[level-membership-for-neurology-category]
CHAPTER 51 GENETICS OF EPILEPSY
Epilepsy is a heterogeneous group of conditions, with a very broad range of possible causes, manifesting as recurrent, unprovoked, clinical seizures, sometimes with additional neurological or extraneural features. It is the most common serious neurological disorder, affecting 5 per every 1000 people, and is associated with increased rates of mortality and morbidity in almost every sphere of life for affected patients. The types of epilepsy can be subdivided in a number of ways; classification schemes focus variously on seizure types, natural history, or etiology. Although there are many causes of epilepsy, it is likely that an individual’s genetic makeup contributes to or modulates, to some degree, the risk of developing epilepsy in response to another insult to the brain, whatever the nature of the insult. Genetic differences between individuals take a number of forms: there are rare variants, called mutations, each of which occurs in less than 1% of the population and usually in a far smaller percentage; there are more common variants, the most common type of which is known as single-nucleotide polymorphism (SNP), a variation occurring at a single nucleotide and seen in greater than 1% (and usually greater than 5%) of the population (Fig. 51-1).
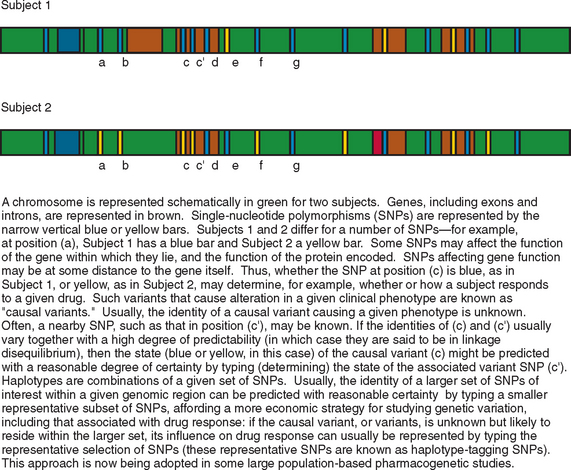
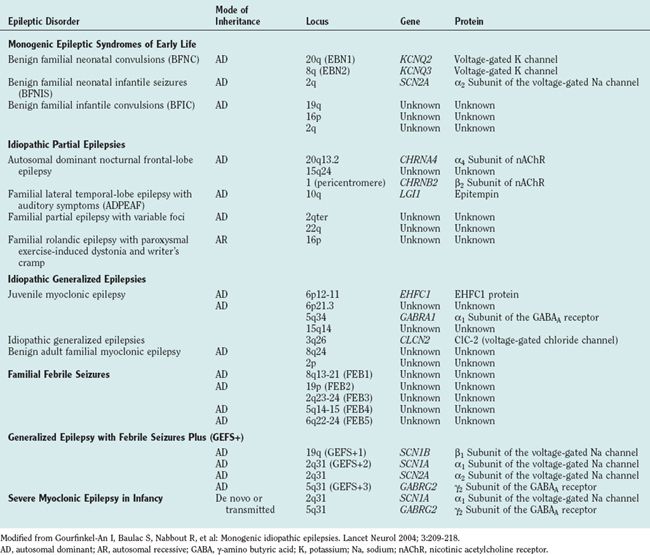
For most people with epilepsy, any genetic contribution to etiology is likely to arise from the additive, individually small effects of SNPs in a number of genes; understanding these effects requires analysis of large numbers of homogeneous groupings of patients and constitutes the field of population genetics. However, this area is only now beginning to be addressed by researchers in epilepsy. Most research to date on epilepsy genetics has concentrated on epilepsy caused by genetic mutations; almost by definition, such types of epilepsy are individually rare and even grouped together are unlikely to account for more than a small proportion of cases of epilepsy. The hope of research on these rare types of epilepsy caused by mendelian genetics is not only to inform disease management in affected individuals but also potentially to cast light on the genetics, pathophysiology, and management of epilepsies more broadly. In fact, this latter aim has yet to be realized, but certainly many epilepsies caused by single gene mutations are now known (Table 51-1) and account for a majority of the disorders covered in this chapter. Most of the genes implicated encode neuronal ion channels that directly affect neuronal excitability, or they function through alteration in ion flux; the finding that channelopathies cause seizures was a surprise initially, but it stands to reason because excitation and inhibition are fundamental neuronal properties, and seizures are generally held to arise from a systems imbalance between excitation and inhibition.
There are more than 200 genetically mediated conditions in which epilepsy is part of a broader phenotype. These conditions are not considered here; this chapter illustrates concepts of epilepsy genetics, drawing from genetic disorders in which the major and often sole manifestation is epilepsy. A glossary is given in Table 51-2.
TABLE 51-2 Glossary of Epilepsy Genetics Terms
| SCN1A | α1 Subunit of the voltage-gated sodium channel |
| SCN1B | β1 Subunit of the voltage-gated sodium channel |
| SCN2A | α2 Subunit of the voltage-gated sodium channel |
| GABA | γ-Amino butyric acid |
| KCNQ | Voltage-gated potassium channel |
| CLCN | Voltage-gated chloride channel |
| CHRNA4 | α4 Subunit of the nicotinic acetylcholine receptor |
| LGI1 | Leucine-rich gene, glioma inactivated |
| Gene | A segment of DNA that normally specifies a functional polypeptide or gene product |
| Allele | One of several alternative forms of a gene or DNA sequence at a specific locus |
| SNP | Single-nucleotide polymorphism, where more than one variant (allele) occurs at a locus with a frequency greater than 1% |
| Mutation | Change within a gene or DNA sequence due to base substitution, deletion or insertion occurring at a frequency < 1% and usually much less |
| Autosome | Any chromosome other than the sex chromosomes X and Y |
| Locus | A unique chromosomal location |
| Linkage | The tendency of genes to be inherited together as a consequence of their physical proximity |
| GEFS+ | Generalized epilepsy with febrile seizures plus |
| SMEI | Severe myoclonic epilepsy of infancy |
| JME | Juvenile myoclonic epilepsy |
| BFNC | Benign familial neonatal convulsions |
| ICCA | Infantile convulsions and choreoathetosis |
| ADNFLE | Autosomal-dominant nocturnal frontal lobe epilepsy |
| BFNIS | Benign familial neonatal-infantile seizures |
| ADPEAF | Autosomal dominant familial lateral temporal lobe epilepsy with auditory features |
| ARFGEF2 | Adenosine diphosphate (ADP)–ribosylation factor guanine nucleotide exchange factor 2 |
| BIG2 | Brefeldin A (BFA): inhibited GEF2 protein |
| GPCR | G protein–coupled receptor |
MONOGENIC INHERITED EPILEPSIES
Generalized Epilepsies
Juvenile Myoclonic Epilepsy
This syndrome is characterized by the occurrence of myoclonic jerks, generalized tonic-clonic seizures, and absence seizures with onset in the early teens, normal magnetic resonance imaging findings, and a characteristic electroencephalographic pattern. Familial cases occur, and despite widespread acceptance that there is, even in sporadic cases, strong genetic susceptibility to juvenile myoclonic epilepsy (JME), only two responsible genes are known.1,2
Linkage has been described for JME in two regions of the short arm of chromosome 6 (6p12-11 and 6p21.3).3,4 Suzuki and associates (2004)2 examined 18 genes encoded in the linked 6p12-11 region. They discovered mutations in only one gene, EHFC1. They described five missense mutations in EHFC1, which encodes a protein with an EF-hand motif that cosegregated with epilepsy or electroencephalographic polyspike and wave activity in six unrelated families and was not detected in 382 control individuals. However, mutations were detected in only 6 of 44 families with JME examined. Although mutations in EHFC1 are the first described for JME, this study also highlights the underlying genetic heterogeneity of this condition.
Genetic heterogeneity may also explain the variable mode of inheritance for JME. Elmslie and colleagues described linkage of JME to 15q14.5 Cossette and associates1 described autosomal dominant inheritance in a French-Canadian family with linkage to 5q34. This region includes the genes for a number of γ-amino butyric acid (GABA) receptor subunits and has since been shown to contain a causative mutation in the GABRA1 gene.
Generalized Epilepsy with Febrile Seizures Plus (GEFS+)
Investigators have described families in which some members have febrile seizures persisting beyond the usually accepted upper age limit for the diagnosis of such seizures (6 years) and are thus denoted “febrile seizures plus”; other members of the same family may have generalized epilepsy. Within such families there can be wide phenotypic variability; febrile seizures, febrile seizures plus, generalized epilepsy, hemiconvulsive seizures, and temporal lobe or frontal lobe seizures may occur. Some members with generalized or partial epilepsy may not have had febrile seizures or suffer from a number of different seizure types, and some members may have febrile seizures but never suffer another seizure beyond the age of 6. Generalized epilepsy with febrile seizures plus (GEFS+) has an autosomal dominant pattern of inheritance with reduced (70% to 80%) penetrance. GEFS+ displays genetic heterogeneity, inasmuch as mutations in different ion channel genes can give rise to similar phenotypes. The most frequent site of mutation in patients with febrile seizures plus (57%) is the in the SCN1A gene6; some mutations probably result in defective fast inactivation of channel gating and neuronal hyperexcitability. Mutations causing GEFS+ have also been described in SCN1B, SCN2A, and the GABA receptor gene GABRG2 (see Table 51-1).
GABAA receptors contain an integral chloride channel that mediates synaptic inhibition. There are specific binding sites for GABA, barbiturates, benzodiazepines, and steroids. Bowser and associates reported altered kinetics and benzodiazepine sensitivity of a GABA receptor subunit mutation, GABRG2 R43Q, in a family with childhood absence epilepsy and febrile seizures.7
GEFS+ may possibly be more correctly termed autosomal dominant epilepsy with febrile seizures plus because of the large degree of phenotypic variability within families.8
Severe Myoclonic Epilepsy in Infancy (Dravet’s Syndrome)
Severe myoclonic epilepsy in infancy (SMEI), or Dravet’s syndrome, is rare. Individuals with previously normal development develop severe epilepsy in their first year of life, initially with myoclonic seizures, cognitive decline, and ataxia; subsequently, myoclonic seizures may abate and other seizure types predominate. Patients frequently suffer febrile seizures and status epilepticus. Mutations in SCN1A, accounting for 35% of sporadic cases9 and GABRG2 in only one familial case,10 have been discovered to underlie SMEI. Of interest is that different mutations in the same gene (SCN1A) can cause both familial epilepsy with a relatively mild phenotype and sporadic epilepsy with a devastating one.
Focal Epilepsies
Benign Familial Neonatal Convulsions
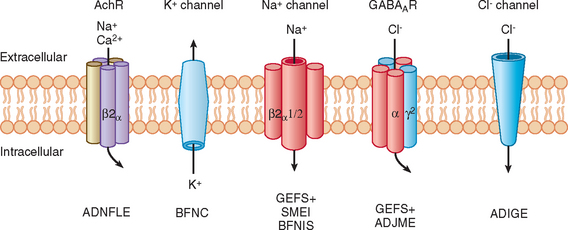
This is an autosomal dominant condition with high penetrance. It is characterized by the development of multiple seizure types on day 2 or 3 after birth, usually remitting spontaneously by 8 months of age, and is not associated with any psychomotor impairment. However, approximately 11% of these patients subsequently develop epilepsy. It was the first idiopathic epilepsy to be genetically linked. Causative mutations have been identified in voltage-gated potassium channel genes (KCNQ2 and KCNQ3) (Fig. 51-2).11–16 Of interest is that mutations in the homologous KCNQ1 gene result in the long QT and Jervell–Lange-Nielsen cardioauditory syndromes.17,18
Benign Familial Infantile Convulsions
An autosomal dominant disorder, this is characterized by convulsions beginning at 3 to 12 months of age, with a very good prognosis. There has been strong linkage to chromosomal locus 19q in five Italian families.19 Benign familial infantile convulsion has been also been linked to chromosomal locus 2q20 and is reported with paroxysmal dyskinesias in another condition, infantile convulsions and choreoathetosis, which has been linked to chromosomal region 16p12.21
Benign Familial Neonatal-Infantile Convulsions
Mutations in SCN2A have been described in two families22 with convulsions commencing at 2 days to 3.5 months of age.
Autosomal Dominant Nocturnal Frontal Lobe Epilepsy
In this disorder, seizures usually occur during sleep, often commencing in childhood, and are often misdiagnosed as a movement disorder. Frequently, it can be successfully treated with carbamazepine, but sometimes there is pharmacoresistance. The seizures have prominent motor manifestations, including hypermotor activity and dystonic posturing. Penetrance is high (∼70%). Mutations in genes encoding subunits of the neuronal nicotinic acetylcholine receptor (CHRNA4, CHRNB2)23,24 have been described in some but not all kindreds, which again illustrates the genetic heterogeneity that may underlie an apparently simple and homogeneous phenotype.
Familial Lateral Temporal Lobe Epilepsy
The most common form of epilepsy of temporal lobe origin arises from the mesial temporal lobe and is caused by hippocampal sclerosis (see “Complex Inheritance of Epilepsy” section). Epilepsy arising from the lateral temporal lobe is less common. In either case, it is usually sporadic, with no indication of an underlying causative genetic mutation. Autosomal dominant partial epilepsy with auditory features is a rare, familial lateral temporal lobe epilepsy syndrome with penetrance of approximately 60% to 70%. Brief auditory hallucinations are often described in its phenotypic spectrum but are not invariably present. Although it is a genetically heterogeneous condition, underlying mutations in LGI1, which encodes the leucine-rich glioma-inactivated factor 1 (also referred to as epitempin), have been discovered.25 This finding is of particular interest because the gene does not encode a channel and is the first to be associated with idiopathic focal epilepsy.
Progressive Myoclonus Epilepsies
Progressive myoclonus epilepsies are a heterogeneous group of conditions characterized by the triad of worsening myoclonic seizures, generalized tonic-clonic seizures, and progressive intellectual decline; in many subtypes, additional features are observed (e.g., photosensitivity, mitochondrial cytopathic manifestations). Many underlying genetic mutations have now been identified (Table 51-3).
| Disorder | Gene | Protein |
|---|---|---|
| Neuronal ceroid lipofuscinosis | ||
| Infantile | CLN1 | Palmitoyl-protein thioesterase 1 (PPT1) |
| Late infantile | CLN2 | Tripeptidyl peptidase 1 (TPP1) |
| Finnish variant late infantile | CLN5 | Novel membrane protein |
| Variant late infantile | CLN6 | Novel membrane protein |
| Juvenile | CLN3 | Novel membrane protein |
| Northern epilepsy | CLN8 | Novel membrane protein |
| Adult (Kufs disease) | — | — |
| Lafora’s disease | EPM2A | Laforin |
| EPM2B (NHLRC1) | Malin | |
| Sialidosis | NEU1 | Neuraminidase 1 |
| Unverricht-Lundborg disease | CSTB (EPM1) | Cystatin B |
| EPM1B | Unknown | |
| Juvenile GM2 gangliosidosis type III | β-N-acetylhexosaminidase A (deficiency) | |
| Myoclonus epilepsy with ragged red fibers (MERRF) | MTTK | tRNALys |
tRNALys, transfer ribonucleic acid that codes for lysine.
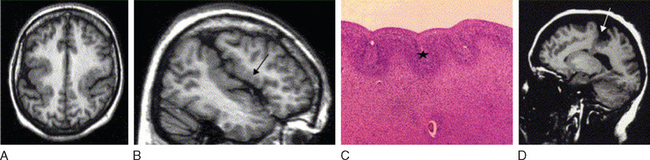
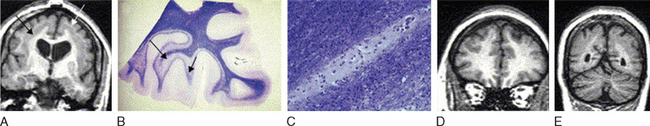
Focal Epilepsies Caused by Malformations of Cortical Development
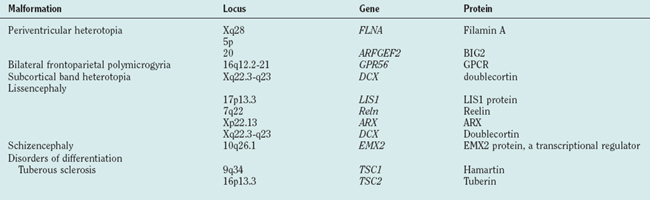
Abnormal development of the brain is an important cause of epilepsy, particularly epilepsy that is refractory to drug treatment. Such malformations can often be identified by magnetic resonance imaging in vivo (Figs. 51-3 to 51-5). In fact, magnetic resonance imaging can be used as a phenotyping tool, allowing the identification of other family members with a malformation who might not have epilepsy. In turn, this allows traditional linkage genetic studies to be undertaken. This strategy has revealed a group of gene mutations that are now known to cause brain malformation that may manifest with epilepsy; of additional importance, these genes are clearly also implicated in normal brain development, and much has been learned about human brain development from such studies (Table 51-4).
Febrile Seizures
Simple febrile seizures occur at 6 months to 6 years of age with fever, typically greater than 38.5° C, without evidence of central nervous system infection. They consist of brief generalized, clonic, tonic-clonic, or atonic seizures and generally do not necessitate prophylactic antiepileptic treatment. Estimates of the prevalence of febrile seizures vary from 2% to 5% in North America and Europe26,27 to 7% to 14% in Japan.28 However, prospective follow-up studies have shown that patients who suffer febrile seizures, especially complex febrile seizures, have a greater risk of developing epilepsy than do children who do not suffer such seizures. Epilepsy has a prevalence of 0.5% to 1% in the population, but afebrile seizures occur in 2% to 7% of patients who had febrile seizures.29,30 It is estimated that 11% of patients with generalized epilepsy and 5% to 6% with partial epilepsies other than temporal lobe epilepsy have suffered febrile seizures.31 Some studies suggest that 25% of patients with temporal lobe epilepsy have had febrile seizures.31 Chromosomal studies of large families with febrile seizures, with or without additional epilepsy, have identified four loci with autosomal dominant inheritance and incomplete penetrance: FEB1 on 8q13-21,32 FEB2 on 19p,33,34 FEB3 on 2q23-24,35 and FEB5 on 6q22-24.36 A locus for FEB4 was found on 5q14-15 from a nonparametric analysis of 47 families.37 No underlying mutations have yet been identified.
COMPLEX INHERITANCE OF EPILEPSY
Pal and associates38 postulated that changes in the BRD2 (RING3) gene may alter neural development and that subsequent interaction with other susceptibility genes for JME may provide a framework for inheritance of JME as a complex disease.
Mesial temporal lobe epilepsy that is refractory to drug treatment is often associated with hippocampal sclerosis and a history of febrile seizures. SNPs in many genes (e.g., the interleukin-1β gene,39 the prodynorphin gene,40 GABBR1,41 the prion protein gene42) have been reported as associated with temporal lobe epilepsy, hippocampal sclerosis, or febrile seizures. However, none of these data have yet reliably survived attempts to replicate initial reports.43 This demonstrates the major difficulty with such population genetic association studies.
One of the major difficulties in epilepsy is understanding why patients with apparently the same type of epilepsy show major differences in pharmacosensitivity. Population genetics studies facilitate pharmacogenetics, in which the genetic basis for individual response to drugs is examined. Pharmacoresistance may be dependent on the type of epilepsy, any underlying structural abnormality (e.g., brain malformation or lesion), drug absorption, drug metabolism, or the presence of drug transporter proteins. Successes with pharmacogenetics have highlighted associations between pharmacoresistance and common genetic variation in the gene ABCB1 encoding P-glycoprotein, a broad-spectrum drug transport protein.44 In another illustration of the complexity of such work, a large replication study has failed to support this intriguing finding.45
CONCLUSIONS
Baulac S, Gourfinkel-An I, Nabbout R, et al. Fever, genes and epilepsy. Lancet Neurol. 2004;3:421-430.
Cavalleri GL, Lynch JM, Depondt C, et al. Failure to replicate previously reported genetic associations with sporadic temporal lobe epilepsy: where to from here? Brain. 2005;128:1832-1840.
Gourfinkel-An I, Baulac S, Nabbout R, et al. Monogenic idiopathic epilepsies. Lancet Neurol. 2004;3:209-218.
Gutierrez-Delicado E, Serratosa JM. Genetics of the epilepsies. Curr Opin Neurol. 2004;17:145-147.
Sisodiya SM. Malformations of cortical development: burdens and insights from important causes of human epilepsy. Lancet Neurol. 2004;3:29-38.
1 Cossette P, Liu L, Brisebois K, et al. Mutation of GABRA1 in an autosomal dominant form of juvenile myoclonic epilepsy. Nat Genet. 2002;31:184-189.
2 Suzuki T, Delgado-Escueta AV, Aguan K, et al. Mutations in EHFC1 cause juvenile myoclonic epilepsy. Nat Genet. 2004;36:842-849.
3 Liu AW, Delgado-Escueta AV, Gee MN, et al. Juvenile myoclonic epilepsy in chromosome 6p12-p11: locus heterogeneity and recombinations. Am J Med Genet. 1996;63:438-446.
4 Sander T, Bockenkamp B, Hildman T, et al. Refined mapping of the epilepsy susceptibility locus EJM1 on chromosome 6. Neurology. 1997;49:842-847.
5 Elmslie FV, Rees M, Williamson MP, et al. Genetic mapping of a major susceptibility locus for juvenile myoclonic epilepsy on chromosome 15q. Hum Mol Genet. 1997;6:1329-1334.
6 Baulac S, Gourfinkel-An I, Nabbout R, et al. Fever genes and epilepsy. Lancet Neurol. 2004;3:421-430.
7 Bowser DN, Wagner DA, Czajkowski C, et al. Altered kinetics of a GABAA receptor subunit mutation [gamma 2(R43Q)] found in human epilepsy. Proc Natl Acad Sci U S A. 2002;99:15170-15175.
8 Ito M, Nagafuji H, Okazawa H, et al. Autosomal dominant epilepsy with febrile seizures plus with missense mutations of the Na+-channel α1 subunit gene, SCN1A.. Epilepsy Res. 2002;48:15-23.
9 Nabbout R, Gennaro E, Dalla Bernardina B, et al. Spectrum of SCN1A mutations in severe myoclonic epilepsy of infancy. Neurology. 2003;60:1961-1967.
10 Harkin LA, Bowser DN, Dibbens LM, et al. Truncation of the GABA(A)-receptor γ2 subunit in a family with generalized epilepsy with febrile seizures plus. Am J Hum Genet. 2002;70:530-536.
11 Singh NA, Charlier C, Stauffer D, et al. A novel potassium channel gene, KCNQ2, is mutated in an inherited epilepsy of newborns. Nat Genet. 1998;18:25-29.
12 Biervert C, Schroeder BC, Kubisch C, et al. A potassium channel mutation in neonatal human epilepsy. Science. 1998;279:403-406.
13 Lerche H, Bievert C, Alekov AK, et al. A reduced K+ current due to a novel mutation in KCNQ2 causes neonatal convulsions. Ann Neurol. 1999;46:305-312.
14 Dedek K, Fusco L, Teloy N, et al. Neonatal convulsions and epileptic encephalopathy in an Italian family with a missense mutation in the fifth transmembrane region of KCNQ2.. Epilepsy Res. 2003;54:21-27.
15 Charlier C, Singh NA, Ryan SG, et al. A pore mutation in a novel KQT-like potassium channel gene in an idiopathic epilepsy family. Nat Genet. 1998;18:53-55.
16 Hirose S, Zenri F, Akiyoshi H, et al. A novel mutation of KCNQ3 (c.925T→C) in a Japanese family with benign familial neonatal convulsions. Ann Neurol. 2000;47:822-826.
17 Wang Q, Curran ME, Splawski I, et al. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat Genet. 1996;12:17-23.
18 Neyroud N, Tesson F, I Denjoy I, et al. A novel mutation in the potassium channel gene KVLQT1 causes the Jervell and Lange-Nielsen cardioauditory syndrome. Nat Genet. 1997;15:186-189.
19 Guipponi M, Rivier F, Vigevano F, et al. Linkage mapping of benign familial infantile convulsions (BFIC) to chromosome 19q. Hum Mol Genet. 1997;6:473-477.
20 Malacarne M, Gennaro E, Madia F, et al. Benign familial infantile convulsions: mapping of a novel locus on chromosome 2q24 and evidence for genetic heterogeneity. Am J Hum Genet. 2001;68:1521-1526.
21 Caraballo R, Pavek SS, Lemainque A, et al. Linkage of benign familial infantile convulsions to chromosome 16p12-q12 suggests allelism to the infantile convulsions and choreoathetosis syndrome. Am J Hum Genet. 2001;68:788-794.
22 Heron SE, Crossland KM, Andermann E, et al. Sodium-channel defects in benign familial neonatal-infantile seizures. Lancet. 2002;360:851-852.
23 Steinlein OK, Mulley JC, Propping P, et al. A missense mutation in the neuronal nicotinic acetylcholine receptor α4 subunit is associated with autosomal dominant nocturnal frontal lobe epilepsy. Nat Genet. 1995;11:201-203.
24 Fusco MD, Becchetti AA, Patrignani A, et al. The nicotinic receptor β2 subunit is mutant in nocturnal frontal lobe epilepsy. Nat Genet. 2000;26:275-276.
25 Kalachikov S, Evgrafov O, Ross B, et al. Mutations in LGI1 cause autosomal-dominant partial epilepsy with auditory features. Nat Genet. 2002;30:335-341.
26 Hauser WA. The prevalence and incidence of convulsive disorders in children. Epilepsia. 1994;35(Suppl 2):S1-S6.
27 Hauser WA, Annegers JF, Rocca WA. Descriptive epidemiology of epilepsy: contributions of population-based studies from Rochester, Minnesota. Mayo Clin Proc. 1996;71:576-586.
28 Tsuboi T. Epidemiology of febrile and afebrile convulsions in children in Japan. Neurology. 1984;34:175-181.
29 Annegers JF, Hauser WA, Elveback LR, et al. The risk of epilepsy following febrile convulsions. Neurology. 1979;29:297-303.
30 Verity CM, Golding J. Risk of epilepsy after febrile convulsions: a national cohort study. BMJ. 1991;303:1373-1376. [Erratum in BMJ 1992; 304:147].
31 Hamati-Haddad A, Abou-Khalil B. Epilepsy diagnosis and localization in patients with antecedent childhood febrile convulsions. Neurology. 1998;50:917-922.
32 Wallace RH, Berkovic SF, Howell RA, et al. Suggestion of a major gene for familial febrile convulsions mapping to 8q13–21. J Med Genet. 1996;33:308-312.
33 Johnson EW, Dubovsky J, Rich SS, et al. Evidence for a novel gene for familial febrile convulsions, FEB2, linked to chromosome 19p in an extended family from the Midwest. Hum Mol Genet. 1998;7:63-67.
34 Kugler SL, Stenroos ES, Mandelbaum DE, et al. Hereditary febrile seizures: phenotype and evidence for a chromosome 19p locus. Am J Med Genet. 1998;79:354-361.
35 Peiffer A, Thompson J, Charlier C, et al. A locus for febrile seizures (FEB3) maps to chromosome 2q23–24. Ann Neurol. 1999;46:671-678.
36 Nabbout R, Prud’homme JF, Herman A, et al. A locus for simple pure febrile seizures maps to chromosome 6q22-q24. Brain. 2002;125:2668-2680.
37 Nakayama J, Hamano K, Iwasaki N, et al. Significant evidence for linkage of febrile seizures to chromosome 5q14-q15. Hum Mol Genet. 2000;9:87-91.
38 Pal DK, Evgrafov OV, Tabares P, et al. BRD2 (RING3) is a probable major susceptibility gene for common juvenile myoclonic epilepsy. Am J Hum Genet. 2003;73:261-270.
39 Kanemoto K, Kawasaki J, Yuasa S, et al. Increased frequency of interleukin-1β–511T allele in patients with temporal lobe epilepsy, hippocampal sclerosis, and prolonged febrile convulsion. Epilepsia. 2003;44:796-799.
40 Stogmann E, Zimprich A, Baumgartner C, et al. A functional polymorphism in the prodynorphin gene promotor is associated with temporal lobe epilepsy. Ann Neurol. 2002;51:260-263.
41 Gambardella A, Manna I, Labate A, et al. GABA(B) receptor 1 polymorphism (G1465A) is associated with temporal lobe epilepsy. Neurology. 2003;60:560-563.
42 Walz R, Castro RM, Velasco TR, et al. Surgical outcome in mesial temporal sclerosis correlates with prion protein gene variant. Neurology. 2003;61:1204-1210.
43 Cavalleri GL, Lynch JM, Depondt C, et al. Failure to replicate previously reported genetic associations with sporadic temporal lobe epilepsy: where to from here? Brain. 2005;128:1832-1840.
44 Siddiqui A, Kerb R, Weale ME, et al. Association of multidrug resistance in epilepsy with a polymorphism in the drug-transporter gene ABCB1.. N Engl J Med. 2003;348:1442-1448.
45 Tan NC, Heron SE, Scheffer IE, et al. Failure to confirm association of a polymorphism in ABCB1 with multidrug-resistant epilepsy. Neurology. 2004;63:1090-1092.
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
CHAPTER 51 GENETICS OF EPILEPSY
Epilepsy is a heterogeneous group of conditions, with a very broad range of possible causes, manifesting as recurrent, unprovoked, clinical seizures, sometimes with additional neurological or extraneural features. It is the most common serious neurological disorder, affecting 5 per every 1000 people, and is associated with increased rates of mortality and morbidity in almost every sphere of life for affected patients. The types of epilepsy can be subdivided in a number of ways; classification schemes focus variously on seizure types, natural history, or etiology. Although there are many causes of epilepsy, it is likely that an individual’s genetic makeup contributes to or modulates, to some degree, the risk of developing epilepsy in response to another insult to the brain, whatever the nature of the insult. Genetic differences between individuals take a number of forms: there are rare variants, called mutations, each of which occurs in less than 1% of the population and usually in a far smaller percentage; there are more common variants, the most common type of which is known as single-nucleotide polymorphism (SNP), a variation occurring at a single nucleotide and seen in greater than 1% (and usually greater than 5%) of the population (Fig. 51-1).
For most people with epilepsy, any genetic contribution to etiology is likely to arise from the additive, individually small effects of SNPs in a number of genes; understanding these effects requires analysis of large numbers of homogeneous groupings of patients and constitutes the field of population genetics. However, this area is only now beginning to be addressed by researchers in epilepsy. Most research to date on epilepsy genetics has concentrated on epilepsy caused by genetic mutations; almost by definition, such types of epilepsy are individually rare and even grouped together are unlikely to account for more than a small proportion of cases of epilepsy. The hope of research on these rare types of epilepsy caused by mendelian genetics is not only to inform disease management in affected individuals but also potentially to cast light on the genetics, pathophysiology, and management of epilepsies more broadly. In fact, this latter aim has yet to be realized, but certainly many epilepsies caused by single gene mutations are now known (Table 51-1) and account for a majority of the disorders covered in this chapter. Most of the genes implicated encode neuronal ion channels that directly affect neuronal excitability, or they function through alteration in ion flux; the finding that channelopathies cause seizures was a surprise initially, but it stands to reason because excitation and inhibition are fundamental neuronal properties, and seizures are generally held to arise from a systems imbalance between excitation and inhibition.
There are more than 200 genetically mediated conditions in which epilepsy is part of a broader phenotype. These conditions are not considered here; this chapter illustrates concepts of epilepsy genetics, drawing from genetic disorders in which the major and often sole manifestation is epilepsy. A glossary is given in Table 51-2.
TABLE 51-2 Glossary of Epilepsy Genetics Terms
| SCN1A | α1 Subunit of the voltage-gated sodium channel |
| SCN1B | β1 Subunit of the voltage-gated sodium channel |
| SCN2A | α2 Subunit of the voltage-gated sodium channel |
| GABA | γ-Amino butyric acid |
| KCNQ | Voltage-gated potassium channel |
| CLCN | Voltage-gated chloride channel |
| CHRNA4 | α4 Subunit of the nicotinic acetylcholine receptor |
| LGI1 | Leucine-rich gene, glioma inactivated |
| Gene | A segment of DNA that normally specifies a functional polypeptide or gene product |
| Allele | One of several alternative forms of a gene or DNA sequence at a specific locus |
| SNP | Single-nucleotide polymorphism, where more than one variant (allele) occurs at a locus with a frequency greater than 1% |
| Mutation | Change within a gene or DNA sequence due to base substitution, deletion or insertion occurring at a frequency < 1% and usually much less |
| Autosome | Any chromosome other than the sex chromosomes X and Y |
| Locus | A unique chromosomal location |
| Linkage | The tendency of genes to be inherited together as a consequence of their physical proximity |
| GEFS+ | Generalized epilepsy with febrile seizures plus |
| SMEI | Severe myoclonic epilepsy of infancy |
| JME | Juvenile myoclonic epilepsy |
| BFNC | Benign familial neonatal convulsions |
| ICCA | Infantile convulsions and choreoathetosis |
| ADNFLE | Autosomal-dominant nocturnal frontal lobe epilepsy |
| BFNIS | Benign familial neonatal-infantile seizures |
| ADPEAF | Autosomal dominant familial lateral temporal lobe epilepsy with auditory features |
| ARFGEF2 | Adenosine diphosphate (ADP)–ribosylation factor guanine nucleotide exchange factor 2 |
| BIG2 | Brefeldin A (BFA): inhibited GEF2 protein |
| GPCR | G protein–coupled receptor |
MONOGENIC INHERITED EPILEPSIES
Generalized Epilepsies
Juvenile Myoclonic Epilepsy
This syndrome is characterized by the occurrence of myoclonic jerks, generalized tonic-clonic seizures, and absence seizures with onset in the early teens, normal magnetic resonance imaging findings, and a characteristic electroencephalographic pattern. Familial cases occur, and despite widespread acceptance that there is, even in sporadic cases, strong genetic susceptibility to juvenile myoclonic epilepsy (JME), only two responsible genes are known.1,2
Linkage has been described for JME in two regions of the short arm of chromosome 6 (6p12-11 and 6p21.3).3,4 Suzuki and associates (2004)2 examined 18 genes encoded in the linked 6p12-11 region. They discovered mutations in only one gene, EHFC1. They described five missense mutations in EHFC1, which encodes a protein with an EF-hand motif that cosegregated with epilepsy or electroencephalographic polyspike and wave activity in six unrelated families and was not detected in 382 control individuals. However, mutations were detected in only 6 of 44 families with JME examined. Although mutations in EHFC1 are the first described for JME, this study also highlights the underlying genetic heterogeneity of this condition.
Genetic heterogeneity may also explain the variable mode of inheritance for JME. Elmslie and colleagues described linkage of JME to 15q14.5 Cossette and associates1 described autosomal dominant inheritance in a French-Canadian family with linkage to 5q34. This region includes the genes for a number of γ-amino butyric acid (GABA) receptor subunits and has since been shown to contain a causative mutation in the GABRA1 gene.
Generalized Epilepsy with Febrile Seizures Plus (GEFS+)
Investigators have described families in which some members have febrile seizures persisting beyond the usually accepted upper age limit for the diagnosis of such seizures (6 years) and are thus denoted “febrile seizures plus”; other members of the same family may have generalized epilepsy. Within such families there can be wide phenotypic variability; febrile seizures, febrile seizures plus, generalized epilepsy, hemiconvulsive seizures, and temporal lobe or frontal lobe seizures may occur. Some members with generalized or partial epilepsy may not have had febrile seizures or suffer from a number of different seizure types, and some members may have febrile seizures but never suffer another seizure beyond the age of 6. Generalized epilepsy with febrile seizures plus (GEFS+) has an autosomal dominant pattern of inheritance with reduced (70% to 80%) penetrance. GEFS+ displays genetic heterogeneity, inasmuch as mutations in different ion channel genes can give rise to similar phenotypes. The most frequent site of mutation in patients with febrile seizures plus (57%) is the in the SCN1A gene6; some mutations probably result in defective fast inactivation of channel gating and neuronal hyperexcitability. Mutations causing GEFS+ have also been described in SCN1B, SCN2A, and the GABA receptor gene GABRG2 (see Table 51-1).
GABAA receptors contain an integral chloride channel that mediates synaptic inhibition. There are specific binding sites for GABA, barbiturates, benzodiazepines, and steroids. Bowser and associates reported altered kinetics and benzodiazepine sensitivity of a GABA receptor subunit mutation, GABRG2 R43Q, in a family with childhood absence epilepsy and febrile seizures.7
GEFS+ may possibly be more correctly termed autosomal dominant epilepsy with febrile seizures plus because of the large degree of phenotypic variability within families.8
Severe Myoclonic Epilepsy in Infancy (Dravet’s Syndrome)
Severe myoclonic epilepsy in infancy (SMEI), or Dravet’s syndrome, is rare. Individuals with previously normal development develop severe epilepsy in their first year of life, initially with myoclonic seizures, cognitive decline, and ataxia; subsequently, myoclonic seizures may abate and other seizure types predominate. Patients frequently suffer febrile seizures and status epilepticus. Mutations in SCN1A, accounting for 35% of sporadic cases9 and GABRG2 in only one familial case,10 have been discovered to underlie SMEI. Of interest is that different mutations in the same gene (SCN1A) can cause both familial epilepsy with a relatively mild phenotype and sporadic epilepsy with a devastating one.
Focal Epilepsies
Benign Familial Neonatal Convulsions
This is an autosomal dominant condition with high penetrance. It is characterized by the development of multiple seizure types on day 2 or 3 after birth, usually remitting spontaneously by 8 months of age, and is not associated with any psychomotor impairment. However, approximately 11% of these patients subsequently develop epilepsy. It was the first idiopathic epilepsy to be genetically linked. Causative mutations have been identified in voltage-gated potassium channel genes (KCNQ2 and KCNQ3) (Fig. 51-2).11–16 Of interest is that mutations in the homologous KCNQ1 gene result in the long QT and Jervell–Lange-Nielsen cardioauditory syndromes.17,
[/not-level-membership-for-neurology-category]
