118 Glomerulonephritis and Interstitial Nephritis
Over half of all critically ill patients develop some degree of acute kidney injury (AKI), and nearly 5% require renal replacement therapy (RRT). For those patients with severe AKI requiring. RRT, mortality can be as high as 70%, and up to 30% of surviving patients remain dialysis dependent.1–6 AKI may be a consequence of prerenal causes resulting in hypoperfusion of the kidneys, intrinsic renal causes, and postrenal or obstructive causes. In critically ill patients, the majority of AKI is related to ischemic or toxic acute tubular injury, which is treated supportively and is often reversible. AKI related to acute glomerulonephritis (GN) and acute interstitial nephritis (AIN) occurs in a smaller percentage of patients, but the incidence may be as high as 20% of all AKI.7 In addition to supportive care, initiation of correct treatment regimens is paramount for patient and renal survival. The focus in this chapter is on the renal causes of AKI, particularly GN and AIN.
 Glomerulonephritis
Glomerulonephritis
In GN, patients present with nephritic syndrome characterized by hematuria, proteinuria, AKI, edema, and hypertension.8 Hematuria may be microscopic or macroscopic, and urine sediment demonstrates dysmorphic red blood cells (RBC) and RBC casts. Urinary protein excretion typically exceeds 1 gram per day, and the degree of proteinuria can be rapidly assessed using a spot urine protein-to-creatinine ratio. In some instances, patients may have nephrotic-range proteinuria (>3 g/d) with associated clinical manifestations including edema, hypoalbuminemia, and hypercholesterolemia. Leukocyturia with or without white blood cell casts may be observed with GN of inflammatory origin.
In renal biopsy series of patients with unexplained AKI, the most common diagnoses included various forms of GN (pauci-immune GN, immunoglobulin [Ig]A nephropathy, postinfectious GN, lupus nephritis, anti–glomerular basement membrane [anti-GBM] disease) and AIN.7,9–11 Indeed, the third most common cause of end-stage kidney disease (ESKD) in the United States and Europe is GN.8 Distinguishing the type of GN with renal biopsy is critical for diagnosis as well as assessing the degree of acute versus chronic disease, which helps guide treatment and prognosis.
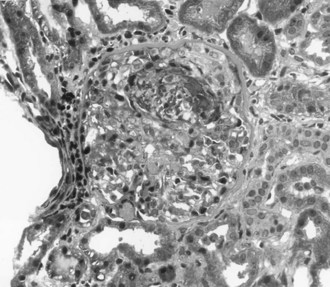
The most aggressive form of GN is described clinically as rapidly progressive glomerulonephritis (RPGN). Rather than a single disease entity, RPGN is the severe form of many of the glomerular diseases that are divided into renal limited etiologies and systemic diseases that involve the kidneys (Table 118-1). RPGN is defined as rapidly declining renal function, progressive oliguria, hematuria, proteinuria, and hypertension.8 Although many critically ill patients may have hematuria associated with infection or trauma, hematuria and AKI should always prompt consideration of acute GN. Renal ultrasound documents normal renal blood flow and normal to slightly enlarged kidneys. Renal biopsy reveals a high degree of glomerular injury with extensive crescent formation (Figure 118-1). Importantly, the transition from an acute cellular crescent to chronic, irreversible injury may occur rapidly over days. The presentation of a patient with RPGN constitutes a need for prompt diagnosis with early intervention and therapy to interrupt a natural progression to chronic renal failure. In adults, the most common cause of RPGN is pauci-immune GN associated with antineutrophil cytoplasmic antibodies (ANCA), and other common causes include Goodpasture’s syndrome (or anti-GBM disease) and immune-complex disease such as lupus nephritis.8,12 Immunohistology of the renal biopsy shows pauci-immune staining in ANCA-associated GN, linear IgG staining of the GBM in Goodpasture’s syndrome, and immune complex deposition in lupus nephritis, IgA nephropathy, and postinfectious GN.
TABLE118-1 Diseases Associated with Rapidly Progressive Glomerulonephritis and Pertinent Laboratory Studies
| Renal Limited | |
| IgA nephropathy | |
| Postinfectious glomerulonephritis | Low complement, streptococcal serologies, bacterial cultures |
| ANCA-associated glomerulonephritis (pauci-immune glomerulonephritis) | ANCA titers |
| Anti-GBM disease (Goodpasture’s syndrome) | Anti-GBM antibodies |
| Systemic Disorders | |
| Lupus nephritis | Low complement, ANA, dsDNA antibodies |
| ANCA-associated small-vessel vasculitis | ANCA titers |
| Anti-GBM disease | Anti-GBM antibodies |
| Henoch-Schönlein purpura | None |
| Cryoglobulinemic vasculitis | Low complement, cryoglobulins, hepatitis C serologies |
ANCA, antineutrophil cytoplasmic antibodies; ANA, antinuclear antibodies; dsDNA, double-stranded DNA; GBM, glomerular basement membrane; IgA, immunoglobulin A.
Pulmonary renal syndrome, characterized by RPGN and diffuse alveolar hemorrhage (DAH), often presents as a medical emergency requiring early aggressive treatment.13–15 It is associated with high mortality rates and rapid progression to ESKD if left untreated. Admission to the intensive care unit (ICU) and mortality are related to both the disease itself and infection. Patients often present with dyspnea, fever, cough, and hemoptysis, with chest radiography documenting pulmonary infiltrates. It may be difficult to distinguish from pneumonia, especially in patients without hemoptysis. Roughly 30% of patients with DAH do not present with hemoptysis. The presence of renal dysfunction and hematuria in patients presenting with these pulmonary symptoms should raise suspicion for a pulmonary renal syndrome. Although Goodpasture’s syndrome was first used in 1958 to describe patients presenting with pulmonary hemorrhage and GN,16 the most common cause of pulmonary renal syndrome is actually ANCA-associated small-vessel vasculitis.8 Goodpasture’s syndrome (also termed anti-GBM disease) now refers to the triad of DAH, RPGN, and the presence of anti-GBM antibodies and is the second most common cause of pulmonary renal syndrome. Much less common causes of pulmonary renal syndromes are SLE (systemic lupus erythematosus), thrombotic microangiopathies, and other systemic vasculitides.
Pauci-Immune Necrotizing Glomerulonephritis
Mortality of untreated disease is roughly 90% at 2 years following disease onset.17 However, systematic studies of different treatment regimens have led to significant progress in this field and improved patient outcomes.18 Treatment consists of pulse intravenous (IV) methylprednisolone followed by oral corticosteroids and IV cyclophosphamide.14,19–21 Even patients who are dialysis dependent on presentation often recover renal function with appropriate treatment. Poor prognostic indicators for patient and renal survival are the presence of DAH, severity of renal injury at diagnosis, degree of glomerular injury, extent of tubulointerstitial lesions on biopsy, and older age.22–26 Patients with DAH have a high mortality rate, and plasma exchange improves patient survival.17,27,28 For severe pulmonary disease, a few patients have been successfully treated with ECMO (extracorporeal membrane oxygenation).29,30 Additionally, patients with severe renal disease have an increased likelihood of renal recovery when treated with plasma exchange.23,25,31–33 With appropriate treatment, roughly 80% to 90% of patients achieve remission.17,21,27,34,35 Treatment resistance is more common in females, African Americans, and patients with severe renal disease. Relapse is more common in patients with anti-PR3 antibodies and involvement of the pulmonary and upper respiratory systems. The ANCA-associated small-vessel vasculitides follow a remitting and relapsing course, making long-term monitoring a key component to patient and kidney survival.
Anti-Glomerular Basement Membrane Glomerulonephritis
Goodpasture’s syndrome or anti-GBM disease presents as DAH and RPGN with evidence of anti-GBM antibodies on serologic testing. However, roughly 30% to 40% of patients present with renal limited disease without pulmonary involvement. It commonly affects Caucasians in a bimodal age distribution with peaks during the third and sixth decades.14,36–38 Renal biopsy shows linear deposition of antibodies, most commonly IgG and C3, along the GBM and glomerular crescent formation.
Untreated disease is highly fatal, and death is usually due to pulmonary hemorrhage or infection. Treatment with plasma exchange, cytotoxic agents, and corticosteroids was introduced in the 1970s, resulting in improved patient and renal survival.39 Plasma exchange is crucial for rapid clearance of anti-GBM antibodies40 and should be continued daily until antibodies are undetectable.38 Long-term outcomes are related to the degree of pulmonary compromise and renal dysfunction at presentation. With appropriate treatment, survival rates may exceed 90% for acute disease, but patients requiring RRT on initial presentation have lower survival rates.37,38,41 For those patients, only a very few recover renal function despite treatment with plasma exchange, corticosteroids, and cyclophosphamide. In contrast, those patients with creatinine (Cr) below 5.7 on presentation demonstrated 100% 1-year patient survival and 95% renal recovery in one study.37,41 In addition to dialysis dependence and elevated creatinine, predictors of poor renal outcome include oligoanuria, high anti-GBM antibody titers, and high percentage of glomeruli with crescent formation and extensive tubulointerstitial disease on renal biopsy.36,40,42,43 Although patient and renal survival is generally worse with anti-GBM disease than with ANCA-associated disease, late recurrence of anti-GBM disease is much rarer than recurrence of ANCA-associated disease.12,38
Both ANCA-associated vasculitis and anti-GBM disease are rare diseases, and interestingly, a subset of patients actually demonstrates both types of antibodies on serologic studies. Roughly 15% to 30% of patients with ANCA-associated disease also have anti-GBM antibodies, while only 5% to 10% of patients with anti-GBM antibodies also have detectable ANCA titers.12,37,38,44–46 Although outcome data are limited in this small group of patients, the outcomes of these patients may be better than patients with only anti-GBM antibodies.
Lupus Nephritis
Lupus nephritis occurs in 40% to 70% of patients with SLE and often occurs in the first 2 years following diagnosis.47–49 Less than 5% of patients present with RPGN or pulmonary renal syndrome. However, 10% to 20% of patients with lupus nephritis ultimately progress to ESKD. In addition to history and physical examination, evaluation includes analysis of urine sediment (because lupus nephritis may present as nephritic or nephrotic syndrome), assessment of proteinuria, complement levels, and serologies for ANA and anti-dsDNA antibodies. Cellular casts or proteinuria over 0.5 g/d is consistent with the diagnosis of lupus nephritis. Renal biopsy is critical for diagnosis, prognosis, and guiding treatment.
Renal biopsy is used to classify lupus nephritis into six categories: class I (minimal mesangial lupus GN), class II (mesangial proliferative lupus GN), class III (focal proliferative lupus GN), class IV (diffuse proliferative lupus GN), class V (membranous lupus GN), and class VI (advanced sclerosis).50–52 The most severe classes are the proliferative lesions of lupus nephritis (classes III and IV) and have poor renal survival without aggressive treatment. These classes often present with hematuria, proteinuria, hypertension, and AKI. Patients who present with RPGN are likely to have class IV lupus nephritis on renal biopsy. Sclerosing lupus nephritis is a chronic lesion that carries a poor prognosis.
Treatment of the more severe forms of lupus nephritis includes pulse methylprednisolone followed by oral corticosteroids and IV cyclophosphamide.48,53–57 Similar to treatment of pauci-immune GN, pulse IV cyclophosphamide is preferred over oral cyclophosphamide. Over 80% of patients respond to treatment.47,48 Importantly, about 5% to 10% of patients who require RRT initially recover enough renal function to become dialysis independent following treatment.49 Recent studies suggest that mycophenolate mofetil (MMF) is similar to cyclophosphamide in inducing remission; however, relapse appears more common in patients treated with MMF.58–62 Induction should be followed by maintenance therapy; the optimal maintenance regimen remains under intense investigation. Options for maintenance include additional cyclophosphamide, azathioprine, and MMF.48,57,63,64
Poor prognostic indicators at the beginning of treatment include male gender, African American race, severe hypertension, antiphospholipid syndrome (APS), and delayed initiation of immunosuppressive therapy. Following induction treatment, poor prognostic indicators are failure to achieve remission at 6 months and uncontrolled hypertension.48,49 Roughly one-third to half of patients will have relapse of disease. In some patients, recurrence of disease may be preceded by falling complement levels and rising anti-dsDNA titers. However, some patients with severe lupus nephritis have negative titers.47,65 Patients with only partial remission often recur sooner than patients with complete remission, and they are more likely to progress to ESKD.66 All patients with a history of lupus nephritis should be carefully monitored for recurrence of disease, and repeat renal biopsy is often needed to guide treatment decisions with relapsed disease.
Postinfectious Glomerulonephritis
Postinfectious glomerulonephritis (PIGN) presents as a classic nephritic syndrome occurring about 1 to 3 weeks after a group A β-hemolytic Streptococcus infection.67–69 It commonly occurs in children following a skin or pharyngeal infection. Although PIGN remains the most common cause of acute nephritic syndrome in the pediatric population in developing countries, the incidence of this disease has declined dramatically in the industrialized world. Children present with a classic nephritic syndrome with hematuria, proteinuria, hypertension, edema, and mild renal impairment. Severe hypertension with encephalopathy and seizures is uncommon and may require admission to the ICU.69–71 Laboratory findings demonstrate depressed complement levels (CH50 and C3) consistent with activation of the alternate complement cascade; levels return to normal by 8 to 12 weeks. Serologic studies may be used to confirm recent streptococcal infection, particularly with recent pharyngitis.69,72,73 Renal biopsy demonstrates endocapillary proliferation and granular deposition of immune complexes by immunohistology.67,72,74–76
The acute nephritic syndrome usually resolves in 7 days, and the prognosis of children with PIGN is excellent. However, roughly 10% to 20% of children have persistent urinary abnormalities including proteinuria and hematuria.68,69,73,77–79 Treatment is generally supportive with antihypertensives and diuretics as needed in the acute phase. Active infections should be treated, and prophylactic antibiotics are often indicated in endemic situations and for household contacts in regions with high prevalence of disease.
In contrast to children, outcomes for PIGN in adults in the industrialized world are much worse, particularly for patients with underlying chronic disease.67,68,80–82 PIGN can be associated with almost any infection, including most streptococcal and staphylococcal strains, gram-negative bacteria, mycobacteria, viruses, fungi, and parasites. Elderly patients often present with AKI, congestive heart failure, and nephrotic-range proteinuria. Up to half of these patients have underlying chronic diseases or risk factors including diabetes mellitus, liver disease or alcoholism, cancer, and IV drug use.80–83 Some patients demonstrate skin or pharyngeal infections, but many have other infections such as endocarditis and pulmonary infections. Streptococcal and staphylococcal infections account for only half of cases. Treatment consists of supportive care and eradication of infection. Although recent studies of adults with PIGN remain small, one-quarter to one-half of patients have persistent renal dysfunction, and as high as 15% may progress to ESKD.80–8284 In one small study, patients with underlying diabetic nephropathy had an extremely poor prognosis, with 81% progressing to ESKD.82
IgA Nephropathy
IgA nephropathy (IgAN) is an extremely common form of GN worldwide. However, IgAN is a renal-limited disease, with only 3% of patients presenting with AKI85 and most patients diagnosed in the outpatient setting. It commonly presents in the second or third decade of life and affects males more often than females.86 The majority of patients present with macroscopic or microscopic hematuria. Many patients have episodic hematuria, often associated with a concurrent upper respiratory tract or gastrointestinal infection. Patients may develop hypertension and varying degrees of proteinuria. Crescentic IgAN is associated with nephrotic-range proteinuria, severe hypertension, and rapidly declining renal function.87 No specific laboratory study to date can establish the diagnosis; renal biopsy is required. The extent of changes by light microscopy is variable, and the diagnosis is based on the demonstration of mesangial IgA deposits by immunohistology.
The long-term prognosis of patients with IgA nephropathy is highly variable, but many patients develop progressive renal failure. Between 15% and 40% of patients reach ESKD within 10 to 20 years of diagnosis.88,89 No consensus on the optimum treatment of IgAN is available owing to the lack of well-designed controlled trials. Progress is hampered by the fact that renal failure develops slowly over decades, and short clinical trials have limited usefulness.74,88,90,91 In all patients, hypertension should be aggressively treated with renin-angiotensin blockade. Patients with significant proteinuria and declining renal function may benefit from corticosteroids or immunosuppressive agents. Corticosteroids appear to reduce the risk of progression to ESKD and decrease proteinuria in selected patients.89,90,92 The small percentage of patients presenting with RPGN and crescentic GN are usually treated with pulse corticosteroids and cyclosphosphamide.87,88 Predictors of disease progression include renal dysfunction at diagnosis, significant proteinuria, hypertension, and evidence of chronic disease by renal biopsy.86,88,93,94
Henoch-Schönlein Purpura
On renal biopsy, Henoch-Schönlein purpura (HSP) is indistinguishable from IgAN. However, HSP is a systemic disease characterized by a distinct purpuric rash and gastrointestinal involvement. It occurs in children much more commonly than adults. The classic presentation is sudden onset of rash, progressing from nonblanching erythematous macules to urticarial papules to purpura with a symmetrical distribution on the extensor surfaces of the distal extremities and buttocks.95,96 Children present more frequently with gastrointestinal manifestations and fevers, whereas adults often have more severe renal involvement as well as joint symptoms.97,98 Renal involvement occurs in roughly one-third of children and two-thirds of adults.99,100
Renal involvement in HSP is usually more severe at presentation than IgAN, but most children completely recover.97,101,102 Estimates of recovery and chronic kidney disease vary widely, but the prognosis for renal recovery is worse in adults. Poor prognostic indicators include renal dysfunction and significant proteinuria at presentation, hypertension, and extensive glomerular disease by renal biopsy.98,99,102,103 Treatment is primarily supportive care, and trials to date do not support any specific treatment regimen.104 Corticosteroids may be useful in the short term, but there is no clear evidence that prednisone prevents serious long-term renal disease.100,105 Recently, two adults with severe systemic manifestations refractory to corticosteroids and immunosuppressive agents were treated with plasmapheresis with subsequent improvement.106
Thrombotic Microangiopathies
Thrombotic microangiopathy (TMA) is characterized by widespread thrombosis of arterioles and capillaries, with intraluminal platelet aggregation and vessel wall thickening.107–109 The underlying pathophysiologic cause of TMA is endothelial damage due to a variety of insults. The classic diseases associated with TMA are thrombotic thrombocytopenic purpura (TTP) and hemolytic uremic syndrome (HUS), but it may also be seen with catastrophic antiphospholipid syndrome (APS) and scleroderma renal crisis. TMA is also associated with a variety of medications including chemotherapeutic agents, calcineurin inhibitors (cyclosporine and tacrolimus), antiplatelet agents (ticlopidine and clopidogrel), and quinine.107 The classic pentad of findings in TTP includes microangiopathic hemolytic anemia, thrombocytopenia, neurologic symptoms and signs, impaired renal function, and fevers.107–109 Neurologic symptoms may dominate, presenting as confusion, headache, seizures, and coma. The renal manifestations are usually more prominent in HUS, and the typical presentation in children includes microangiopathic hemolytic anemia, thrombocytopenia, and AKI. Laboratory hallmarks include microangiopathic hemolytic anemia with schistocytes on peripheral smear, elevated lactate dehydrogenase levels, and thrombocytopenia, with platelets usually less than 60,000.
Remarkable progress has been made in elucidating the molecular basis for TTP and HUS. TTP occurs as familial and acquired forms, and both forms are related to abnormalities in the function of a zinc metalloprotease, ADAMTS 13 (a disintegrin and metalloprotease with thrombospondin type 1 motif 13).108–110 This protein is involved in the cleavage of von Willebrand factor (vWF), and deficiency of ADAMTS 13 leads to the accumulation of large multimers of vWF which bind platelets, leading to microvascular thrombosis. Familial TTP is associated with mutations of ADAMTS 13 leading to decreased or undetectable activity, and acquired TTP is caused by antibodies which inhibit its activity. Historically, untreated TTP had a mortality rate of over 90%. However, mortality has fallen to 10% to 20% with the advent of treatment using plasma exchange. Familial forms require chronic treatment with fresh frozen plasma or cryosupernatant that contains the active metalloprotease.
Hemolytic uremic syndrome is the most common cause of AKI in children and presents with hemolytic anemia, thrombocytopenia, and AKI.107,111–113 The classic or diarrheal form of HUS (D+ HUS) occurs most commonly following diarrheal infection with Shiga-like toxin–producing Escherichia coli (SLTEC). The peak incidence occurs in children younger than 5 years of age, and outbreaks often occur in association with E. coli O157:H7. The illness begins with abdominal cramps and nonbloody diarrhea, following by hemorrhagic diarrhea in 70% of patients. Within days, patients develop severe renal failure, anemia, and thrombocytopenia. These children are often critically ill, and roughly one-half to two-thirds of patients require RRT. About 70% of patients will require RBC transfusions, and 25% will have neurologic involvement. Over the last few decades, mortality rates have fallen from roughly 40% to 50% to 3% to 5%, primarily due to aggressive supportive care with red blood cell transfusions and RRT as needed. Numerous therapies for HUS have been investigated without clear benefit, and treatment remains largely supportive. Treatment of the diarrheal illness associated with E. coli O157:H7 with antibiotics is associated with increased risk of developing HUS. Spontaneous resolution occurs 1 to 3 weeks following disease onset, and the majority of patients demonstrate renal recovery. Unfortunately, some children develop ESKD, and up to 40% have long-term sequelae including chronic kidney disease, persistent proteinuria, and hypertension. Non–diarrheal associated HUS occurs in a minority of patients and may be associated with other infections such as Streptococcus pneumoniae.
A small percentage of patients with HUS have sporadic or familial forms. These patients have defects in the alternative complement pathway, and mutations have been described in complement factor H, complement factor I, and membrane cofactor protein.108–110 Mortality rates are over 50%, and most survivors progress to ESKD. Therapies with fresh frozen plasma, plasma exchange, and monoclonal antibodies targeting the alternative complement pathway are under investigation for treatment of this devastating disease.
A small percentage of patients with antiphospholipid syndrome (APS) present with “catastrophic” APS characterized by acute TMA involving the small vessels of multiple organs.114 The disease progresses over days to weeks and commonly affects the kidneys, lungs, central nervous system, heart, and skin. The kidney is the most common organ affected, with renal involvement in over 70% of patients. Renal disease manifests as malignant hypertension and AKI, with 25% of patients requiring RRT. Mortality is estimated at 50% of patients, and treatment based on case reports includes anticoagulation, corticosteroids, plasmapheresis, and intravenous immunoglobulin.
Scleroderma renal crisis presents as accelerated hypertension, and AKI and may be accompanied by encephalopathy with seizures or flash pulmonary edema.115 Roughly 10% of patients develop scleroderma renal crisis, usually occurring within 4 years of disease onset. The risk is greatest with diffuse cutaneous disease, and antecedent treatment with high-dose corticosteroids increases the risk of scleroderma renal crisis.116,117 Patients demonstrate microangiopathic hemolytic anemia, thrombocytopenia, proteinuria, microscopic hematuria, and marked increases in plasma renin. In the past, untreated disease had a dismal prognosis with less than 10% survival. The use of angiotensin-converting enzyme (ACE) inhibitors has revolutionized treatment; acute mortality rates are now below 25% with appropriate treatment.115,118,119 About half to two-thirds of patients will require RRT, but half of those patients recover enough renal function to become dialysis independent. Poor outcomes are associated with Cr above 3 at the initiation of ACE inhibitor therapy, poor blood pressure control, male gender, older age, and congestive heart failure. Patients with scleroderma renal crisis who do not require RRT have 90% survival rates at 5 years. In contrast, patients who become dialysis dependent have only 40% survival at 5 years. Early recognition and treatment are critical for both patient and renal outcomes. ACE inhibitors should be initiated rapidly and continued even if patients develop progressive renal failure or require RRT.
 Interstitial Nephritis
Interstitial Nephritis
Acute interstitial nephritis (AIN) demonstrates inflammation of both the renal interstitium and tubules, being more properly described as acute tubulointerstitial nephritis (Figure 118-2). This disorder reflects a hypersensitivity reaction, commonly induced by medications or infections.120,121 AIN accounts for 2% to 6% of renal biopsies, but the incidence may be as high as 25% in patients with unexplained AKI.120,122–124 Many critically ill patients are treated with medications commonly associated with AIN, such as antibiotics, proton pump inhibitors, and diuretics (Table 118-2).125,126 AIN is important to recognize early so that the cause may be identified and the medication discontinued to minimize renal damage.
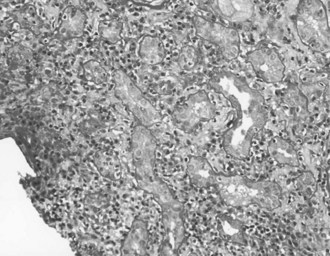
TABLE118-2 Common Medications Associated with Acute Interstitial Nephritis
The classic example of AIN is described for methicillin. The majority of patients developed fevers, eosinophilia, pyuria, and hematuria a few weeks following exposure. About half of patients developed AKI for a duration of several weeks, followed by full recovery in 90%.125 Unfortunately, the classic triad of fever, rash, and eosinophilia occurs in only 10% to 15% of patients.120–122125 Patients may present with mild renal impairment or severe AKI requiring RRT. Urine sediment may be bland or demonstrate sterile pyuria, white blood cell casts, and hematuria. In most cases, patients have subnephrotic-range proteinuria (<3 grams per day), but two-thirds of patients with NSAID-induced AIN present with nephrotic syndrome.121,125 Eosinophiluria, based on Wright or Hansel stain, is suggestive of acute AIN but is neither sensitive nor specific for this disorder.125,127,128 Eosinophiluria is also found in a variety of other disorders including pyelonephritis, cystitis, prostatitis, acute tubular necrosis, and glomerulonephritis.
Drug-induced AIN usually presents a few weeks following initiation of the medication, but it may take months to develop. With removal of the offending agent, the duration of AKI is quite variable from a few weeks to months. Medications account for over two-thirds of AIN, and the remainder of cases are associated with infection and other diseases, particularly autoimmune diseases.120,121,126 A few cases of acute AIN are associated with Chinese herbal remedies.129
The initial management of a patient with AIN is largely supportive, with dialysis as indicated. Identification of all candidate etiologic agents, elimination of potentially causative medications, and control of potential infectious causes are fundamental to the control of AIN.121,125 When replacing medications, it is important to choose medications that are not likely to cross-react with the original agent. The use of corticosteroid therapy remains controversial, and no large randomized trials have thoroughly examined the effectiveness of corticosteroid therapy in AIN.125,130 One small study in which the majority of patients developed AIN from antibiotics or NSAIDs demonstrated improved renal recovery with early steroid use.131 Another study demonstrated that the majority of patients improve with medication withdrawal, but those patients who do not respond after a few weeks may subsequently benefit from corticosteroid administration.132 In another retrospective study in which over 90% of patients had drug-induced AIN and 60% of patients received corticosteroids, there was no difference in renal outcomes.123 Recently, MMF has been used successfully in patients who did not initially respond to corticosteroids.133 Early improvement of renal impairment and patchy infiltrates on biopsy are prognostic indicators for improved renal outcomes.121,134 Poor prognostic indicators include advanced age, prolonged renal impairment, and degree of chronic tubulointerstitial changes on renal biopsy. Roughly 30% to 40% of patients will have some degree of long-term renal impairment.121,125,126
Key Points
Appel GB, Contreras G, Dooley MA, Ginzler EM, Isenberg D, Jayne D, et al. Mycophenolate mofetil versus cyclophosphamide for induction treatment of lupus nephritis. J Am Soc Nephrol. 2009;20:1103-1112.
De Groot K, Harper L, Jayne DR, Florez Suarez LF, Gregorini G, Gross WL, et al. Pulse versus daily oral cyclophosphamide for induction of remission in antineutrophil cytoplasmic antibody-associated vasculitis: a randomized trial. Ann Intern Med. 2009;150:670-680.
Levy JB, Turner AN, Rees AJ, Pusey CD. Long-term outcome of anti-glomerular basement membrane antibody disease treated with plasma exchange and immunosuppression. Ann Intern Med. 2001;134:1033-1042.
Nasr SH, Markowitz GS, Stokes MB, Said SM, Valeri AM, D’Agati VD. Acute postinfectious glomerulonephritis in the modern era: experience with 86 adults and review of the literature. Medicine (Baltimore). 2008;87:21-32.
Schwarz A, Krause PH, Kunzendorf U, Keller F, Distler A. The outcome of acute interstitial nephritis: risk factors for the transition from acute to chronic interstitial nephritis. Clin Nephrol. 2000;54:179-190.
Steen VD, Medsger TAJr. Long-term outcomes of scleroderma renal crisis. Ann Intern Med. 2000;133:600-603.
1 Barrantes F, Tian J, Vazquez R, Amoateng-Adjepong Y, Manthous CA. Acute kidney injury criteria predict outcomes of critically ill patients. Crit Care Med. 2008;36:1397-1403.
2 Bell M. Acute kidney injury: new concepts, renal recovery. Nephron Clin Pract. 2008;109:c224-c228.
3 Hoste EA, Schurgers M. Epidemiology of acute kidney injury: how big is the problem? Crit Care Med. 2008;36:S146-S151.
4 Mehta RL, Pascual MT, Soroko S, et al. Spectrum of acute renal failure in the intensive care unit: the PICARD experience. Kidney Int. 2004;66:1613-1621.
5 Uchino S, Kellum JA, Bellomo R, et al. Acute renal failure in critically ill patients: a multinational, multicenter study. JAMA. 2005;294:813-818.
6 Waikar SS, Liu KD, Chertow GM. Diagnosis, epidemiology and outcomes of acute kidney injury. Clin J Am Soc Nephrol. 2008;3:844-861.
7 Prakash J, Sen D, Kumar NS, Kumar H, Tripathi LK, Saxena RK. Acute renal failure due to intrinsic renal diseases: review of 1122 cases. Ren Fail. 2003;25:225-233.
8 Couser WG. Glomerulonephritis. Lancet. 1999;353:1509-1515.
9 Farrington K, Levison DA, Greenwood RN, Cattell WR, Baker LR. Renal biopsy in patients with unexplained renal impairment and normal kidney size. Q J Med. 1989;70:221-233.
10 Haas M, Spargo BH, Wit EJ, Meehan SM. Etiologies and outcome of acute renal insufficiency in older adults: a renal biopsy study of 259 cases. Am J Kidney Dis. 2000;35:433-447.
11 Schena FP. Survey of the Italian Registry of Renal Biopsies. Frequency of the renal diseases for 7 consecutive years. The Italian Group of Renal Immunopathology. Nephrol Dial Transplant. 1997;12:418-426.
12 Jennette JC. Rapidly progressive crescentic glomerulonephritis. Kidney Int. 2003;63:1164-1177.
13 Collard HR, Schwarz MI. Diffuse alveolar hemorrhage. Clin Chest Med. 2004;25:583-592. vii
14 Papiris SA, Manali ED, Kalomenidis I, Kapotsis GE, Karakatsani A, Roussos C. Bench-to-bedside review: pulmonary-renal syndromes–an update for the intensivist. Crit Care. 2007;11:213.
15 Semple D, Keogh J, Forni L, Venn R. Clinical review: Vasculitis on the intensive care unit–part 2: treatment and prognosis. Crit Care. 2005;9:193-197.
16 Stanton MC, Tange JD. Goodpasture’s syndrome (pulmonary haemorrhage associated with glomerulonephritis). Australas Ann Med. 1958;7:132-144.
17 Kamesh L, Harper L, Savage CO. ANCA-positive vasculitis. J Am Soc Nephrol. 2002;13:1953-1960.
18 Tesar V, Rihova Z, Jancova E, Rysava R, Merta M. Current treatment strategies in ANCA-positive renal vasculitis-lessons from European randomized trials. Nephrol Dial Transplant. 2003;18(Suppl 5):v2-v4.
19 De Groot K, Rasmussen N, Bacon PA, et al. Randomized trial of cyclophosphamide versus methotrexate for induction of remission in early systemic antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheum. 2005;52:2461-2469.
20 de Groot K, Harper L, Jayne DR, et al. Pulse versus daily oral cyclophosphamide for induction of remission in antineutrophil cytoplasmic antibody-associated vasculitis: a randomized trial. Ann Intern Med. 2009;150:670-680.
21 Jayne D, Rasmussen N, Andrassy K, et al. A randomized trial of maintenance therapy for vasculitis associated with antineutrophil cytoplasmic autoantibodies. N Engl J Med. 2003;349:36-44.
22 Gallagher H, Kwan JT, Jayne DR. Pulmonary renal syndrome: a 4-year, single-center experience. Am J Kidney Dis. 2002;39:42-47.
23 Gaskin G, Pusey CD. Plasmapheresis in antineutrophil cytoplasmic antibody-associated systemic vasculitis. Ther Apher. 2001;5:176-181.
24 de Lind van Wijngaarden RA, Hauer HA, Wolterbeek R, et al. Chances of renal recovery for dialysis-dependent ANCA-associated glomerulonephritis. J Am Soc Nephrol. 2007;18:2189-2197.
25 de Lind van Wijngaarden RA, Hauer HA, Wolterbeek R, et al. Clinical and histologic determinants of renal outcome in ANCA-associated vasculitis: A prospective analysis of 100 patients with severe renal involvement. J Am Soc Nephrol. 2006;17:2264-2274.
26 Hedger N, Stevens J, Drey N, Walker S, Roderick P. Incidence and outcome of pauci-immune rapidly progressive glomerulonephritis in Wessex, UK: a 10-year retrospective study. Nephrol Dial Transplant. 2000;15:1593-1599.
27 Hogan SL, Falk RJ, Chin H, et al. Predictors of relapse and treatment resistance in antineutrophil cytoplasmic antibody-associated small-vessel vasculitis. Ann Intern Med. 2005;143:621-631.
28 Booth AD, Almond MK, Burns A, et al. Outcome of ANCA-associated renal vasculitis: a 5-year retrospective study. Am J Kidney Dis. 2003;41:776-784.
29 Ahmed SH, Aziz T, Cochran J, Highland K. Use of extracorporeal membrane oxygenation in a patient with diffuse alveolar hemorrhage. Chest. 2004;126:305-309.
30 Matsumoto T, Ueki K, Tamura S, et al. Extracorporeal membrane oxygenation for the management of respiratory failure due to ANCA-associated vasculitis. Scand J Rheumatol. 2000;29:195-197.
31 Walters G, Willis NS, Craig JC. Interventions for renal vasculitis in adults. Cochrane Database Syst Rev 2008:CD003232.
32 Jayne DR, Gaskin G, Rasmussen N, et al. Randomized trial of plasma exchange or high-dosage methylprednisolone as adjunctive therapy for severe renal vasculitis. J Am Soc Nephrol. 2007;18:2180-2188.
33 Klemmer PJ, Chalermskulrat W, Reif MS, Hogan SL, Henke DC, Falk RJ. Plasmapheresis therapy for diffuse alveolar hemorrhage in patients with small-vessel vasculitis. Am J Kidney Dis. 2003;42:1149-1153.
34 Hogan SL, Nachman PH, Wilkman AS, Jennette JC, Falk RJ. Prognostic markers in patients with antineutrophil cytoplasmic autoantibody-associated microscopic polyangiitis and glomerulonephritis. J Am Soc Nephrol. 1996;7:23-32.
35 de Groot K, Jayne D. What is new in the therapy of ANCA-associated vasculitides? Take home messages from the 12th workshop on ANCA and systemic vasculitides. Clin Nephrol. 2005;64:480-484.
36 Bolton WK. Goodpasture’s syndrome. Kidney Int. 1996;50:1753-1766.
37 Pusey CD. Anti-glomerular basement membrane disease. Kidney Int. 2003;64:1535-1550.
38 Salama AD, Levy JB, Lightstone L, Pusey CD. Goodpasture’s disease. Lancet. 2001;358:917-920.
39 Lockwood CM, Rees AJ, Pearson TA, Evans DJ, Peters DK, Wilson CB. Immunosuppression and plasma-exchange in the treatment of Goodpasture’s syndrome. Lancet. 1976;1:711-715.
40 Johnson JP, Moore JJr, Austin HA3rd, Balow JE, Antonovych TT, Wilson CB. Therapy of anti-glomerular basement membrane antibody disease: analysis of prognostic significance of clinical, pathologic and treatment factors. Medicine (Baltimore). 1985;64:219-227.
41 Levy JB, Turner AN, Rees AJ, Pusey CD. Long-term outcome of anti-glomerular basement membrane antibody disease treated with plasma exchange and immunosuppression. Ann Intern Med. 2001;134:1033-1042.
42 Herody M, Bobrie G, Gouarin C, Grunfeld JP, Noel LH. Anti-GBM disease: predictive value of clinical, histological and serological data. Clin Nephrol. 1993;40:249-255.
43 Savage CO, Pusey CD, Bowman C, Rees AJ, Lockwood CM. Antiglomerular basement membrane antibody mediated disease in the British Isles 1980-4. Br Med J (Clin Res Ed). 1986;292:301-304.
44 Niles JL, Bottinger EP, Saurina GR, et al. The syndrome of lung hemorrhage and nephritis is usually an ANCA-associated condition. Arch Intern Med. 1996;156:440-445.
45 Levy JB, Hammad T, Coulthart A, Dougan T, Pusey CD. Clinical features and outcome of patients with both ANCA and anti-GBM antibodies. Kidney Int. 2004;66:1535-1540.
46 Lindic J, Vizjak A, Ferluga D, et al. Clinical outcome of patients with coexistent antineutrophil cytoplasmic antibodies and antibodies against glomerular basement membrane. Ther Apher Dial. 2009;13:278-281.
47 Balow JE. Clinical presentation and monitoring of lupus nephritis. Lupus. 2005;14:25-30.
48 Boumpas DT, Sidiropoulos P, Bertsias G. Optimum therapeutic approaches for lupus nephritis: what therapy and for whom? Nat Clin Pract Rheumatol. 2005;1:22-30.
49 Tassiulas IU, Boumpas DT. clinical features and treatment of systemic lupus erythematosus. In: Firestein GS, Budd RC, Harris ED, McInnes IB, Ruddy S, Sergent JS, editors. Kelley’s textbook of rheumatology. 8th ed. Saunders Elsevier; 2009:1263-1300.
50 Weening JJ, D’Agati VD, Schwartz MM, et al. The classification of glomerulonephritis in systemic lupus erythematosus revisited. J Am Soc Nephrol. 2004;15:241-250.
51 Appel GB, Valeri A. The course and treatment of lupus nephritis. Annu Rev Med. 1994;45:525-537.
52 Jacobsen S, Starklint H, Petersen J, et al. Prognostic value of renal biopsy and clinical variables in patients with lupus nephritis and normal serum creatinine. Scand J Rheumatol. 1999;28:288-299.
53 Austin HA3rd, Klippel JH, Balow JE, et al. Therapy of lupus nephritis. Controlled trial of prednisone and cytotoxic drugs. N Engl J Med. 1986;314:614-619.
54 Boumpas DT, Austin HA3rd, Vaughn EM, et al. Controlled trial of pulse methylprednisolone versus two regimens of pulse cyclophosphamide in severe lupus nephritis. Lancet. 1992;340:741-745.
55 Gourley MF, Austin HA3rd, Scott D, et al. Methylprednisolone and cyclophosphamide, alone or in combination, in patients with lupus nephritis. A randomized, controlled trial. Ann Intern Med. 1996;125:549-557.
56 Illei GG, Austin HA, Crane M, et al. Combination therapy with pulse cyclophosphamide plus pulse methylprednisolone improves long-term renal outcome without adding toxicity in patients with lupus nephritis. Ann Intern Med. 2001;135:248-257.
57 Houssiau FA. Management of lupus nephritis: an update. J Am Soc Nephrol. 2004;15:2694-2704.
58 Chan TM, Tse KC, Tang CS, Mok MY, Li FK. Long-term study of mycophenolate mofetil as continuous induction and maintenance treatment for diffuse proliferative lupus nephritis. J Am Soc Nephrol. 2005;16:1076-1084.
59 Ginzler EM, Dooley MA, Aranow C, et al. Mycophenolate mofetil or intravenous cyclophosphamide for lupus nephritis. N Engl J Med. 2005;353:2219-2228.
60 Hu W, Liu Z, Chen H, et al. Mycophenolate mofetil vs cyclophosphamide therapy for patients with diffuse proliferative lupus nephritis. Chin Med J (Engl). 2002;115:705-709.
61 Appel GB, Contreras G, Dooley MA, et al. Mycophenolate mofetil versus cyclophosphamide for induction treatment of lupus nephritis. J Am Soc Nephrol. 2009;20:1103-1112.
62 Ginzler EM, Wofsy D, Isenberg D, Gordon C, Lisk L, Dooley MA. Nonrenal disease activity following mycophenolate mofetil or intravenous cyclophosphamide as induction treatment for lupus nephritis: findings in a multicenter, prospective, randomized, open-label, parallel-group clinical trial. Arthritis Rheum. 2010;62:211-221.
63 Contreras G, Pardo V, Leclercq B, et al. Sequential therapies for proliferative lupus nephritis. N Engl J Med. 2004;350:971-980.
64 Moroni G, Doria A, Mosca M, et al. A randomized pilot trial comparing cyclosporine and azathioprine for maintenance therapy in diffuse lupus nephritis over four years. Clin J Am Soc Nephrol. 2006;1:925-932.
65 Hahn BH. Antibodies to DNA. N Engl J Med. 1998;338:1359-1368.
66 Illei GG, Takada K, Parkin D, et al. Renal flares are common in patients with severe proliferative lupus nephritis treated with pulse immunosuppressive therapy: long-term followup of a cohort of 145 patients participating in randomized controlled studies. Arthritis Rheum. 2002;46:995-1002.
67 Kanjanabuch T, Kittikowit W, Eiam-Ong S. An update on acute postinfectious glomerulonephritis worldwide. Nat Rev Nephrol. 2009;5:259-269.
68 Rodriguez-Iturbe B, Musser JM. The current state of poststreptococcal glomerulonephritis. J Am Soc Nephrol. 2008;19:1855-1864.
69 Holm SE, Normstrand A, Stevens DL, Norgren M. Acute poststreptococcal glomerulonephritis. In: Stevens DL, Kaplan EL, editors. Streptococcal infections: clinical aspects, microbiology, and molecular pathogenesis. New York: Oxford University Press; 2000:152-162.
70 Wong W, Morris MC. Cerebral vasculitis in a child following post-streptococcal glomerulonephritis. J Paediatr Child Health. 2001;37:597-599.
71 Kaplan RA, Zwick DL, Hellerstein S, Warady BA, Alon U. Cerebral vasculitis in acute post-streptococcal glomerulonephritis. Pediatr Nephrol. 1993;7:194-195.
72 Richards J. Acute post-streptococcal glomerulonephritis. W V Med J. 1991;87:61-65.
73 Berrios X, Lagomarsino E, Solar E, Sandoval G, Guzman B, Riedel I. Post-streptococcal acute glomerulonephritis in Chile–20 years of experience. Pediatr Nephrol. 2004;19:306-312.
74 Wyatt RJ, Forristal J, West CD, Sugimoto S, Curd JG. Complement profiles in acute post-streptococcal glomerulonephritis. Pediatr Nephrol. 1988;2:219-223.
75 Matsell DG, Roy S3rd, Tamerius JD, Morrow PR, Kolb WP, Wyatt RJ. Plasma terminal complement complexes in acute poststreptococcal glomerulonephritis. Am J Kidney Dis. 1991;17:311-316.
76 Madaio MP, Harrington JT. Current concepts. The diagnosis of acute glomerulonephritis. N Engl J Med. 1983;309:1299-1302.
77 Wallace MR. Acute glomerulonephritis in childhood: a prospective study of hospital admissions. N Z Med J. 1981;94:134-137.
78 Kasahara T, Hayakawa H, Okubo S, et al. Prognosis of acute poststreptococcal glomerulonephritis (APSGN) is excellent in children, when adequately diagnosed. Pediatr Int. 2001;43:364-367.
79 Drachman R, Aladjem M, Vardy PA. Natural history of an acute glomerulonephritis epidemic in children. An 11- to 12-year follow-up. Isr J Med Sci. 1982;18:603-607.
80 Montseny JJ, Meyrier A, Kleinknecht D, Callard P. The current spectrum of infectious glomerulonephritis. Experience with 76 patients and review of the literature. Medicine (Baltimore). 1995;74:63-73.
81 Moroni G, Pozzi C, Quaglini S, et al. Long-term prognosis of diffuse proliferative glomerulonephritis associated with infection in adults. Nephrol Dial Transplant. 2002;17:1204-1211.
82 Nasr SH, Markowitz GS, Stokes MB, Said SM, Valeri AM, D’Agati VD. Acute postinfectious glomerulonephritis in the modern era: experience with 86 adults and review of the literature. Medicine (Baltimore). 2008;87:21-32.
83 Keller CK, Andrassy K, Waldherr R, Ritz E. Postinfectious glomerulonephritis–is there a link to alcoholism? Q J Med. 1994;87:97-102.
84 Sesso R, Pinto SW. Five-year follow-up of patients with epidemic glomerulonephritis due to Streptococcus zooepidemicus. Nephrol Dial Transplant. 2005;20:1808-1812.
85 Packham DK, Hewitson TD, Yan HD, Elliott CE, Nicholls K, Becker GJ. Acute renal failure in IgA nephropathy. Clin Nephrol. 1994;42:349-353.
86 Nachman PH, Jennette JC, Falk RJ. Primary glomerular disease. In: Brenner BM, editor. Brenner and Rector’s: the kidney. 8th ed. Saunders Elsevier; 2008:987-1066.
87 Tumlin JA, Hennigar RA. Clinical presentation, natural history, and treatment of crescentic proliferative IgA nephropathy. Semin Nephrol. 2004;24:256-268.
88 Barratt J, Feehally J. Treatment of IgA nephropathy. Kidney Int. 2006;69:1934-1938.
89 Samuels JA, Strippoli GF, Craig JC, Schena FP, Molony DA. Immunosuppressive treatments for immunoglobulin A nephropathy: a meta-analysis of randomized controlled trials. Nephrology. 2004;9:177-185.
90 Ballardie FW. Quantitative appraisal of treatment options for IgA nephropathy. J Am Soc Nephrol. 2007;18:2806-2809.
91 Kanno Y, Okada H, Saruta T, Suzuki H. Blood pressure reduction associated with preservation of renal function in hypertensive patients with IgA nephropathy: a 3-year follow-up. Clin Nephrol. 2000;54:360-365.
92 Ballardie FW, Roberts IS. Controlled prospective trial of prednisolone and cytotoxics in progressive IgA nephropathy. J Am Soc Nephrol. 2002;13:142-148.
93 Bartosik LP, Lajoie G, Sugar L, Cattran DC. Predicting progression in IgA nephropathy. Am J Kidney Dis. 2001;38:728-735.
94 Donadio JV, Bergstralh EJ, Grande JP, Rademcher DM. Proteinuria patterns and their association with subsequent end-stage renal disease in IgA nephropathy. Nephrol Dial Transplant. 2002;17:1197-1203.
95 Cameron JS. Henoch-Schonlein purpura: clinical presentation. Contrib Nephrol. 1984;40:246-249.
96 Allen DM, Diamond LK, Howell DA. Anaphylactoid purpura in children (Schonlein-Henoch syndrome): review with a follow-up of the renal complications. AMA J Dis Child. 1960;99:833-854.
97 Blanco R, Martinez-Taboada VM, Rodriguez-Valverde V, Garcia-Fuentes M, Gonzalez-Gay MA. Henoch-Schonlein purpura in adulthood and childhood: two different expressions of the same syndrome. Arthritis Rheum. 1997;40:859-864.
98 Pillebout E, Thervet E, Hill G, Alberti C, Vanhille P, Nochy D. Henoch-Schonlein purpura in adults: outcome and prognostic factors. J Am Soc Nephrol. 2002;13:1271-1278.
99 Rieu P, Noel LH. Henoch-Schonlein nephritis in children and adults. Morphological features and clinicopathological correlations. Ann Med Interne (Paris). 1999;150:151-159.
100 Kaku Y, Nohara K, Honda S. Renal involvement in Henoch-Schonlein purpura: a multivariate analysis of prognostic factors. Kidney Int. 1998;53:1755-1759.
101 Yoshikawa N, Ito H, Yoshiya K, et al. Henoch-Schoenlein nephritis and IgA nephropathy in children: a comparison of clinical course. Clin Nephrol. 1987;27:233-237.
102 Kawasaki Y, Suzuki J, Sakai N, et al. Clinical and pathological features of children with Henoch-Schoenlein purpura nephritis: risk factors associated with poor prognosis. Clin Nephrol. 2003;60:153-160.
103 Goldstein AR, White RH, Akuse R, Chantler C. Long-term follow-up of childhood Henoch-Schonlein nephritis. Lancet. 1992;339:280-282.
104 Zaffanello M, Fanos V. Treatment-based literature of Henoch-Schonlein purpura nephritis in childhood. Pediatr Nephrol. 2009;24:1901-1911.
105 Chartapisak W, Opastirakul S, Hodson EM, Willis NS, Craig JC. Interventions for preventing and treating kidney disease in Henoch-Schonlein Purpura (HSP). Cochrane Database Syst Rev 2009:CD005128.
106 Donghi D, Schanz U, Sahrbacher U, et al. Life-threatening or organ-impairing Henoch-Schonlein purpura: plasmapheresis may save lives and limit organ damage. Dermatology. 2009;219:167-170.
107 Ruggenenti P, Noris M, Remuzzi G. Thrombotic microangiopathy, hemolytic uremic syndrome, and thrombotic thrombocytopenic purpura. Kidney Int. 2001;60:831-846.
108 Tsai HM. Current concepts in thrombotic thrombocytopenic purpura. Annu Rev Med. 2006;57:419-436.
109 Moake JL. Thrombotic microangiopathies. N Engl J Med. 2002;347:589-600.
110 Tsai HM. The molecular biology of thrombotic microangiopathy. Kidney Int. 2006;70:16-23.
111 Andreoli SP, Trachtman H, Acheson DW, Siegler RL, Obrig TG. Hemolytic uremic syndrome: epidemiology, pathophysiology, and therapy. Pediatr Nephrol. 2002;17:293-298.
112 Noris M, Remuzzi G. Hemolytic uremic syndrome. J Am Soc Nephrol. 2005;16:1035-1050.
113 Scheiring J, Rosales A, Zimmerhackl LB. Clinical practice. Today’s understanding of the haemolytic uraemic syndrome. Eur J Pediatr. 2010;169:7-13.
114 Levine JS, Branch DW, Rauch J. The antiphospholipid syndrome. N Engl J Med. 2002;346:752-763.
115 Steen VD. Scleroderma renal crisis. Rheum Dis Clin North Am. 2003;29:315-333.
116 Kohno K, Katayama T, Majima K, et al. A case of normotensive scleroderma renal crisis after high-dose methylprednisolone treatment. Clin Nephrol. 2000;53:479-482.
117 Steen VD, Medsger TAJr. Case-control study of corticosteroids and other drugs that either precipitate or protect from the development of scleroderma renal crisis. Arthritis Rheum. 1998;41:1613-1619.
118 Steen VD, Medsger TAJr. Long-term outcomes of scleroderma renal crisis. Ann Intern Med. 2000;133:600-603.
119 Penn H, Howie AJ, Kingdon EJ, et al. Scleroderma renal crisis: patient characteristics and long-term outcomes. QJM. 2007;100:485-494.
120 John R, Herzenberg AM. Renal toxicity of therapeutic drugs. J Clin Pathol. 2009;62:505-515.
121 Baker RJ, Pusey CD. The changing profile of acute tubulointerstitial nephritis. Nephrol Dial Transplant. 2004;19:8-11.
122 Abdel-Kader K, Palevsky PM. Acute kidney injury in the elderly. Clin Geriatr Med. 2009;25:331-358.
123 Clarkson MR, Giblin L, O’Connell FP, et al. Acute interstitial nephritis: clinical features and response to corticosteroid therapy. Nephrol Dial Transplant. 2004;19:2778-2783.
124 Davison AM, Jones CH. Acute interstitial nephritis in the elderly: a report from the UK MRC Glomerulonephritis Register and a review of the literature. Nephrol Dial Transplant. 1998;13(Suppl 7):12-16.
125 Rossert J. Drug-induced acute interstitial nephritis. Kidney Int. 2001;60:804-817.
126 Schwarz A, Krause PH, Kunzendorf U, Keller F, Distler A. The outcome of acute interstitial nephritis: risk factors for the transition from acute to chronic interstitial nephritis. Clin Nephrol. 2000;54:179-190.
127 Fletcher A. Eosinophiluria and acute interstitial nephritis. N Engl J Med. 2008;358:1760-1761.
128 Nolan CR3rd, Anger MS, Kelleher SP. Eosinophiluria–a new method of detection and definition of the clinical spectrum. N Engl J Med. 1986;315:1516-1519.
129 Debelle FD, Vanherweghem JL, Nortier JL. Aristolochic acid nephropathy: a worldwide problem. Kidney Int. 2008;74:158-169.
130 Appel GB. The treatment of acute interstitial nephritis: More data at last. Kidney Int. 2008;73:905-907.
131 Gonzalez E, Gutierrez E, Galeano C, et al. Early steroid treatment improves the recovery of renal function in patients with drug-induced acute interstitial nephritis. Kidney Int. 2008;73:940-946.
132 Buysen JG, Houthoff HJ, Krediet RT, Arisz L. Acute interstitial nephritis: a clinical and morphological study in 27 patients. Nephrol Dial Transplant. 1990;5:94-99.
133 Preddie DC, Markowitz GS, Radhakrishnan J, et al. Mycophenolate mofetil for the treatment of interstitial nephritis. Clin J Am Soc Nephrol. 2006;1:718-722.
134 Laberke HG, Bohle A. Acute interstitial nephritis: correlations between clinical and morphological findings. Clin Nephrol. 1980;14:263-273.
