[level-membership-for-pediatrics-category]
Genetics
• common, with 2% of live-born babies having a significant congenital malformation and about 5% a genetic disorder
• burdensome to the affected individual, family and society, as many are associated with severe and permanent disability.
There has recently been an unprecedented growth in knowledge about the genetic basis of diseases :
• The Human Genome Project resulted in the first publication of the human genome sequence in 2001.
• It is now estimated that the human genome contains 20 000–25 000 genes, although the function of many of them remains unknown. Greater diversity and complexity at the protein level is achieved by alternative mRNA splicing and post-translational modification of gene products.
• Microarray techniques and high throughput sequencing are increasing the volume and speed of genetic investigations and reducing their costs, leading to a greater understanding of the impact of genetics on health and disease.
• Access to genome browser databases containing DNA sequence and protein structure has greatly enhanced progress in scientific research and the interpretation of clinical test results (Fig. 8.1).
• Genetic databases are available on thousands of multiple congenital anomaly syndromes, on chromosomal variations and disease phenotypes and on all Mendelian disorders.
• Clinical application of these advances is available to families through specialist genetic centres that offer investigation, diagnosis, counselling and antenatal diagnosis for an ever-widening range of disorders.
• Gene-based knowledge is entering mainstream medical and paediatric practice, especially in diagnosis and in therapeutic guidance, such as for the treatment of malignancies.
Genetically determined diseases include those resulting from:
Chromosomal abnormalities
Down syndrome (trisomy 21)
Clinical features
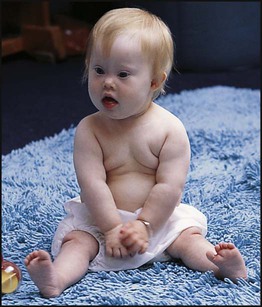
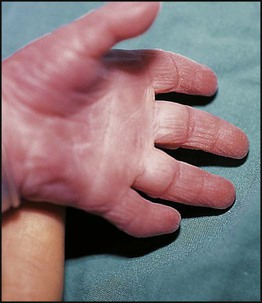
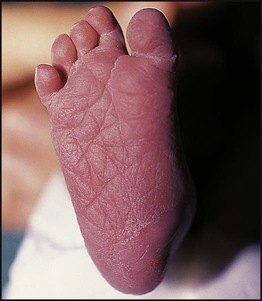
Down syndrome is usually suspected at birth because of the baby’s facial appearance. Most affected infants are hypotonic and other useful clinical signs include a flat occiput, single palmar creases, incurved fifth finger and wide ‘sandal’ gap between the big and second toe (Fig. 8.2a–c, Box 8.1). The diagnosis can be difficult to make when relying on clinical signs alone and a suspected diagnosis should be confirmed by a senior paediatrician. Before blood is sent for analysis, parents should be informed that a test for Down syndrome is being performed. The results may take 1–2 days, using rapid FISH (fluorescent in situ hybridisation) techniques. Parents need information about the short- and long-term implications of the diagnosis. They are also likely, at some stage in the future, to appreciate the opportunity to discuss how and why the condition has arisen, the risk of recurrence and the possibility of antenatal diagnosis in future pregnancies.
Cytogenetics
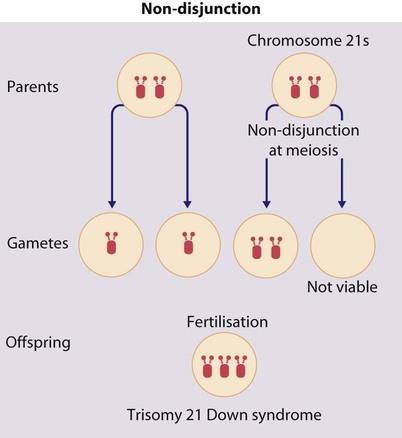
The extra chromosome 21 may result from meiotic non-disjunction, translocation or mosaicism.
Meiotic non-disjunction (94%)
In non-disjunction trisomy 21:
• most cases result from an error at meiosis
• the pair of chromosome 21s fails to separate, so that one gamete has two chromosome 21s and one has none (Fig. 8.3)
• fertilisation of the gamete with two chromosome 21s gives rise to a zygote with trisomy 21
The incidence of trisomy 21 due to non-disjunction is related to maternal age (Table 8.1). However, as the proportion of pregnancies in older mothers is small, most affected babies are born to younger mothers. Furthermore, meiotic non-disjunction can occur in spermatogenesis so that the extra 21 can be of paternal origin. All pregnant women are now offered screening tests measuring biochemical markers in blood samples and often also nuchal thickening on ultrasound (thickening of the soft tissues at the back of the neck) to identify an increased risk of Down syndrome in the fetus. When an increased risk is identified, amniocentesis is offered to check the fetal karyotype. After having one child with trisomy 21 due to non-disjunction, the risk of recurrence of Down syndrome is given as 1 in 200 for mothers under the age of 35 years, but remains similar to their age-related population risk for those over the age of 35 years.
Table 8.1
Risk of Down syndrome (live births) with maternal age at delivery, prior to screening in pregnancy
| Maternal age (years) | Risk of Down syndrome |
| All ages | 1 in 650 |
| 20 | 1 in 1530 |
| 30 | 1 in 900 |
| 35 | 1 in 385 |
| 37 | 1 in 240 |
| 40 | 1 in 110 |
| 44 | 1 in 37 |
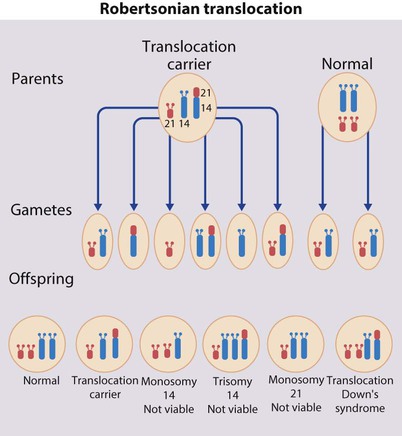
Translocation (5%)
When the extra chromosome 21 is joined onto another chromosome (usually chromosome 14, but occasionally chromosome 15, 22 or 21), this is known as a Robertsonian translocation. This may be present in a phenotypically normal carrier with 45 chromosomes (two being ‘joined together’) or in someone with Down syndrome and a set of 46 chromosomes but with three copies of chromosome 21 material. In this situation, parental chromosomal analysis is recommended, since one of the parents may well carry the translocation in balanced form (in 25% of cases) (Fig. 8.4).
In translocation Down syndrome:
• The risk of recurrence is 10–15% if the mother is the translocation carrier and about 2.5% if the father is the carrier.
• If a parent carries the rare 21 : 21 translocation, all the offspring will have Down syndrome.
• If neither parent carries a translocation (75% of cases), the risk of recurrence is <1%.
Edwards syndrome (trisomy 18) and Patau syndrome (trisomy 13)
Although rarer than Down syndrome (1 in 8000 and 1 in 14 000 live births, respectively), particular constellations of severe multiple abnormalities suggest these diagnoses at birth; most affected babies die in infancy (Fig. 8.5, Boxes 8.2 and 8.3). The diagnosis is confirmed by chromosome analysis. Many affected fetuses are detected by ultrasound scan during the second trimester of pregnancy and diagnosis can be confirmed antenatally by amniocentesis and chromosome analysis. Recurrence risk is low, except when the trisomy is due to a balanced chromosome rearrangement in one of the parents.
Turner syndrome (45, X)
Usually (>95%), Turner syndrome results in early miscarriage and is increasingly detected by ultrasound antenatally when fetal oedema of the neck, hands or feet or a cystic hygroma may be identified. In live-born females, the incidence is about 1 in 2500. Figure 8.6 and Box 8.4 show the clinical features of Turner syndrome, although short stature may be the only clinical abnormality in children.
• Oestrogen replacement for development of secondary sexual characteristics at the time of puberty (but infertility persists).
The incidence does not increase with maternal age and risk of recurrence is very low.

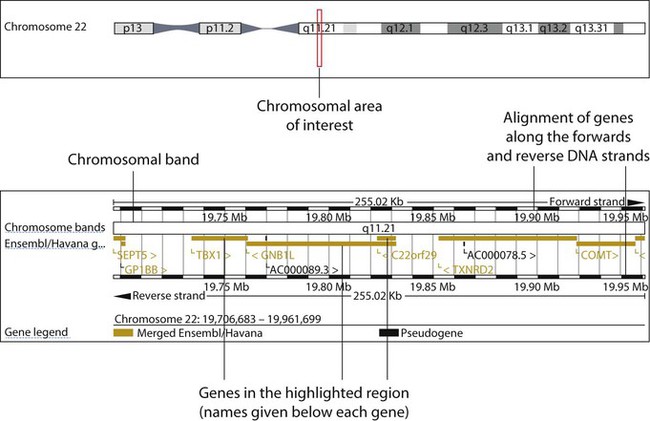
Deletions
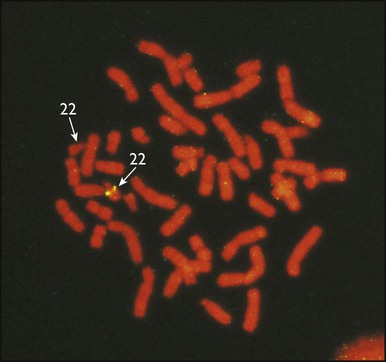
DiGeorge syndrome is associated with a deletion of band q11 on chromosome 22 (i.e. 22q11) (Fig. 8.7). Williams syndrome is another example of a microdeletion syndrome due to loss of chromosomal material at band q11 on the long arm of chromosome 7 (i.e. 7q11) (Fig. 8.18, see also Box 8.12).
Mendelian inheritance
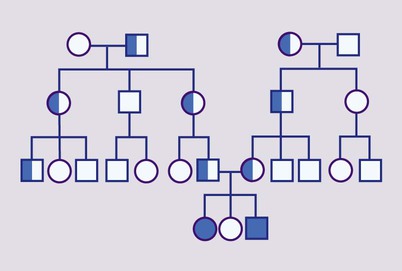
Mendelian inheritance, described by Mendel in garden peas in 1866, is the transmission of inherited traits or diseases caused by variation in a single gene in a characteristic pattern. These Mendelian traits or disorders are individually rare but collectively numerous and important: over 6000 have been described. For many disorders, the Mendelian pattern of inheritance is known. If the diagnosis of a condition is uncertain, its pattern of inheritance may be evident on drawing a family tree (pedigree), which is an essential part of genetic evaluation (Fig. 8.8).
Autosomal dominant inheritance
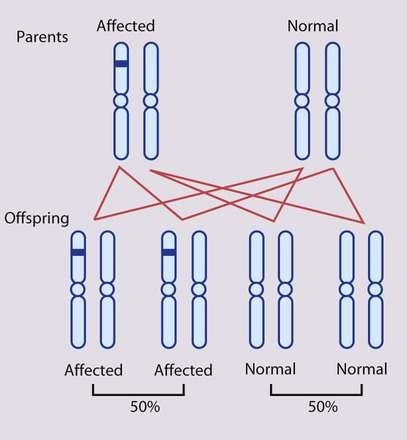
This is the most common mode of Mendelian inheritance (Box 8.6). Autosomal dominant conditions are caused by alterations in only one copy of a gene pair, i.e. the condition occurs in the heterozygous state despite the presence of an intact copy of the relevant gene. Autosomal dominant genes are located on the autosomes (chromosomes 1–22) so males and females are equally affected. Each child from an affected parent has a 1 in 2 (50%) chance of inheriting the abnormal gene (Fig. 8.9a, b). This appears to be straightforward, but complicating factors include the following factors.
No family history of the disorder
• A new mutation in one of the gametes leading to the conception of the affected person. This is the most common reason for absence of a family history in dominant disorders, e.g. >80% of individuals with achondroplasia have unaffected parents.
• Parental mosaicism – very occasionally a healthy parent harbours the mutation only in some of their cells, e.g. in their gonads. This can account for recurrences of autosomal dominant disorders in siblings born to apparently unaffected parents. It has been described in congenital lethal osteogenesis imperfecta.
• Non-paternity – if the apparent father is not the biological father.
Autosomal recessive inheritance
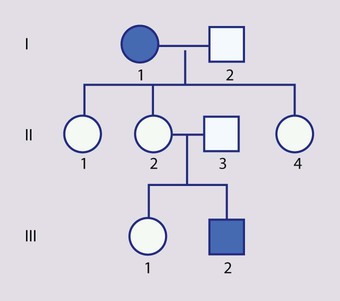
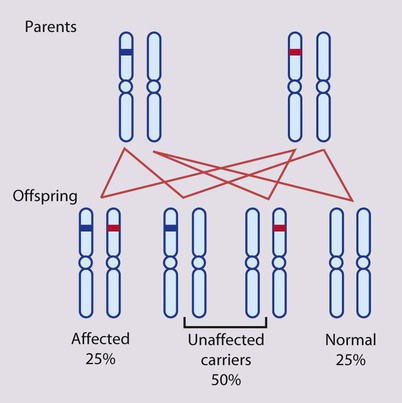
An affected individual is homozygous for the abnormal gene, having inherited an abnormal allele from each parent, both of whom are unaffected heterozygous carriers (Box 8.7). For two carrier parents, the risk of each child, male or female, being affected is 1 in 4 (25%) (Fig. 8.11a,b). All offspring of affected individuals will be carriers.
Autosomal recessive inheritance
X-linked inheritance
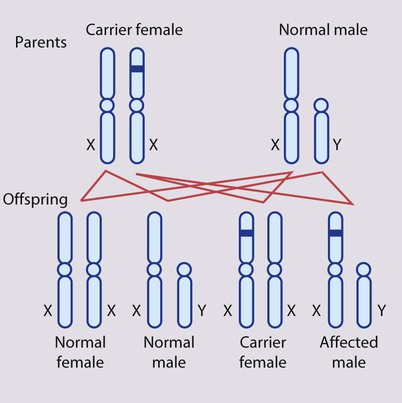
In X-linked recessive inheritance (Box 8.8, Fig. 8.12a,b):
• Female carriers are usually healthy
• Occasionally a female carrier shows mild signs of the disease (manifesting carrier)
• Each son of a female carrier has a 1 in 2 (50%) risk of being affected
• Each daughter of a female carrier has a 1 in 2 (50%) risk of being a carrier
• Daughters of affected males will all be carriers
• Sons of affected males will not be affected, since a man passes a Y chromosome to his son.
Unusual genetic mechanisms
Trinucleotide repeat expansion mutations
Fragile X syndrome
The prevalence of significant learning difficulties in males due to fragile X syndrome is about 1 in 4000 (Fig. 8.13 and Box 8.9). This condition was initially diagnosed on the basis of the appearance of an apparent gap or break (a fragile site) in the distal part of the long arm of the X chromosome. Diagnosis is now achieved by molecular analysis of the CGG trinucleotide repeat expansion in the relevant gene (FMR1).
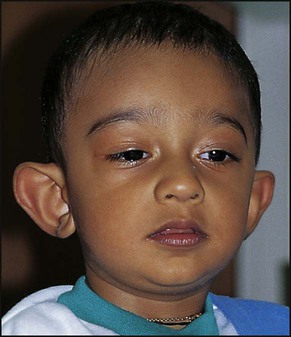
 Fragile X syndrome is the commonest familial form of learning difficulties and the second most common genetic cause of severe learning difficulties after Down syndrome.
Fragile X syndrome is the commonest familial form of learning difficulties and the second most common genetic cause of severe learning difficulties after Down syndrome.Imprinting and uniparental disomy
• De novo deletion (Fig. 8.14). Parental chromosomes are normal, and a deletion occurs as a new mutation in the child. If the deletion occurs on the paternal chromosome 15, the child has Prader–Willi syndrome. If the deletion affects the maternal chromosome 15, the child has Angelman syndrome.
• Uniparental disomy (Fig. 8.15). This is when a child inherits two copies of a chromosome from one parent and none from the other parent. In Prader–Willi syndrome the affected child has no paternal (but two maternal) copies of chromosome 15q11–13. In Angelman syndrome, the affected child has no maternal (but two paternal) copies of chromosome 15q11–13. This can be detected with DNA analysis.
• There exist other mechanisms that can lead to these conditions.
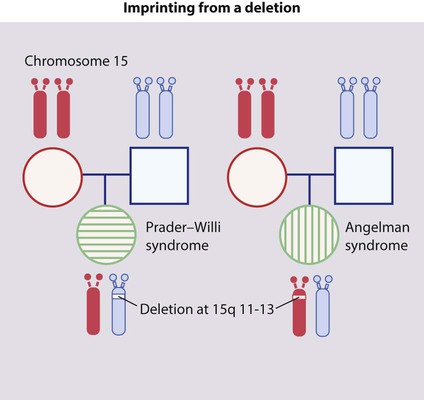
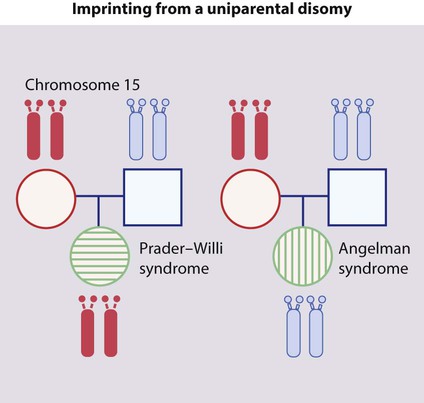
 Imprinting is the unusual property of some genes that express only the copy derived from the parent of a given sex.
Imprinting is the unusual property of some genes that express only the copy derived from the parent of a given sex.
Polygenic, multifactorial or complex inheritance
There is a spectrum in the aetiology of disease, from environmental factors (e.g. trauma) at one end to purely genetic causes (e.g. Mendelian disorder) at the other. Between these two extremes are many disorders which result from the interacting effects of several genes (hence the term polygenic) with or without the influence of environmental or other unknown factors, including chance (multifactorial or complex). The terms are used interchangeably (Box 8.10).
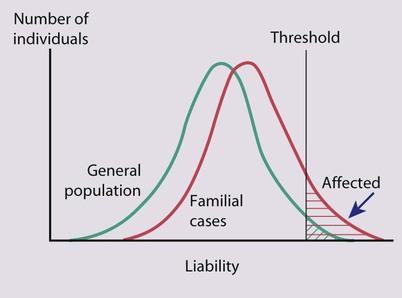
Normal quantitative traits such as height and intelligence are inherited in this fashion, with many relevant influences including genetic constitution, environmental exposures and early life (including intrauterine) experiences. These parameters show a Gaussian (or ‘normal’) distribution in the population. Similarly, the liability of an individual to develop a disease of multifactorial or polygenic aetiology has a Gaussian distribution. The condition occurs when a certain threshold level of liability is exceeded. Relatives of an affected person show an increased liability due to inheritance of genes conferring susceptibility, and so a greater proportion of them than in the general population will fall beyond the threshold and will manifest the disorder (Fig. 8.16). The risk of recurrence of a polygenic disorder in a family is usually low and is most significant for first-degree relatives. Empirical recurrence risk data are used for genetic counselling. They are derived from family studies that have reported the frequency at which various family members are affected. Factors that increase the risk to relatives are:
• having a more severe form of the disorder, e.g. the risk of recurrence to siblings is greater in bilateral cleft lip and palate than in unilateral cleft lip alone
• close relationship to the affected person, e.g. overall risk to siblings or children is greater than to more distant relatives
• multiple affected family members, e.g. the more siblings already affected, the greater the risk of recurrence
• sex difference in prevalence, with the recurrence risk greater in the more commonly affected sex and if the affected individual is of the less commonly affected sex.
Dysmorphology
Syndrome diagnosis
• likely complications which can be sought and perhaps treated successfully if detected early
• the avoidance of unnecessary investigations
• experience and information which parents can share with other affected families through family support groups.
Examples of syndromes recognisable by facial appearance are shown in Figures 8.17–8.19 (see also Boxes 8.11–8.13). The importance and impact of syndrome diagnosis is demonstrated in Case History 8.1. Databases are available to assist with the recognition of thousands of multiple congenital anomaly syndromes (e.g. London Dysmorphology Database (LDDB) & POSSUM).
Syndromes recognised by ‘Gestalt’ (clinical recognition)
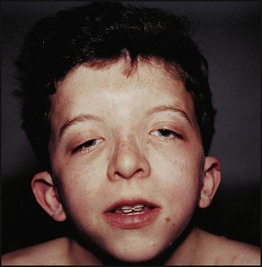
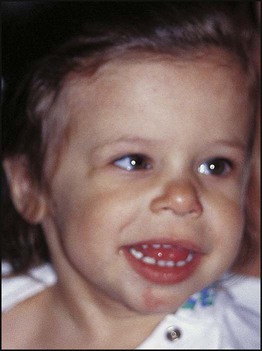
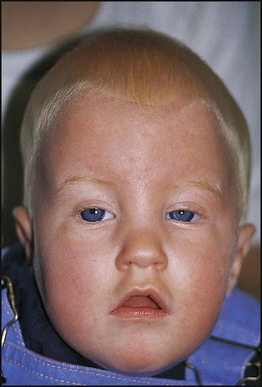
Case History
8.1 Syndrome diagnosis and genetic counselling
The parents asked about recurrence risk for congenital heart disease and were referred to the genetic clinic. At that time, Sean was thriving and early developmental progress appeared normal. On examination, there were minor dysmorphic features, including a short philtrum, thin upper lip and prominent ears (Fig. 8.20). There was no family history of congenital heart disease or other significant problems and no abnormalities were detected on examination of the parents.
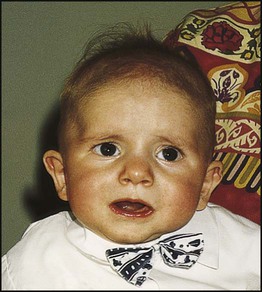
Gene-based therapies
The treatment of most genetic disorders is based on conventional therapeutic approaches.
• Suppressors of nonsense (stop codon, chain-terminating) mutations, which are under trial in patients with cystic fibrosis and Duchenne muscular dystrophy caused by the appropriate mutations.
• Antisense oligo-DNA molecules that cause the skipping of specific exons in the Duchenne muscular dystrophy gene, i.e. complementary DNA molecules that bind and block exons around the faulty part of the gene (often deletions or duplications in Duchenne muscular dystrophy). This approach can restore the reading frame of the message downstream of the causal deletion; it is hoped that this will result in a milder form of the disease.
Genetic services
Genetic investigations
• Confirmation of a clinical diagnosis of an increasing number of single gene disorders
• Detection of female carriers in X-linked disorders, e.g. Duchenne and Becker muscular dystrophies, haemophilia A and B
• Carrier detection in autosomal recessive disorders, e.g. cystic fibrosis
• Presymptomatic diagnosis in autosomal dominant disorders, e.g. Huntington disease, myotonic dystrophy, familial cancer syndromes
• Antenatal diagnosis of an increasing number of Mendelian conditions.
These are accomplished by the following.
Mutation analysis
• Deletions: large deletion mutations are common in a variety of disorders including Duchenne and Becker muscular dystrophies, alpha-thalassaemia and 21-hydroxylase deficiency (congenital adrenal hyperplasia).
• Point mutations and small deletions: these can be readily identified if the same mutation causes all cases of the disorder, as in sickle cell disease. For most disorders, however, there is a spectrum of mutations. About 78% of cystic fibrosis carriers in the UK possess the ΔF508 mutation, but over 900 other mutations have been identified. Most laboratories test for a certain number of the most common mutations in the population they serve.
• Trinucleotide repeat expansion mutations: these are readily tested for because the mutation in a given disease is virtually always at the same site and can be amplified from the same oligo-DNA primers used in the amplification by PCR: the only difference is the size of the repeat sequence, which can be determined from the size of the DNA fragment containing the repeat.
Genetic linkage
Some of the genetic investigations now available are summarised in Table 8.2.
Table 8.2
| Investigation | Application |
| Cytogenetic analysis – karyotype | Chromosomes stained and visualised under a microscope |
| Detects alterations in chromosome number and structural rearrangements; this method is being replaced by molecular methods such as CGH. | |
| Molecular cytogenetic analysis – FISH (fluorescent in situ hybridisation) | Fluorescent-labelled DNA probes to detect the presence, number and chromosomal location of specific chromosomal sequences |
| Useful for microdeletion syndromes | |
| Microarray comparative genomic hybridisation (aCGH) | Detects chromosomal imbalances using thousands of DNA probes to investigate a whole genome with much greater sensitivity than cytogenetic methods |
| DNA analysis | Polymerase chain reaction (PCR) to amplify the DNA and determine the sequence of the relevant gene |
| High throughput DNA sequencing | Rapid sequencing of whole genomes or many loci within the genome |
| Linkage disequilibrium and genome-wide association studies (GWAS) | Comparing the frequency of combinations of alleles at nearby loci in a given population to identify genetic variants associated with complex diseases |

Genetic counselling
• To understand their situation
• To make their own decisions about managing the disease or risk of disease, including decisions about genetic testing and reproduction
• To adjust to their situation of being affected by or at risk of the genetic condition.
• Listening to the questions and concerns of the patient, client or family.
• Establishing the correct diagnosis. This involves detailed history, examination and appropriate investigations that may include chromosome or DNA or other molecular genetic analysis, biochemical tests, X-rays and clinical photographs. Despite extensive investigation, including searching databases, the diagnosis may remain unknown, e.g. in children with learning disability and mild or non-specific dysmorphic features.
• Risk estimation. This requires both diagnostic and pedigree information. Drawing a pedigree of three generations is an essential part of a clinical genetic assessment. The mode of inheritance may be apparent from the pedigree even when the precise diagnosis is not known. In some cases it may not be possible to define a precise recurrence risk and uncertainty may remain, e.g. conditions that only affect one member of a family and are known to follow autosomal dominant inheritance in some families and autosomal recessive inheritance in others (genetic heterogeneity).
• Communication. Information must be presented in an understandable and unbiased way. Families often find written information helpful to refer back to and diagrams are often used to explain patterns of inheritance. The impact of saying ‘the recurrence risk is 5% or 1 in 20’ may be different from saying ‘the chance of an unaffected child is 95% or 19 out of 20’, and so both should be presented.
• Discussing options for management and prevention. If there appears to be a risk to offspring, all reproductive options should be discussed. These include not having (any more) children, reducing intended family size, taking the risk and proceeding with pregnancy or having antenatal diagnosis and selective termination of an affected fetus. For some couples donor insemination or ovum donation may be appropriate and for others achieving a pregnancy through IVF (in vitro fertilisation) and preimplantation diagnosis may be possible.
Counselling should be non-directive, but should also assist in the decision-making process (Box 8.14). Information from lay support groups may also be helpful.
 Genetic counselling aims to allow parents greater autonomy and choice in reproductive decisions.
Genetic counselling aims to allow parents greater autonomy and choice in reproductive decisions.
 Pre-symptomatic testing of disorders which manifest in adult life should not be performed until the individual can give informed consent.
Pre-symptomatic testing of disorders which manifest in adult life should not be performed until the individual can give informed consent.[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
Genetics
• common, with 2% of live-born babies having a significant congenital malformation and about 5% a genetic disorder
• burdensome to the affected individual, family and society, as many are associated with severe and permanent disability.
There has recently been an unprecedented growth in knowledge about the genetic basis of diseases :
• The Human Genome Project resulted in the first publication of the human genome sequence in 2001.
• It is now estimated that the human genome contains 20 000–25 000 genes, although the function of many of them remains unknown. Greater diversity and complexity at the protein level is achieved by alternative mRNA splicing and post-translational modification of gene products.
• Microarray techniques and high throughput sequencing are increasing the volume and speed of genetic investigations and reducing their costs, leading to a greater understanding of the impact of genetics on health and disease.
• Access to genome browser databases containing DNA sequence and protein structure has greatly enhanced progress in scientific research and the interpretation of clinical test results (Fig. 8.1).
• Genetic databases are available on thousands of multiple congenital anomaly syndromes, on chromosomal variations and disease phenotypes and on all Mendelian disorders.
• Clinical application of these advances is available to families through specialist genetic centres that offer investigation, diagnosis, counselling and antenatal diagnosis for an ever-widening range of disorders.
• Gene-based knowledge is entering mainstream medical and paediatric practice, especially in diagnosis and in therapeutic guidance, such as for the treatment of malignancies.
Genetically determined diseases include those resulting from:
Chromosomal abnormalities
Down syndrome (trisomy 21)
Clinical features
Down syndrome is usually suspected at birth because of the baby’s facial appearance. Most affected infants are hypotonic and other useful clinical signs include a flat occiput, single palmar creases, incurved fifth finger and wide ‘sandal’ gap between the big and second toe (Fig. 8.2a–c, Box 8.1). The diagnosis can be difficult to make when relying on clinical signs alone and a suspected diagnosis should be confirmed by a senior paediatrician. Before blood is sent for analysis, parents should be informed that a test for Down syndrome is being performed. The results may take 1–2 days, using rapid FISH (fluorescent in situ hybridisation) techniques. Parents need information about the short- and long-term implications of the diagnosis. They are also likely, at some stage in the future, to appreciate the opportunity to discuss how and why the condition has arisen, the risk of recurrence and the possibility of antenatal diagnosis in future pregnancies.
Cytogenetics
The extra chromosome 21 may result from meiotic non-disjunction, translocation or mosaicism.
Meiotic non-disjunction (94%)
In non-disjunction trisomy 21:
• most cases result from an error at meiosis
• the pair of chromosome 21s fails to separate, so that one gamete has two chromosome 21s and one has none (Fig. 8.3)
• fertilisation of the gamete with two chromosome 21s gives rise to a zygote with trisomy 21
The incidence of trisomy 21 due to non-disjunction is related to maternal age (Table 8.1). However, as the proportion of pregnancies in older mothers is small, most affected babies are born to younger mothers. Furthermore, meiotic non-disjunction can occur in spermatogenesis so that the extra 21 can be of paternal origin. All pregnant women are now offered screening tests measuring biochemical markers in blood samples and often also nuchal thickening on ultrasound (thickening of the soft tissues at the back of the neck) to identify an increased risk of Down syndrome in the fetus. When an increased risk is identified, amniocentesis is offered to check the fetal karyotype. After having one child with trisomy 21 due to non-disjunction, the risk of recurrence of Down syndrome is given as 1 in 200 for mothers under the age of 35 years, but remains similar to their age-related population risk for those over the age of 35 years.
Table 8.1
Risk of Down syndrome (live births) with maternal age at delivery, prior to screening in pregnancy
| Maternal age (years) | Risk of Down syndrome |
| All ages | 1 in 650 |
| 20 | 1 in 1530 |
| 30 | 1 in 900 |
| 35 | 1 in 385 |
| 37 | 1 in 240 |
| 40 | 1 in 110 |
| 44 | 1 in 37 |
Translocation (5%)
When the extra chromosome 21 is joined onto another chromosome (usually chromosome 14, but occasionally chromosome 15, 22 or 21), this is known as a Robertsonian translocation. This may be present in a phenotypically normal carrier with 45 chromosomes (two being ‘joined together’) or in someone with Down syndrome and a set of 46 chromosomes but with three copies of chromosome 21 material. In this situation, parental chromosomal analysis is recommended, since one of the parents may well carry the translocation in balanced form (in 25% of cases) (Fig. 8.4).
In translocation Down syndrome:
• The risk of recurrence is 10–15% if the mother is the translocation carrier and about 2.5% if the father is the carrier.
• If a parent carries the rare 21 : 21 translocation, all the offspring will have Down syndrome.
• If neither parent carries a translocation (75% of cases), the risk of recurrence is <1%.
Edwards syndrome (trisomy 18) and Patau syndrome (trisomy 13)
Although rarer than Down syndrome (1 in 8000 and 1 in 14 000 live births, respectively), particular constellations of severe multiple abnormalities suggest these diagnoses at birth; most affected babies die in infancy (Fig. 8.5, Boxes 8.2 and 8.3). The diagnosis is confirmed by chromosome analysis. Many affected fetuses are detected by ultrasound scan during the second trimester of pregnancy and diagnosis can be confirmed antenatally by amniocentesis and chromosome analysis. Recurrence risk is low, except when the trisomy is due to a balanced chromosome rearrangement in one of the parents.
Turner syndrome (45, X)
Usually (>95%), Turner syndrome results in early miscarriage and is increasingly detected by ultrasound antenatally when fetal oedema of the neck, hands or feet or a cystic hygroma may be identified. In live-born females, the incidence is about 1 in 2500. Figure 8.6 and Box 8.4 show the clinical features of Turner syndrome, although short stature may be the only clinical abnormality in children.
• Oestrogen replacement for development of secondary sexual characteristics at the time of puberty (but infertility persists).
The incidence does not increase with maternal age and risk of recurrence is very low.
Deletions
DiGeorge syndrome is associated with a deletion of band q11 on chromosome 22 (i.e. 22q11) (Fig. 8.7). Williams syndrome is another example of a microdeletion syndrome due to loss of chromosomal material at band q11 on the long arm of chromosome 7 (i.e. 7q11) (Fig. 8.18, see also Box 8.12).
Mendelian inheritance
Mendelian inheritance, described by Mendel in garden peas in 1866, is the transmission of inherited traits or diseases caused by variation in a single gene in a characteristic pattern. These Mendelian traits or disorders are individually rare but collectively numerous and important: over 6000 have been described. For many disorders, the Mendelian pattern of inheritance is known. If the diagnosis of a condition is uncertain, its pattern of inheritance may be evident on drawing a family tree (pedigree), which is an essential part of genetic evaluation (Fig. 8.8).
