1.Ehmer, U, et al. Rapid allelic discrimination by TaqMan PCR for the detection of the Gilbert’s syndrome marker UGT1A1*28. J Mol Diagn. 2008; 10(6):549–552.
2.Costa, E. Hematologically important mutations: bilirubin UDP-glucuronosyltransferase gene mutations in Gilbert and Crigler-Najjar syndromes. Blood Cells Mol Dis. 2006; 36(1):77–80.
3.Hallal, H, et al. A shortened, 2-hour rifampin test: a useful tool in Gilbert’s syndrome. Gastroenterol Hepatol. 2006; 29(2):63–65.
4.Erdil, A, et al. Rifampicin test in the diagnosis of Gilbert’s syndrome. Int J Clin Pract. 2001; 55(2):81–83.
5.Ishihara, T, et al. Role of UGT1A1 mutation in fasting hyperbilirubinemia. J Gastroenterol Hepatol. 2001; 16(6):678–682.
6.Kadakol, A, et al. Genetic lesions of bilirubin uridine-diphosphoglucuronate glucuronosyltransferase (UGT1A1) causing Crigler-Najjar and Gilbert syndromes: correlation of genotype to phenotype. Hum Mutat. 2000; 16(4):297–306.
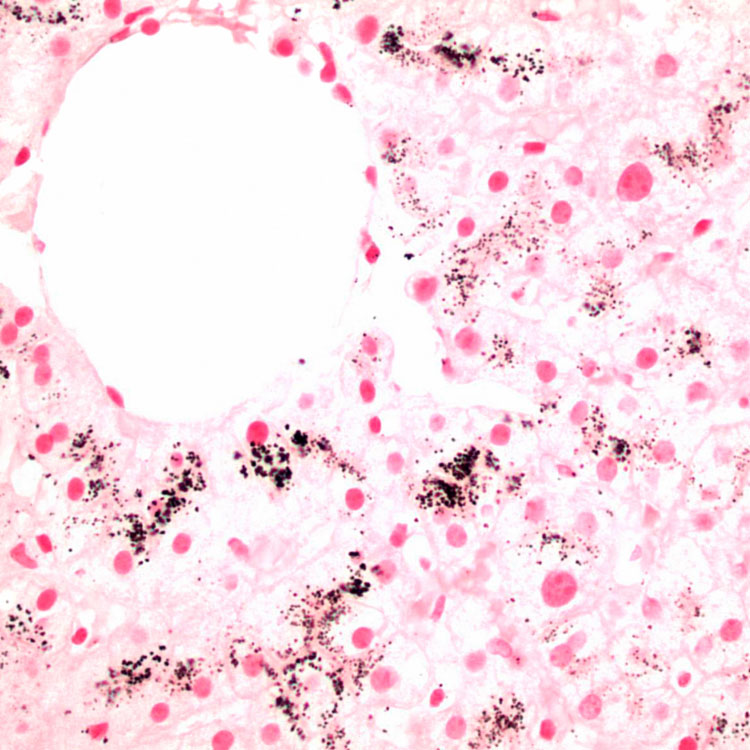
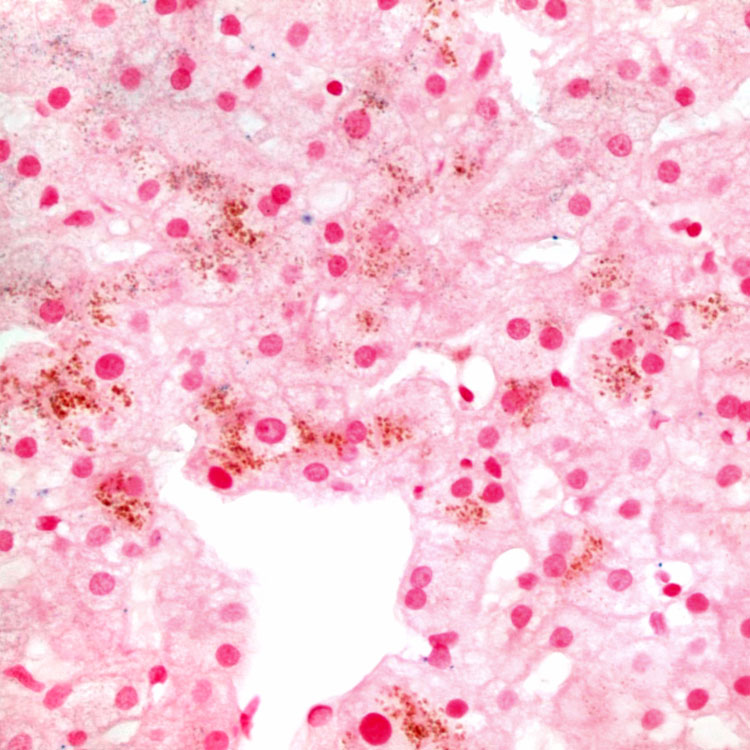
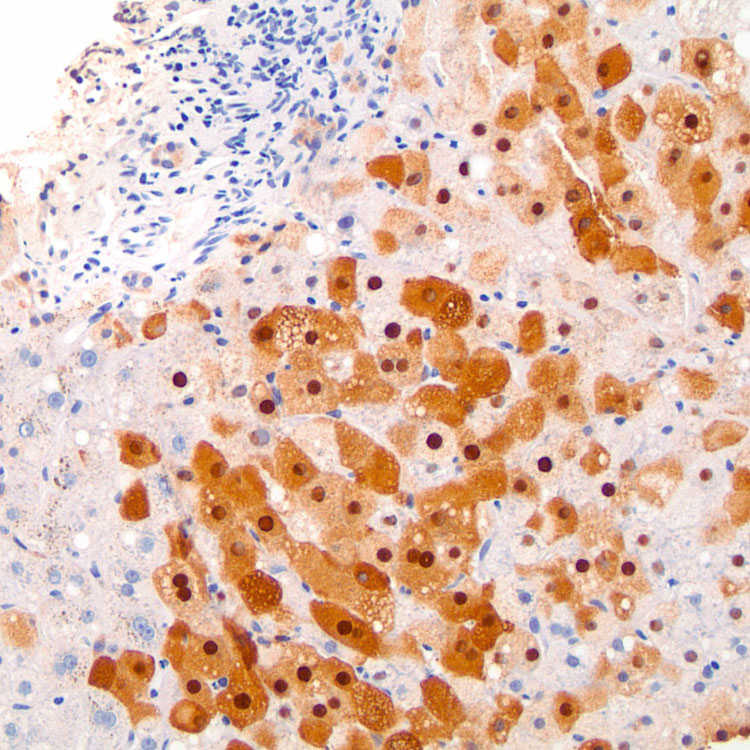
 in centrizonal hepatocytes.
in centrizonal hepatocytes.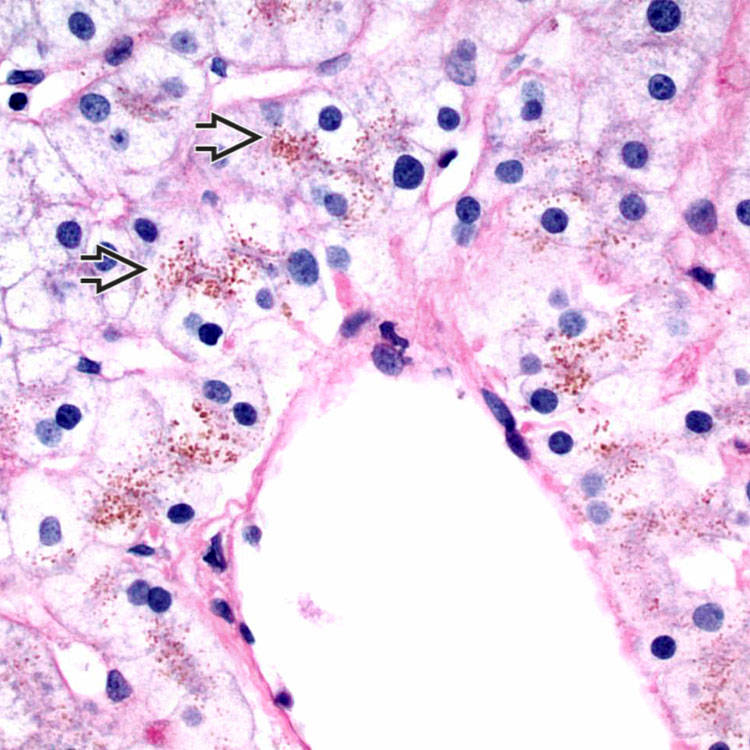
 , even though the pigment is not PAS-D positive.
, even though the pigment is not PAS-D positive.