Genetic disorders: A pediatric perspective
SANDRA G. BELLAMY, PT, MS, DPT, PCS and EUNICE YU CHIU SHEN, PT, PhD, DPT, PCS
After reading this chapter the student or therapist will be able to:
1. Describe the main types of genetic disorders and give examples of each type.
2. Differentiate between genetic disorders diagnosed with clinical versus laboratory methods.
3. Describe three modes of inheritance for single-gene disorders.
4. Recognize key impairments that are common to many genetic conditions in pediatric clients.
5. Explain the physical or occupational therapist’s role in the recognition, referral, and multidisciplinary management of genetic conditions in pediatric clients.
6. Identify resources and strategies for accessing information and increasing knowledge about genetic disorders for use in clinical decision making.
7. Explain why it is important to include family members in the planning and development of therapy programs for children with genetic disorders.
8. Describe and give examples of three types of assessment tools, and state the intended purpose of each.
9. Describe the importance of developing therapy programs for children that are outcome focused on functional skills in natural environments.
The Human Genome Project, completed in 2003,1 expanded knowledge about the genetic basis for disease and congenital malformations. The impact of this project is just being realized, with new research into diagnostic techniques and treatment options for genetic disorders. Pediatric health care professionals will be faced with questions from families who, in seeking diagnostic and prognostic information, are accessing the wealth of information both in the lay scientific press and on the World Wide Web (Box 13-1 and Table 13-1).
BOX 13-1  RESOURCES ON TOPICS IN GENETICS FOR CLINICIANS AND FAMILIES
RESOURCES ON TOPICS IN GENETICS FOR CLINICIANS AND FAMILIES
http://geneticalliance.org/sites/default/files/ksc_assets/pdfs/manual_all2.pdf
Free pdf-format primer on basics of human genetics for health professionals and families.
2. Human Genome Project Information
www.ornl.gov/sci/techresources/Human_Genome/home.shtml
Links to research; teacher and student tools; gene testing; summary of current scientific knowledge in genomics; and fundamentals of genetics including interactive map of each human chromosome and associated diseases and disorders.
3. Genetics Fact Sheets, Centre for Genetics Education
www.genetics.com/au/pdf/factsheets/fs01.pdf
Consumer-friendly pdf-format modules on basic genetics with simple illustrations for difficult-to-understand scientific concepts.
4. Genetics Home Reference, National Library of Medicine (Bethesda, Md), 1993-2008
Website for children and adults with disabilities and their families, friends, and allies.
Covers various diagnoses, assistive technology, legal rights and legislation, special education, and leisure activities.
6. Making Sense of Your Genes: A Guide to Genetic Counseling
7. American College of Medical Genetics
TABLE 13-1 
| RESOURCE | WEBSITE | INFORMATION |
| AbleData | www.abledata.com | Information about assistive technology (AT) products and rehabilitation equipment |
| AccessIT | www.washington.edu/accessit | National Center on Accessible Information Technology in Education |
| Alliance for Technology Access | www.ataccess.org | Public education, information, referral; network of technology resources |
| Assistive Technology Industry Association | www.atia.org/i4a/pages/index.cfm | Information on products and services |
| Assistive Technology Partners | www.assistivetechnologypartners.com | Information to assist persons with cognitive, sensory, and/or physical disabilities |
| Assistive Technology Training Online Project | http://atto.buffalo.edu | AT applications that help students with disabilities learn in elementary classrooms |
| Family Center on Technology and Disability | www.fctd.info | Provides guide to AT and transition planning |
| National Public Website on Assistive Technology | www.assistivetech.net | Features products by related functional area or disability, by activity, and by vendor |
| Protection and Advocacy for Assistive Technology Program | www.workworld.org/wwwebhelp/protection_and_advocacy_systems _overview.htm | Provides protection and advocacy services to help individuals with disabilities of all ages acquire, use, and maintain AT services or devices; website identifies each state’s program |
| Rehabtool.com | www.rehabtool.com | Information on AT products by categories |
| National Institute of Standards and Technology (Standards.gov) | http://standards.gov/standards_gov/index.cfm | Authoritative information and guidance on measurement and standards for all industry sectors |
An accurate diagnosis of a specific genetic disorder (syndrome or disease) is necessary for a prognosis to be provided, for eligibility for therapy and education services to be determined, and as a basis for genetic counseling for the child’s family.2 The diagnostic process for genetic disorders includes a combination of clinical assessments by the physician who collects the child’s medical history and a clinical geneticist who may construct a family history or “pedigree” to recognize disorders with familiar inheritance patterns. Molecular studies may confirm a clinical diagnosis, differentiate between diagnoses with similar clinical presentation, and identify the genetic cause of the disorder. Some genetic disorders are not easily identified, and laboratory testing can be extensive, prolonged, and often inconclusive; therefore pediatricians may refer children to occupational and physical therapy before the nature of their condition is fully known.2,3 Although sometimes far removed from the hospitals and specialized centers that perform genetic testing and diagnosis, the pediatric therapist is often able to contribute clinical evidence that will assist the diagnostic process.4–6 Furthermore, many genetic diseases and syndromes are increasingly survivable into adulthood; thus it is vital that physical and occupational therapists achieve competence in genetics and genomics in order to deliver care throughout the patient’s life span.6,7 An overview of the general categories and subtypes of genetic disorders is presented first. Specific examples of each type are given, along with a brief description of key diagnostic features and issues commonly addressed with medical and therapeutic intervention. A summary of impairments common to many pediatric genetic disorders is presented in the second section. The third section includes a discussion of the medical management of genetic disorders, genetic counseling, and the ethical implications of genetic screening and testing. The final section focuses on the physical or occupational therapist’s role in the clinical management of children with genetic disorders. The therapist’s role and responsibilities in developing competence in recognition, referral, and clinical practice when working with patients and families affected by a genetic disorder are discussed. Evaluation procedures, treatment goals and objectives, and general treatment principles and strategies are discussed from a family-centered perspective. A list of educational resources for clinicians and families is provided.
An overview: clinical diagnosis and types of genetic disorders with representative clinical examples
Genetic disorders are typically divided into four categories: chromosomal, single-gene, mitochondrial, and multi-factorial. Chromosomal disorders arise when there is an alteration in either the number or structure of chromosomes that exist in either autosomal or sex (X, Y) chromosomes.8 Numerical or large structural chromosomal abnormalities can be seen through a microscope; therefore a sample of the patient’s peripheral blood can be used in detection of disorders such as Down syndrome. When there is a suspicion of a clinical spectrum associated with some of the known chromosomal microdeletions, translocations, or inversions, direct deoxyribonucleic acid (DNA) analysis techniques such as fluorescence in situ hybridization (FISH) with use of specific sequence DNA probes can confirm a specific suspected diagnosis. Indirect DNA analysis techniques such as linkage analysis can be performed to confirm single-gene disorders when the gene or genomic region associated with the disorder is unknown.9
Of our 20,000 to 25,000 protein-coding genes,1 a single gene may be responsible for approximately 6000 known genetic traits. Approximately 4000 of these known traits are diseases or disorders.10 Single-gene disorders may be transmitted through three different patterns: autosomal dominant, autosomal recessive, and sex linked. Dominant refers to the case in which a mutated gene from one parent is sufficient to produce the disorder in offspring. Recessive refers to the case in which the disorder will not be expressed unless offspring inherited a mutated copy of that gene from both parents. It is incorrect to say that a gene is recessive or dominant; rather the trait, or disorder, is dominant or recessive.7
It is important to understand how a disorder was acquired, because the relative risks to other offspring for the disorder vary according to mode of transmission. For example, the risk of having another child with the same genetic disorder that occurred as a result of a spontaneous mutation is low. However, when one parent is affected by an inherited dominant mutation, the risk of passing that faulty gene onto each child is 50%.8
Most congenital malformations and many serious diseases that have an onset in childhood or adulthood are not caused by single genes or chromosomal defects; these are called multifactorial disorders.7,8
Mitochondrial disorders are caused by alterations in maternally inherited cytoplasmic mitochondrial DNA (mtDNA). The clinical manifestations of mtDNA-related disorders are extremely variable,11 and the occurrence is reportedly rare (5.0 per 100,000)8,12; however, collectively as a group of neuromuscular disorders, they account for substantial use of health care resources.12
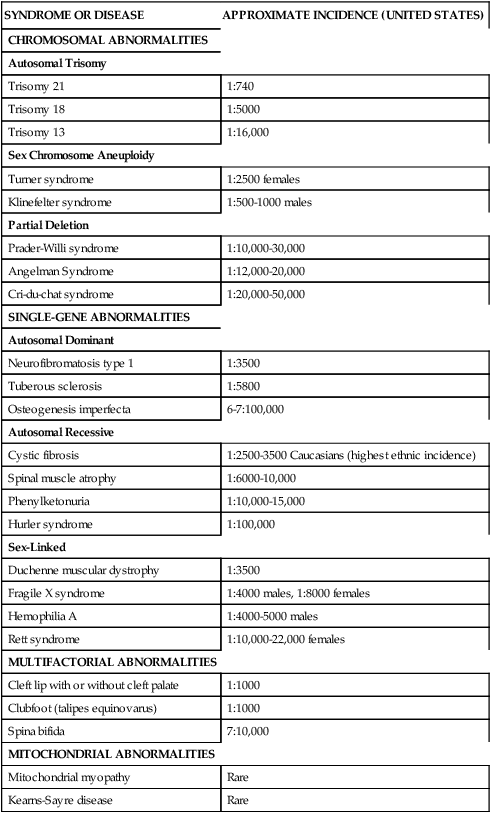
Currently there are over 1000 genetic tests available in the United States.1 Specific DNA testing may soon be able to identify nearly all human genetic disorders. This not only allows for accurate and more complete diagnosis but should pave the way for the development of mechanisms for treatment, cure, and prevention of certain genetic conditions.4,5,8,9 Table 13-2 lists examples of specific disorders in categories of the most common pattern of inheritance by which each occurs.
TABLE 13-2 
PARTIAL LISTING OF PEDIATRIC GENETIC CONDITIONS
| SYNDROME OR DISEASE | APPROXIMATE INCIDENCE (UNITED STATES) |
| CHROMOSOMAL ABNORMALITIES | |
| Autosomal Trisomy | |
| Trisomy 21 | 1:740 |
| Trisomy 18 | 1:5000 |
| Trisomy 13 | 1:16,000 |
| Sex Chromosome Aneuploidy | |
| Turner syndrome | 1:2500 females |
| Klinefelter syndrome | 1:500-1000 males |
| Partial Deletion | |
| Prader-Willi syndrome | 1:10,000-30,000 |
| Angelman Syndrome | 1:12,000-20,000 |
| Cri-du-chat syndrome | 1:20,000-50,000 |
| SINGLE-GENE ABNORMALITIES | |
| Autosomal Dominant | |
| Neurofibromatosis type 1 | 1:3500 |
| Tuberous sclerosis | 1:5800 |
| Osteogenesis imperfecta | 6-7:100,000 |
| Autosomal Recessive | |
| Cystic fibrosis | 1:2500-3500 Caucasians (highest ethnic incidence) |
| Spinal muscle atrophy | 1:6000-10,000 |
| Phenylketonuria | 1:10,000-15,000 |
| Hurler syndrome | 1:100,000 |
| Sex-Linked | |
| Duchenne muscular dystrophy | 1:3500 |
| Fragile X syndrome | 1:4000 males, 1:8000 females |
| Hemophilia A | 1:4000-5000 males |
| Rett syndrome | 1:10,000-22,000 females |
| MULTIFACTORIAL ABNORMALITIES | |
| Cleft lip with or without cleft palate | 1:1000 |
| Clubfoot (talipes equinovarus) | 1:1000 |
| Spina bifida | 7:10,000 |
| MITOCHONDRIAL ABNORMALITIES | |
| Mitochondrial myopathy | Rare |
| Kearns-Sayre disease | Rare |
Chromosomal disorders
Cytogenics is the study of chromosomal abnormalities. A karyotype is prepared that displays the 46 chromosomes—22 pairs of autosomes arranged according to length, and then the two sex chromosomes that determine male or female sex. Modern methods of staining karyotypes enable analysis of the various numerical and structural abnormalities that can occur. Most chromosomal abnormalities appear as numerical abnormalities (aneuploidy) such as one missing chromosome (monosomy) or an additional chromosome, as in trisomy 21 (Down syndrome).8 Structural abnormalities occur in many forms. They include a missing or “extra portion” of a chromosome or a translocation error, which is an interchange of genetic material between nonhomologous chromosomes. The incidence of chromosomal abnormalities among spontaneously aborted fetuses may be as high as 60%.8,13 About one in 150 live-born infants have a detectable chromosomal abnormality; and in about half of these cases the chromosomal abnormality is accompanied by congenital anomalies, intellectual disability, or phenotypical changes that manifest later in life.8 Of the fetuses with abnormal chromosomes that survive to term, about half have sex chromosome abnormalities and the other half have autosomal trisomies.8
Autosomal trisomies
Trisomy is the condition of a single extranuclear chromosome. Trisomies occur frequently among live births, usually as a result of the failure of the parental chromosomes to disjoin normally during meiosis. Trisomy can occur in autosomal or sex cells. Trisomies 21, 18, and 13 are the most frequently occurring trisomies; however, few children with trisomy 18 and 13 survive beyond 1 year of age.1
Trisomy 21 (down syndrome).
Trisomy 21 occurs in approximately one in every 740 live births,14 and its incidence is distributed equally between the sexes.10 The pathophysiological features of Down syndrome are caused by an overexpression of genes on human chromosome 21. Ninety-five percent of individuals have an extra copy in all of their body’s cells. The remaining 5% have the mosaic and translocation forms.15 In the United States the incidence of Down syndrome increases with advanced maternal age.10 Detection of Down syndrome is possible with various prenatal tests, and the diagnosis is confirmed by the presence of characteristic physical features present in the infant at birth.16 Down syndrome is the most common chromosomal cause of moderate to severe intellectual disability.15 The typical phenotypical features observable from birth are hypotonia, epicanthic folds, flat nasal bridge, upward slanting palpebral fissures, small mouth, excessive skin at the nape of the neck, and a single transverse palmar crease (Figure 13-1).
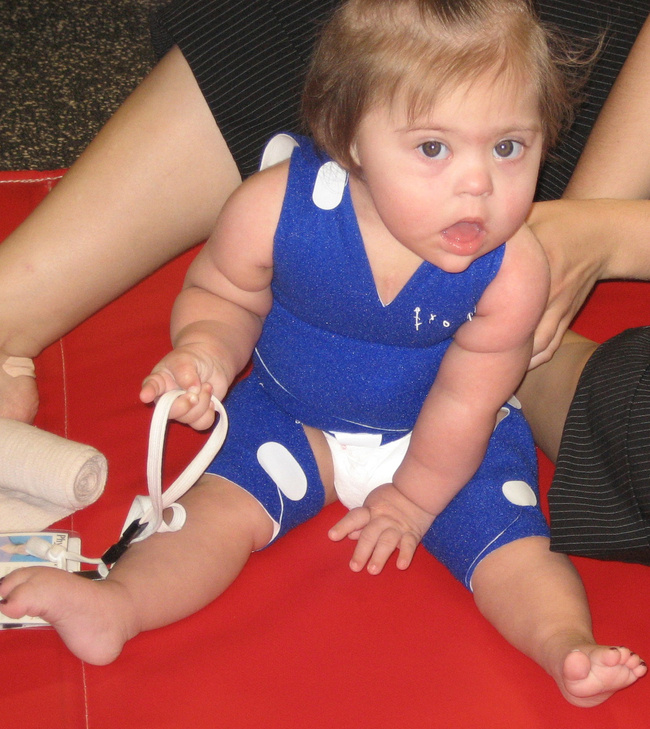
 Ten-month-old girl with Down syndrome.
Ten-month-old girl with Down syndrome.Information compiled by the Centers for Disease Control and Prevention for years 1968 through 1997 indicates that the median survival age of individuals with Down syndrome is 49 years, compared with 1 year in 1968. Improvements in the median survival age were less in races other than white, although the reasons for this remain unclear.14 Half of all children with Down syndrome have congenital heart defects.16 Congenital heart problems, respiratory infection, and leukemia are the most common factors associated with morbidity and mortality in childhood,17 whereas a possible increased tendency for premature cellular aging and Alzheimer disease may account for higher mortality rates later in life.18
Impairments of visual and sensory systems are also common in individuals with Down syndrome. As many as 77% of children with Down syndrome have a refractive error (myopia, hyperopia), astigmatism, or problems in accommodation.19 Hearing losses that interfere with language development are reportedly present in 80% of children with Down syndrome. In most cases the hearing loss is conductive; in up to 20% of cases the loss is sensorineural or mixed.16,20 Obstructive sleep apnea has been reported to exist frequently in young children21,22 and adults with Down syndrome.23 Craniofacial impairments such as a shortened palate and midface hypoplasia, along with oral hypotonia, tongue thrusting, and poor lip closure, frequently result in feeding difficulties at birth.24 Bell and colleagues studied the prevalence of obesity in adults with Down syndrome and reported it in 70% of male subjects and 95% of female subjects.25 Children with Down syndrome also appear to have a higher risk of being overweight or obese,26–28 which may be, in part, a result of the retarded growth and endocrine and metabolic disorders associated with trisomy 21.28 In a small population study of children with Down syndrome, Dyken and co-workers29 reported that there was a high prevalence of obstructive sleep apnea associated with a higher body mass index.
Children with Down syndrome may have musculoskeletal anomalies such as metatarsus primus varus, pes planus, thoracolumbar scoliosis, and patellar instability and have an increased risk for atlantoaxial dislocation,30–32 which has been observed through radiography in up to 10% to 30% of individuals with this syndrome30,31 with and without neurological compromise.33 There is some controversy in the medical community as to the necessity and efficacy of radiographic screening for the instability.31,32 Proponents of radiographic screening argue that neurological symptoms of atlantoaxial instability may often go undetected in this population because symptoms are often masked by the wide-based gait and motor dysfunction already associated with the disorder. If the child is unable to verbalize complaints or the child is uncooperative with physical and neurological examinations, symptoms may be missed. There is particular concern about cervical instability if these children undergo surgical procedures requiring general anesthesia32 and participate in recreational sports such as the Special Olympics.31 Symptomatic instability can result in spinal cord compression leading to myelopathy with leg weakness, decreased walking ability,33 spasticity, or incontinence. Although reportedly rare, there have been cases where atlantoaxial dislocation has resulted in quadriplegia.30
Several researchers have explored the neuropathology associated with Down syndrome. Changes in brain shape, size, weight, and function occur during prenatal and infant development of babies with Down syndrome, with important differences apparent by 6 months of age.34 The relatively small size of the cerebellum and brain stem was reported by Crome and Stern in the 1970s.35 Marin-Padilla36 studied the neuronal organization of the motor cortex of a 19-month-old child with Down syndrome and found various structural abnormalities in the dendritic spines of the pyramidal neurons of the motor cortex. He suggested that these structural differences may underlie the motor incoordination and intellectual disability characteristic of individuals with Down syndrome. Loesch-Mdzewska37 also found neurological abnormalities of the corticospinal system (in addition to reduced brain weight) in his neuropathological study of 123 individuals with Down syndrome aged 3 to 62 years. Crome38 reported lesser brain weight in comparison with normal persons. Finally, Benda39 noted a lack of myelinization of the nerve fibers in the precentral area, frontal lobe, and cerebellum of infants with Down syndrome. As McGraw40 has pointed out, the amount of myelin in the brain reflects the stage of developmental maturation. The delayed myelinization characteristic of neonates and infants with Down syndrome is thought to be a contributing factor to the generalized hypotonicity and persistence of primitive reflexes characteristic of this syndrome.41
Trisomy 18.
Trisomy 18, or Edwards syndrome, is the second most common of the trisomic syndromes to occur in term deliveries, although it is far less prevalent than Down syndrome. It occurs in one in 5000 newborns, and approximately 80% of affected infants are female.42 As with Down syndrome, advanced maternal age is positively correlated with trisomy 18. Most cases of Edwards syndrome occur as random events during the formation of reproductive cells; fewer cases occur as errors in cell division during early fetal development; and inherited, translocation forms rarely occur.42 Only 10% of infants born with trisomy 18 survive past the first year of life; female and non-Caucasian children survive longest.43 The survival of girls averages 7 months; the survival of boys averages 2 months.43 Individuals surviving past infancy most often have the mosaic form, and there is high variance in phenotype (Figure 13-2).44
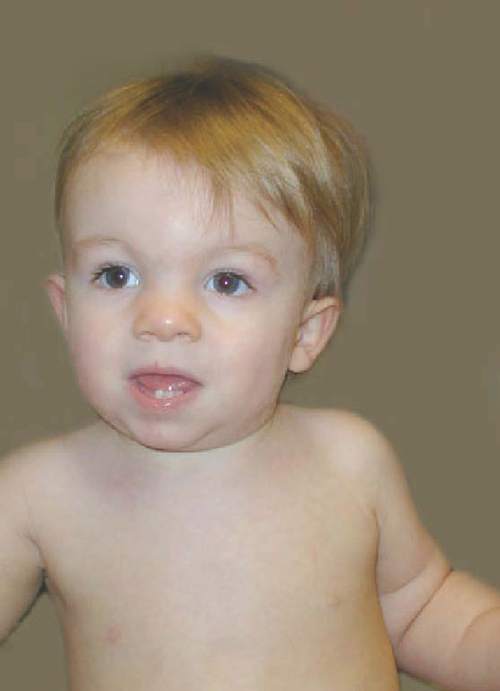

Individuals with trisomy 18 generally have far more serious organic malformations than seen in those with Down syndrome.45 Typical malformations affect the cardiovascular, gastrointestinal, urogenital, and skeletal systems. Infants with trisomy 18 have low birth weight and small stature, with a long narrow skull, low-set ears, flexion deformities of the fingers, and rocker-bottom feet. Muscle tone is initially hypotonic, but it becomes hypertonic in children with longer than typical life span.45 The period of hypertonicity in the early years may change to low tone and joint hyperextensibility by preschool and school age. Microcephaly, abnormal gyri, cerebellar anomalies, myelomeningocele, hydrocephaly, and corpus callosum defects have been reported in individuals with trisomy 18.46
Common skeletal malformations that may warrant attention from the developmental physical or occupational therapist include scoliosis,46 limited hip abduction, flexion contractures of the fingers, rocker-bottom feet, and talipes equinovarus.45 Infants with trisomy 18 may also have feeding difficulties as a result of a poor suck.47 Profound intellectual disability is another clinical factor that will affect the developmental therapy programs for children with trisomy 18.46,47
Trisomy 13.
Trisomy 13, also commonly called Patau syndrome, is the least common of the three major autosomal trisomies, with an incidence of one in 10,000 to 20,000 live births.8,42 As in the other trisomic syndromes, advanced maternal age is correlated with the incidence of trisomy 13.48 Fewer than 10% of individuals with trisomy 13 survive past the first year of life42,43; girls and non-Caucasian infants appear to survive longer.42,43 Individuals surviving past infancy most often have the mosaic form, and there is high variance in phenotype.43 As with Edwards syndrome, most cases of Patau syndrome occur as random events during the formation of eggs and sperm, such as nondisjunction errors during cell division.48
Trisomy 13 is characterized by microcephaly, deafness, anophthalmia or microphthalmia, coloboma, and cleft lip and palate.48 As in trisomy 18, infants with trisomy 13 frequently have serious cardiovascular and urogenital malformations and typically have severe to profound intellectual disability.49 Skeletal deformities and anomalies include flexion contractures of the fingers and polydactyly of the hands and feet.10 Rocker-bottom feet also have been reported, although less frequently than in individuals with trisomy 18. Reported central nervous system (CNS) malformations include arhinencephalia, cerebellar anomalies, defects of the corpus callosum, and hydrocephaly.50
Sex chromosome aneuploidy
The human X chromosome is large, containing approximately 5% of a human’s nuclear DNA. The Y chromosome, much smaller, contains few known genes.8 Females, with genotype XX, are mosaic for the X chromosome, meaning that one copy of their X chromosome is inactive in a given cell; some cell types will have a paternally derived active chromosome, and others a maternally derived X chromosome. Males, genotype XY, have only one copy of the X chromosome; therefore diseases caused by genes on the X chromosome, called X-linked diseases (see section on sex-linked disorders), can be devastating to males and less severe in females.8 In the presence of abnormal numbers of sex chromosomes, neither male nor female individuals will be phenotypically normal.8 Two of the most prevalent sex chromosome anomalies are Turner syndrome and Klinefelter syndrome.
Turner syndrome.
Turner syndrome affects females with monosomy of the X chromosome. The syndrome, also known as gonadal dysgenesis, occurs in one in 2500 live female births.51,52 Turner syndrome is the most common chromosomal anomaly among spontaneous abortions.53,54 Most infants who survive to term have the mosaic form of this syndrome, with a mix of cell karyotypes, 45,X and 46,XX. The SHOX gene, found on both the X and Y chromosomes, codes for proteins essential to skeletal development. Deficiency of the SHOX gene in females accounts for most of the characteristic abnormalities of this disorder.52,55 Three characteristic impairments of the syndrome are sexual infantilism, a congenital webbed neck, and cubitus valgus.56 Other clinical characteristics noted at birth include dorsal edema of hands and feet, hypertelorism, epicanthal folds, ptosis of the upper eyelids, elongated ears, and shortening of all the hand bones.51,57 Growth retardation is particularly noticeable after the age of 5 or 6 years, and sexual infantilism, characterized by primary amenorrhea, lack of breast development, and scanty pubic and axillary hair, is apparent during the pubertal years. Ovarian development is severely deficient, as is estrogen production.10,58 Congenital heart disease is present in 20% to 30% of individuals with Turner syndrome,57 with a fewer number of cardiovascular malformations in individuals with the mosaic form59; 33% to 60% of individuals with Turner syndrome have kidney malformations.51 Hypertension is common even in the absence of cardiac or renal malformations.57,60
There are numerous incidences of skeletal anomalies, some of which may be significant enough to require the attention of a pediatric therapist. Included among these are hip dislocation, pes planus and pes equinovarus, dislocated patella,51 deformity of the medial tibial condyles,46 idiopathic scoliosis,57 and deformities resulting from osteoporosis.10,57
Sensory impairments include decrease in gustatory and olfactory sensitivity61,62 and deficits in spatial perception and orientation,61 and up to 90% of adult females have moderate sensorineural hearing loss. Recurrent ear infections are common and may result in future conductive hearing loss.60 Although the average intellect of individuals with Turner syndrome is within normal limits, the incidence of intellectual disability is higher than in the general population.45 Noonan syndrome, once thought to be a variant of Turner syndrome, has several common clinical characteristics; however, advancements in genetics research have shown that the syndromes have different genetic causes.63,64
Klinefelter syndrome.
Klinefelter syndrome is an example of aneuploidy with an excessive number of chromosomes that occurs in males. The most common type, 47,XXY, is usually not clinically apparent until puberty, when the testes fail to enlarge and gynecomastia occurs.65 Nearly 90% of males with Klinefelter syndrome possess a karyotype of 47,XXY, and the other 10% of patients are variants.66 The incidence of Klinefelter syndrome (XXY) is about one in 500 to 1000 males, and an estimated half of 47,XXY conceptions are spontaneously aborted.8 The extra X chromosome(s) can be derived from either the mother or the father, with nearly equal occurrence.67 Advanced maternal age is widely accepted as a causal factor.8,66 FISH analysis of spermatozoa from fathers of boys with Klinefelter syndrome suggests that advanced paternal age increases the frequency of aneuploid offspring.68–70
Most individuals with karyotype XXY have normal intelligence, a somewhat passive personality, and a reduced libido. Eighty-five percent of individuals having the nonmosaic karyotype are sterile. Individuals with the karyotypes 48,XXXY and 49,XXXXY tend to display a more severe clinical picture. Individuals with 48,XXXY usually have severe intellectual disability, with multiple congenital anomalies, including microcephaly, hypertelorism, strabismus, and cleft palate.10,65 Skeletal anomalies include radioulnar synostosis, genu valgum, malformed cervical vertebrae, and pes planus.10 A 2010 systematic review of literature71 on neurocognitive outcomes of persons with Klinefelter syndrome concluded that problems of delayed walking in children and persistent deficits in fine and gross motor development, and problems in motor planning.71,72 Giedd and co-workers published the results of a case-control study examining brain magnetic resonance imaging (MRI) scans of 42 males with Klinefelter syndrome and reported cortical thinning in the motor strip associated with impaired control of the upper trunk, shoulders, and muscles involved in speech production.73
Partial deletion disorders
Cri-du-chat syndrome.
Cri-du-chat syndrome, also referred to as cat-cry syndrome, and 5p minus syndrome results from a partial deletion of the short arm of chromosome 5. Example nomenclature for a female with this syndrome is (46,XX,del[5p]). The incidence of the syndrome is estimated to be one case per 20,000 to 50,000 live births.10,74 Although approximately 70% of individuals with cri-du-chat syndrome are female, there is an unexplained higher prevalence of older males with this disorder.75 Advanced parental age is not a causal factor. A study completed in 1978 indicated that life expectancy was 1 year for 90% of infants born with this disorder,76 but now life expectancy is nearly normal with routine medical care.77
Primary identifying characteristics at birth include a definitive high-pitched catlike cry, microcephaly, evidence of intrauterine growth retardation, and subsequent low birth weight.10,76,78 Abnormal laryngeal development accounts for the characteristic cry, which is present in most individuals and disappears in the first few years of life.76 Other features of individuals with this syndrome include hypertelorism, strabismus, “moon face,” and low-set ears.10,76,78 Associated musculoskeletal deformities include scoliosis, hip dislocations, clubfeet, and hyperextensibility of fingers and toes. Muscular hypotonicity is associated with this syndrome, although cases with hypertonicity have also been noted.79 Severe respiratory and feeding problems have also been reported.77 Postnatal growth retardation has been documented, with the median near the 5th percentile of the normal growth curve.80
Although intellectual disability and physical deformities are more severe with larger deletions,74 there is evidence that with early developmental intervention these children can develop language, functional ambulation, and self-care skills.81,82
Prader-willi syndrome and angelman syndrome.
Prader-Willi syndrome (PWS) and Angelman syndrome (AS) are discussed together because they result from a structural or functional loss of the PWS and AS region of chromosome 15 (15q11-13), which can occur by one of several genetic mechanisms.83,84 PWS has an incidence of one in 15,000 to 30,00083 and AS has an incidence of one in 12,000 to 20,000.83,84 These two syndromes illustrate the effect of genomic imprinting, which is the differential activation of genes of the same chromosome and location, depending on the sex of the parent of origin (Figure 13-3).8
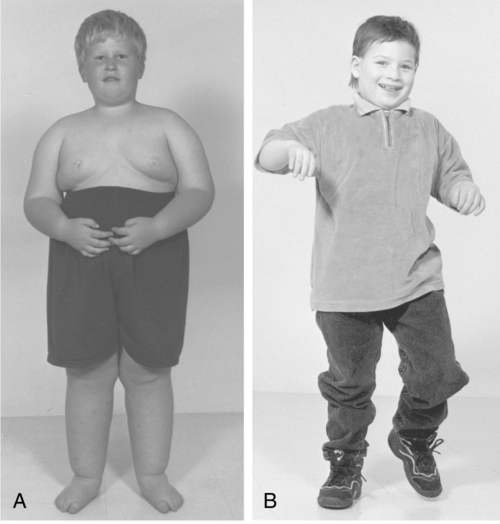

PWS results from a failure of expression of paternally inherited genes in the PWS region of chromosome 15.83 Conversely, AS results when the maternal contribution in the 15q11.2-q13 region is lost.84 OCA2 is a gene located within the PWS and AS region of chromosome 15 that codes for the protein involved in melanin production. With loss of one copy of this gene, individuals with PWS or AS will have light hair and fair skin. In the rare case that both copies of the gene are lost, these individuals may have a condition called oculocutaneous albinism, type 2, which causes severe vision problems.84
Characteristics of PWS in infancy include hypotonia, poor feeding, lethargy, and hypogonadism.85,86 Developmental milestones in the first 2 years of life are not acquired until approximately twice the normal age.87,88 Between 1 and 4 years of age, hyperphagia is apparent and if uncontrolled will lead to morbid obesity and its associated health complications.86,87,89 Most individuals with PWS have mild to moderate intellectual disability, although some individuals have IQ scores within normal limits.90 Maladaptive behaviors such as temper tantrums, aggression, self-abuse, and emotional lability have been reported.91 As a result of extreme obesity, many individuals with PWS have impaired breathing that can produce sleepiness, cyanosis, cor pulmonale, and heart failure.91 Scoliosis is common but does not appear to be related to obesity.92
Clinical diagnosis is confirmed by laboratory genetic testing techniques including DNA-based methylation testing, FISH probe, and pyrosequencing assays.86,93 Most cases of PWS are caused by random mutations in parental reproductive cells.87 Other cases may result from translocation errors.86,94 Parental studies are important in translocation cases because 20% of cases cited in the literature involved familial rearrangements, which may significantly increase the risk of recurrence.95
Angelman syndrome, named after Dr. Harry Angelman, who first described children with AS in 1965, is characterized by developmental delay or intellectual disability, seizures, ataxia, progressive microcephaly, and severe speech impairments. Tongue thrusting, drooling, and sucking and swallowing disorders occur in 20% to 80% of children. Individuals often display spontaneous bouts of laughter accompanied by hand-flapping movements and a characteristic walking posture of arms overhead and flexed elbows.8,84,96 Infants appear normal at birth, but severe developmental delay becomes apparent by 6 to 12 months of age. More unique features of the disorder do not appear until after 1 year of age. Children with AS typically have structurally normal brains on MRI and computed tomography (CT) scans, but electroencephalogram (EEG) findings are often abnormal, showing a characteristic pattern that may assist with diagnosis before other clinical symptoms emerge,84,97 and molecular studies can also confirm the disorder before all of the clinical criteria for this diagnosis are met.84
Most cases of AS occur as a result of mutations involving deletion or deficient function of the maternally inherited UBE3A gene. This gene codes for an enzyme, ubiquitin protein ligase, involved in the normal process of removing damaged or unnecessary proteins in healthy cells. In most of the body’s tissues except the brain, both copies (maternal and paternal) of the UBE3A gene are active. Only the maternal copy of the gene is normally active in the brain, so if this copy is absent or deficient, the normal cellular housekeeping process breaks down.84 The risk of having another child with AS can vary from 1% to 50% depending on which of the six known genetic mechanisms is responsible for the disorder.
Translocation disorders
Translocation errors have been identified in many childhood hematologic cancers and sarcomas.98,99 Translocation errors are also commonly seen in couples with infertility.100 Translocation abnormalities occur when genetic material is exchanged and rearranged between two nonhomologous chromosomes (those not in the same numbered pair). The structural abnormality can result in the loss or gain of chromosomal material (an unbalanced arrangement) or no loss or gain of material (a balanced arrangement). Unbalanced arrangements can produce serious disease or deformity in individuals or their offspring. Carriers of balanced arrangements—estimated at one in 500 individuals—often have a normal phenotype, but their offspring may have an abnormal phenotype.8 There are two basic types of translocations: reciprocal translocation and robertsonian translocation. Reciprocal translocations occur when two different chromosomes break and the genetic material is mutually exchanged. A robertsonian translocation occurs when there is a break in a portion of two different chromosomes, with the longest remaining portions of both chromosomes forming a single chromosome. The shorter portions that broke away usually do not contain vital genetic information; therefore the individual may be phenotypically normal.8 An example notation of a reciprocal translocation is 46,XY,t(7;9)(q36;q34). This individual is male with a normal number of chromosomes but with a translocation of genetic material on chromosomes 7 and 9; “q” refers to the short arm of these chromosomes, and the numbers “36” and “34” refer to the location.
Translocations occur in children seen in therapy settings, including about 3% to 5% of children with Down syndrome,10 and translocations are found in 40% of all cases of acute lymphoblastic leukemia (ALL).101
Acute lymphoblastic leukemia.
ALL accounts for one fourth of all childhood cancers, and it is the most common type of childhood cancer.102–104 ALL occurs when the DNA of immature lymphoblasts is altered and they reproduce in abnormal numbers, crowding out the formation of normal cells in the bone marrow.102,105 Sixty percent of cases of ALL occur in children, with the peak incidence in the first 5 years of life. A rise in the incidence of ALL has been reported during major periods of industrialization worldwide105,106 and is hypothesized to be associated with exposure to radiation107 and other environmental teratogens108,109 in the preconception, gestational, and postpregnancy periods.103,106
With advancements in medical treatment protocols for pediatric patients, 5-year survival rates have improved to 80%.104 Children aged 1 to 9 years at diagnosis have a better prognosis than infants, adolescents, or adults diagnosed with ALL.103 There are numerous forms of translocation mutations associated with ALL. Some translocation forms of ALL do not respond well to combination chemotherapy treatment; an example is the translocation that occurs between chromosomes 9 and 22, known as the “Philadelphia chromosome.”104,110 Other translocations that result in hyperdiploidy (more than 50 chromosomes), in particular within chromosomes 4, 10, and 17, may confer a more favorable outcome.111
Frequently, diagnosis is made when a physician relates the child’s history of a persistent viral respiratory infection with other characteristic clinical signs and symptoms consistent with hematopoietic leukemia. The key symptoms of ALL are pallor, poor appetite, lethargy, easy fatigue and bruising, fever, mucosal bleeding, and bone pain.99 A complete blood count will show a shortage of all types of blood cells, including red, white, and platelets. Diagnosis is confirmed by the presence of lymphoblasts in bone marrow. Radiographs may be necessary to determine metastases, and cerebrospinal fluid will be examined because early involvement of the CNS has important prognostic implications.106 Cytogenetic studies will be performed to aid in selection of treatment protocols and prognosis.104
Referral to physical and occupational therapists is made for other common problems such as muscle cramps, muscle weakness, impaired gross and fine motor performance, decreased energy expenditure, osteopenia, and osteoporosis.112
Single-gene disorders
Autosomal dominant disorders
Mutations on one of the 22 numbered pairs of autosomes may result in isolated anomalies that occur in otherwise normal individuals, such as extra digits or short fingers. Each child of a parent with an autosomal dominant trait has a 50:50 chance of inheriting that trait.8 Other autosomal dominant disorders include syndromes characterized by profound musculoskeletal and neurological impairments that may require intervention from a physical or an occupational therapist. Three examples of autosomal dominant disorders are osteogenesis imperfecta (OI), tuberous sclerosis, and neurofibromatosis (NFM).
Osteogenesis imperfecta.
OI is a spectrum of diseases that results from deficits in collagen synthesis associated with single-gene defects, most commonly of COL1A1 and COL1A2, located on chromosomes 17 and 7, respectively.10,113 OI is characterized by brittle bones resulting from impaired quality, quantity, and geometry of bone material and hyperextensible ligaments.114,115 Deafness, resulting from otosclerosis, is found in 35% of individuals by the third decade of life.45 New knowledge about this disease from molecular genetic studies and bone histomorphometry has expanded the classification subtypes of OI into types I through VII.113,116 These classifications are helpful in determining prognosis and management, although there is a continuum of severity of clinical features and much overlap in the features among the different classifications.116 Types I, IV, V, and VI occur in the autosomal dominant pattern; whereas type VII occurs as a recessive trait, and types II and III can occur as either dominant or recessive traits.113 OI types V and VI account for only 5% of cases, and type VII has been found to date only in a Native Canadian population.116 This section will compare and contrast only types I through IV.
The overall incidence of OI is one in 10,000 live births in the United States, with types I and IV accounting for almost 95% of all patients with OI.113 Ninety percent of dominant forms of OI can be confirmed by DNA analysis.117 Type I is the least severe form, followed by types IV and III, with type II being the most severe.
Type I is characterized by blue sclera, mild to moderate bone fragility, joint hyperextensibility, and hearing loss in young adulthood.117 There are no significant deformities; individuals with this type may not sustain fracture until ambulatory, and incidence of fractures decreases with age.117 Type IV OI is characterized by more severe bone fragility and joint hyperextensibility than is type I. Bowing of long bones, scoliosis, dentinogenesis imperfecta, and short stature are common.114,116,118
Children with type IV OI are often ambulatory but may require splinting or crutches.114
Children with type III OI have severe bone fragility and osteoporosis; often there are fractures in utero. Type III occurs primarily in autosomal dominant inheritance in North Americans and Europeans.116 The less frequent, autosomal recessive form of OI, type III is characterized by progressive skeletal deformity, scoliosis, triangular facies, large skull, normal cognitive ability, short stature, and limited ambulatory ability.114,116,119 The long bones of the lower extremities are most susceptible to fractures, particularly between the ages of 2 to 3 years and 10 to 15 years,45 with the frequency of fractures diminishing with age. Intramedullary rods inserted in the tibia or femur may minimize recurrent fractures.36
Type II, the most severe form, is most often lethal before or shortly after birth, although there are a few cases of children living to 3 years.116,119 Infants with type II OI have multiple fractures, often in utero, and underdeveloped lungs and thorax; therefore many die from respiratory complications after birth. Most type II cases are the result of spontaneous mutations; because only one copy of the gene is sufficient to cause the disorder, it is still commonly classified as an autosomal dominant condition. There are fewer cases of autosomal recessive inheritance.8
Prevention of fractures is an important goal in working with individuals with OI, but fear of handling and overprotection by caregivers may limit a child’s optimal functional independence. Caregiver education in careful handling and positioning should begin in the patient’s early infancy, and training in the use of protective orthoses and assistive devices is appropriate from the period of crawling through ambulation.115,120,121 Aquatic therapy can be a valuable treatment strategy for children with OI.120,121
Tuberous sclerosis complex.
Tuberous sclerosis complex (TSC) is characterized by a triad of impairments: seizures, intellectual disability, and sebaceous adenomas; however, there is wide variability in expression, with some individuals displaying skin lesions only.122 Infants are frequently normal in appearance at birth, but 70% of those who go on to show the complete triad of symptoms display seizures during the first year of life. Although tuberous sclerosis is inherited as an autosomal dominant trait, 86% of cases occur as spontaneous mutations, with older paternal age a contributing factor. TSC affects both sexes equally, with a frequency of one in 5800 births.123 Mutations in the TSCI and TSC2 genes are known to cause tuberous sclerosis.10 The normal function of these genes is to regulate cell growth; if these genes are defective, cellular overgrowth and noncancerous tumor formation can occur.123 Tumor formation in the CNS is responsible for most of the morbidity and mortality with TSC,123 followed by renal disease associated with formation of benign angiomyolipomas.122 Diagnostic criteria for TSC have been established, and the determination can be made clinically; results of genetic testing are currently viewed as corroborative.122 Hypopigmented macules are often the initial finding. These lesions vary in number and are small and ovoid. Larger lesions, known as leaf spots, may have jagged edges.123 Sebaceous adenomas first appear at age 4 to 5 years, with early individual brown, yellow, or red lesions of firm consistency in the nose and upper lips. These isolated lesions may later coalesce to form a characteristic butterfly pattern on the cheeks. Known also as hamartomas (tumor-like nodules of superfluous tissue), the skin lesions are present in 83% of individuals with tuberous sclerosis.45
Delayed development is another characteristic during infancy,124 particularly in the achievement of motor and speech milestones. Cerebral cortical tubers are present in over 80% of patients and account for cognitive disability including autism.122 Ultimately, 93% of individuals who are severely affected will have seizures, usually of the myoclonic type, in early life, progressing in later life to grand mal seizures. Seizure development is the result of formation of nodular lesions in the cerebral cortex and white matter.45 Tumors are also found in the walls of the ventricles. Neurocytological examination reveals a decreased number of neurons and an increased number of glial cells and enlarged nerve cells with abnormally shaped cell bodies.10 Surgical excision of seizure-producing tumors has been successful in some cases.122
Other associated impairments include retinal tumors and hemorrhages, glaucoma, and corneal opacities.123 Cyst formation in the long bones and in the bones of the fingers and toes contributes to osteoporosis. Cardiac and lung tissues are also affected by TSC, and these effects are included in the major diagnostic criteria.122
Neurofibromatosis.
There are two recognized forms of NFM: neurofibromatosis 1 (NFM1) and neurofibromatosis 2 (NFM2).125–127 Neurofibromas, or connective tissue tumors of the nerve fiber fasciculus, impede the development and growth of neural cell tissues126,127 and are the hallmark feature of NFM1. Neurofibromas are noncancerous, and malignant changes are rare in children128 but an increased risk of malignancy has been observed in adult patients with NFM1 and is a major contributor to decreased life expectancy by approximately 15 years.129 Tumors typically increase in number with increasing age. About half of all cases of NFM are caused by sporadic mutation in parental germ cells or during fetal development.125–127 Schwannomas are the main tumor type of NFM2 and classically appear bilaterally on the vestibular nerves.127,130 NFM1 is also known as von Recklinghausen disease. Compared with type II, type I is more common (one per 3000 births)10,126 and usually identified in younger children. It is associated with mutations in the NF1 gene, which produces a protein, neurofibromin, the complete function of which is not yet understood but which is suspected to be a tumor suppressor. Diagnostic criteria for NFM1 include the presence of two of the following features: six or more café-au-lait spots, two or more fibromas, freckling in the axillary or inguinal region, optic pathway glioma, two or more Lisch nodules, specific osseous lesions, and a first-degree relative with NFM1.126 Infants usually appear normal at birth, but initial café-au-lait spots appear by age 3 years in 95% of individuals (Figure 13-4).131 Cognitive impairment is the most common neurological complication of NFM1131 and is postulated to be caused by altered expression of neurofibromin in the brain and/or hyperintense lesions in the brain seen on MRI.131 These focal areas of high signal intensity on T2-weighted MRI, known as unidentified bright objects (UBOs), are seen in 60% of children and young adults with NFM1. The lesions, commonly found in the basal ganglia, internal capsule, thalamus, cerebellum, and brain stem,128,132 tend to disappear in adulthood and often do not cause other overt neurological symptoms.128 Fewer than 10% of individuals are mentally retarded, but about 30% to 60% of affected children have learning disabilities that are mild and nonprogressive.128,133 Poorer social skills and differences in personality, behavior, and quality-of-life perception have been reported in children with NFM1 compared with children without the disorder.126
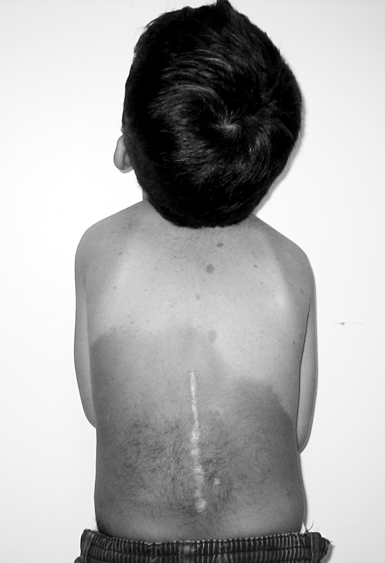
 Four-year-old boy with neurofibromatosis and characteristic café-au-lait spots on trunk.
Four-year-old boy with neurofibromatosis and characteristic café-au-lait spots on trunk.In older children and adolescents, pain, itching, and stinging can occur from cutaneous neurofibromas, and in approximately half of all patients, neurological motor deficits occur from plexiform neurofibromas when the growth puts pressure on peripheral nerves, spinal nerve roots, and the spinal cord.131 One percent to 5% of children aged 0 to 6 years develop symptoms associated with optic pathway glioma.126,128 Neurofibromatous vasculopathy interferes with arterial and venous circulation in the brain.126,131,134 Hydrocephalus occurs in some individuals.126,128 Hypertension is common and may develop at any age,126 and cardiovascular disease is a major cause of premature death.129,131,135 Headaches are a commonly reported symptom in children, adolescents, and adults.126,128,136,137
Scoliosis may develop in 10% of patients and is rapidly progressive from ages 6 to 10 years, or it may manifest in a milder form without vertebral anomalies during adolescence.126 Other skeletal deformities include pseudarthrosis of the tibia and fibula, tibial bowing, craniofacial and vertebral dysplasia, rib fusion, and dislocation of the radius and ulna.126 Differences in leg length126 also have been noted and may contribute to scoliosis. NFM2 occurs less frequently than type I (one in 25,000 to 40,000 births)10 and is caused by a mutation in the gene encoding the protein neurofibromin 2, also called Merlin.10 Merlin is produced in the nervous system, particularly in Schwann cells that surround and insulate the nerve cells of the brain and spinal cord. Although type II shares characteristics with type I, it is commonly characterized by tumors of the eighth cranial nerve (usually bilateral), meningiomas of the brain, and schwannomas of the dorsal roots of the spinal cord.10 Contrary to first descriptions of NFM1 and NFM2, café-au-lait spots are seldom a singular feature of NFM2127; rather, signs and symptoms of tinnitus, hearing loss, and balance dysfunction usually appear during adolescence or in the person’s early 20s.125,127 Problems with visual acuity caused by strabismus and refractive errors are common in young children.138 NFM2 may be underrecognized in children up to 10 years old because early hearing loss and tinnitus are present in only 20% of cases and otherwise only singular features of the condition are observed. Infants may have cataracts, and children may demonstrate unilateral facial paralysis, eye squinting, mononeuropathy (foot or hand drop), meningioma, spinal tumor, or cutaneous tumor. It is recommended that children of parents with NFM2 should be considered to be at 50% risk for NFM2 and screened from birth.130
Autosomal recessive disorders
An unaffected carrier of a disease-causing trait is heterozygous for the abnormal gene (possessing one normal and one mutated copy of the gene). If both parents are unaffected carriers of the gene, each of their offspring has a 25% risk of exhibiting the disorder.8 Consanguinity involving close relatives increases the chance of passing on autosomal recessive traits.8 Certain types of limb defects, familial microcephaly, and a variety of syndromes are passed on through autosomal recessive genes. Four examples of autosomal recessive disorders affecting children in therapy settings are presented in this section: cystic fibrosis (CF), Hurler syndrome, phenylketonuria (PKU), and spinal muscle atrophy (SMA).
Cystic fibrosis.
CF is one of the most common autosomal recessive disorders and is more common in Caucasians, affecting one in 2000 to 4000.10 The CF gene has been mapped to chromosome 7, and its protein product, CF transmembrane regulator (CFTR).8 CFTR is involved in the regulation of chloride channels of the bowel and lung, which is dysfunctional in patients with CF. Although CF has markedly variable expression, the overall median survival time has improved from about 6 years of age in the 1940s to an average of 36 years of age in 2006.139 In addition to the phenotypical features of CF, diagnosis of CF is made when two or more disease-causing mutations exist on the CTFR gene.139 Newborn screening tests for CF are required in all states in the United States.140
Fibrotic lesions of the pancreas cause pancreatic insufficiency in the majority of patients, which leads to chronic malnutrition. Ten percent to 20% of newborn infants with CF also have intestinal tract involvement with a meconium ileus. The sweat glands are commonly affected; high levels of chloride found in the sweat is the basis for the sweat chloride test used in diagnosis. The most serious impairment in CF is the obstruction of the lungs by thick mucus, which leads to chronic pulmonary obstruction, infection that destroys lung tissue, and eventual death from pulmonary disease in 90% of individuals.8
Improved survival rate in recent decades is a result of improved antibiotic management, aggressive chest physical therapy, and pancreatic replacement therapy. Postural drainage, percussion, vibration, and breathing exercises are key components of the management program provided by the therapist and caregivers.141 Modern and less labor-intensive devices such as those that provide positive expiratory pressure may not be as effective at clearing secretions as conventional chest physiotherapy,141 but patient and caregiver compliance with a regular program may be improved with them.142 Attention to diet is important, and every attempt should be made to maintain a routine exercise program with a goal of helping the children be more active to improve their respiratory status and to prevent secondary impairments of adolescence and adulthood such as stress incontinence in young women caused by excessive coughing,143,144 chronic back pain, and osteopenia and osteoporosis.145
Massery143 describes the relationship among respiration, postural control, and secondary impairments that develop in individuals with CF in the musculoskeletal and neuromuscular systems. She addresses the threefold problem faced by individuals with CF: (1) lung dysfunction leading to increased respiratory demand; (2) increased workload of respiration as a deforming force on the immature musculo-skeletal frame; and (3) resultant impaired motor strategies for postural control during physical activity. Patients were once widely cautioned to avoid overexertion and fatigue with exercise, but as patients are living longer, more evidence supports the benefits of regular, even vigorous exercise for children and adults with CF. Guidelines for exercise frequency and intensity have been published by the Association of Chartered Physiotherapists in Cystic Fibrosis.146
Hurler syndrome (mucopolysaccharidosis I, severe type).
Hurler syndrome is an inborn error of metabolism that results in abnormal storage of mucopolysaccharides in many different tissues of the body.97 The incidence is estimated to be one in 100,000 live births for the severe forms10 and one in 500,000 for milder forms.147 IDUA is the only gene currently known to be associated with this multisystem disorder.147
Infants born with Hurler syndrome are usually normal in appearance at birth, may have inguinal or umbilical hernias,147 and may have higher birth weights than their siblings. Symptoms of this progressively deteriorating disease usually appear during the latter half of the first year of life,147 with the full disease picture apparent by 2 to 3 years of age.10,147 Diagnosis is made by identification of deficiency in lysosomal enzymes.147,148 Premature death, usually from cardiorespiratory failure, occurs within the first 10 years of life.147
Characteristic physical features are caused by storage of glycosaminoglycans (GAGs)147 and include a large skull with frontal bossing, heavy eyebrows, edematous eyelids, corneal clouding, a small upturned nose with a flat nasal bridge, thick lips, low-set ears, hirsutism, and gargoyle-like facial features. Growth retardation results in characteristic dwarfism.147 Some individuals with the physical characteristics of Hurler syndrome have normal intelligence, but most have progressive and profound intellectual disability.10,147
Spastic paraparesis or paraplegia and ataxia have been observed in individuals with Hurler syndrome.8 Commonly reported orthopedic deformities include flexion contractures of the extremities, thoracolumbar kyphosis, genu valgum, pes cavus, hip dislocation, and claw hands as a result of joint deformities.45 Defective ossification centers of the vertebral bodies results in spinal deformity, complications of nerve entrapment, atlanto-occipital instability, and restricted cervical range of motion.147 Conductive and sensorineural hearing loss is common.147 Delayed motor milestones have been noted as early as 10 months of age,148 with severe disabilities occurring with increasing age. Adaptive equipment often is needed, and most children with Hurler syndrome become wheelchair users in their later years.148
Phenylketonuria.
PKU is the result of one of the more common inborn errors of metabolism. Mutations of the PAH gene located on chromosome 12 cause a deficiency in the production of phenylalanine hydroxylase.149 Without this enzyme, there is no conversion of phenylalanine to tyrosine, resulting in an abnormally excessive accumulation of phenylalanine in the blood and other body fluids.149,150 If untreated, this metabolic error results in mental and growth retardation, seizures, and pigment deficiency of hair and skin.151 PKU is most prevalent among individuals of northern European ancestry, with a frequency of one in 10,000 to 15,000 births in the United States.149 It is estimated that one of every 50 individuals is heterozygous for PKU.8
Children born with PKU are usually normal in appearance, with microcephaly and delayed development becoming apparent toward the end of the first year. Parents usually become concerned with the child’s slow development during the preschool years.151 If PKU is untreated, the affected child may go on to develop hypertonicity (75%), hyperactive reflexes (66%), hyperkinesis (50%), or tremors (30%),152 in addition to intellectual disability. IQ levels generally fall between 10 and 50, although there have been reported rare cases of untreated individuals with normal intelligence.151
A simple blood plasma analysis, which is mandatory for newborn infants in all 50 U.S. states,140 can detect the presence of elevated phenylalanine levels in nearly 100% of cases.150 This test is ideally performed when the infant is at least 72 hours old. If elevated phenylalanine levels are found, the test is repeated, and further diagnostic procedures are performed. Placing the infant on a low phenylalanine diet (low protein) can prevent the intellectual disability and other neurological sequelae characteristic of this disorder.151 Follow-up management by an interdisciplinary team consisting of a nutritionist, psychologist, and appropriate medical personnel is advised in addition to the special diet. Individuals with poor compliance with the recommended diet have a greater risk of osteopenia in adulthood.150
Spinal muscle atrophy.
SMA (5q SMA) is characterized by progressive muscle weakness because of degeneration and loss of the anterior horn cells in the spinal cord and brain stem nuclei.153,154 Diagnosis of SMA is based on molecular genetic testing for deletion of the SMN1 gene (named for “survival of motor neuron 1”), location 5q13. Another gene, SMA2, can modify the course of SMA. Individuals with multiple copies of SMA2 can have less severe symptoms or symptoms that appear later in life as the number of copies of the SMN2 gene increases.155 The overall disease incidence of SMA is five in 100,000 live births.155
The clinical classifications of SMA are still evolving.153,154,156 At present, four subtypes (types I to IV) are well accepted, and a fifth, type 0, is being explored. The subtypes are based on age at symptom onset and expectations for maximum physical function, the latter being more closely related to life expectancy.156
SMA type 0 is characterized by extreme muscle weakness apparent before 6 months of age that likely had a prenatal onset.153,154 Some infants have a prenatal history of decreased fetal movements during the third trimester.153
SMA I, otherwise known as Werdnig-Hoffmann disease or acute infantile SMA,10 has an onset before 6 months of age.153,156 Incidence is estimated to be one in 20,000 live births.10 It is characterized clinically by severe hypotonicity, generalized symmetrical muscle weakness, absent deep tendon reflexes, and markedly delayed motor development. Intellect, sensation, and sphincter functioning, however, are normal.153 Children usually cannot sit without support and have poor head control.156 They have a weak cry and cough and problems with swallowing, feeding, and handling oral secretions.154 The diaphragm is spared, but combined with weakness in intercostal muscles, infants exhibit paradoxical breathing, abdominal protrusion, and a bell-shaped trunk with chest wall collapse.154 Overall, this pattern of chest wall weakness and poor respiratory function contributes to the greatly increased susceptibility to pulmonary infection, which usually results in death before the age of 2 years.10,154,155
SMA II, otherwise known as intermediate or chronic infantile SMA, has an onset at age 6 to 18 months and is associated with delayed motor milestones.156 Seventy percent of children diagnosed with SMA II are alive at 25 years of age.153
Children with SMA II can usually sit independently if placed but never stand unsupported.154 Bulbar weakness with swallowing difficulties, poor weight gain, and diaphragmatic breathing are common.155 Finger trembling is almost always present.153,154 Joint contractures are present in most individuals. Kyphoscoliosis of severity to require bracing and/or surgery often develops, but patients are at risk of postanesthesia complications.154 Respiratory failure is the major cause of morbidity and mortality. Nocturnal oxygen desaturation and hypoventilation occur before daytime hypercarbia and are early indications of need for ventilator support.154
SMA III is characterized by onset of symptoms in childhood after 18 months.153 It is also known as juvenile SMA or Kugelberg-Welander syndrome.10 These individuals have a normal life span and usually attain independent ambulation and maintain it until the third or fourth decade of life.153 Lower extremities are often more severely affected than the arms. Strength is often not sufficient for stair climbing, and balance problems are common.153 Muscle aches and joint overuse symptoms are frequently reported.154
SMA IV typically has an onset at older than 10 years of age and is associated with a normal life expectancy and no respiratory complications.154,156 Individuals maintain ambulation during the adult years.154
Variants of SMA occur in individuals with similar phenotypes and clinical diagnostic features of electromyography (EMG) that are not associated with deletion of SMN1.156 Genetic testing for SMN gene deletion achieves up to 95% sensitivity and nearly 100% specificity.154 For cases that remain unclear, a clinical diagnosis may be accomplished through EMG and muscle biopsy, which reveal neurogenic atrophy. Key physical signs are common: symmetrical weakness in the more proximal musculature versus distal, and lower extremity weakness that is greater than in the arms.154 Traditional strength measurements are not practical for children with SMA. The Gross Motor Function Measure157 has excellent reliability in studies of gross motor evaluation in this population.154,158 Consensus guidelines on pulmonary care including assessment, monitoring, and treatment; feeding and swallowing, gastrointestinal dysfunction and nutrition; and orthopedic management have been published by the Standard of Care Committee for Spinal Muscle Atrophy.154 Currently there are no efficacious drugs to effectively treat the symptoms of SMA.160,161
Sex-linked disorders
The third mechanism for transmission of specific gene defects is through sex-linked inheritance. In most sex-linked disorders, the abnormal gene is carried on the X chromosome. Female individuals carrying one abnormal gene usually do not display the trait because of the presence of a normal copy on the other X chromosome. Each son born to a carrier mother, however, has a 50:50 chance of inheriting the abnormal gene and thus exhibiting the disorder. Each daughter of a carrier mother has a 50:50 chance of becoming a carrier of the trait.8 Four syndromes that result in disability are discussed in this section: hemophilia, fragile X syndrome (FXS), Lesch-Nyhan syndrome (LNS), and Rett syndrome (RS).
Hemophilia.
Hemophilia is a bleeding disorder caused by a deficient clotting process. Affected individuals will have hemorrhage into joints and muscles, easy bruising, and prolonged bleeding from wounds. The term hemophilia refers to hemophilia A (coagulation factor VIII deficiency) and hemophilia B or Christmas disease (coagulation factor IX deficiency). There are numerous other clotting diseases, and some that were once referred to as hemophilia are now genomically distinguished. For example, von Willebrand disease has a distinctly different genetic basis from hemophilia; it follows an autosomal recessive or autosomal dominant pattern and involves mutation of the von Willebrand factor (VWF) gene, located on chromosome 12. VWF plays a role in stabilizing blood coagulation factor VIII.162 Hemophilia A and B occur as X-linked recessive traits owing to mutations of genes F8 and F9, respectively, both of which are located on the X chromosome.163,164
Hemophilia A is reported to affect one in 4000 to 5000 males worldwide.163 Hemophilia B is less common, affecting one in 20,000 males worldwide.164 Hemophilia can affect females, though in milder form. The severity and frequency of bleeding in hemophilia A are inversely related to the amount of residual factor VIII (less than 1%, severe; 2% to 5%, moderate; and 6% to 35%, mild).163 The proportions of cases that are severe, moderate, and mild are about 50%, 10%, and 40%, respectively.165 The joints (ankles, knees, hips, and elbows) are frequently affected, causing swelling, pain, decreased function, and degenerative arthritis. Similarly, muscle hemorrhage can cause necrosis, contractures, and neuropathy by entrapment. Hematuria and intracranial hemorrhage, although uncommon, can occur after even mild trauma. Bleeding from tongue or lip lacerations is often persistent.8
Hemophilia is usually diagnosed during childhood, with the most severe cases diagnosed in the first year of life: bleeding from minor mouth injuries and large “goose eggs” from minor head bumps are the most frequent presenting signs in untreated children.163 Children are especially vulnerable to bleeding episodes owing to the nature of their physical activity combined with periods of rapid growth.163
Treatment includes guarding against trauma and replacement with factor VIII derived from human plasma or recombinant techniques.8 In the late 1970s to mid 1980s it was estimated that half of the affected individuals in the United States contracted hepatitis B or C or human immunodeficiency virus (HIV) infection when treated with donor-derived factor VIII. The initiation of donor blood screening and use of heat treatment of donor-derived factor VIII has almost completely eliminated the threat of infection.8 Although replacement therapy is effective in most cases, 30% of treated individuals with hemophilia A and 3% of individuals with hemophilia B have neutralizing antibodies that decrease its effectiveness.163,164 Before treatment with clotting factor concentrates was available, the average life expectancy was 11 years163; currently, excluding death from HIV, life expectancy for those with severe hemophilia who receive adequate treatment is 63 years.163 Factor replacement therapy is credited for increasing the ease and safety of vigorous exercise and sports participation for individuals.166 The benefits of regular exercise are the same as for unaffected individuals and outweigh the risks in treated persons.167 A 2002 pilot study by Tiktinsky and colleagues167 found decreased episodes of bleeding in a population of young adults with a long-term history of resistance training that began in adolescence.
Fragile X syndrome.
FXS is the most common sex-linked inherited cause of intellectual disability, affecting one in 4000 males and one in 8000 females.168 Males manifest a more severe form than females. A fragile site on the long arm of an X chromosome is present, with breaks or gaps shown on chromosome analysis. A region of the X chromosome, named FMR1, normally codes for proteins that may play a role in the development of synapses in the brain. Mutations of this region are errors of trinucleotide repeats, in which the number of CGG triplets at this region is expanded, thereby making the gene segment unable to produce the necessary protein.168
Developmental milestones are slightly delayed in affected males.168 Eighty percent of males are reported to have intellectual disability, with IQs of 30 to 50 being common but ranging up to the mildly retarded to borderline range.168 Penetrance (the proportion of individuals with a mutation that actually exhibit clinical symptoms) in the female is reported to be only 30%.8 Other impairments include epilepsy, emotional lability, attention-deficit/hyperactivity disorder (ADHD), and clinical autistic disorder in 30% of males.168,169 Life span is normal for individuals with this condition.168
Lesch-nyhan syndrome.
Also known as hereditary choreoathetosis,170 LNS leads to profound neurological deterioration. First described in 1964 by Lesch and Nyhan,171 it is associated with a mutation in the HPRT1 gene on the X chromosome. This gene codes for an enzyme, hypoxanthine guanine phosphoribosyltransferase, which allows cells to recycle purines, some of the building blocks of DNA and ribonucleic acid (RNA).172 Without this gene’s normal function, there is an overproduction of uric acid (hyperuricemia),172 which accumulates in the body. High uric acid levels are thought to cause neurological damage.170,172
The prevalence of LNS is one in 380,000 individuals.172 Females born to carrier mothers have a 25% chance of inheriting the mutation. There are rare reports of females demonstrating this syndrome as a result of X chromosome inactivation. Most female carriers are considered to be asymptomatic, but some may have symptoms of hyperuricemia in adulthood.172
LNS is detectable through amniocentesis, and genetic counseling is advisable for parents who have already given birth to an affected son.173
The prenatal and perinatal course is typical for affected individuals. Hypotonia and delayed motor skills are noticeable by age 3 to 6 months.172 Dystonia, choreoathetosis, and opisthotonus indicative of extrapyramidal involvement emerge during the first few years of life.172 Many children are initially diagnosed with athetoid cerebral palsy when pyramidal signs such as spasticity, hyperreflexia, and abnormal plantar reflexes emerge.172 Most children never walk. A hallmark of the disease is severe and frequent self-injurious behaviors such as lip and finger biting, which emerge in almost all affected children by their third birthday.172 Because of the extreme self-mutilation that characterizes this disorder, it has been questioned whether these children have normal pain perception.174 Although these children have the abnormal catecholamine metabolism seen in other patients with congenital pain insensitivity,175 behaviors documented in children with LNS suggest that they do sense pain, demonstrated by their apparent relief when they are restrained from hurting themselves. Children may actually request the restraining device176 even when the device may be one that would not physically prevent biting, such as a glove or bandaid.172 A reported survey of parents of children with LNS indicated that parents often find behavioral programming techniques helpful in modifying aggression toward self or others.176 However, there is no consensus on the best kind of behavioral treatments, as any reward, either positive or negative, may increase the frequency of self-injury.177 Some parents have reported that they elected tooth extraction as a means to prevent biting. Other impairments in children with LNS include severe dysarthria and dysphagia. Bilateral dislocation of the hips may occur as a result of the spasticity.172 Growth retardation is also apparent, as well as moderate to severe intellectual disability.10 Individuals may have gouty arthritis and kidney and bladder stones.
Blood and urine levels of uric acid have been decreased successfully through the administration of allopurinol, with a resultant decrease in kidney damage. With current management techniques, most individuals survive into their second or third decade of life.172
Rett syndrome.
RS is inherited in an X-linked pattern and it affects females almost exclusively, as it is most often lethal in boys before age 2 years. Males may inherit RS with an extra X chromosome in many or all of the body’s cells.178–181 The estimated incidence is one in 15,000 to 20,000 females.10,182 It has been reported that 99% of all cases of RS are the result of sporadic mutations.181,183 Most cases of RS, called classic RS, are caused by mutations in the MECP2 gene, which is responsible for directing proteins critical for normal synaptic development; however, it is unclear how these mutations lead to all the signs and symptoms of the syndrome.181,184 Several variants of RS exist; they have overlapping features with classic RS but may have a much milder or more severe course.181
Classic RS is characterized by apparently normal development during the first 6 months of life, followed by a short period of developmental plateau, and then rapid deterioration of language and motor skills typically occurring at 6 to 18 months of age.181,185 Most girls survive into adulthood.181 The hallmark of the syndrome is that during the period of regression, previously acquired purposeful hand skills are also lost and replaced by stereotypical hand movements. These nonspecific hand movements have been described as hand wringing, clapping, waving, or mouthing. Virtually all language ability is lost, although some children may produce echolalic sounds and learn simple manual signing. Evidence of minimal receptive language skills may be observed. Autistic behaviors, inconsolable crying and screaming, and bruxism are common features of individuals with RS.181 Almost all individuals with RS function in the range of severe to profound intellectual disabilities.
Head circumference is normal at birth, and its increase may decelerate in early childhood, but microcephaly is not a consistent feature of RS.181 Retarded growth and muscle wasting are observed in most girls, likely associated with poor food intake and gastrointestinal problems.181
Almost one fourth of girls with RS never develop independent ambulation skills; otherwise the onset of walking is usually delayed until about 19 months of age.186 Initially hypotonia may be evident, but with advancing age, spasticity of the extremities develops.187 Increased muscle tone is usually observed first in the lower extremities, with continued greater involvement than in the upper extremities. Peripheral vasomotor disturbances, especially in the lower limbs, are often noted.181
Scoliosis, which is often severe enough to require surgical correction, occurs in most girls by adolescence, characterized by a long C-shaped thoracolumbar curve, kyphoscoliosis, and an early onset of posterior pelvic tilt and abducted shoulder girdles.186,188–192 Heel cord tightening, and hip instability have also been identified as areas of potential concern.188 Abnormal EEG and seizures occur in 70% of individuals with RS in the first 5 years of life. Cranial CT results are normal or show mild generalized atrophy. Breathing dysfunction, including wake apnea and intermittent hyperventilation,186 is also associated with RS. Interventions reported in the literature have focused on splinting,193 behavioral modification techniques to teach self-feeding skills,194 aquatic therapy,195 occupational therapy,196 music therapy,197 physical therapy,181,190,191,197 and the first two combined in a dual-intervention approach.198
Mitochondrial DNA disorders
In addition to the nuclear genome, humans have another set of genetic information within their mitochondria. Nuclear genes exist in pairs of one maternal and one paternal allele. In contrast, there are hundreds or thousands of copies of mtDNA in every cell. mtDNA is small, circular, and double stranded. It has been well studied and was mapped long before the human nuclear genome. mtDNA contains 37 genes responsible for normal function of the mitochondria in all body cells.198a Humans inherit mtDNA maternally. mtDNA is highly susceptible to mutation, and the molecule has limited ability to repair itself. Tissues that have a high demand for oxidative energy metabolism, such as brain and muscle, appear to be most vulnerable to mtDNA mutations.11 Normal and mutated versions of mtDNA can coexist within a patient’s body, but when a certain critical number of mutations exist, the body’s tissues will show clinical signs of dysfunction. These disorders affect the metabolic functions of the mitochondria, such as the generation of the body’s energy currency, adenosine triphosphate. Many patients with point mutations of mtDNA exhibit symptoms in early childhood; these mutations may be the most frequent cause of metabolic abnormality in children.11 The minimum birth prevalence of childhood mitochondrial respiratory chain disorders is reported to be 6.2 per 100,000.12,199–201 Medical intervention for mitochondrial encephalomyopathies cannot treat the underlying disease, but the value of rehabilitative therapies has been reported.202,203 An example of a childhood disorder that can result from an mtDNA mutation is Leigh syndrome.
Leigh syndrome
Leigh syndrome, or subacute necrotizing encephalomyopathy, may also be transmitted by X-linked recessive and autosomal recessive inheritance. Approximately 20% of all cases of Leigh syndrome are caused by mitochondrial mutations.204 The discussion in this section will focus on characteristics of mtDNA-associated Leigh syndrome.
Leigh syndrome has an onset in infancy, typically at 3 to 12 months of age. Initial features may be nonspecific, such as a failure to thrive and persistent vomiting.204,205 It is a progressive disorder caused by lesions that can occur in the brain stem, thalamus, basal ganglia, cerebellum, and spinal cord. Common clinical features include seizures, epilepsy, muscle weakness, peripheral neuropathy, speech and feeding difficulties, gastrointestinal and digestive problems, and heart problems. Most affected children have hypotonia, movement disorders such as chorea, and ataxia. Life expectancy is 2 to 3 years; death most often results from respiratory or cardiac failure.204
Multifactorial disorders
Multifactorial disorders are believed to be a result of the combined effects of mutations in multiple genes and environmental factors.8 Environmental factors may be those that have an impact on a developing fetus, such as prenatal diet, or those that have an impact on humans as we age, such as cigarette smoking. Disorders in this category can result in congenital malformations such as spina bifida and clubfoot. An in-depth discussion of spina bifida can be found in Chapter 15. Management information on clubfoot can be found in pediatric textbooks that include orthopedic information.206–209
Many diseases such as cancer can result when the environment interacts with genetic variations that exist in all humans.8 Scientists are exploring genetic contributions to premature births.210,211 Premature birth is the leading cause of infant mortality and morbidity,212,213 and it most likely is a result of multiple genetic and environmental determinants that tend to run in families.210,211 Premature infants are at higher risk of neurological, musculoskeletal, and respiratory problems than term infants are. Management of infants with low birth weight is discussed in Chapter 11 of this text.
Body structure and function problems common to many pediatric genetic disorders
Specific examples of genetic disorders in children were presented in the foregoing section. Although there are many disorders that physical and occupational therapists may see often, others are rare and may only be suspected based on clustered problems of body structure, function, and activity limitations. Decisions about which interventions to implement and the expected outcomes will be largely influenced by the diagnosis of the specific disorder, once attained. Many genetic conditions share in common a short list of primary problems that will negatively affect the child’s physical movement and daily activities and participation, both immediately and in the long term. Table 13-3 summarizes the problems common to many genetic disorders that are most relevant for physical or occupational therapists.
TABLE 13-3 
CHARACTERISTIC FEATURES OF SELECTED GENETIC CONDITIONS
| GENETIC CONDITION | TYPICAL AGE AT DIAGNOSIS | CRANIOFACIAL DYSMORPHISM | MUSCULOSKELETAL INVOLVEMENT | NEUROMUSCULAR SYSTEM INVOLVEMENT AND TONE | CARDIO-PULMONARY INVOLVEMENT | OTHER SYSTEM INVOLVEMENT | OTHER DISEASE PROCESSES | SENSORY DYSFUNCTION | MOTOR DELAY | COGNITIVE DELAY | HALLMARK FEATURE(S) |
| Trisomy 21 | Prenatal or infancy | Yes | Joint laxity and instability | Hypotonia | Yes | Alzheimer disease, sleep apnea, obesity | Vision, hearing | Yes | Yes | Facial features, simian crease | |
| Trisomy 18 | Prenatal, neonatal | Yes | Yes | Hypotonia | Yes | Yes | Yes | Yes | Yes | Life span < 1 year | |
| Turner syndrome | Infancy, adolescence; girls | Yes | Short stature, hip dislocation, scoliosis | Yes | Distal lymphedema; reproductive system | Hearing | Webbed neck appearance and short stature | ||||
| Klinefelter syndrome | Adolescence; adulthood; males | Yes | Yes | Varies with age | Endocrine and reproductive system | Yes | Yes | Intellectual disability; course of gonadal development | |||
| Cri-du-chat syndrome | Infancy | Yes | Yes | Hypotonia | Yes | Vision | “Cat cry” | ||||
| Prader-Willi syndrome | Infancy | Yes | Scoliosis | Hypotonia | Secondary to obesity | Reproductive system | Morbid obesity | Vision | Yes | Yes | Hypotonia, obesity |
| Angelman syndrome | Infancy | Microcephaly | Hypotonia and seizures, ataxia | Yes | Yes | Happy demeanor, sleep disorders | |||||
| Acute lymphoblastic leukemia | Childhood | Bone pain and muscle cramps | Osteopenia, osteoporosis | Lethargy, fever, respiratory infections | |||||||
| Osteogenesis imperfecta | Varies with type and severity | Multiple fractures and muscle weakness | Yes | Dentinogenesis | Hearing (type I) | Multiple fractures | |||||
| Tuberous sclerosis complex | Infancy, early childhood | Yes, cyst formation | Hypertonia; seizures | Yes, cyst formation | Skin lesions, adenomas; renal disease | Vision | Yes | Yes | Sebaceous adenomas, seizures | ||
| Neurofibromatosis type 1 | Infancy, childhood | Yes | Café-au-lait spots and cutaneous neurofibromas; vasculopathy | Vision | Café-au-lait spots, neurofibromas | ||||||
| Cystic fibrosis | Infancy | Chest wall deformity, muscle and bone pain | Yes | Gastrointestinal system; urinary stress incontinence in females | Osteoporosis, osteopenia | Lung and digestive dysfunction | |||||
| Hurler syndrome | Infancy | Yes | Yes | Yes | Yes | Yes | Vision, hearing | Yes | Yes | Progressive craniofacial abnormalities and developmental deterioration | |
| Spinal muscle atrophy type II | Infancy | Yes | Hypotonia | Yes | Yes | Yes | Progressive loss of peripheral motor function | ||||
| Hemophilia type A | Childhood or earlier if severe | Joint pain and hemarthroses | Skin bruising | Bleeding, bruising, joint pain, and loss of motion | |||||||
| Fragile X syndrome | Childhood | Yes | Joint laxity | Yes | Yes | Yes | Intellectual disability, autism | ||||
| Lesch-Nyhan syndrome | Infancy | Gouty arthritis | Varies | Urogenital system | Yes | Yes | Yes | Self-injurious behavior, gouty arthritis | |||
| Rett syndrome | Late infancy to early childhood | Scoliosis | Hypotonia, ataxia, seizures | Yes | Yes | Yes | Regressive developmental delay; stereotypical, purposeless hand movements |
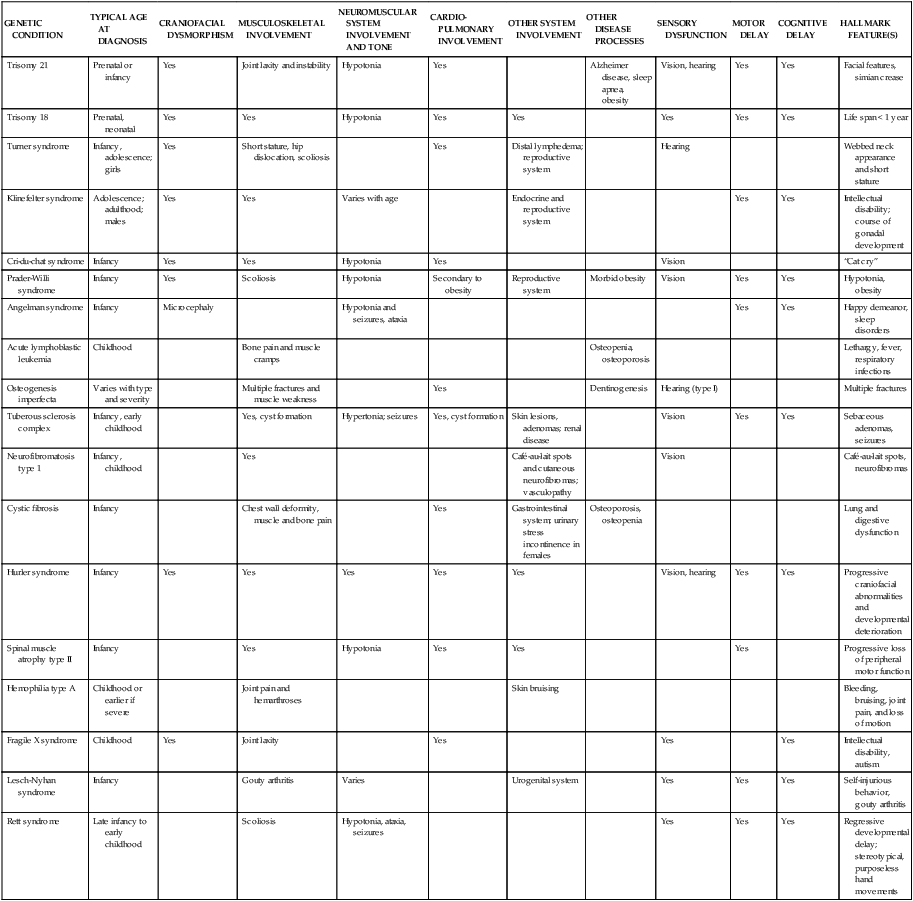
Hypertonicity
Abnormalities of tone may result from dysgenesis or injury to developing motor pathways. Hypertonia is common to many motor disorders and is defined as “abnormally increased resistance to externally imposed movement about a joint.”214 Examples of externally imposed movement are passive movement by the therapist or changes in ankle and knee position resulting from ground reaction forces during ambulation. Children with hypertonus generally display stiff or jerky movements that are limited in variety, speed, and coordination. Controlled, voluntary movements tend to be limited to the middle ranges of a joint. Total patterns of flexion or extension may dominate, with limited ability for selective joint movements. Motor development of children with hypertonicity may be further complicated by the retention of primitive reflexes, which can result in stereotyped movements associated with sensory input.214
Sanger and colleagues214 proposed a classification system to objectively define and distinguish different types of hypertonicity. Three general types of hypertonicity—spasticity, dystonia, and rigidity—may occur alone or in combination. Spasticity is a velocity-dependent, increased resistance to muscle stretch that may occur above a given threshold of speed and/or joint angle and may depend on the direction of joint movement. Dystonia is also an involuntary alteration in the pattern of muscle activation during voluntary movement or maintenance of posture. The observable disorder is demonstrated by intermittent muscle contractions causing twisting or repetitive movement, postures, or both.214 Dystonia may be triggered by attempts at voluntary movement or to prevent the movement of a joint (e.g., to prevent knee buckling in stance). The pattern and magnitude of the abnormal muscle activity may change with the child’s arousal, emotional state, and tactile contact.214 Rigidity is a form of hypertonus in which the speed of movement and joint angle do not affect the movement quality. Stiffness caused by diminished tissue length and extensibility of muscles and connective tissue is not included in the recent definitions of hypertonus (but may exist alongside hypertonicity).214,215
Children may learn to use stereotypical patterns of movement and hypertonus to achieve functional goals by activating the muscle synergies of a reflex without sensory feedback.216 If a goal of therapy is to facilitate functional movement that is not dominated by persistent reflexes, it is critical to practice new motor patterns to accomplish the functional activity for which that reflex is being used. The focus of therapy activities needs to be on active movement of the child and not on passive inhibition techniques of abnormal reflexes for the sake of “normalization” of tone and movement.216–219
Hypotonicity
In contrast to hypertonia, low tone or hypotonia is not clearly defined. The “floppy” infant or child is commonly characterized as having hypermobility of joints that lack resistance to passive movement, with diminished antigravity movement and postural stability. Hypotonia may occur because of central or peripheral nervous system dysfunction, as is the case in newborns with PWS and toddlers with SMA type II, respectively. Many genetic disorders are revealed in newborns based on the common features of severe, global hypotonia and low Apgar scores.220 Retrospective studies of newborns report key features of absence of antigravity movements and decreased reflexes. The presence of fetal hypokinesia and/or polyhydramnios is reported to be predictive of neonatal hypotonia in many cases.221 In full-term neonates with hypotonia, studies report that 30% to 60% of cases are associated with a genetic disorder. A clinical neurological examination such as described by Dubowitz and the use of dysmorphic data bases can identify the majority of cases.221,222 First-line genetic testing is indicated in neonates with hypotonia plus facial dysmorphism or signs of peripheral hypotonia (e.g., as seen in SMA).221
Martin and colleagues223 surveyed physical and occupational pediatric therapists and reported that the majority of therapists do not use formal examination methods to quantify hypotonia directly, but rather use measurements for various expressions of hypotonia, most often muscle strength and developmental milestones. This study also confirmed that most therapists agree that children with hypotonicity have diminished postural control and thus tend to lean on supports to maintain a position. Examples of this behavior are locking out weight-bearing joints and assuming positions that provide a broad base of support to maximize their stability (Figure 13-5). Although retention of primitive reflexes is less likely in children with hypotonia compared with those with hypertonia, delays in the development of postural reactions are a major concern. Limited strength and lack of endurance are often concerns with children who have hypotonicity. Hypotonicity and joint laxity are often associated with motor delay; however, therapists should not assume that hypotonia and joint laxity are absolutely predicative of persistent motor delay.224 For example, many premature infants, with or without a genetic disorder, have global hypotonia at birth that resolves and does not cause long-term functional impairment.224,225 Hypotonicity is a persistent problem in many children with developmental delay. Therapists may address hypotonia and problems of postural control with a variety of treatment modalities and techniques including aquatic therapy,226 hippotherapy,227 and neurodevelopmental therapy.228


Hyperextensible joints
Hyperextensible joints are commonly observed in children with hypotonicity and are noted in many children with a variety of genetic disorders, representing different underlying organ structure problems. Activities should be modified to avoid undue stress to these joints and the surrounding ligaments, tendons, and fascia. For example, positions that allow the knee or elbow joints to lock into extension should be modified so that weight bearing occurs through more neutral alignment. Varying the placement of toys and support surfaces, providing physical assistance, and using adaptive equipment can help modify weight-bearing forces to achieve more neutral alignment.229 For example, if hyperextensibility of ligaments leads to excessive pronation in stance (Figure 13-6), the use of ankle-foot orthoses may provide enough support to the structures to allow functional activities in standing (see the discussion of orthotics in Chapter 34). For a child who stands with knee hyperextension, a vertical stander may allow that child to stand and play at a water table with her or his classmates for extended periods with the knees in a more neutral position. Rather than restricting a child’s repertoire of upright positions, it is preferable to modify an activity or provide external support to enable a child to participate fully (Figure 13-7).230



Contractures and musculoskeletal deformities
Conditions that cause hypertonicity or spasticity are well known to place children at risk for joint contracture.231 Children with hemophilia are at great risk of joint contractures associated with hemarthroses and intramuscular hemorrhages.232 Spinal deformities, such as lumbar lordosis and thoracic kyphosis and scoliosis, are also common concerns in children with hypertonia or hypotonia. Although joint contractures are less likely to occur in a child with hypotonicity, habitual positioning may lead to soft tissue restrictions. For example, children with hypotonia often adopt a constant position of wide abduction, external rotation, and flexion at the hips (“frog” or “reverse W” position)233; in these children, soft tissue contractures can develop at the hips and knees. Children whose hips are maintained in a position of adduction, flexion, and internal rotation are at risk for hip subluxation or dislocation.233
Deformation of the anterior chest wall and shoulder girdles may result from primary problems in the cardiopulmonary system such as in children with CF. When coupled with hypotonicity and poor postural control, the anterior chest wall muscles tighten as a result of the long-term rounded, internally rotated shoulders and protracted scapulae, in which case therapeutic interventions should target improving chest wall and scapulothoracic mobility as well as strengthening postural muscles (Figure 13-8).234–236 In general, contractures and deformities are a concern for most children who display a limited variety of postures and movements. Therapists should consider the nature of the disorder that places the child at risk for contractures when choosing treatment techniques; disorder-specific techniques can be found in pediatric occupational and physical therapy textbooks.206,237

Respiratory problems
A genetic risk for respiratory distress in infancy has been suggested by reports of family clusters.238 Furthermore, comparison of short- and long-term respiratory function in infants with respiratory distress syndrome suggests that if all other factors of nutrition, previous mechanical ventilation, and gestational development are comparable, genetic risk may account for cases of chronic and potentially irreversible respiratory failure.238
Respiratory problems are often observed in children with limited mobility. If the mobility impairments are the result of hypotonicity or hypertonicity, impaired respiration may be a result of chest and skeletal deformities. Many infants with genetic disorders are born prematurely and are more susceptible to respiratory problems than infants born at full term.210,211,233 Prolonged mechanical ventilation and other medical procedures may increase the time neonates spend in the supine position, thus increasing the risk of gravity-induced deformity of the rib cage and inefficiency of the respiratory musculature.234,239,240
Some children may find it difficult to tolerate one position for an extended time owing to respiratory difficulties. For these children, frequent changes of position and use of adapted positioning devices may be necessary. Premature infants in the neonatal intensive care unit may benefit from regular prone positioning to facilitate restorative sleep,240–242 improved arterial oxygen saturation,243 and improved respiratory synchrony.244 Children with respiratory problems may require mobilization techniques, deep breathing, chest expansion exercises, and postural drainage. In the case of children with CF, a comprehensive program of respiratory care is the primary therapy goal.245
Developmental delay
Developmental delay is typified by the failure to meet expected age-related milestones in one or more of five areas: physical, social and emotional, intellectual, speech and language, and adaptive life skills. Developmental milestones that are typically assessed in the first 5 years of life can be found in Box 13-2.
BOX 13-2  DEVELOPMENTAL MILESTONES
DEVELOPMENTAL MILESTONES
Lower jaw trembles constantly even when infant is not crying or excited
Does not respond to loud sounds or bright light
Does not focus on and follow a nearby object moving side to side
Failure to develop heel-toe walking pattern after several months of walking
Does not speak at least 15 words by 18 months
Does not use two-word sentences by 2 years
Does not know the function of common objects (brush, telephone, spoon) by 15 months
Does not imitate actions or words; does not follow simple instructions
Cannot jump in place with both feet
Cannot grasp a crayon between thumb and fingers; cannot scribble
Resists dressing, sleeping, using the toilet
Does not use sentences of more than three words; does not use “me” and “you” appropriately
Ignores other children or people outside the family
Does not engage in a variety of physical activities
Has trouble eating, sleeping, using the toilet
Cannot differentiate between fantasy and reality
Seems unusually passive or aloof with others
Cannot correctly give her or his first and last names
Does not use plurals or past tense when speaking
Does not talk about daily experiences
Does not understand two-part commands
Cannot brush teeth efficiently
Cannot build a tower of six to eight blocks
Physical and occupational therapists can observe the interaction among each of the five areas of development in an infant or child. For example, a child with severe hypotonia who has limited movement experiences will not develop a well adapted sensory system. Children with problems processing sensory information often withdraw from social interaction through which they would otherwise find opportunities to develop speech, language, and social skills. Dynamical systems theory246 explains this relationship among all of the developing components in a child; language does not develop independently of gross motor skills, and the ability to feed or dress oneself is as related to social, emotional, and intellectual development as it is to fine motor skills.
Suspicion of developmental delay often leads to physician referral. An accurate medical diagnosis is important in that it facilitates knowledgeable surveillance for potentially associated health problems. A delayed diagnosis can preclude timely implementation of beneficial medical, therapeutic, and educational services. Children who are identified to be at risk for developmental delay may be referred to early intervention programs. Examples of assessment techniques and interventions for children with developmental delay can be found in pediatric physical therapy textbooks.247,248
Behavioral phenotypes in genetic syndromes
Study into the cognitive and behavioral aspects of individuals with certain genetic syndromes has given rise to the term behavioral phenotype. Certain clusters of behavior that characterize a given syndrome can aid in the early recognition and diagnosis of a syndrome and can guide intervention choices. Example aspects of behavioral phenotypes include social interaction, sleeping patterns, mood, attention, motivation, adaptive and maladaptive strategies, intellect, and memory.249–253
Down syndrome, PWS, AS, FXS, and LNS are examples of genetic disorders discussed in this chapter with delineated behavioral phenotypes.250,251 Compulsive overeating in children with PWS, sleep disturbances in children with AS, and self-injury in children with LNS are behavioral problems that can have significant negative impact on quality of life for children and their families. Although children with Down syndrome have fewer maladaptive behaviors than most children with intellectual disabilities,249,251,253 they have been shown to abandon challenging tasks sooner than other children at similar developmental levels in exchange for peer social interaction.252 Furthermore, this strength of sociability in children with Down syndrome contributes to the child’s learning through modeling and peer collaboration. A knowledgeable, observant therapist can use peer groups to motivate and model for a child with Down syndrome but should also recognize that the child may be distracted by other children and default to a social strategy and avoid the task at hand.252