336 |
Dialysis in the Treatment of Renal Failure |
Dialysis may be required for the treatment of either acute or chronic kidney disease. The use of continuous renal replacement therapies (CRRTs) and slow low-efficiency dialysis (SLED) is specific to the management of acute renal failure and is discussed in Chap. 334. These modalities are performed continuously (CRRT) or over 6–12 h per session (SLED), in contrast to the 3–4 h of an intermittent hemodialysis session. Advantages and disadvantages of CRRT and SLED are discussed in Chap. 334.
Peritoneal dialysis is rarely used in developed countries for the treatment of acute renal failure because of the increased risk of infection and (as will be discussed in more detail below) less efficient clearance per unit of time. The focus of this chapter will be on the use of peritoneal and hemodialysis for end-stage renal disease (ESRD).
With the widespread availability of dialysis, the lives of hundreds of thousands of patients with ESRD have been prolonged. In the United States alone, there are now approximately 615,000 patients with ESRD, the vast majority of whom require dialysis. The incidence rate for ESRD is 357 cases per million population per year. The incidence of ESRD is disproportionately higher in African Americans (940 per million population per year) as compared with white Americans (280 per million population per year). In the United States, the leading cause of ESRD is diabetes mellitus, currently accounting for nearly 45% of newly diagnosed cases of ESRD. Approximately 30% of patients have ESRD that has been attributed to hypertension, although it is unclear whether in these cases hypertension is the cause or a consequence of vascular disease or other unknown causes of kidney failure. Other prevalent causes of ESRD include glomerulonephritis, polycystic kidney disease, and obstructive uropathy.
![]() Globally, mortality rates for patients with ESRD are lowest in Europe and Japan but very high in the developing world because of the limited availability of dialysis. In the United States, the mortality rate of patients on dialysis has decreased slightly but remains extremely high, with a 5-year survival rate of approximately 35–40%. Deaths are due mainly to cardiovascular diseases and infections (approximately 40 and 10% of deaths, respectively). Older age, male sex, nonblack race, diabetes mellitus, malnutrition, and underlying heart disease are important predictors of death.
Globally, mortality rates for patients with ESRD are lowest in Europe and Japan but very high in the developing world because of the limited availability of dialysis. In the United States, the mortality rate of patients on dialysis has decreased slightly but remains extremely high, with a 5-year survival rate of approximately 35–40%. Deaths are due mainly to cardiovascular diseases and infections (approximately 40 and 10% of deaths, respectively). Older age, male sex, nonblack race, diabetes mellitus, malnutrition, and underlying heart disease are important predictors of death.
TREATMENT OPTIONS FOR ESRD PATIENTS
Commonly accepted criteria for initiating patients on maintenance dialysis include the presence of uremic symptoms, the presence of hyperkalemia unresponsive to conservative measures, persistent extracellular volume expansion despite diuretic therapy, acidosis refractory to medical therapy, a bleeding diathesis, and a creatinine clearance or estimated glomerular filtration rate (GFR) below 10 mL/min per 1.73 m2 (see Chap. 335 for estimating equations). Timely referral to a nephrologist for advanced planning and creation of a dialysis access, education about ESRD treatment options, and management of the complications of advanced chronic kidney disease (CKD), including hypertension, anemia, acidosis, and secondary hyperparathyroidism, are advisable. Recent data have suggested that a sizable fraction of ESRD cases result following episodes of acute renal failure, particularly among persons with underlying CKD. Furthermore, there is no benefit to initiating dialysis preemptively at a GFR of 10–14 mL/min per 1.73 m2 compared to initiating dialysis for symptoms of uremia.
In ESRD, treatment options include hemodialysis (in center or at home); peritoneal dialysis, as either continuous ambulatory peritoneal dialysis (CAPD) or continuous cyclic peritoneal dialysis (CCPD); or transplantation (Chap. 337). Although there are significant geographic variations and differences in practice patterns, hemodialysis remains the most common therapeutic modality for ESRD (>90% of patients) in the United States. In contrast to hemodialysis, peritoneal dialysis is continuous, but much less efficient, in terms of solute clearance. Although no large-scale clinical trials have been completed comparing outcomes among patients randomized to either hemodialysis or peritoneal dialysis, outcomes associated with both therapies are similar in most reports, and the decision of which modality to select is often based on personal preferences and quality-of-life considerations.
HEMODIALYSIS
Hemodialysis relies on the principles of solute diffusion across a semipermeable membrane. Movement of metabolic waste products takes place down a concentration gradient from the circulation into the dialysate. The rate of diffusive transport increases in response to several factors, including the magnitude of the concentration gradient, the membrane surface area, and the mass transfer coefficient of the membrane. The latter is a function of the porosity and thickness of the membrane, the size of the solute molecule, and the conditions of flow on the two sides of the membrane. According to laws of diffusion, the larger the molecule, the slower is its rate of transfer across the membrane. A small molecule, such as urea (60 Da), undergoes substantial clearance, whereas a larger molecule, such as creatinine (113 Da), is cleared less efficiently. In addition to diffusive clearance, movement of waste products from the circulation into the dialysate may occur as a result of ultrafiltration. Convective clearance occurs because of solvent drag, with solutes being swept along with water across the semipermeable dialysis membrane.
THE DIALYZER
There are three essential components to hemodialysis: the dialyzer, the composition and delivery of the dialysate, and the blood delivery system (Fig. 336-1). The dialyzer is a plastic chamber with the ability to perfuse blood and dialysate compartments simultaneously at very high flow rates. The hollow-fiber dialyzer is the most common in use in the United States. These dialyzers are composed of bundles of capillary tubes through which blood circulates while dialysate travels on the outside of the fiber bundle. The majority of dialyzers now manufactured in the United States are “biocompatible” synthetic membranes derived from polysulfone or related compounds (versus older cellulose “bioincompatible” membranes that activated the complement cascade). The frequency of reprocessing and reuse of hemodialyzers and blood lines varies across the world. In general, as the cost of disposable supplies has decreased, their use has increased. Formaldehyde, peracetic acid–hydrogen peroxide, glutaraldehyde, and bleach have all been used as reprocessing agents.
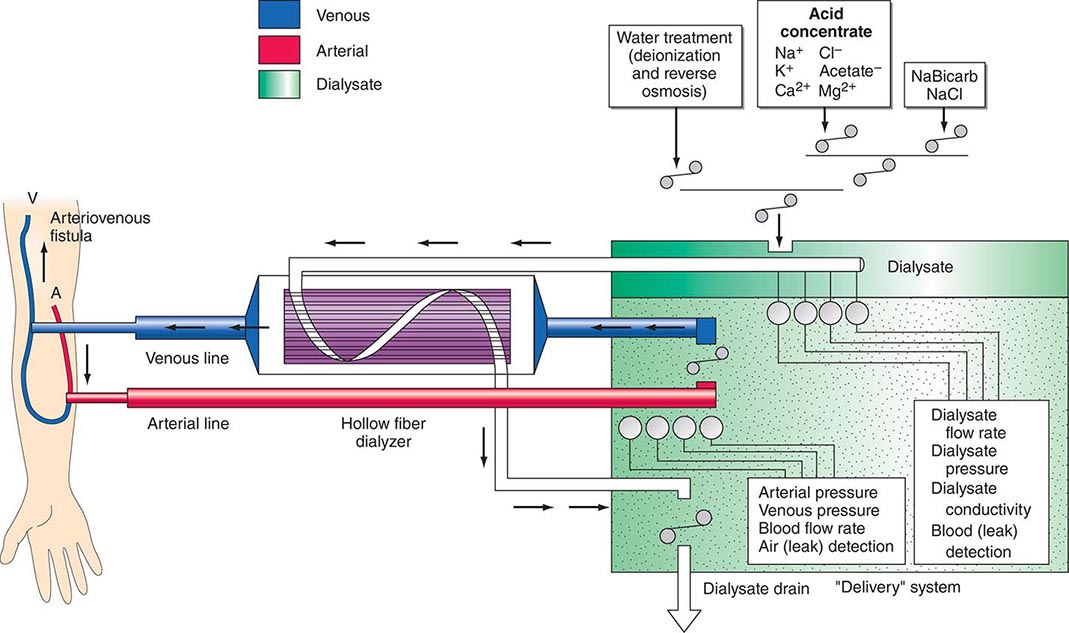
FIGURE 336-1 Schema for hemodialysis. A, artery; V, vein.
DIALYSATE
The potassium concentration of dialysate may be varied from 0 to 4 mmol/L depending on the predialysis serum potassium concentration. The usual dialysate calcium concentration is 1.25 mmol/L (2.5 meq/L), although modification may be required in selected settings (e.g., higher dialysate calcium concentrations may be used in patients with hypocalcemia associated with secondary hyperparathyroidism or following parathyroidectomy). The usual dialysate sodium concentration is 136–140 mmol/L. In patients who frequently develop hypotension during their dialysis run, “sodium modeling” to counterbalance urea-related osmolar gradients is often used. With sodium modeling, the dialysate sodium concentration is gradually lowered from the range of 145–155 mmol/L to isotonic concentrations (136–140 mmol/L) near the end of the dialysis treatment, typically declining either in steps or in a linear or exponential fashion. Higher dialysate sodium concentrations and sodium modeling may predispose patients to positive sodium balance and increased thirst; thus, these strategies to ameliorate intradialytic hypotension may be undesirable in hypertensive patients or in patients with large interdialytic weight gains. Because patients are exposed to approximately 120 L of water during each dialysis treatment, water used for the dialysate is subjected to filtration, softening, deionization, and, ultimately, reverse osmosis to remove microbiologic contaminants and dissolved ions.
BLOOD DELIVERY SYSTEM
The blood delivery system is composed of the extracorporeal circuit and the dialysis access. The dialysis machine consists of a blood pump, dialysis solution delivery system, and various safety monitors. The blood pump moves blood from the access site, through the dialyzer, and back to the patient. The blood flow rate may range from 250–500 mL/min, depending on the type and integrity of the vascular access. Negative hydrostatic pressure on the dialysate side can be manipulated to achieve desirable fluid removal or ultrafiltration. Dialysis membranes have different ultrafiltration coefficients (i.e., mL removed/min per mmHg) so that along with hydrostatic changes, fluid removal can be varied. The dialysis solution delivery system dilutes the concentrated dialysate with water and monitors the temperature, conductivity, and flow of dialysate.
DIALYSIS ACCESS
The fistula, graft, or catheter through which blood is obtained for hemodialysis is often referred to as a dialysis access. A native fistula created by the anastomosis of an artery to a vein (e.g., the Brescia-Cimino fistula, in which the cephalic vein is anastomosed end-to-side to the radial artery) results in arterialization of the vein. This facilitates its subsequent use in the placement of large needles (typically 15 gauge) to access the circulation. Although fistulas have the highest long-term patency rate of all dialysis access options, fistulas are created in a minority of patients in the United States. Many patients undergo placement of an arteriovenous graft (i.e., the interposition of prosthetic material, usually polytetrafluoroethylene, between an artery and a vein) or a tunneled dialysis catheter. In recent years, nephrologists, vascular surgeons, and health care policy makers in the United States have encouraged creation of arteriovenous fistulas in a larger fraction of patients (the “fistula first” initiative). Unfortunately, even when created, arteriovenous fistulas may not mature sufficiently to provide reliable access to the circulation, or they may thrombose early in their development.
Grafts and catheters tend to be used among persons with smaller-caliber veins or persons whose veins have been damaged by repeated venipuncture, or after prolonged hospitalization. The most important complication of arteriovenous grafts is thrombosis of the graft and graft failure, due principally to intimal hyperplasia at the anastomosis between the graft and recipient vein. When grafts (or fistulas) fail, catheter-guided angioplasty can be used to dilate stenoses; monitoring of venous pressures on dialysis and of access flow, although not routinely performed, may assist in the early recognition of impending vascular access failure. In addition to an increased rate of access failure, grafts and (in particular) catheters are associated with much higher rates of infection than fistulas.
Intravenous large-bore catheters are often used in patients with acute and chronic kidney disease. For persons on maintenance hemodialysis, tunneled catheters (either two separate catheters or a single catheter with two lumens) are often used when arteriovenous fistulas and grafts have failed or are not feasible due to anatomic considerations. These catheters are tunneled under the skin; the tunnel reduces bacterial translocation from the skin, resulting in a lower infection rate than with nontunneled temporary catheters. Most tunneled catheters are placed in the internal jugular veins; the external jugular, femoral, and subclavian veins may also be used.
Nephrologists, interventional radiologists, and vascular surgeons generally prefer to avoid placement of catheters into the subclavian veins; while flow rates are usually excellent, subclavian stenosis is a frequent complication and, if present, will likely prohibit permanent vascular access (i.e., a fistula or graft) in the ipsilateral extremity. Infection rates may be higher with femoral catheters. For patients with multiple vascular access complications and no other options for permanent vascular access, tunneled catheters may be the last “lifeline” for hemodialysis. Translumbar or transhepatic approaches into the inferior vena cava may be required if the superior vena cava or other central veins draining the upper extremities are stenosed or thrombosed.
GOALS OF DIALYSIS
The hemodialysis procedure consists of pumping heparinized blood through the dialyzer at a flow rate of 300–500 mL/min, while dialysate flows in an opposite counter-current direction at 500–800 mL/min. The efficiency of dialysis is determined by blood and dialysate flow through the dialyzer as well as dialyzer characteristics (i.e., its efficiency in removing solute). The dose of dialysis, which is currently defined as a derivation of the fractional urea clearance during a single treatment, is further governed by patient size, residual kidney function, dietary protein intake, the degree of anabolism or catabolism, and the presence of comorbid conditions.
Since the landmark studies of Sargent and Gotch relating the measurement of the dose of dialysis using urea concentrations with morbidity in the National Cooperative Dialysis Study, the delivered dose of dialysis has been measured and considered as a quality assurance and improvement tool. Although the fractional removal of urea nitrogen and derivations thereof are considered to be the standard methods by which “adequacy of dialysis” is measured, a large multicenter randomized clinical trial (the HEMO Study) failed to show a difference in mortality associated with a large difference in urea clearance. Current targets include a urea reduction ratio (the fractional reduction in blood urea nitrogen per hemodialysis session) of >65–70% and a body water–indexed clearance × time product (KT/V) above 1.2 or 1.05, depending on whether urea concentrations are “equilibrated.” For the majority of patients with ESRD, between 9 and 12 h of dialysis are required each week, usually divided into three equal sessions. Several studies have suggested that longer hemodialysis session lengths may be beneficial (independent of urea clearance), although these studies are confounded by a variety of patient characteristics, including body size and nutritional status. Hemodialysis “dose” should be individualized, and factors other than the urea nitrogen should be considered, including the adequacy of ultrafiltration or fluid removal and control of hyperkalemia, hyperphosphatemia, and metabolic acidosis. A recent randomized clinical trial (the Frequent Hemodialysis Network Trial) demonstrated improved control of hypertension and hyperphosphatemia, reduced left ventricular mass, and improved self-reported physical health with six times per week hemodialysis compared to the usual three times per week therapy. A companion trial in which frequent nocturnal hemodialysis was compared to conventional hemodialysis at home showed no significant effect on left ventricular mass or self-reported physical health. Finally, an evaluation of the U.S. Renal Data System registry showed a significant increase in mortality and hospitalization for heart failure after the longer interdialytic interval that occurs over the dialysis “weekend.”
COMPLICATIONS DURING HEMODIALYSIS
Hypotension is the most common acute complication of hemodialysis, particularly among patients with diabetes mellitus. Numerous factors appear to increase the risk of hypotension, including excessive ultrafiltration with inadequate compensatory vascular filling, impaired vasoactive or autonomic responses, osmolar shifts, overzealous use of antihypertensive agents, and reduced cardiac reserve. Patients with arteriovenous fistulas and grafts may develop high-output cardiac failure due to shunting of blood through the dialysis access; on rare occasions, this may necessitate ligation of the fistula or graft. Because of the vasodilatory and cardiodepressive effects of acetate, its use as the buffer in dialysate was once a common cause of hypotension. Since the introduction of bicarbonate-containing dialysate, dialysis-associated hypotension has become less common. The management of hypotension during dialysis consists of discontinuing ultrafiltration, the administration of 100–250 mL of isotonic saline or 10 mL of 23% saturated hypertonic saline, or administration of salt-poor albumin. Hypotension during dialysis can frequently be prevented by careful evaluation of the dry weight and by ultrafiltration modeling, such that more fluid is removed at the beginning rather than the end of the dialysis procedure. Additional maneuvers include the performance of sequential ultrafiltration followed by dialysis, cooling of the dialysate during dialysis treatment, and avoiding heavy meals during dialysis. Midodrine, an oral selective α1 adrenergic agent, has been advocated by some practitioners, although there is insufficient evidence of its safety and efficacy to support its routine use.
Muscle cramps during dialysis are also a common complication. The etiology of dialysis-associated cramps remains obscure. Changes in muscle perfusion because of excessively rapid volume removal (e.g., >10–12 mL/kg per hour) or targeted removal below the patient’s estimated dry weight often precipitate dialysis-associated cramps. Strategies that may be used to prevent cramps include reducing volume removal during dialysis, ultrafiltration profiling, and the use of sodium modeling (see above).
Anaphylactoid reactions to the dialyzer, particularly on its first use, have been reported most frequently with the bioincompatible cellulosic-containing membranes. Dialyzer reactions can be divided into two types, A and B. Type A reactions are attributed to an IgE-mediated intermediate hypersensitivity reaction to ethylene oxide used in the sterilization of new dialyzers. This reaction typically occurs soon after the initiation of a treatment (within the first few minutes) and can progress to full-blown anaphylaxis if the therapy is not promptly discontinued. Treatment with steroids or epinephrine may be needed if symptoms are severe. The type B reaction consists of a symptom complex of nonspecific chest and back pain, which appears to result from complement activation and cytokine release. These symptoms typically occur several minutes into the dialysis run and typically resolve over time with continued dialysis.
PERITONEAL DIALYSIS
In peritoneal dialysis, 1.5–3 L of a dextrose-containing solution is infused into the peritoneal cavity and allowed to dwell for a set period of time, usually 2–4 h. As with hemodialysis, toxic materials are removed through a combination of convective clearance generated through ultrafiltration and diffusive clearance down a concentration gradient. The clearance of solutes and water during a peritoneal dialysis exchange depends on the balance between the movement of solute and water into the peritoneal cavity versus absorption from the peritoneal cavity. The rate of diffusion diminishes with time and eventually stops when equilibration between plasma and dialysate is reached. Absorption of solutes and water from the peritoneal cavity occurs across the peritoneal membrane into the peritoneal capillary circulation and via peritoneal lymphatics into the lymphatic circulation. The rate of peritoneal solute transport varies from patient to patient and may be altered by the presence of infection (peritonitis), drugs, and physical factors such as position and exercise.
FORMS OF PERITONEAL DIALYSIS
Peritoneal dialysis may be carried out as CAPD, CCPD, or a combination of both. In CAPD, dialysate is manually infused into the peritoneal cavity and exchanged three to five times during the day. A nighttime dwell is frequently instilled at bedtime and remains in the peritoneal cavity through the night. In CCPD, exchanges are performed in an automated fashion, usually at night; the patient is connected to an automated cycler that performs a series of exchange cycles while the patient sleeps. The number of exchange cycles required to optimize peritoneal solute clearance varies by the peritoneal membrane characteristics; as with hemodialysis, solute clearance should be tracked to ensure dialysis “adequacy.”
Peritoneal dialysis solutions are available in volumes typically ranging from 1.5 to 3 L. The major difference between the dialysate used for peritoneal dialysis rather than hemodialysis is that the hypertonicity of peritoneal dialysis solutions drives solute and fluid removal, whereas solute removal in hemodialysis depends on concentration gradients, and fluid removal requires transmembrane pressure. Typically, dextrose at varying concentrations contributes to the hypertonicity of peritoneal dialysate. Icodextrin is a nonabsorbable carbohydrate that can be used in place of dextrose. Studies have demonstrated more efficient ultrafiltration with icodextrin than with dextrose-containing solutions. Icodextrin is typically used as the “last fill” for patients on CCPD or for the longest dwell in patients on CAPD. The most common additives to peritoneal dialysis solutions are heparin to prevent obstruction of the dialysis catheter lumen with fibrin and antibiotics during an episode of acute peritonitis. Insulin may also be added in patients with diabetes mellitus.
ACCESS TO THE PERITONEAL CAVITY
Access to the peritoneal cavity is obtained through a peritoneal catheter. Catheters used for maintenance peritoneal dialysis are flexible, being made of silicone rubber with numerous side holes at the distal end. These catheters usually have two Dacron cuffs. The scarring that occurs around the cuffs anchors the catheter and seals it from bacteria tracking from the skin surface into the peritoneal cavity; it also prevents the external leakage of fluid from the peritoneal cavity. The cuffs are placed in the preperitoneal plane and ~2 cm from the skin surface.
The peritoneal equilibrium test is a formal evaluation of peritoneal membrane characteristics that measures the transfer rates of creatinine and glucose across the peritoneal membrane. Patients are classified as low, low–average, high–average, and high transporters. Patients with rapid equilibration (i.e., high transporters) tend to absorb more glucose and lose efficiency of ultrafiltration with long daytime dwells. High transporters also tend to lose larger quantities of albumin and other proteins across the peritoneal membrane. In general, patients with rapid transporting characteristics require more frequent, shorter dwell time exchanges, nearly always obligating use of a cycler. Slower (low and low–average) transporters tend to do well with fewer exchanges. The efficiency of solute clearance also depends on the volume of dialysate infused. Larger volumes allow for greater solute clearance, particularly with CAPD in patients with low and low–average transport characteristics.
As with hemodialysis, the optimal dose of peritoneal dialysis is unknown. Several observational studies have suggested that higher rates of urea and creatinine clearance (the latter generally measured in liters per week) are associated with lower mortality rates and fewer uremic complications. However, a randomized clinical trial (Adequacy of Peritoneal Dialysis in Mexico [ADEMEX]) failed to show a significant reduction in mortality or complications with a relatively large increment in urea clearance. In general, patients on peritoneal dialysis do well when they retain residual kidney function. The rates of technique failure increase with years on dialysis and have been correlated with loss of residual function to a greater extent than loss of peritoneal membrane capacity. For some patients in whom CCPD does not provide sufficient solute clearance, a hybrid approach can be adopted where one or more daytime exchanges are added to the CCPD regimen. Although this approach can enhance solute clearance and prolong a patient’s capacity to remain on peritoneal dialysis, the burden of the hybrid approach can be overwhelming.
COMPLICATIONS DURING PERITONEAL DIALYSIS
The major complications of peritoneal dialysis are peritonitis, catheter-associated nonperitonitis infections, weight gain and other metabolic disturbances, and residual uremia (especially among patients with no residual kidney function).
Peritonitis typically develops when there has been a break in sterile technique during one or more of the exchange procedures. Peritonitis is usually defined by an elevated peritoneal fluid leukocyte count (100/μL, of which at least 50% are polymorphonuclear neutrophils); these cutoffs are lower than in spontaneous bacterial peritonitis because of the presence of dextrose in peritoneal dialysis solutions and rapid bacterial proliferation in this environment without antibiotic therapy. The clinical presentation typically consists of pain and cloudy dialysate, often with fever and other constitutional symptoms. The most common culprit organisms are gram-positive cocci, including Staphylococcus, reflecting the origin from the skin. Gram-negative rod infections are less common; fungal and mycobacterial infections can be seen in selected patients, particularly after antibacterial therapy. Most cases of peritonitis can be managed either with intraperitoneal or oral antibiotics, depending on the organism; many patients with peritonitis do not require hospitalization. In cases where peritonitis is due to hydrophilic gram-negative rods (e.g., Pseudomonas sp.) or yeast, antimicrobial therapy is usually not sufficient, and catheter removal is required to ensure complete eradication of infection. Nonperitonitis catheter-associated infections (often termed tunnel infections) vary widely in severity. Some cases can be managed with local antibiotic or silver nitrate administration, whereas others are severe enough to require parenteral antibiotic therapy and catheter removal.
Peritoneal dialysis is associated with a variety of metabolic complications. Albumin and other proteins can be lost across the peritoneal membrane in concert with the loss of metabolic wastes. Hypoproteinemia obligates a higher dietary protein intake in order to maintain nitrogen balance. Hyperglycemia and weight gain are also common complications of peritoneal dialysis. Several hundred calories in the form of dextrose are absorbed each day, depending on the concentration employed. Peritoneal dialysis patients, particularly those with diabetes mellitus, are then prone to other complications of insulin resistance, including hypertriglyceridemia. On the positive side, the continuous nature of peritoneal dialysis usually allows for a more liberal diet, due to continuous removal of potassium and phosphorus—two major dietary components whose accumulation can be hazardous in ESRD.
LONG-TERM OUTCOMES IN ESRD
Cardiovascular disease constitutes the major cause of death in patients with ESRD. Cardiovascular mortality and event rates are higher in dialysis patients than in patients after transplantation, although rates are extraordinarily high in both populations. The underlying cause of cardiovascular disease is unclear but may be related to shared risk factors (e.g., diabetes mellitus, hypertension, atherosclerotic and arteriosclerotic vascular disease), chronic inflammation, massive changes in extracellular volume (especially with high interdialytic weight gains), inadequate treatment of hypertension, dyslipidemia, anemia, dystrophic vascular calcification, hyperhomocysteinemia, and, perhaps, alterations in cardiovascular dynamics during the dialysis treatment. Few studies have targeted cardiovascular risk reduction in ESRD patients; none have demonstrated consistent benefit. Two clinical trials of statin agents in ESRD demonstrated significant reductions in low-density lipoprotein (LDL) cholesterol concentrations, but no significant reductions in death or cardiovascular events (Die Deutsche Diabetes Dialyse Studie [4D] and A Study to Evaluate the Use of Rosuvastatin in Subjects on Regular Hemodialysis [AURORA]). The Study of Heart and Renal Protection (SHARP), which included patients on dialysis- and non-dialysis-requiring CKD, showed a 17% reduction in the rate of major cardiovascular events or cardiovascular death with simvastatin-ezetimibe treatment. Most experts recommend conventional cardioprotective strategies (e.g., lipid-lowering agents, aspirin, inhibitors of the renin-angiotensin-aldosterone system, and β-adrenergic antagonists) in dialysis patients based on the patients’ cardiovascular risk profile, which appears to be increased by more than an order of magnitude relative to persons unaffected by kidney disease. Other complications of ESRD include a high incidence of infection, progressive debility and frailty, protein-energy malnutrition, and impaired cognitive function.
GLOBAL PERSPECTIVE
![]() The incidence of ESRD is increasing worldwide with longer life expectancies and improved care of infectious and cardiovascular diseases. The management of ESRD varies widely by country and within country by region, and it is influenced by economic and other major factors. In general, peritoneal dialysis is more commonly performed in poorer countries owing to its lower expense and the high cost of establishing in-center hemodialysis units.
The incidence of ESRD is increasing worldwide with longer life expectancies and improved care of infectious and cardiovascular diseases. The management of ESRD varies widely by country and within country by region, and it is influenced by economic and other major factors. In general, peritoneal dialysis is more commonly performed in poorer countries owing to its lower expense and the high cost of establishing in-center hemodialysis units.
337 |
Transplantation in the Treatment of Renal Failure |
Transplantation of the human kidney is the treatment of choice for advanced chronic renal failure. Worldwide, tens of thousands of these procedures have been performed with more than 180,000 patients bearing functioning kidney transplants in the United States today. When azathioprine and prednisone initially were used as immunosuppressive drugs in the 1960s, the results with properly matched familial donors were superior to those with organs from deceased donors: 75–90% compared with 50–60% graft survival rates at 1 year. During the 1970s and 1980s, the success rate at the 1-year mark for deceased-donor transplants rose progressively. Currently, deceased-donor grafts have a 92% 1-year survival and living-donor grafts have a 96% 1-year survival. Although there has been improvement in long-term survival, it has not been as impressive as the short-term survival, and currently the “average” (t1/2) life expectancy of a living-donor graft is around 20 years and that of a deceased-donor graft is close to 14 years.
Mortality rates after transplantation are highest in the first year and are age-related: 2% for ages 18–34 years, 3% for ages 35–49 years, and 6.8% for ages ≥50–60 years. These rates compare favorably with those in the chronic dialysis population even after risk adjustments for age, diabetes, and cardiovascular status. Although the loss of kidney transplant due to acute rejection is currently rare, most allografts succumb at varying rates to a chronic process consisting of interstitial fibrosis, tubular atrophy, vasculopathy, and glomerulopathy, the pathogenesis of which is incompletely understood. Overall, transplantation returns most patients to an improved lifestyle and an improved life expectancy compared with patients on dialysis.
RECENT ACTIVITY AND RESULTS
In 2011, there were more than 11,835 deceased-donor kidney transplants and 5772 living-donor transplants in the United States, with the ratio of deceased to living donors remaining stable over the last few years. The backlog of patients with end-stage renal disease (ESRD) has been increasing every year, and it always lags behind the number of available donors. As the number of patients with end-stage kidney disease increases, the demand for kidney transplants continues to increase. In 2011, there were 55,371 active adult candidates on the waiting list, and less than 18,000 patients were transplanted. This imbalance is set to worsen over the coming years with the predicted increased rates of obesity and diabetes worldwide. In an attempt to increase utilization of deceased-donor kidneys and reduce discard rates of organs, criteria for the use of so-called expanded criteria donor (ECD) kidneys and kidneys from donors after cardiac death (DCD) have been developed (Table 337-1). ECD kidneys are usually used for older patients who are expected to fare less well on dialysis.
|
DEFINITION OF AN EXPANDED CRITERIA DONOR AND A NON-HEART-BEATING DONOR (DONATION AFTER CARDIAC DEATH) |
aKidneys can be used for transplantation from categories II–V but are commonly only used from categories III and IV. The survival of these kidneys has not been shown to be inferior to that of deceased-donor kidneys.
Note: Kidneys can be both ECD and DCD. ECD kidneys have been shown to have a poorer survival, and there is a separate shorter waiting list for ECD kidneys. They are generally used for patients for whom the benefits of being transplanted earlier outweigh the associated risks of using an ECD kidney.
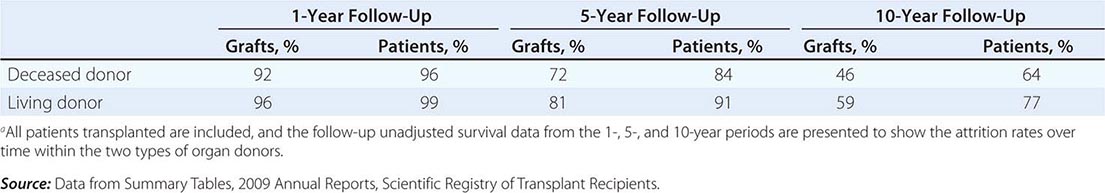
The overall results of transplantation are presented in Table 337-2 as the survival of grafts and of patients. At the 1-year mark, graft survival is higher for living-donor recipients, most likely because those grafts are not subject to as much ischemic injury. The more effective drugs now in use for immunosuppression have almost equalized the risk of graft rejection in all patients for the first year. At 5 and 10 years, however, there has been a steeper decline in survival of those with deceased-donor kidneys.
|
MEAN RATES OF GRAFT AND PATIENT SURVIVAL FOR KIDNEYS TRANSPLANTED IN THE UNITED STATES FROM 1998 TO 2008a |
RECIPIENT SELECTION
There are few absolute contraindications to renal transplantation. The transplant procedure is relatively noninvasive, as the organ is placed in the inguinal fossa without entering the peritoneal cavity. Recipients without perioperative complications often can be discharged from the hospital in excellent condition within 5 days of the operation.
Virtually all patients with ESRD who receive a transplant have a higher life expectancy than do risk-matched patients who remain on dialysis. Even though diabetic patients and older candidates have a higher mortality rate than other transplant recipients, their survival is improved with transplantation compared with those remaining on dialysis. This global benefit of transplantation as a treatment modality poses substantial ethical issues for policy makers, as the number of deceased kidneys available is far from sufficient to meet the current needs of the candidates. The current standard of care is that the candidate should have a life expectancy of >5 years to be put on a deceased organ wait list. Even for living donation, the candidate should have >5 years of life expectancy. This standard has been established because the benefits of kidney transplantation over dialysis are realized only after a perioperative period in which the mortality rate is higher in transplanted patients than in dialysis patients with comparable risk profiles.
All candidates must have a thorough risk-versus-benefit evaluation before being approved for transplantation. In particular, an aggressive approach to diagnosis of correctable coronary artery disease, presence of latent or indolent infection (HIV, hepatitis B or C, tuberculosis), and neoplasm should be a routine part of the candidate workup. Most transplant centers consider overt AIDS and active hepatitis absolute contraindications to transplantation because of the high risk of opportunistic infection. Some centers are now transplanting individuals with hepatitis and even HIV infection under strict protocols to determine whether the risks and benefits favor transplantation over dialysis.
Among the few absolute “immunologic” contraindications to transplantation is the presence of antibodies against the donor kidney at the time of the anticipated transplant that can cause hyperacute rejection. Those harmful antibodies include natural antibodies against the ABO blood group antigens and antibodies against human leukocyte antigen (HLA) class I (A, B, C) or class II (DR) antigens. These antibodies are routinely excluded by proper screening of the candidate’s ABO compatibility and direct cytotoxic cross-matching of candidate serum with lymphocytes of the donor.
TISSUE TYPING AND CLINICAL IMMUNOGENETICS
Matching for antigens of the HLA major histocompatibility gene complex (Chap. 373e) is an important criterion for selection of donors for renal allografts. Each mammalian species has a single chromosomal region that encodes the strong, or major, transplantation antigens, and this region on the human sixth chromosome is called HLA. HLA antigens have been classically defined by serologic techniques, but methods to define specific nucleotide sequences in genomic DNA are increasingly being used. Other “minor” antigens may play crucial roles, in addition to the ABH(O) blood groups and endothelial antigens that are not shared with lymphocytes. The Rh system is not expressed on graft tissue. Evidence for designation of HLA as the genetic region that encodes major transplantation antigens comes from the success rate in living related donor renal and bone marrow transplantation, with superior results in HLA-identical sibling pairs. Nevertheless, 5% of HLA-identical renal allografts are rejected, often within the first weeks after transplantation. These failures represent states of prior sensitization to non-HLA antigens. Non-HLA minor antigens are relatively weak when initially encountered and are, therefore, suppressible by conventional immunosuppressive therapy. Once priming has occurred, however, secondary responses are much more refractory to treatment.
DONOR SELECTION
Donors can be deceased or volunteer living donors. When first-degree relatives are donors, graft survival rates at 1 year are 5–7% greater than those for deceased-donor grafts. The 5-year survival rates still favor a partially matched (3/6 HLA mismatched) family donor over a randomly selected cadaver donor. In addition, living donors provide the advantage of immediate availability. For both living and deceased donors, the 5-year outcomes are poor if there is a complete (6/6) HLA mismatch.
The survival rate of living unrelated renal allografts is as high as that of perfectly HLA-matched cadaver renal transplants and comparable to that of kidneys from living relatives. This outcome is probably a consequence of both short cold ischemia time and the extra care taken to document that the condition and renal function of the donor are optimal before proceeding with a living unrelated donation. It is illegal in the United States to purchase organs for transplantation.
Living volunteer donors should be cleared of any medical conditions that may cause morbidity and mortality after kidney transplantation. Concern has been expressed about the potential risk to a volunteer kidney donor of premature renal failure after several years of increased blood flow and hyperfiltration per nephron in the remaining kidney. There are a few reports of the development of hypertension, proteinuria, and even lesions of focal segmental sclerosis in donors over long-term follow-up. It is also desirable to consider the risk of development of type 1 diabetes mellitus in a family member who is a potential donor to a diabetic renal failure patient. Anti-insulin and anti-islet cell antibodies should be measured and glucose tolerance tests should be performed in such donors to exclude a prediabetic state. Selective renal arteriography should be performed on donors to rule out the presence of multiple or abnormal renal arteries, because the surgical procedure is difficult and the ischemic time of the transplanted kidney is long when there are vascular abnormalities. Transplant surgeons are now using a laparoscopic method to isolate and remove the living donor’s kidney. This operation has the advantage of less evident surgical scars, and, because there is less tissue trauma, the laparoscopic donors have a substantially shorter hospital stay and less discomfort than those who have the traditional surgery.
Deceased donors should be free of malignant neoplastic disease, hepatitis, and HIV due to possible transmission to the recipient, although there is increasing interest in using hepatitis C– and HIV-positive organs in previously infected recipients. Increased risk of graft failure exists when the donor is elderly or has renal failure and when the kidney has a prolonged period of ischemia and storage.
In the United States, there is a coordinated national system of regulations, allocation support, and outcomes analysis for kidney transplantation called the Organ Procurement Transplant Network. It is now possible to remove deceased-donor kidneys and maintain them for up to 48 h on cold pulsatile perfusion or with simple flushing and cooling. This approach permits adequate time for typing, cross-matching, transportation, and selection problems to be solved.
PRESENSITIZATION
A positive cytotoxic cross-match of recipient serum with donor T lymphocytes indicates the presence of preformed donor-specific anti-HLA class I antibodies and is usually predictive of an acute vasculitic event termed hyperacute rejection. This finding, along with ABO incompatibility, represents the only absolute immunologic contraindication for kidney transplantation. Recently, more tissue typing laboratories have shifted to a flow cytometric–based cross-match assay, which detects the presence of anti-HLA antibodies that are not necessarily detected on a cytotoxic cross-match assay and may not be an absolute contraindication to transplantation. The known sources of such sensitization are blood transfusion, a prior transplant, pregnancy, and vaccination/infection. Patients sustained by dialysis often show fluctuating antibody titers and specificity patterns. At the time of assignment of a cadaveric kidney, cross-matches are performed with at least a current serum. Previously analyzed antibody specificities and additional cross-matches are performed accordingly. Flow cytometry detects binding of anti-HLA antibodies of a candidate’s serum by a recipient’s lymphocytes. This highly sensitive test can be useful for avoidance of accelerated, and often untreatable, early graft rejection in patients receiving second or third transplants.
For the purposes of cross-matching, donor T lymphocytes, which express class I but not class II antigens, are used as targets for detection of anti–class I (HLA-A and -B) antibodies that are expressed on all nucleated cells. Preformed anti–class II (HLA-DR and -DQ) antibodies against the donor also carry a higher risk of graft loss, particularly in recipients who have suffered early loss of a prior kidney transplant. B lymphocytes, which express both class I and class II antigens, are used as targets in these assays.
Some non-HLA antigens restricted in expression to endothelium and monocytes have been described, but clinical relevance is not well established. A series of minor histocompatibility antigens do not elicit antibodies, and sensitization to these antigens is detectable only by cytotoxic T cells, an assay too cumbersome for routine use.
Desensitization before transplantation by reducing the level of antidonor antibodies using plasmapheresis and administration of pooled immunoglobulin, or both, has been useful in reducing the risk of hyperacute rejection following transplantation.
IMMUNOLOGY OF REJECTION
Both cellular and humoral (antibody-mediated) effector mechanisms can play roles in kidney transplant rejection.
Cellular rejection is mediated by lymphocytes that respond to HLA antigens expressed within the organ. The CD4+ lymphocyte responds to class II (HLA-DR) incompatibility by proliferating and releasing proinflammatory cytokines that augment the proliferative response of the immune system. CD8+ cytotoxic lymphocyte precursors respond primarily to class I (HLA-A, -B) antigens and mature into cytotoxic effector cells that cause organ damage through direct contact and lysis of donor target cells. Full T cell activation requires not only T cell receptor binding to the alloantigens presented by self or donor HLA molecules (indirect and direct presentation, respectively), but also engaging costimulatory molecules such as CD28 on T cells and CD80 and CD86 ligands on antigen-presenting cells (Fig. 337-1). Signaling through both of these pathways induces activation of the kinase activity of calcineurin, which, in turn, activates transcription factors, leading to upregulation of multiple genes, including interleukin 2 (IL-2) and interferon gamma. IL-2 signals through the target of rapamycin (TOR) to induce cell proliferation in an autocrine fashion. There is evidence that non-HLA antigens can also play a role in renal transplant rejection episodes. Recipients who receive a kidney from an HLA-identical sibling can have rejection episodes and require maintenance immunosuppression, whereas identical twin transplants require no immunosuppression. There are documented non-HLA antigens, such as an endothelial-specific antigen system with limited polymorphism and a tubular antigen, which can act as targets of humoral or cellular rejection responses, respectively.
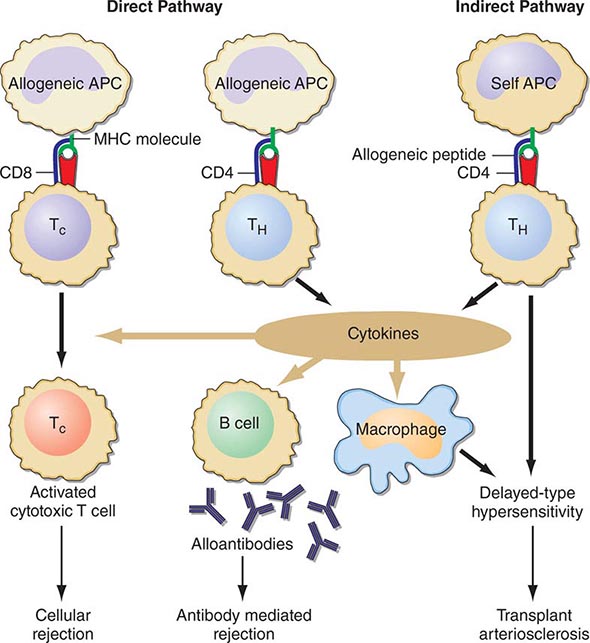
FIGURE 337-1 Recognition pathways for major histocompatibility complex (MHC) antigens. Graft rejection is initiated by CD4 helper T lymphocytes (TH) having antigen receptors that bind to specific complexes of peptides and MHC class II molecules on antigen-presenting cells (APC). In transplantation, in contrast to other immunologic responses, there are two sets of T cell clones involved in rejection. In the direct pathway, the class II MHC of donor allogeneic APCs is recognized by CD4 TH cells that bind to the intact MHC molecule, and class I MHC allogeneic cells are recognized by CD8 T cells. The latter generally proliferate into cytotoxic cells (TC). In the indirect pathway, the incompatible MHC molecules are processed into peptides that are presented by the self-APCs of the recipient. The indirect, but not the direct, pathway is the normal physiologic process in T cell recognition of foreign antigens. Once TH cells are activated, they proliferate and, by secretion of cytokines and direct contact, exert strong helper effects on macrophages, TC, and B cells. (From MH Sayegh, LH Turka: N Engl J Med, 338:1813, 1998. Copyright 1998, Massachusetts Medical Society. All rights reserved.)
IMMUNOSUPPRESSIVE TREATMENT
Immunosuppressive therapy, as currently available, generally suppresses all immune responses, including those to bacteria, fungi, and even malignant tumors. In general, all clinically useful drugs are more selective to primary than to memory immune responses. Agents to suppress the immune response are classically divided into induction and maintenance agents and will be discussed in the following paragraphs. Those currently in clinical use are listed in Table 337-3.
|
MAINTENANCE IMMUNOSUPPRESSIVE DRUGS |

INDUCTION THERAPY
Induction therapy is currently given to most kidney transplant recipients in the United States at the time of transplant to reduce the risk of early acute rejection and to minimize or eliminate the use of either steroids or calcineurin inhibitors and their associated toxicities. Induction therapy consists of antibodies that could be monoclonal or polyclonal and depletional or nondepletional.
Depleting Agents Peripheral human lymphocytes, thymocytes, or lymphocytes from spleens or thoracic duct fistulas are injected into horses, rabbits, or goats to produce antilymphocyte serum, from which the globulin fraction is then separated, resulting in antithymocyte globulin. Those polyclonal antibodies induce lymphocyte depletion, and the immune system may take several months to recover.
Monoclonal antibodies against defined lymphocyte subsets offer a more precise and standardized form of therapy. Alemtuzumab is directed to the CD52 protein, widely distributed on immune cells such as B and T cells, natural killer cells, macrophages, and some granulocytes.
Nondepleting Agents Another approach to more selective therapy is to target the 55-kDa alpha chain of the IL-2 receptor, which is expressed only on T cells that have been recently activated. This approach is used as prophylaxis for acute rejection in the immediate posttransplant period and is effective at decreasing the early acute rejection rate with few adverse side effects.
The next step in the evolution of this therapeutic strategy, which has already been achieved in the short term in small numbers of immunologically well-matched patients, is the elimination of all maintenance immunosuppression therapy.
MAINTENANCE THERAPY
All kidney transplant recipients should receive maintenance immunosuppressive therapies except identical twins. The most frequently used combination is triple therapy with prednisone, a calcineurin inhibitor, and an antimetabolite; mammalian TOR (mTOR) inhibitors can replace one of the last two agents. More recently, the U.S. Food and Drug Administration (FDA) approved a new costimulatory blocking antibody, belatacept, as a new strategy to prevent long-term calcineurin inhibitor toxicity.
Antimetabolites Azathioprine, an analogue of mercaptopurine, was for two decades the keystone to immunosuppressive therapy in humans, but has given way to more effective agents. This agent can inhibit synthesis of DNA, RNA, or both. Azathioprine is administered in doses of 1.5–2 mg/kg per day. Reduction in the dose is required because of leukopenia and occasionally thrombocytopenia. Excessive amounts of azathioprine may also cause jaundice, anemia, and alopecia. If it is essential to administer allopurinol concurrently, the azathioprine dose must be reduced. Because inhibition of xanthine oxidase delays degradation, this combination is best avoided.
Mycophenolate mofetil or mycophenolate sodium, both of which are metabolized to mycophenolic acid, is now used in place of azathioprine in most centers. It has a similar mode of action and a mild degree of gastrointestinal toxicity but produces less bone marrow suppression. Its advantage is its increased potency in preventing or reversing rejection.
Steroids Glucocorticoids are important adjuncts to immunosuppressive therapy. Among all the agents employed, prednisone has effects that are easiest to assess, and in large doses it is usually effective for the reversal of rejection. In general, 200–300 mg prednisone is given immediately before or at the time of transplantation, and the dose is reduced to 30 mg within a week. The side effects of the glucocorticoids, particularly impairment of wound healing and predisposition to infection, make it desirable to taper the dose as rapidly as possible in the immediate postoperative period. Many centers now have protocols for early discontinuance or avoidance of steroids because of long-term adverse effects on bone, skin, and glucose metabolism. For treatment of acute rejection, methylprednisolone, 0.5–1 g IV, is administered immediately upon diagnosis of beginning rejection and continued once daily for 3 days. Such “pulse” doses are not effective in chronic rejection. Most patients whose renal function is stable after 6 months or a year do not require large doses of prednisone; maintenance doses of 5–10 mg/d are the rule. A major effect of steroids is preventing the release of IL-6 and IL-1 by monocytes-macrophages.
Calcineurin Inhibitors Cyclosporine is a fungal peptide with potent immunosuppressive activity. It acts on the calcineurin pathway to block transcription of mRNA for IL-2 and other proinflammatory cytokines, thereby inhibiting T cell proliferation. Although it works alone, cyclosporine is more effective in conjunction with glucocorticoids and mycophenolate. Clinical results with tens of thousands of renal transplants have been impressive. Among its toxic effects (nephrotoxicity, hepatotoxicity, hirsutism, tremor, gingival hyperplasia, diabetes), only nephrotoxicity presents a serious management problem and is further discussed below.
Tacrolimus (previously called FK506) is a fungal macrolide that has the same mode of action as cyclosporine as well as a similar side effect profile; it does not, however, produce hirsutism or gingival hyperplasia. De novo diabetes mellitus is more common with tacrolimus. The drug was first used in liver transplantation and may substitute for cyclosporine entirely or as an alternative in renal patients whose rejections are poorly controlled by cyclosporine.
mTOR Inhibitors Sirolimus (previously called rapamycin) is another fungal macrolide but has a different mode of action; i.e., it inhibits T cell growth factor signaling pathways, preventing the response to IL-2 and other cytokines. Sirolimus can be used in conjunction with cyclosporine or tacrolimus, or with mycophenolic acid, to avoid the use of calcineurin inhibitors.
Everolimus is another mTOR inhibitor with similar mechanism of action as sirolimus but with better bioavailability.
Belatacept Belatacept is a fusion protein that binds costimulatory ligands (CD80 and CD86) present on antigen-presenting cells, interrupting their binding to CD28 on T cells. This inhibition leads to T cell anergy and apoptosis. Belatacept is FDA approved for kidney transplant recipients and is given monthly as an intravenous infusion.
CLINICAL COURSE AND MANAGEMENT OF THE RECIPIENT
Adequate hemodialysis should be performed within 48 h of surgery, and care should be taken that the serum potassium level is not markedly elevated so that intraoperative cardiac arrhythmias can be averted. The diuresis that commonly occurs postoperatively must be carefully monitored. In some instances, it may be massive, reflecting the inability of ischemic tubules to regulate sodium and water excretion; with large diureses, massive potassium losses may occur. Most chronically uremic patients have some excess of extracellular fluid, and it is useful to maintain an expanded fluid volume in the immediate postoperative period. Acute tubular necrosis (ATN) due to ischemia may cause immediate oliguria or may follow an initial short period of graft function. Recovery usually occurs within 3 weeks, although periods as long as 6 weeks have been reported. Superimposition of rejection on ATN is common, and the differential diagnosis may be difficult without a graft biopsy. Cyclosporine therapy prolongs ATN, and some patients do not diurese until the dose is reduced drastically. Many centers avoid starting cyclosporine for the first several days, using antilymphocyte globulin (ALG) or a monoclonal antibody along with mycophenolic acid and prednisone until renal function is established. Figure 337-2 illustrates an algorithm followed by many transplant centers for early posttransplant management of recipients at high or low risk of early renal dysfunction.
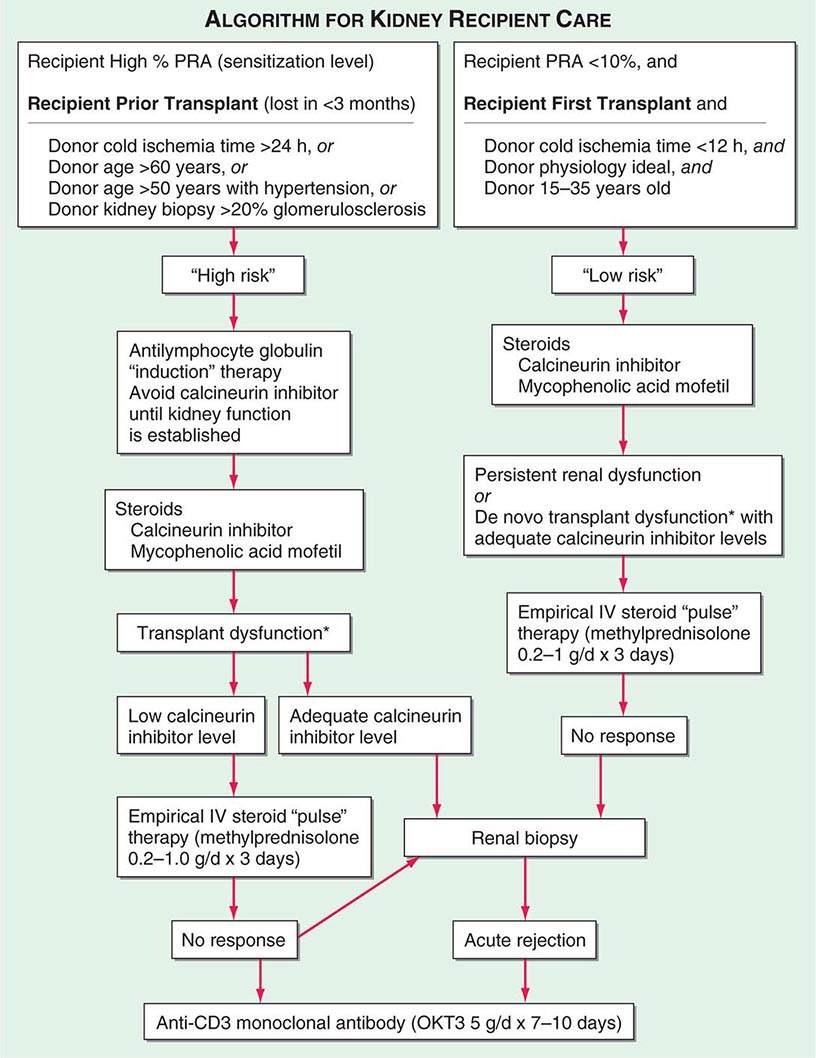
FIGURE 337-2 A typical algorithm for early posttransplant care of a kidney recipient. If any of the recipient or donor “high-risk” factors exist, more aggressive management is called for. Low-risk patients can be treated with a standard immunosuppressive regimen. Patients at higher risk of rejection or early ischemic and nephrotoxic transplant dysfunction are often induced with an antilymphocyte globulin to provide more potent early immunosuppression or to spare calcineurin nephrotoxicity. *When there is early transplant dysfunction, prerenal, obstructive, and vascular causes must be ruled out by ultrasonographic examination. The panel reactive antibody (PRA) is a quantitation of how much antibody is present in a candidate against a panel of cells representing the distribution of antigens in the donor pool.
THE REJECTION EPISODE
Early diagnosis of rejection allows prompt institution of therapy to preserve renal function and prevent irreversible damage. Clinical evidence of rejection is rarely characterized by fever, swelling, and tenderness over the allograft. Rejection may present only with a rise in serum creatinine, with or without a reduction in urine volume. The focus should be on ruling out other causes of functional deterioration.
Doppler ultrasonography may be useful in ascertaining changes in the renal vasculature and in renal blood flow. Thrombosis of the renal vein occurs rarely; it may be reversible if it is caused by technical factors and intervention is prompt. Diagnostic ultrasound is the procedure of choice to rule out urinary obstruction or to confirm the presence of perirenal collections of urine, blood, or lymph. A rise in the serum creatinine level is a late marker of rejection, but it may be the only sign. Novel biomarkers are needed for early noninvasive detection of allograft rejection.
Calcineurin inhibitors (cyclosporine and tacrolimus) have an afferent arteriolar constrictor effect on the kidney and may produce permanent vascular and interstitial injury after sustained high-dose therapy. This action will lead to a deterioration in renal function difficult to distinguish from rejection without a renal biopsy. Interstitial fibrosis, isometric tubular vacuolization, and thickening of arteriolar walls are suggestive of this side effect, but not diagnostic. Hence, if no rejection is detected on the biopsy, serum creatinine may respond to a reduction in dose. However, if rejection activity is present in the biopsy, appropriate therapy is indicated. The first rejection episode is usually treated with IV administration of methylprednisolone, 500–1000 mg daily for 3 days. Failure to respond is an indication for antibody therapy, usually with antithymocyte globulin.
Evidence of antibody-mediated injury is present when endothelial injury and deposition of complement component c4d is detected by fluorescence labeling. This is usually accompanied by detection of the antibody in the recipient blood. The prognosis is poor, and aggressive use of plasmapheresis, immunoglobulin infusions, anti-CD20 monoclonal antibody (rituximab) to target B lymphocytes, bortezomib to target antibody-producing plasma cells, and eculizumab to inhibit complement is indicated.
MANAGEMENT PROBLEMS
The typical times after transplantation when the most common opportunistic infections occur are shown in Table 337-4. Prophylaxis for cytomegalovirus (CMV) and Pneumocystis jiroveci pneumonia is given for 6–12 months after transplantation.
|
THE MOST COMMON OPPORTUNISTIC INFECTIONS IN RENAL TRANSPLANT RECIPIENTS |
The signs and symptoms of infection may be masked or distorted. Fever without obvious cause is common, and only after days or weeks may it become apparent that it has a viral or fungal origin. Bacterial infections are most common during the first month after transplantation. The importance of blood cultures in such patients cannot be overemphasized because systemic infection without obvious foci is common. Particularly ominous are rapidly occurring pulmonary lesions, which may result in death within 5 days of onset. When these lesions become apparent, immunosuppressive agents should be discontinued, except for maintenance doses of prednisone.
Aggressive diagnostic procedures, including transbronchial and open-lung biopsy, are frequently indicated. In the case of P. jiroveci (Chap. 244) infection, trimethoprim-sulfamethoxazole (TMP-SMX) is the treatment of choice; amphotericin B has been used effectively in systemic fungal infections. Prophylaxis against P. jiroveci with daily or alternate-day low-dose TMP-SMX is very effective. Involvement of the oropharynx with Candida (Chap. 240) may be treated with local nystatin. Tissue-invasive fungal infections require treatment with systemic agents such as fluconazole. Small doses (a total of 300 mg) of amphotericin given over a period of 2 weeks may be effective in fungal infections refractory to fluconazole. Macrolide antibiotics, especially ketoconazole and erythromycin, and some calcium channel blockers (diltiazem, verapamil) compete with calcineurin inhibitors for P450 catabolism and cause elevated levels of these immunosuppressive drugs. Analeptics, such as phenytoin and carbamazepine, will increase catabolism to result in low levels. Aspergillus (Chap. 241), Nocardia (Chap. 199), and especially CMV (Chap. 219) infections also occur.
CMV is a common and dangerous DNA virus in transplant recipients. It does not generally appear until the end of the first posttransplant month. Active CMV infection is sometimes associated, or occasionally confused, with rejection episodes. Patients at highest risk for severe CMV disease are those without anti-CMV antibodies who receive a graft from a CMV antibody–positive donor (15% mortality). Valganciclovir is a cost-effective and bioavailable oral form of ganciclovir that has been proved effective in both prophylaxis and treatment of CMV disease. Early diagnosis in a febrile patient with clinical suspicion of CMV disease can be made by determining CMV viral load in the blood. A rise in IgM antibodies to CMV is also diagnostic. Culture of CMV from blood may be less sensitive. Tissue invasion of CMV is common in the gastrointestinal tract and lungs. CMV retinopathy occurs late in the course, if untreated. Treatment of active CMV disease with valganciclovir is always indicated. In many patients immune to CMV, viral activation can occur with major immunosuppressive regimens.
The polyoma group (BK, JC, SV40) is another class of DNA viruses that can become dormant in kidneys and can be activated by immunosuppression. When reactivation occurs with BK, there is a 50% chance of progressive fibrosis and loss of the graft within 1 year by the activated virus. Risk of infection is associated with the overall degree of immunosuppression rather than the individual immunosuppressive drugs used. Renal biopsy is necessary for the diagnosis. There have been variable results with leflunomide, cidofovir, and quinolone antibiotics (which are effective against polyoma helicase), but it is most important to reduce the immunosuppressive load.
The complications of glucocorticoid therapy are well known and include gastrointestinal bleeding, impairment of wound healing, osteoporosis, diabetes mellitus, cataract formation, and hemorrhagic pancreatitis. The treatment of unexplained jaundice in transplant patients should include cessation or reduction of immunosuppressive drugs if hepatitis or drug toxicity is suspected. Therapy in such circumstances often does not result in rejection of a graft, at least for several weeks. Acyclovir is effective in therapy for herpes simplex virus infections.
CHRONIC LESIONS OF THE TRANSPLANTED KIDNEY
Although 1-year transplant survival is excellent, most recipients experience progressive decline in kidney function over time thereafter. Chronic renal transplant dysfunction can be caused by recurrent disease, hypertension, cyclosporine or tacrolimus nephrotoxicity, chronic immunologic rejection, secondary focal glomerulosclerosis, or a combination of these pathophysiologies. Chronic vascular changes with intimal proliferation and medial hypertrophy are commonly found. Control of systemic and intrarenal hypertension with angiotensin-converting enzyme (ACE) inhibitors is thought to have a beneficial influence on the rate of progression of chronic renal transplant dysfunction. Renal biopsy can distinguish subacute cellular rejection from recurrent disease or secondary focal sclerosis.
MALIGNANCY
The incidence of tumors in patients on immunosuppressive therapy is 5–6%, or approximately 100 times greater than that in the general population in the same age range. The most common lesions are cancer of the skin and lips and carcinoma in situ of the cervix, as well as lymphomas such as non-Hodgkin’s lymphoma. The risks are increased in proportion to the total immunosuppressive load administered and the time elapsed since transplantation. Surveillance for skin and cervical cancers is necessary.
OTHER COMPLICATIONS
Both chronic dialysis and renal transplant patients have a higher incidence of death from myocardial infarction and stroke than does the population at large, and this is particularly true in diabetic patients. Contributing factors are the use of glucocorticoids and sirolimus and hypertension. Recipients of renal transplants have a high prevalence of coronary artery and peripheral vascular diseases. The percentage of deaths from these causes has been slowly rising as the numbers of transplanted diabetic patients and the average age of all recipients increase. More than 50% of renal recipient mortality is attributable to cardiovascular disease. In addition to strict control of blood pressure and blood lipid levels, close monitoring of patients for indications of further medical or surgical intervention is an important part of management.
Hypertension may be caused by (1) native kidney disease, (2) rejection activity in the transplant, (3) renal artery stenosis if an end-to-end anastomosis was constructed with an iliac artery branch, and (4) renal calcineurin inhibitor toxicity, which may improve with reduction in dose. Whereas ACE inhibitors may be useful, calcium channel blockers are more frequently used initially. Amelioration of hypertension to the range of 120–130/70–80 mmHg should be the goal in all patients.
Hypercalcemia after transplantation may indicate failure of hyperplastic parathyroid glands to regress. Aseptic necrosis of the head of the femur is probably due to preexisting hyperparathyroidism, with aggravation by glucocorticoid treatment. With improved management of calcium and phosphorus metabolism during chronic dialysis, the incidence of parathyroid-related complications has fallen dramatically. Persistent hyperparathyroid activity may require subtotal parathyroidectomy.
Although most transplant patients have robust production of erythropoietin and normalization of hemoglobin, anemia is commonly seen in the posttransplant period. Often the anemia is attributable to bone marrow–suppressant immunosuppressive medications such as azathioprine, mycophenolic acid, and sirolimus. Gastrointestinal bleeding is a common side effect of high-dose and long-term steroid administration. Many transplant patients have creatinine clearances of 30–50 mL/min and can be considered in the same way as other patients with chronic renal insufficiency for anemia management, including supplemental erythropoietin.
Chronic hepatitis, particularly when due to hepatitis B virus, can be a progressive, fatal disease over a decade or so. Patients who are persistently hepatitis B surface antigen–positive are at higher risk, according to some studies, but the presence of hepatitis C virus is also a concern when one embarks on a course of immunosuppression in a transplant recipient.
338 |
Glomerular Diseases |
Two human kidneys harbor nearly 1.8 million glomerular capillary tufts. Each glomerular tuft resides within Bowman’s space. The capsule circumscribing this space is lined by parietal epithelial cells that transition into tubular epithelia forming the proximal nephron or migrate into the tuft to replenish podocytes. The glomerular capillary tuft derives from an afferent arteriole that forms a branching capillary bed embedded in mesangial matrix (Fig. 338-1). This capillary network funnels into an efferent arteriole, which passes filtered blood into cortical peritubular capillaries or medullary vasa recta that supply and exchange with a folded tubular architecture. Hence the glomerular capillary tuft, fed and drained by arterioles, represents an arteriolar portal system. Fenestrated endothelial cells resting on a glomerular basement membrane (GBM) line glomerular capillaries. Delicate foot processes extending from epithelial podocytes shroud the outer surface of these capillaries, and podocytes interconnect to each other by slit-pore membranes forming a selective filtration barrier.
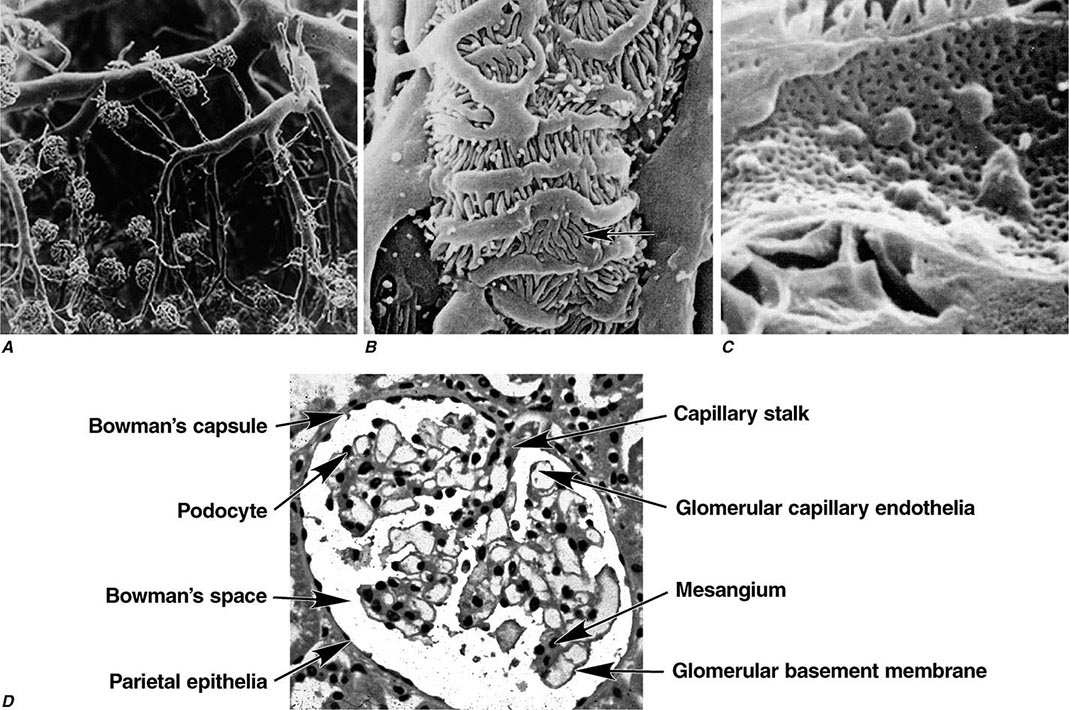
FIGURE 338-1 Glomerular architecture. A. The glomerular capillaries form from a branching network of renal arteries, arterioles, leading to an afferent arteriole, glomerular capillary bed (tuft), and a draining efferent arteriole. (From VH Gattone II et al: Hypertension 5:8, 1983.) B. Scanning electron micrograph of podocytes that line the outer surface of the glomerular capillaries (arrow shows foot process). C. Scanning electron micrograph of the fenestrated endothelia lining the glomerular capillary. D. The various normal regions of the glomerulus on light microscopy. (A–C: Courtesy of Dr. Vincent Gattone, Indiana University; with permission.)
The glomerular capillaries filter 120–180 L/d of plasma water containing various solutes for reclamation or discharge by downstream tubules. Most large proteins and all cells are excluded from filtration by a physicochemical barrier governed by pore size and negative electrostatic charge. The mechanics of filtration and reclamation are quite complicated for many solutes (Chap. 325). For example, in the case of serum albumin, the glomerulus is an imperfect barrier. Although albumin has a negative charge, which would tend to repel the negatively charged GBM, it only has a physical radius of 3.6 nm, while pores in the GBM and slit-pore membranes have a radius of 4 nm. Consequently, variable amounts of albumin inevitably cross the filtration barrier to be reclaimed by megalin and cubilin receptors along the proximal tubule. Remarkably, humans with normal nephrons excrete on average 8–10 mg of albumin in daily voided urine, approximately 20–60% of total excreted protein. This amount of albumin, and other proteins, can rise to gram quantities following glomerular injury.
The breadth of diseases affecting the glomerulus is expansive because the glomerular capillaries can be injured in a variety of ways, producing many different lesions. Some order to this vast subject is brought by grouping all of these diseases into a smaller number of clinical syndromes.
PATHOGENESIS OF GLOMERULAR DISEASE
There are many forms of glomerular disease with pathogenesis variably linked to the presence of genetic mutations, infection, toxin exposure, autoimmunity, atherosclerosis, hypertension, emboli, thrombosis, or diabetes mellitus. Even after careful study, however, the cause often remains unknown, and the lesion is called idiopathic. Specific or unique features of pathogenesis are mentioned with the description of each of the glomerular diseases later in this chapter.
![]() Some glomerular diseases result from genetic mutations producing familial disease or a founder effect: congenital nephrotic syndrome from mutations in NPHS1 (nephrin) and NPHS2 (podocin) affect the slit-pore membrane at birth, and TRPC6 cation channel mutations produce focal segmental glomerulosclerosis (FSGS) in adulthood; polymorphisms in the gene encoding apolipoprotein L1, APOL1, are a major risk for nearly 70% of African Americans with nondiabetic end-stage renal disease, particularly FSGS; mutations in complement factor H associate with membranoproliferative glomerulonephritis (MPGN) or atypical hemolytic uremic syndrome (aHUS), type II partial lipodystrophy from mutations in genes encoding lamin A/C, or PPARγ cause a metabolic syndrome associated with MPGN, which is sometimes accompanied by dense deposits and C3 nephritic factor; Alport’s syndrome, from mutations in the genes encoding for the α3, α4, or α5 chains of type IV collagen, produces split-basement membranes with glomerulosclerosis; and lysosomal storage diseases, such as α-galactosidase A deficiency causing Fabry’s disease and N -acetylneuraminic acid hydrolase deficiency causing nephrosialidosis, produce FSGS.
Some glomerular diseases result from genetic mutations producing familial disease or a founder effect: congenital nephrotic syndrome from mutations in NPHS1 (nephrin) and NPHS2 (podocin) affect the slit-pore membrane at birth, and TRPC6 cation channel mutations produce focal segmental glomerulosclerosis (FSGS) in adulthood; polymorphisms in the gene encoding apolipoprotein L1, APOL1, are a major risk for nearly 70% of African Americans with nondiabetic end-stage renal disease, particularly FSGS; mutations in complement factor H associate with membranoproliferative glomerulonephritis (MPGN) or atypical hemolytic uremic syndrome (aHUS), type II partial lipodystrophy from mutations in genes encoding lamin A/C, or PPARγ cause a metabolic syndrome associated with MPGN, which is sometimes accompanied by dense deposits and C3 nephritic factor; Alport’s syndrome, from mutations in the genes encoding for the α3, α4, or α5 chains of type IV collagen, produces split-basement membranes with glomerulosclerosis; and lysosomal storage diseases, such as α-galactosidase A deficiency causing Fabry’s disease and N -acetylneuraminic acid hydrolase deficiency causing nephrosialidosis, produce FSGS.
Systemic hypertension and atherosclerosis can produce pressure stress, ischemia, or lipid oxidants that lead to chronic glomerulosclerosis. Malignant hypertension can quickly complicate glomerulosclerosis with fibrinoid necrosis of arterioles and glomeruli, thrombotic microangiopathy, and acute renal failure. Diabetic nephropathy is an acquired sclerotic injury associated with thickening of the GBM secondary to the long-standing effects of hyperglycemia, advanced glycosylation end products, and reactive oxygen species.
Inflammation of the glomerular capillaries is called glomerulonephritis. Most glomerular or mesangial antigens involved in immune-mediated glomerulonephritis are unknown (Fig. 338-2). Glomerular epithelial or mesangial cells may shed or express epitopes that mimic other immunogenic proteins made elsewhere in the body. Bacteria, fungi, and viruses can directly infect the kidney producing their own antigens. Autoimmune diseases like idiopathic membranous glomerulonephritis (MGN) or MPGN are confined to the kidney, whereas systemic inflammatory diseases like lupus nephritis or granulomatosis with polyangiitis (Wegener’s) spread to the kidney, causing secondary glomerular injury. Antiglomerular basement membrane disease producing Goodpasture’s syndrome primarily injures both the lung and kidney because of the narrow distribution of the α3 NC1 domain of type IV collagen that is the target antigen.
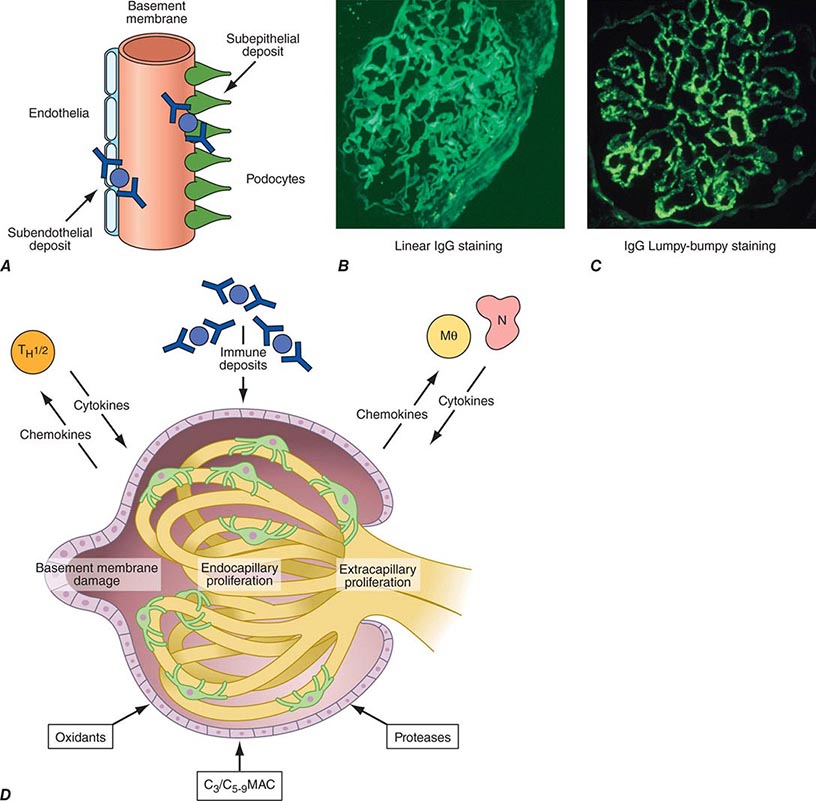
FIGURE 338-2 The glomerulus is injured by a variety of mechanisms. A. Preformed immune deposits can precipitate from the circulation and collect along the glomerular basement membrane (GBM) in the subendothelial space or can form in situ along the subepithelial space. B. Immunofluorescent staining of glomeruli with labeled anti-IgG demonstrating linear staining from a patient with anti-GBM disease or immune deposits from a patient with membranous glomerulonephritis. C. The mechanisms of glomerular injury have a complicated pathogenesis. Immune deposits and complement deposition classically draw macrophages and neutrophils into the glomerulus. T lymphocytes may follow to participate in the injury pattern as well. D. Amplification mediators as locally derived oxidants and proteases expand this inflammation, and, depending on the location of the target antigen and the genetic polymorphisms of the host, basement membranes are damaged with either endocapillary or extracapillary proliferation.
Local activation of Toll-like receptors on glomerular cells, deposition of immune complexes, or complement injury to glomerular structures induces mononuclear cell infiltration, which subsequently leads to an adaptive immune response attracted to the kidney by local release of chemokines. Neutrophils, macrophages, and T cells are drawn by chemokines into the glomerular tuft, where they react with antigens and epitopes on or near somatic cells or their structures, producing more cytokines and proteases that damage the mesangium, capillaries, and/or the GBM. While the adaptive immune response is similar to that of other tissues, early T cell activation plays an important role in the mechanism of glomerulonephritis. Antigens presented by class II major histocompatibility complex (MHC) molecules on macrophages and dendritic cells in conjunction with associative recognition molecules engage the CD4/8 T cell repertoire.
Mononuclear cells by themselves can injure the kidney, but autoimmune events that damage glomeruli classically produce a humoral immune response. Poststreptococcal glomerulonephritis, lupus nephritis, and idiopathic membranous nephritis typically are associated with immune deposits along the GBM, while anti-GBM antibodies produce the linear binding of anti-GBM disease. Preformed circulating immune complexes can precipitate along the subendothelial side of the GBM, while other immune deposits form in situ on the subepithelial side. These latter deposits accumulate when circulating autoantibodies find their antigen trapped along the subepithelial edge of the GBM. Immune deposits in the glomerular mesangium may result from the deposition of preformed circulating complexes or in situ antigen-antibody interactions. Immune deposits stimulate the release of local proteases and activate the complement cascade, producing C5–9 attack complexes. In addition, local oxidants damage glomerular structures, producing proteinuria and effacement of the podocytes. Overlapping etiologies or pathophysiologic mechanisms can produce similar glomerular lesions, suggesting that downstream molecular and cellular responses often converge toward common patterns of injury.
PROGRESSION OF GLOMERULAR DISEASE
Persistent glomerulonephritis that worsens renal function is always accompanied by interstitial nephritis, renal fibrosis, and tubular atrophy (see Fig. 62e-27). What is not so obvious, however, is that renal failure in glomerulonephritis best correlates histologically with the appearance of tubulointerstitial nephritis rather than with the type of inciting glomerular injury.
Loss of renal function due to interstitial damage is explained hypothetically by several mechanisms. The simplest explanation is that urine flow is impeded by tubular obstruction as a result of interstitial inflammation and fibrosis. Thus, obstruction of the tubules with debris or by extrinsic compression results in aglomerular nephrons. A second mechanism suggests that interstitial changes, including interstitial edema or fibrosis, alter tubular and vascular architecture and thereby compromise the normal tubular transport of solutes and water from tubular lumen to vascular space. This failure increases the solute and water content of the tubule fluid, resulting in isosthenuria and polyuria. Adaptive mechanisms related to tubuloglomerular feedback also fail, resulting in a reduction of renin output from the juxtaglomerular apparatus trapped by interstitial inflammation. Consequently, the local vasoconstrictive influence of angiotensin II on the glomerular arterioles decreases, and filtration drops owing to a generalized decrease in arteriolar tone. A third mechanism involves changes in vascular resistance due to damage of peritubular capillaries. The cross-sectional volume of these capillaries is decreased by interstitial inflammation, edema, or fibrosis. These structural alterations in vascular resistance affect renal function through two mechanisms. First, tubular cells are very metabolically active, and, as a result, decreased perfusion leads to ischemic injury. Second, impairment of glomerular arteriolar outflow leads to increased intraglomerular hypertension in less-involved glomeruli; this selective intraglomerular hypertension aggravates and extends mesangial sclerosis and glomerulosclerosis to less-involved glomeruli. Regardless of the exact mechanism, early acute tubulointerstitial nephritis (see Fig. 62e-27) suggests potentially recoverable renal function, whereas the development of chronic interstitial fibrosis prognosticates permanent loss (see Fig. 62e-30).
Persistent damage to glomerular capillaries spreads to the tubulointerstitium in association with proteinuria. There is a hypothesis that efferent arterioles leading from inflamed glomeruli carry forward inflammatory mediators, which induces downstream interstitial nephritis, resulting in fibrosis. Glomerular filtrate from injured glomerular capillaries adherent to Bowman’s capsule may also be misdirected to the periglomerular interstitium. Most nephrologists believe, however, that proteinuric glomerular filtrate forming tubular fluid is the primary route to downstream tubulointerstitial injury, although none of these hypotheses are mutually exclusive.
The simplest explanation for the effect of proteinuria on the development of interstitial nephritis is that increasingly severe proteinuria, carrying activated cytokines and lipoproteins producing reactive oxygen species, triggers a downstream inflammatory cascade in and around epithelial cells lining the tubular nephron. These effects induce T lymphocyte and macrophage infiltrates in the interstitial spaces along with fibrosis and tubular atrophy.
Tubules disaggregate following direct damage to their basement membranes, leading to epithelial-mesenchymal transitions forming more interstitial fibroblasts at the site of injury. Transforming growth factor β (TGF-β), fibroblast growth factor 2 (FGF-2), hypoxemia-inducible factor 1α (HIF-1α), and platelet-derived growth factor (PDGF) are particularly active in this transition. With persistent nephritis, fibroblasts multiply and lay down tenascin and a fibronectin scaffold for the polymerization of new interstitial collagen types I/III. These events form scar tissue through a process called fibrogenesis. In experimental studies, bone morphogenetic protein 7 and hepatocyte growth factor can reverse early fibrogenesis and preserve tubular architecture. When fibroblasts outdistance their survival factors, apoptoses occurs, and the permanent renal scar becomes acellular, leading to irreversible renal failure.
APPROACH TO THE PATIENT:
Glomerular Disease
HEMATURIA, PROTEINURIA, AND PYURIA
Patients with glomerular disease usually have some hematuria with varying degrees of proteinuria. Hematuria is typically asymptomatic. As few as three to five red blood cells in the spun sediment from first-voided morning urine is suspicious. The diagnosis of glomerular injury can be delayed because patients will not realize they have microscopic hematuria, and only rarely with the exception of IgA nephropathy and sickle cell disease is gross hematuria present. When working up microscopic hematuria, perhaps accompanied by minimal proteinuria (<500 mg/24 h), it is important to exclude anatomic lesions, such as malignancy of the urinary tract, particularly in older men. Microscopic hematuria may also appear with the onset of benign prostatic hypertrophy, interstitial nephritis, papillary necrosis, hypercalciuria, renal stones, cystic kidney diseases, or renal vascular injury. However, when red blood cell casts (see Fig. 62e-34) or dysmorphic red blood cells are found in the sediment, glomerulonephritis is likely.
Sustained proteinuria >1–2 g/24 h is also commonly associated with glomerular disease. Patients often will not know they have proteinuria unless they become edematous or notice foaming urine on voiding. Sustained proteinuria has to be distinguished from lesser amounts of so-called benign proteinuria in the normal population (Table 338-1). This latter class of proteinuria is nonsustained, generally <1 g/24 h, and is sometimes called functional or transient proteinuria. Fever, exercise, obesity, sleep apnea, emotional stress, and congestive heart failure can explain transient proteinuria. Proteinuria only seen with upright posture is called orthostatic proteinuria and has a benign prognosis. Isolated proteinuria sustained over multiple clinic visits is found in many glomerular lesions. Proteinuria in most adults with glomerular disease is nonselective, containing albumin and a mixture of other serum proteins, whereas in children with minimal change disease, the proteinuria is selective and composed largely of albumin.
|
URINE ASSAYS FOR ALBUMINURIA/PROTEINURIA |
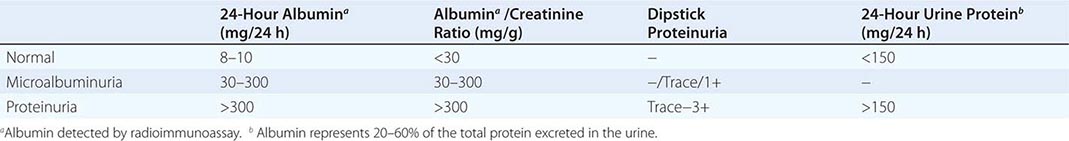
Some patients with inflammatory glomerular disease, such as acute poststreptococcal glomerulonephritis or MPGN, have pyuria characterized by the presence of considerable numbers of leukocytes. This latter finding has to be distinguished from urine infected with bacteria.
CLINICAL SYNDROMES
Various forms of glomerular injury can also be parsed into several distinct syndromes on clinical grounds (Table 338-2). These syndromes, however, are not always mutually exclusive. There is an acute nephritic syndrome producing 1–2 g/24 h of proteinuria, hematuria with red blood cell casts, pyuria, hypertension, fluid retention, and a rise in serum creatinine associated with a reduction in glomerular filtration. If glomerular inflammation develops slowly, the serum creatinine will rise gradually over many weeks, but if the serum creatinine rises quickly, particularly over a few days, acute nephritis is sometimes called rapidly progressive glomerulonephritis (RPGN); the histopathologic term crescentic glomerulonephritis is the pathologic equivalent of the clinical presentation of RPGN. When patients with RPGN present with lung hemorrhage from Goodpasture’s syndrome, antineutrophil cytoplasmic antibodies (ANCA)-associated small-vessel vasculitis, lupus erythematosus, or cryoglobulinemia, they are often diagnosed as having a pulmonary-renal syndrome. Nephrotic syndrome describes the onset of heavy proteinuria (>3.0 g/24 h), hypertension, hypercholesterolemia, hypoalbuminemia, edema/anasarca, and microscopic hematuria; if only large amounts of proteinuria are present without clinical manifestations, the condition is sometimes called nephrotic-range proteinuria. The glomerular filtration rate (GFR) in these patients may initially be normal or, rarely, higher than normal, but with persistent hyperfiltration and continued nephron loss, it typically declines over months to years. Patients with a basement membrane syndrome either have genetically abnormal basement membranes (Alport’s syndrome) or an autoimmune response to basement membrane collagen IV (Goodpasture’s syndrome) associated with microscopic hematuria, mild to heavy proteinuria, and hypertension with variable elevations in serum creatinine. Glomerular–vascular syndrome describes patients with vascular injury producing hematuria and moderate proteinuria. Affected individuals can have vasculitis, thrombotic microangiopathy, antiphospholipid syndrome, or, more commonly, a systemic disease such as atherosclerosis, cholesterol emboli, hypertension, sickle cell anemia, and autoimmunity. Infectious disease–associated syndrome is most important if one has a global perspective. Save for subacute bacterial endocarditis in the Western Hemisphere, malaria and schistosomiasis may be the most common causes of glomerulonephritis throughout the world, closely followed by HIV and chronic hepatitis B and C. These infectious diseases produce a variety of inflammatory reactions in glomerular capillaries, ranging from nephrotic syndrome to acute nephritic injury, and urinalyses that demonstrate a combination of hematuria and proteinuria.
|
PATTERNS OF CLINICAL GLOMERULONEPHRITIS |
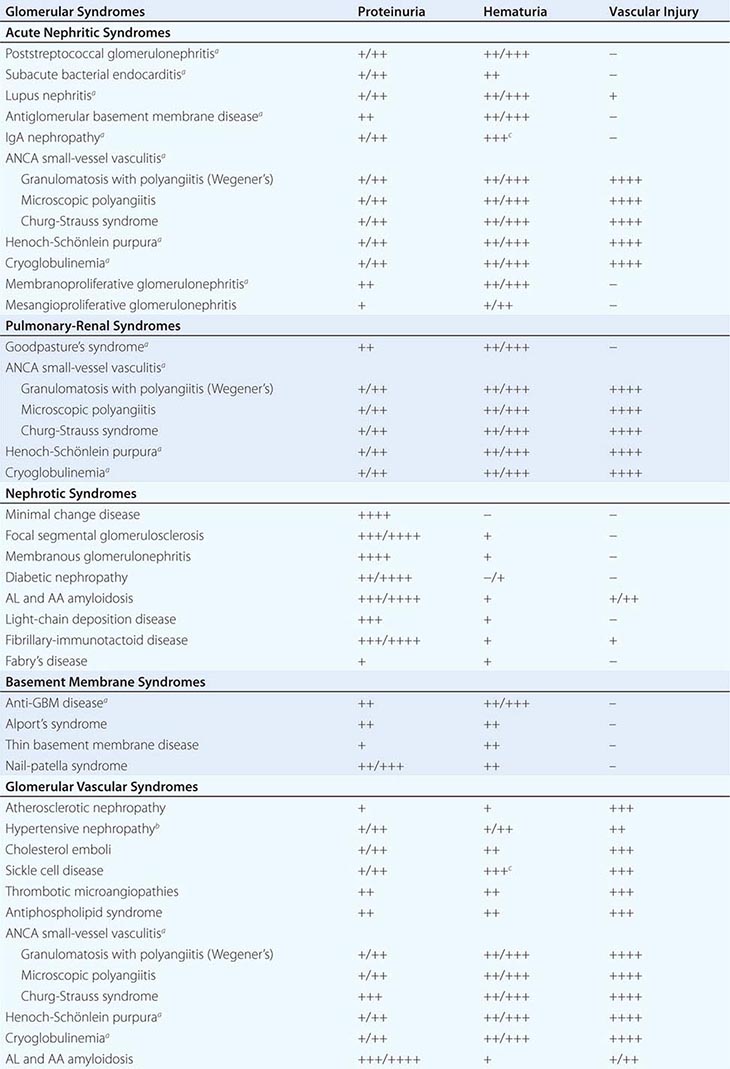
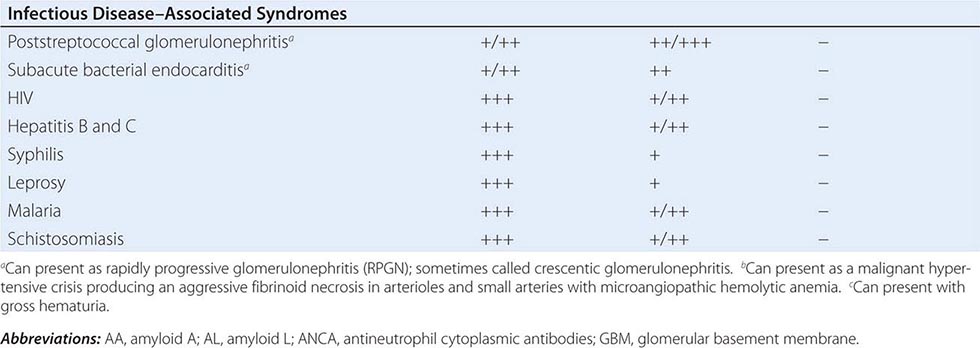
These six general categories of syndromes are usually determined at the bedside with the help of a history and physical examination, blood chemistries, renal ultrasound, and urinalysis. These initial studies help frame further diagnostic workup that typically involves testing of the serum for the presence of various proteins (HIV and hepatitis B and C antigens), antibodies (anti-GBM, antiphospholipid, antistreptolysin O [ASO], anti-DNAse, antihyaluronidase, ANCA, anti-DNA, cryoglobulins, anti-HIV, and anti-hepatitis B and C antibodies) or depletion of complement components (C3 and C4). The bedside history and physical examination can also help determine whether the glomerulonephritis is isolated to the kidney (primary glomerulonephritis) or is part of a systemic disease (secondary glomerulonephritis).
When confronted with an abnormal urinalysis and elevated serum creatinine, with or without edema or congestive heart failure, one must consider whether the glomerulonephritis is acute or chronic. This assessment is best made by careful history (last known urinalysis or serum creatinine during pregnancy or insurance physical, evidence of infection, or use of medication or recreational drugs); the size of the kidneys on renal ultrasound examination; and how the patient feels at presentation. Chronic glomerular disease often presents with decreased kidney size. Patients who quickly develop renal failure are fatigued and weak and often have uremic symptoms associated with nausea, vomiting, fluid retention, and somnolence. Primary glomerulonephritis presenting with renal failure that has progressed slowly, however, can be remarkably asymptomatic, as are patients with acute glomerulonephritis without much loss in renal function. Once this initial information is collected, selected patients who are clinically stable, have adequate blood clotting parameters, and are willing and able to receive treatment are encouraged to have a renal biopsy.
RENAL PATHOLOGY
A renal biopsy in the setting of glomerulonephritis quickly identifies the type of glomerular injury and often suggests a course of treatment. The biopsy is processed for light microscopy using stains for hematoxylin and eosin (H&E) to assess cellularity and architecture, periodic acid–Schiff (PAS) to stain carbohydrate moieties in the membranes of the glomerular tuft and tubules, Jones-methenamine silver to enhance basement membrane structure, Congo red for amyloid deposits, and Masson’s trichrome to identify collagen deposition and assess the degree of glomerulosclerosis and interstitial fibrosis. Biopsies are also processed for direct immunofluorescence using conjugated antibodies against IgG, IgM, and IgA to detect the presence of “lumpy-bumpy” immune deposits or “linear” IgG or IgA antibodies bound to GBM, antibodies against trapped complement proteins (C3 and C4), or specific antibodies against a relevant antigen. High-resolution electron microscopy can clarify the principal location of immune deposits and the status of the basement membrane.
Each region of a renal biopsy is assessed separately. By light microscopy, glomeruli (at least 10 and ideally 20) are reviewed individually for discrete lesions; <50% involvement is considered focal, and >50% is diffuse. Injury in each glomerular tuft can be segmental, involving a portion of the tuft, or global, involving most of the glomerulus. Glomeruli having proliferative characteristics show increased cellularity. When cells in the capillary tuft proliferate, it is called endocapillary, and when cellular proliferation extends into Bowman’s space, it is called extracapillary. Synechiae are formed when epithelial podocytes attach to Bowman’s capsule in the setting of glomerular injury; crescents, which in some cases may be the extension of synechiae, develop when fibrocellular/fibrin collections fill all or part of Bowman’s space; and sclerotic glomeruli show acellular, amorphous accumulations of proteinaceous material throughout the tuft with loss of functional capillaries and normal mesangium. Since age-related glomerulosclerosis is common in adults, one can estimate the background percentage of sclerosis by dividing the patient’s age in half and subtracting 10. Immunofluorescent and electron microscopy can detect the presence and location of subepithelial, subendothelial, or mesangial immune deposits, or reduplication or splitting of the basement membrane. In the other regions of the biopsy, the vasculature surrounding glomeruli and tubules can show angiopathy, vasculitis, the presence of fibrils, or thrombi. The tubules can be assessed for adjacency to one another; separation can be the result of edema, tubular dropout, or collagen deposition resulting from interstitial fibrosis. Interstitial fibrosis is an ominous sign of irreversibility and progression to renal failure.
ACUTE NEPHRITIC SYNDROMES
Acute nephritic syndromes classically present with hypertension, hematuria, red blood cell casts, pyuria, and mild to moderate proteinuria. Extensive inflammatory damage to glomeruli causes a fall in GFR and eventually produces uremic symptoms with salt and water retention, leading to edema and hypertension.
POSTSTREPTOCOCCAL GLOMERULONEPHRITIS
Poststreptococcal glomerulonephritis is prototypical for acute endocapillary proliferative glomerulonephritis. The incidence of poststreptococcal glomerulonephritis has dramatically decreased in developed countries and in these locations is typically sporadic. Acute poststreptococcal glomerulonephritis in underdeveloped countries is epidemic and usually affects children between the ages of 2 and 14 years, but in developed countries is more typical in the elderly, especially in association with debilitating conditions. It is more common in males, and the familial or cohabitant incidence is as high as 40%. Skin and throat infections with particular M types of streptococci (nephritogenic strains) antedate glomerular disease; M types 47, 49, 55, 2, 60, and 57 are seen following impetigo and M types 1, 2, 4, 3, 25, 49, and 12 with pharyngitis. Poststreptococcal glomerulonephritis due to impetigo develops 2–6 weeks after skin infection and 1–3 weeks after streptococcal pharyngitis.
The renal biopsy in poststreptococcal glomerulonephritis demonstrates hypercellularity of mesangial and endothelial cells, glomerular infiltrates of polymorphonuclear leukocytes, granular subendothelial immune deposits of IgG, IgM, C3, C4, and C5–9, and subepithelial deposits (which appear as “humps”) (see Fig. 62e-6). (See Glomerular Schematic 1.) Poststreptococcal glomerulonephritis is an immune-mediated disease involving putative streptococcal antigens, circulating immune complexes, and activation of complement in association with cell-mediated injury. Many candidate antigens have been proposed over the years; candidates from nephritogenic streptococci of interest at the moment are: a cationic cysteine proteinase known as streptococcal pyrogenic exotoxin B (SPEB) that is generated by proteolysis of a zymogen precursor (zSPEB), and NAPlr, the nephritis-associated plasmin receptor. These two antigens have biochemical affinity for plasmin, bind as complexes facilitated by this relationship, and activate the alternate complement pathway. The nephritogenic antigen, SPEB, has been demonstrated inside the subepithelial “humps” on biopsy.
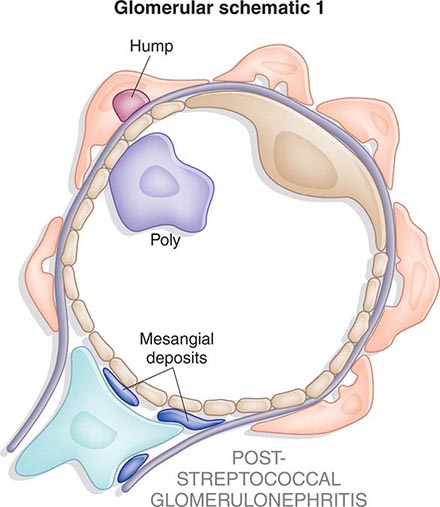
The classic presentation is an acute nephritic picture with hematuria, pyuria, red blood cell casts, edema, hypertension, and oliguric renal failure, which may be severe enough to appear as RPGN. Systemic symptoms of headache, malaise, anorexia, and flank pain (due to swelling of the renal capsule) are reported in as many as 50% of cases. Five percent of children and 20% of adults have proteinuria in the nephrotic range. In the first week of symptoms, 90% of patients will have a depressed CH50 and decreased levels of C3 with normal levels of C4. Positive rheumatoid factor (30–40%), cryoglobulins and circulating immune complexes (60–70%), and ANCA against myeloperoxidase (10%) are also reported. Positive cultures for streptococcal infection are inconsistently present (10–70%), but increased titers of ASO (30%), anti-DNAse, (70%), or antihyaluronidase antibodies (40%) can help confirm the diagnosis. Consequently, the diagnosis of poststreptococcal glomerulonephritis rarely requires a renal biopsy. A subclinical disease is reported in some series to be four to five times as common as clinical nephritis, and these latter cases are characterized by asymptomatic microscopic hematuria with low serum C3 complement levels.
Treatment is supportive, with control of hypertension, edema, and dialysis as needed. Antibiotic treatment for streptococcal infection should be given to all patients and their cohabitants. There is no role for immunosuppressive therapy, even in the setting of crescents. Recurrent poststreptococcal glomerulonephritis is rare despite repeated streptococcal infections. Early death is rare in children but does occur in the elderly. Overall, the prognosis is good, with permanent renal failure being very uncommon, less than 1% in children. Complete resolution of the hematuria and proteinuria in the majority of children occurs within 3–6 weeks of the onset of nephritis but 3–10% of children may have persistent microscopic hematuria, nonnephrotic proteinuria, or hypertension. The prognosis in elderly patients is worse with a high incidence of azotemia (up to 60%), nephrotic-range proteinuria, and end-stage renal disease.
SUBACUTE BACTERIAL ENDOCARDITIS
Endocarditis-associated glomerulonephritis is typically a complication of subacute bacterial endocarditis, particularly in patients who remain untreated for a long time, have negative blood cultures, or have right-sided endocarditis. Glomerulonephritis is unusual in acute bacterial endocarditis because it takes 10–14 days to develop immune complex–mediated injury, by which time the patient has been treated, often with emergent surgery. Grossly, the kidneys in subacute bacterial endocarditis have subcapsular hemorrhages with a “flea-bitten” appearance, and microscopy on renal biopsy reveals focal proliferation around foci of necrosis associated with abundant mesangial, subendothelial, and subepithelial immune deposits of IgG, IgM, and C3. Patients who present with a clinical picture of RPGN have crescents. Embolic infarcts or septic abscesses may also be present. The pathogenesis hinges on the renal deposition of circulating immune complexes in the kidney with complement activation. Patients present with gross or microscopic hematuria, pyuria, and mild proteinuria or, less commonly, RPGN with rapid loss of renal function. A normocytic anemia, elevated erythrocyte sedimentation rate, hypocomplementemia, high titers of rheumatoid factor, type III cryoglobulins, circulating immune complexes, and ANCAs may be present. Levels of serum creatinine may be elevated at diagnosis, but with modern therapy there is little progression to chronic renal failure. Primary treatment is eradication of the infection with 4–6 weeks of antibiotics, and if accomplished expeditiously, the prognosis for renal recovery is good. ANCA-associated vasculitis sometimes accompanies or is confused with subacute bacterial endocarditis (SBE) and should be ruled out, as the treatment is different.
As variants of persistent bacterial infection in blood-associated glomerulonephritis, postinfectious glomerulonephritis can occur in patients with ventriculoatrial and ventriculoperitoneal shunts; pulmonary, intraabdominal, pelvic, or cutaneous infections; and infected vascular prostheses. In developed countries, a significant proportion of cases afflict adults, especially the immunocompromised, and the predominant organism is Staphylococcus. The clinical presentation of these conditions is variable and includes proteinuria, microscopic hematuria, acute renal failure, and hypertension. Serum complement levels are low, and there may be elevated levels of C-reactive proteins, rheumatoid factor, antinuclear antibodies, and cryoglobulins. Renal lesions include membranoproliferative glomerulonephritis (MPGN), diffuse proliferative and exudative glomerulonephritis (DPGN), or mesangioproliferative glomerulonephritis, sometimes leading to RPGN. Treatment focuses on eradicating the infection, with most patients treated as if they have endocarditis. The prognosis is guarded.
LUPUS NEPHRITIS
Lupus nephritis is a common and serious complication of systemic lupus erythematosus (SLE) and most severe in African-American female adolescents. Thirty to 50% of patients will have clinical manifestations of renal disease at the time of diagnosis, and 60% of adults and 80% of children develop renal abnormalities at some point in the course of their disease. Lupus nephritis results from the deposition of circulating immune complexes, which activate the complement cascade leading to complement-mediated damage, leukocyte infiltration, activation of procoagulant factors, and release of various cytokines. In situ immune complex formation following glomerular binding of nuclear antigens, particularly necrotic nucleosomes, also plays a role in renal injury. The presence of antiphospholipid antibodies may also trigger a thrombotic microangiopathy in a minority of patients.
The clinical manifestations, course of disease, and treatment of lupus nephritis are closely linked to renal pathology. The most common clinical sign of renal disease is proteinuria, but hematuria, hypertension, varying degrees of renal failure, and active urine sediment with red blood cell casts can all be present. Although significant renal pathology can be found on biopsy even in the absence of major abnormalities in the urinalysis, most nephrologists do not biopsy patients until the urinalysis is convincingly abnormal. The extrarenal manifestations of lupus are important in establishing a firm diagnosis of systemic lupus because, while serologic abnormalities are common in lupus nephritis, they are not diagnostic. Anti-dsDNA antibodies that fix complement correlate best with the presence of renal disease. Hypocomplementemia is common in patients with acute lupus nephritis (70–90%) and declining complement levels may herald a flare. Although urinary biomarkers of lupus nephritis are being identified to assist in predicting renal flares, renal biopsy is the only reliable method of identifying the morphologic variants of lupus nephritis.
The World Health Organization (WHO) workshop in 1974 first outlined several distinct patterns of lupus-related glomerular injury; these were modified in 1982. In 2004 the International Society of Nephrology in conjunction with the Renal Pathology Society again updated the classification. This latest version of lesions seen on biopsy (Table 338-3) best defines clinicopathologic correlations, provides valuable prognostic information, and forms the basis for modern treatment recommendations. Class I nephritis describes normal glomerular histology by any technique or normal light microscopy with minimal mesangial deposits on immunofluorescent or electron microscopy. Class II designates mesangial immune complexes with mesangial proliferation. Both class I and II lesions are typically associated with minimal renal manifestation and normal renal function; nephrotic syndrome is rare. Patients with lesions limited to the renal mesangium have an excellent prognosis and generally do not need therapy for their lupus nephritis.
|
CLASSIFICATION FOR LUPUS NEPHRITIS |
The subject of lupus nephritis is presented under acute nephritic syndromes because of the aggressive and important proliferative lesions seen in class III–V renal disease. Class III describes focal lesions with proliferation or scarring, often involving only a segment of the glomerulus (see Fig. 62e-12). Class III lesions have the most varied course. Hypertension, an active urinary sediment, and proteinuria are common with nephrotic-range proteinuria in 25–33% of patients. Elevated serum creatinine is present in 25% of patients. Patients with mild proliferation involving a small percentage of glomeruli respond well to therapy with steroids alone, and fewer than 5% progress to renal failure over 5 years. Patients with more severe proliferation involving a greater percentage of glomeruli have a far worse prognosis and lower remission rates. Treatment of those patients is the same as that for class IV lesions. Many nephrologists believe that class III lesions are simply an early presentation of class IV disease. Others believe severe class III disease is a discrete lesion requiring aggressive therapy. Class IV describes global, diffuse proliferative lesions involving the vast majority of glomeruli. Patients with class IV lesions commonly have high anti-DNA antibody titers, low serum complement, hematuria, red blood cell casts, proteinuria, hypertension, and decreased renal function; 50% of patients have nephrotic-range proteinuria. Patients with crescents on biopsy often have a rapidly progressive decline in renal function (see Fig. 62e-12). Without treatment, this aggressive lesion has the worst renal prognosis. However, if a remission—defined as a return to near-normal renal function and proteinuria ≤330 mg/dL per day—is achieved with treatment, renal outcomes are excellent. Current evidence suggests that inducing a remission with administration of high-dose steroids and either cyclophosphamide or mycophenolate mofetil for 2–6 months, followed by maintenance therapy with lower doses of steroids and mycophenolate mofetil or azathioprine, best balances the likelihood of successful remission with the side effects of therapy. There is no consensus on use of high-dose intravenous methylprednisolone versus oral prednisone, monthly intravenous cyclophosphamide versus daily oral cyclophosphamide, or other immunosuppressants such as cyclosporine, tacrolimus, rituximab, or belimumab. Nephrologists tend to avoid prolonged use of cyclophosphamide in patients of childbearing age without first banking eggs or sperm.
The class V lesion describes subepithelial immune deposits producing a membranous pattern; a subcategory of class V lesions is associated with proliferative lesions and is sometimes called mixed membranous and proliferative disease (see Fig. 62e-11); this category of injury is treated like class IV glomerulonephritis. Sixty percent of patients present with nephrotic syndrome or lesser amounts of proteinuria. Patients with lupus nephritis class V, like patients with idiopathic membranous nephropathy, are predisposed to renal-vein thrombosis and other thrombotic complications. A minority of patients with class V will present with hypertension and renal dysfunction. There are conflicting data on the clinical course, prognosis, and appropriate therapy for patients with class V disease, which may reflect the heterogeneity of this group of patients. Patients with severe nephrotic syndrome, elevated serum creatinine, and a progressive course will probably benefit from therapy with steroids in combination with other immunosuppressive agents. Therapy with inhibitors of the renin-angiotensin system also may attenuate the proteinuria. Antiphospholipid antibodies present in lupus may result in glomerular microthromboses and complicate the course in up to 20% of lupus nephritis patients. The renal prognosis is worse even with anticoagulant therapy.
Patients with any of the above lesions also can transform to another lesion; hence patients often require reevaluation, including repeat renal biopsy. Lupus patients with class VI lesions have greater than 90% sclerotic glomeruli and end-stage renal disease with interstitial fibrosis. As a group, approximately 20% of patients with lupus nephritis will reach end-stage disease, requiring dialysis or transplantation. Systemic lupus tends to become quiescent once there is renal failure, perhaps due to the immunosuppressant effects of uremia. However, patients with lupus nephritis have a markedly increased mortality compared with the general population. Renal transplantation in renal failure from lupus, usually performed after approximately 6 months of inactive disease, results in allograft survival rates comparable to patients transplanted for other reasons.
ANTIGLOMERULAR BASEMENT MEMBRANE DISEASE
Patients who develop autoantibodies directed against glomerular basement antigens frequently develop a glomerulonephritis termed antiglomerular basement membrane (anti-GBM) disease. When they present with lung hemorrhage and glomerulonephritis, they have a pulmonary-renal syndrome called Goodpasture’s syndrome. The target epitopes for this autoimmune disease lie in the quaternary structure of α3 NC1 domain of collagen IV. Indeed, anti-GBM disease may be considered an autoimmune “conformeropathy” that involves the perturbation of quaternary structure of the α 345NC1 hexamer. MHC-restricted T cells initiate the autoantibody response because humans are not tolerant to the epitopes created by this quaternary structure. The epitopes are normally sequestered in the collagen IV hexamer and can be exposed by infection, smoking, oxidants, or solvents. Goodpasture’s syndrome appears in two age groups: in young men in their late twenties and in men and women in their sixties and seventies. Disease in the younger age group is usually explosive, with hemoptysis, a sudden fall in hemoglobin, fever, dyspnea, and hematuria. Hemoptysis is largely confined to smokers, and those who present with lung hemorrhage as a group do better than older populations who have prolonged, asymptomatic renal injury; presentation with oliguria is often associated with a particularly bad outcome. The performance of an urgent kidney biopsy is important in suspected cases of Goodpasture’s syndrome to confirm the diagnosis and assess prognosis. Renal biopsies typically show focal or segmental necrosis that later, with aggressive destruction of the capillaries by cellular proliferation, leads to crescent formation in Bowman’s space (see Fig. 62e-14). As these lesions progress, there is concomitant interstitial nephritis with fibrosis and tubular atrophy.
The presence of anti-GBM antibodies and complement is recognized on biopsy by linear immunofluorescent staining for IgG (rarely IgA). In testing serum for anti-GBM antibodies, it is particularly important that the α3 NC1 domain of collagen IV alone be used as the target. This is because nonnephritic antibodies against the α1 NC1 domain are seen in paraneoplastic syndromes and cannot be discerned from assays that use whole basement membrane fragments as the binding target. Between 10 and 15% of sera from patients with Goodpasture’s syndrome also contain ANCA antibodies against myeloperoxidase. This subset of patients has a vasculitis-associated variant, which has a surprisingly good prognosis with treatment. Prognosis at presentation is worse if there are >50% crescents on renal biopsy with advanced fibrosis, if serum creatinine is >5–6 mg/dL, if oliguria is present, or if there is a need for acute dialysis. Although frequently attempted, most of these latter patients will not respond to plasmapheresis and steroids. Patients with advanced renal failure who present with hemoptysis should still be treated for their lung hemorrhage, as it responds to plasmapheresis and can be lifesaving. Treated patients with less severe disease typically respond to 8–10 treatments of plasmapheresis accompanied by oral prednisone and cyclophosphamide in the first 2 weeks. Kidney transplantation is possible, but because there is risk of recurrence, experience suggests that patients should wait for 6 months and until serum antibodies are undetectable.
IgA NEPHROPATHY
Berger first described the glomerulonephritis now termed IgA nephropathy. It is classically characterized by episodic hematuria associated with the deposition of IgA in the mesangium. IgA nephropathy is one of the most common forms of glomerulonephritis worldwide. There is a male preponderance, a peak incidence in the second and third decades of life, and rare familial clustering. There are geographic differences in the prevalence of IgA nephropathy, with 30% prevalence along the Asian and Pacific Rim and 20% in southern Europe, compared to a much lower prevalence in northern Europe and North America. It was initially hypothesized that variation in detection, in part, accounted for regional differences. With clinical care in nephrology becoming more uniform, this variation in prevalence more likely reflects true differences among racial and ethnic groups.
IgA nephropathy is predominantly a sporadic disease but susceptibility to it has been shown uncommonly to have a genetic component depending on geography and the existence of “founder effects.” Familial forms of IgA nephropathy are more common in northern Italy and eastern Kentucky. No single causal gene has been identified. Clinical and laboratory evidence suggests close similarities between Henoch-Schönlein purpura and IgA nephropathy. Henoch-Schönlein purpura is distinguished clinically from IgA nephropathy by prominent systemic symptoms, a younger age (<20 years old), preceding infection, and abdominal complaints. Deposits of IgA are also found in the glomerular mesangium in a variety of systemic diseases, including chronic liver disease, Crohn’s disease, gastrointestinal adenocarcinoma, chronic bronchiectasis, idiopathic interstitial pneumonia, dermatitis herpetiformis, mycosis fungoides, leprosy, ankylosing spondylitis, relapsing polychondritis, and Sjögren’s syndrome. IgA deposition in these entities is not usually associated with clinically significant glomerular inflammation or renal dysfunction and thus is not called IgA nephropathy.
IgA nephropathy is an immune complex–mediated glomerulonephritis defined by the presence of diffuse mesangial IgA deposits often associated with mesangial hypercellularity. (See Glomerular Schematic 2.) IgM, IgG, C3, or immunoglobulin light chains may be codistributed with IgA. IgA deposited in the mesangium is typically polymeric and of the IgA1 subclass, the pathogenic significance of which is not clear. Abnormalities have been described in IgA production by plasma cells, particularly secretory IgA; in IgA clearance, predominately by the liver; in mesangial IgA clearance and receptors for IgA; and in growth factor and cytokine-mediated events. Currently, however, abnormalities in the O -glycosylation of the hinge region of IgA seem to best account for the pathogenesis of sporadic IgA nephropathy. Despite the presence of elevated serum IgA levels in 20–50% of patients, IgA deposition in skin biopsies in 15–55% of patients, or elevated levels of secretory IgA and IgA-fibronectin complexes, a renal biopsy is necessary to confirm the diagnosis. Although the immunofluorescent pattern of IgA on renal biopsy defines IgA nephropathy in the proper clinical context, a variety of histologic lesions may be seen on light microscopy (see Fig. 62e-8), including DPGN; segmental sclerosis; and, rarely, segmental necrosis with cellular crescent formation, which typically presents as RPGN.
The two most common presentations of IgA nephropathy are recurrent episodes of macroscopic hematuria during or immediately following an upper respiratory infection often accompanied by proteinuria or persistent asymptomatic microscopic hematuria. Nephrotic syndrome is uncommon. Proteinuria can also first appear late in the course of the disease. Rarely patients present with acute renal failure and a rapidly progressive clinical picture. IgA nephropathy is a benign disease for the majority of patients, and 5–30% of patients may go into a complete remission, with others having hematuria but well preserved renal function. In the minority of patients who have progressive disease, progression is slow, with renal failure seen in only 25–30% of patients with IgA nephropathy over 20–25 years. This risk varies considerably among populations. Cumulatively, risk factors for the loss of renal function identified thus far account for less than 50% of the variation in observed outcome but include the presence of hypertension or proteinuria, the absence of episodes of macroscopic hematuria, male sex, older age of onset, and extensive glomerulosclerosis or interstitial fibrosis on renal biopsy. Several analyses in large populations of patients found persistent proteinuria for 6 months or longer to have the greatest predictive power for adverse renal outcomes.
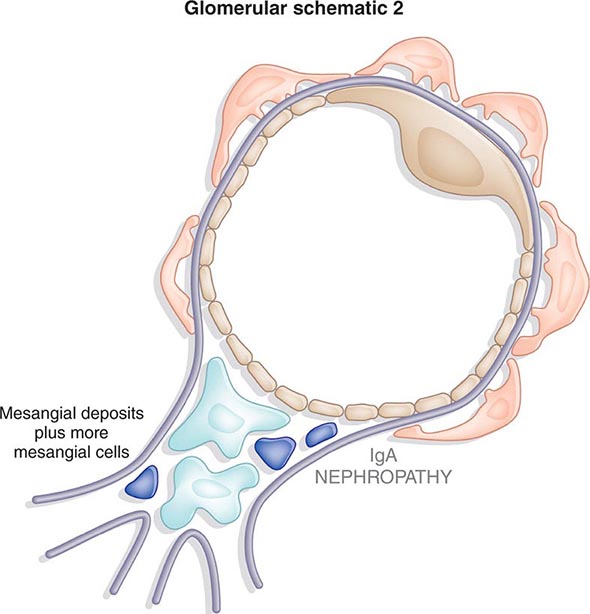
There is no agreement on optimal treatment. Both large studies that include patients with multiple glomerular diseases and small studies of patients with IgA nephropathy support the use of angiotensin-converting enzyme (ACE) inhibitors in patients with proteinuria or declining renal function. Tonsillectomy, steroid therapy, and fish oil have all been suggested in small studies to benefit select patients with IgA nephropathy. When presenting as RPGN, patients typically receive steroids, cytotoxic agents, and plasmapheresis.
ANCA SMALL-VESSEL VASCULITIS
A group of patients with small-vessel vasculitis (arterioles, capillaries, and venules; rarely small arteries) and glomerulonephritis have serum ANCA; the antibodies are of two types, anti-proteinase 3 (PR3) or anti-myeloperoxidase (MPO) (Chap. 385); Lamp-2 antibodies have also been reported experimentally as potentially pathogenic. ANCA are produced with the help of T cells and activate leukocytes and monocytes, which together damage the walls of small vessels. Endothelial injury also attracts more leukocytes and extends the inflammation. Granulomatosis with polyangiitis, microscopic polyangiitis, and Churg-Strauss syndrome belong to this group because they are ANCA-positive and have a pauci-immune glomerulonephritis with few immune complexes in small vessels and glomerular capillaries. Patients with any of these three diseases can have any combination of the above serum antibodies, but anti-PR3 antibodies are more common in granulomatosis with polyangiitis and anti-MPO antibodies are more common in microscopic polyangiitis or Churg-Strauss. Although each of these diseases has some unique clinical features, most features do not predict relapse or progression, and as a group, they are generally treated in the same way. Since mortality is high without treatment, virtually all patients receive urgent treatment. Induction therapy usually includes some combination of plasmapheresis, methylprednisolone, and cyclophosphamide. Monthly “pulse” IV cyclophosphamide to induce remission of ANCA-associated vasculitis is as effective as daily oral cyclophosphamide but may be associated with increased relapses. Steroids are tapered soon after acute inflammation subsides, and patients are maintained on cyclophosphamide or azathioprine for up to a year to minimize the risk of relapse. Benefit with using mycophenolate mofetil or rituximab has not been proven.
Granulomatosis with Polyangiitis Patients with this disease classically present with fever, purulent rhinorrhea, nasal ulcers, sinus pain, polyarthralgias/arthritis, cough, hemoptysis, shortness of breath, microscopic hematuria, and 0.5–1 g/24 h of proteinuria; occasionally there may be cutaneous purpura and mononeuritis multiplex. Presentation without renal involvement is termed limited granulomatosis with polyangiitis, although some of these patients will show signs of renal injury later. Chest x-ray often reveals nodules and persistent infiltrates, sometimes with cavities. Biopsy of involved tissue will show a small-vessel vasculitis and adjacent noncaseating granulomas. Renal biopsies during active disease demonstrate segmental necrotizing glomerulonephritis without immune deposits (see Fig. 62e-13). The disease is more common in patients exposed to silica dust and those with α1-antitrypsin deficiency, which is an inhibitor of PR3. Relapse after achieving remission is common and is more common in patients with granulomatosis with polyangiitis than the other ANCA-associated vasculitis, necessitating diligent follow-up care. Although associated with an unacceptable high mortality rate without treatment, the greatest threat to patients, especially elderly patients in the first year of therapy, is from adverse events, which are often secondary to treatment, rather than active vasculitis.
Microscopic Polyangiitis Clinically, these patients look somewhat similar to those with granulomatosis with polyangiitis, except they rarely have significant lung disease or destructive sinusitis. The distinction is made on biopsy, where the vasculitis in microscopic polyangiitis is without granulomas. Some patients will also have injury limited to the capillaries and venules.
Churg-Strauss Syndrome When small-vessel vasculitis is associated with peripheral eosinophilia, cutaneous purpura, mononeuritis, asthma, and allergic rhinitis, a diagnosis of Churg-Strauss syndrome is considered. Hypergammaglobulinemia, elevated levels of serum IgE, or the presence of rheumatoid factor sometimes accompanies the allergic state. Lung inflammation, including fleeting cough and pulmonary infiltrates, often precedes the systemic manifestations of disease by years; lung manifestations are rarely absent. A third of patients may have exudative pleural effusions associated with eosinophils. Small-vessel vasculitis and focal segmental necrotizing glomerulonephritis can be seen on renal biopsy, usually absent eosinophils or granulomas. The cause of Churg-Strauss syndrome is autoimmune, but the inciting factors are unknown.
MEMBRANOPROLIFERATIVE GLOMERULONEPHRITIS
MPGN is sometimes called mesangiocapillary glomerulonephritis or lobar glomerulonephritis. It is an immune-mediated glomerulonephritis characterized by thickening of the GBM with mesangioproliferative changes; 70% of patients have hypocomplementemia. MPGN is rare in African Americans, and idiopathic disease usually presents in childhood or young adulthood. MPGN is subdivided pathologically into type I, type II, and type III disease. Type I MPGN is commonly associated with persistent hepatitis C infections, autoimmune diseases like lupus or cryoglobulinemia, or neoplastic diseases (Table 338-4). Types II and III MPGN are usually idiopathic, except in patients with complement factor H deficiency, in the presence of C3 nephritic factor and/or in partial lipodystrophy producing type II disease, or complement receptor deficiency in type III disease. MPGN has been proposed to be reclassified into immunoglobulin-mediated disease (driven by the classical complement pathway) and non–immunoglobulin-mediated disease (driven by the alternative complement pathway).
|
MEMBRANOPROLIFERATIVE GLOMERULONEPHRITIS |
Type I MPGN, the most proliferative of the three types, shows mesangial proliferation with lobular segmentation on renal biopsy and mesangial interposition between the capillary basement membrane and endothelial cells, producing a double contour sometimes called tram-tracking (see Fig. 62e-9). (See Glomerular Schematic 3.) Subendothelial deposits with low serum levels of C3 are typical, although 50% of patients have normal levels of C3 and occasional intramesangial deposits. Low serum C3 and a dense thickening of the GBM containing ribbons of dense deposits and C3 characterize type II MPGN, sometimes called dense deposit disease (see Fig. 62e-10). Classically, the glomerular tuft has a lobular appearance; intramesangial deposits are rarely present and subendothelial deposits are generally absent. Proliferation in type III MPGN is less common than the other two types and is often focal; mesangial interposition is rare, and subepithelial deposits can occur along widened segments of the GBM that appear laminated and disrupted.
Type I MPGN is secondary to glomerular deposition of circulating immune complexes or their in situ formation. Types II and III MPGN may be related to “nephritic factors,” which are autoantibodies that stabilize C3 convertase and allow it to activate serum C3. MPGN can also result from acquired or genetic abnormalities in the alternative complement pathway. Patients with MPGN present with proteinuria, hematuria, and pyuria (30%); systemic symptoms of fatigue and malaise that are most common in children with type I disease; or an acute nephritic picture with RPGN and a speedy deterioration in renal function in up to 25% of patients. Low serum C3 levels are common. Fifty percent of patients with MPGN develop end-stage disease 10 years after diagnosis, and 90% have renal insufficiency after 20 years. Nephrotic syndrome, hypertension, and renal insufficiency all predict poor outcome. In the presence of proteinuria, treatment with inhibitors of the renin-angiotensin system is prudent. Evidence for treatment with dipyridamole, Coumadin (warfarin), or cyclophosphamide is not strongly established. There is some evidence supporting the efficacy of treatment of primary MPGN with steroids, particularly in children, as well as reports of efficacy with plasma exchange and other immunosuppressive drugs. If defects in the complement pathway are found, treatment with eculizumab is of hypothetical but unproven benefit. In secondary MPGN, treating the associated infection, autoimmune disease, or neoplasms is of demonstrated benefit. In particular, pegylated interferon and ribavirin are useful in reducing viral load. Although all primary renal diseases can recur over time in transplanted renal allografts, patients with MPGN are well known to be at risk for not only a histologic recurrence but also a clinically significant recurrence with loss of graft function.
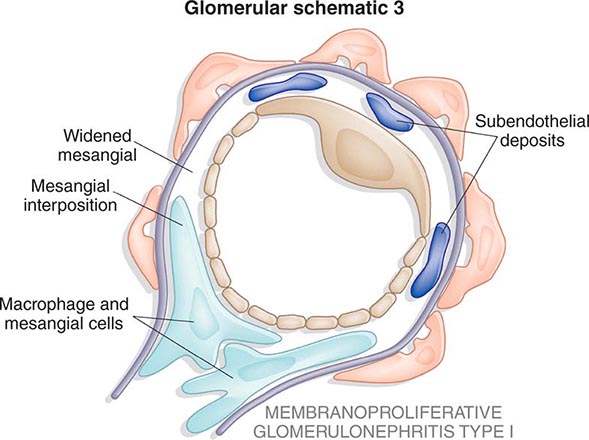
MESANGIOPROLIFERATIVE GLOMERULONEPHRITIS
Mesangioproliferative glomerulonephritis is characterized by expansion of the mesangium, sometimes associated with mesangial hypercellularity; thin, single contoured capillary walls; and mesangial immune deposits. Clinically, it can present with varying degrees of proteinuria and, commonly, hematuria. Mesangioproliferative disease may be seen in IgA nephropathy, Plasmodium falciparum malaria, resolving postinfectious glomerulonephritis, and class II nephritis from lupus, all of which can have a similar histologic appearance. With these secondary entities excluded, the diagnosis of primary mesangioproliferative glomerulonephritis is made in less than 15% of renal biopsies. As an immune-mediated renal lesion with deposits of IgM, C1q, and C3, the clinical course is variable. Patients with isolated hematuria may have a very benign course, and those with heavy proteinuria occasionally progress to renal failure. There is little agreement on treatment, but some clinical reports suggest benefit from use of inhibitors of the renin-angiotensin system, steroid therapy, and even cytotoxic agents.
NEPHROTIC SYNDROME
Nephrotic syndrome classically presents with heavy proteinuria, minimal hematuria, hypoalbuminemia, hypercholesterolemia, edema, and hypertension. If left undiagnosed or untreated, some of these syndromes will progressively damage enough glomeruli to cause a fall in GFR, producing renal failure. Multiple studies have noted that the higher the 24-h urine protein excretion, the more rapid is the decline in GFR.
Therapies for various causes of nephrotic syndrome are noted under individual disease headings below. In general, all patients with hypercholesterolemia secondary to nephrotic syndrome should be treated with lipid-lowering agents because they are at increased risk for cardiovascular disease. Edema secondary to salt and water retention can be controlled with the judicious use of diuretics, avoiding intravascular volume depletion. Venous complications secondary to the hypercoagulable state associated with nephrotic syndrome can be treated with anticoagulants. The losses of various serum binding proteins, such as thyroid-binding globulin, lead to alterations in functional tests. Lastly, proteinuria itself is hypothesized to be nephrotoxic, and treatment of proteinuria with inhibitors of the renin-angiotensin system can lower urinary protein excretion.
MINIMAL CHANGE DISEASE
Minimal change disease (MCD), sometimes known as nil lesion, causes 70–90% of nephrotic syndrome in childhood but only 10–15% of nephrotic syndrome in adults. Minimal change disease usually presents as a primary renal disease but can be associated with several other conditions, including Hodgkin’s disease, allergies, or use of nonsteroidal anti-inflammatory agents; significant interstitial nephritis often accompanies cases associated with nonsteroidal drug use. Minimal change disease on renal biopsy shows no obvious glomerular lesion by light microscopy and is negative for deposits by immunofluorescent microscopy, or occasionally shows small amounts of IgM in the mesangium (see Fig. 62e-1). (See Glomerular Schematic 4.) Electron microscopy, however, consistently demonstrates an effacement of the foot process supporting the epithelial podocytes with weakening of slit-pore membranes. The pathophysiology of this lesion is uncertain. Most agree there is a circulating cytokine, perhaps related to a T cell response that alters capillary charge and podocyte integrity. The evidence for cytokine-related immune injury is circumstantial and is suggested by the presence of preceding allergies, altered cell-mediated immunity during viral infections, and the high frequency of remissions with steroids.
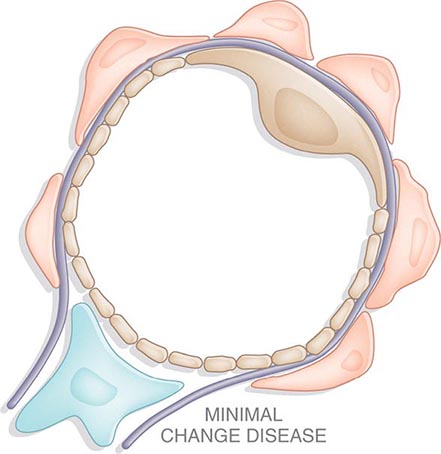
Minimal change disease presents clinically with the abrupt onset of edema and nephrotic syndrome accompanied by acellular urinary sediment. Average urine protein excretion reported in 24 h is 10 g with severe hypoalbuminemia. Less common clinical features include hypertension (30% in children, 50% in adults), microscopic hematuria (20% in children, 33% in adults), atopy or allergic symptoms (40% in children, 30% in adults), and decreased renal function (<5% in children, 30% in adults). The appearance of acute renal failure in adults is often seen more commonly in patients with low serum albumin and intrarenal edema (nephrosarca) that is responsive to intravenous albumin and diuretics. This presentation must be distinguished from acute renal failure secondary to hypovolemia. Acute tubular necrosis and interstitial inflammation are also reported. In children, the abnormal urine principally contains albumin with minimal amounts of higher-molecular-weight proteins, and is sometimes called selective proteinuria. Although up to 30% of children have a spontaneous remission, all children today are treated with steroids; only children who are nonresponders are biopsied in this setting. Primary responders are patients who have a complete remission (<0.2 mg/24 h of proteinuria) after a single course of prednisone; steroid-dependent patients relapse as their steroid dose is tapered. Frequent relapsers have two or more relapses in the 6 months following taper, and steroid-resistant patients fail to respond to steroid therapy. Adults are not considered steroid-resistant until after 4 months of therapy. Ninety to 95% of children will develop a complete remission after 8 weeks of steroid therapy, and 80–85% of adults will achieve complete remission, but only after a longer course of 20–24 weeks. Patients with steroid resistance may have FSGS on repeat biopsy. Some hypothesize that if the first renal biopsy does not have a sample of deeper corticomedullary glomeruli, then the correct diagnosis of FSGS may be missed.
Relapses occur in 70–75% of children after the first remission, and early relapse predicts multiple subsequent relapses, as do high levels of basal proteinuria. The frequency of relapses decreases after puberty. There is an increased risk of relapse following the rapid tapering of steroids in all groups. Relapses are less common in adults but are more resistant to subsequent therapy. Prednisone is first-line therapy, either given daily or on alternate days. Other immunosuppressive drugs, such as cyclophosphamide, chlorambucil, and mycophenolate mofetil, are saved for frequent relapsers, steroid-dependent patients, or steroid-resistant patients. Cyclosporine can induce remission, but relapse is also common when cyclosporine is withdrawn. The long-term prognosis in adults is less favorable when acute renal failure or steroid resistance occurs.
FOCAL SEGMENTAL GLOMERULOSCLEROSIS
Focal segmental glomerulosclerosis (FSGS) refers to a pattern of renal injury characterized by segmental glomerular scars that involve some but not all glomeruli; the clinical findings of FSGS largely manifest as proteinuria. When the secondary causes of FSGS are eliminated (Table 338-5), the remaining patients are considered to have primary FSGS. The incidence of this disease is increasing, and it now represents up to one-third of cases of nephrotic syndrome in adults and one-half of cases of nephrotic syndrome in African Americans, in whom it is seen more commonly. The pathogenesis of FSGS is probably multifactorial. Possible mechanisms include a T cell–mediated circulating permeability factor, increased soluble urokinase receptor levels, TGF-β–mediated cellular proliferation and matrix synthesis, and podocyte abnormalities associated with genetic mutations. Risk polymorphisms at the APOL1 locus encoding apolipoprotein L1 expressed in podocytes substantially explain the increased burden of FSGS among African Americans with or without HIV-associated disease.
|
FOCAL SEGMENTAL GLOMERULOSCLEROSIS |
The pathologic changes of FSGS are most prominent in glomeruli located at the corticomedullary junction (see Fig. 62e-2), so if the renal biopsy specimen is from superficial tissue, the lesions can be missed, which sometimes leads to a misdiagnosis of MCD. In addition to focal and segmental scarring, other variants have been described, including cellular lesions with endocapillary hypercellularity and heavy proteinuria; collapsing glomerulopath y (see Fig. 62e-3) with segmental or global glomerular collapse and a rapid decline in renal function; a hilar stalk lesion (see Fig. 62e-4); or the glomerular tip lesion (see Fig. 62e-5), which may have a better prognosis. (See Glomerular Schematic 5.)
FSGS can present with hematuria, hypertension, any level of proteinuria, or renal insufficiency. Nephrotic-range proteinuria, African-American race, and renal insufficiency are associated with a poor outcome, with 50% of patients reaching renal failure in 6–8 years. FSGS rarely remits spontaneously, but treatment-induced remission of proteinuria significantly improves prognosis. Treatment of patients with primary FSGS should include inhibitors of the renin-angiotensin system. Based on retrospective studies, patients with nephrotic-range proteinuria can be treated with steroids but respond far less often and after a longer course of therapy than patients with MCD. Proteinuria remits in only 20–45% of patients receiving a course of steroids over 6–9 months. Limited evidence suggests the use of cyclosporine in steroid-responsive patients helps ensure remissions. Relapse frequently occurs after cessation of cyclosporine therapy, and cyclosporine itself can lead to a deterioration of renal function due to its nephrotoxic effects. A role for other agents that suppress the immune system has not been established. Primary FSGS recurs in 25–40% of patients given allografts at end-stage disease, leading to graft loss in half of those cases. The treatment of secondary FSGS typically involves treating the underlying cause and controlling proteinuria. There is no role for steroids or other immunosuppressive agents in secondary FSGS.
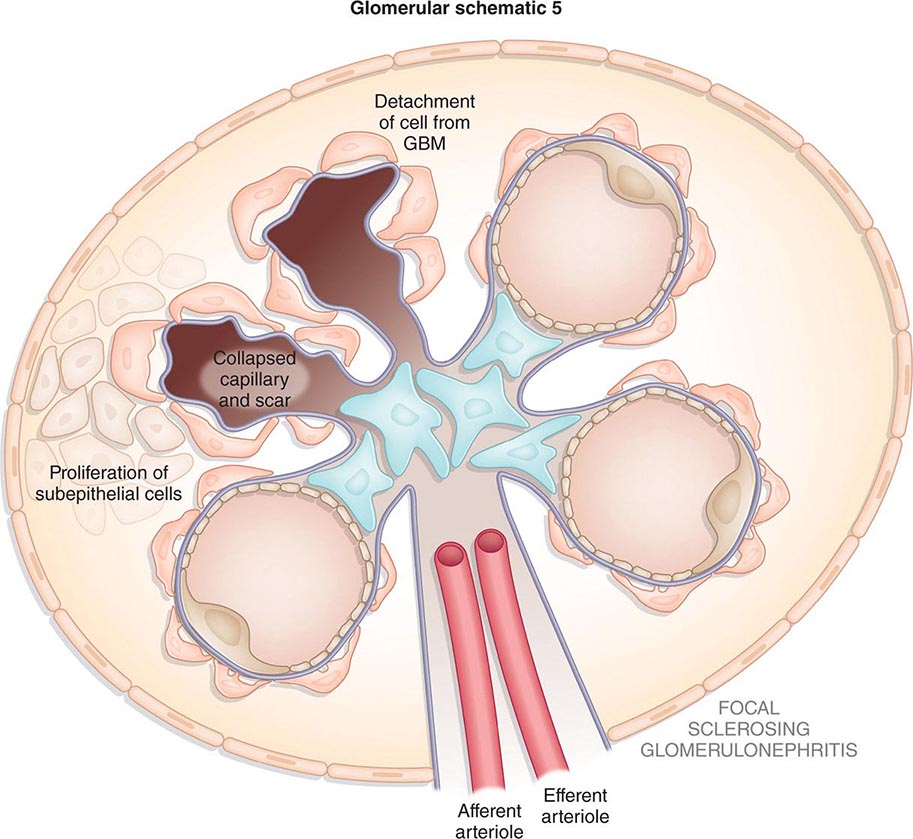
MEMBRANOUS GLOMERULONEPHRITIS
Membranous glomerulonephritis (MGN), or membranous nephropathy as it is sometimes called, accounts for approximately 30% of cases of nephrotic syndrome in adults, with a peak incidence between the ages of 30 and 50 years and a male to female ratio of 2:1. It is rare in childhood and the most common cause of nephrotic syndrome in the elderly. In 25–30% of cases, MGN is associated with a malignancy (solid tumors of the breast, lung, colon), infection (hepatitis B, malaria, schistosomiasis), or rheumatologic disorders like lupus or rarely rheumatoid arthritis (Table 338-6).
|
MEMBRANOUS GLOMERULONEPHRITIS |
Uniform thickening of the basement membrane along the peripheral capillary loops is seen by light microscopy on renal biopsy (see Fig. 62e-7); this thickening needs to be distinguished from that seen in diabetes and amyloidosis. (See Glomerular Schematic 6.) Immunofluorescence demonstrates diffuse granular deposits of IgG and C3, and electron microscopy typically reveals electron-dense subepithelial deposits. While different stages (I–V) of progressive membranous lesions have been described, some published analyses indicate the degree of tubular atrophy or interstitial fibrosis is more predictive of progression than is the stage of glomerular disease. The presence of subendothelial deposits or the presence of tubuloreticular inclusions strongly points to a diagnosis of membranous lupus nephritis, which may precede the extrarenal manifestations of lupus. Work in Heyman nephritis, an animal model of MGN, suggests that glomerular lesions result from in situ formation of immune complexes with megalin receptor–associated protein as the putative antigen. This antigen is not found in human podocytes. Human antibodies have been described against neutral endopeptidase expressed by podocytes in infants whose mothers lack this protein. In most adults, autoantibodies against the M-type phospholipase A2 receptor (PLA2R) circulate and bind to a conformational epitope present in the receptor on human podocytes, producing in situ deposits characteristic of idiopathic membranous nephropathy. Other renal diseases and secondary membranous nephropathy do not appear to involve such autoantibodies and levels of these autoantibodies have correlated with the severity of MGN. Eighty percent of patients with MGN present with nephrotic syndrome and nonselective proteinuria. Microscopic hematuria is seen but less commonly than in IgA nephropathy or FSGS. Spontaneous remissions occur in 20–33% of patients and often occur late in the course after years of nephrotic syndrome, which make treatment decisions difficult. One-third of patients continue to have relapsing nephrotic syndrome but maintain normal renal function, and approximately another third of patients develop renal failure or die from the complications of nephrotic syndrome. Male gender, older age, hypertension, and the persistence of proteinuria are associated with worse prognosis. Although thrombotic complications are a feature of all nephrotic syndromes, MGN has the highest reported incidences of renal vein thrombosis, pulmonary embolism, and deep vein thrombosis. Prophylactic anticoagulation is controversial but has been recommended for patients with severe or prolonged proteinuria in the absence of risk factors for bleeding.
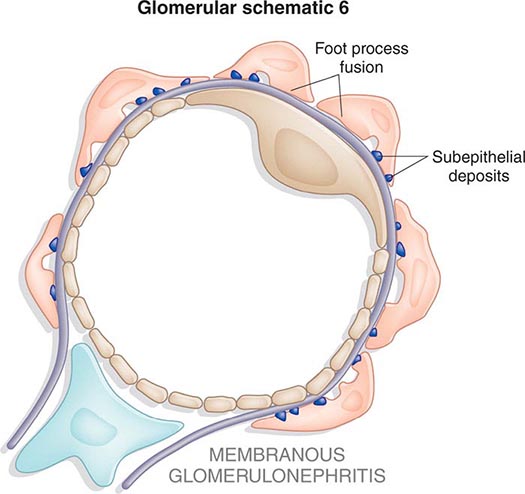
In addition to the treatment of edema, dyslipidemia, and hypertension, inhibition of the renin-angiotensin system is recommended. Therapy with immunosuppressive drugs is also recommended for patients with primary MGN and persistent proteinuria (>3.0 g/24 h). The choice of immunosuppressive drugs for therapy is controversial, but current recommendations are to treat with steroids and cyclophosphamide, chlorambucil, mycophenolate mofetil, or cyclosporine. In patients who relapse or fail to respond to this therapy, the use of rituximab, an anti-CD20 antibody directed at B cells, or synthetic adrenocorticotropic hormone may be considered.
DIABETIC NEPHROPATHY
Diabetic nephropathy is the single most common cause of chronic renal failure in the United States, accounting for 45% of patients receiving renal replacement therapy, and is a rapidly growing problem worldwide. The dramatic increase in the number of patients with diabetic nephropathy reflects the epidemic increase in obesity, metabolic syndrome, and type 2 diabetes mellitus. Approximately 40% of patients with types 1 or 2 diabetes develop nephropathy, but due to the higher prevalence of type 2 diabetes (90%) compared to type 1 (10%), the majority of patients with diabetic nephropathy have type 2 disease. Renal lesions are more common in African-American, Native American, Polynesian, and Maori populations. Risk factors for the development of diabetic nephropathy include hyperglycemia, hypertension, dyslipidemia, smoking, a family history of diabetic nephropathy, and gene polymorphisms affecting the activity of the renin-angiotensin-aldosterone axis.
Within 1–2 years after the onset of clinical diabetes, morphologic changes appear in the kidney. Thickening of the GBM is a sensitive indicator for the presence of diabetes but correlates poorly with the presence or absence of clinically significant nephropathy. The composition of the GBM is altered notably with a loss of heparan sulfate moieties that form the negatively charged filtration barrier. This change results in increased filtration of serum proteins into the urine, predominately negatively charged albumin. The expansion of the mesangium due to the accumulation of extracellular matrix correlates with the clinical manifestations of diabetic nephropathy (see stages in Fig. 62e-20). This expansion in mesangial matrix is associated with the development of mesangial sclerosis. Some patients also develop eosinophilic, PAS+ nodules called nodular glomerulosclerosis or Kimmelstiel-Wilson nodules. Immunofluorescence microscopy often reveals the nonspecific deposition of IgG (at times in a linear pattern) or complement staining without immune deposits on electron microscopy. Prominent vascular changes are frequently seen with hyaline and hypertensive arteriosclerosis. This is associated with varying degrees of chronic glomerulosclerosis and tubulointerstitial changes. Renal biopsies from patients with types 1 and 2 diabetes are largely indistinguishable.
These pathologic changes are the result of a number of postulated factors. Multiple lines of evidence support an important role for increases in glomerular capillary pressure (intraglomerular hypertension) in alterations in renal structure and function. Direct effects of hyperglycemia on the actin cytoskeleton of renal mesangial and vascular smooth-muscle cells as well as diabetes-associated changes in circulating factors such as atrial natriuretic factor, angiotensin II, and insulin-like growth factor (IGF) may account for this. Sustained glomerular hypertension increases matrix production, alterations in the GBM with disruption in the filtration barrier (and hence proteinuria), and glomerulosclerosis. A number of factors have also been identified that alter matrix production, including the accumulation of advanced glycosylation end products, circulating factors including growth hormone, IGF-I, angiotensin II, connective tissue growth factor, TGF-β, and dyslipidemia.
The natural history of diabetic nephropathy in patients with types 1 and 2 diabetes is similar. However, since the onset of type 1 diabetes is readily identifiable and the onset of type 2 diabetes is not, a patient newly diagnosed with type 2 diabetes may present with advanced diabetic nephropathy. At the onset of diabetes, renal hypertrophy and glomerular hyperfiltration are present. The degree of glomerular hyperfiltration correlates with the subsequent risk of clinically significant nephropathy. In the approximately 40% of patients with diabetes who develop diabetic nephropathy, the earliest manifestation is an increase in albuminuria detected by sensitive radioimmunoassay (Table 338-1). Albuminuria in the range of 30–300 mg/24 h is called microalbuminuria. Microalbuminuria appears 5–10 years after the onset of diabetes. It is currently recommended to test patients with type 1 disease for microalbuminuria 5 years after diagnosis of diabetes and yearly thereafter and, because the time of onset of type 2 diabetes is often unknown, to test type 2 patients at the time of diagnosis of diabetes and yearly thereafter.
Patients with small increases in albuminuria increase their levels of urinary albumin excretion, typically reaching dipstick positive levels of proteinuria (>300 mg albuminuria) 5–10 years after the onset of early albuminuria. Microalbuminuria is a potent risk factor for cardiovascular events and death in patients with type 2 diabetes. Many patients with type 2 diabetes and microalbuminuria succumb to cardiovascular events before they progress to proteinuria or renal failure. Proteinuria in frank diabetic nephropathy can be variable, ranging from 500 mg to 25 g/24 h, and is often associated with nephrotic syndrome. More than 90% of patients with type 1 diabetes and nephropathy have diabetic retinopathy, so the absence of retinopathy in type 1 patients with proteinuria should prompt consideration of a diagnosis other than diabetic nephropathy; only 60% of patients with type 2 diabetes with nephropathy have diabetic retinopathy. There is a significant correlation between the presence of retinopathy and the presence of Kimmelstiel-Wilson nodules (see Fig. 62e-20). Also, characteristically, patients with advanced diabetic nephropathy have normal to enlarged kidneys, in contrast to other glomerular diseases where kidney size is usually decreased. Using the above epidemiologic and clinical data, and in the absence of other clinical or serologic data suggesting another disease, diabetic nephropathy is usually diagnosed without a renal biopsy. After the onset of proteinuria, renal function inexorably declines, with 50% of patients reaching renal failure over another 5–10 years; thus, from the earliest stages of microalbuminuria, it usually takes 10–20 years to reach end-stage renal disease. Once renal failure appears, however, survival on dialysis is shorter for patients with diabetes compared to other dialysis patients. Survival is best for patients with type 1 diabetes who receive a transplant from a living related donor.
Good evidence supports the benefits of blood sugar and blood pressure control as well as inhibition of the renin-angiotensin system in retarding the progression of diabetic nephropathy. In patients with type 1 diabetes, intensive control of blood sugar clearly prevents the development or progression of diabetic nephropathy. The evidence for benefit of intensive blood glucose control in patients with type 2 diabetes is less certain, with current studies reporting conflicting results.
Controlling systemic blood pressure decreases renal and cardiovascular adverse events in this high-risk population. The vast majority of patients with diabetic nephropathy require three or more antihypertensive drugs to achieve this goal. Drugs that inhibit the renin-angiotensin system, independent of their effects on systemic blood pressure, have been shown in numerous large clinical trials to slow the progression of diabetic nephropathy at early (microalbuminuria) and late (proteinuria with reduced glomerular filtration) stages, independent of any effect they may have on systemic blood pressure. Since angiotensin II increases efferent arteriolar resistance and, hence, glomerular capillary pressure, one key mechanism for the efficacy of ACE inhibitors or angiotensin receptor blockers (ARBs) is reducing glomerular hypertension. Patients with type 1 diabetes for 5 years who develop albuminuria or declining renal function should be treated with ACE inhibitors. Patients with type 2 diabetes and microalbuminuria or proteinuria may be treated with ACE inhibitors or ARBs. Evidence suggests increased risk for cardiovascular adverse events in some patients with a combination of two drugs (ACE inhibitors, ARBs, renin inhibitors, or aldosterone antagonists) that suppress several components of the renin-angiotensin system.
GLOMERULAR DEPOSITION DISEASES
Plasma cell dyscrasias producing excess light chain immunoglobulin sometimes lead to the formation of glomerular and tubular deposits that cause heavy proteinuria and renal failure; the same is true for the accumulation of serum amyloid A protein fragments seen in several inflammatory diseases. This broad group of proteinuric patients has glomerular deposition disease.
Light Chain Deposition Disease The biochemical characteristics of nephrotoxic light chains produced in patients with light chain malignancies often confer a specific pattern of renal injury; that of either cast nephropathy (see Fig. 62e-17), which causes renal failure but not heavy proteinuria or amyloidosis, or light chain deposition disease (see Fig. 62e-16), which produces nephrotic syndrome with renal failure. These latter patients produce kappa light chains that do not have the biochemical features necessary to form amyloid fibrils. Instead, they self-aggregate and form granular deposits along the glomerular capillary and mesangium, tubular basement membrane, and Bowman’s capsule. When predominant in glomeruli, nephrotic syndrome develops, and about 70% of patients progress to dialysis. Light-chain deposits are not fibrillar and do not stain with Congo red, but they are easily detected with anti–light chain antibody using immunofluorescence or as granular deposits on electron microscopy. A combination of the light chain rearrangement, self-aggregating properties at neutral pH, and abnormal metabolism probably contribute to the deposition. Treatment for light chain deposition disease is treatment of the primary disease and, if possible, autologous stem cell transplantation.
Renal Amyloidosis Most renal amyloidosis is either the result of primary fibrillar deposits of immunoglobulin light chains known as amyloid L (AL), or secondary to fibrillar deposits of serum amyloid A (AA) protein fragments (Chap. 137). Even though both occur for different reasons, their clinicopathophysiology is quite similar and will be discussed together. Amyloid infiltrates the liver, heart, peripheral nerves, carpal tunnel, upper pharynx, and kidney, producing restrictive cardiomyopathy, hepatomegaly, macroglossia, and heavy proteinuria sometimes associated with renal vein thrombosis. In systemic AL amyloidosis, also called primary amyloidosis, light chains produced in excess by clonal plasma cell dyscrasias are made into fragments by macrophages so they can self-aggregate at acid pH. A disproportionate number of these light chains (75%) are of the lambda class. About 10% of these patients have overt myeloma with lytic bone lesions and infiltration of the bone marrow with >30% plasma cells; nephrotic syndrome is common, and about 20% of patients progress to dialysis. AA amyloidosis is sometimes called secondary amyloidosis and also presents as nephrotic syndrome. It is due to deposition of β-pleated sheets of serum amyloid A protein, an acute phase reactant whose physiologic functions include cholesterol transport, immune cell attraction, and metalloproteases activation. Forty percent of patients with AA amyloid have rheumatoid arthritis, and another 10% have ankylosing spondylitis or psoriatic arthritis; the rest derive from other lesser causes. Less common in Western countries but more common in Mediterranean regions, particularly in Sephardic and Iraqi Jews, is familial Mediterranean fever (FMF). FMF is caused by a mutation in the gene encoding pyrin, whereas Muckle-Wells syndrome, a related disorder, results from a mutation in cryopyrin; both proteins are important in the apoptosis of leukocytes early in inflammation; such proteins with pyrin domains are part of a new pathway called the inflammasome. Receptor mutations in tumor necrosis factor receptor 1 (TNFR1)-associated periodic syndrome also produce chronic inflammation and secondary amyloidosis. Fragments of serum amyloid A protein increase and self-aggregate by attaching to receptors for advanced glycation end products in the extracellular environment; nephrotic syndrome is common, and about 40–60% of patients progress to dialysis. AA and AL amyloid fibrils are detectable with Congo red or in more detail with electron microscopy (see Fig. 62e-15). Currently developed serum free light chain nephelometry assays are useful in the early diagnosis and follow-up of disease progression. Biopsy of involved liver or kidney is diagnostic 90% of the time when the pretest probability is high; abdominal fat pad aspirates are positive about 70% of the time, but apparently less so when looking for AA amyloid. Amyloid deposits are distributed along blood vessels and in the mesangial regions of the kidney. The treatment for primary amyloidosis, melphalan and autologous hematopoietic stem cell transplantation, can delay the course of disease in about 30% of patients. Secondary amyloidosis is also relentless unless the primary disease can be controlled. Some new drugs in development that disrupt the formation of fibrils may be available in the future.
Fibrillary-Immunotactoid Glomerulopathy Fibrillary-immunotactoid glomerulopathy is a rare (<1.0% of renal biopsies), morphologically defined disease characterized by glomerular accumulation of nonbranching randomly arranged fibrils. Some classify amyloid and nonamyloid fibril-associated renal diseases all as fibrillary glomerulopathies with immunotactoid glomerulopathy reserved for nonamyloid fibrillary disease not associated with a systemic illness. Others define fibrillary glomerulonephritis as a nonamyloid fibrillary disease with fibrils 12–24 nm and immunotactoid glomerulonephritis with fibrils >30 nm. In either case, fibrillar/microtubular deposits of oligoclonal or oligotypic immunoglobulins and complement appear in the mesangium and along the glomerular capillary wall. Congo red stains are negative. The cause of this “nonamyloid” glomerulopathy is mostly idiopathic; reports of immunotactoid glomerulonephritis describe an occasional association with chronic lymphocytic leukemia or B cell lymphoma. Both disorders appear in adults in the fourth decade with moderate to heavy proteinuria, hematuria, and a wide variety of histologic lesions, including DPGN, MPGN, MGN, or mesangioproliferative glomerulonephritis. Nearly half of patients develop renal failure over a few years. There is no consensus on treatment of this uncommon disorder. The disease has been reported to recur following renal transplantation in a minority of cases.
FABRY’S DISEASE
Fabry’s disease is an X-linked inborn error of globotriaosylceramide metabolism secondary to deficient lysosomal α-galactosidase A activity, resulting in excessive intracellular storage of globotriaosylceramide. Affected organs include the vascular endothelium, heart, brain, and kidneys. Classically, Fabry’s disease presents in childhood in males with acroparesthesias, angiokeratoma, and hypohidrosis. Over time male patients develop cardiomyopathy, cerebrovascular disease, and renal injury, with an average age of death around 50 years of age. Hemizygotes with hypomorphic mutations sometimes present in the fourth to sixth decade with single-organ involvement. Rarely, dominant-negative α-galactosidase A mutations or female heterozygotes with unfavorable × inactivation present with mild single-organ involvement. Rare females develop severe manifestations including renal failure but do so later in life than males. Renal biopsy reveals enlarged glomerular visceral epithelial cells packed with small clear vacuoles containing globotriaosylceramide; vacuoles may also be found in parietal and tubular epithelia (see Fig. 62e-18). These vacuoles of electron-dense materials in parallel arrays (zebra bodies) are easily seen on electron microscopy. Ultimately, renal biopsies reveal FSGS. The nephropathy of Fabry’s disease typically presents in the third decade as mild to moderate proteinuria, sometimes with microscopic hematuria or nephrotic syndrome. Urinalysis may reveal oval fat bodies and birefringent glycolipid globules under polarized light (Maltese cross). Renal biopsy is necessary for definitive diagnosis. Progression to renal failure occurs by the fourth or fifth decade. Treatment with inhibitors of the renin-angiotensin system is recommended. Treatment with recombinant α-galactosidase A clears microvascular endothelial deposits of globotriaosylceramide from the kidneys, heart, and skin. In patients with advanced organ involvement, progression of disease occurs despite enzyme replacement therapy. Variable responses to enzyme therapy may be due to the occurrence of neutralizing antibodies or differences in uptake of the enzyme. Graft and patient survival following renal transplantation in patients with Fabry’s are similar to other causes of end-stage renal disease.
PULMONARY-RENAL SYNDROMES
Several diseases can present with catastrophic hemoptysis and glomerulonephritis associated with varying degrees of renal failure. The usual causes include Goodpasture’s syndrome, granulomatosis with polyangiitis, microscopic polyangiitis, Churg-Strauss vasculitis, and, rarely, Henoch-Schönlein purpura or cryoglobulinemia. Each of these diseases can also present without hemoptysis and are discussed in detail earlier in “Acute Nephritic Syndromes.” (See Glomerular Schematic 7.) Pulmonary bleeding in this setting is life-threatening and often results in airway intubation, and acute renal failure requires dialysis. Diagnosis is difficult initially because biopsies and serologic testing take time. Treatment with plasmapheresis and methylprednisolone is often empirical and temporizing until results of testing are available.
BASEMENT MEMBRANE SYNDROMES
All kidney epithelia, including podocytes, rest on basement membranes assembled into a planar surface through the interweaving of collagen IV with laminins, nidogen, and sulfated proteoglycans. Structural abnormalities in GBM associated with hematuria are characteristic of several familial disorders related to the expression of collagen IV genes. The extended family of collagen IV contains six chains, which are expressed in different tissues at different stages of embryonic development. All epithelial basement membranes early in human development are composed of interconnected triple-helical protomers rich in α1.α1.α2(IV) collagen. Some specialized tissues undergo a developmental switch replacing α1.α1.α2(IV) protomers with an α3.α4.α5(IV) collagen network; this switch occurs in the kidney (glomerular and tubular basement membrane), lung, testis, cochlea, and eye, while an α5.α5.α6(IV) network appears in skin, smooth muscle, and esophagus and along Bowman’s capsule in the kidney. This switch probably occurs because the α3.α4.α5(IV) network is more resistant to proteases and ensures the structural longevity of critical tissues. When basement membranes are the target of glomerular disease, they produce moderate proteinuria, some hematuria, and progressive renal failure.
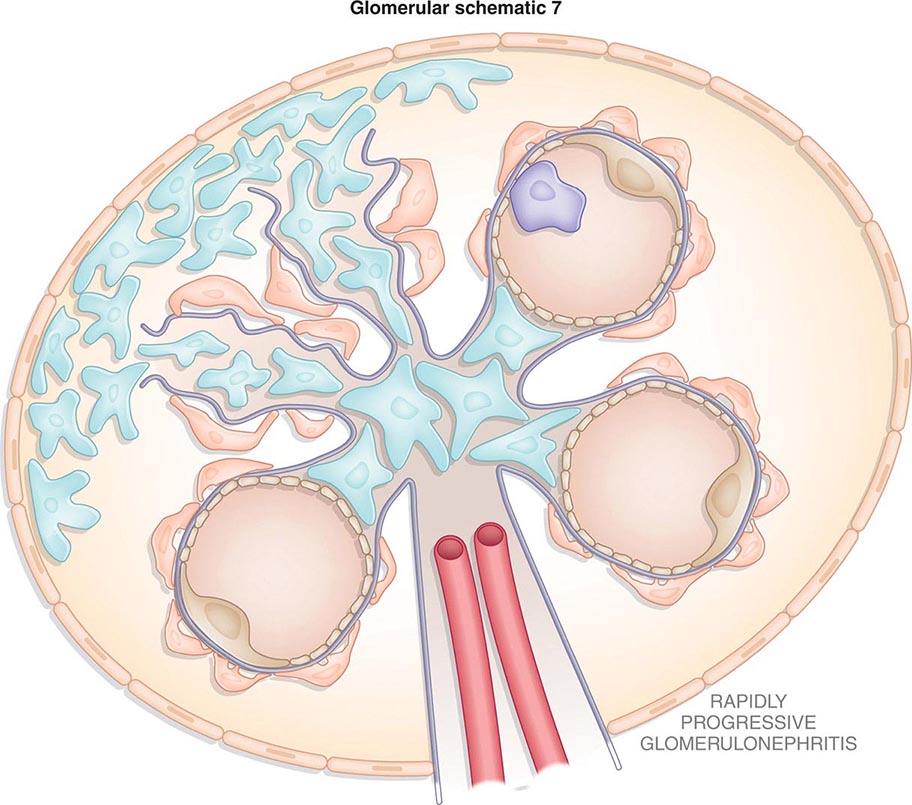
ANTI-GBM DISEASE
Autoimmune disease where antibodies are directed against the α3 NC1 domain of collagen IV produces an anti-GBM disease often associated with RPGN and/or a pulmonary-renal syndrome called Goodpasture’s syndrome. Discussion of this disease is covered earlier in “Acute Nephritic Syndromes.”
ALPORT’S SYNDROME
Classically, patients with Alport’s syndrome develop hematuria, thinning and splitting of the GBMs, mild proteinuria (<1–2 g/24 h), which appears late in the course, followed by chronic glomerulosclerosis leading to renal failure in association with sensorineural deafness. Some patients develop lenticonus of the anterior lens capsule, “dot and fleck” retinopathy, and rarely, mental retardation or leiomyomatosis. Approximately 85% of patients with Alport’s syndrome have an X-linked inheritance of mutations in the α5(IV) collagen chain on chromosome Xq22–24. Female carriers have variable penetrance depending on the type of mutation or the degree of mosaicism created by × inactivation. Fifteen percent of patients have autosomal recessive disease of the α3(IV) or α4(IV) chains on chromosome 2q35–37. Rarely, some kindred have an autosomal dominant inheritance of dominant-negative mutations in α3(IV) or α4(IV) chains.
Pedigrees with the X-linked syndrome are quite variable in their rate and frequency of tissue damage leading to organ failure. Seventy percent of patients have the juvenile form with nonsense or missense mutations, reading frame shifts, or large deletions and generally develop renal failure and sensorineural deafness by age 30. Patients with splice variants, exon skipping, or missense mutations of α-helical glycines generally deteriorate after the age of 30 (adult form) with mild or late deafness. Early severe deafness, lenticonus, or proteinuria suggests a poorer prognosis. Usually females from X-linked pedigrees have only microhematuria, but up to 25% of carrier females have been reported to have more severe renal manifestations. Pedigrees with the autosomal recessive form of the disease have severe early disease in both females and males with asymptomatic parents.
Clinical evaluation should include a careful eye examination and hearing tests. However, the absence of extrarenal symptoms does not rule out the diagnosis. Since α5(IV) collagen is expressed in the skin, some X-linked Alport’s patients can be diagnosed with a skin biopsy revealing the lack of the α5(IV) collagen chain on immunofluorescent analysis. Patients with mutations in α3(IV) or α4(IV) require a renal biopsy. Genetic testing can be used for the diagnosis of Alport’s syndrome and the demonstration of the mode of inheritance. Early in their disease, Alport’s patients typically have thin basement membranes on renal biopsy (see Fig. 62e-19), which thicken over time into multilamellations surrounding lucent areas that often contain granules of varying density—the so-called split basement membrane. In any Alport’s kidney, there are areas of thinning mixed with splitting of the GBM. Tubules drop out, glomeruli scar, and the kidney eventually succumbs to interstitial fibrosis. All affected members of a family with X-linked Alport’s syndrome should be identified and followed, including mothers of affected males. Primary treatment is control of systemic hypertension and use of ACE inhibitors to slow renal progression. Although patients who receive renal allografts usually develop anti-GBM antibodies directed toward the collagen epitopes absent in their native kidney, overt Goodpasture’s syndrome is rare and graft survival is good.
THIN BASEMENT MEMBRANE DISEASE
Thin basement membrane disease (TBMD) characterized by persistent or recurrent hematuria is not typically associated with proteinuria, hypertension, or loss of renal function or extrarenal disease. Although not all cases are familial (perhaps a founder effect), it usually presents in childhood in multiple family members and is also called benign familial hematuria. Cases of TBMD have genetic defects in type IV collagen but in contrast to Alport behave as an autosomal dominant disorder that in ~40% of families segregates with the COL(IV) α3/COL(IV) α4 loci. Mutations in these loci can result in a spectrum of disease ranging from TBMD to autosomal dominant or recessive Alport’s. The GBM shows diffuse thinning compared to normal values for the patient’s age in otherwise normal biopsies (see Fig. 62e-19). The vast majority of patients have a benign course.
NAIL-PATELLA SYNDROME
Patients with nail-patella syndrome develop iliac horns on the pelvis and dysplasia of the dorsal limbs involving the patella, elbows, and nails, variably associated with neural-sensory hearing impairment, glaucoma, and abnormalities of the GBM and podocytes, leading to hematuria, proteinuria, and FSGS. The syndrome is autosomal dominant, with haploinsufficiency for the LIM homeodomain transcription factor LMX1B; pedigrees are extremely variable in the penetrance for all features of the disease. LMX1B regulates the expression of genes encoding α3 and α4 chains of collagen IV, interstitial type III collagen, podocin, and CD2AP that help form the slit-pore membranes connecting podocytes. Mutations in the LIM domain region of LMX1B associate with glomerulopathy, and renal failure appears in as many as 30% of patients. Proteinuria or isolated hematuria is discovered throughout life, but usually by the third decade, and is inexplicably more common in females. On renal biopsy there is lucent damage to the lamina densa of the GBM, an increase in collagen III fibrils along glomerular capillaries and in the mesangium, and damage to the slit-pore membrane, producing heavy proteinuria not unlike that seen in congenital nephrotic syndrome. Patients with renal failure do well with transplantation.
GLOMERULAR-VASCULAR SYNDROMES
A variety of diseases result in classic vascular injury to the glomerular capillaries. Most of these processes also damage blood vessels elsewhere in the body. The group of diseases discussed here lead to vasculitis, renal endothelial injury, thrombosis, ischemia, and/or lipid-based occlusions.
ATHEROSCLEROTIC NEPHROPATHY
Aging in the developed world is commonly associated with the occlusion of coronary and systemic blood vessels. The reasons for this include obesity, insulin resistance, smoking, hypertension, and diets rich in lipids that deposit in the arterial and arteriolar circulation, producing local inflammation and fibrosis of small blood vessels. When the renal arterial circulation is involved, the glomerular microcirculation is damaged, leading to chronic nephrosclerosis. Patients with GFRs <60 mL/min have more cardiovascular events and hospitalizations than those with higher filtration rates. Several aggressive lipid disorders can accelerate this process, but most of the time atherosclerotic progression to chronic nephrosclerosis is associated with poorly controlled hypertension. Approximately 10% of glomeruli are normally sclerotic by age 40, rising to 20% by age 60 and 30% by age 80. Serum lipid profiles in humans are greatly affected by apolipoprotein E polymorphisms; the E4 allele is accompanied by increases in serum cholesterol and is more closely associated with atherogenic profiles in patients with renal failure. Mutations in E2 alleles, particularly in Japanese patients, produce a specific renal abnormality called lipoprotein glomerulopathy associated with glomerular lipoprotein thrombi and capillary dilation.
HYPERTENSIVE NEPHROSCLEROSIS
Uncontrolled systemic hypertension causes permanent damage to the kidneys in about 6% of patients with elevated blood pressure. As many as 27% of patients with end-stage kidney disease have hypertension as a primary cause. Although there is not a clear correlation between the extent or duration of hypertension and the risk of end-organ damage, hypertensive nephrosclerosis is fivefold more frequent in African Americans than whites. Risk alleles associated with APOL1, a functional gene for apolipoprotein L1 expressed in podocytes substantially explains the increased burden of end-stage renal disease among African Americans. Associated risk factors for progression to end-stage kidney disease include increased age, male gender, race, smoking, hypercholesterolemia, duration of hypertension, low birth weight, and preexisting renal injury. Kidney biopsies in patients with hypertension, microhematuria, and moderate proteinuria demonstrate arteriolosclerosis, chronic nephrosclerosis, and interstitial fibrosis in the absence of immune deposits (see Fig. 62e-21). Today, based on a careful history, physical examination, urinalysis, and some serologic testing, the diagnosis of chronic nephrosclerosis is usually inferred without a biopsy. Treating hypertension is the best way to avoid progressive renal failure; most guidelines recommend lowering blood pressure to <130/80 mmHg if there is preexisting diabetes or kidney disease. In the presence of kidney disease, most patients begin antihypertensive therapy with two drugs, classically a thiazide diuretic and an ACE inhibitor; most will require three drugs. There is strong evidence in African Americans with hypertensive nephrosclerosis that therapy initiated with an ACE inhibitor can slow the rate of decline in renal function independent of effects on systemic blood pressure. Malignant acceleration of hypertension complicates the course of chronic nephrosclerosis, particularly in the setting of scleroderma or cocaine use (see Fig. 62e-24). The hemodynamic stress of malignant hypertension leads to fibrinoid necrosis of small blood vessels, thrombotic microangiography, a nephritic urinalysis, and acute renal failure. In the setting of renal failure, chest pain, or papilledema, the condition is treated as a hypertensive emergency. Slightly lowering the blood pressure often produces an immediate reduction in GFR that improves as the vascular injury attenuates and autoregulation of blood vessel tone is restored.
CHOLESTEROL EMBOLI
Aging patients with clinical complications from atherosclerosis sometimes shower cholesterol crystals into the circulation—either spontaneously or, more commonly, following an endovascular procedure with manipulation of the aorta—or with use of systemic anticoagulation. Spontaneous emboli may shower acutely or shower subacutely and somewhat more silently. Irregular emboli trapped in the microcirculation produce ischemic damage that induces an inflammatory reaction. Depending on the location of the atherosclerotic plaques releasing these cholesterol fragments, one may see cerebral transient ischemic attacks; livedo reticularis in the lower extremities; Hollenhorst plaques in the retina with visual field cuts; necrosis of the toes; and acute glomerular capillary injury leading to focal segmental glomerulosclerosis sometimes associated with hematuria, mild proteinuria, and loss of renal function, which typically progresses over a few years. Occasional patients have fever, eosinophilia, or eosinophiluria. A skin biopsy of an involved area may be diagnostic. Since tissue fixation dissolves the cholesterol, one typically sees only residual, biconvex clefts in involved vessels (see Fig. 62e-22). There is no therapy to reverse embolic occlusions, and steroids do not help. Controlling blood pressure and lipids and cessation of smoking are usually recommended for prevention.
SICKLE CELL DISEASE
Although individuals with SA-hemoglobin are usually asymptomatic, most will gradually develop hyposthenuria due to subclinical infarction of the renal medulla, thus predisposing them to volume depletion. There is an unexpectedly high prevalence of sickle trait among dialysis patients who are African American. Patients with homozygous SS-sickle cell disease develop chronic vasoocclusive disease in many organs. Polymers of deoxygenated SS-hemoglobin distort the shape of red blood cells. These cells attach to endothelia and obstruct small blood vessels, producing frequent and painful sickle cell crises over time. Vessel occlusions in the kidney produce glomerular hypertension, FSGS, interstitial nephritis, and renal infarction associated with hyposthenuria, microscopic hematuria, and even gross hematuria; some patients also present with MPGN. Renal function can be overestimated due to the increased tubular secretion of creatinine seen in many patients with SS-sickle cell. By the second or third decade of life, persistent vasoocclusive disease in the kidney leads to varying degrees of renal failure, and some patients end up on dialysis. Treatment is directed to reducing the frequency of painful crises and administering ACE inhibitors in the hope of delaying a progressive decline in renal function. In sickle cell patients undergoing renal transplantation, renal graft survival is comparable to African Americans in the general transplant population.
THROMBOTIC MICROANGIOPATHIES
Thrombotic thrombocytopenic purpura (TTP) and hemolytic-uremic syndrome (HUS) represent a spectrum of thrombotic microangiopathies. Thrombotic thrombocytopenic purpura and hemolytic-uremic syndrome share the general features of idiopathic thrombocytopenic purpura, hemolytic anemia, fever, renal failure, and neurologic disturbances. When patients, particularly children, have more evidence of renal injury, their condition tends to be called HUS. In adults with neurologic disease, it is considered to be TTP. In adults there is often a mixture of both, which is why they are often referred to as having TTP/HUS. On examination of kidney tissue, there is evidence of glomerular capillary endotheliosis associated with platelet thrombi, damage to the capillary wall, and formation of fibrin material in and around glomeruli (see Fig. 62e-23). These tissue findings are similar to what is seen in preeclampsia/HELLP (hemolysis, elevated liver enzymes, and low platelet count syndrome), malignant hypertension, and the antiphospholipid syndrome. TTP/HUS is also seen in pregnancy; with the use of oral contraceptives or quinine; in renal transplant patients given OKT3 for rejection; in patients taking the calcineurin inhibitors, cyclosporine and tacrolimus, or in patients taking the antiplatelet agents, ticlopidine and clopidogrel; or following HIV infection.
Although there is no agreement on how much they share a final common pathophysiology, two general groups of patients are recognized: childhood HUS associated with enterohemorrhagic diarrhea and TTP/HUS in adults. Childhood HUS is caused by a toxin released by Escherichia coli 0157:H7 and occasionally by Shigella dysenteriae. This shiga toxin (verotoxin) directly injures endothelia, enterocytes, and renal cells, causing apoptosis, platelet clumping, and intravascular hemolysis by binding to the glycolipid receptors (Gb3). These receptors are more abundant along endothelia in children compared to adults. Shiga toxin also inhibits the endothelial production of ADAMTS13. In familial cases of adult TTP/HUS, there is a genetic deficiency of the ADAMTS13 metalloprotease that cleaves large multimers of von Willebrand’s factor. Absent ADAMTS13, these large multimers cause platelet clumping and intravascular hemolysis. An antibody to ADAMTS13 is found in many sporadic cases of adult TTP/HUS, but not all; many patients also have antibodies to the thrombospondin receptor on selected endothelial cells in small vessels or increased levels of plasminogen-activator inhibitor 1 (PAI-1). Some children with complement protein deficiencies express atypical HUS (aHUS), which can be treated with liver transplant. The treatment of adult TTP/HUS is daily plasmapheresis, which can be lifesaving. Plasmapheresis is given until the platelet count rises, but in relapsing patients it normally is continued well after the platelet count improves, and in resistant patients twice-daily exchange may be helpful. Most patients respond within 2 weeks of daily plasmapheresis. Since TTP/HUS often has an autoimmune basis, there is an anecdotal role in relapsing patients for using splenectomy, steroids, immunosuppressive drugs, bortezomib, or rituximab, an anti-CD20 antibody. Patients with childhood HUS from infectious diarrhea are not given antibiotics, because antibiotics are thought to accelerate the release of the toxin and the diarrhea is usually self-limited. No intervention appears superior to supportive therapy in children with postdiarrheal HUS.
ANTIPHOSPHOLIPID ANTIBODY SYNDROME (SEE CHAP. 379)
GLOBAL CONSIDERATIONS
INFECTIOUS DISEASE–ASSOCIATED SYNDROMES
![]() A number of infectious diseases will injure the glomerular capillaries as part of a systemic reaction producing an immune response or from direct infection of renal tissue. Evidence of this immune response is collected by glomeruli in the form of immune deposits that damage the kidney, producing moderate proteinuria and hematuria. A high prevalence of many of these infectious diseases in undeveloped countries results in infection-associated renal disease being the most common cause of glomerulonephritis in many parts of the world.
A number of infectious diseases will injure the glomerular capillaries as part of a systemic reaction producing an immune response or from direct infection of renal tissue. Evidence of this immune response is collected by glomeruli in the form of immune deposits that damage the kidney, producing moderate proteinuria and hematuria. A high prevalence of many of these infectious diseases in undeveloped countries results in infection-associated renal disease being the most common cause of glomerulonephritis in many parts of the world.
Poststreptococcal Glomerulonephritis This form of glomerulonephritis is one of the classic complications of streptococcal infection. The discussion of this disease can be found earlier, in the section “Acute Nephritic Syndromes.”
Subacute Bacterial Endocarditis Renal injury from persistent bacteremia absent the continued presence of a foreign body, regardless of cause, is treated presumptively as if the patient has endocarditis. The discussion of this disease can be found earlier, in the section “Acute Nephritic Syndromes.”
Human Immunodeficiency Virus Renal disease is an important complication of HIV disease. The risk of development of end-stage renal disease is much higher in HIV-infected African Americans than in HIV-infected whites. About 50% of HIV-infected patients with kidney disease have HIV-associated nephropathy (HIVAN) on biopsy. The lesion in HIVAN is FSGS, characteristically revealing a collapsing glomerulopathy (see Fig. 62e-3) with visceral epithelial cell swelling, microcystic dilatation of renal tubules, and tubuloreticular inclusion. Renal epithelial cells express replicating HIV virus, but host immune responses also play a role in the pathogenesis. MPGN and DPGN have also been reported but more commonly in HIV-infected whites and in patients coinfected with hepatitis B or C. HIV-associated TTP has also been reported. Other renal lesions include DPGN, IgA nephropathy, and MCD. Renal biopsy may be indicated to distinguish between these lesions.
HIV patients with FSGS typically present with nephrotic-range proteinuria and hypoalbuminemia, but unlike patients with other etiologies for nephrotic syndrome, they do not commonly have hypertension, edema, or hyperlipidemia. Renal ultrasound also reveals large, echogenic kidneys despite the finding that renal function in some patients declines rapidly. Treatment with inhibitors of the renin-angiotensin system decreases the proteinuria. Effective antiretroviral therapy benefits both the patient and the kidney and improves survival of HIV-infected patients with chronic kidney disease (CKD) or end-stage renal disease. In HIV-infected patients not yet on therapy, the presence of HIVAN is an indication to initiate therapy. Following the introduction of antiretroviral therapy, survival on dialysis for the HIV-infected patient has improved dramatically. Renal transplantations in HIV-infected patients without detectable viral loads or histories of opportunistic infections provide a better survival benefit over dialysis. Following transplantation, patient and graft survival are similar to the general transplant population despite frequent rejections.
Hepatitis B and C Typically infected patients present with microscopic hematuria, nonnephrotic or nephrotic-range proteinuria, and hypertension. There is a close association between hepatitis B infection and polyarteritis nodosa with vasculitis appearing generally in the first 6 months following infection. Renal manifestations include renal artery aneurysms, renal infarction, and ischemic scars. Alternatively, the hepatitis B carrier state can produce a MGN that is more common in children than adults, or MPGN that is more common in adults than in children. Renal histology is indistinguishable from idiopathic MGN or type I MPGN. Viral antigens are found in the renal deposits. There are no good treatment guidelines, but interferon α-2b and lamivudine have been used to some effect in small studies. Children have a good prognosis, with 60–65% achieving spontaneous remission within 4 years. In contrast, 30% of adults have renal insufficiency and 10% have renal failure 5 years after diagnosis.
Up to 30% of patients with chronic hepatitis C infection have some renal manifestations. Patients often present with type II mixed cryoglobulinemia, nephrotic syndrome, microscopic hematuria, abnormal liver function tests, depressed C3 levels, anti–hepatitis C virus (HCV) antibodies, and viral RNA in the blood. The renal lesions most commonly seen, in order of decreasing frequency, are cryoglobulinemic glomerulonephritis, MGN, and type I MPGN. Treatment with pegylated interferon and ribavirin is typical to reduce the viral load.
Other Viruses Other viral infections are occasionally associated with glomerular lesions, but cause and effect are not well established. These viral infections and their respective glomerular lesions include: cytomegalovirus producing MPGN; influenza and anti-GBM disease; measles-associated endocapillary proliferative glomerulonephritis, with measles antigen in the capillary loops and mesangium; parvovirus causing mild proliferative or mesangioproliferative glomerulonephritis or FSGS; mumps and mesangioproliferative glomerulonephritis; Epstein-Barr virus producing MPGN, diffuse proliferative nephritis, or IgA nephropathy; dengue hemorrhagic fever causing endocapillary proliferative glomerulonephritis; and coxsackievirus producing focal glomerulonephritis or DPGN.
Syphilis Secondary syphilis, with rash and constitutional symptoms, develops weeks to months after the chancre first appears and occasionally presents with the nephrotic syndrome from MGN caused by subepithelial immune deposits containing treponemal antigens. Other lesions have also rarely been described including interstitial syphilitic nephritis. The diagnosis is confirmed with nontreponemal and treponemal tests for Treponema pallidum. The renal lesion responds to treatment with penicillin or an alternative drug, if allergic. Additional testing for other sexually transmitted diseases is an important part of disease management.
Leprosy Despite aggressive eradication programs, approximately 400,000 new cases of leprosy appear annually worldwide. The diagnosis is best made in patients with multiple skin lesions accompanied by sensory loss in affected areas, using skin smears showing paucibacillary or multibacillary infection (WHO criteria). Leprosy is caused by infection with Mycobacterium leprae and can be classified by Ridley-Jopling criteria into various types: tuberculoid, borderline tuberculoid, mid-borderline and borderline lepromatous, and lepromatous. Renal involvement in leprosy is related to the quantity of bacilli in the body, and the kidney is one of the target organs during splanchnic localization. In some series, all cases with borderline lepromatous and lepromatous types of leprosy have various forms of renal involvement including FSGS, mesangioproliferative glomerulonephritis, or renal amyloidosis; much less common are the renal lesions of DPGN and MPGN. Treatment of the infection can cause remission of the renal disease.
Malaria There are 300–500 million incident cases of malaria each year worldwide, and the kidney is commonly involved. Glomerulonephritis is due to immune complexes containing malarial antigens that are implanted in the glomerulus. In malaria from P. falciparum, mild proteinuria is associated with subendothelial deposits, mesangial deposits, and mesangioproliferative glomerulonephritis that usually resolve with treatment. In quartan malaria from infection with Plasmodium malariae, children are more commonly affected and renal involvement is more severe. Transient proteinuria and microscopic hematuria can resolve with treatment of the infection. However, resistant nephrotic syndrome with progression to renal failure over 3–5 years does happen, as <50% of patients respond to steroid therapy. Affected patients with nephrotic syndrome have thickening of the glomerular capillary walls, with subendothelial deposits of IgG, IgM, and C3 associated with a sparse membranoproliferative lesion. The rare mesangioproliferative glomerulonephritis reported with Plasmodium vivax or Plasmodium ovale typically has a benign course.
Schistosomiasis Schistosomiasis affects more than 300 million people worldwide and primarily involves the urinary and gastrointestinal tracts. Glomerular involvement varies with the specific strain of schistosomiasis; Schistosoma mansoni is most commonly associated with clinical renal disease, and the glomerular lesions can be classified: Class I is a mesangioproliferative glomerulonephritis; class II is an extracapillary proliferative glomerulonephritis; class III is a membranoproliferative glomerulonephritis; class IV is a focal segmental glomerulonephritis; and class V is amyloidosis. Classes I–II often remit with treatment of the infection, but classes III and IV lesions are associated with IgA immune deposits and progress despite antiparasitic and/or immunosuppressive therapy.
Other Parasites Renal involvement with toxoplasmosis infections is rare. When it occurs, patients present with nephrotic syndrome and have a histologic picture of MPGN. Fifty percent of patients with leishmaniasis will have mild to moderate proteinuria and microscopic hematuria, but renal insufficiency is rare. Acute DPGN, MGN, and mesangioproliferative glomerulonephritis have all been observed on biopsy. Filariasis and trichinosis are caused by nematodes and are sometimes associated with glomerular injury presenting with proteinuria, hematuria, and a variety of histologic lesions that typically resolve with eradication of the infection.




