[level-membership-for-cardiovascular-category]49
Genetics of Atrial Fibrillation
Heritability of AF
Reports of familial clustering of AF date back to the early 1940s.1,2 Familial AF was generally regarded as a rare condition for many years thereafter. However, over the past decade a major paradigm shift occurred with widespread recognition of the heritability underlying AF.3–7 In the community-based Framingham Heart Study, 27% of individuals with AF had a first-degree relative with AF confirmed by electrocardiography.7 Familial AF was associated with a 40% increased risk of AF for another family member over a subsequent 8-year period (Figure 49-1). The risk associated with familial AF remained even after adjustment for established clinical risk factors for AF. A study from Denmark demonstrated that AF was more common among monozygotic twins as compared with dizygotic twins, implicating a genetic predisposition to the arrhythmia even among those raised with shared environmental exposures.6
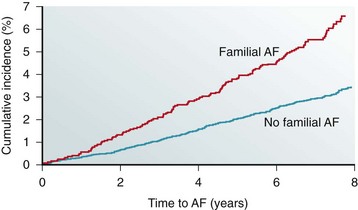
Figure 49-1 The risk of atrial fibrillation is increased in individuals with a first-degree relative with antecedent atrial fibrillation in the Framingham Heart Study. The estimated 8-year risk of atrial fibrillation (AF) is increased by 40% among individuals with a first-degree relative with atrial fibrillation as compared with those without familial atrial fibrillation. (Adapted with permission from Lubitz et al.7)
In numerous reports, the heritability of AF appears to be greatest among younger individuals3,5,7 and those without structural heart disease.3,4 Premature familial AF, or that occurring in family members ≤65 years of age, was associated with a 2-fold increase in the risk of AF compared with individuals without familial AF in the Framingham Heart Study.7 Nevertheless, data from Framingham provide evidence that the heritability of AF is present in the elderly as well.7
Genetic Mapping of AF
Linkage Analysis and Candidate Gene Resequencing
The first genetic locus for AF was described in 1997 by Brugada et al, who identified an AF susceptibility region on chromosome 10 in a large family with autosomal dominant AF (Table 49-1).8 Since this initial report, linkage analysis has repeatedly been used to identify genetic mutations underlying AF in a number of large families with AF.
Table 49-1
Atrial Fibrillation Susceptibility Loci Identified by Linkage Analysis or Candidate Gene Association Studies
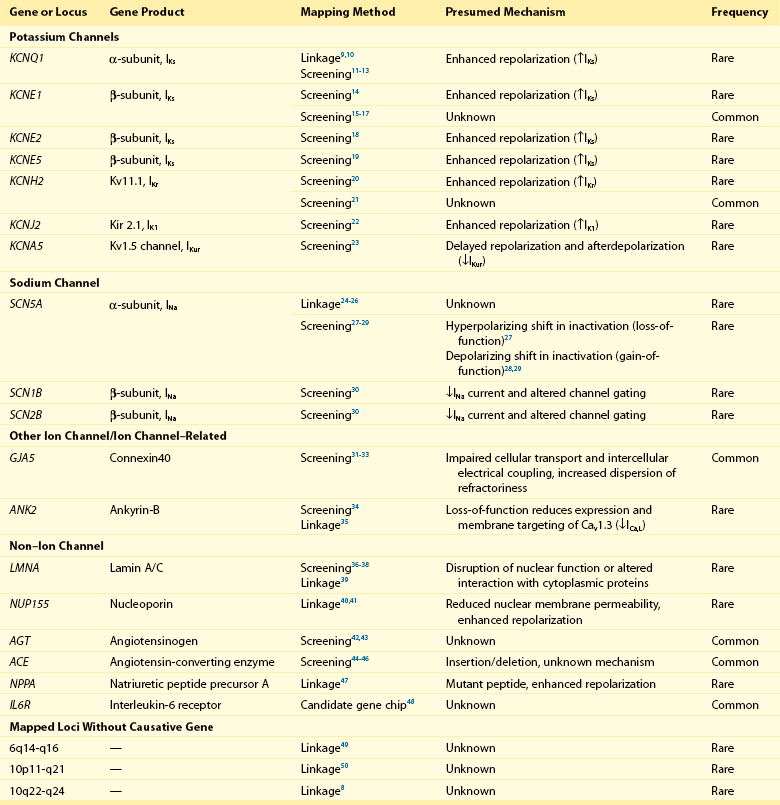
Chen et al narrowed an AF susceptibility locus to a region on chromosome 11p15 in a family with autosomal dominant AF spanning four generations.9 Investigators extended their analysis by sequencing KCNQ1, a candidate gene in the region that encodes a potassium channel α-subunit. In the first transmembrane segment of the channel, they discovered a highly conserved serine residue that was mutated to a glycine in all affected family members. Subsequent characterization revealed that the mutant protein increases the repolarizing IKs current density when expressed with the KCNE1-subunit. This gain-of-function mutation is expected to result in shortened atrial refractory periods and thereby promote reentry, a well-founded mechanism underlying AF.51 Our group used a similar approach to map a mutation in the third transmembrane domain of KCNQ1.10 The S209P variant demonstrated a similar gain-of-function effect again implicating this as a disease susceptibility gene for AF.
Linkage analysis also has identified mutations in SCN5A that segregate with AF, conduction abnormalities, cardiomyopathy, and possibly early-onset ischemic stroke.24–26 SCN5A encodes a sodium channel α-subunit responsible for the depolarizing INa current. The D1275N mutation that results in an aspartic acid for asparagine substitution was independently identified by different investigators,24–26 and associates with atrial standstill when co-segregating with connexin-40 (GJA5) mutations.52
In yet another large multigenerational family with prevalent AF, Hodgson-Zingman et al mapped a frameshift mutation to NPPA, which encodes atrial natriuretic peptide.47 The two–base-pair deletion eliminates a stop codon, resulting in an additional 12 amino acids at the carboxy terminus of the mature 28-residue long atrial natriuretic peptide (ANP) protein. Ex vivo rabbit hearts bathed in the mutant peptide demonstrated significantly shortened atrial effective refractory periods as compared with those bathed with a wild-type atrial natriuretic peptide, again consistent with reentry as a pathogenic model for AF.
In aggregate, linkage analysis has implicated potassium (KCNQ19,10) and sodium (SCN5A24–26) channel mutations, a nuclear envelope protein (NUP15540,41), NPPA,47 and loci on chromosomes 649 and 10.8,50 Newer genetic techniques such as exome and whole genome sequencing have emerged in recent years that make mapping of these families even more efficient. However, the large, multigenerational families needed for linkage analysis remain rare. More often AF is observed in smaller families and thus is it is often hard to establish the causality of apparently disease-causing mutations with such limited genetic information.
Candidate Gene Association Studies
In contrast to linkage mapping, in which inferences about recombination are made on the basis of segregation of markers in a pedigree, investigators have also selected and screened candidate genes for mutations in cohorts of patients with and without AF. As an extrapolation of initial studies on KCNQ1, numerous additional candidate gene association studies have focused on cardiac ion channels (see Table 49-1).
In one report, genetic variants in KCNQ1 were identified that predisposed to AF in a stretch-sensitive fashion,12 illustrating the potential for a concealed predisposition for AF to be elicited by an acquired exposure (e.g., valvular disease, heart failure). In another report implicating KCNQ1, a mutation was identified in a patient with long QT syndrome.13 Diverging effects on IKs were observed depending on whether the mutant protein was expressed with KCNE1 or KCNE2 β-subunits, each of which are differentially expressed in the atria and ventricles, leading the authors to speculate that the mutation was the cause of both long QT syndrome and AF. Gain-of-function mutations in both KCNQ111 and KCNH220 have been discovered in AF in the context of short QT syndrome.
In contrast to the enhanced atrial repolarization mechanism invoked by most discovered potassium channel mutations, a nucleotide substitution resulting in a premature stop codon in KCNA5 has been described that manifested a loss-of-function of the Kv1.5 channel protein.23 The mutation effectively abolished IKur and prolonged the action potential duration, increasing susceptibility to early afterdepolarization-induced atrial tachycardia and fibrillation. This mechanism has been described as “atrial torsades,” and observations from this report underscore the heterogeneity of mechanisms that can lead to AF.
Candidate gene association studies in patients with AF also have identified variants in genes encoding sodium channel subunits. Both loss-of-function27 and gain-of-function28,29 mutations in SCN5A have been identified in patients with lone AF. Mutations in sodium channel β-subunits, SCN1B and SCN2B, have been described that decrease sodium current amplitude and alter channel gating kinetics when coexpressed with SCN5A.30 These mutations are speculated to predispose to AF through shortening of the atrial action potential duration or by conduction slowing, both of which may facilitate reentry.
Mutations in GJA5, which encodes the gap junction connexin-40, have been described in 4 of 15 patients with AF screened in one candidate gene association study.32 The GJA5 mutations in three of the four patients were present in cardiac tissue but not in lymphocyte specimens, suggesting that these mutations were acquired or somatic rather than inherited or germline mutations. Other candidate gene association studies have linked common genetic variation in GJA5 to AF.31,33
Non-ion channel variants also have been discovered in candidate gene association studies. In a recent large-scale cardiovascular candidate gene association study, an intronic variant in the IL6R receptor was associated with AF.48 Mutations in LMNA, which encodes the nuclear envelope proteins lamin A and lamin C, have been identified in patients with cardiomyopathy and AF.36,37 Mutations in these proteins underlie a diverse spectrum of disorders that include Emery-Dreifuss syndrome, Charcot-Marie-Tooth disease, and premature aging syndromes. Nevertheless, LMNA mutations appear to be rare causes of AF.38
Recently, loss-of-function variants in ANK2 have been identified in kindreds with early-onset AF, in which family members had frequently progressed to permanent AF.34 ANK2 encodes ankyrin-B, which was previously implicated in the long QT syndrome in a family in which a substantial proportion of members was affected by AF.35,53 Loss-of-function mutations in ANK2 appear to decrease the expression and trafficking of CaV 1.3 channels, resulting in decreased ICa,L current.34 Heterozygous ANK2 knockout mice exhibit a lack of discrete P waves and increased susceptibility to pacing-induced atrial arrhythmias, including AF.
Similar to linkage mapping, candidate gene association studies have provided insight into the pathogenic mechanisms of AF. These studies demonstrate that mutations in ion channels and genes hypothesized to be involved in AF are rare.54–56 Furthermore, like linkage analysis, candidate gene association studies have generally identified rare mutations underlying monogenic forms of AF that are private to individual families. The validity and generalizability of more common variants identified in some candidate gene association studies have been called into question owing to the lack of replication of many associations.57
Nevertheless, the potential importance of such rare variants was underscored in a recent analysis, in which sequencing for potassium channel mutations in a sample of 80 probands with AF and 240 controls demonstrated an excess number of nonsynonymous variants among those with AF.58 Despite their rarity, modeling of the effects of multiple potassium channel mutations suggested that the combination of such variants could result in substantial changes in action potential duration and dispersion of repolarization, potentially establishing a substrate for AF.
Genome-Wide Association Studies
Over the past decade, systematic efforts to examine human genetic variation such as the international HapMap project59 have resulted in appreciation of genetic diversity in different ancestral groups. The HapMap project revealed the presence of about 10 million common genetic variants occurring with a frequency of 5% or greater in the general population, most of which were single-nucleotide polymorphisms (SNPs). Such efforts provided a reference against which observed genetic variation could be compared in cohorts of individuals assembled for investigation. High-density chips were developed that allowed the simultaneous genotyping of hundreds of thousands of SNPs across the genome, enabling efficient genetic profiling of individuals.
Unique study design and interpretation considerations are applicable to genome-wide association studies. First, owing to the massive number of tests performed in each genome-wide association study, stringent significance thresholds (e.g., P < 5 × 10−8) are used to guard against false-positive test results that occur by chance with multiple hypothesis testing. Second, common genetic variants associated with disease are expected to confer small or modest disease risks, in contrast to rare and large-effect genetic variants observed in monogenic forms of AF. Third, the stringent significance thresholds used and the modest relative risks expected in genome-wide association studies necessitate extremely large sample sizes to adequately power genetic variant discovery. By convention genome-wide associations require replication in independent samples to claim validity, making these studies some of the largest human scientific experiments ever conducted. Diverse international collaborations have formed over the past several years and have contributed to the success of genome-wide association studies. As of 2011, 1617 genome-wide associations encompassing 249 human traits had been published.60
Genome-wide association studies have identified nine genomic regions associated with AF (Table 49-2 and Figure 49-2).61–65 Association signals at these loci often span tens of thousands of base pairs and encompass both genic and nongenic regions. Associated SNPs are not interpreted as causal genetic variants but rather as markers that tag causal genetic elements located nearby. To date, causal variants for AF have not been discovered. Nevertheless, candidate genes at these loci frequently exist, and have implicated transcription factors involved in cardiac and pulmonary development, ion channels, and cell signaling molecules (Figure 49-3). Functional work has begun to explore the mechanisms by which these genomic regions potentially underlie AF.
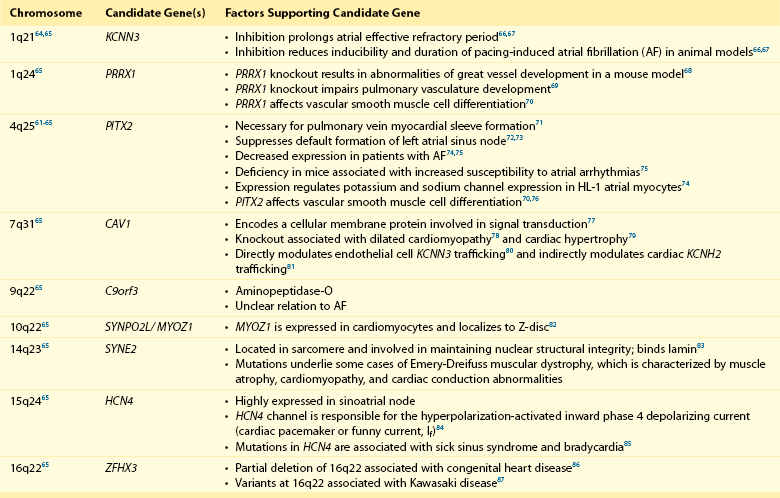
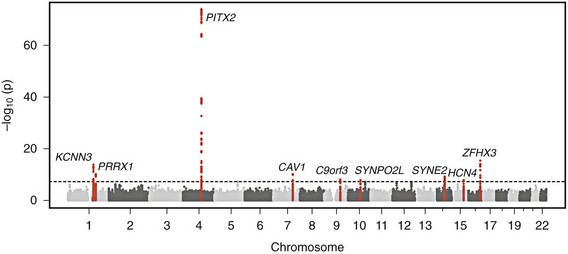
Figure 49-2 Nine Genetic Loci Associated With Atrial Fibrillation Discovered in Genome-Wide Association Studies
The chromosome and position of each of 2.2 million tested single-nucleotide polymorphisms are plotted on the x-axis, and the −log10 (P value) is plotted on the y-axis. Peaks above the dotted horizontal line are significantly associated with atrial fibrillation at P < 5 × 10−8. The closest gene at each associated locus is indicated. (Adapted with permission from Ellinor et al.65)
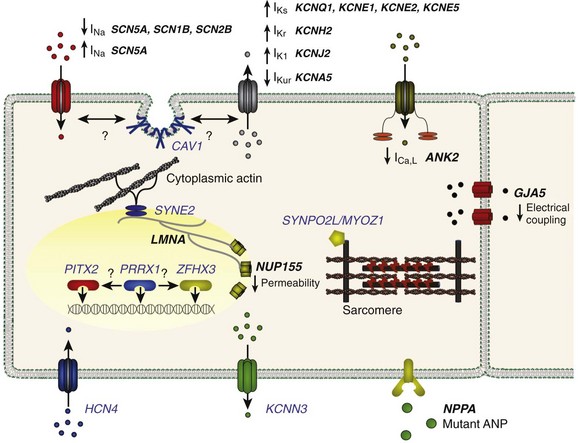
Figure 49-3 Genes Implicated in the Pathogenesis of Atrial Fibrillation
A depiction of a myocyte indicates the putative location and function of each implicated gene product. Genes associated with atrial fibrillation mapped via linkage or candidate gene testing are depicted in black. Potential candidate genes associated with atrial fibrillation mapped via genome-wide association studies are depicted in blue. (Figure courtesy Saagar Mahida, MB, ChB.)
The most significantly associated variants associated with AF exist at the chromosome 4q25 locus, and were reported in the first genome-wide association study for AF in 2007.61 Review of this discovery provides an example of the power of genome-wide association studies and the follow-up work involved in identifying causal elements at a particular susceptibility locus.
Investigators first identified genetic variants at the chromosome 4q25 locus that were highly associated with AF in approximately 550 individuals with and 4500 without AF from Iceland.61 The association between SNPs at chromosome 4q25 and AF has been widely replicated in samples of European,61,88–91 Han Chinese,61,92 African American,48,93 and Japanese65 ancestry, demonstrating the fundamental role of this locus in the pathophysiology of AF.
The mechanisms linking chromosome 4q25 to AF remain unclear. The top variants associated with AF lie in a region of the genome with no known genes, and are approximately 150 thousand base-pairs away from the nearest gene at the locus, the paired-like homeodomain 2 transcription factor (PITX2). Among the genes at 4q25, PITX2 is a plausible candidate for involvement in AF. PITX2 encodes a transcription factor that is involved in cardiac and pulmonary development. Mice deficient in one isoform of the protein that is expressed in the heart, Pitx2c, do not form myocardial sleeves in the pulmonary veins.71 Given the importance of pulmonary vein ectopic foci in the pathogenesis of AF,94 the relations between PITX2 and pulmonary vein myocardial cell development are particularly intriguing. Additionally, Pitx2c is responsible for suppressing default formation of a sinus node in the developing left atrial region.72,73
Decreased expression of Pitx2c was noted in patients with AF as compared with those with sinus rhythm,74,75 suggesting that Pitx2c deficiency or downregulation may predispose to AF. Increased inducibility of AF was observed in heterozygous Pitx2c knockout mice subjected to programmed atrial stimulation.75 Pitx2c expression also appears to regulate expression of certain sodium and potassium channels in HL-1 atrial cardiomyocytes, demonstrating a direct role for Pitx2c in cellular electrophysiology.74
How genetic variants at chromosome 4q25 associate with AF is unknown, but in light of evidence of long-range enhancer activity that has been oberved,95–97 it is possible that variants exert regulatory effects on distant gene targets. Indeed, independent susceptibility signals for AF at the chromosome 4q25 locus have been identified in conserved noncoding regions of chromosome 4q25.98 The number of risk alleles at three SNPs tagging these independent signals correlated with increased risk of AF in a sample of about 6000 individuals with AF and 32,000 without AF. Consideration of genotypes at these three SNPs identified a subset of 12% of individuals in the sample who had at least a 2-fold increased risk for AF, and 1% with at least a 6-fold increased risk for AF relative to those with the most common genotypes at each of the three SNPs.
Despite data implicating PITX2 in the pathogenesis of AF, genetic variants in PITX2 have not been identified through sequencing in cohorts of patients with AF.99 Mutations in PITX2 are found in Axenfeld-Rieger syndrome100 and Peters anomaly,101 which are characterized by ocular abnormalities but not AF.
After the discovery of AF-associated variants on chromosome 4q25, genome-wide association studies demonstrated additional associations on chromosome 16q2262,63 and 1q21.64 In 2012, the largest meta-analysis of genome-wide association studies of AF was published, which included 6707 individuals with and 52,426 individuals without AF.65 In addition to the three previously identified AF susceptibility loci, six novel loci for AF were discovered.65 Although causal variants at these loci have not been identified, candidate genes exist at most of these loci and are summarized in Table 49-2.
Future Directions
Understanding of the genetic basis of AF has improved rapidly in the past several years, yet numerous knowledge gaps still exist. The heritability underlying AF is incompletely explained by top variants at genetic loci.7 Despite associations between variation at some loci and AF across different ancestral groups, significant undiscovered genetic differences may underlie differences in AF prevalence between races.102 It is important to note that causal variants at established loci remain undiscovered.
References
1. Levy, RL. Paroxysmal auricular fibrillation and flutter without signs of organic cardiac disease in two brothers. J Mt Sinai Hosp. 1942; 8:765–770.
2. Wolff, L. Familial auricular fibrillation. N Engl J Med. 1993; 229:396–398.
3. Fox, CS, Parise, H, D’Agostino, RB, Sr., et al. Parental atrial fibrillation as a risk factor for atrial fibrillation in offspring. JAMA. 2004; 291:2851–2855.
4. Ellinor, PT, Yoerger, DM, Ruskin, JN, et al. Familial aggregation in lone atrial fibrillation. Hum Genet. 2005; 118:179–184.
5. Arnar, DO, Thorvaldsson, S, Manolio, TA, et al. Familial aggregation of atrial fibrillation in Iceland. Eur Heart J. 2006; 27:708–712.
6. Christophersen, IE, Ravn, LS, Budtz-Joergensen, E, et al. Familial aggregation of atrial fibrillation: A study in Danish twins. Circ Arrhythm Electrophysiol. 2009; 2:378–383.
7. Lubitz, SA, Yin, X, Fontes, JD, et al. Association between familial atrial fibrillation and risk of new-onset atrial fibrillation. JAMA. 2010; 304:2263–2269.
8. Brugada, R, Tapscott, T, Czernuszewicz, GZ, et al. Identification of a genetic locus for familial atrial fibrillation. N Engl J Med. 1997; 336:905–911.
9. Chen, YH, Xu, SJ, Bendahhou, S, et al. KCNQ1 gain-of-function mutation in familial atrial fibrillation. Science. 2003; 299:251–254.
10. Das, S, Makino, S, Melman, YF, et al. Mutation in the S3 segment of KCNQ1 results in familial lone atrial fibrillation. Heart Rhythm. 2009; 6:1146–1153.
11. Hong, K, Piper, DR, Diaz-Valdecantos, A, et al. De novo KCNQ1 mutation responsible for atrial fibrillation and short QT syndrome in utero. Cardiovasc Res. 2005; 68:433–440.
12. Otway, R, Vandenberg, JI, Guo, G, et al. Stretch-sensitive KCNQ1 mutation: A link between genetic and environmental factors in the pathogenesis of atrial fibrillation? J Am Coll Cardiol. 2007; 49:578–586.
13. Lundby, A, Ravn, LS, Svendsen, JH, et al. KCNQ1 mutation Q147R is associated with atrial fibrillation and prolonged QT interval. Heart Rhythm. 2007; 4:1532–1541.
14. Olesen, MS, Bentzen, BH, Nielsen, JB, et al. Mutations in the potassium channel subunit KCNE1 are associated with early-onset familial atrial fibrillation. BMC Med Genet. 2012; 13:24.
15. Lai, LP, Su, MJ, Yeh, HM, et al. Association of the human minK gene 38G allele with atrial fibrillation: Evidence of possible genetic control on the pathogenesis of atrial fibrillation. Am Heart J. 2002; 144:485–490.
16. Fatini, C, Sticchi, E, Genuardi, M, et al. Analysis of minK and eNOS genes as candidate loci for predisposition to non-valvular atrial fibrillation. Eur Heart J. 2006; 27:1712–1718.
17. Prystupa, A, Dzida, G, Myslinski, W, et al. MinK gene polymorphism in the pathogenesis of lone atrial fibrillation. Kardiol Pol. 2006; 64:1205–1211.
18. Yang, Y, Xia, M, Jin, Q, et al. Identification of a KCNE2 gain-of-function mutation in patients with familial atrial fibrillation. Am J Hum Genet. 2004; 75:899–905.
19. Ravn, LS, Aizawa, Y, Pollevick, GD, et al. Gain of function in IKs secondary to a mutation in KCNE5 associated with atrial fibrillation. Heart Rhythm. 2008; 5:427–435.
20. Hong, K, Bjerregaard, P, Gussak, I, et al. Short QT syndrome and atrial fibrillation caused by mutation in KCNH2. J Cardiovasc Electrophysiol. 2005; 16:394–396.
21. Sinner, MF, Pfeufer, A, Akyol, M, et al. The non-synonymous coding IKr-channel variant KCNH2-K897T is associated with atrial fibrillation: Results from a systematic candidate gene-based analysis of KCNH2 (HERG). Eur Heart J. 2008; 29:907–914.
22. Xia, M, Jin, Q, Bendahhou, S, et al. A Kir2. 1 gain-of-function mutation underlies familial atrial fibrillation. Biochem Biophys Res Commun. 2005; 332:1012–1019.
23. Olson, TM, Alekseev, AE, Liu, XK, et al. Kv1. 5 channelopathy due to KCNA5 loss-of-function mutation causes human atrial fibrillation. Hum Mol Genet. 2006; 15:2185–2191.
24. McNair, WP, Ku, L, Taylor, MR, et al. SCN5A mutation associated with dilated cardiomyopathy, conduction disorder, and arrhythmia. Circulation. 2004; 110:2163–2167.
25. Olson, TM, Michels, VV, Ballew, JD, et al. Sodium channel mutations and susceptibility to heart failure and atrial fibrillation. JAMA. 2005; 293:447–454.
26. Laitinen-Forsblom, PJ, Makynen, P, Makynen, H, et al. SCN5A mutation associated with cardiac conduction defect and atrial arrhythmias. J Cardiovasc Electrophysiol. 2006; 17:480–485.
27. Ellinor, PT, Nam, EG, Shea, MA, et al. Cardiac sodium channel mutation in atrial fibrillation. Heart Rhythm. 2008; 5:99–105.
28. Makiyama, T, Akao, M, Shizuta, S, et al. A novel SCN5A gain-of-function mutation M1875T associated with familial atrial fibrillation. J Am Coll Cardiol. 2008; 52:1326–1334.
29. Li, Q, Huang, H, Liu, G, et al. Gain-of-function mutation of Nav1. 5 in atrial fibrillation enhances cellular excitability and lowers the threshold for action potential firing. Biochem Biophys Res Commun. 2009; 380:132–137.
30. Watanabe, H, Darbar, D, Kaiser, DW, et al. Mutations in sodium channel beta1- and beta2-subunits associated with atrial fibrillation. Circ Arrhythm Electrophysiol. 2009; 2:268–275.
31. Firouzi, M, Ramanna, H, Kok, B, et al. Association of human connexin40 gene polymorphisms with atrial vulnerability as a risk factor for idiopathic atrial fibrillation. Circ Res. 2004; 95:e29–e33.
32. Gollob, MH, Jones, DL, Krahn, AD, et al. Somatic mutations in the connexin 40 gene (GJA5) in atrial fibrillation. N Engl J Med. 2006; 354:2677–2688.
33. Juang, JM, Chern, YR, Tsai, CT, et al. The association of human connexin 40 genetic polymorphisms with atrial fibrillation. Int J Cardiol. 2007; 116:107–112.
34. Cunha, SR, Hund, TJ, Hashemi, S, et al. Defects in ankyrin-based membrane protein targeting pathways underlie atrial fibrillation. Circulation. 2011; 124:1212–1222.
35. Schott, JJ, Charpentier, F, Peltier, S, et al. Mapping of a gene for long QT syndrome to chromosome 4q25-27. Am J Hum Genet. 1995; 57:1114–1122.
36. Fatkin, D, MacRae, C, Sasaki, T, et al. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N Engl J Med. 1999; 341:1715–1724.
37. Sebillon, P, Bouchier, C, Bidot, LD, et al. Expanding the phenotype of LMNA mutations in dilated cardiomyopathy and functional consequences of these mutations. J Med Genet. 2003; 40:560–567.
38. Brauch, KM, Chen, LY, Olson, TM. Comprehensive mutation scanning of LMNA in 268 patients with lone atrial fibrillation. Am J Cardiol. 2009; 103:1426–1428.
39. Pan, H, Richards, AA, Zhu, X, et al. A novel mutation in LAMIN A/C is associated with isolated early-onset atrial fibrillation and progressive atrioventricular block followed by cardiomyopathy and sudden cardiac death. Heart Rhythm. 2009; 6:707–710.
40. Oberti, C, Wang, L, Li, L, et al. Genome-wide linkage scan identifies a novel genetic locus on chromosome 5p13 for neonatal atrial fibrillation associated with sudden death and variable cardiomyopathy. Circulation. 2004; 110:3753–3759.
41. Zhang, X, Chen, S, Yoo, S, et al. Mutation in nuclear pore component NUP155 leads to atrial fibrillation and early sudden cardiac death. Cell. 2008; 135:1017–1027.
42. Tsai, CT, Lai, LP, Lin, JL, et al. Renin-angiotensin system gene polymorphisms and atrial fibrillation. Circulation. 2004; 109:1640–1646.
43. Wang, QS, Li, YG, Chen, XD, et al. Angiotensinogen polymorphisms and acquired atrial fibrillation in Chinese. J Electrocardiol. 2010; 43:373–377.
44. Bedi, M, McNamara, D, London, B, et al. Genetic susceptibility to atrial fibrillation in patients with congestive heart failure. Heart Rhythm. 2006; 3:808–812.
45. Fatini, C, Sticchi, E, Gensini, F, et al. Lone and secondary nonvalvular atrial fibrillation: Role of a genetic susceptibility. Int J Cardiol. 2007; 120:59–65.
46. Watanabe, H, Kaiser, DW, Makino, S, et al. ACE I/D polymorphism associated with abnormal atrial and atrioventricular conduction in lone atrial fibrillation and structural heart disease: Implications for electrical remodeling. Heart Rhythm. 2009; 6:1327–1332.
47. Hodgson-Zingman, DM, Karst, ML, Zingman, LV, et al. Atrial natriuretic peptide frameshift mutation in familial atrial fibrillation. N Engl J Med. 2008; 359:158–165.
48. Schnabel, RB, Kerr, KF, Lubitz, SA, et al. Large-scale candidate gene analysis in whites and African Americans identifies IL6R polymorphism in relation to atrial fibrillation: The National Heart, Lung, and Blood Institute’s Candidate Gene Association Resource (CARe) project. Circ Cardiovasc Genet. 2011; 4:557–564.
49. Ellinor, PT, Shin, JT, Moore, RK, et al. Locus for atrial fibrillation maps to chromosome 6q14-16. Circulation. 2003; 107:2880–2883.
50. Volders, PG, Zhu, Q, Timmermans, C, et al. Mapping a novel locus for familial atrial fibrillation on chromosome 10p11-q21. Heart Rhythm. 2007; 4:469–475.
51. Nattel, S. New ideas about atrial fibrillation 50 years on. Nature. 2002; 415:219–226.
52. Groenewegen, WA, Firouzi, M, Bezzina, CR, et al. A cardiac sodium channel mutation cosegregates with a rare connexin40 genotype in familial atrial standstill. Circ Res. 2003; 92:14–22.
53. Mohler, PJ, Schott, J-J, Gramolini, AO, et al. Ankyrin-B mutation causes type 4 long-QT cardiac arrhythmia and sudden cardiac death. Nature. 2003; 421:634–639.
54. Ellinor, PT, MacRae, CA. Ion channel mutations in AF: Signal or noise? Heart Rhythm. 2008; 5:436–437.
55. Ellinor, PT, Moore, RK, Patton, KK, et al. Mutations in the long QT gene, KCNQ1, are an uncommon cause of atrial fibrillation. Heart. 2004; 90:1487–1488.
56. Ellinor, PT, Petrov-Kondratov, VI, Zakharova, E, et al. Potassium channel gene mutations rarely cause atrial fibrillation. BMC Med Genet. 2006; 7:70.
57. Sinner, MF, Lubitz, SA, Pfeufer, A, et al. Lack of replication in polymorphisms reported to be associated with atrial fibrillation. Heart Rhythm. 2011; 8:403–409.
58. Mann, SA, Otway, R, Guo, G, et al. Epistatic effects of potassium channel variation on cardiac repolarization and atrial fibrillation risk. J Am Coll Cardiol. 2012; 59:1017–1025.
59. The International HapMap Project. Nature. 2003; 426:789–796.
60. Hindorff, LA, Junkins, HA, Mehta, JP, et al. A catalog of published genome-wide association studies. http://www.genome.gov/gwastudies, 2011. [Accessed August 2].
61. Gudbjartsson, DF, Arnar, DO, Helgadottir, A, et al. Variants conferring risk of atrial fibrillation on chromosome 4q25. Nature. 2007; 448:353–357.
62. Benjamin, EJ, Rice, KM, Arking, DE, et al. Variants in ZFHX3 are associated with atrial fibrillation in individuals of European ancestry. Nat Genet. 2009; 41:879–881.
63. Gudbjartsson, DF, Holm, H, Gretarsdottir, S, et al. A sequence variant in ZFHX3 on 16q22 associates with atrial fibrillation and ischemic stroke. Nat Genet. 2009; 41:876–878.
64. Ellinor, PT, Lunetta, KL, Glazer, NL, et al. Common variants in KCNN3 are associated with lone atrial fibrillation. Nat Genet. 2010; 42:240–244.
65. Ellinor, PT, Lunetta, KL, Albert, CM, et al. Meta-analysis identifies six new susceptibility loci for atrial fibrillation. Nat Genet. 2012; 44:670–675.
66. Diness, JG, Sorensen, US, Nissen, JD, et al. Inhibition of small-conductance Ca2+-activated K+ channels terminates and protects against atrial fibrillation. Circ Arrhythm Electrophysiol. 2010; 3:380–390.
67. Skibsbye, L, Diness, JG, Sorensen, US, et al. The duration of pacing-induced atrial fibrillation is reduced in vivo by inhibition of small conductance Ca(2+)-activated K(+) channels. J Cardiovasc Pharmacol. 2011; 57:672–681.
68. Bergwerff, M, Gittenberger-de Groot, AC, Wisse, LJ, et al. Loss of function of the Prx1 and Prx2 homeobox genes alters architecture of the great elastic arteries and ductus arteriosus. Virchows Arch. 2000; 436:12–19.
69. Ihida-Stansbury, K, McKean, DM, Gebb, SA, et al. Paired-related homeobox gene Prx1 is required for pulmonary vascular development. Circ Res. 2004; 94:1507–1514.
70. Shang, Y, Yoshida, T, Amendt, BA, et al. Pitx2 is functionally important in the early stages of vascular smooth muscle cell differentiation. J Cell Biol. 2008; 181:461–473.
71. Mommersteeg, MT, Brown, NA, Prall, OW, et al. Pitx2c and Nkx2-5 are required for the formation and identity of the pulmonary myocardium. Circ Res. 2007; 101:902–909.
72. Wang, J, Klysik, E, Sood, S, et al. Pitx2 prevents susceptibility to atrial arrhythmias by inhibiting left-sided pacemaker specification. Proc Natl Acad Sci U S A. 2010; 107:9753–9758.
73. Mommersteeg, MT, Hoogaars, WM, Prall, OW, et al. Molecular pathway for the localized formation of the sinoatrial node. Circ Res. 2007; 100:354–362.
74. Chinchilla, A, Daimi, H, Lozano-Velasco, E, et al. PITX2 insufficiency leads to atrial electrical and structural remodeling linked to arrhythmogenesis. Circ Cardiovasc Genet. 2011; 4:269–279.
75. Kirchhof, P, Kahr, PC, Kaese, S, et al. PITX2c is expressed in the adult left atrium, and reducing Pitx2c expression promotes atrial fibrillation inducibility and complex changes in gene expression. Circ Cardiovasc Genet. 2011; 4:123–133.
76. Yoshida, T, Hoofnagle, MH, Owens, GK. Myocardin and Prx1 contribute to angiotensin II-induced expression of smooth muscle alpha-actin. Circ Res. 2004; 94:1075–1082.
77. Gratton, JP, Bernatchez, P, Sessa, WC. Caveolae and caveolins in the cardiovascular system. Circ Res. 2004; 94:1408–1417.
78. Zhao, YY, Liu, Y, Stan, RV, et al. Defects in caveolin-1 cause dilated cardiomyopathy and pulmonary hypertension in knockout mice. Proc Natl Acad Sci U S A. 2002; 99:11375–11380.
79. Cohen, AW, Park, DS, Woodman, SE, et al. Caveolin-1 null mice develop cardiac hypertrophy with hyperactivation of p42/44 MAP kinase in cardiac fibroblasts. Am J Physiol Cell Physiol. 2003; 284:C457–C474.
80. Lin, MT, Adelman, JP, Maylie, J. Modulation of endothelial SK3 channel activity by Ca2+-dependent caveolar trafficking. Am J Physiol Cell Physiol. 2012; 303:C318–C327.
81. Lin, J, Lin, S, Choy, PC, et al. The regulation of the cardiac potassium channel (HERG) by caveolin-1. Biochem Cell Biol. 2008; 86:405–415.
82. Frey, N, Olson, EN. Calsarcin-3, a novel skeletal muscle-specific member of the calsarcin family, interacts with multiple Z-disc proteins. J Biol Chem. 2002; 277:13998–14004.
83. Zhang, Q, Ragnauth, CD, Skepper, JN, et al. Nesprin-2 is a multi-isomeric protein that binds lamin and emerin at the nuclear envelope and forms a subcellular network in skeletal muscle. J Cell Sci. 2005; 118(Pt 4):673–687.
84. DiFrancesco, D. The role of the funny current in pacemaker activity. Circ Res. 2010; 106:434–446.
85. Dobrzynski, H, Boyett, MR, Anderson, RH. New insights into pacemaker activity: Promoting understanding of sick sinus syndrome. Circulation. 2007; 115:1921–1932.
86. Yamamoto, T, Dowa, Y, Ueda, H, et al. Tetralogy of Fallot associated with pulmonary atresia and major aortopulmonary collateral arteries in a patient with interstitial deletion of 16q21-q22. 1. Am J Med Genet A. 2008; 146A:1575–1580.
87. Burgner, D, Davila, S, Breunis, WB, et al. A genome-wide association study identifies novel and functionally related susceptibility loci for Kawasaki disease. PLoS Genet. 2009; 5:e1000319.
88. Kaab, S, Darbar, D, van Noord, C, et al. Large scale replication and meta-analysis of variants on chromosome 4q25 associated with atrial fibrillation. Eur Heart J Apr. 2009; 30:813–819.
89. Body, SC, Collard, CD, Shernan, SK, et al. Variation in the 4q25 chromosomal locus predicts atrial fibrillation after coronary artery bypass graft surgery. Circ Cardiovasc Genet. 2009; 2:499–506.
90. Kiliszek, M, Franaszczyk, M, Kozluk, E, et al. Association between variants on chromosome 4q25, 16q22 and 1q21 and atrial fibrillation in the Polish population. PLoS ONE. 2011; 6:e21790.
91. Viviani Anselmi, C, Novelli, V, Roncarati, R, et al. Association of rs2200733 at 4q25 with atrial flutter/fibrillation diseases in an Italian population. Heart. 2008; 94:1394–1396.
92. Shi, L, Li, C, Wang, C, et al. Assessment of association of rs2200733 on chromosome 4q25 with atrial fibrillation and ischemic stroke in a Chinese Han population. Hum Genet. 2009; 126:843–849.
93. Delaney, JT, Jeff, JM, Brown, NJ, et al. Characterization of genome-wide association-identified variants for atrial fibrillation in African Americans. PLoS ONE. 2012; 7:e32338.
94. Haissaguerre, M, Jais, P, Shah, DC, et al. Spontaneous initiation of atrial fibrillation by ectopic beats originating in the pulmonary veins. N Engl J Med. 1998; 339:659–666.
95. Nobrega, MA, Ovcharenko, I, Afzal, V, et al. Scanning human gene deserts for long-range enhancers. Science. 2003; 302:413.
96. Lettice, LA, Heaney, SJ, Purdie, LA, et al. A long-range Shh enhancer regulates expression in the developing limb and fin and is associated with preaxial polydactyly. Hum Mol Genet. 2003; 12:1725–1735.
97. Kleinjan, DA, van Heyningen, V. Long-range control of gene expression: Emerging mechanisms and disruption in disease. Am J Hum Genet. 2005; 76:8–32.
98. Lubitz, SA, Sinner, MF, Lunetta, KL, et al. Independent susceptibility markers for atrial fibrillation on chromosome 4q25. Circulation. 2010; 122:976–984.
99. Boldt, LH, Posch, MG, Perrot, A, et al. Mutational analysis of the PITX2 and NKX2-5 genes in patients with idiopathic atrial fibrillation. Int J Cardiol. 2010; 145:316–317.
100. Semina, EV, Reiter, R, Leysens, NJ, et al. Cloning and characterization of a novel bicoid-related homeobox transcription factor gene, RIEG, involved in Rieger syndrome. Nat Genet. 1996; 14:392–399.
101. Doward, W, Perveen, R, Lloyd, IC, et al. A mutation in the RIEG1 gene associated with Peters’ anomaly. J Med Genet. 1999; 36:152–155.
102. Marcus, GM, Alonso, A, Peralta, CA, et al. European ancestry as a risk factor for atrial fibrillation in African Americans. Circulation. 2010; 122:2009–2015.
[/level-membership-for-cardiovascular-category][not-level-membership-for-cardiovascular-category]49
Genetics of Atrial Fibrillation
Heritability of AF
Reports of familial clustering of AF date back to the early 1940s.1,2 Familial AF was generally regarded as a rare condition for many years thereafter. However, over the past decade a major paradigm shift occurred with widespread recognition of the heritability underlying AF.3–7 In the community-based Framingham Heart Study, 27% of individuals with AF had a first-degree relative with AF confirmed by electrocardiography.7 Familial AF was associated with a 40% increased risk of AF for another family member over a subsequent 8-year period (Figure 49-1). The risk associated with familial AF remained even after adjustment for established clinical risk factors for AF. A study from Denmark demonstrated that AF was more common among monozygotic twins as compared with dizygotic twins, implicating a genetic predisposition to the arrhythmia even among those raised with shared environmental exposures.6
Figure 49-1 The risk of atrial fibrillation is increased in individuals with a first-degree relative with antecedent atrial fibrillation in the Framingham Heart Study. The estimated 8-year risk of atrial fibrillation (AF) is increased by 40% among individuals with a first-degree relative with atrial fibrillation as compared with those without familial atrial fibrillation. (Adapted with permission from Lubitz et al.7)
In numerous reports, the heritability of AF appears to be greatest among younger individuals3,5,7 and those without structural heart disease.3,4 Premature familial AF, or that occurring in family members ≤65 years of age, was associated with a 2-fold increase in the risk of AF compared with individuals without familial AF in the Framingham Heart Study.7 Nevertheless, data from Framingham provide evidence that the heritability of AF is present in the elderly as well.7
Genetic Mapping of AF
Linkage Analysis and Candidate Gene Resequencing
The first genetic locus for AF was described in 1997 by Brugada et al, who identified an AF susceptibility region on chromosome 10 in a large family with autosomal dominant AF (Table 49-1).8 Since this initial report, linkage analysis has repeatedly been used to identify genetic mutations underlying AF in a number of large families with AF.
Table 49-1
Atrial Fibrillation Susceptibility Loci Identified by Linkage Analysis or Candidate Gene Association Studies
Chen et al narrowed an AF susceptibility locus to a region on chromosome 11p15 in a family with autosomal dominant AF spanning four generations.9 Investigators extended their analysis by sequencing KCNQ1, a candidate gene in the region that encodes a potassium channel α-subunit. In the first transmembrane segment of the channel, they discovered a highly conserved serine residue that was mutated to a glycine in all affected family members. Subsequent characterization revealed that the mutant protein increases the repolarizing IKs current density when expressed with the KCNE1-subunit. This gain-of-function mutation is expected to result in shortened atrial refractory periods and thereby promote reentry, a well-founded mechanism underlying AF.51 Our group used a similar approach to map a mutation in the third transmembrane domain of KCNQ1.10 The S209P variant demonstrated a similar gain-of-function effect again implicating this as a disease susceptibility gene for AF.
Linkage analysis also has identified mutations in SCN5A that segregate with AF, conduction abnormalities, cardiomyopathy, and possibly early-onset ischemic stroke.24–26 SCN5A encodes a sodium channel α-subunit responsible for the depolarizing INa current. The D1275N mutation that results in an aspartic acid for asparagine substitution was independently identified by different investigators,24–26 and associates with atrial standstill when co-segregating with connexin-40 (GJA5) mutations.52
In yet another large multigenerational family with prevalent AF, Hodgson-Zingman et al mapped a frameshift mutation to NPPA, which encodes atrial natriuretic peptide.47 The two–base-pair deletion eliminates a stop codon, resulting in an additional 12 amino acids at the carboxy terminus of the mature 28-residue long atrial natriuretic peptide (ANP) protein. Ex vivo rabbit hearts bathed in the mutant peptide demonstrated significantly shortened atrial effective refractory periods as compared with those bathed with a wild-type atrial natriuretic peptide, again consistent with reentry as a pathogenic model for AF.
In aggregate, linkage analysis has implicated potassium (KCNQ19,10) and sodium (SCN5A24–26) channel mutations, a nuclear envelope protein (NUP15540,41), NPPA,47 and loci on chromosomes 649 and 10.8,50 Newer genetic techniques such as exome and whole genome sequencing have emerged in recent years that make mapping of these families even more efficient. However, the large, multigenerational families needed for linkage analysis remain rare. More often AF is observed in smaller families and thus is it is often hard to establish the causality of apparently disease-causing mutations with such limited genetic information.
Candidate Gene Association Studies
In contrast to linkage mapping, in which inferences about recombination are made on the basis of segregation of markers in a pedigree, investigators have also selected and screened candidate genes for mutations in cohorts of patients with and without AF. As an extrapolation of initial studies on KCNQ1, numerous additional candidate gene association studies have focused on cardiac ion channels (see Table 49-1).
In one report, genetic variants in KCNQ1 were identified that predisposed to AF in a stretch-sensitive fashion,12 illustrating the potential for a concealed predisposition for AF to be elicited by an acquired exposure (e.g., valvular disease, heart failure). In another report implicating KCNQ1, a mutation was identified in a patient with long QT syndrome.13 Diverging effects on IKs were observed depending on whether the mutant protein was expressed with KCNE1 or KCNE2 β-subunits, each of which are differentially expressed in the atria and ventricles, leading the authors to speculate that the mutation was the cause of both long QT syndrome and AF. Gain-of-function mutations in both KCNQ111 and KCNH220 have been discovered in AF in the context of short QT syndrome.
In contrast to the enhanced atrial repolarization mechanism invoked by most discovered potassium channel mutations, a nucleotide substitution resulting in a premature stop codon in KCNA5 has been described that manifested a loss-of-function of the Kv1.5 channel protein.23 The mutation effectively abolished IKur and prolonged the action potential duration, increasing susceptibility to early afterdepolarization-induced atrial tachycardia and fibrillation. This mechanism has been described as “atrial torsades,” and observations from this report underscore the heterogeneity of mechanisms that can lead to AF.
Candidate gene association studies in patients with AF also have identified variants in genes encoding sodium channel subunits. Both loss-of-function27 and gain-of-function28,29 mutations in SCN5A have been identified in patients with lone AF. Mutations in sodium channel β-subunits, SCN1B and SCN2B, have been described that decrease sodium current amplitude and alter channel gating kinetics when coexpressed with SCN5A.30 These mutations are speculated to predispose to AF through shortening of the atrial action potential duration or by conduction slowing, both of which may facilitate reentry.
Mutations in GJA5, which encodes the gap junction connexin-40, have been described in 4 of 15 patients with AF screened in one candidate gene association study.32 The GJA5 mutations in three of the four patients were present in cardiac tissue but not in lymphocyte specimens, suggesting that these mutations were acquired or somatic rather than inherited or germline mutations. Other candidate gene association studies have linked common genetic variation in GJA5 to AF.31,33
Non-ion channel variants also have been discovered in candidate gene association studies. In a recent large-scale cardiovascular candidate gene association study, an intronic variant in the IL6R receptor was associated with AF.48 Mutations in LMNA, which encodes the nuclear envelope proteins lamin A and lamin C, have been identified in patients with cardiomyopathy and AF.36,37 Mutations in these proteins underlie a diverse spectrum of disorders that include Emery-Dreifuss syndrome, Charcot-Marie-Tooth disease, and premature aging syndromes. Nevertheless, LMNA mutations appear to be rare causes of AF.38
Recently, loss-of-function variants in ANK2 have been identified in kindreds with early-onset AF, in which family members had frequently progressed to permanent AF.34 ANK2 encodes ankyrin-B, which was previously implicated in the long QT syndrome in a family in which a substantial proportion of members was affected by AF.35,53 Loss-of-function mutations in ANK2 appear to decrease the expression and trafficking of CaV 1.3 channels, resulting in decreased ICa,L current.34 Heterozygous ANK2 knockout mice exhibit a lack of discrete P waves and increased susceptibility to pacing-induced atrial arrhythmias, including AF.
Similar to linkage mapping, candidate gene association studies have provided insight into the pathogenic mechanisms of AF. These studies demonstrate that mutations in ion channels and genes hypothesized to be involved in AF are rare.54–56 Furthermore, like linkage analysis, candidate gene association studies have generally identified rare mutations underlying monogenic forms of AF that are private to individual families. The validity and generalizability of more common variants identified in some candidate gene association studies have been called into question owing to the lack of replication of many associations.57
Nevertheless, the potential importance of such rare variants was underscored in a recent analysis, in which sequencing for potassium channel mutations in a sample of 80 probands with AF and 240 controls demonstrated an excess number of nonsynonymous variants among those with AF.58 Despite their rarity, modeling of the effects of multiple potassium channel mutations suggested that the combination of such variants could result in substantial changes in action potential duration and dispersion of repolarization, potentially establishing a substrate for AF.
Genome-Wide Association Studies
Over the past decade, systematic efforts to examine human genetic variation such as the international HapMap project59 have resulted in appreciation of genetic diversity in different ancestral groups. The HapMap project revealed the presence of about 10 million common genetic variants occurring with a frequency of 5% or greater in the general population, most of which were single-nucleotide polymorphisms (SNPs). Such efforts provided a reference against which observed genetic variation could be compared in cohorts of individuals assembled for investigation. High-density chips were developed that allowed the simultaneous genotyping of hundreds of thousands of SNPs across the genome, enabling efficient genetic profiling of individuals.
[/not-level-membership-for-cardiovascular-category]
