[level-membership-for-neurology-category]
CHAPTER 22 GENETIC CAUSES OF BLINDNESS
The study of genetic diseases offers an opportunity to understand the pathophysiology at the molecular level. Identification of genetic defects that lead to clinical syndromes and how a syndrome can be caused by a variety of genetic defects offer powerful insight. In addition, such insights offer a rationale for therapeutic target development.
A variety of genetic diseases may lead to blindness by affecting the entire globe, primarily the anterior segment (cornea and lens), or primarily the posterior segment (retina and optic nerve) of the eye. Disorders of the globe are often caused by abnormal closure of the fetal fissure resulting in colobomatous malformations and microphthalmia. Nanophthalmos refers to a small but normally formed eye. Anophthalmia, or absence of the eye, results from failure of outgrowth of the primary optic vesicle. Congenital genetic blinding disorders of the anterior segment include congenital cataracts; the Axenfeld-Riger spectrum, which encompasses a variety of anterior segment malformations involving the cornea, anterior chamber angle, and the iris; and Peter’s anomaly, consisting of a central corneal leukoma with varying amounts of iris and lens attachments. Progressive genetic disorders affecting the anterior segment include a variety of corneal dystrophies with gradual deposition of amyloid, mucopolysaccharide, or other components into the cornea. Neural genetic blindness arises from disorders affecting the retina and optic nerve. To a lesser extent, retrogeniculate genetic disorders may affect the vision but often have other neurological manifestations as well. Retina and optic nerve disorders are the focus of this chapter. A highly recommended and thorough discussion of ophthalmic genetics is available in Traboulsi (1998).
RETINAL DISEASES—CONGENITAL
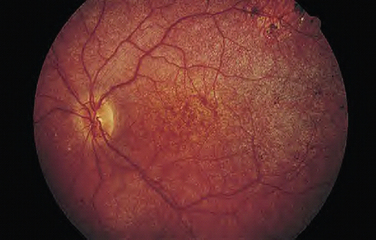
LCA is an autosomal recessive syndrome characterized by significantly reduced vision before age one, nystagmus, paradoxical pupillary reactivity, and retinal degeneration. This syndrome has a prevalence of 3:100,000 children and is a common cause of congenital nystagmus. Six LCA-causing genes have been identified, which account for approximately one half of the cases.1 These genes are expressed preferentially in the retina or the retinal pigment epithelium. Their putative functions are quite diverse and include retinal embryonic development (CRX), photoreceptor cell structure (CRB1), phototransduction (GUCY2D), protein trafficking (AIPL1, RPGRIP1), and vitamin A metabolism (RPE65). The clinical appearance is varying with fundus findings ranging from a retinitis pigmentosa picture with bony spicules to a salt and pepper appearance (Fig. 22-1). Electroretinography (ERG) demonstrates a markedly reduced or nonrecordable scotopic and photopic response, confirming the diagnosis. Although no therapy is presently available, promising gene-based interventions have demonstrated long-term rescue of vision as assessed by psychophysical, behavioral, and molecular biology studies. In a naturally occurring LCA animal model, the RPE65-/- dog, recombinant adenoassociated virus carrying wild-type RPE65 successfully restored visual function.2
Achromatopsia is a rare retinal disorder characterized by a complete absence of cone photoreceptor function. In accordance with the trichromatic theory of vision, individuals with normal color vision can match any color with a combination of three primary colors: red, green, and blue. Dichromats who are missing either the red (protanopes) or green (deuteranopes), but retain the blue cone function, can only match colors with two primary colors. The red (OPN1LW, opsin 1, long wave sensitive) and green (OPN1MW, opsin 1 medium wave sensitive) photopigments are encoded on the long arm of the X chromosome and, as such, these disorders are transmitted in a pattern of X-linked inheritance. The blue (OPN1SW, opsin 1, short wave sensitive) photopigment is encoded on chromosome 7. Achromats present in infancy with reduced vision, photophobia, total color blindness, nystagmus, and a normal-appearing retina. Achromatopsia refers to a spectrum of disease encompassing complete achromatopsia (rod monochromacy), in which there are no cones; atypical rod monochromacy, in which there are some functioning cones; and blue cone monochromacy, where the red and green photopigments are absent but the blue photopigment is functional. Psychophysical testing in such patients must be performed after age 10 in order to get reproducible results. Genes associated with achromatopsia include CNGA33 and CNGB3,4 encoding the α and β subunits of the cone cyclic nucleotide-gated cation channel, which generates the light-evoked electrical responses of cone photoreceptors. A third gene identified in achromatopsia is GNAT2,5 encoding the cone specific α unit of transducin, a G protein of the phototransduction cascade.
Aniridia is a syndrome in which the most prominent manifestation is absence or hypoplasia of the iris. Importantly, visual acuity is reduced due to hypoplasia of the fovea, macula, or optic nerve. Patients present with reduced visual acuity, elevated intraocular pressure, and nystagmus and may develop cataract, glaucoma, keratopathy, strabismus, and amblyopia. Aniridia is caused by mutations in PAX6, a homeobox gene on chromosome 11.6 The homeobox encodes the homeodomain, a protein domain that binds DNA and regulates the transcription of other genes. Aniridia is inherited as an autosomal dominant disorder. WAGR syndrome consists of Wilm’s tumor, aniridia, genitourinary abnormalities, and retardation, resulting from a deletion on chromosome 11p.
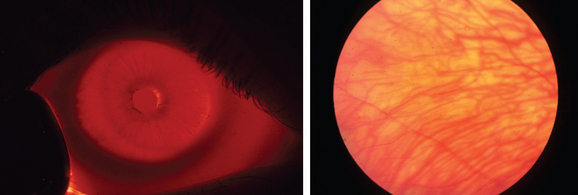
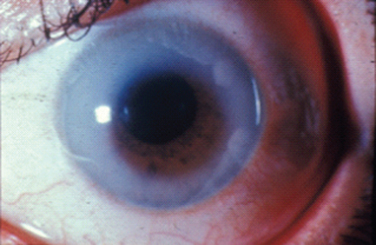
Albinism is traditionally divided into oculocutaneous albinism and ocular albinism. Oculocutaneous albinism is autosomal recessive and has been divided into tyrosinase-positive and -negative forms. Tyrosinase catalyzes three steps in a series of reactions in the melanosome that lead to the formation of melanin from its precursor tyrosine. Major oculocutaneous albinism syndromes include Hermansky-Pudlak syndrome and Chediak-Higashi syndrome, which are inherited in autosomal recessive manner. Nettleship-Falls ocular albinism is an X-linked recessive disorder characterized by reduced visual acuity, congenital nystagmus, transillumination defects of the iris (Fig. 22-2), hypopigmentation of the uveal tract and retinal pigment epithelium, hypoplasia of the fovea, and abnormal decussation of optic nerve fibers through the chiasm. Strabismus and refractive abnormalities are common. Ocular albinism is caused by mutations in OA1, a member of the G protein-coupled receptor superfamily.7
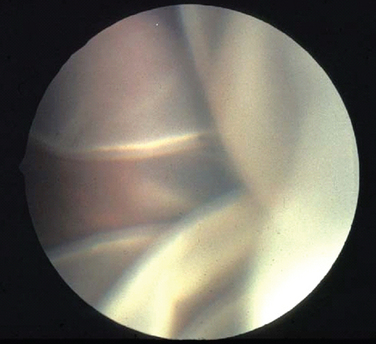
Hereditary vitreoretinopathies are characterized by degenerative changes involving the vitreous and retina. These include familial exudative vitreoretinopathy, Goldmann-Favre syndrome, Stickler’s syndrome, Knobloch’s syndrome, and Norrie disease. Familial exudative vitreoretinopathy (FEVR) has features similar to retinopathy of prematurity but without premature birth or supplemental oxygen. This autosomal dominant disorder is caused by mutations in the frizzled-4 gene (FZD4)8 and is characterized by peripheral retinal vascular nonperfusion, exudative retinal detachment, and proliferative, cicatricial vitreoretinopathy. With severe loss of vision, patients develop nystagmus and strabismus. Goldmann-Favre syndrome is an autosomal recessive disorder caused by mutations in the nuclear receptor gene NR2E3 with characteristic features of retinitis pigmentosa along with central and peripheral retinoschisis, a splitting of the retina. Stickler’s syndrome, a progressive hereditary arthro-ophthalmopathy, is characterized by high myopia, vitreous degeneration, and retinal detachment (Fig. 22-3) in association with orofacial abnormalities such as Pierre-Robin sequence and musculoskeletal abnormalities such as arthritis, scoliosis, and arachnodactyly. It is inherited as an autosomal dominant disorder and caused by mutations in type II collagen (COL2A1). Knobloch’s syndrome is characterized by high myopia, vitroretinal degeneration with retinal detachment, and occipital encephalocele and is caused by a mutation in collagen XVIII (COL18A1). Norrie’s disease is characterized by mental retardation and bilateral retinal detachment presenting early in life. It is inherited as an X-linked disorder caused by mutations in the Norrie gene, which is thought to interact with the FZD4 gene.
RETINAL DISEASES—ONSET IN CHILDHOOD AND ADULTHOOD
The most common hereditary form of RP is autosomal recessive (60%), followed by autosomal dominant (10% to 25%) and X-linked (5% to 18%). The X-linked and recessive forms are more severe than autosomal dominant RP. The first gene determined to be mutated in RP was rhodopsin.9 Other genes include peripherin, tissue inhibitor of metalloproteinase, and geronyl-geronyl transferase. More than 20 genes causing RP have been identified.
Juvenile retinoschisis is an X-linked recessive disorder that manifests in childhood with reduction in visual acuity. The characteristic macular abnormality is a cystlike appearance with spoke like extensions from the fovea. It is highly penetrant in males, whereas carrier females rarely show macular pathology. It is caused by mutations that lead to the pathological development of a schisis or splitting of the retina in the nerve fiber layer. The RS gene is implicated in cell-cell adhesion and phospholipid binding.10
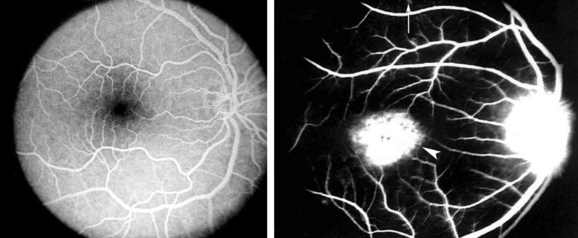
Stargardt’s disease is a storage disease of the retinal pigment epithelium that leads to bilateral progressive loss of central visual acuity. Stargardt’s disease is an autosomal recessive disorder caused by mutations in the ABCR4 gene, encoding an ATP-binding cassette (ABC) transporter.11 Patients often present in the second decade of life with unexplained reduction in visual acuity. Features include subretinal yellow pisiform flecks, referred to as fundus flavimaculatus, and macular changes including increased granularity and a “beaten metal” appearance. ERG shows a moderately reduced photopic response and a nearly normal scotopic response. Fluorescein angiography is important in establishing the diagnosis, demonstrating a “silent choroid sign,” which refers to the darkened appearance of the choroid due to blockage by diffuse storage of material at the RPE (Fig. 22-4).
Best’s vitelliform macular dystrophy is an autosomal dominant disease characterized by the development of an egg-yellow, slightly raised lesion in the macula that is usually 1 to 3 disc diameters in size. Patients may experience blurred central vision and metamorphopsia. Although the fundus appearance is often dramatic, the visual acuity is often better than 20/40. An abnormal electro-oculogram, which measures the electrical potential across the retinal pigment epithelium, is particularly helpful in the diagnosis of patients. The macular abnormality evolves with time from an “egg yolk” appearance to a “scrambled egg” appearance to a late cicatricial stage. Mutations in VMD2, a gene encoding the bestrophin protein, have been associated with Best’s disease. Bestrophin localizes to the basolateral plasma membrane of RPE cells12 and is likely involved in chloride ion conductance.
Congenital stationary night blindness (CSNB) describes a group of retinal diseases characterized by nyctalopia without progressive retinal degeneration. CSNB may be inherited in an autosomal dominant or X-linked pattern. Two types of ERG abnormalities that may be observed in different subtypes of CSNB are (1) a reduced scotopic ERG waveform in the dark adapted ERG or (2) absence of the b-wave on a dark-adapted bright-flash ERG referred to as a “negative” waveform. Autosomal dominant CSNB has been associated with mutations in either the α or β subunit of rod cGMP phosphodiesterase as well as the rhodopsin gene. X-linked CSNB patients have a myopic tigroid-appearing fundus and congenital nystagmus. The visual acuity is typically better than 20/40. It is caused by mutations in NYX, encoding nyctalopin, and a retina-specific calcium channel α1 subunit gene (CACNA1F).13 Although the typical CSNB fundus doe not have any significant pathologicalfeatures, two types of CSNB stand out. Fundus albipunctatus is a type of CSNB inherited in an autosomal recessive pattern that shows distinct, impressive white, round flecks scattered throughout the fundus. Oguchi’s disease is inherited as an autosomal recessive disorder in which the macular retina has an abnormally dark appearance compared with the rest of the fundus, an appearance that disappears with dark adaptation. It is caused by mutations in the arrestin gene.
OPTIC NERVE DISEASES—CONGENITAL
Optic nerve hypoplasia may be observed with normal visual acuity in association with a subtle visual field defect or manifest with profound visual loss (Fig. 22-5). In childhood, optic nerve hypoplasia may manifest as a unilateral decrease in vision diagnosed initially as amblyopia or bilateral decreased vision in infancy diagnosed initially as congenital nystagmus. In these instances, it is important to recognize its association with midline forebrain abnormalities, which can result in pituitary hormone deficiencies and even sudden death.
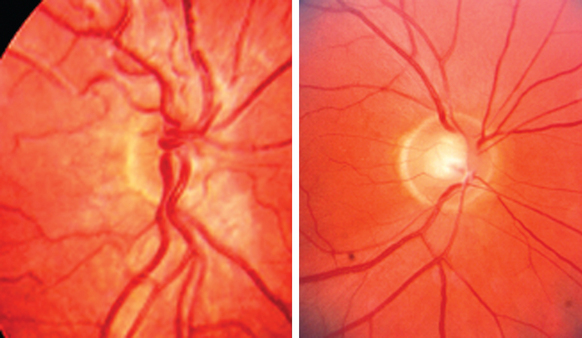
Both teratogenic and genetic etiologies have been described. Recognized teratogens associated with optic nerve hypoplasia include alcohol, quinine, and anticonvulsants. Maternal insulin-dependent diabetes mellitus is associated with superior segmental optic nerve hypoplasia as well. Optic nerve hypoplasia has also been observed in association with many ocular and systemic syndromes including chromosomal duplications and deletions. Septo-optic dysplasia (de Morsier’s syndrome) refers to the association of hypoplasia of the anterior visual pathways, absence of the septum pellucidum, and thinning or agenesis of the corpus callosum. Although familial cases have been reported, most cases are sporadic. Mutations in the homeobox containing transcription factor, HESX1, have been implicated with homozygous inheritance, causing the more severe phenotype, and heterozygous inheritance, causing a mild phenotype.14,15 Mutations in PAX6 have been observed with a variety of optic nerve abnormalities including coloboma, morning glory disc anomaly, optic-nerve hypoplasia/aplasia, and persistent hyperplastic primary vitreous.16
Papillorenal syndrome (renal-coloboma syndrome) is a primary dysgenesis that causes vascular abnormalities predominantly affecting the eye, kidney, and urinary tract. The characteristic optic nerve finding in papillorenal syndrome is an absence or attenuation of the central retinal vessels within the optic nerves, with multiple compensatory cilioretinal vessels. Although the abnormality in these patients has been referred to as a coloboma, it is not a true coloboma arising from failure of closure of the optic nerve fissure with superonasal displacement of the central retinal vessels.17,18 Papillorenal syndrome is inherited in an autosomal dominant pattern. Mutations in PAX2 have been identified in papillorenal syndrome,19 but it is a heterogeneous disease. Patients should undergo renal function testing including serum creatinine and urea nitrogen measurements, urinalysis to test for microalbuminuria, and renal ultrasound.
OPTIC NERVE—ONSET IN CHILDHOOD AND ADULTHOOD
Autosomal dominant optic atrophy is characterized by bilateral insidious vision loss often manifesting in the first or second decade of life. It is inherited in an autosomal dominant pattern with high penetrance. OPA1, located on the long arm of chromosome 3, accounts for the majority of cases, although there is evidence of genetic heterogeneity. The protein is a dynamin-related GTPase targeted to mitochondria, further demonstrating a role for mitochondria in retinal ganglion cell pathophysiology.20,21
At presentation, the visual acuity is typically 20/40 to 20/60, bilateral, and symmetrical. There is an insidious progression of vision loss, although final visual acuity may vary from 20/20 to no light perception. Most individuals retain a visual acuity of 20/40 to 20/200. Color vision testing has demonstrated a characteristic tritanopic-type deficiency, although a generalized dyschromatopsia is most common. Visual field defects include central and cecocentral scotomas. Optic atrophy is present, often localized to the temporal portion of the optic nerve (Fig. 22-6). Other than sensineural hearing loss, neurological or systemic findings are uncommon.
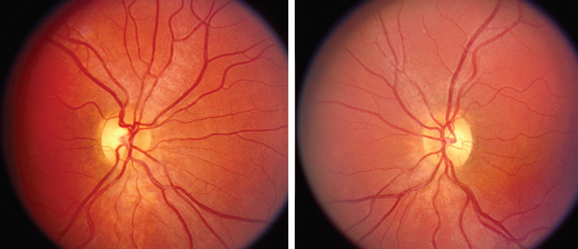
The differential diagnosis includes nutritional deficiency, toxic optic neuropathy, and macular dystrophy. Diagnosis is based on family history and clinical examination. Genetic testing has not become widely available for this disorder. Unfortunately, no treatments are available at the present time to prevent vision loss, arrest the progression of vision loss, or restore vision.
Wolfram’s disease is an autosomal recessive disease caused by mutations in WFS1 which encodes an integral membrane glycoprotein that localizes primarily in the endoplasmic reticulum.22 The most consistent criteria for diagnosis of this syndrome are juvenile-onset diabetes mellitus and optic atrophy. However, other findings include diabetes insipidus and sensory neural hearing loss. The constellation of findings has led to the acronym DIDMOAD: Diabetes Insipidus, Diabetes Mellitus, Optic Atrophy, Deafness. Less commonly recognized features include central apnea and neurogenic upper airway collapse, together precipitating primary respiratory failure, startle myoclonus, axial rigidity, and Parinaud’s syndrome.23
Behr’s disease is an autosomal recessive syndrome of optic atrophy, beginning in the first decade of life, associated with pyramidal tract signs, ataxia, mental retardation, nystagmus, urinary incontinence, and pes cavus. Behr’s disease may represent a phenotype that is common to several genetic disorders that are likely metabolic in origin. Methylglutaconic aciduria, diagnosed by increased amounts of 3-methylglutaconic and 3-methylglutaric acid in urine, may manifest with this constellation of findings.24
X-linked optic atrophy patients present with vision loss in early childhood, which may be progressive. This rare disease is often associated with other neurological findings, including ataxia, tremor, sensineural deafness, and polyneuropathy.25
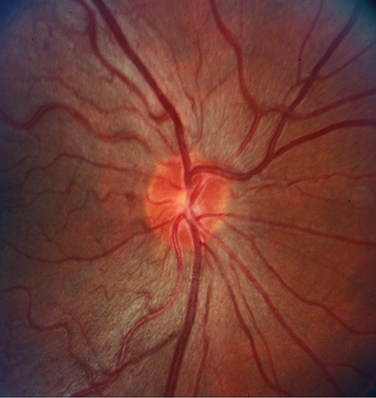
LHON is caused by point mutations in the mitochondrial genome at nucleotide positions 3460,26 11778,27,28 and 1448429 in genes encoding subunits of complex I of the respiratory chain, with the 11778 mutation accounting for the majority of cases. As mitochondria are only transmitted by the mother to all offspring, the typical rules of mendelian inheritance do not apply. All children of maternal carriers are at risk of vision loss, although male children are at greater risk of vision loss than their female siblings. The offspring of male carriers are not at risk for vision loss.
Visual acuity is commonly worse than 20/200 in each eye with bilateral central scotomas. On ophthalmoscopy, the optic nerve may appear normal or have a characteristic abnormality that has been described as a triad of circumpapillary telangiectasia (Fig. 22-7), swelling of the nerve fiber layer around the disc, and absence of leakage on fluorescein angiography. The optic nerve progresses to optic atrophy with nonglaucomatous cupping, pallor, and arteriole attenuation.
RETINOPATHY AND OPTIC NEUROPATHY ASSOCIATED WITH SYSTEMIC AND NEURODEGENERATIVE DISEASE
Disorders of amino acid, protein, and lipoprotein metabolism may lead to blindness from retinal degeneration. Gyrate atrophy of the retina and choroid is an autosomal recessive disease due to a defect in ornithine aminotransferase leading to serum hyperornithemia. It leads to geographic and round-shaped areas of chorioretinal atrophy that begin peripherally and progress centrally. Cystinosis is an autosomal recessive disease characterized by progressive renal failure, pigmentary retinopathy, and growth retardation due to a deposition of cysteine crystals throughout the body. It is caused by a lysosomal defect preventing transport of cysteine crystals from the lysosome to the cytosol. Cysteine crystals are also deposited in the cornea leading to significant photophobia. Methylmalonic aciduria and homocystinuria result in a pigmentary retinopathy and optic nerve pallor due to an abnormality in cobalamin metabolism. Abetalipoproteinemia is an autosomal recessive disorder associated with a pigmentary retinopathy in which patients have fat malabsorption, progressive ataxia, and abnormal plasma lipids due to deficient beta lipoproteins and chylomicons.
Lysosomal storage diseases are caused by enzymatic defects that lead to an accumulation of partially degraded intermediates in cells, tissues, and organs leading to dysfunction. They are generally inherited in an autosomal recessive manner. Mucopolysaccharidoses are caused by defects in specific lysosomal enzymes involved in the degradation of glycosaminoglycans or mucopolysaccharides. General features include facial dysmorphic changes, mental retardation, corneal clouding (Fig. 22-8), and retinal degeneration. Optic disc swelling and optic atrophy are also features of mucopolysaccharidoses. Mucopolysaccharidoses associated with ophthalmological features include Hurler’s syndrome, Scheie’s syndrome, Hunter’s syndrome, Sanfilippo’s syndrome, and Maroteaux-Lamy syndrome. Sialadoses are characterized by the progressive lysosomal storage of sialidated glycopeptides and oligosaccharides caused by a deficiency of the enzyme neuraminidase. There is a progressive accumulation and excretion of sialic acid. Patients develop corneal clouding and a cherry-red spot in the macula. Mucolipidoses have similar features to mucopolysaccharidoses but without mucopolysacchariduria. Patients have a Hurler-like facies, hepatosplenomegaly, and a thickened skull. Major subtypes of mucolipidoses include mucolipidosis II (I cell disease), mucolipidosis III, and mucolipidosis IV. Mucolipidosis IV has the most prominent ocular features including corneal clouding and retinal degeneration in addition to hypotonia and psychomotor retardation. Sphingolipidoses are due to an accumulation of glycosphingolipids, an abundant component of neurons. These disorders include the gangliosidoses (GM2 gangliosidoses [type 1: Tay-Sachs disease; type 2: Sandhoff’s disease] and GM1 gangliosidoses [Gaucher’s disease, Farber’s disease, Fabry’s disease, and Neimann-Pick disease]). Tay-Sachs and Sandhoff diseases are notable for the development of a cherry-red spot in the macula in which there is a deep red spot in the fovea surrounded by a ring of opacified retina. In addition, patients have hepatosplenomegaly and skeletal dysostosis.
Metachromatic leukodystrophy (MLD) is a lipid metabolic disorder caused by mutations in the arylsulfatase A gene. There are five allelic forms, including late infantile, adult partial cerebroside sulfate deficiency, and pseudo-arylsulfatase deficiency.30 On histopathological staining, there is a metachromatic staining of abnormally stored galactosphingosulfatides in central nervous system white matter. The late infantile form is the most common form of MLD, manifesting in the second year of life with gait disturbance and muscle rigidity. This is followed by progressive mental deterioration and convulsions. A cherry-red spot may be present in the macula and optic atrophy may develop.
A second group of peroxisomal disorders are due to gene defects that result in abnormal peroxisome function without affecting its assembly. This group includes entities such as X-linked adrenoleukodystrophy, primary hyperoxaluria type 1, and classic Refsum’s disease. XLA is characterized by an accumulation of very-long-chain fatty acids of 22 to 30 carbons. X-linked adrenoleukodystrophy may manifest in childhood with gait disturbance and intellectual deterioration between the ages of 5 and 8. There is impressive inflammation of white matter and demyelination. In latter stages of the disease, patients develop vision loss with optic atrophy. The adult-onset variety of X-linked adrenoleukodystrophy is referred to as adrenomyeloneuropathy. X-linked adrenoleukodystrophy is caused by mutations in ABCD1, a member of the ATP-binding cassette (ABC) transporter superfamily that contains membrane proteins that translocate a wide variety of substrates across extracellular and intracellular membranes. Primary hyperoxaluria type 1 is an autosomal recessive disorder caused by a defect in the enzyme alanine glyoxylate aminotransferase and characterized by renal failure and elevated intracranial pressure. Patients develop a hyperplasia of the retinal pigment epithelium due to deposition of calcium oxalate crystals. Classic Refsum’s disease is characterized by a pigmentary retinopathy, polyneuropathy, hearing loss, icthyosis, and ataxia due to a defect in phytanoyl-CoA hydroxylase (PAHX) or peroxin-7 (PEX7), which impair the degradation of phytanic acid.
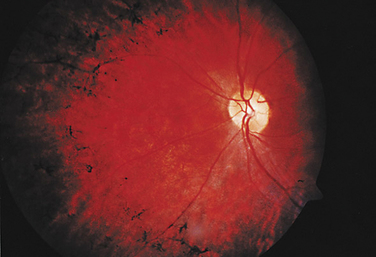
Mitochondrial disorders in addition to LHON include NARP, MELAS, and Kearns-Sayre syndrome. NARP (neurogenic muscle weakness, ataxia, and retinitis pigmentosa) is caused by a T-to-G point mutation in nucleotide 8993 of mtDNA, which results in a substitution of a highly conserved leucine by an arginine residue in the mitochondrial ATPase 6 gene.31 As the acronym implies, clinical features include migraine, sensory neuropathy, proximal muscle weakness, ataxia, seizures, dementia, and pigmentary retinopathy (Fig. 22-9). The retinal degeneration in NARP may manifest as a cone-rod dystrophy, a progressive cone dystrophy, a bull’s eye maculopathy, or rod-cone type of retinal dystrophy. The severity of NARP appears to correlate with the burden of mutated mitochondria within the population of mitochondria in the cell. The 8993 mutation also causes maternally inherited Leigh disease.32
Kearns-Sayre syndrome (KSS) is characterized by the triad of external ophthalmoplegia (CPEO), pigmentary retinopathy, and heart block. Onset is in the first or second decades of life. CPEO is characterized by a bilateral symmetrical ptosis associated with ophthalmoplegia and orbicularis oculi weakness. The pigmentary retinopathy of KSS differs from RP in that the macula is often the first part of the retina to be affected, followed by the retinal periphery. Visual acuity, visual fields, and ERG are usually only mildly affected. Neurological manifestations in KSS may include cerebellar ataxia, hearing loss, dementia, and weakness of facial, pharyngeal, trunk, and extremity muscles. Heart block is a characteristic finding. Skeletal muscle biopsy demonstrates ragged-red fibers. KSS is caused by mitochondrial DNA deletions, usually 1.3 to 7.6kb in size, affecting 45% to 75% of total mtDNA.33 KSS may be maternally inherited or sporadic.
RETINOPATHY AND OPTIC NEUROPATHY ASSOCIATED WITH DERMATOLOGICAL DISEASE, SKELETAL ANOMALIES, HEARING LOSS, OR RENAL DISEASE
Cockayne’s syndrome (CS) is an autosomal recessive disorder whose features include dwarfism, precociously senile appearance, pigmentary retinal degeneration, optic atrophy, deafness, marble epiphyses in some digits, photosensitivity, and mental retardation. Magnetic resonance imaging demonstrates hypomyelination, cerebellar atrophy, and basal ganglia calcification. CS cells are abnormally sensitive to ultraviolet radiation and are defective in the repair of transcriptionally active genes. The CSA and CSB genes are involved with DNA repair.
Usher’s syndrome comprises a group of autosomal recessive disorders that are characterized by autosomal recessive inheritance, congenital sensorineural hearing loss, and retinitis pigmentosa. In some varieties, vestibular function is not significantly affected. Usher’s syndrome is caused by at least 12 loci with several identified genes: USH2A (encoding usherin), MYO7A (encoding myosin VIIa), CDH23 (encoding cadherin 23), PCDH15 (encoding protocadherin 15), USH1C (encoding harmonin), USH3A (encoding clarin 1), and USH1G (encoding SANS).34
Newman NJ. Hereditary optic neuropathies. Miller NR, Newman NJ, Biousse V, Kerrison JB, editors. Walsh and Hoyt’s Clinical Neuro-Ophthalmology, 6th ed., Vol 1. Baltimore: Lippincott, Willians, and Wilkins, 2004;465-502.
Repka MX. Degenerative and metabolic diseases in infants and children. Miller NR, Newman NJ, Biousse V, Kerriscn JB, editors. Walsh and Hoyt’s Clinical Neuro-Ophthalmology, 6th ed., Vol 2. Baltmore: Lippincott, Willians, and Wilkins, 2004;2469-2512.
Traboulsi EI, editor. Genetic Diseases of the Eye. New York: Oxford University Press, 1998. Online Mendelian Inheritance in Man. available at http://www.ncbi.nlm.nih.gov.
1 Cremers FP, van den Hurk JA, den Hollander AI. Molecular genetics of Leber congenital amaurosis. Hum Mol Genet. 2002;11:1169-1176.
2 Acland GM, Aguirre GD, Ray J, et al. Gene therapy restores vision in a canine model of childhood blindness. Nat Genet. 2001;28:92-95.
3 Kohl S, Marx T, Giddings I, et al. Total colour blindness is caused by mutations in the gene encoding the alpha-subunit of the cone photoreceptor cGMP-gated cation channel. Nat Genet. 1998;19:257-259.
4 Sundin OH, Yang JM, Li Y, et al. Genetic basis of total colour blindness among the Pingelapese islanders. Nat Genet. 2000;25:289-293.
5 Kohl S, Baumann B, Rosenberg T, et al. Mutations in the cone photoreceptor G-protein alpha-subunit gene GNAT2 in patients with achromatopsia. Am J Hum Genet. 2002;71:422-425.
6 Davis A, Cowell JK. Mutations in the PAX6 gene in patients with hereditary aniridia. Hum Mol Genet. 1993;2:2093-2097.
7 Bassi MT, Schiaffino MV, Renieri A, et al. Cloning of the gene for ocular albinism type 1 from the distal short arm of the X chromosome. Nat Genet. 1995;10:13-19.
8 Robitaille J, MacDonald ML, Kaykas A, et al. Mutant frizzled-4 disrupts retinal angiogenesis in familial exudative vitreoretinopathy. Nat Genet. 2002;32:326-330.
9 Dryja TP, McGee TL, Hahn LB, et al. Mutations within the rhodopsin gene in patients with autosomal dominant retinitis pigmentosa. N Engl J Med. 1990;323:1302-1307.
10 Sauer CG, Gehrig A, Warneke-Wittstock R, et al. Positional cloning of the gene associated with X-linked juvenile retinoschisis. 1997;17:164-170.
11 Allikmets R, Singh N, Sun H, et al. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat Genet. 1997;15:236-246.
12 Marmorstein AD, Marmorstein LY, Rayborn M, et al. Bestrophin, the product of the Best vitelliform macular dystrophy gene (VMD2), localizes to the basolateral plasma membrane of the retinal pigment epithelium. Proc Natl Acad Sci U S A. 2000;97:12758-12763.
13 Strom TM, Nyakatura G, Apfelstedt-Sylla E, et al. An L-type calcium-channel gene mutated in incomplete X-linked congenital stationary night blindness. Nat Genet. 1998;19:260-263.
14 Thomas PQ, Dattani MT, Brickman JM, et al. Heterozygous HESX1 mutations associated with isolated congenital pituitary hypoplasia and septo-optic dysplasia. Hum Mol Genet. 2001;10:39-45.
15 Tajima T, Hattorri T, Nakajima T, et al. Sporadic heterozygous frameshift mutation of HESX1 causing pituitary and optic nerve hypoplasia and combined pituitary hormone deficiency in a Japanese patient. J Clin Endocrinol Metab. 2003;88:45-50.
16 Azuma N, Yamaguchi Y, Handa H, et al. Mutations of the PAX6 gene detected in patients with a variety of optic-nerve malformations. Am J Hum Genet. 2003;72:1565-1570.
17 Parsa CF, Goldberg MF, Hunter DG. Papillorenal syndrome in a Brazilian family. Arch Ophthalmol. 2002;120:1772-1773. author reply 1773.
18 Parsa CF, Goldberg MF, Hunter DG. Papillorenal (“renal coloboma”) syndrome. Am J Ophthalmol. 2002;134:300-301. author reply 301.
19 Chung GW, Edwards AO, Schimmenti LA, et al. Renalcoloboma syndrome: report of a novel PAX2 gene mutation. Am J Ophthalmol. 2001;132:910-914.
20 Alexander C, Votruba M, Pesch UE, et al. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat Genet. 2000;26:211-215.
21 Delettre C, Lenaers G, Griffoin JM, et al. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat Genet. 2000;26:207-210.
22 Inoue H, Tanizawa Y, Wasson J, et al. A gene encoding a transmembrane protein is mutated in patients with diabetes mellitus and optic atrophy (Wolfram syndrome). Nat Genet. 1998;20:143-148.
23 Scolding NJ, Kellar-Wood HF, Shaw C, et al. Wolfram syndrome: hereditary diabetes mellitus with brainstem and optic atrophy. Ann Neurol. 1996;39:352-360.
24 Sheffer RN, Zlotogora J, Elpeleg ON, et al. Behr’s syndrome and 3-methylglutaconic aciduria. Am J Ophthalmol. 1992;114:494-497.
25 Assink JJ, Tijmes NT, ten Brink JB, et al. A gene for X-linked optic atrophy is closely linked to the Xp11.4-Xp11.2 region of the X chromosome. Am J Hum Genet. 1997;61:934-939.
26 Johns DR, Smith KH, Miller NR. Leber’s hereditary optic neuropathy: clinical manifestations of the 3460 mutation. Arch Ophthalmol. 1992;110:1577-1581.
27 Wallace DC, Singh G, Lott MT, et al. Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy. Science. 1988;242:1427-1430.
28 Newman NJ, Lott MT, Wallace DC. The clinical characteristics of pedigrees of Leber’s hereditary optic neuropathy with the 11778 mutation. Am J Ophthalmol. 1991;111:750-762.
29 Johns DR, Heher KL, Miller NR, et al. Leber’s hereditary optic neuropathy: clinical manifestations of the 14484 mutation. Arch Ophthalmol. 1993;111:495-498.
30 Polten A, Fluharty AL, Fluharty CB, et al. Molecular basis of different forms of metachromatic leukodystrophy. N Engl J Med. 1991;324:18-22.
31 Holt IJ, Harding AE, Petty RK, et al. A new mitochondrial disease associated with mitochondrial DNA heteroplasmy. Am J Hum Genet. 1990;46:428-433.
32 Tatuch Y, Christodoulou J, Feigenbaum A, et al. Heteroplasmic mtDNA mutation (T-G) at 8993 can cause Leigh disease when the percentage of abnormal mtDNA is high. Am J Hum Genet. 1992;50:852-858.
33 Zeviani M, Moraes CT, DiMauro S, et al. Deletions of mitochondrial DNA in Kearns-Sayre syndrome. Neurology. 1988;38:1339-1346.
34 Ahmed Z, Riazuddin S, Wilcox E. The molecular genetics of Usher syndrome. Clin Genet. 2003;63:431-444.
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
CHAPTER 22 GENETIC CAUSES OF BLINDNESS
The study of genetic diseases offers an opportunity to understand the pathophysiology at the molecular level. Identification of genetic defects that lead to clinical syndromes and how a syndrome can be caused by a variety of genetic defects offer powerful insight. In addition, such insights offer a rationale for therapeutic target development.
A variety of genetic diseases may lead to blindness by affecting the entire globe, primarily the anterior segment (cornea and lens), or primarily the posterior segment (retina and optic nerve) of the eye. Disorders of the globe are often caused by abnormal closure of the fetal fissure resulting in colobomatous malformations and microphthalmia. Nanophthalmos refers to a small but normally formed eye. Anophthalmia, or absence of the eye, results from failure of outgrowth of the primary optic vesicle. Congenital genetic blinding disorders of the anterior segment include congenital cataracts; the Axenfeld-Riger spectrum, which encompasses a variety of anterior segment malformations involving the cornea, anterior chamber angle, and the iris; and Peter’s anomaly, consisting of a central corneal leukoma with varying amounts of iris and lens attachments. Progressive genetic disorders affecting the anterior segment include a variety of corneal dystrophies with gradual deposition of amyloid, mucopolysaccharide, or other components into the cornea. Neural genetic blindness arises from disorders affecting the retina and optic nerve. To a lesser extent, retrogeniculate genetic disorders may affect the vision but often have other neurological manifestations as well. Retina and optic nerve disorders are the focus of this chapter. A highly recommended and thorough discussion of ophthalmic genetics is available in Traboulsi (1998).
RETINAL DISEASES—CONGENITAL
LCA is an autosomal recessive syndrome characterized by significantly reduced vision before age one, nystagmus, paradoxical pupillary reactivity, and retinal degeneration. This syndrome has a prevalence of 3:100,000 children and is a common cause of congenital nystagmus. Six LCA-causing genes have been identified, which account for approximately one half of the cases.1 These genes are expressed preferentially in the retina or the retinal pigment epithelium. Their putative functions are quite diverse and include retinal embryonic development (CRX), photoreceptor cell structure (CRB1), phototransduction (GUCY2D), protein trafficking (AIPL1, RPGRIP1), and vitamin A metabolism (RPE65). The clinical appearance is varying with fundus findings ranging from a retinitis pigmentosa picture with bony spicules to a salt and pepper appearance (Fig. 22-1). Electroretinography (ERG) demonstrates a markedly reduced or nonrecordable scotopic and photopic response, confirming the diagnosis. Although no therapy is presently available, promising gene-based interventions have demonstrated long-term rescue of vision as assessed by psychophysical, behavioral, and molecular biology studies. In a naturally occurring LCA animal model, the RPE65-/- dog, recombinant adenoassociated virus carrying wild-type RPE65 successfully restored visual function.2
Achromatopsia is a rare retinal disorder characterized by a complete absence of cone photoreceptor function. In accordance with the trichromatic theory of vision, individuals with normal color vision can match any color with a combination of three primary colors: red, green, and blue. Dichromats who are missing either the red (protanopes) or green (deuteranopes), but retain the blue cone function, can only match colors with two primary colors. The red (OPN1LW, opsin 1, long wave sensitive) and green (OPN1MW, opsin 1 medium wave sensitive) photopigments are encoded on the long arm of the X chromosome and, as such, these disorders are transmitted in a pattern of X-linked inheritance. The blue (OPN1SW, opsin 1, short wave sensitive) photopigment is encoded on chromosome 7. Achromats present in infancy with reduced vision, photophobia, total color blindness, nystagmus, and a normal-appearing retina. Achromatopsia refers to a spectrum of disease encompassing complete achromatopsia (rod monochromacy), in which there are no cones; atypical rod monochromacy, in which there are some functioning cones; and blue cone monochromacy, where the red and green photopigments are absent but the blue photopigment is functional. Psychophysical testing in such patients must be performed after age 10 in order to get reproducible results. Genes associated with achromatopsia include CNGA33 and CNGB3,4 encoding the α and β subunits of the cone cyclic nucleotide-gated cation channel, which generates the light-evoked electrical responses of cone photoreceptors. A third gene identified in achromatopsia is GNAT2,5 encoding the cone specific α unit of transducin, a G protein of the phototransduction cascade.
Aniridia is a syndrome in which the most prominent manifestation is absence or hypoplasia of the iris. Importantly, visual acuity is reduced due to hypoplasia of the fovea, macula, or optic nerve. Patients present with reduced visual acuity, elevated intraocular pressure, and nystagmus and may develop cataract, glaucoma, keratopathy, strabismus, and amblyopia. Aniridia is caused by mutations in PAX6, a homeobox gene on chromosome 11.6 The homeobox encodes the homeodomain, a protein domain that binds DNA and regulates the transcription of other genes. Aniridia is inherited as an autosomal dominant disorder. WAGR syndrome consists of Wilm’s tumor, aniridia, genitourinary abnormalities, and retardation, resulting from a deletion on chromosome 11p.
Albinism is traditionally divided into oculocutaneous albinism and ocular albinism. Oculocutaneous albinism is autosomal recessive and has been divided into tyrosinase-positive and -negative forms. Tyrosinase catalyzes three steps in a series of reactions in the melanosome that lead to the formation of melanin from its precursor tyrosine. Major oculocutaneous albinism syndromes include Hermansky-Pudlak syndrome and Chediak-Higashi syndrome, which are inherited in autosomal recessive manner. Nettleship-Falls ocular albinism is an X-linked recessive disorder characterized by reduced visual acuity, congenital nystagmus, transillumination defects of the iris (Fig. 22-2), hypopigmentation of the uveal tract and retinal pigment epithelium, hypoplasia of the fovea, and abnormal decussation of optic nerve fibers through the chiasm. Strabismus and refractive abnormalities are common. Ocular albinism is caused by mutations in OA1, a member of the G protein-coupled receptor superfamily.7
Hereditary vitreoretinopathies are characterized by degenerative changes involving the vitreous and retina. These include familial exudative vitreoretinopathy, Goldmann-Favre syndrome, Stickler’s syndrome, Knobloch’s syndrome, and Norrie disease. Familial exudative vitreoretinopathy (FEVR) has features similar to retinopathy of prematurity but without premature birth or supplemental oxygen. This autosomal dominant disorder is caused by mutations in the frizzled-4 gene (FZD4)8 and is characterized by peripheral retinal vascular nonperfusion, exudative retinal detachment, and proliferative, cicatricial vitreoretinopathy. With severe loss of vision, patients develop nystagmus and strabismus. Goldmann-Favre syndrome is an autosomal recessive disorder caused by mutations in the nuclear receptor gene NR2E3 with characteristic features of retinitis pigmentosa along with central and peripheral retinoschisis, a splitting of the retina. Stickler’s syndrome, a progressive hereditary arthro-ophthalmopathy, is characterized by high myopia, vitreous degeneration, and retinal detachment (Fig. 22-3) in association with orofacial abnormalities such as Pierre-Robin sequence and musculoskeletal abnormalities such as arthritis, scoliosis, and arachnodactyly. It is inherited as an autosomal dominant disorder and caused by mutations in type II collagen (COL2A1). Knobloch’s syndrome is characterized by high myopia, vitroretinal degeneration with retinal detachment, and occipital encephalocele and is caused by a mutation in collagen XVIII (COL18A1). Norrie’s disease is characterized by mental retardation and bilateral retinal detachment presenting early in life. It is inherited as an X-linked disorder caused by mutations in the Norrie gene, which is thought to interact with the FZD4 gene.
RETINAL DISEASES—ONSET IN CHILDHOOD AND ADULTHOOD
The most common hereditary form of RP is autosomal recessive (60%), followed by autosomal dominant (10% to 25%) and X-linked (5% to 18%). The X-linked and recessive forms are more severe than autosomal dominant RP. The first gene determined to be mutated in RP was rhodopsin.9 Other genes include peripherin, tissue inhibitor of metalloproteinase, and geronyl-geronyl transferase. More than 20 genes causing RP have been identified.
In contrast to retinitis pigmentosa, patients with cone dystrophies
[/not-level-membership-for-neurology-category]
