIEGATE PORPHYRIA (VP)
 VP is an autosomal dominant hepatic porphyria that results from the deficient activity of PROTO oxidase, the seventh enzyme in the heme biosynthetic pathway, and can present with neurologic symptoms, photosensitivity, or both. VP is particularly common in South Africa, where 3 of every 1000 whites have the disorder. Most are descendants of a couple who emigrated from Holland to South Africa in 1688. In other countries, VP is less common than AIP. Rare cases of homozygous dominant VP, presenting in childhood with cutaneous symptoms, also have been reported.
VP is an autosomal dominant hepatic porphyria that results from the deficient activity of PROTO oxidase, the seventh enzyme in the heme biosynthetic pathway, and can present with neurologic symptoms, photosensitivity, or both. VP is particularly common in South Africa, where 3 of every 1000 whites have the disorder. Most are descendants of a couple who emigrated from Holland to South Africa in 1688. In other countries, VP is less common than AIP. Rare cases of homozygous dominant VP, presenting in childhood with cutaneous symptoms, also have been reported.
Clinical Features VP can present with skin photosensitivity, acute neurovisceral crises, or both. In two large studies of VP patients, 59% had only skin lesions, 20% had only acute attacks, and 22% had both. Acute attacks are identical to those in AIP and are precipitated by the same factors as AIP (see above). Blistering skin manifestations are similar to those in PCT, but are more difficult to treat and usually are of longer duration. Homozygous VP is associated with photosensitivity, neurologic symptoms, and developmental disturbances, including growth retardation, in infancy or childhood; all cases had increased erythrocyte levels of zinc protoporphyrin, a characteristic finding in all homozygous porphyrias so far described.
Diagnosis Urinary ALA and PBG levels are increased during acute attacks, but may return to normal more quickly than in AIP. Increases in fecal protoporphyrin and COPRO III and in urinary COPRO III are more persistent. Plasma porphyrin levels also are increased, particularly when there are cutaneous lesions. VP can be distinguished rapidly from all other porphyrias by examining the fluorescence emission spectrum of porphyrins in plasma since VP has a unique fluorescence peak at neutral pH.
Assays of PROTO oxidase activity in cultured fibroblasts or lymphocytes are not widely available. Over 174 mutations have been identified in the PPOX gene from unrelated VP patients (Human Gene Mutation Database; www.hgmd.org). The missense mutation R59W is the common mutation in most South Africans with VP of Dutch descent. Five missense mutations were common in English and French VP patients; however, most mutations have been found in only one or two families.
|
TREATMENT |
VARIEGATE PORPHYRIA |
Acute attacks are treated as in AIP, and hemin should be started early in most cases. Other than avoiding sun exposure, there are few effective measures for treating the skin lesions. β-Carotene, phlebotomy, and chloroquine are not helpful.
THE ERYTHROPOIETIC PORPHYRIAS
In the erythropoietic porphyrias, excess porphyrins from bone marrow erythrocyte precursors are transported via the plasma to the skin and lead to cutaneous photosensitivity.
X-LINKED SIDEROBLASTIC ANEMIA (XLSA)
XLSA results from the deficient activity of the erythroid form of ALA synthase (ALA synthase 2) and is associated with ineffective erythropoiesis, weakness, and pallor.
Clinical Features Typically, males with XLSA develop refractory hemolytic anemia, pallor, and weakness during infancy. They have secondary hypersplenism, become iron overloaded, and can develop hemosiderosis. The severity depends on the level of residual erythroid ALA synthase activity and on the responsiveness of the specific mutation to pyridoxal 5′-phosphate supplementation (see below). Peripheral blood smears reveal a hypochromic, microcytic anemia with striking anisocytosis, poikilocytosis, and polychromasia; the leukocytes and platelets appear normal. Hemoglobin content is reduced, and the mean corpuscular volume and mean corpuscular hemoglobin concentration are decreased. Patients with milder, late-onset disease have been reported recently.
Diagnosis Bone marrow examination reveals hypercellularity with a left shift and megaloblastic erythropoiesis with an abnormal maturation. A variety of Prussian blue-staining sideroblasts are observed. Levels of urinary porphyrin precursors and of both urinary and fecal porphyrins are normal. The activity of erythroid ALA synthase 2 is decreased in bone marrow, but this enzyme is difficult to measure in the presence of the normal ALA synthase 1 housekeeping enzyme. Definitive diagnosis requires the demonstration of mutations in the erythroid ALAS2 gene.
|
TREATMENT |
X-LINKED SIDEROBLASTIC ANEMIA |
The severe anemia may respond to pyridoxine supplementation. This cofactor is essential for ALA synthase activity, and mutations in the pyridoxine binding site of the enzyme have been found in several responsive patients. Cofactor supplementation may make it possible to eliminate or reduce the frequency of transfusion. Unresponsive patients may be transfusion-dependent and require chelation therapy.
CONGENITAL ERYTHROPOIETIC PORPHYRIA (CEP)
CEP, also known as Günther’s disease, is an autosomal recessive disorder. It is due to the markedly deficient, but not absent, activity of URO synthase and the resultant accumulation of URO I and COPRO I isomers. CEP is associated with hemolytic anemia and cutaneous lesions.
Clinical Features Severe cutaneous photosensitivity typically begins in early infancy. The skin over light-exposed areas is friable, and bullae and vesicles are prone to rupture and infection. Skin thickening, focal hypo- and hyperpigmentation, and hypertrichosis of the face and extremities are characteristic. Secondary infection of the cutaneous lesions can lead to disfigurement of the face and hands. Porphyrins are deposited in teeth and in bones. As a result, the teeth are brownish and fluoresce on exposure to long-wave ultraviolet light. Hemolysis is probably due to the marked increase in erythrocyte porphyrins and leads to splenomegaly. Adults with a milder later-onset form of the disease also have been described.
Diagnosis URO and COPRO (mostly type I isomers) accumulate in the bone marrow, erythrocytes, plasma, urine, and feces. The predominant porphyrin in feces is COPRO I. The diagnosis of CEP can be confirmed by demonstration of markedly deficient URO synthase activity and/or by the identification of specific mutations in the UROS gene. The disease can be detected in utero by measuring porphyrins in amniotic fluid and URO synthase activity in cultured amniotic cells or chorionic villi, or by the detection of the family’s specific gene mutations. Molecular analyses of the mutant alleles from unrelated patients have revealed the presence of over 48 mutations in the UROS gene, including four in the erythroid-specific promoter of the UROS gene. Genotype/phenotype correlations can predict the severity of the disease. The CEP phenotype may be modulated by sequence variations in the erythroid specific ALA synthase 2, mutation of which typically causes XLP. One mutation (p.ArgR216WTrp) in GATA1, encoding the X-linked erythroid-specific transcription factor GATA binding protein 1 (GATA1), has been identified in an individual with CEP, thrombocytopenia, and β thalassemia.
|
TREATMENT |
CONGENITAL ERYTHROPOIETIC PORPHYRIA |
Severe cases often require transfusions for anemia. Chronic transfusions of sufficient blood to suppress erythropoiesis are effective in reducing porphyrin production but result in iron overload. Splenectomy may reduce hemolysis and decrease transfusion requirements. Protection from sunlight and from minor skin trauma is important. β-Carotene may be of some value. Complicating bacterial infections should be treated promptly. Recently, bone marrow and cord blood transplantation has proven curative in several transfusion-dependent children, providing the rationale for stem cell gene therapy.
ERYTHROPOIETIC PROTOPORPHYRIA (EPP)
EPP is an inherited disorder resulting from the deficient activity of ferrochelatase (FECH), the last enzyme in the heme biosynthetic pathway. EPP is the most common erythropoietic porphyria in children and, after PCT, the second most common porphyria in adults. EPP patients have FECH activities as low as 15–25% of normal in lymphocytes and cultured fibroblasts. Protoporphyrin accumulates in bone marrow reticulocytes and then appears in plasma, is taken up in the liver, and is excreted in bile and feces. Protoporphyrin transported to the vessels in the skin causes the nonblistering photosensitivity. In most symptomatic patients (~90%) with this autosomal recessive disorder, a mutation in one FECH allele is inherited with a relatively common (~10% of normal whites) intronic 3 (IVS3) alteration (IVS3–48T>C) that results in the low expression of the normal enzyme. In about 10% of EPP families, two FECH mutations have been found. Recently, deletion mutations in exon 11 of the ALAS2 gene have been described, causing XLP that is clinically indistinguishable from EPP. The deletion of the C-terminal amino acids of ALAS2 results in its increased activity and the accumulation of protoporphyrin. XLP accounts for approximately 2–10% of cases with the EPP phenotype in Europe and North America.
Clinical Features Skin photosensitivity, which differs from that in other porphyrias, usually begins in childhood and consists of pain, redness, and itching occurring within minutes of sunlight exposure (Fig. 430-4). Photosensitivity is associated with substantial elevations in erythrocyte protoporphyrin and occurs only in patients with genotypes that result in ferrochelatase activities below ~35% of normal. Vesicular lesions are uncommon. Redness, swelling, burning, and itching can develop shortly after sun exposure and resemble angioedema. Pain symptoms may seem out of proportion to the visible skin involvement. Sparse vesicles and bullae occur in ~10% of cases. Chronic skin changes may include lichenification, leathery pseudovesicles, labial grooving, and nail changes. Severe scarring is rare, as are pigment changes, friability, and hirsutism. Unless hepatic or other complications develop, protoporphyrin levels and symptoms of photosensitivity remain remarkably stable over many years in most patients. Factors that exacerbate the hepatic porphyrias play little or no role in EPP.
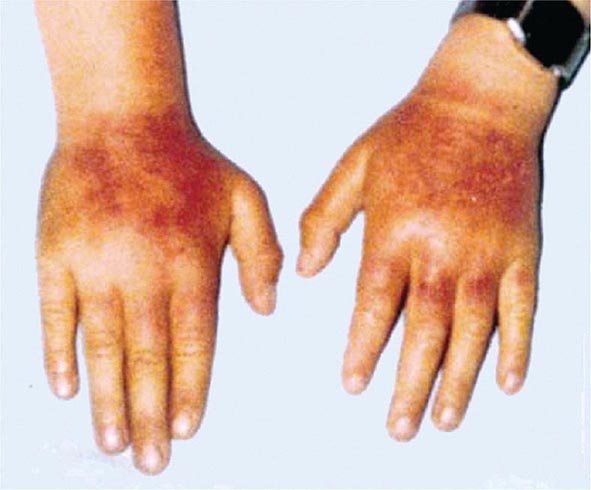
FIGURE 430-4 Erythema and edema of the hands due to acute photosensitivity in a 10-year-old boy with erythropoietic protoporphyria. (From P Poblette-Gutierrez et al: Eur J Dermatol 16:230, 2006.)
The primary source of excess protoporphyrin is the bone marrow reticulocytes. Erythrocyte protoporphyrin is free (not complexed with zinc) and is mostly bound to hemoglobin. In plasma, protoporphyrin is bound to albumin. Hemolysis and anemia are usually absent or mild.
Although EPP is an erythropoietic porphyria, up to 20% of EPP patients may have minor abnormalities of liver function, and in about 5% of these patients the accumulation of protoporphyrins causes chronic liver disease that can progress to liver failure and death. Protoporphyrin is insoluble, and excess amounts form crystalline structures in liver cells (Fig. 430-4) and can decrease hepatic bile flow. Studies in the mouse model of EPP have shown that the bile duct epithelium may be damaged by toxic bile, leading to biliary fibrosis. Thus, rapidly progressive liver disease appears to be related to the cholestatic effects of protoporphyrins and is associated with increasing hepatic protoporphyrin levels due to impaired hepatobiliary excretion and increased photosensitivity. The hepatic complications also are often characterized by increasing levels of protoporphyrins in erythrocytes and plasma as well as severe abdominal and back pains, especially in the right upper quadrant. Gallstones composed at least in part of protoporphyrin occur in some patients. Hepatic complications appear to be higher in autosomal recessive EPP due to two FECH mutations and in XLP.
Diagnosis A substantial increase in erythrocyte protoporphyrin, which is predominantly free and not complexed with zinc, is the hallmark of EPP. Protoporphyrin levels are also variably increased in bone marrow, plasma, bile, and feces. Erythrocyte protoporphyrin concentrations are increased in other conditions such as lead poisoning, iron deficiency, various hemolytic disorders, all homozygous forms of other porphyrias, and sometimes even in acute porphyrias. In all these conditions, however, in contrast to EPP, protoporphyrin is complexed with zinc. Therefore, after an increase in erythrocyte protoporphyrin is found in a suspected EPP patient, it is important to confirm the diagnosis by an assay that distinguishes free and zinc-complexed protoporphyrin. Erythrocytes in EPP also exhibit red fluorescence under a fluorescence microscopy at 620 nm. Urinary levels of porphyrins and porphyrin precursors are normal. Ferrochelatase activity in cultured lymphocytes or fibroblasts is decreased. DNA diagnosis by mutation analysis is recommended to detect the causative FECH mutation(s) and/or the presence of the IVS3–48T>C low expression allele. To date, over 190 mutations have been identified in the FECH gene, many of which result in an unstable or absent enzyme protein (null alleles) (Human Gene Mutation Database; www.hgmd.org). Studies suggest that EPP patients with a null allele (and the IVS3–48T>C low expression allele) have a greater risk for developing severe liver complications.
In XLP, the erythrocyte protoporphyrin levels appear to be higher than other forms of EPP and the proportions of free and zinc protopophyrins may reach 50%. To date, four ALAS2 mutations, three deletions of one to four bases, and one novel nonsense mutation have been described, which markedly increase ALA synthase 2 activity and cause XLP. XLP accounts for about 2% of patients with the EPP phenotype in Western Europe. Recent studies show that about 10% of North American patients with the EPP phenotype have XLP.
|
TREATMENT |
ERYTHROPOIETIC PROTOPORPHYRIA |
Avoiding sunlight exposure and wearing clothing designed to provide protection for conditions with chronic photosensitivity are essential. Oral β-carotene (120–180 mg/dL) may improve tolerance to sunlight in some patients. The beneficial effects of β-carotene may involve quenching of singlet oxygen or free radicals. The dosage may need to be adjusted to maintain serum carotene levels in the recommended range of 10–15 mmol/L (600–800 mg/dL). Mild skin discoloration due to carotenemia is the only significant side effect. Afamelanotide, an α-melanocyte-stimulating hormone (MSH) analogue has completed phase III clinical trials in the United States for patients with EPP and XLP.
Treatment of hepatic complications, which may be accompanied by motor neuropathy, is difficult. Cholestyramine and other porphyrin absorbents such as activated charcoal may interrupt the enterohepatic circulation of protoporphyrin and promote its fecal excretion, leading to some improvement. Splenectomy may be helpful when the disease is accompanied by hemolysis and significant splenomegaly. Plasmapheresis and intravenous hemin are sometimes beneficial.
Liver transplantation has been carried out in some EPP and XLP patients with severe liver complications and is often successful in the short term. However, the disease often recurs in the transplanted liver due to continued bone marrow production of excess protoporphyrin. In a retrospective study of 17 liver-transplanted EPP patients, 11 (65%) had recurrent EPP liver disease. Posttransplantation treatment with hematin and plasmapheresis should be considered to prevent the recurrence of liver disease. However, bone marrow transplantation, which has been successful in human EPP and which prevented liver disease in a mouse model, should be considered after liver transplantation, if a suitable donor can be found.
ACKNOWLEDGMENT
The authors thank Dr. Karl E. Anderson for his review of the manuscript and helpful comments and suggestions. This work is supported in part by the Porphyrias Consortium (U54 DK083909), a part of the National Institutes of Health (NIH) Rare Disease Clinical Research Network (RDCRN), supported through collaboration between the NIH Office of Rare Diseases Research (ORDR) at the National Center for Advancing Translational Science (NCATS), and the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. MB is supported by a career development award K23-DK-095946
431e |
Disorders of Purine and Pyrimidine Metabolism |
Purines (adenine and guanine) and pyrimidines (cytosine, thymine, uracil) serve fundamental roles in the replication of genetic material, gene transcription, protein synthesis, and cellular metabolism. Disorders that involve abnormalities of nucleotide metabolism range from relatively common diseases such as hyperuricemia and gout, in which there is increased production or impaired excretion of a metabolic end product of purine metabolism (uric acid), to rare enzyme deficiencies that affect purine and pyrimidine synthesis or degradation. Understanding these biochemical pathways has led, in some instances, to the development of specific forms of treatment, such as the use of allopurinol and febuxostat to reduce uric acid production.
URIC ACID METABOLISM
Uric acid is the final breakdown product of purine degradation in humans. It is a weak diprotic acid with pKa values of 5.75 and 10.3. Urates, the ionized forms of uric acid, predominate in plasma, extracellular fluid, and synovial fluid, with ~98% existing as monosodium urate at pH 7.4.
Plasma is saturated with monosodium urate at a concentration of 405 μmol/L (6.8 mg/dL) at 37°C. At higher concentrations, plasma is therefore supersaturated—a situation that creates the potential for urate crystal precipitation. However, plasma urate concentrations can reach 4800 μmol/L (80 mg/dL) without precipitation, perhaps because of the presence of solubilizing substances.
The pH of urine greatly influences the solubility of uric acid. At pH 5.0, urine is saturated with uric acid at concentrations ranging from 360 to 900 μmol/L (6–15 mg/dL). At pH 7, saturation is reached at concentrations from 9840 to 12,000 μmol/L (158–200 mg/dL). Ionized forms of uric acid in urine include monosodium, disodium, potassium, ammonium, and calcium urates.
Although purine nucleotides are synthesized and degraded in all tissues, urate is produced only in tissues that contain xanthine oxidase, primarily the liver and small intestine. Urate production varies with the purine content of the diet and with rates of purine biosynthesis, degradation, and salvage (Fig. 431e-1). Normally, two-thirds to three-fourths of urate is excreted by the kidneys, and most of the remainder is eliminated through the intestines.
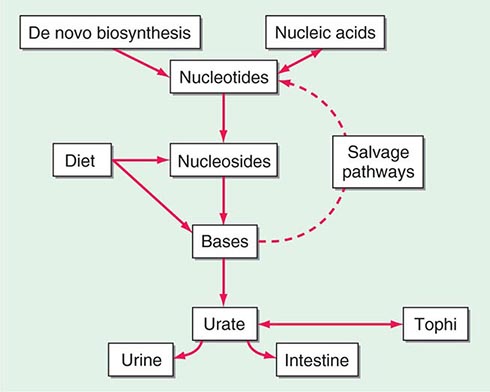
FIGURE 431e-1 The total-body urate pool is the net result between urate production and excretion. Urate production is influenced by dietary intake of purines and the rates of de novo biosynthesis of purines from nonpurine precursors, nucleic acid turnover, and salvage by phosphoribosyltransferase activities. The formed urate is normally excreted by urinary and intestinal routes. Hyperuricemia can result from increased production, decreased excretion, or a combination of both mechanisms. When hyperuricemia exists, urate can precipitate and deposit in tissues as tophi.
The kidneys clear urate from the plasma and maintain physiologic balance by utilizing specific organic anion transporters (OATs), including urate transporter 1 (URAT1, SLC22A12) (Fig. 431e-2). In humans, OAT1 (SLC22A6), OAT2 (SLC22A7), and OAT3 (SLC22A8) are located on the basolateral membrane of renal proximal tubule cells. OAT4 (SLC22A11), OAT10 (SLC22A13), and URAT1 are located on the apical brush-border membrane of these cells. The latter transporters carry urate and other organic anions into the tubular cells from the lumen in exchange for intracellular organic anions. Once inside the cell, urate must pass to the basolateral side of the lumen in a process controlled by voltage-dependent carriers, including glucose transporter 9 (GLUT9, SLC2A9). Uricosuric compounds (Table 431e-1) directly inhibit URAT1 on the apical side of the tubular cell (so-called cis-inhibition). In contrast, antiuricosuric compounds (those that promote hyperuricemia), such as nicotinate, pyrazinoate, lactate, and other aromatic organic acids, serve as the exchange anion inside the cell, thereby stimulating anion exchange and urate reabsorption (trans-stimulation). The activities of URAT1, other OATs, and sodium anion transporters result in excretion of 8–12% of the filtered urate as uric acid.
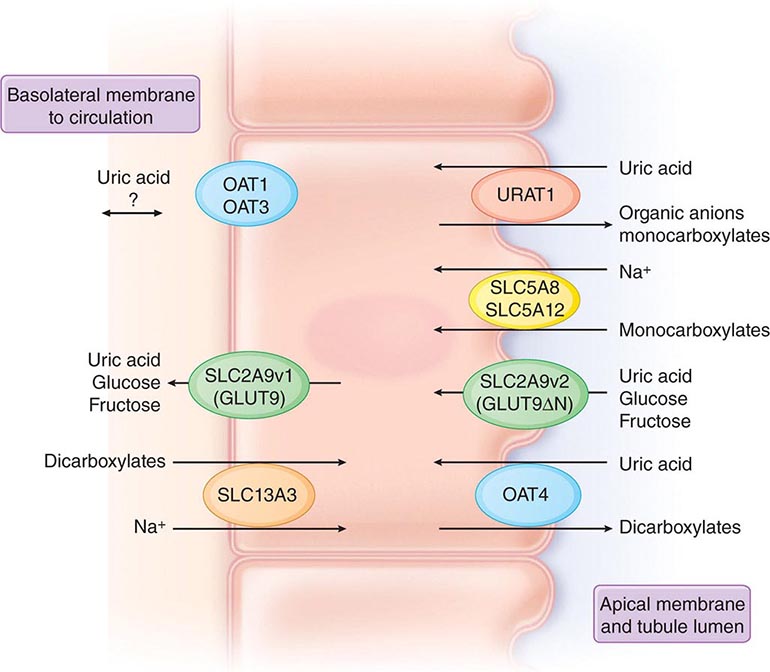
FIGURE 431e-2 Schematic for handling of uric acid by the kidney. A complex interplay of transporters on both the apical and basolateral aspects of the renal tubule epithelial cell is involved in the reabsorption of uric acid. See text for details. Most uricosuric compounds inhibit URAT1 on the apical side, as well as OAT1, OAT3, and GLUT9 on the basolateral side.
|
MEDICATIONS WITH URICOSURIC ACTIVITY |
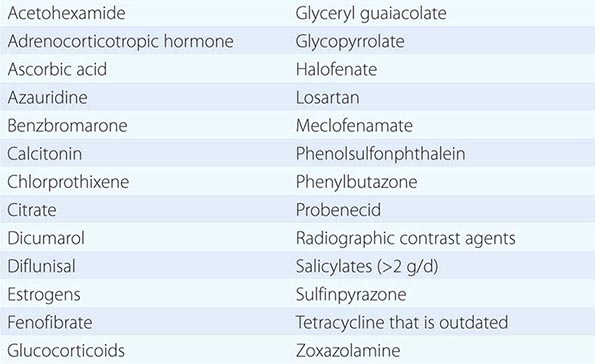
Most children have serum urate concentrations of 180–240 μmol/L (3–4 mg/dL). Levels begin to rise in males during puberty but remain low in females until menopause. The most recent mean serum urate values for men and premenopausal women in the United States are 415 and 360 μmol/L (6.14 and 4.87 mg/dL), respectively, according to National Health and Nutrition Evaluation Survey (NHANES) data for 2007–2008. After menopause, values for women increase to approximately those for men. In adulthood, concentrations rise steadily over time and vary with height, body weight, blood pressure, renal function, and alcohol intake.
HYPERURICEMIA
Hyperuricemia can result from increased production or decreased excretion of uric acid or from a combination of the two processes. Sustained hyperuricemia predisposes some individuals to develop clinical manifestations including gouty arthritis (Chap. 395), urolithiasis, and renal dysfunction (see below).
In general, hyperuricemia is defined as a plasma (or serum) urate concentration >405 μmol/L (>6.8 mg/dL). The risk of developing gouty arthritis or urolithiasis increases with higher urate levels and escalates in proportion to the degree of elevation. The prevalence of hyperuricemia is increasing among ambulatory adults and even more markedly among hospitalized patients. The prevalence of gout in the United States more than doubled between the 1960s and the 1990s. Based on NHANES data from 2007–2008, these trends continue, with an approximate prevalence of gout among men of 5.9% (6.1 million) and among women of 2.0% (2.2 million). Mean serum urate levels rose to 6.14 mg/dL among men and 4.87 mg/dL among women, with consequent hyperuricemia prevalences of 21.2% and 21.6%, respectively (with hyperuricemia defined as a serum urate level of >7.0 mg/dL [415 μmol/L] for men and >5.7 mg/dL [340 μmol/L] for women). These numbers represent a 1.2% increase in the prevalence of gout, a 0.15-mg/dL increase in the serum urate level, and a 3.2% increase in the prevalence of hyperuricemia over figures reported in NHANES-III (1988–1994). These rises are thought to be driven by increased obesity and hypertension and perhaps also by better medical care and increased longevity.
CAUSES OF HYPERURICEMIA
Hyperuricemia may be classified as primary or secondary, depending on whether the cause is innate or an acquired disorder. However, it is more useful to classify hyperuricemia in relation to the underlying pathophysiology—i.e., whether it results from increased production, decreased excretion, or a combination of the two (Fig. 431e-1, Table 431e-2).
|
CLASSIFICATION OF HYPERURICEMIA BY PATHOPHYSIOLOGY |
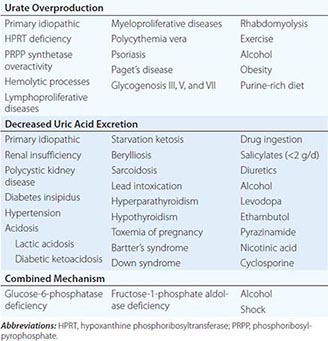
Increased Urate Production Diet contributes to the serum urate concentration in proportion to its purine content. Strict restriction of purine intake reduces the mean serum urate level by ~60 μmol/L (~1 mg/dL) and urinary uric acid excretion by ~1.2 mmol/d (~200 mg/d). Foods high in nucleic acid content include liver, “sweetbreads” (i.e., thymus and pancreas), kidney, and anchovy.
Endogenous sources of purine production also influence the serum urate level (Fig. 431e-3). De novo purine biosynthesis is a multistep process that forms inosine monophosphate (IMP). The rates of purine biosynthesis and urate production are predominantly determined by amidophosphoribosyltransferase (amidoPRT), which combines phosphoribosylpyrophosphate (PRPP) and glutamine. A secondary regulatory pathway is the salvage of purine bases by hypoxanthine phosphoribosyltransferase (HPRT). HPRT catalyzes the combination of the purine bases hypoxanthine and guanine with PRPP to form the respective ribonucleotides IMP and guanosine monophosphate (GMP).
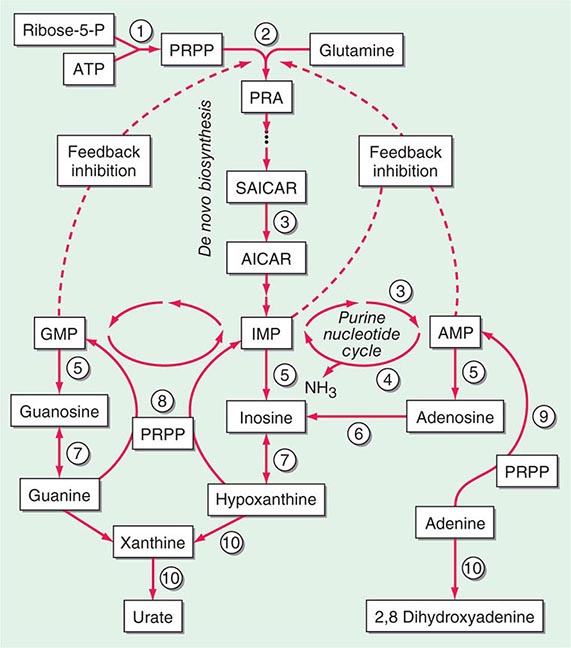
FIGURE 431e-3 Abbreviated scheme of purine metabolism. (1) Phosphoribosylpyrophosphate (PRPP) synthetase, (2) amidophosphoribosyltransferase (amidoPRT), (3) adenylosuccinate lyase, (4) (myo-)adenylate (AMP) deaminase, (5) 5′-nucleotidase, (6) adenosine deaminase, (7) purine nucleoside phosphorylase, (8) hypoxanthine phosphoribosyltransferase (HPRT), (9) adenine phosphoribosyltransferase (APRT), and (10) xanthine oxidase. PRA, phosphoribosylamine; SAICAR, succinylaminoimidazole carboxamide ribotide; AICAR, aminoimidazole carboxamide ribotide; GMP, guanylate; IMP, inosine monophosphate; ATP, adenosine triphosphate.
Serum urate levels are closely coupled to the rates of de novo purine biosynthesis, which is driven in part by the level of PRPP, as evidenced by two X-linked inborn errors of purine metabolism (Table 431e-3). Both increased PRPP synthetase activity and HPRT deficiency are associated with overproduction of purines, hyperuricemia, and hyperuricaciduria (see below for clinical descriptions).
|
INBORN ERRORS OF PURINE METABOLISM |
Accelerated purine nucleotide degradation can also cause hyperuricemia—i.e., with conditions of rapid cell turnover, proliferation, or cell death, as in leukemic blast crises, cytotoxic therapy for malignancy, hemolysis, or rhabdomyolysis. Hyperuricemia can result from excessive degradation of skeletal muscle ATP after strenuous physical exercise or status epilepticus and in glycogen storage disease types III, V, and VII (Chap. 433e). The hyperuricemia of myocardial infarction, smoke inhalation, and acute respiratory failure may also be related to accelerated breakdown of ATP.
Decreased Uric Acid Excretion More than 90% of individuals with sustained hyperuricemia have a defect in the renal handling of uric acid. For any given plasma urate concentration, patients who have gout excrete ~40% less uric acid than those who do not. When plasma urate levels are raised by purine ingestion or infusion, uric acid excretion increases in patients with and without gout; however, in those with gout, plasma urate concentrations must be 60–120 μmol/L (1–2 mg/dL) higher than normal to achieve equivalent uric acid excretion rates.
Diminished uric acid excretion could theoretically result from decreased glomerular filtration, decreased tubular secretion, or enhanced tubular reabsorption. Decreased urate filtration does not appear to cause primary hyperuricemia but does contribute to the hyperuricemia of renal insufficiency. Although hyperuricemia is invariably present in chronic renal disease, the correlation among serum creatinine, urea nitrogen, and urate concentrations is poor. Extrarenal clearance of uric acid increases as renal damage becomes more severe.
Many agents that cause hyperuricemia exert their effects by stimulating reabsorption rather than inhibiting secretion. This stimulation appears to occur through a process of “priming” renal urate reabsorption through the sodium-dependent loading of proximal tubular epithelial cells with anions capable of trans-stimulating urate reabsorption. The sodium-coupled monocarboxyl transporters SMCT1 and 2 (SLC5A8, SLC5A12) in the brush border of the proximal tubular cells mediate sodium-dependent loading of these cells with monocarboxylates. A similar transporter, SLC13A3, mediates sodium-dependent influx of dicarboxylates into the epithelial cell from the basolateral membrane. Some of these carboxylates are well known to cause hyperuricemia, including pyrazinoate (from pyrazinamide treatment), nicotinate (from niacin therapy), and the organic acids lactate, β-hydroxybutyrate, and acetoacetate. The mono- and divalent anions then become substrates for URAT1 and OAT4, respectively, and are exchanged for uric acid from the proximal tubule. Increased blood levels of these anions result in their increased glomerular filtration and greater reabsorption by proximal tubular cells. The increased intraepithelial cell concentrations lead to increased uric acid reabsorption by promoting URAT1-, OAT4-, and OAT10-dependent anion exchange. Low doses of salicylates also promote hyperuricemia by this mechanism. Sodium loading of proximal tubular cells also provokes urate retention by reducing extracellular fluid volume and increasing angiotensin II, insulin, and parathyroid hormone release. Additional organic anion transporters OAT1, OAT2, and OAT3 are involved in the movement of uric acid through the basolateral membrane, although the detailed mechanisms are still being elucidated.
GLUT9 (SLC2A9) is an electrogenic hexose transporter with splicing variants that mediate co-reabsorption of uric acid along with glucose and fructose at the apical membrane (GLUT9ΔN/SLC2A9v2) as well as through the basolateral membrane (SLC2A9v1) and thus into the circulation. GLUT9 has recently been identified as a high-capacity urate transporter, with rates 45–60 times faster than its glucose/fructose transport activity. GLUT9 may be responsible for the observed association of the consumption of fructose-sweetened soft drinks with an increased risk of hyperuricemia and gout. Genome-wide association scanning suggests that polymorphisms in SLC2A9 may play an important role in susceptibility to gout in the Caucasian population. The presence of one predisposing variant allele increases the relative risk of developing gout by 30–70%, most likely by increasing expression of the shorter isoform, SLC2A9v2 (GLUT9ΔN). Notably, though, genetic polymorphisms explain only ~6% of the differences in serum uric acid levels in Caucasians. Clearly, gout is polygenic and complex, and at this time the utility of genetic testing for relevant polymorphisms remains investigational and of no clinical utility.
Alcohol promotes hyperuricemia because of increased urate production and decreased uric acid excretion. Excessive alcohol consumption accelerates hepatic breakdown of ATP to increase urate production. Alcohol consumption can also induce hyperlacticacidemia, which blocks uric acid secretion. The higher purine content in some alcoholic beverages may also be a factor. Consumption of beer confers a greater risk of gout than liquor, and moderate wine intake does not increase gout risk. Intake of red meat and fructose increases the risk of gout, whereas intake of low-fat dairy products, purine-rich vegetables, whole grains, nuts and legumes, less sugary fruits, coffee, and vitamin C reduces the risk.
EVALUATION
Hyperuricemia does not necessarily represent a disease, nor is it a specific indication for therapy. The decision to treat depends on the cause and the potential consequences of hyperuricemia in each individual.
Quantification of uric acid excretion can be used to determine whether hyperuricemia is caused by overproduction or decreased excretion. On a purine-free diet, men with normal renal function excrete <3.6 mmol/d (600 mg/d). Thus, the hyperuricemia of individuals who excrete uric acid above this level while on a purine-free diet is due to purine overproduction; for those who excrete lower amounts on the purine-free diet, it is due to decreased excretion. If the assessment is performed while the patient is on a regular diet, the level of 4.2 mmol/d (800 mg/d) can be used as the discriminating value.
COMPLICATIONS
The most recognized complication of hyperuricemia is gouty arthritis. NHANES 2007–2008 found a prevalence of gout among U.S. adults of 3.9%, with figures of ~6% for men and ~2% for women. The higher the serum urate level, the more likely an individual is to develop gout. In one study, the incidence of gout was 4.9% among individuals with serum urate concentrations >540 μmol/L (>9.0 mg/dL) as opposed to only 0.5% among those with values between 415 and 535 μmol/L (7.0 and 8.9 mg/dL). The complications of gout correlate with both the duration and the severity of hyperuricemia. For further discussion of gout, see Chap. 395.
Hyperuricemia also causes several renal problems: (1) nephrolithiasis; (2) urate nephropathy, a rare cause of renal insufficiency attributed to monosodium urate crystal deposition in the renal interstitium; and (3) uric acid nephropathy, a reversible cause of acute renal failure resulting from deposition of large amounts of uric acid crystals in the renal collecting ducts, pelvis, and ureters.
Nephrolithiasis Uric acid nephrolithiasis occurs most commonly, but not exclusively, in individuals with gout. In gout, the prevalence of nephrolithiasis correlates with the serum and urinary uric acid levels, reaching ~50% with serum urate levels of 770 μmol/L (13 mg/dL) or urinary uric acid excretion >6.5 mmol/d (1100 mg/d).
Uric acid stones can develop in individuals with no evidence of arthritis, only 20% of whom are hyperuricemic. Uric acid can also play a role in other types of kidney stones. Some individuals who do not have gout but have calcium oxalate or calcium phosphate stones have hyperuricemia or hyperuricaciduria. Uric acid may act as a nidus on which calcium oxalate can precipitate or lower the formation product for calcium oxalate crystallization.
Urate Nephropathy Urate nephropathy, sometimes referred to as urate nephrosis, is a late manifestation of severe gout and is characterized histologically by deposits of monosodium urate crystals surrounded by a giant-cell inflammatory reaction in the medullary interstitium and pyramids. The disorder is now rare and cannot be diagnosed in the absence of gouty arthritis. The lesions may be clinically silent or cause proteinuria, hypertension, and renal insufficiency.
Uric Acid Nephropathy This reversible cause of acute renal failure is due to precipitation of uric acid in renal tubules and collecting ducts that obstructs urine flow. Uric acid nephropathy develops following sudden urate overproduction and marked hyperuricaciduria. Factors that favor uric acid crystal formation include dehydration and acidosis. This form of acute renal failure occurs most often during an aggressive “blastic” phase of leukemia or lymphoma prior to or coincident with cytolytic therapy but has also been observed in individuals with other neoplasms, following epileptic seizures, and after vigorous exercise with heat stress. Autopsy studies have demonstrated intraluminal precipitates of uric acid, dilated proximal tubules, and normal glomeruli. The initial pathogenic events are believed to include obstruction of collecting ducts with uric acid and obstruction of the distal renal vasculature.
If recognized, uric acid nephropathy is potentially reversible. Appropriate therapy has reduced the mortality rate from ~50% to practically nil. Serum levels cannot be relied on for diagnosis because this condition has developed in the presence of urate concentrations varying from 720 to 4800 μmol/L (12–80 mg/dL). The distinctive feature is the urinary uric acid concentration. In most forms of acute renal failure with decreased urine output, urinary uric acid content is either normal or reduced, and the ratio of uric acid to creatinine is <1. In acute uric acid nephropathy, the ratio of uric acid to creatinine in a random urine sample or a 24-h specimen is >1, and a value that high is essentially diagnostic.
HYPERURICEMIA AND METABOLIC SYNDROME
Metabolic syndrome (Chap. 422) is characterized by abdominal obesity with visceral adiposity, impaired glucose tolerance due to insulin resistance with hyperinsulinemia, hypertriglyceridemia, increased low-density lipoprotein cholesterol, decreased high-density lipoprotein cholesterol, and hyperuricemia. Hyperinsulinemia reduces the renal excretion of uric acid and sodium. Not surprisingly, hyperuricemia resulting from euglycemic hyperinsulinemia may precede the onset of type 2 diabetes, hypertension, coronary artery disease, and gout in individuals with metabolic syndrome.
HYPOURICEMIA
Hypouricemia, defined as a serum urate concentration <120 μmol/L (<2.0 mg/dL), can result from decreased production of urate, increased excretion of uric acid, or a combination of both mechanisms. This condition occurs in <0.2% of the general population and <0.8% of hospitalized individuals. Hypouricemia causes no symptoms or pathology and therefore requires no therapy.
Most hypouricemia results from increased renal uric acid excretion. The finding of normal amounts of uric acid in a 24-h urine collection from an individual with hypouricemia is evidence for a renal cause. Medications with uricosuric properties (Table 431e-1) include aspirin (at doses >2.0 g/d), losartan, fenofibrate, x-ray contrast materials, and glyceryl guaiacolate. Total parenteral hyperalimentation can also cause hypouricemia, possibly a result of the high glycine content of the infusion formula. Other causes of increased urate clearance include conditions such as neoplastic disease, hepatic cirrhosis, diabetes mellitus, and inappropriate secretion of vasopressin; defects in renal tubular transport such as primary Fanconi syndrome and Fanconi syndromes caused by Wilson’s disease, cystinosis, multiple myeloma, and heavy metal toxicity; and isolated congenital defects in the bidirectional transport of uric acid. Hypouricemia can be a familial disorder that is generally inherited in an autosomal recessive manner. Most cases are caused by a loss of function mutation in SLC22A12, the gene that encodes URAT-1, resulting in increased renal urate clearance. Individuals with normal SLC22A12 most likely have a defect in other urate transporters. Although hypouricemia is usually asymptomatic, some patients suffer from urate nephrolithiasis or exercise-induced renal failure.
SELECTED INBORN ERRORS OF PURINE AND PYRIMIDINE METABOLISM
(See also Table 431e-3, Table 431e-4, Fig. 431e-3, and Fig. 431e-4) More than 30 defects in human purine and pyrimidine metabolic pathways have been identified thus far. Many are benign, but about half are associated with clinical manifestations, some causing major morbidity and mortality. Advances in genetics, along with high-performance liquid chromatography and tandem mass spectrometry, have facilitated diagnosis.
|
INBORN ERRORS OF PYRIMIDINE METABOLISM |
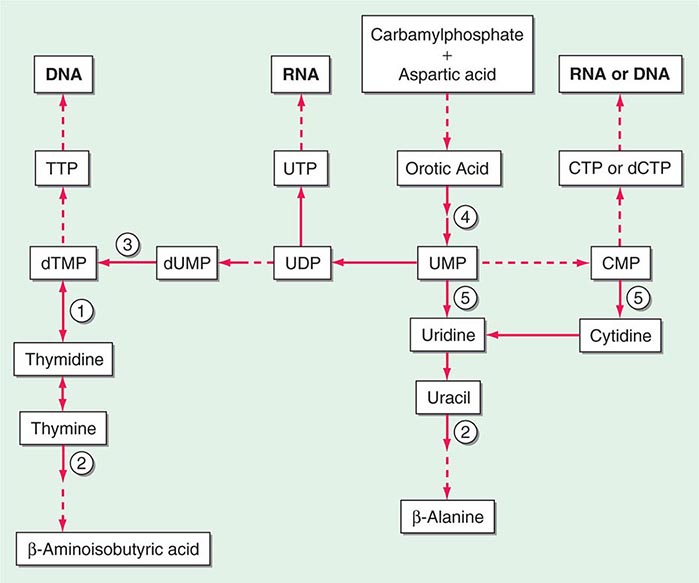
FIGURE 431e-4 Abbreviated scheme of pyrimidine metabolism. (1) Thymidine kinase, (2) dihydropyrimidine dehydrogenase, (3) thymidylate synthase, (4) UMP synthase, (5) 5′-nucleotidase. CMP, cytidine-5′-monophosphate; UMP, uridine-5′-monophosphate; UDP, uridine-5′-diphosphate; dUMP, deoxyuridine-5′-monophosphate; dTMP, deoxythymidine-5′-monophosphate; TTP, thymidine triphosphate; UTP, uridine triphosphate.
PURINE DISORDERS
HPRT Deficiency The HPRT gene is located on the × chromosome. Affected males are hemizygous for the mutant gene; carrier females are asymptomatic. A complete deficiency of HPRT, the Lesch-Nyhan syndrome, is characterized by hyperuricemia, self-mutilative behavior, choreoathetosis, spasticity, and mental retardation. A partial deficiency of HPRT, the Kelley-Seegmiller syndrome, is associated with hyperuricemia but no central nervous system manifestations. In both disorders, the hyperuricemia results from urate overproduction and can cause uric acid crystalluria, nephrolithiasis, obstructive uropathy, and gouty arthritis. Early diagnosis and appropriate therapy with allopurinol can prevent or eliminate all the problems attributable to hyperuricemia without affecting behavioral or neurologic abnormalities.
Increased PRPP Synthetase Activity Like the HPRT deficiency states, PRPP synthetase overactivity is X-linked and results in gouty arthritis and uric acid nephrolithiasis. Nerve deafness occurs in some families.
Adenine Phosphoribosyltransferase (APRT) Deficiency APRT deficiency is inherited as an autosomal recessive trait. Affected individuals develop kidney stones composed of 2,8-dihydroxyadenine. Caucasians with the disorder have a complete deficiency (type I), whereas Japanese individuals have some measurable enzyme activity (type II). Expression of the defect is similar in the two populations, as is the frequency of the heterozygous state (0.4–1.1 per 100). Allopurinol treatment prevents stone formation.
Hereditary Xanthinuria A deficiency of xanthine oxidase causes all purine in the urine to occur in the form of hypoxanthine and xanthine. About two-thirds of deficient individuals are asymptomatic. The remainder develop kidney stones composed of xanthine.
Myoadenylate Deaminase Deficiency Primary (inherited) and secondary (acquired) forms of myoadenylate deaminase deficiency have been described. The primary form is inherited as an autosomal recessive trait. Clinically, some persons may have relatively mild myopathic symptoms with exercise or other triggers, but most individuals with this defect are asymptomatic. Therefore, another explanation for the myopathy should be sought in symptomatic patients with this deficiency. The acquired deficiency occurs in association with a wide variety of neuromuscular diseases, including muscular dystrophies, neuropathies, inflammatory myopathies, and collagen vascular diseases.
Adenylosuccinate Lyase Deficiency Deficiency of this enzyme is due to an autosomal recessive trait and causes profound psychomotor retardation, seizures, and other movement disorders. All individuals with this deficiency are mentally retarded, and most are autistic.
Adenosine Deaminase Deficiency and Purine Nucleoside Phosphorylase Deficiency See Chap. 374.
PYRIMIDINE DISORDERS
The pyrimidine cytidine is found in both DNA and RNA; it is a complementary base pair for guanine. Thymidine is found only in DNA, where it is paired with adenine. Uridine is found only in RNA and can pair with either adenine or guanine in RNA secondary structures. Pyrimidines can be synthesized by a de novo pathway (Fig. 431e-4) or reused in a salvage pathway. Although more than 25 different enzymes are involved in pyrimidine metabolism, disorders of these pathways are rare. Seven disorders of pyrimidine metabolism have been discovered (Table 431e-4), three of which are discussed below.
Orotic Aciduria Hereditary orotic aciduria is caused by mutations in a bifunctional enzyme, uridine-5′-monophosphate (UMP) synthase, which converts orotic acid to UMP in the de novo synthesis pathway (Fig. 431e-4). The disorder is characterized by hypochromic megaloblastic anemia that is unresponsive to vitamin B12 and folic acid, growth retardation, and neurologic abnormalities. Increased excretion of orotic acid causes crystalluria and obstructive uropathy. Replacement of uridine (100–200 mg/kg per day) corrects anemia, reduces orotic acid excretion, and improves the other sequelae of the disorder.
Pyrimidine 5′-nucleotidase Deficiency Pyrimidine 5′-nucleotidase catalyzes the removal of the phosphate group from pyrimidine ribonucleoside monophosphates (cytidine-5′-monophosphate or UMP) (Fig. 431e-4). An inherited deficiency of this enzyme causes hemolytic anemia with prominent basophilic stippling of erythrocytes. The accumulation of pyrimidines or cytidine diphosphate choline is thought to induce hemolysis. There is no specific treatment. Acquired pyrimidine 5′-nucleotidase deficiency has been reported in lead poisoning and in thalassemia.
Dihydropyrimidine Dehydrogenase Deficiency Dihydropyrimidine dehydrogenase is the rate-limiting enzyme in the pathway of uracil and thymine degradation (Fig. 431e-4). Deficiency of this enzyme causes excessive urinary excretion of uracil and thymine. In addition, this deficiency causes nonspecific cerebral dysfunction with convulsive disorders, motor retardation, and mental retardation. No specific treatment is available.
Medication Effect on Pyrimidine Metabolism A variety of medications can influence pyrimidine metabolism. The anticancer agents fluorodeoxyuridine and 5-fluorouracil and the antimicrobial agent fluorocytosine cause cytotoxicity when converted to fluorodeoxyuridylate, a specific suicide inhibitor of thymidylate synthase. Fluorocytosine must be converted to 5-fluorouracil to be effective. This conversion is catalyzed by cytosine deaminase activity. Fluorocytosine’s action is selective because cytosine deaminase is present in bacteria and fungi but not in human cells. Dihydropyrimidine dehydrogenase is involved in the degradation of 5-fluorouracil. Consequently, deficiency of this enzyme is associated with 5-fluorouracil neurotoxicity.
Leflunomide, which is used to treat rheumatoid arthritis, inhibits de novo pyrimidine synthesis by inhibiting dihydroorotate dehydrogenase, resulting in an antiproliferative effect on T cells. Allopurinol, which inhibits xanthine oxidase in the purine metabolic pathway, also inhibits the activity of orotidine-5′-phosphate decarboxylase, a step in UMP synthesis. Consequently, allopurinol use is associated with increased excretion of orotidine and orotic acid. There are no known clinical effects of this inhibition.
432e |
Lysosomal Storage Diseases |
Lysosomes are heterogeneous subcellular organelles containing specific hydrolyses that allow selective processing or degradation of proteins, nucleic acids, carbohydrates, and lipids. There are more than 40 different lysosomal storage diseases (LSDs), classified according to the nature of the stored material (Table 432e-1). Several of the most prevalent disorders are reviewed here: Tay-Sachs disease, Fabry disease, Gaucher disease, Niemann-Pick disease, lysosomal acid lipase deficiencies, the mucopolysaccharidoses, and Pompe disease. LSDs should be considered in the differential diagnosis of patients with neurologic, renal, or muscular degeneration and/or unexplained hepatomegaly, splenomegaly, cardiomyopathy, or skeletal dysplasias and deformations. Physical findings are disease specific, and enzyme assays or genetic testing can be used to make a definitive diagnosis. Although the nosology of LSDs segregates the variants into distinct phenotypes, these are heuristic; in the clinic, each disease exhibits—to some degree—a continuous spectrum of manifestations, from severe to attenuated variants.
|
SELECTED LYSOSOMAL STORAGE DISEASES |
PATHOGENESIS
Lysosomal biogenesis involves ongoing synthesis of lysosomal hydrolases, membrane constitutive proteins, and new membranes. Lysosomes originate from the fusion of trans-Golgi network vesicles with late endosomes. Progressive vesicular acidification accompanies the maturation of these vesicles; this gradient facilitates the pH-dependent dissociation of receptors and ligands and also activates lysosomal hydrolases. Lysosomes are components of the lysosome/autophagy/mitophagy system, which can be disrupted in the LSDs.
Abnormalities at any biosynthetic step can impair enzyme activation and lead to a lysosomal storage disorder. After leader sequence clipping, remodeling of complex oligosaccharides (including the lysosomal targeting ligand mannose-6-phosphate as well as high-mannose oligosaccharide chains of many soluble lysosomal hydrolases) occurs during transit through the Golgi. Lysosomal integral or associated membrane proteins are sorted to the membrane or interior of the lysosome by several different peptide signals. Phosphorylation, sulfation, additional proteolytic processing, and macromolecular assembly of heteromers occur concurrently. Such posttranslational modifications are critical to enzyme function, and defects can result in multiple enzyme/protein deficiencies.
The final common pathway for LSDs is the accumulation of specific macromolecules within tissues and cells that normally have a high flux of these substrates. The majority of lysosomal enzyme deficiencies result from point mutations or genetic rearrangements at a locus that encodes a single lysosomal hydrolase. However, some mutations cause deficiencies of several different lysosomal hydrolases by alteration of the enzymes/proteins involved in targeting, active site modifications, or macromolecular association or trafficking. All LSDs are inherited as autosomal recessive disorders except for Hunter (mucopolysaccharidosis type II), Danon, and Fabry diseases, which are X-linked. Substrate accumulation leads to lysosomal distortion, which has significant pathologic consequences. In addition, abnormal amounts of metabolites may also have pharmacologic effects important to disease pathophysiology and propagation.
For many LSDs, the accumulated substrates are endogenously synthesized within particular tissue sites of pathology. Other diseases have greater exogenous substrate supplies. For example, they are delivered by low-density lipoprotein receptor–mediated uptake in Fabry and cholesteryl ester storage diseases (CESDs) or by phagocytosis in Gaucher disease type 1. The threshold hypothesis refers to a level of enzyme activity below which disease develops; small changes in enzyme activity near the threshold can lead to or prevent disease. A critical element of this model is that enzymatic activity can be challenged by changes in substrate flux based on genetic background, cell turnover, recycling, or metabolic demands. Thus, a set level of residual enzyme may be adequate for substrate in some tissues or cells but not in others. In addition, several variants of each LSD exist at a clinical level. These disorders therefore represent a continuum of manifestations that are not easily dissociated into discrete entities. The bases for such variations have not been elucidated in any detail.
SELECTED DISORDERS
TAY-SACHS DISEASE
About 1 in 30 Ashkenazi Jews is a carrier for Tay-Sachs disease, which is caused by total hexosaminidase A (Hex A) deficiency resulting from defective α-chains. The infantile form is a fatal neurodegenerative disease with macrocephaly, loss of motor skills, increased startle reaction, and a macular cherry red spot. The juvenile-onset form presents as ataxia and dementia, with death by age 10–15 years. The adult-onset disorder is characterized by clumsiness in childhood; progressive motor weakness in adolescence; and additional spinocerebellar and lower-motor-neuron symptoms and dysarthria in adulthood. Intelligence declines slowly, and psychosis is common. Screening for Tay-Sachs disease carriers is recommended in the Ashkenazi Jewish population. Sandhoff disease, due to a deficiency in both Hex A and Hex B resulting from defective β-chains, is phenotypically similar to Tay-Sachs disease but also includes hepatosplenomegaly and bony dysplasias.
FABRY DISEASE
Fabry disease is an X-linked disorder that results from mutations in the α-galactosidase A gene. The estimated prevalence of hemizygous males ranges from 1 in 40,000 to 1 in 3500 in selected populations. Clinically, the disease manifests with angiokeratomas (telangiectatic skin lesions), hypohidrosis, corneal and lenticular opacities, acroparesthesia; and progressive small-vessel disease of the kidney, heart, and brain.
The angiokeratomas and acroparesthesias may appear in childhood. Angiokeratomas are punctate, dark red to blue-black, flat or slightly raised, and usually symmetric; they do not blanch with pressure. They range from barely visible to several millimeters in diameter and have a tendency to increase in size and number with age. They usually are most dense between the umbilicus and the knees—the “bathing suit area”—but may occur anywhere, including the mucosal surfaces. Angiokeratomas also occur in several other very rare LSDs. Corneal and lenticular lesions, detectable on slit-lamp examination, may help in establishing a diagnosis of Fabry disease. Debilitating episodic burning pain of the hands, feet, and proximal extremities (acroparesthesia) can last from minutes to days and can be precipitated by changes in temperature, exercise, fatigue, or fever. Abdominal pain can resemble that from appendicitis or renal colic. Proteinuria, isosthenuria, and progressive renal dysfunction occur in the second to fourth decades; ~5% of male patients with idiopathic renal failure have α-galactosidase A mutations. Hypertension, left ventricular hypertrophy, anginal chest pain, and congestive heart failure can occur in the third to fourth decades. About 1–3% of patients with idiopathic hypertrophic myocardiopathy have Fabry disease. Similarly, ~3–5% of male patients with idiopathic stroke at 35–50 years of age have α-galactosidase A mutations. Leg lymphedema without hypoproteinemia and episodic diarrhea also occur. Death is due to renal failure or cardiovascular or cerebrovascular disease in untreated male patients. Variants with residual α-galactosidase A activity may have late-onset manifestations that are limited to the cardiovascular system and resemble hypertrophic cardiomyopathy. Variants with predominant cardiac, renal, or central nervous system (CNS) manifestations are becoming better defined. Up to 70% of heterozygous females may exhibit clinical manifestations. However, in females, heart disease is the most common life-threatening manifestation, followed in frequency by stroke and then renal disease.
Phenytoin and carbamazepine diminish chronic and episodic acroparesthesia. Chronic hemodialysis or kidney transplantation can be lifesaving in patients with renal failure. Enzyme therapy clears stored lipids from a variety of cells, particularly those of the renal, cardiac, and skin vascular endothelium. Renal insufficiency appears to be irreversible. Early institution of enzyme therapy may prevent or slow the progression of life-threatening complications.
GAUCHER DISEASE
Gaucher disease is an autosomal recessive disorder that results from defective activity of acid β-glucosidase; ~400 mutations have been described at the GBA1 locus of such patients. Disease variants are classified by the absence or presence and progression of neuronopathic involvement.
Gaucher disease type 1 is a nonneuronopathic disease that can present in childhood to adulthood as slowly to rapidly progressive visceral disease. About 55–60% of patients are diagnosed at <20 years of age in white populations and at even younger ages in other groups. This pattern of presentation is distinctly bimodal, with peaks at <10–15 years and at ~25 years. Younger patients tend to have a greater degree of hepatosplenomegaly and accompanying blood cytopenias. In contrast, the older group has a greater tendency for chronic bone disease. Hepatosplenomegaly occurs in virtually all symptomatic patients and can be minor or massive. Accompanying anemia and thrombocytopenia are variable and are not directly related to liver or spleen volumes. Severe liver dysfunction is unusual. Splenic infarctions can resemble an acute abdomen. Pulmonary hypertension and alveolar Gaucher cell accumulation are uncommon but life-threatening and can occur at any age. GBA1 mutations in the hetero- or homozygous state are a significant risk factor for early-onset or more rapidly progressive Parkinson disease.
All patients with Gaucher disease have nonuniform infiltration of bone marrow by lipid-laden macrophages termed Gaucher cells. This phenomenon can lead to marrow packing with subsequent infarction, ischemia, necrosis, and cortical bone destruction. Bone marrow involvement spreads from proximal to distal in the limbs and can involve the axial skeleton extensively, causing vertebral collapse. In addition to bone marrow involvement, bone remodeling is defective, with loss of total bone calcium leading to osteopenia, osteonecrosis, avascular infarction, and vertebral compression fractures and spinal cord involvement. Aseptic necrosis of the femoral head is common, as is fracture of the femoral neck. The mechanism by which diseased bone marrow macrophages interact with osteoclasts and/or osteoblasts to cause bone disease is not well understood. Chronic, ill-defined bone pain can be debilitating and poorly correlated with radiographic findings. “Bone crises” are associated with localized excruciating pain and, on occasion, local erythema, fever, and leukocytosis. These crises represent acute infarctions of bone, as evidenced in nuclear scans by localized absent uptake of pyrophosphate agents. Decreased acid β-glucosidase activity (0–20% of normal) in nucleated cells establishes the diagnosis. The enzyme is not present in bodily fluids. The sensitivity of enzyme testing is poor for heterozygote detection; molecular testing by GBA1 sequencing is preferred. The disease frequency varies from about 1 in 1000 among Ashkenazi Jews to <1 in 100,000 in other populations; ~1 in 12–15 Ashkenazi Jews carries a Gaucher disease allele. Four common mutations account for ~85% of the mutations in that population of affected patients: N370S (1226G), 84GG (a G insertion at cDNA position 84), L444P (1448C), and IVS-2 (an intron 2 splice junction mutation).
Genotype/phenotype studies indicate a significant, though not absolute, correlation between disease type and severity and the GBA1 genotype. The most common mutation in the Ashkenazi Jewish population (N370S) shares a 100% association with nonneuronopathic or type 1 Gaucher disease. The N370S/N370S and N370S/other mutant allele genotypes are associated with later-onset/less severe disease and with earlier-onset/severe disease, respectively. As many as 50–60% of individuals with the N370S/N370S genotype are asymptomatic. Other alleles include L444P (very low activity), 84GG (null), or IVS-2 (null) and rare/private or uncharacterized alleles. The L444P/L444P patients almost always have life-threatening to very severe/early-onset disease, and many, though not all, develop CNS involvement in the first two decades of life.
Symptom-based treatment of blood cytopenias and joint replacement surgeries continue to have important roles in management. However, regular intravenous enzyme therapy is currently the treatment of choice in significantly affected patients and is highly efficacious and safe in diminishing hepatosplenomegaly and improving hematologic values. Bone disease is decreased but not eliminated by enzyme therapy. Adult patients may benefit from adjunctive treatment with bisphosphonates to improve bone density. Patients who cannot be treated with enzyme, either because it is not effective or because they have an allergy or other hypersensitivities, may be receive substrate reduction therapy with medications that decrease the production of the complex lipid molecules that are broken down by acid β-glucosidase.
Gaucher disease type 2 is a rare, severe, progressive CNS disease that leads to death by 2 years of age. Gaucher disease type 3 has highly variable manifestations in the CNS and viscera. It can present in early childhood with rapidly progressive, massive visceral disease and slowly progress to static CNS involvement; in adolescence with dementia; or in early adulthood with rapidly progressive, uncontrollable myoclonic seizures and mild visceral disease. Visceral disease in type 3 is nearly identical to that in type 1 but is generally more severe. Early CNS findings may be limited to defects in lateral gaze tracking, which may remain static for decades. Mental retardation can be slowly progressive or static. This variant is most frequent among individuals of Swedish descent. Visceral—but not CNS—involvement responds to enzyme therapy.
NIEMANN-PICK DISEASE
Niemann-Pick diseases are autosomal recessive disorders that result from defects in acid sphingomyelinase. Types A and B are distinguished by the early age of onset and progressive CNS disease in type A. Type A typically has its onset in the first 6 months of life, with rapidly progressive CNS deterioration, spasticity, failure to thrive, and massive hepatosplenomegaly. Type B has a later, more variable onset and is characterized by a progression of hepatosplenomegaly, with eventual development of cirrhosis and hepatic replacement by foam cells. Affected patients develop progressive pulmonary disease with dyspnea, hypoxemia, and a reticular infiltrative pattern on chest x-ray. Foam cells are present in alveoli, lymphatic vessels, and pulmonary arteries. Progressive hepatic or lung disease leads to death in adolescence or early adulthood.
The diagnosis is established by markedly decreased (1–10% of normal) sphingomyelinase activity in nucleated cells. There is no specific treatment for Niemann-Pick disease. The efficacy of hepatic or bone marrow transplantation has not been clearly established. Clinical trials of enzyme therapy are in phases 2 and 3.
Niemann-Pick C diseases are progressive CNS diseases due to mutations in either NPC1 or NPC2. They present with liver or splenic disease, but their major manifestations are progressive CNS disease over one to two decades. Treatment with substrate inhibition agents (e.g., Miglustat) and substrate depletion with cyclodextrin have shown promise.
LYSOSOMAL ACID LIPASE
Lysosomal acid lipase deficiency may result in Wolman disease (severe deficiency) or CESD, which presents later and has some (3–10%) residual enzyme activity. Wolman disease presents in early infancy with hepatosplenomegaly, diarrhea, vomiting, and abdominal distention, sometimes accompanied by adrenal calcification, anemia, and mixed hyperlipidemia. Death occurs before the age of 1 year and is often due to severe intestinal malabsorption. CESD is heterogeneous and presents with hepatomegaly and hepatosteatosis at any age in childhood or adulthood. CESD should be included in the differential diagnosis for all patients with isolated hypercholesterolemia. The disease may progress to hepatic fibrosis, cirrhosis, and liver failure. In addition, patients often develop very early-onset atherosclerotic vascular disease, which may be life-threatening in childhood. Preliminary results of treatment with enzyme replacement therapy are promising for Wolman disease and CESD.
MUCOPOLYSACCHARIDOSES
Mucopolysaccharidosis type I (MPS I) is an autosomal recessive disorder caused by deficiency of α-L-iduronidase. The continuum of involvement traditionally has been divided into three categories: (1) Hurler disease (MPS I H) for severe deficiency with neurodegeneration, (2) Scheie disease (MPS I S) for later-onset disease without neurologic involvement and with relatively less severe disease in other organ systems, and (3) Hurler-Scheie syndrome (MPS I H/S) for patients intermediate between these extremes. MPS I H/S is characterized by severe somatic disease, usually without overt neurologic deterioration.
MPS I often presents in infancy or early childhood as chronic rhinitis, clouding of the corneas, and hepatosplenomegaly. As the disease progresses, nearly every organ system can be affected. In the more severe forms, cardiac and respiratory diseases become life-threatening in childhood. Skeletal disease can be quite severe, resulting in very limited mobility.
There are two current treatments for the MPS I diseases. Hematopoietic stem cell transplantation (HSCT) is the standard treatment for patients presenting at <2 years of age who appear to have or are at risk for neurologic degeneration. HSCT results in stabilization of CNS disease and reverses hepatosplenomegaly. It also beneficially affects cardiac and respiratory disease. HSCT does not eliminate corneal disease or result in the resolution of progressive skeletal disease. Enzyme therapy effectively addresses hepatosplenomegaly and alleviates cardiac and respiratory disease. The enzyme does not effectively penetrate the CNS and does not directly affect CNS disease. Enzyme therapy and HSCT appear to have similar effects on visceral signs and symptoms. Enzyme therapy poses a lower risk of life-threatening complications and may therefore be advantageous for patients who have attenuated manifestations without CNS disease. A combination of enzyme therapy and HSCT has been used, with enzyme therapy initiated prior to transplantation in an attempt to reduce the disease burden. The experience with this approach is not well documented, but it appears to have advantages over HSCT alone.
Enzyme therapy for Maroteaux-Lamy disease (MPS VI) has received U.S. Food and Drug Administration (FDA) approval. This very rare autosomal recessive disorder is characterized by hepatosplenomegaly, bone disease, heart disease, and respiratory compromise similar to those seen in MPS I; however, MPS VI is due to deficiency of arylsulfatase B and is not associated with neurologic degeneration.
Hunter disease (MPS II) is an X-linked disorder due to deficiency in iduronate sulfate sulfatase and has manifestations similar to those of MPS I, including neurologic degeneration. There is no corneal clouding or other eye disease. Like MPS I, MPS II is clinically variable, with CNS and non-CNS variants. HSCT has not been successful in treating CNS disease associated with MPS II. The FDA and the European Medicines Agency (EMA) have approved enzyme therapy for the visceral manifestations of MPS II.
POMPE DISEASE
Acid maltase (acid α-glucosidase, GAA) deficiency, also called Pompe disease, is the only glycogen LSD. The classic severe infantile form presents with hypotonia, myocardiopathy, and hepatosplenomegaly. This variant is rapidly progressive and generally results in death in the first year of life. However, as with other LSDs, there are early- and late-onset forms of this disorder. The late-onset variants may be as common as 1 in 40,000; patients typically present with a slowly progressive myopathy that may resemble limb-girdle muscular dystrophy. Respiratory insufficiency may be the presenting sign or may develop with advancing disease. In late stages of the disease, patients may require mechanical ventilation, report swallowing difficulties, and experience loss of bowel and bladder control. Myocardiopathy is not usually seen in late-onset variants of Pompe disease.
The FDA and EMA have approved enzyme therapy for Pompe disease. This treatment clearly prolongs life in the infantile form, consistently resulting in improved cardiac function. Respiratory function is also improved in most treated infants. Some infants demonstrate marked improvement in motor functions, while others have minor changes in muscle tone or strength. Prevention of deterioration has been shown with GAA enzyme therapy in the late-onset forms. Early intervention with GAA enzyme therapy in such patients may limit or prevent deterioration, but very advanced disease will have significant irreversible components.
433e |
Glycogen Storage Diseases and Other Inherited Disorders of Carbohydrate Metabolism |
Carbohydrate metabolism plays a vital role in cellular function by providing the energy required for most metabolic processes. The relevant biochemical pathways involved in the metabolism of these carbohydrates are shown in Fig. 433e-1. Glucose is the principal substrate of energy metabolism in humans. Metabolism of glucose generates ATP through glycolysis and mitochondrial oxidative phosphorylation. The body obtains glucose through the ingestion of polysaccharides (primarily starch) and disaccharides (e.g., lactose, maltose, and sucrose). Galactose and fructose are two other monosaccharides that serve as sources of fuel for cellular metabolism; however, their role as fuel sources is much less significant than that of glucose. Galactose is derived from lactose (galactose + glucose), which is found in milk products, and is an important component of certain glycolipids, glycoproteins, and glycosaminoglycans. Fructose is found in fruits, vegetables, and honey. Sucrose (fructose + glucose) is another dietary source of fructose and is a commonly used sweetener.
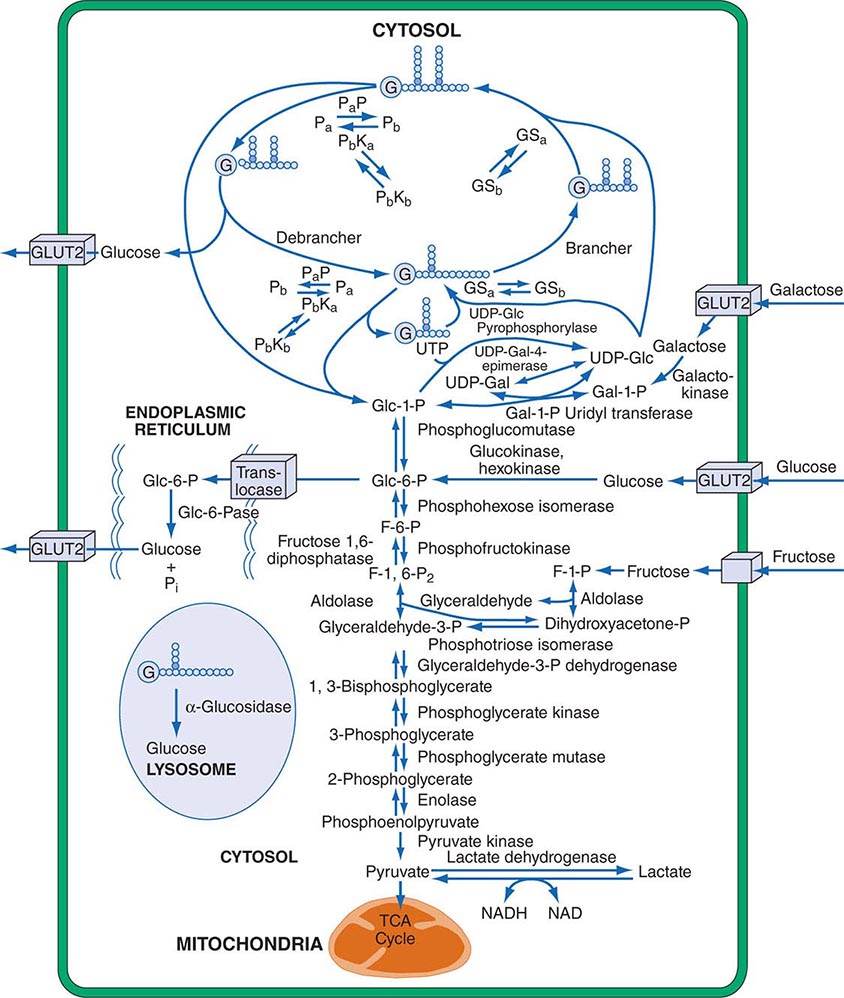
FIGURE 433e-1 Metabolic pathways related to glycogen storage diseases and galactose and fructose disorders. Nonstandard abbreviations are as follows: GSa, active glycogen synthase; GSb, inactive glycogen synthase; Pa, active phosphorylase; Pb, inactive phosphorylase; PaP, phosphorylase α phosphatase; PbKa, active phosphorylase β kinase; PbKb, inactive phosphorylase β kinase; G, glycogenin, the primer protein for glycogen synthesis. (Modified from AR Beaudet, in KJ Isselbacher et al [eds]: Harrison’s Principles of Internal Medicine, 13th ed., New York, McGraw-Hill, 1994, p 1855.)
Glycogen, the storage form of glucose in animal cells, is composed of glucose residues joined in straight chains by α1-4 linkages and branched at intervals of 4–10 residues by α1-6 linkages. Glycogen forms a treelike molecule and can have a molecular weight of many millions. Glycogen may aggregate to form structures recognizable by electron microscopy. With the exception of type 0 disease, defects in glycogen metabolism typically cause an accumulation of glycogen in the tissues—hence the designation glycogen storage diseases (GSDs). The structure of stored glycogen can be normal or abnormal in the various disorders. Defects in gluconeogenesis or glycolytic pathways, including galactose and fructose metabolism, usually do not result in glycogen accumulation.
Clinical manifestations of the various disorders of carbohydrate metabolism differ markedly. The symptoms range from harmless to lethal. Unlike disorders of lipid metabolism, mucopolysaccharidoses, or other storage diseases, many carbohydrate disorders have been effectively managed with dietary therapy. All of the genes responsible for inherited defects of carbohydrate metabolism have been cloned, and mutations have been identified. Advances in our understanding of the molecular basis of these diseases are being used to improve diagnosis and management. Some of these disorders are candidates for enzyme replacement therapy, substrate reduction therapy, and early trials of gene therapy.
Historically, the GSDs were categorized numerically in the order in which the enzymatic defects were identified. They are also classified by the organs involved (liver, muscle, and/or heart) and clinical manifestations. The latter is the system followed in this chapter (Table 433e-1). The overall frequency of all forms of GSD is ~1 in 20,000 live births. Most are inherited as autosomal recessive traits; however, phosphoglycerate kinase deficiency—one form of liver phosphorylase kinase (PhK) deficiency—and lysosomal-associated membrane protein 2 (LAMP2) deficiency are X-linked disorders. The most common childhood disorders are glucose-6-phosphatase deficiency (type I), lysosomal acid α-glucosidase deficiency (type II), debrancher deficiency (type III), and liver PhK deficiency (type IX). The most common adult disorder is myophosphorylase deficiency (type V, or McArdle disease).
|
FEATURES OF GLYCOGEN STORAGE DISEASES AND GALACTOSE AND FRUCTOSE DISORDERS |
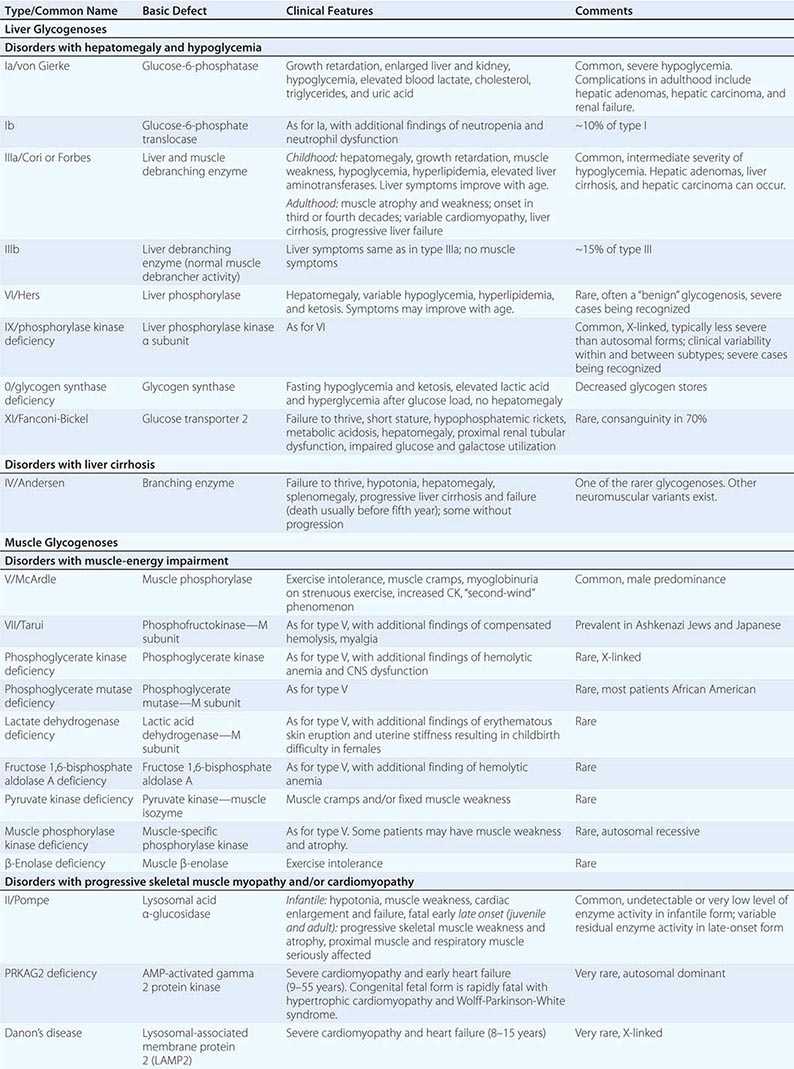

SELECTED LIVER GLYCOGENOSES
DISORDERS WITH HEPATOMEGALY AND HYPOGLYCEMIA
Type I GSD (Glucose-6-Phosphatase or Translocase Deficiency, Von Gierke’s Disease) Type I GSD is an autosomal recessive disorder caused by glucose-6-phosphatase deficiency in liver, kidney, and intestinal mucosa. There are two subtypes of GSD I: type Ia, in which the glucose-6-phosphatase enzyme is defective, and type Ib, in which the translocase that transports glucose-6-phosphate across the microsomal membrane is defective. The defects in both subtypes lead to inadequate conversion of glucose-6-phosphate to glucose in the liver and thus make affected individuals susceptible to fasting hypoglycemia.
CLINICAL AND LABORATORY FINDINGS Persons with type I GSD may develop hypoglycemia and lactic acidosis during the neonatal period; however, more commonly, they exhibit hepatomegaly at 3–4 months of age. Hypoglycemia, hypoglycemic seizures, and lactic acidosis can develop after a short fast. These children usually have doll-like faces with fat cheeks, relatively thin extremities, short stature, and a protuberant abdomen that is due to massive hepatomegaly. The kidneys are enlarged, but the spleen and heart are of normal size. The hepatocytes are distended by glycogen and fat, with large and prominent lipid vacuoles. Despite hepatomegaly, liver enzyme levels are usually normal or near normal. Easy bruising and epistaxis are associated with a prolonged bleeding time as a result of impaired platelet aggregation/adhesion. Hyperuricemia is present. Hyperlipidemia includes elevation of triglycerides, low-density lipoproteins, and phospholipids. Type Ib patients have additional findings of neutropenia and impaired neutrophil function, which result in recurrent bacterial infections and oral and intestinal mucosal ulceration. GSD I patients may experience intermittent diarrhea, which can worsen with age. In GSD Ib, diarrhea is largely due to loss of mucosal barrier function caused by inflammation.
LONG-TERM COMPLICATIONS Gout usually becomes symptomatic at puberty as a result of long-term hyperuricemia. Puberty is often delayed. Nearly all female patients have ultrasound findings consistent with polycystic ovaries; however, the other clinical features of polycystic ovary syndrome, such as acne and hirsutism, are not seen. Several reports of successful pregnancy in women with GSD I suggest that fertility is not affected. Increased bleeding during menstrual cycles, including life-threatening menorrhagia, has been reported. Secondary to lipid abnormalities, there is an increased risk of pancreatitis. Patients with GSD I may be at increased risk for cardiovascular disease. In adult patients, frequent fractures can occur and radiographic evidence of osteopenia/osteoporosis can be found; in prepubertal patients, radial bone mineral content is significantly reduced. Pulmonary hypertension—although rare—has been reported. By the second or third decade of life, many patients with type I GSD develop hepatic adenomas that can hemorrhage and, in some cases, become malignant. Renal disease is a serious late complication. Almost all patients older than 20 years have proteinuria, and many have hypertension, kidney stones, nephrocalcinosis, and altered creatinine clearance. Laboratory findings include abnormally high levels of blood lactate, triglycerides, cholesterol, and uric acid. In some patients, renal function deteriorates and progresses to complete failure, requiring dialysis or transplantation.
DIAGNOSIS Clinical presentation and abnormal plasma lactate and lipid values suggest that a patient may have GSD I, but gene-based mutation analysis provides a noninvasive means of reaching a definitive diagnosis for most patients with types Ia and Ib disease. Before the glucose-6-phosphatase and glucose-6-phosphate translocase genes were cloned, a definitive diagnosis required a liver biopsy to demonstrate a deficiency.
Type III GSD (Debrancher Deficiency, Limit Dextrinosis) Type III GSD is an autosomal recessive disorder caused by a deficiency of glycogen debranching enzyme. Debranching and phosphorylase enzymes are responsible for complete degradation of glycogen. When the debranching enzyme is defective, glycogen breakdown is incomplete. Abnormal glycogen accumulates with short outer chains and resembles dextrin.
CLINICAL AND LABORATORY FINDINGS Deficiency of glycogen debranching enzyme causes hepatomegaly, hypoglycemia, short stature, variable skeletal myopathy, and cardiomyopathy. The disorder usually involves both liver and muscle and, in such cases, is termed type IIIa GSD. However, in ~15% of patients, the disease appears to involve only the liver and is classified as type IIIb. Hypoglycemia and hyperlipidemia occur in children. In type III disease (as opposed to type I disease), fasting ketosis can be prominent, aminotransferase levels are elevated, and blood lactate and uric acid concentrations are usually normal. Serum creatine kinase (CK) levels can sometimes be used to identify patients with muscle involvement, but normal levels do not rule out muscle enzyme deficiency. In most patients with type III disease, hepatomegaly improves with age; however, liver cirrhosis and hepatocellular carcinoma may occur in late adulthood. Hepatic adenomas may occur, although less commonly than in GSD I. Left ventricular hypertrophy and life-threatening arrhythmias have been reported. Patients with type IIIa disease may experience muscle weakness in childhood that can become severe after the third or fourth decade of life. Polycystic ovaries are common in GSD III, and some patients develop features of polycystic ovarian syndrome, such as hirsutism and irregular menstrual cycles. Reports of successful pregnancy in women with GSD III suggest that fertility is normal.
DIAGNOSIS In type IIIa GSD, deficient debranching enzyme activity can be demonstrated in liver, skeletal muscle, and heart. In contrast, patients with type IIIb have debranching enzyme deficiency in the liver but not in muscle. The liver has distended hepatocytes due to glycogen buildup; areas of fibrosis are also noted very early in the disease course. In the past, definitive assignment of subtype required enzyme assays in both liver and muscle. DNA-based analyses now provide a noninvasive way of subtyping these disorders in most patients. However, the large size of the gene and the distribution of private mutations across it pose challenges in DNA-based analysis.
Type IX GSD (Liver Phosphorylase Kinase Deficiency) Defects of PhK cause a heterogeneous group of glycogenoses. The PhK enzyme complex consists of four subunits (α, β, γ, and δ). Each subunit is encoded by different genes (X chromosome as well as autosomes) that are differentially expressed in various tissues. PhK deficiency can be divided into several subtypes on the basis of the gene/subunit involved, the tissues primarily affected, and the mode of inheritance. The most common subtype is X-linked liver PhK deficiency, which is also one of the most common liver glycogenoses. PhK activity may also be deficient in erythrocytes and leukocytes but is normal in muscle. Typically, a child between the ages of 1 year and 5 years presents with growth retardation and hepatomegaly. Children tend eventually to exhibit normal growth patterns initiated by a delayed growth spurt during puberty. Liver fibrosis has been identified in some patients, including children. Levels of cholesterol, triglycerides, and liver enzymes are mildly elevated. Fasting ketosis is another feature of the disease. Lactic and uric acid levels are usually normal. Hypoglycemia is typically mild; however, phenotypic variability is being increasingly recognized, with significant involvement in some cases of the X-linked form. The accumulated glycogen in liver (β particles, rosette form) has a frayed or burst appearance and is less compact than the glycogen seen in type I or type III GSD. Hepatomegaly and abnormal blood chemistries gradually return to normal with age. Most adults reach a normal final height and are practically asymptomatic, despite persistent PhK deficiency. The prognosis is usually good, and adult patients have minimal hepatomegaly. Some patients have significant ketosis and progressive liver disease that can advance to fibrosis and liver failure. It is recommended that adult patients be monitored for hepatic complications with regular CT or MRI scans.
Treatment is symptom based. A high-carbohydrate diet and frequent feedings are effective in preventing hypoglycemia. Some patients require no specific treatment. Recent studies have shown that instituting a treatment regimen of cornstarch and protein feedings early, even in seemingly stable patients, may prevent long-term complications. Blood ketones and glucose should be evaluated during times of stress.
Other subtypes of type IX GSD include an autosomal recessive form of liver and muscle PhK deficiency, an autosomal recessive form of liver PhK deficiency that often develops into liver cirrhosis, a muscle-specific PhK deficiency that causes cramps and myoglobinuria with exercise, and a cardiac-specific PhK deficiency that is lethal during infancy because of massive glycogen deposition in the myocardium. The finding of PhK deficiency in the cardiac-specific PhK deficiency may be a secondary phenomenon, as a subset of these patients have mutations in PRKAG2.
Other Liver Glycogenoses with Hepatomegaly and Hypoglycemia These disorders include hepatic phosphorylase deficiency (Hers disease, type VI) and hepatic glycogenosis with renal Fanconi syndrome (type XI). Patients with GSD type VI can have growth retardation, hyperlipidemia, and hyperketosis in addition to hepatomegaly and hypoglycemia. Some patients have a benign clinical course. GSD XI is caused by defects in the facilitative glucose transporter 2 (GLUT-2), which transports glucose and galactose in and out of hepatocytes, pancreatic cells, and the basolateral membranes of intestinal and renal epithelial cells. The disease is characterized by proximal renal tubular dysfunction, impaired glucose and galactose utilization, and accumulation of glycogen in liver and kidney.
SELECTED MUSCLE GLYCOGENOSES
DISORDERS WITH MUSCLE-ENERGY IMPAIRMENT
Type V GSD (Muscle Phosphorylase Deficiency, McArdle Disease) Type V GSD is an autosomal recessive disorder caused by deficiency of muscle phosphorylase. McArdle disease is a prototypical muscle-energy disorder as the enzyme deficiency limits ATP generation by glycogenolysis and results in glycogen accumulation.
CLINICAL AND LABORATORY FINDINGS Usually, symptoms first develop in adulthood and involve exercise intolerance with muscle cramps. Two types of activity tend to cause symptoms: (1) brief exercise of great intensity, such as sprinting or carrying heavy loads; and (2) less intense but sustained activity, such as climbing stairs or walking uphill. Most patients can engage in moderate exercise, such as walking on level ground, for long periods. Patients often exhibit the “second-wind” phenomenon, in which, after a short break from the initiation of strenuous physical effort, they are able to continue the activity without pain. Although most patients experience episodic muscle pain and cramping as a result of exercise, 35% report permanent pain that seriously affects sleep and other activities. About half of patients report burgundy-colored urine after exercise; this coloration results from myoglobinuria secondary to rhabdomyolysis. Intense myoglobinuria after vigorous exercise can lead to renal failure. Clinical heterogeneity is uncommon; however, there are cases with symptom onset as late as the eighth decade and cases that present early with hypotonia, generalized muscle weakness, and progressive respiratory insufficiency, which is often fatal.
Although cardiac involvement is not usually associated with muscle phosphorylase deficiency, hypertrophic cardiomyopathy has been observed in an adult patient with GSD V. In rare cases, electromyographic findings may suggest inflammatory myopathy, a diagnosis that may be confused with polymyositis. These patients may be at risk for statin-induced myopathy and rhabdomyolysis.
During rest, the serum CK level is usually elevated; after exercise, the CK level increases even more. Exercise also increases levels of blood ammonia, inosine, hypoxanthine, and uric acid; these abnormalities reflect residues of accelerated muscle purine nucleotide recycling as a result of insufficient ATP production. NADH is underproduced during physical exertion.
DIAGNOSIS Lack of an increase in blood lactate and exaggerated blood ammonia elevations after an ischemic exercise test are indicative of a muscle glycogenosis and suggest a defect in the conversion of glycogen or glucose to lactate. This abnormal exercise response, however, can also occur with other defects in glycogenolysis or glycolysis, such as deficiencies of muscle phosphofructokinase or debranching enzyme (when the test is done after fasting). The cycle test detects the hallmark heart rate observed during the second-wind phenomenon. A definitive diagnosis is made by enzymatic assay in muscle tissue or by mutation analysis of the myophosphorylase gene.
DISORDERS WITH PROGRESSIVE SKELETAL MUSCLE MYOPATHY AND/OR CARDIOMYOPATHY
Pompe Disease, Type II GSD (Acid α-1,4 Glucosidase Deficiency) Pompe disease is an autosomal recessive disorder caused by a deficiency of lysosomal acid α-1,4 glucosidase, an enzyme responsible for the degradation of glycogen in the lysosomes. This disease is characterized by the accumulation of glycogen in the lysosomes as opposed to accumulation in cytoplasm (as in the other glycogenoses).
CLINICAL AND LABORATORY FINDINGS The disorder encompasses a range of phenotypes. Each includes myopathy but differs in the age of onset, extent of organ involvement, and clinical severity. The most severe is the infantile form, with cardiomegaly, hypotonia, and death before the age of 1 year. Infants may appear normal at birth but soon develop generalized muscle weakness with feeding difficulties, macroglossia, hepatomegaly, and congestive heart failure due to hypertrophic cardiomyopathy.
The late-onset form (juvenile/late-childhood or adult form) is characterized by skeletal muscle manifestations, usually with minimal or no cardiac involvement, and a more slowly progressive course. The juvenile form typically presents as delayed motor milestones (if age of onset is early enough) and difficulty in walking. With disease progression, patients often develop swallowing difficulties, proximal muscle weakness, and respiratory muscle involvement. Death may occur before the end of the second decade.
Adults typically present between the second and seventh decades with slowly progressive myopathy without overt cardiac involvement. The clinical picture is dominated by slowly progressive proximal limb girdle muscle weakness. The pelvic girdle, paraspinal muscles, and diaphragm are most seriously affected. Respiratory symptoms include somnolence, morning headache, orthopnea, and exertional dyspnea. In rare instances, patients present with respiratory insufficiency as the initial symptom. Basilar artery aneurysms and dilation of the ascending aorta have been observed in patients with Pompe disease. Ptosis, lingual weakness, gastrointestinal dysmotility, and incontinence due to poor sphincter tone are now being recognized as part of the clinical spectrum. Individuals with advanced disease often require some form of ventilation and are dependent on a walking aid or wheelchair.
Laboratory findings include elevated levels of serum CK, aspartate aminotransferase, and lactate dehydrogenase. Levels of urine glucose tetrasaccharide (Hex4), a breakdown product of glycogen, are elevated, especially on the severe end of the disease spectrum. In infants, chest x-ray shows massive cardiomegaly, and electrocardiographic findings include a high-voltage QRS complex and a shortened PR interval. Muscle biopsy shows vacuoles that stain positive for glycogen; the muscle acid phosphatase level is increased, presumably from a compensatory increase of lysosomal enzymes. Electromyography reveals myopathic features, with irritability of muscle fibers and pseudomyotonic discharges. Serum CK is not always elevated in adults, and, depending on the muscle biopsied or tested, muscle histology or electromyography may not be abnormal. The affected muscle should be examined.
DIAGNOSIS The confirmatory step for a diagnosis of Pompe disease is enzyme assay demonstrating deficient acid α-glucosidase or a gene sequence with two pathogenic mutations in the GAA gene. Enzyme activity can be measured in muscle, cultured skin fibroblasts, or blood. Deficiency is usually more severe in the infantile form. Early diagnosis is the key to treatment efficacy.
GSD Mimicking Hypertrophic Cardiomyopathy Deficiency of LAMP2—also called Danon’s disease—or of the protein kinase, AMP-activated gamma 2 noncatalytic subunit (PRKAG2), results in the accumulation of glycogen in the heart and skeletal muscle. LAMP2 deficiency is X-linked, whereas PRKAG2 deficiency is autosomal dominant. Clinically, both subsets of patients present primarily with hypertrophic cardiomyopathy. Their electrophysiologic abnormalities, particularly ventricular preexcitation and conduction defects, can distinguish them from patients with hypertrophic cardiomyopathy resulting from defects in sarcomere-protein genes. In patients with LAMP2 deficiency, mental delays are common and the onset of cardiac symptoms, including chest pain, palpitation, syncope, and cardiac arrest, can occur between the ages of 8 and 15 years—i.e., earlier than the average age of 33 years for patients with PRKAG2 deficiency. Patients as young as 9 years old have presented with PRKAG2 deficiency. A rapidly fatal congenital form of PRKAG2 presents in early infancy with severe hypertrophic cardiomyopathy and Wolff-Parkinson-White syndrome. In these patients, levels of PhK have been found to be low. The prognosis for LAMP2 deficiency is poor, with progressive end-stage heart failure early in adulthood. By contrast, except in the fatal congenital form, long-term survival is possible for patients with cardiomyopathy due to PRKAG2 mutations. Some patients may require the implantation of a pacemaker and aggressive control of arrhythmias. Congestive heart failure has been documented in patients with PRKAG2 deficiency.
SELECTED DISORDERS OF GALACTOSE METABOLISM
“Classic” galactosemia is caused by galactose 1-phosphate uridyltransferase (GALT) deficiency. It is a serious disease with an incidence of 1 in 60,000 and an early onset of symptoms. The newborn infant normally receives up to 40% of caloric intake as lactose (glucose + galactose). Without the transferase, the infant is unable to metabolize galactose 1-phosphate (Fig. 433e-1), which consequently accumulates, resulting in injury to parenchymal cells of the kidney, liver, and brain. After the first feeding, infants can present with vomiting, diarrhea, hypotonia, jaundice, and hepatomegaly. Patients with galactosemia are at increased risk for Escherichia coli neonatal sepsis; the onset of sepsis often precedes the diagnosis of galactosemia.
Widespread newborn screening for galactosemia has identified these infants early and allowed them to be placed on dietary restriction. Elimination of galactose from the diet reverses growth failure as well as renal and hepatic dysfunction, improving the prognosis. However, on long-term follow-up, some patients still have ovarian failure manifesting as primary or secondary amenorrhea as well as developmental delays and learning disabilities that increase in severity with age. Of women with classic galactosemia, 80–90% or more report hypergonadotropic hypogonadism. While most female patients are infertile when they reach childbearing age, a few have given birth. Several mutations appear to be protective, particularly the p.Ser135Leu mutation, which is more common in the African-American population. Methods for fertility preservation, such as cryopreservation, are still in the experimental stages. In addition, most patients have speech disorders, and a smaller proportion demonstrate poor growth and impaired motor function and balance (with or without overt ataxia). Adults on dairy-free diets have developed cataracts, tremors, and low bone density. The treatment of galactosemia to prevent long-term complications remains a challenge.
Deficiency of galactokinase (Fig. 433e-1) causes cataracts. Deficiency of uridine diphosphate galactose 4-epimerase can be benign when the enzyme deficiency is limited to blood cells but can be as severe as classic galactosemia when the enzyme deficiency is generalized.
SELECTED DISORDERS OF FRUCTOSE METABOLISM
Fructokinase deficiency, or essential fructosemia (Fig. 433e-1), causes a benign condition that is usually an incidental finding made through the detection of fructose as a reducing substance in the urine.
Deficiency of fructose 1,6-bisphosphate aldolase (aldolase B; hereditary fructose intolerance) is a serious disease in infants. These patients are healthy and asymptomatic until fructose or sucrose (table sugar) is ingested (usually from fruit, sweetened cereal, or sucrose-containing formula). Clinical manifestations may include jaundice, hepatomegaly, vomiting, lethargy, irritability, and convulsions. The incidence of celiac disease is higher among patients with hereditary fructose intolerance (>10%) than in the general population (1–3%). Laboratory findings show prolonged clotting time, hypoalbuminemia, elevation of bilirubin and aminotransferase levels, and proximal renal tubular dysfunction. If the disease is not diagnosed and intake of the noxious sugar continues, hypoglycemic episodes recur, and liver and kidney failure progresses, eventually leading to death. Treatment requires the elimination of all sources of sucrose, fructose, and sorbitol from the diet. Through this treatment, liver and kidney dysfunction improve, and catch-up growth is common; intellectual development is usually unimpaired. Over time, the patient’s symptoms become milder, even after fructose ingestion, and the long-term prognosis is good.
Fructose 1,6-diphosphatase deficiency is characterized by childhood life-threatening episodes of acidosis, hypoglycemia, hyperventilation, convulsions, and coma. These episodes are triggered by febrile infections and gastroenteritis when oral food intake decreases. Laboratory findings show low blood glucose levels, high lactate and uric acid levels, and metabolic acidosis. Unlike hereditary fructose intolerance, this deficiency usually is not associated with an aversion to sweets, and renal tubular and liver functions are normal. Treatment of acute attacks requires the correction of hypoglycemia and acidosis by IV infusion. Later, avoidance of fasting and elimination of fructose and sucrose from the diet prevent further episodes. A slowly released carbohydrate such as cornstarch is useful for the long-term prevention of hypoglycemia. The prognosis is good, as patients who survive childhood develop normally.
GLOBAL CONSIDERATIONS
 The GSDs and other inherited disorders of carbohydrate metabolism, although rare, have been reported in most ethnic populations. The prevalent genetic mutations for each disease may vary in different ethnic populations, but clinical symptoms are remarkably similar and treatment guidelines apply to all populations. The practice of newborn screening should be considered worldwide to intercept the rapid progression of many of these disorders.
The GSDs and other inherited disorders of carbohydrate metabolism, although rare, have been reported in most ethnic populations. The prevalent genetic mutations for each disease may vary in different ethnic populations, but clinical symptoms are remarkably similar and treatment guidelines apply to all populations. The practice of newborn screening should be considered worldwide to intercept the rapid progression of many of these disorders.
434e |
Inherited Disorders of Amino Acid Metabolism in Adults |
Amino acids are not only the building blocks of proteins but also serve as neurotransmitters (glycine, glutamate, γ-aminobutyric acid) or as precursors of hormones, coenzymes, pigments, purines, or pyrimidines. Eight amino acids, referred to as essential, cannot be synthesized by humans and must be obtained from dietary sources. The others are formed endogenously. Each amino acid has a unique degradative pathway by which its nitrogen and carbon components are used for the synthesis of other amino acids, carbohydrates, and lipids. Disorders of amino acid metabolism and transport (Chap. 435e) are individually rare—the incidences range from 1 in 10,000 for cystinuria or phenylketonuria to 1 in 200,000 for homocystinuria or alkaptonuria—but collectively, they affect perhaps 1 in 1000 newborns. Almost all are transmitted as autosomal recessive traits.
The features of inherited disorders of amino acid catabolism are summarized in Table 434e-1. In general, these disorders are named for the compound that accumulates to highest concentration in blood (-emias) or urine (-urias). In the aminoacidopathies, the parent amino acid is found in excess, whereas products in the catabolic pathway accumulate in organic acidemias. Which compound(s) accumulates depends on the site of the enzymatic block, the reversibility of the reactions proximal to the lesion, and the availability of alternative pathways of metabolic “runoff.” Biochemical and genetic heterogeneity are common. Five distinct forms of hyperphenylalaninemia, nine forms of homocystinuria, and methylmalonic acidemia are recognized. Such heterogeneity reflects the presence of a large array of molecular defects.
|
INHERITED DISORDERS OF AMINO ACID METABOLISM |
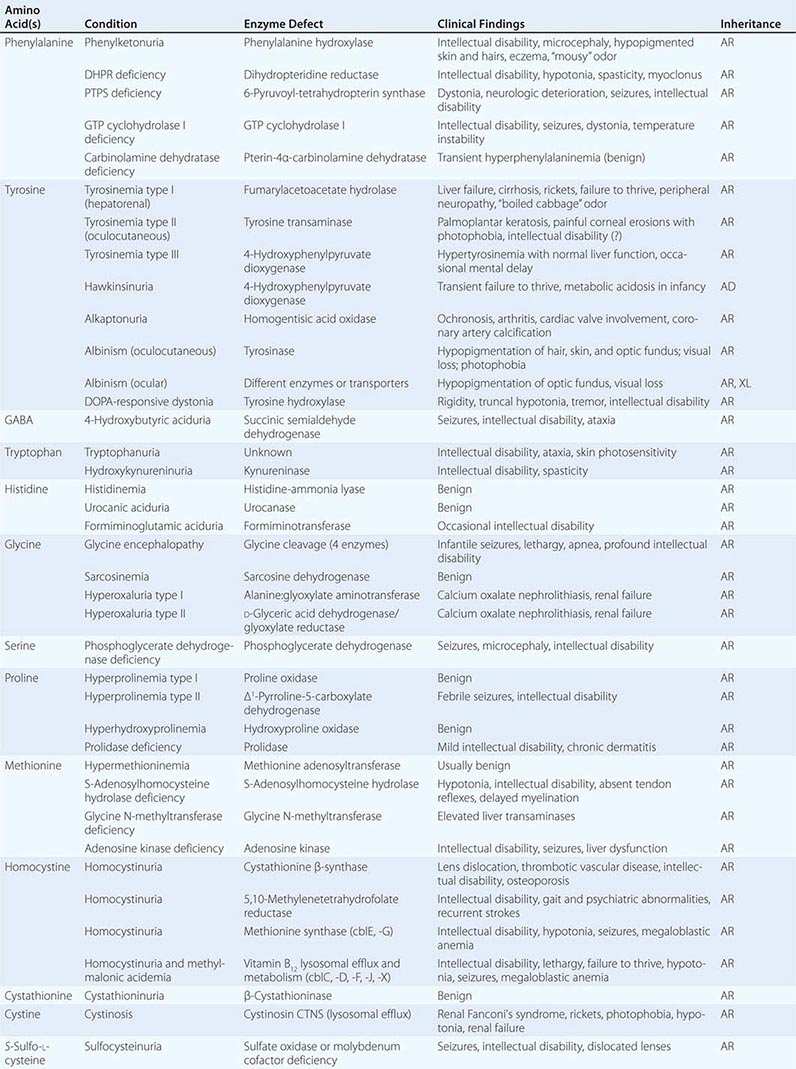
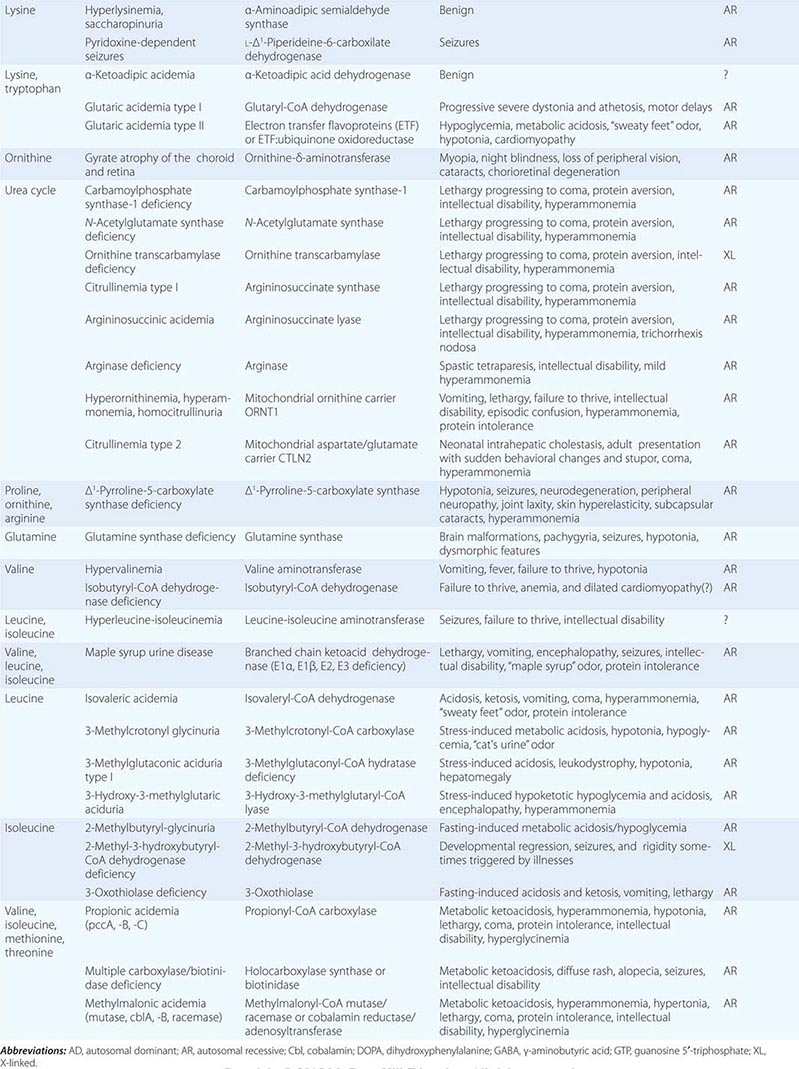
The manifestations of these conditions differ widely (Table 434e-1). Some, such as sarcosinemia, produce no clinical consequences. At the other extreme, complete deficiency of ornithine transcarbamylase is lethal in the untreated neonate. Central nervous system (CNS) dysfunction, in the form of developmental retardation, seizures, alterations in sensorium, or behavioral disturbances, is present in more than half the disorders. Protein-induced vomiting, neurologic dysfunction, and hyperammonemia occur in many disorders of urea cycle intermediates. Metabolic ketoacidosis, often accompanied by hyperammonemia, is a frequent presenting finding in disorders of branched-chain amino acid metabolism. Some disorders produce focal tissue or organ involvement such as liver disease, renal failure, cutaneous abnormalities, or ocular lesions.
The analysis of plasma amino acids (by ion-exchange chromatography), urine organic acids (by gas chromatography/mass spectrometry), and plasma acylcarnitine profile (by tandem mass spectrometry) is commonly used to diagnose and monitor most of these disorders. The diagnosis is confirmed by enzyme assay on cells or tissues from the patients or by DNA testing. The clinical manifestations in many of these conditions can be prevented or mitigated if a diagnosis is achieved early and appropriate treatment (e.g., dietary protein or amino acid restriction or vitamin supplementation) is instituted promptly. For this reason, newborn screening programs seek to identify several of these disorders. Infants with a positive screening test need additional metabolic testing (usually suggested by the newborn screening program) to confirm or exclude the diagnosis. Confirmed cases should be referred to a metabolic center for initiation of therapy. The parents need to be counseled about the recurrence risk of the disease in future pregnancies. In some cases, parents need testing to exclude metabolic alterations seen in carriers for some of these disorders (such as some forms of homocystinuria) or because they might have a disorder themselves (such as glutaric acidemia type 1, methylcrotonyl coenzyme A carboxylase deficiency, or fatty acid oxidation defects). Some metabolic disorders can remain asymptomatic until adult age, presenting only when fasting or severe stress require full activity of affected metabolic pathways to provide energy.
Selected disorders that illustrate the principles, properties, and problems presented by the disorders of amino acid metabolism are discussed in this chapter.
THE HYPERPHENYLALANINEMIAS
The hyperphenylalaninemias (Table 434e-1) result from impaired conversion of phenylalanine to tyrosine. The most common and clinically important is phenylketonuria (frequency 1:10,000), which is an autosomal recessive disorder characterized by an increased concentration of phenylalanine and its by-products in body fluids and by severe mental retardation if untreated in infancy. It results from reduced activity of phenylalanine hydroxylase. The accumulation of phenylalanine inhibits the transport of other amino acids required for protein or neurotransmitter synthesis, reduces synthesis and increases degradation of myelin, and leads to inadequate formation of norepinephrine and serotonin. Phenylalanine is a competitive inhibitor of tyrosinase, a key enzyme in the pathway of melanin synthesis, and accounts for the hypopigmentation of hair and skin. Untreated children with classic phenylketonuria are normal at birth but fail to attain early developmental milestones, develop microcephaly, and demonstrate progressive impairment of cerebral function. Hyperactivity, seizures, and severe intellectual disability are major clinical problems later in life. Electroencephalographic abnormalities; “mousy” odor of skin, hair, and urine (due to phenylacetate accumulation); and a tendency to develop hypopigmentation and eczema complete the devastating clinical picture. In contrast, affected children who are detected and treated at birth show none of these abnormalities.
|
TREATMENT |
PHENYLKETONURIA |
To prevent intellectual disability, diagnosis and initiation of dietary treatment of classic phenylketonuria must occur before the child is 2 weeks of age. For this reason, most newborns in North America, Australia, and Europe are screened by determinations of blood phenylalanine levels. Abnormal values are confirmed using quantitative analysis of plasma amino acids. Dietary phenylalanine restriction is usually instituted if blood phenylalanine levels are >360 μmol/L (6 mg/dL). Treatment consists of a special diet low in phenylalanine and supplemented with tyrosine, since tyrosine becomes an essential amino acid in phenylalanine hydroxylase deficiency. With therapy, plasma phenylalanine concentrations should be maintained between 120 and 360 μmol/L (2 and 6 mg/dL). Dietary restriction should be continued and monitored indefinitely. Some patients with milder forms of phenylketonuria (phenylalanine <1200 μm at presentation) show increased tolerance to dietary proteins and improved metabolic control when treated with tetrahydrobiopterin (5–20 mg/kg per day), an essential cofactor of phenylalanine hydroxylase.
A number of women with phenylketonuria who have been treated since infancy will reach adulthood and become pregnant. If maternal phenylalanine levels are not strictly controlled before and during pregnancy, their offspring are at increased risk for congenital defects and microcephaly (maternal phenylketonuria). After birth, these children have severe intellectual disability and growth retardation. Pregnancy risks can be minimized by continuing lifelong phenylalanine-restricted diets and assuring strict phenylalanine restriction 2 months prior to conception and throughout gestation.
THE HOMOCYSTINURIAS (HYPERHOMOCYSTEINEMIAS)
The homocystinurias are nine biochemically and clinically distinct disorders (Table 434e-1) characterized by increased concentration of the sulfur-containing amino acid homocystine in blood and urine.
Classic homocystinuria, the most common (frequency 1:200,000), results from reduced activity of cystathionine β-synthase (Fig. 434e-1), the pyridoxal phosphate–dependent enzyme that condenses homocysteine with serine to form cystathionine. Most patients present between 3 and 5 years of age with dislocated optic lenses and intellectual disability (in about half of cases). Some patients develop a marfanoid habitus and radiologic evidence of osteoporosis.
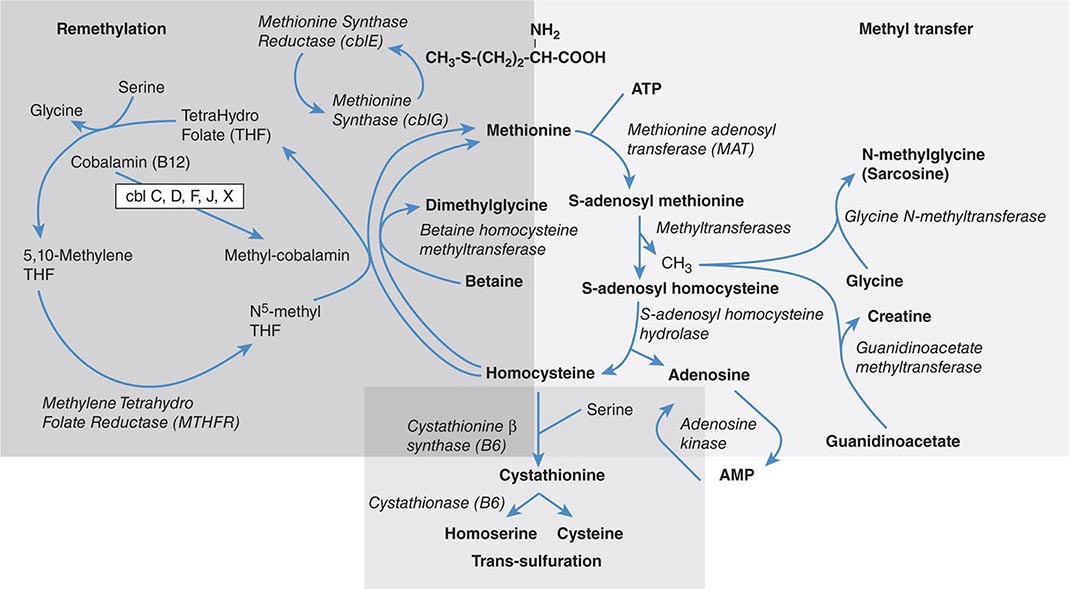
FIGURE 434e-1 Pathways, enzymes, and coenzymes involved in the homocystinurias. Methionine transfers a methyl group during its conversion to homocysteine. Defects in methyl transfer or in the subsequent metabolism of homocysteine by the pyridoxal phosphate (vitamin B6)-dependent cystathionine β-synthase increase plasma methionine levels. Homocysteine is transformed into methionine via remethylation. This occurs through methionine synthase, a reaction requiring methylcobalamin and folic acid. Deficiencies in these enzymes or lack of cofactors is associated with decreased or normal methionine levels. In an alternative pathway, homocysteine can be remethylated by betaine:homocysteine methyl transferase.
Life-threatening vascular complications (affecting coronary, renal, and cerebral arteries) can occur during the first decade of life and are the major cause of morbidity and mortality. Classic homocystinuria can be diagnosed with analysis of plasma amino acids, showing elevated methionine and presence of free homocystine. Total plasma homocysteine is also extremely elevated (usually >100 μM). Treatment consists of a special diet restricted in protein and methionine and supplemented with cystine. In approximately half of patients, oral pyridoxine (25–500 mg/d) produces a fall in plasma methionine and homocystine concentration in body fluids. Folate and vitamin B12 deficiency should be prevented by adequate supplementation. Betaine is also effective in reducing homocystine levels in pyridoxine-unresponsive patients.
The other forms of homocystinuria are the result of impaired remethylation of homocysteine to methionine. This can be caused by defective methionine synthase or reduced availability of two essential cofactors, 5-methyltetrahydrofolate and methylcobalamin (methyl-vitamin B12).
Hyperhomocysteinemia refers to increased total plasma concentration of homocysteine with or without an increase in free homocystine (disulfide form). Hyperhomocysteinemia, in the absence of significant homocystinuria, is found in some heterozygotes for the genetic defects noted above or in homozygotes for milder variants. Changes of homocysteine levels are also observed with increasing age; with smoking; in postmenopausal women; in patients with renal failure, hypothyroidism, leukemias, inflammatory bowel disease, or psoriasis; and during therapy with drugs such as methotrexate, nitrous oxide, isoniazid, and some antiepileptic agents. Homocysteine acts as an atherogenic and thrombophilic agent. An increase in total plasma homocysteine is an independent risk factor for coronary, cerebrovascular, and peripheral arterial disease as well as for deep vein thrombosis (Chap. 291e). Homocysteine is synergistic with hypertension and smoking, and it is additive with other risk factors that predispose to peripheral arterial disease. In addition, hyperhomocysteinemia and folate and vitamin B12 deficiency have been associated with an increased risk of neural tube defects in pregnant women. Vitamin supplements are effective in reducing plasma homocysteine levels in these cases, although there are limited effects on cardiovascular disease.
ALKAPTONURIA
Alkaptonuria is a rare (frequency 1:200,000) disorder of tyrosine catabolism in which deficiency of homogentisate 1,2-dioxygenase (also known as homogentisic acid oxidase) leads to excretion of large amounts of homogentisic acid in urine and accumulation of oxidized homogentisic acid pigment in connective tissues (ochronosis). Alkaptonuria may go unrecognized until middle life, when degenerative joint disease develops. Prior to this time, about half of patients might be diagnosed for the presence of dark urine. Foci of gray-brown scleral pigment and generalized darkening of the concha, anthelix, and, finally, helix of the ear usually develop after age 30. Low back pain usually starts between 30 and 40 years of age. Ochronotic arthritis is heralded by pain, stiffness, and some limitation of motion of the hips, knees, and shoulders. Acute arthritis may resemble rheumatoid arthritis, but small joints are usually spared. Pigmentation of heart valves, larynx, tympanic membranes, and skin occurs, and occasional patients develop pigmented renal or prostatic calculi. Pigment deposition in the heart and blood vessels leads to aortic stenosis necessitating valve replacement, especially after 60 years of age. The diagnosis should be suspected in a patient whose urine darkens to blackness. Homogentisic acid in urine is identified by urine organic acid analysis. Ochronotic arthritis is treated symptomatically with pain medications, spinal surgery, and arthroplasty (Chap. 394). Ascorbic acid and protein restriction are not effective in reducing homogentisic acid production. By contrast, nitisone (2-[2-nitro-4-trifluoromethylbenzoyl]-1,3-cyclohexanedione), a drug used in tyrosinemia type I, reduces urinary excretion of homogentisic acid and, in conjunction with a low-protein diet, might prevent the long-term complications of alkaptonuria.
UREA CYCLE DEFECTS
Excess ammonia generated from protein nitrogen is removed by the urea cycle, a process mediated by several enzymes and transporters (Table 434e-1). Complete absence of any of these enzymes usually causes severe hyperammonemia in newborns, while milder variants can be seen in adults. The accumulation of ammonia and glutamine leads to brain edema and direct neuronal toxicity. Deficiencies in urea cycle enzymes are individually rare, but as a group, they affect about 1:25,000 individuals. They are all transmitted as autosomal recessive traits, with the exception of ornithine transcarbamylase deficiency, which is X-linked. Hepatocytes of females with ornithine transcarbamylase deficiency express either the normal or the mutant allele due to random X-inactivation and may be unable to remove excess ammonia if mutant cells are predominant.
Infants with classic urea cycle defects present at 1–4 days of life with refusal to eat and lethargy progressing to coma and death. Milder enzyme deficiencies present with protein avoidance, recurrent vomiting, migraine, mood swings, chronic fatigue, irritability, and disorientation that can progress to coma. Females with ornithine transcarbamylase deficiency can present at time of childbirth due to the combination of involuntary fasting and stress that favors catabolism. The diagnosis requires measurement of plasma ammonia, plasma amino acids, and urine orotic acid, useful for differentiating ornithine transcarbamylase deficiency from carbamyl phosphate synthase-1 and N-acetylglutamate synthase deficiency. Hyperammonemia can also be caused by liver disease from any cause and several organic acidemias and fatty acid oxidation defects (the latter two excluded by the analysis of urine organic acids and plasma acylcarnitine profile).
|
TREATMENT |
UREA CYCLE DEFECTS |
Therapy is aimed at stopping catabolism and ammonia production by providing adequate calories (as IV glucose and lipids in the comatose patient) and, if needed, insulin. Excess nitrogen is removed by IV phenylacetate and benzoate (0.25 g/kg for the priming dose and subsequently as an infusion over 24 h) that conjugate with glutamine and glycine, respectively, to form phenylacetylglutamine and hippuric acid, water-soluble molecules efficiently excreted in urine. Arginine (200 mg/kg per day) becomes an essential amino acid (except in arginase deficiency) and should be provided intravenously to resume protein synthesis. If these measures fail to reduce ammonia, hemodialysis should be initiated promptly. Chronic therapy consists of a protein-restricted diet, phenylbutyrate, glycerol phenylbutyrate (a liquid drug better tolerated by most patients), arginine, or citrulline supplements, depending on the specific diagnosis. Liver transplantation should be considered in patients with severe urea cycle defects that are difficult to control medically.
Hyperammonemia due to a functional deficiency of glutamine synthase can occur in patients receiving chemotherapy for different malignancies or undergoing solid organ transplants. It can also be seen with hepatic cirrhosis. Several of these patients have been successfully rescued from hyperammonemia using the protocol described above for urea cycle defects.
435e |
Inherited Defects of Membrane Transport |
Specific membrane transporters mediate the passage of a wide variety of substances across cellular membranes. Classes of substrates include amino acids, sugars, cations, anions, vitamins, and water. The number of inherited disorders of membrane transport continues to increase with the identification of new transporters on the plasma membrane or intracellular organelles and the clarification of the molecular basis of diseases with previously unknown pathophysiology. The first transport disorders identified affected the gut or the kidney, but transport processes are now proving essential for the normal function of every organ. Mutations in transporter molecules cause disorders of the heart, muscle, brain, and endocrine and sensory organs (Table 435e-1). Inherited defects impairing the transport of selected amino acids that can present in adults are discussed here as examples of the abnormalities encountered; others are considered elsewhere in this text.
|
GENETIC DISORDERS OF MEMBRANE TRANSPORT (SELECTED EXAMPLES) |
CYSTINURIA
Cystinuria (frequency of 1 in 10,000 to 1 in 15,000) is an autosomal recessive disorder caused by defective transporters in the apical brush border of proximal renal tubule and small intestinal cells. It is characterized by impaired reabsorption and excessive urinary excretion of the dibasic amino acids lysine, arginine, ornithine, and cystine. Because cystine is poorly soluble, its excess excretion predisposes to the formation of renal, ureteral, and bladder stones. Such stones are responsible for the signs and symptoms of the disorder.
There are two variants of cystinuria. Homozygotes for both variants have high urinary excretion of cystine, lysine, arginine, and ornithine. Type I heterozygotes usually have normal urinary amino acid excretion, whereas most non–type I (formerly type II and type III) heterozygotes have moderately increased urinary excretion of each of the four amino acids. The gene for type I cystinuria (SLC3A1, chromosome 2p16.3) encodes a membrane glycoprotein. Non–type I cystinuria is caused by mutations in SLC7A9 (chromosome 19q13) that encodes the b0,+ amino acid transporter. The glycoprotein encoded by SLC3A1 favors the correct processing of the b0,+ membrane transporter and explains why mutations in two different genes cause a similar disease.
Cystine stones account for 1–2% of all urinary tract calculi but are the most common cause of stones in children. Cystinuria homozygotes regularly excrete 2400–7200 μmol (600–1800 mg) of cystine daily. Since the maximum solubility of cystine in the physiologic urinary pH range of 4.5–7.0 is about 1200 μmol/L (300 mg/L), cystine needs to be diluted to 2.5–7 L of water to prevent crystalluria. Stone formation usually manifests in the second or third decade but may occur in the first year of life. Symptoms and signs are those typical of urolithiasis: hematuria, flank pain, renal colic, obstructive uropathy, and infection (Chap. 342). Recurrent urolithiasis may lead to progressive renal insufficiency.
Cystinuria is suspected after observing typical hexagonal crystals in the sediment of acidified, concentrated, chilled urine or after performing a urinary nitroprusside test. Quantitative urine amino acid analysis confirms the diagnosis of cystinuria by showing selective overexcretion of cystine, lysine, arginine, and ornithine. Quantitative measurements are important for differentiating heterozygotes from homozygotes and for following free cystine excretion during therapy.
Management is aimed at preventing cystine crystal formation by increasing urinary volume and by maintaining an alkaline urine pH. Fluid ingestion in excess of 4 L/d is essential, and 5–7 L/d is optimal. Urinary cystine concentration should be <1000 μmol/L (250 mg/L). The daily fluid ingestion necessary to maintain this dilution of excreted cystine should be spaced over 24 h, with one-third of the total volume ingested between bedtime and 3 A.M. Cystine solubility rises sharply above pH 7.5, and urinary alkalinization (with bicarbonate or potassium citrate) can be therapeutic. Penicillamine (1–3 g/d) and tiopronin (α-mercaptopropionylglycine, 800–1200 mg/d in four divided doses) undergo sulfhydryl-disulfide exchange with cystine to form mixed disulfides. Because these disulfides are much more soluble than cystine, pharmacologic therapy can prevent and promote dissolution of calculi. Penicillamine can have significant side effects and should be reserved for patients who fail to respond to hydration alone or who are in a high-risk category (e.g., one remaining kidney, renal insufficiency). When medical management fails, shock wave lithotripsy, ureteroscopy, and percutaneous nephrolithotomy are effective for most stones. Open urologic surgery is considered for complex staghorn stones or when the patient has concomitant renal or ureteral abnormalities. Occasional patients progress to renal failure and require kidney transplantation.
DIBASIC AMINOACIDURIA (LYSINURIC PROTEIN INTOLERANCE)
This disorder is characterized by a defect in renal tubular reabsorption of the three dibasic amino acids lysine, arginine, and ornithine but not cystine (lysinuric protein intolerance). Homozygotes show defective intestinal transport of dibasic amino acids as well as exaggerated renal losses. Lysinuric protein intolerance is most common in Finland (1 in 60,000), southern Italy, and Japan, but is rare elsewhere. The transport defect affects basolateral rather than luminal membrane transport and is associated with impairment of the urea cycle. The defective gene (SLC7A7, chromosome 14q11.2) encodes the y+LAT membrane transporter, which associates with the cell-surface glycoprotein 4F2 heavy chain to form the complete sodium-independent transporter y+L.
Manifestations are related to impairment of the urea cycle and to immune dysfunction potentially attributable to nitric oxide overproduction secondary to arginine intracellular trapping. Affected patients present in childhood with hepatosplenomegaly, protein intolerance, and episodic ammonia intoxication. Older patients may present with severe osteoporosis, impairment of kidney function, pulmonary alveolar proteinosis, various autoimmune disorders, and an incompletely characterized immune deficiency. Plasma concentrations of lysine, arginine, and ornithine are reduced, whereas urinary excretion of lysine and orotic acid are increased. Hyperammonemia may develop after the ingestion of protein loads or with infections, probably because of insufficient amounts of arginine and ornithine to maintain proper function of the urea cycle. Therapy consists of dietary protein restriction and supplementation of citrulline (2–8 g/d), a neutral amino acid that fuels the urea cycle when metabolized to arginine and ornithine. Pulmonary disease responds to glucocorticoids or bronchoalveolar lavage in some patients. Women with lysinuric protein intolerance who become pregnant have an increased risk of anemia, toxemia, and bleeding complications during delivery. These can be minimized by aggressive nutritional therapy and control of blood pressure. Their infants can have intrauterine growth restriction but have normal neurologic function
CITRULLINEMIA TYPE 2 (CITRIN DEFICIENCY)
Citrullinemia type 2 is a recessive condition caused by deficiency of the mitochondrial aspartate-glutamate carrier AGC2 (citrin). A defect in this transporter reduces the availability of cytoplasmic aspartate to combine with citrulline, impairing the urea cycle and decreasing the transfer of reducing equivalents from the cytosol to the mitochondria through the malate–aspartate NADH shuttle. Mutations in the SLC25A13 gene on chromosome 7q21.3 that encodes for this transporter are rare in Caucasians, but affect about 1:20,000 people with ancestry from Japan, China, and Southeast Asia with variable penetrance.
The disease usually presents with sudden onset between 20 and 50 years of age with recurring episodes of hyperammonemia with associated neuropsychiatric symptoms such as altered mental status, irritability, seizures, or coma resembling hepatic encephalopathy. Some patients might come to medical attention for hypertriglyceridemia, pancreatitis, hepatoma, or fatty liver histologically similar to nonalcoholic steatohepatitis. Without therapy, most patients die with cerebral edema within a few years of onset. Episodes are usually triggered by medications (such as acetaminophen), surgery, alcohol consumption or high sugar intake, the latter conditions causing excess NADH production. NADH is not generated by the metabolism of proteins or fats, and many individuals with citrullinemia type 2 spontaneously prefer foods such as meat, eggs, and fish, and avoid carbohydrates.
Laboratory studies during an acute attack include elevated ammonia, citrulline, and arginine with low or normal levels of glutamine (the latter is usually increased in classic urea cycle defects). The diagnosis is confirmed by demonstrating mutations in the SLC25A13 gene. Liver transplantation prevents progression of the disease and normalizes biochemical parameters. A diet high in fats and proteins and low in carbohydrates with supplements of arginine and pyruvate is also effective in preventing further episodes, at least in the short term.
HARTNUP DISEASE
Hartnup disease (frequency 1 in 24,000) is an autosomal recessive disorder characterized by pellagra-like skin lesions, variable neurologic manifestations, and neutral and aromatic aminoaciduria. Alanine, serine, threonine, valine, leucine, isoleucine, phenylalanine, tyrosine, tryptophan, glutamine, asparagine, and histidine are excreted in urine in quantities 5–10 times greater than normal, and intestinal transport of these same amino acids is defective. The defective neutral amino acid transporter, B°AT1 encoded by the SLC6A19 gene on chromosome 5p15, requires either collectrin or angiotensin-converting enzyme 2 for surface expression in the kidney and intestine, respectively.
The clinical manifestations result from nutritional deficiency of the essential amino acid tryptophan, caused by its intestinal and renal malabsorption, and of niacin, which derives in part from tryptophan metabolism. Only a small fraction of patients with the chemical findings of this disorder develop a pellagra-like syndrome, implying that manifestations depend on other factors in addition to the transport defect. The diagnosis of Hartnup disease should be suspected in any patient with clinical features of pellagra who does not have a history of dietary niacin deficiency (Chap. 96e). The neurologic and psychiatric manifestations range from attacks of cerebellar ataxia to mild emotional lability to frank delirium, and they are usually accompanied by exacerbations of the erythematous, eczematoid skin rash. Fever, sunlight, stress, and sulfonamide therapy provoke clinical relapses. Diagnosis is made by detection of the neutral aminoaciduria, which does not occur in dietary niacin deficiency. Treatment is directed at niacin repletion and includes a high-protein diet and daily nicotinamide supplementation (50–250 mg).
436e |
Atlas of Clinical Manifestations of Metabolic Diseases |
The term metabolism is derived from the Greek metabol, meaning “to change.” This term encompasses the broad array of chemical pathways that are necessary for normal development and homeostasis. In practice, clinicians generally use the term metabolism in reference to energy utilization for anabolism or catabolism. Alternatively, intermediary metabolism describes the myriad cellular pathways that convert energy sources from one form to another (e.g., the citric acid cycle). The emerging field of metabolomics is based on the premise that the identification and measurement of metabolic products will enhance our understanding of physiology and disease.
Over the years, the classification of metabolic diseases has extended beyond traditional pathways involved in fuel metabolism to include disorders such as lysosomal storage diseases and connective tissue diseases. Thus, metabolic diseases really reflect disorders of cell biology, and many have a well-defined genetic basis. For example, lysosomal storage diseases (Chap. 432e) result from a variety of genetic defects, usually in a lysosomal enzyme, causing accumulation of a substrate within the lysosome. Certain lipodystrophies and cardiomyopathies can be caused by mutations in lamin A, a structural protein in the nuclear envelope. Membrane defects (Chap. 435e), usually involving transporters of amino acids, sugars, or ions, cause disorders such as cystinuria, Hartnup’s disease, or Wilson’s disease (Chap. 429). Connective tissue diseases (Chap. 427) frequently involve defects in collagen synthesis or structure (osteogenesis imperfecta, Ehlers-Danlos syndrome, Alport’s syndrome) or in other extracellular matrix structural proteins such as fibrillin (Marfan syndrome). Many metabolic disorders originate from defects in enzymes involved in the synthesis or degradation of amino acids, carbohydrates, lipids, purines, or pyrimidines (Chaps. 431e, 433e, and 434e). Lipoprotein disorders (Chap. 421) are caused by defects in a wide array of cellular pathways including membrane receptors (the low-density lipoprotein receptor), enzyme defects (lipoprotein lipase), carrier proteins (apolipoprotein B100), or transporters (ATP-binding cassette transporter ABCA1). In some instances, metabolic abnormalities induce compensatory physiologic responses that reflect the interactions of multiple metabolic pathways. For example, the metabolic syndrome (Chap. 422), which includes a constellation of clinical features (central obesity, hypertriglyceridemia, low high-density lipoprotein cholesterol, hyperglycemia, and hypertension), likely has multiple genetic and environmental origins. Cushing’s syndrome reflects the metabolic effects of excess cortisol on multiple tissues (Chap. 406).
This broader definition results in a plethora of metabolic diseases, numbering in the thousands. Fortunately, comprehensive reference sources exist, such as the Online Metabolic and Molecular Bases of Inherited Disease (OMMBID) (http://www.ommbid.com/) and the Online Mendelian Inheritance in Man (OMIM) (http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=OMIM). The study of metabolic diseases has been invaluable for advancing our understanding of human genetics by providing insight into principles such as patterns of inheritance, variable expressivity, phenotypic variation, and novel approaches to therapy, including screening programs, blood and organ transplantation, gene therapy, and enzyme replacement (Chap. 82).
This atlas provides a visual survey of selected metabolic disorders with references to the topics elsewhere in the text. The author encourages submission of additional illustrations that might facilitate learning among our peers and thereby enhance the recognition and care of patients with these disorders.
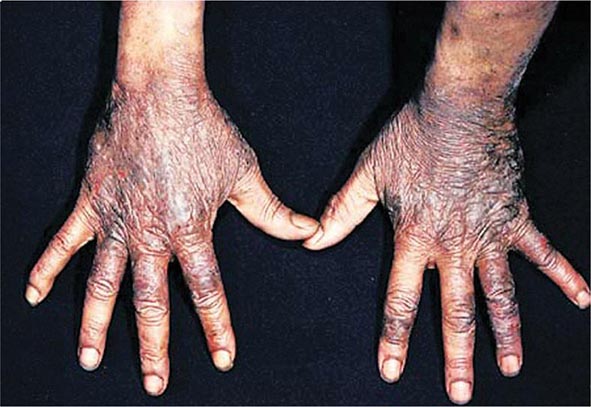
FIGURE 436e-1 “Gauntlet” of pellagra (niacin deficiency). Note indurated, lichenified, pigmented, and scaly skin on the dorsa of the hands. (Source: K Wolff et al: Fitzpatrick’s Color Atlas & Synopsis of Clinical Dermatology, 5th ed. New York, McGraw-Hill, 2005.) See Chap. 96e.
FIGURE 436e-2 Scurvy (vitamin C deficiency). Note perifollicular hemorrhage on the leg. The follicles are often plugged by keratin (perifollicular hyperkeratosis). This eruption occurred in a 46-year-old alcoholic, homeless man who also had bleeding gums and loose teeth. (Source: K Wolff et al: Fitzpatrick’s Color Atlas & Synopsis of Clinical Dermatology, 5th ed. New York, McGraw-Hill, 2005.) See Chap. 96e.
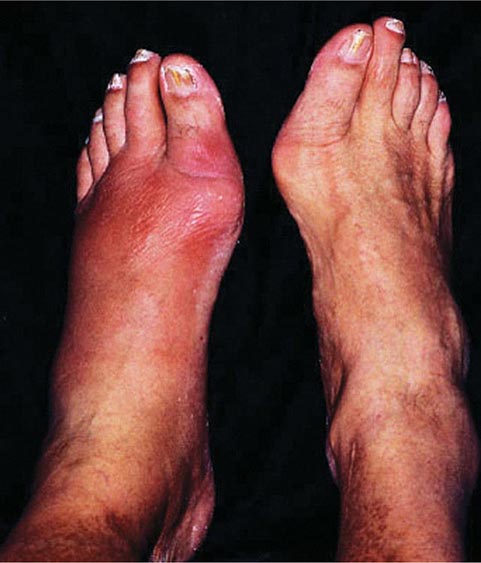
FIGURE 436e-3 Podagra with gouty inflammation of the left first metatarsophalangeal joint. Note swelling and erythema. (From KJ Knoop et al: The Atlas of Emergency Medicine, 2nd ed. New York, McGraw-Hill, 2002. Courtesy of Kevin J. Knoop, MD, MS; with permission.) See Chaps. 395 and 431e.
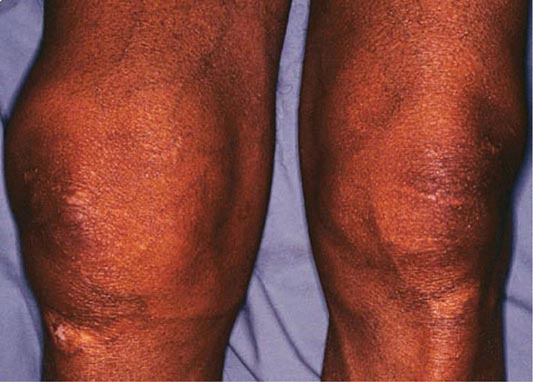
FIGURE 436e-4 Large tophi of gout located in and around the right knee. (Courtesy of Daniel L. Savitt, MD; with permission.) See Chaps. 395 and 431e.
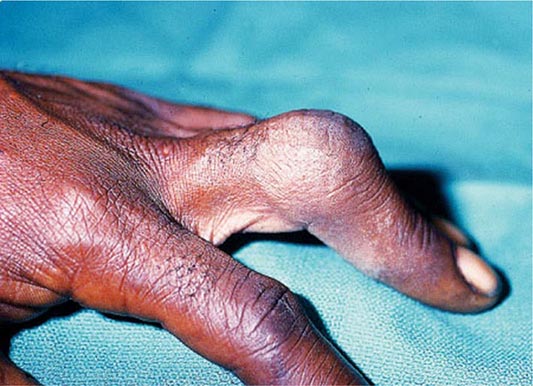
FIGURE 436e-5 Gouty arthritis of the finger. The finger is an unusual site for gouty arthritis. Examination of the synovial fluid confirmed the diagnosis. (Courtesy of Alan B. Storrow, MD; with permission.) See Chaps. 395 and 431e.
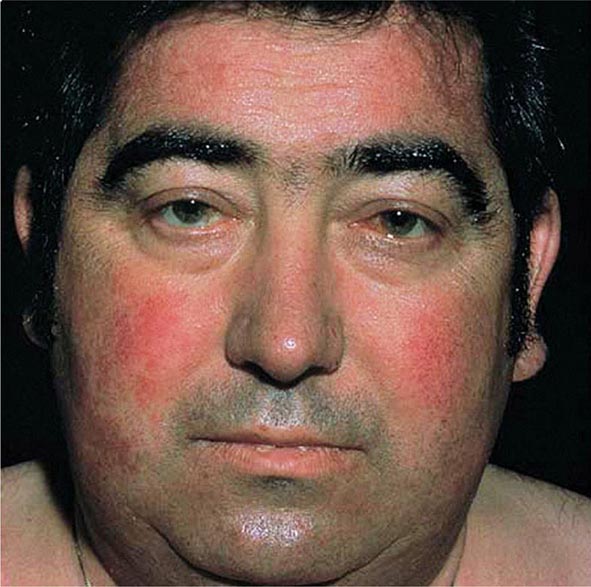
FIGURE 436e-6 Cushing’s syndrome. Note plethoric moon facies with erythema and telangiectases of cheek and forehead. The face and neck show increased deposition of fat, which was also seen in the supraclavicular areas (not depicted here). (Source: K Wolff et al: Fitzpatrick’s Color Atlas & Synopsis of Clinical Dermatology, 5th ed. New York, McGraw-Hill, 2005.) See Chap. 406.
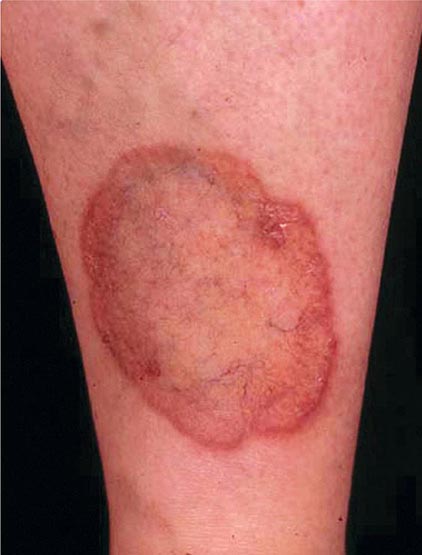
FIGURE 436e-7 Necrobiosis lipoidica diabeticorum. A large symmetric plaque with active tan-pink, well-demarcated, raised, firm borders and a yellow center in the pretibial regions of a 28-year-old diabetic woman is shown. The central parts of the lesion are depressed with atrophic changes of epidermal thinning and telangiectasis against a yellow background. (Source: K Wolff et al: Fitzpatrick’s Color Atlas & Synopsis of Clinical Dermatology, 5th ed. New York, McGraw-Hill, 2005.) See Chap. 417.
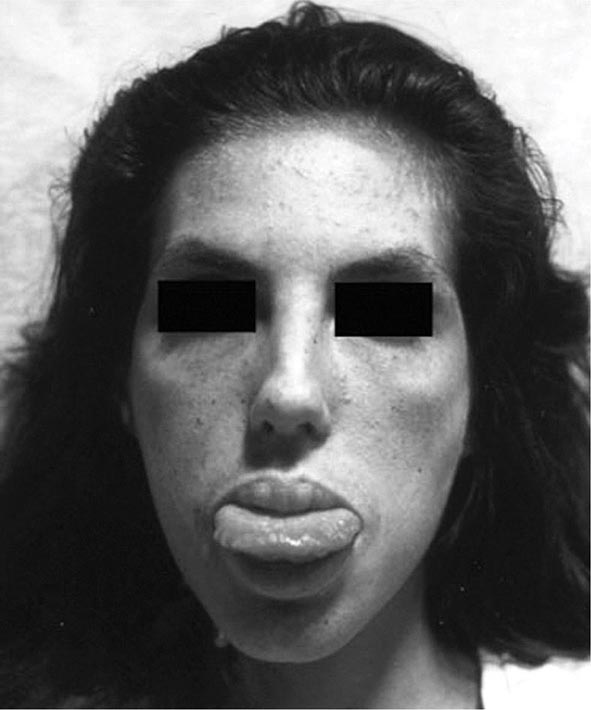
FIGURE 436e-8 Patient with multiple endocrine neoplasia 2B syndrome. Note the multiple neuromas on the lips and tongue and the marfanoid facies. (Source: DG Gardner, D Shoback, eds: Greenspan’s Basic & Clinical Endocrinology, 8th ed. New York, McGraw-Hill, 2006, www.accessmedicine.com.) See Chap. 408.
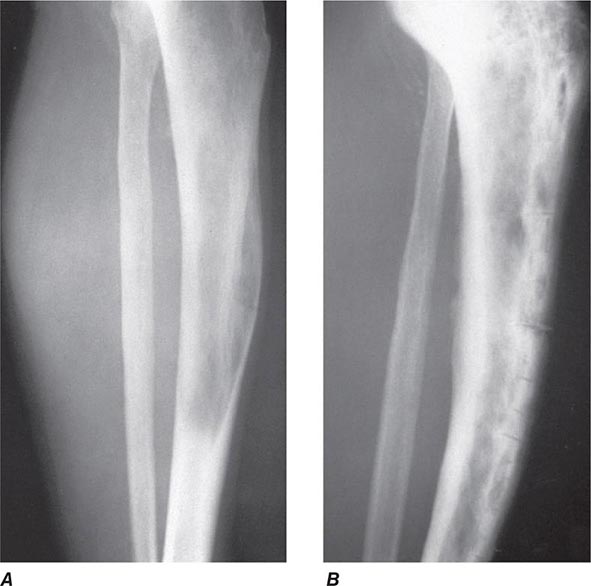
FIGURE 436e-9 Early and late radiographs of Paget’s disease of the tibia of a male patient, taken at 45 (A) and 65 years of age (B). (Source: HB Skinner: Current Diagnosis & Treatment in Orthopedics, 4th ed. New York, McGraw-Hill, 2007, www.accessmedicine.com.) See Chap. 426e.
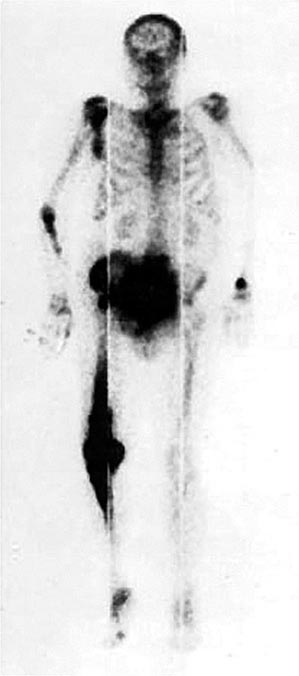
FIGURE 436e-10 Bone scan of a patient with severe Paget’s disease of the skull, ribs, spine, pelvis, right femur, and acetabulum. Note localization of bone-seeking isotope (99mTc-labeled bisphosphonate) in these areas. (Source: DG Gardner, D Shoback, eds: Greenspan’s Basic & Clinical Endocrinology, 8th ed. New York, McGraw-Hill, 2006, www.accessmedicine.com.) See Chap. 426e.
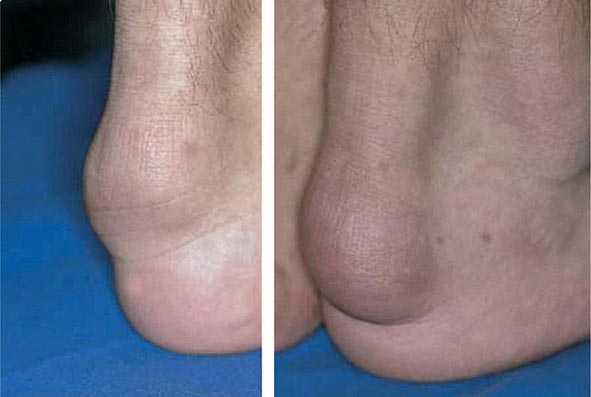
FIGURE 436e-11 Tendinous xanthomas. Large subcutaneous tumors adherent to the Achilles tendons. (Source: K Wolff et al: Fitzpatrick’s Color Atlas & Synopsis of Clinical Dermatology, 5th ed. New York, McGraw-Hill, 2005.) See Chap. 421.
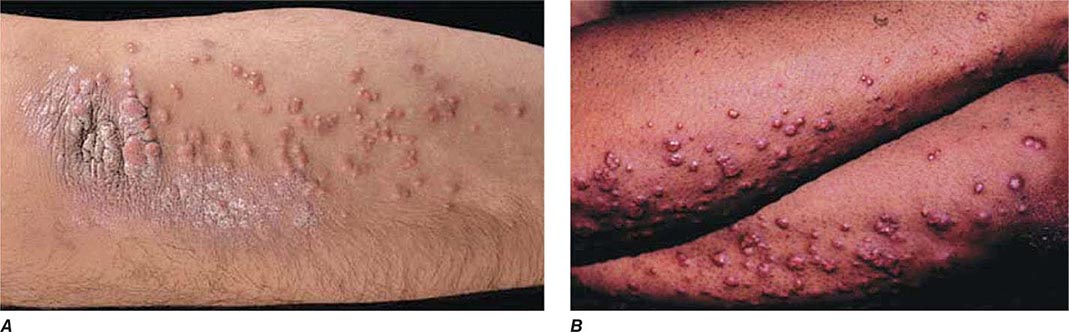
FIGURE 436e-12 Papular eruptive xanthomas. A. Multiple, discrete, red-to-yellow papules becoming confluent on the elbow of a white individual with uncontrolled diabetes mellitus; lesions were present on both elbows and buttocks. B. Papular eruptive xanthomas on the elbows and lower arms of an African-American patient. (Source: K Wolff et al: Fitzpatrick’s Color Atlas & Synopsis of Clinical Dermatology, 5th ed. New York, McGraw-Hill, 2005.) See Chap. 421.
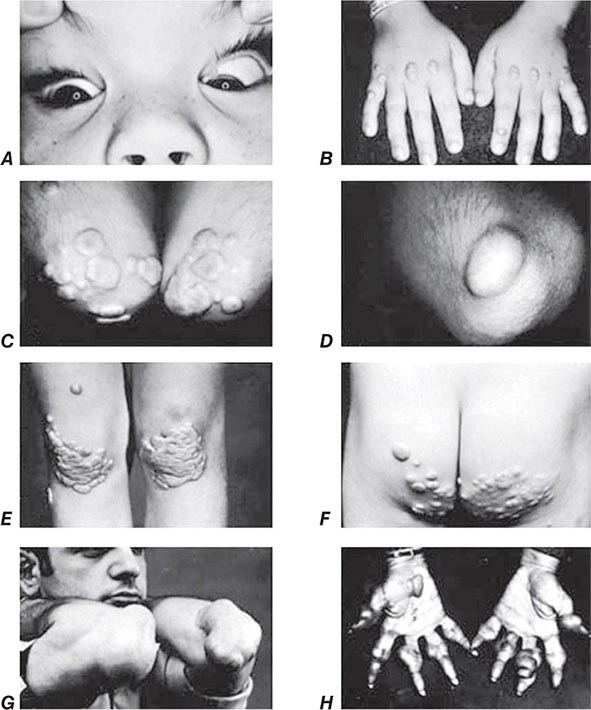
FIGURE 436e-13 Forms of xanthomas and other lipid deposits frequently seen in familial hypercholesterolemia homozygotes. A. Arcus corneae. B, E, and F. Cutaneous planar xanthomas, which usually have a bright orange hue. C, D, and G. Tuberous xanthomas on the elbows. (Source for panels C and D: CR Scriver et al [eds]: The Metabolic and Molecular Bases of Inherited Disease online, 8th ed. New York, McGraw-Hill, www.ommbid.com.) H. Tendon and tuberous xanthomas. (Panel H reproduced through the courtesy of Dr. A. Khachadurian; with permission.) See Chap. 421.
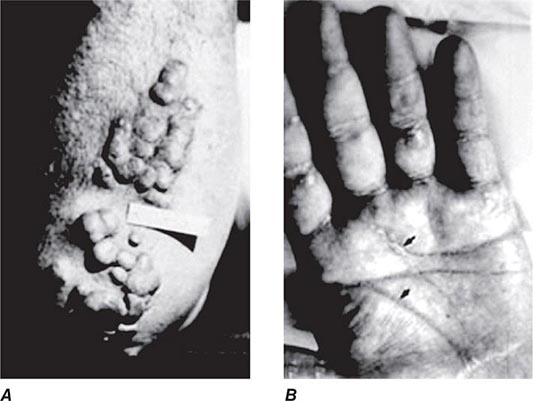
FIGURE 436e-14 Examples of xanthomas in type III hyperlipoproteinemic patients. A. Tuberoeruptive xanthomas of the elbows. B. Tuberous xanthomas of the digits and xanthomas of the palmar creases (xanthoma striata palmaris) (arrows). (Courtesy of Dr. Thomas P. Bersot; with permission.) See Chap. 421.
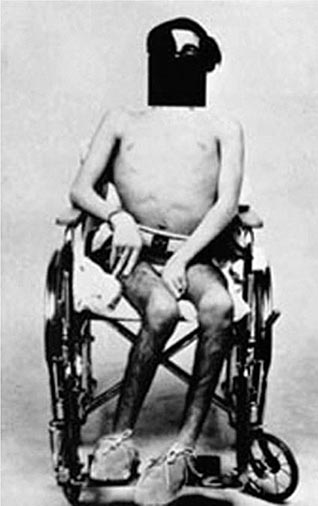
FIGURE 436e-15 A 17-year-old patient with abetalipoproteinemia, with generalized weakness, kyphoscoliosis, and lordosis. (Courtesy of Drs. Peter Herbert, Gerd Assmann, Antonio M. Gotto, Jr., and Donald Fredrickson; with permission.) See Chap. 421.
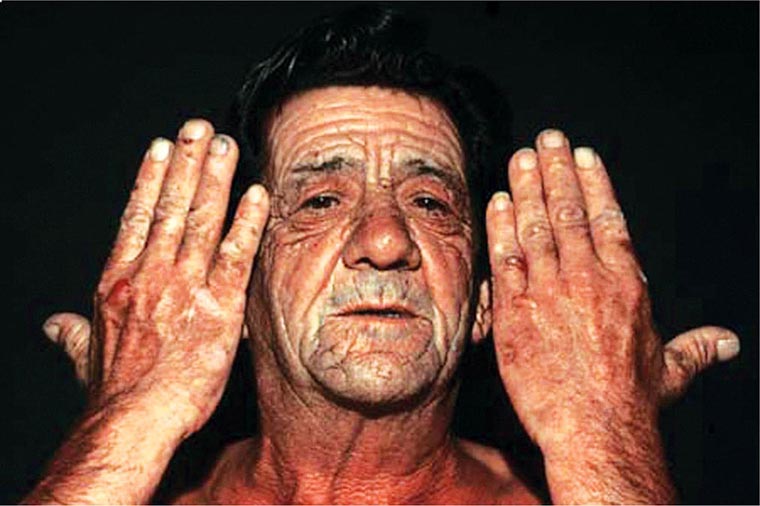
FIGURE 436e-16 Porphyria cutanea tarda. Periorbital and malar violaceous coloration, hyperpigmentation, and hypertrichosis on the face; bullae, crusts, and scars on the dorsa of the hands. (Source: K Wolff et al: Fitzpatrick’s Color Atlas & Synopsis of Clinical Dermatology, 5th ed. New York, McGraw-Hill, 2005.) See Chap. 430.
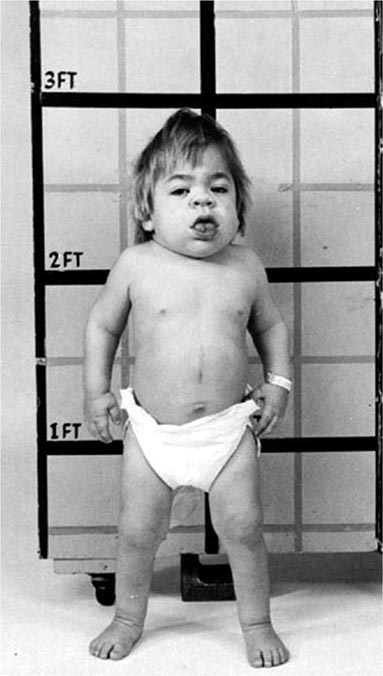
FIGURE 436e-17 Mucopolysaccharidosis type IH (Hurler’s syndrome) in a 4-year-old boy. The diagnosis was made at the age of 15 months, at which time he had developmental delay, hepatomegaly, and skeletal involvement. At the time of the picture, the patient had short stature, an enlarged tongue, persistent nasal discharge, stiff joints, and hydrocephalus. Verbal language skills consisted of four or five words. The patient had a severe hearing loss and wore hearing aids. (Source: CR Scriver et al [eds]: The Metabolic and Molecular Bases of Inherited Disease online, 8th ed. New York, McGraw-Hill, www.ommbid.com.) See Chap. 432e.
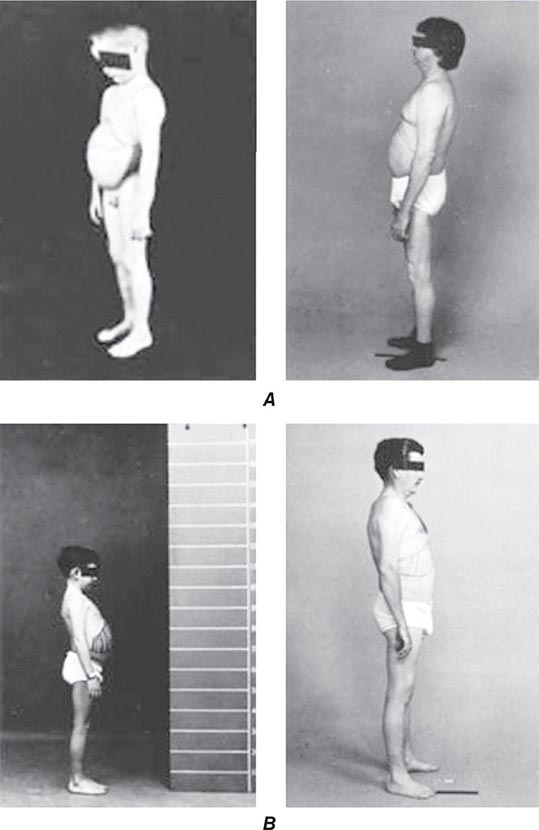
FIGURE 436e-18 Growth and development of two patients with type Ia glycogen storage disease. A. Patient at age 7 years and at age 39 years. B. Another patient at age 10 years and at age 33 years. Both patients survive despite inadequate treatment of their disease. Note that the abdomen is less protuberant with age. Hypoglycemia also improves with age. In adulthood, however, both patients continue to be short, and both have gout, multiple liver adenomas, and progressive renal disease. (Source: CR Scriver et al [eds]: The Metabolic and Molecular Bases of Inherited Disease online, 8th ed. New York, McGraw-Hill, www.ommbid.com.) See Chap. 433e.
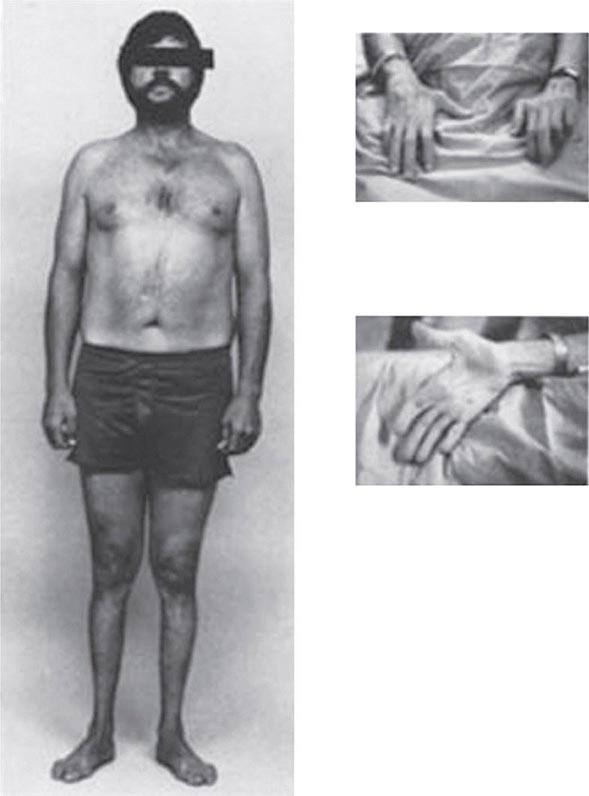
FIGURE 436e-19 Progressive myopathy in a patient with type IIIa glycogen storage disease. The patient has a debrancher deficiency in both liver and muscle (subtype IIIa). As a child, he had hepatomegaly, hypoglycemia, and growth retardation. After puberty, he no longer had hepatomegaly, and his final height is normal. Note the muscle wasting in the lower legs and both hands at age 44 years of age (left panel); this condition progressed to pronounced muscle atrophy at age 53 years (two right panels). (Source: CR Scriver et al [eds]: The Metabolic and Molecular Bases of Inherited Disease online, 8th ed. New York, McGraw-Hill, www.ommbid.com.) See Chap. 433e.
FIGURE 436e-20 Skeletal features of Marfan’s syndrome in a 16-year-old girl. Note the long limbs (associated with disproportionately tall stature), long fingers, scoliosis, and genu valgum. (Source: CR Scriver et al [eds]: The Metabolic and Molecular Bases of Inherited Disease online, 8th ed. New York, McGraw-Hill, www.ommbid.com.) See Chap. 427.
FIGURE 436e-21 Marfan’s syndrome. A. Long, narrow face. B. Arachnodactyly and positive wrist sign. C. High-arched palate. D. Ectopia lentis associated with aortic aneurysm and severe aortic regurgitation in a teenage girl. (Source: V Fuster et al [eds]: Hurst’s The Heart, 11th ed. New York, McGraw-Hill, 2004, www.accessmedicine.com.) See Chap. 427.
FIGURE 436e-22 Ochronotic pigmentation of the femur of a 56-year-old alkaptonuric patient. (Courtesy of Dr. H. W. Edmonds of the Washington Hospital Center, Washington, DC; with permission.) See Chap. 434e.
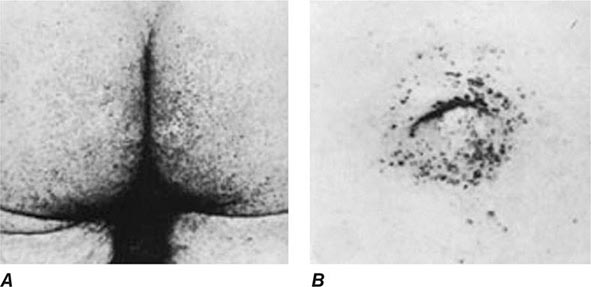
FIGURE 436e-23 Clusters of angiokeratomas (telangiectases) on the buttocks (A) and in the umbilical area (B) of a hemizygote with Fabry disease. (Source: CR Scriver et al [eds]: The Metabolic and Molecular Bases of Inherited Disease online, 8th ed. New York, McGraw-Hill, www.ommbid.com.) See Chap. 432e.
FIGURE 436e-24 Two patients with type B Niemann-Pick disease (NPD). A. A 4.7-year-old patient. (From DS Fredrickson, HR Sloan, in JB Stanbury et al: The Metabolic Basis of Inherited Disease, 3rd ed. New York, McGraw-Hill, 1972. Used by permission.) B. A 44-year-old patient. See Chap. 432e.
FIGURE 436e-25 “Cherry red” spot in the eye of a Tay-Sachs patient. (From http://www.nei.nih.gov/resources/eyegene.asp.) See Chap. 432e.
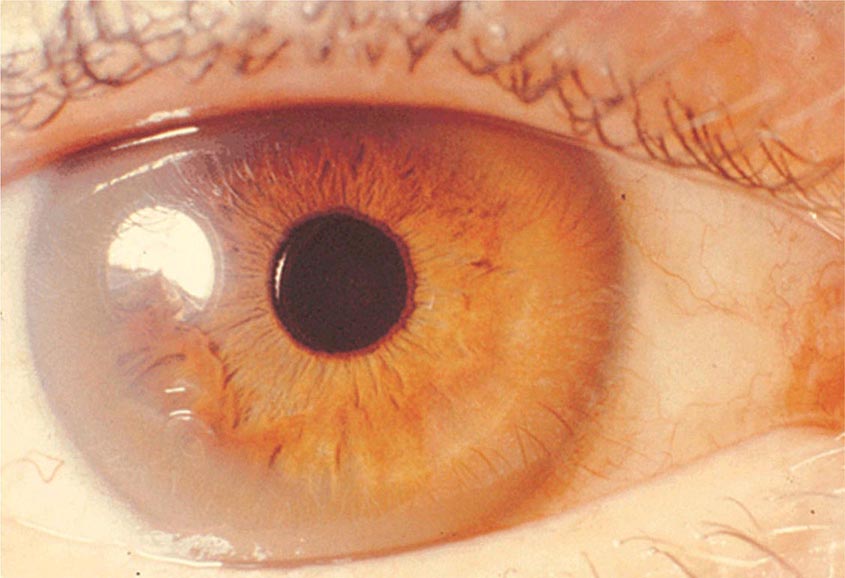
FIGURE 436e-26 Kayser-Fleischer ring. This manifestation develops in Wilson’s disease from copper deposition in Descemet’s membrane, which produces brownish discoloration of the peripheral cornea. It should not be confused with the yellow-white lipid ring of arcus senilis, which is common in the elderly and occasionally signifies hyperlipidemia, especially when it appears at a young age. (Courtesy of Jonathan C. Horton, MD, PhD; with permission.) See Chap. 429.
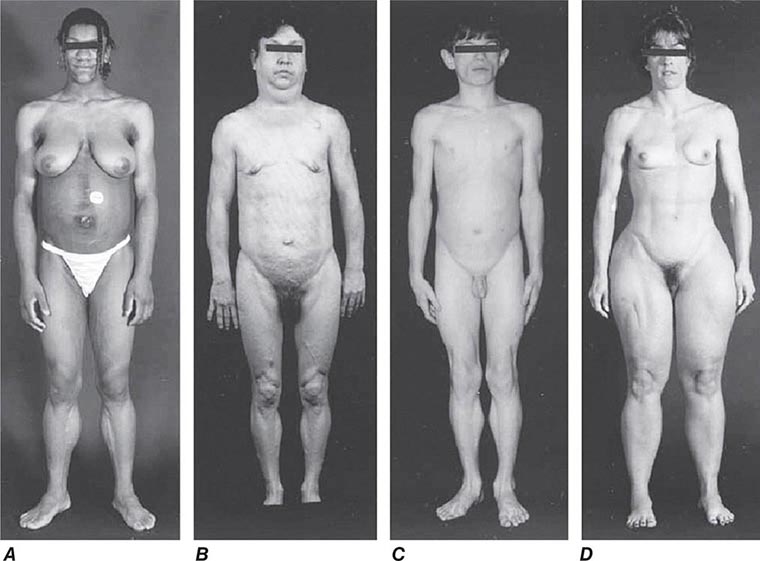
FIGURE 436e-27 Anterior view of patients with different forms of lipodystrophy. A. Congenital generalized lipodystrophy: a 16-year-old girl with generalized loss of fat, acromegaloid features, severe acanthosis nigricans affecting the axillae and abdomen, and umbilical hernia. (From A Garg et al: J Clin Endocrinol Metab 84:3390, 1999; with permission.) B. Familial partial lipodystrophy, Dunnigan variety: a 43-year-old woman with marked loss of subcutaneous fat from both the limbs and the trunk and excess fat deposition in the face, chin, supraclavicular area, and labia majora. (From JM Peters et al: Nat Genet 18;292, 1998; with permission.) C. Acquired generalized lipodystrophy: a 10-year-old boy who developed generalized loss of fat that also affected the palms and soles after panniculitis at the age of 3 months. D. Acquired partial lipodystrophy: a 30-year-old woman with onset of lipodystrophy at age 14 years. Note loss of fat from the face, neck, upper limbs, trunk, and anterior thighs. There is accumulation of excess fat in the hips and other regions of the lower limbs.





