[level-membership-for-neurosurgery-category]
CHAPTER 57 Gene Therapy for Meningiomas
RATIONALE FOR ALTERNATIVE TREATMENT STRATEGIES IN MENINGIOMAS
Surgery is the mainstay of treatment in meningiomas, and most such tumors can be easily and safely resected surgically with an excellent outcome. Despite an overall favorable outcome, treatment attempts fail in some patients due to intrinsic biology of meningiomas or to difficult localization. Most meningiomas recur after resection if followed long enough.1 Adjuvant treatment modalities such as radiosurgery and radiotherapy are effective, however, with limitations and side effects. Chemotherapy is marginally effective and used as a salvage therapy in cases that have failed other available treatment options. Gene therapy is an appealing new strategy for meningioma treatment. Areas of potential use are (1) recurrent meningiomas, (2) meningiomas in difficult to treat localizations, (3) multiple meningiomas, (4) meningiomas in high-risk patients, (5) metastatic cases, and (6) atypical or malignant meningiomas that have not responded to conventional treatment modalities.
INTRODUCTION TO GENE THERAPY
Gene therapy is defined as an experimental treatment that involves introducing genetic material (DNA or RNA) into a person’s cells for therapeutic purposes. This field initially started with efforts to replace or substitute missing or altered genes that caused disease. Advances in vector design and therapeutic virology and an increased understanding of molecular tumor biology have extended this initial concept to a multitude of promising strategies. When broadly classified, current gene therapy applications to treat brain malignancies fall into three categories (Fig. 57-1):
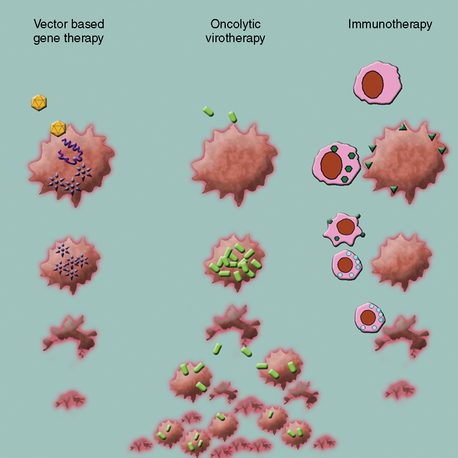
In Vivo Gene Delivery Methods
A wide range of methods are available for delivery of DNA or RNA into target cells. In vitro techniques such as direct microinjection and chemical (calcium phosphate) or physical (electroporation) transfection methods are not practical for in vivo application. This leaves “naked DNA” administration, liposomal gene delivery, and virally mediated gene transfer as the methods of choice for in vivo gene therapy application. Injection of “naked DNA” plasmids has been shown to result in intracranial gene expression.2 However, with current technology, expression efficiency is very low and the practical value of this technique for brain tumor gene therapy remains to be seen. In contrast, liposomal gene transfer has been established as a promising therapeutic modality for brain tumors.3 Good transfer efficiency, safety, simple design, and their unique capability to carry, not only genetic material, but also proteins, chemicals, or physical particles into a cell make these artificial lipid bilayered vesicles useful and exciting therapeutic tools. However, viral-based vectors and therapeutic viruses are the absolute mainstay in the field of gene therapy.
Viral Therapies
Viruses have evolved as nature’s way to deliver genomic material into cells with unsurpassed efficiency, displaying an overwhelming repertoire of mechanisms. Since the early 1990s advances in the field of molecular biology have provided the tools necessary to manipulate viral genomes. The vast majority of viral vectors are composed of recombinant viruses, often engineered to exhibit attenuated phenotypes and harbor nonviral genes of interest. The search for and the design of appropriate vectors are directed by certain paradigms that depend on the clinical context. Simplified, in gene therapy applications for genetic or degenerative diseases the duration of the expression of the therapeutic gene is a foremost priority. In contrast, antitumor gene therapy generally seeks to destroy the target cell, making transduction efficiency much more important, and a short term gene expression appears to be sufficient. For tumor treatment, viruses have both been used as gene delivery vehicles (viral vectors) or as biological agents that attack and kill tumor cells directly (oncolytic viruses). Gene therapy strategies using adenoviral or retroviral vectors to deliver suicide genes to tumor cells have been the forerunners in the field and both have been used in clinical trials against human high-grade gliomas. In vivo transduction efficiency has been the main problem with retroviruses,4 which can deliver genes only to cells that are actively dividing.5 Insertional mutagenesis is a rare but feared side effect and can lead to the formation of secondary tumors resulting from oncogene activation or tumor-suppressor gene silencing due to the uncontrollable random insertion of the viral transgene into the human genome. Low transfection efficiency and lack of any therapeutic effect in phase III trials of RV-HSV-tk in newly diagnosed glioblastoma led to abandonment of retroviral strategies.6 First-generation adenoviruses (AdV) had been deprived of their replicative capabilities by deletion of variable portions (E1 or E3) of the viral genome.7 As these AdV vectors retain large portions of their viral genome, vectors and transduced cells are highly immunogenic once administered into the immunocompetent host. Consequently, therapeutic effects are short lived, though the enhanced immune response may noticeably contribute synergistically in the setting of anticancer therapy. Newer adenovirus strategies use either highly stripped down “gutless” AdV, requiring helper systems for production8 with minimized immune response and prolonged gene expression or conditional replication competent variants with inherent tumor selective affinity.9 An important aspect of antitumor gene therapy is the spatial distribution of the viral therapeutic. The therapeutic activity increases significantly from cell-limited gene expression over the so-called bystander effect of released therapeutic products to the active spread of replication-competent viruses. Replication-incompetent viral vectors have been proven ineffective in vivo, as only a small percentage of tumor cells can be infected under the most optimal circumstances with the delivered viral load.10,11 Whether the viral therapeutic will replicate in the tissue is an important issue: Nonreplicating viruses are most commonly used to deliver toxic or therapeutic genes. Conditionally replicating viruses are based on replicating viruses but one or more of their viral genes are deleted to make them depend on cellular mechanisms, which are present only in tumor cells. Although this is a very attractive strategy, replicative health of most of these viruses is also impaired.6,12 Replicating viruses are used to infect tumor tissues and viral progeny produced inside the target tissue infects surrounding cells to cause “local self-amplification.” A considerably small amount of virus can therefore be delivered to the target tissue to yield a much heavier therapeutic dose inside the tumor. Such a phenomenon does not exist in any other therapeutic modality. Newer, recombinant adenoviruses were engineered to be conditionally replicative in tumor cells or carried transgenes that are expressed only in specific cell types.13 Recombinant herpes viruses have also been commonly used in gene therapy protocols for brain tumors.6 Recombinant viral vectors based on vaccinia have also been used. In contrast to strategies that use viral vectors to transfer genes to the target cell, other researchers have experimented with oncolytic viruses, which preferentially replicate inside the tumor tissue and kill the tumor cells as a direct result of this infection.14 Other viruses including reovirus; Newcastle disease virus; and measles, vesicular stomatitis, and myxoma virus, have shown merit for infecting some types of brain tumor cells.15
In animal experiments viral infection can easily be monitored inside the target tissue or elsewhere using virally expressed fluorescent reporter genes. Fluorescent reporter genes can be engineered into most viruses, and when combined with tumor models that express other fluorescent proteins, temporal and spatial monitoring of both the oncolytic activity and/or side effects is greatly simplified.16
Immunotherapy/Immunogene Therapy Strategies
Cancer immunotherapy has been intensely studied in the field of malignant melanoma therapy, and the promise in that field among others led in October 2000 to the formation of the Immunotherapy Task Force for malignant brain tumors.17 As with gene therapy and oncolytic virotherapy, the focus of current immunotherapy studies of brain tumors is on tumors of neuroectodermal lineage, that is, glioblastoma and anaplastic astrocytoma. Though the transfer of this strategy to meningioma remains hypothetical at this point, a brief discussion of the current studies is merited.
Besides detection of infectious or foreign organisms the immune system has the capability to recognize and even eliminate cancerous cells. For that response to be effective, certain preconditions need to be met. Tumor-associated antigens (TAAs) need to be expressed and the tumor environment needs to be “immune-compatible,” as certain tumors can generate a local and systemic state of immune suppression. Evidence suggests both membrane and intracellular elements can serve as TAAs.18 However; despite its remarkably high selectivity, the natural immune response falls short in terms of efficiency to control advanced, sizable cancers. The limit of a patient’s immune response seems to be in the range of less than a million tumor cells, which is a negligible fraction compared to the billions of cells of an average sized brain tumor.19 In that regard, immunotherapy can be defined as strategies aiming to amplify the natural immune response to cancerous cells. Both arms of the immune response, the initiating cascade and the executing effector phase, can be targeted.
Earlier attempts of immunotherapy included the use of tumor lysates and microbe preparations such as nonspecific immune activators with little success.20 With increasing knowledge of mechanisms underlying antigen presentation and immune modulation, the direction shifted toward highly specific stimulation of the antitumor response. The main strategies comprise the use of cytokines,21 active vaccination with tumor antigen-pulsed dendritic cells,22 and adoptive immunotherapy using cytotoxic T cells raised and expanded ex vivo.23 Among others, cytokines interleuking-2 (IL-2), IL-4, IL-12, IL-18, interferon (IFN), granulocyte-macrophage colony-stimulating factor (GM-CSF), and B7-2 have been used with promising results alone, in combinations, or as adjuvants to cell-based immunotherapy.24 Applications range from direct peripheral infusion and convection-enhanced delivery to in vivo cytokine gene therapy and ex vivo gene transfer to antitumor dendritic or cytotoxic T cells. Active antitumor vaccination involves the collection of blood mononuclear cells and the in vitro isolation and priming of dendritic cells. Considered the most potent antigen presenting cells, dendritic cells pulsed with tumor cells or tumor lysate can effectively prime T-helper cells once reinjected into the patient’s body, leading to an efficient mounting of an antitumor response by cytotoxic T lymphocytes. In contrast, adoptive immunotransfer focuses on ex vivo expansion and reinjection of tumor-specific effector cells. Several cell types have been described with potential for adoptive immunotherapy: lymphokine-activated killer cells, tumor infiltrating lymphocytes, cytotoxic T lymphocytes, and others.
CURRENT GENE THERAPY STRATEGIES FOR MENINGIOMAS
What Has Been Achieved So Far?
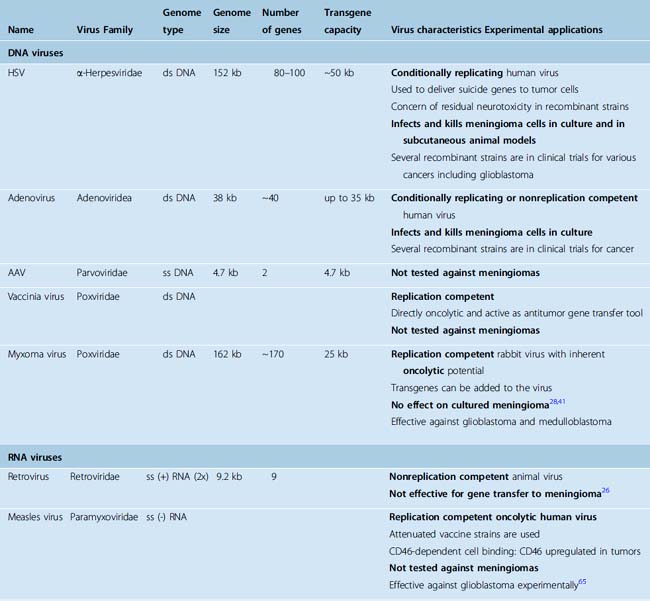
Herpes simplex virus and adenovirus are among the first recombinant virus species used for gene therapy applications. Both viruses have been extensively used both in preclinical and clinical studies against various types of tumors and have shown promise against meningioma. Similarly, poliovirus25 and herpesvirus26 replicons have been found to effectively transduce meningioma cells. Other viruses such as reovirus,27 myxoma virus,28 and retroviruses26 have been reported to be ineffective against meningioma cultures, despite their potential against other kinds of brain tumor.
Herpes simplex virus and adenovirus have been the most common recombinant virus species used for gene therapy attempts. Both viruses have been extensively used both in preclinical and clinical studies against various types of tumors. Herpes simplex was the first oncolytic virus tested against meningiomas.29 Herpes simplex virus is a large, enveloped virus that belongs to the alpha-herpesvirus family and has a 152-kb double-stranded linear DNA genome. The wild-type virus has 80 densely packed genes, approximately 50% of which are necessary for viral replication. Almost 30 kilobases can be replaced with transgenes without affecting viral replication. The virus can infect both replicating and nonreplicating cells. Lytic infection of herpesviruses has three phases (α, β, and γ) and genes taking part in these three phases are responsible for regulating transcription, promoting DNA synthesis and creating a favorable environment for protein synthesis, respectively. Earlier genes in the genome have a suppressor role on late genes. Recombinant HSVs have been designed by making changes in or deleting α-genes (e.g., G47Δ, NV1020), β-genes (DLSPTK, G207), or γ-genes (R3616, G207). Herpes simplex is a large virus with a large transgene capacity, is stable, infects many cell types, and has animal models available for toxicity. Absence of proinflammatory effects and bone marrow toxicity, the availability of antiherpetic chemotherapy, and the absence of genomic integration are advantages of the HSV for gene therapy. In contrast, the large size of the genome, presence of preexisting HSV immunity in humans, and risk of encephalitis from primary or latent infection are the basic concerns associated with HSV. G207, which is the only HSV that was used in experimental studies against meningioma, had been tested in a phase 1 clinical trial against human glioblastoma.30–32 Magnetic resonance imaging (MRI) responses and few long-term survivors have been reported in these glioblastoma trials, with no chronic toxicity observed in survivors.
The first study of viral therapy on meningiomas was reported by Markert and colleagues33 using a thymidine kinase (TK) deleted recombinant HSV-1, called dlsptk. The authors demonstrated that primary cell cultures from three malignant and one atypical meningioma specimen were effectively infected and killed by this recombinant herpesvirus. The authors also tested the virus against malignant meningioma xenografts in the subrenal capsule of nude mice and showed that the tumor growth rate was significantly inhibited by injection of the virus.33 Yazaki and colleagues29 reported that G207 killed three different malignant meningiomas in cell culture at a multiplicity of infection (MOI; infectious particle to target cell ratio) of 0.1. The same study reported that intratumoral injection of G207 prolonged survival of nude mice harboring subcutaneous or intracranial, subdural F5 malignant meningioma xenografts. In a more recent study, Liu and colleagues34 showed that a recombinant herpesvirus, bG47Δ, which also carried a dominant negative FGF receptor, was effective in killing F5 malignant meningioma cells. Such recombinant strains are called “armed” as they provide more than one therapeutic action. FGF has mitogenic, angiogenic, and antiapoptotic properties and the authors showed that this recombinant herpesvirus effectively suppressed the FGF signaling pathway. In an effort to insert the NF2 gene into meningiomas, Ikeda and colleagues26 tested retroviral, adenoviral, and a herpes simplex amplicon vector (a vector that contains only a portion of the viral genome and that is not replication competent) and reported that the herpes simplex vector achieved up to 100% transduction efficiency with documented Merlin expression in three different meningioma cell cultures, which were derived from patient samples. The authors reported that the retroviral and adenoviral vectors were not as effective as the herpes vector. The authors reported that Merlin overexpression significantly inhibited proliferation of meningioma cells when compared to cells transduced with a control vector.
Several studies have reported the use of adenoviruses for experimental meningioma treatment.26,35–38 Adenoviruses are nonenveloped viruses with a 36-kb double-stranded DNA genome and are natural pathogens of the human respiratory epithelium. There are approximately 47 different human specific serotypes and serotypes 2 and 5 have been most commonly used as viral therapeutics. Serotype 5 binds to the coxsackievirus–adenovirus receptor (CAR) on the cell surface and enters the cell through a process mediated by the integrin αvβ5. Adenoviruses do not depend on cell division for transfection and therefore can transduce many cell types including postmitotic neurons. However, host cell specificity is not unlimited and simply depends on the expression of the CAR molecule. After cell entry, the transgene is inserted as an episome to the nucleus and therefore the risk of insertional mutagenesis is low. Replication-competent adenoviruses replicate exclusively inside the nucleus and have early and late transcription phases. The viral immediate-early protein E1 is important and is known to inactivate p53 (via E1B) and retinoblastoma (Rb) proteins (via E1A) to promote viral growth. Adenoviruses are easily produced in the laboratory and high titers can be reached. Replication-deficient strains have a carrying capacity of up to a 8-kbp transgene. The transgene expression may last up to 12 months in the periphery and up to 18 months in the brain. A strong immune response to adenoviruses mounted by the host has been reported as a serious clinical problem and has resulted in termination of early clinical studies.13 Another drawback lies in the fact that adenoviruses do not infect most rodents; therefore such animal models are inadequate to determine viral safety in the human host. There are two classes of adenovirus-based therapeutics: replication deficient and conditionally replicative adenoviruses.14 Replication-deficient vectors have E1 (E1A and/or E1B) gene deleted and therefore do not express virally encoded proteins. However; the transgene is transferred to the nucleus and expressed under an intermediate early cytomegalovirus (CMV) promoter sequence, independent from viral protein synthesis. Conditionally replicative adenoviruses depend on alterations in intracellular pathways, which are present in transformed cells, for their replication; therefore in theory they grow only in transformed cells. These viruses are most commonly used as oncolytic viruses rather than simple vectors. Onyx-O15 is an E1B knockout and selectively replicates in transformed cells with defective p53 pathway. Δ24 is a conditionally replicative virus that has an E1A 24-bp deletion at the Rb binding domain and preferentially grows in Rb-positive cells.
There are only two reports on the use of a replication-deficient adenovirus against meningioma.
As noted in the preceding text, Ikeda and colleagues26 found that the adenoviral vector they used had a low transduction efficiency (mean 8%–37%) against three different meningioma cultures. Chauvet and colleagues38 published a unique, but single, case report of a successful and widespread gene transfer after intra-arterial injection of a replication-deficient adenovirus (with lac-z reporter) to a spontaneous olfactory groove meningioma in a West-highland terrier. Viola and colleagues39 showed that intrathecal injection of a replication deficient lac-z expressing adenoviral vector in rats with meningeal cancer resulted in transduction of the infiltrating tumor in the subarachnoid space when injections were given simultaneously with, or 7 days after, tumor inoculation. The tumor used in this study was not meningioma but the findings indicated that leptomeningeal tumors can be transfected with intrathecal viral vector injections.
Adenoviruses have so far been reported to effectively infect meningioma cells and result in successful gene transfer.26,37 Shu and colleagues,37 in an effort to study the role of RAS activation in meningiomas, used adenoviruses for gene transfer to cultured meningioma cells. The authors documented successful gene transfer in 9 of 10 short-term primary meningioma cultures, indicating that most meningiomas are susceptible to adenovirus infection. The most commonly used serotype, adenovirus type 5, enters the cell by binding to the coxsackie virus–adenovirus receptor (CAR) on the cell surface, the expression of which varies among cells. Expression of the CAR necessary for adenovirus entry was reported both in surgical meningioma samples and in primary cultured meningioma cells.35 Low expression on tumor cells and present expression on normal epithelial cells can result in low efficiency and side effects, respectively. To further facilitate adenoviral entry and increase specificity, targeted recombinant adenoviruses have been introduced. Facilitated entry has also been reported for the recombinant adenoviruses that carry a RGD (arginine–glycine–aspartic acid) motif on the viral surface fiber which targets alpha-v integrins on the cell surface. Recombinant-RGD adenoviruses have been reported to effectively infect meningioma cells and spheroids with increased affinity.35 Some authors have attempted to retarget the virus by replacing the viral surface structure. Successful transfection of human meningioma cell cultures and spheroids has been achieved using targeted adenoviruses.35,36 van Beusechem and colleagues36 tested recombinant adenoviruses double ablated for the CAR and the adenovirus binding capacity against primary human glioma and meningioma spheroids and showed that this strategy can increase the tumor specificity of the adenovirus.
In the molecular biology era, most initial viral therapy efforts for brain tumors were concentrated on herpes and adenoviruses. More recent studies have reported use of a much wider spectrum of viruses against brain tumors, mostly glioblastoma. Viruses that have shown promise against glioblastoma include both DNA viruses (vaccinia and myxoma viruses) and RNA viruses (measles, vesicular stomatitis virus, reovirus, Newcastle disease virus, and polio viruses). Only very few of these viruses have been tested on meningiomas (Table 57-1). Ansardi and colleagues25 showed that replication-incompetent polivirus replicons killed both meningioma cell lines and primary-explant cultures from fresh patient samples in culture. Reovirus27,40 and myxoma viruses28,41 have also been tested against meningioma cells in cultures but were found ineffective.
Biology of Meningiomas and Molecular Targets for Gene Therapy
Meningiomas are extra-axial solid tumors of the central nervous system (CNS) that originate from arachnoid cap cells.42 Most cases are located intradurally. The etiology of meningiomas can be identified only in a minority (such as in the case of radiation induced meningiomas) and the pathogenesis is largely unknown. Accumulating evidence supports the hypothesis that meningiomas form as a result of a series of sequential genetic events. Meningiomas can progress from benign to atypical and malignant forms, and genetic analysis has indicated that such increasingly aggressive behavior is accompanied by sequential accumulation of more complex genetic changes.43
Formation of a tumor cell requires that cells acquire abilities for self-sufficiency in growth signals, insensitivity to antigrowth signals, evading apoptosis, limitless replicative potential, sustained angiogenesis, and tissue invasion and metastasis.44 Currently, very little is known regarding the mechanisms of oncogenesis in meningiomas that result in these capabilities. Epidermal growth factor (EGF) receptors have been documented in meningiomas.45 Activated platelet-derived growth factor (PDGF) receptors and their possible autocrine stimulation have also been reported in meningiomas.46,47 RAS activation may also play a role in meningioma oncogenesis.37 Tumor suppressor gene inactivation also plays an important role in the genesis of meningiomas. Meningioma is the second most common tumor observed in patients with neurofibromatosis type II (NF2). This association is even stronger in pediatric meningiomas, 40% of which are associated with NF2.48 Detailed analysis of these intracellular changes in meningioma cells is of special importance with viral therapies as viruses exploit physiologic and pathologic intracellular mechanisms. To name a few examples, adenoviruses make use of the p53, Rb, BAX, P300/CBP, and herpes simplex viruses of PKR and ribonucleotide reductase; reovirus replicates in RAS transformed cells; Newcastle disease virus and vesicular stomatitis virus selectively replicate in interferon deficient cells; and myxoma virus replicates in STAT-deficient cells.15 Precise definition of oncogenic mechanisms therefore can also guide rational treatment strategies in specific tumors or tumor subgroups. Currently there is no such example in meningiomas. In addition, study of different viral infections in tumor cells may also shed light to definition of oncogenic mechanisms.
Virus–Host Interactions
Healthy individuals mount up an immune response to invading viruses, which adds another dimension to the development and action of viral therapeutics. This may be suppression or potentiation of the anti-tumor action. The virus may be cleared from the circulation by the reticuloendothelial system; the extent of the viral infection may be limited by the innate immune response or the antibody response can interfere with the delivery and action of viruses that are normally human pathogens, such as herpesvirus.11 On the other hand, immune mechanisms may also potentate the effects of viruses, as seen in the example of adenoviruses.11 Newcastle disease virus, which was first used in 1964 by Wheelock and Dingle against leukemia, is also known to induce antitumor immunity.49 Controlled use and manipulation of these effects (with molecules such as interleukins, interferons, tumor necrosis factor-α, manipulated dendritic cells, GM-CSF, sFlt-3-L, B7, and intercellular adhesion molecules [ICAM]) gave birth to the field of immuno-gene therapy.
EXPERIMENTAL MENINGIOMA MODELS AND GENE THERAPY STUDIES
Both immortalized cell lines and low passage primary cultures from meningiomas are used in experimental studies. Several permanent cell lines of meningiomas have been described. Most of these are malignant meningiomas (IOMM-Lee50 and HKBMM,51 F5,52 CH-157-MN,53 HBL-5254). These immortal cultures are most commonly derived from meningiomas with complex genetic defects, and grow both in vitro and in vivo with KI67 labeling indexes around 30%.52,55 Meningioma cell lines are vimentin immunopositive, glial fibrillary acidic protein (GFAP) immunonegative and exhibit ultrastructural findings reminiscent of meningiomas.54,55 The development of cell lines from benign meningiomas was complicated by the onset of replicative senescence. However, this problem has been overcome by introducing activated telomerase into cells cultured from benign meningiomas.56,57 Freshly cultured human tumors cells are also used to test therapeutic activity. Several such studies have been reported in meningiomas, as noted in the preceding text.
The first animal model was that of human meningioma xenografts in guinea pig eyes.58 The subrenal capsule has also been used as an implantation site for human meningioma xenografts.59 Subsequently, the subcutaneous flank tumor model has been introduced as a practical small animal modality.55 With subsequently introduced refinements, approximately 60% of the injections “take” and form subcutaneous tumors.60 The probability of a successful xenograft take is reported to be higher in meningiomas with multiple genetic defects.55 Orthotopic models of meningioma have also been reported.52 IOMM-Lee or F5 malignant meningioma cells injected to the subdural space reportedly form spherical tumors with a mass effect.52 As a model of skull-base meningiomas, IOMM-Lee cells injected into the skull base form tumors, invade both bone and brain, and encase cranial nerves.52 Tumor cells transfected with both green fluorescent protein and luciferase have also been successfully used in the skull base for bioluminescent imaging.61 One drawback of these model systems is the leptomeningeal spread through the subdural and subarachnoid spaces, which complicates quantification of the tumor size.52,62 Human tumor xenografts in rodents are good experimental models but also have major drawbacks. Immunosuppression is absolutely required in these models, and this significantly limits the translation of experimental findings to reality as the immune system plays a very important role in the course of viral infections in the living organism.
Human tumors are genetically and epigenetically more complex than their rodent counterparts,63 so traditionally an effort was made to use tumor cells of human origin in these animal models, rather than animal tumors. However, this requires the use of immunocompromised animals and these are extremely susceptible to infectious agents, including most therapeutically designed replication competent viruses. Newer strategies include transgenic animals that carry human cancer associated mutations and develop “humanized” animal tumors. In the only study reported so far, Kalamarides and colleagues described a transgenic mouse line with CRE-mediated inactivation of the NF2 gene in arachnoidal cells, which resulted in formation of meningiomas resembling human tumors in 30% of the animals.64 The applicability if such meningioma models has not been thoroughly studied.
[1] Simpson D. Recurrence of intracranial meningiomas after surgical treatment. J Neurol Neurosurg Psychiatry. 1957;20:22-39.
[2] Oh S., Pluhar G.E., McNeil E.A., et al. Efficacy of nonviral gene transfer in the canine brain. J Neurosurg. 2007;107:136-144.
[3] Yoshida J., Mizuno M. Clinical gene therapy for brain tumors. Liposomal delivery of anticancer molecule to glioma. J Neurooncol. 2003;65:261-267.
[4] Rainov N.G., Ren H. Clinical trials with retrovirus mediated gene therapy–what have we learned? J Neuro-oncol. 2003;65:227-236.
[5] Humphries E.H., Glover C., Reichmann M.E. Rous sarcoma virus infection of synchronized cells establishes provirus integration during S-phase DNA synthesis prior to cellular division. Proc Natl Acad Sci USA. 1981;78:2601-2605.
[6] Pulkkanen K.J., Yla-Herttuala S. Gene therapy for malignant glioma: current clinical status. Mol Ther. 2005;12:585-598.
[7] Danthinne X., Imperiale M.J. Production of first generation adenovirus vectors: a review. Gene Ther. 2000;7:1707-1714.
[8] Zou L., Zhou H., Pastore L., Yang K. Prolonged transgene expression mediated by a helper-dependent adenoviral vector (hdAd) in the central nervous system. Mol Ther. 2000;2:105-113.
[9] Gomez-Manzano C., Balague C., Alemany R., et al. A novel E1A-E1B mutant adenovirus induces glioma regression in vivo. Oncogene. 2004;23:1821-1828.
[10] Puumalainen A.M., Vapalahti M., Yla-Herttuala S. Gene therapy for malignant glioma patients. Adv Exp Med Biol. 1998;451:505-509.
[11] Rainov N.G., Ren H. Oncolytic viruses for treatment of malignant brain tumours. Acta Neurochir Suppl. 2003;88:113-123.
[12] Khuri F.R., Nemunaitis J., Ganly I., et al. A controlled trial of intratumoral ONYX-015, a selectively-replicating adenovirus, in combination with cisplatin and 5–fluorouracil in patients with recurrent head and neck cancer. Nat Med. 2000;6:879-885.
[13] Rainov N.G., Ren H. Gene therapy for human malignant brain tumors. Cancer J. 2003;9:180-188.
[14] Chiocca E.A. Oncolytic viruses. Nat Rev Cancer. 2002;2:938-950.
[15] Parato K.A., Senger D., Forsyth P.A., Bell J.C. Recent progress in the battle between oncolytic viruses and tumours. Nat Rev Cancer. 2005;5:965-976.
[16] Wollmann G., Tattersall P., van den Pol A.N. Targeting human glioblastoma cells: comparison of nine viruses with oncolytic potential. J Virol. 2005;79:6005-6022.
[17] Glick R.P., Lichtor T. Immunotherapy task force for malignant brain tumors. April 21, 2001, Toronto, Ontario. Neuro Oncol. 2001;3:258-264.
[18] Lampson L.A., Lampson M.A., Dunne A.D. Exploiting the lacZ reporter gene for quantitative analysis of disseminated tumor growth within the brain: use of the lacZ gene product as a tumor antigen, for evaluation of antigenic modulation, and to facilitate image analysis of tumor growth in situ. Cancer Res. 1993;53:176-182.
[19] Croteau D., Mikkelsen T., Rempel S.A., et al. New innovations and developments for glioma treatment. Clin Neurosurg. 2001;48:60-81.
[20] Yang L., Ng K.Y., Lillehei K.O. Cell-mediated immunotherapy: a new approach to the treatment of malignant glioma. Cancer Control. 2003;10:138-147.
[21] Okada H., Pollack I.F. Cytokine gene therapy for malignant glioma. Expert Opin Biol Ther. 2004;4:1609-1620.
[22] Yu J.S., Wheeler C.J., Zeltzer P.M., et al. Vaccination of malignant glioma patients with peptide-pulsed dendritic cells elicits systemic cytotoxicity and intracranial T-cell infiltration. Cancer Res. 2001;61:842-847.
[23] Mitchell D.A., Fecci P.E., Sampson J.H. Adoptive immunotherapy for malignant glioma. Cancer J. 2003;9:157-166.
[24] Sikorski C.W., Lesniak M.S. Immunotherapy for malignant glioma: current approaches and future directions. Neurol Res. 2005;27:703-716.
[25] Ansardi D.C., Porter D.C., Jackson C.A., et al. RNA replicons derived from poliovirus are directly oncolytic for human tumor cells of diverse origins. Cancer Res. 2001;61:8470-8479.
[26] Ikeda K., Saeki Y., Gonzalez-Agosti C., et al. Inhibition of NF2–negative and NF2–positive primary human meningioma cell proliferation by overexpression of merlin due to vector-mediated gene transfer. J Neurosurg. 1999;91:85-92.
[27] Wilcox M.E., Yang W., Senger D., et al. Reovirus as an oncolytic agent against experimental human malignant gliomas. J Natl Cancer Inst. 2001;93:903-912.
[28] Lun X., Yang W., Alain T., et al. Myxoma virus is a novel oncolytic virus with significant antitumor activity against experimental human gliomas. Cancer Res. 2005;65:9982-9990.
[29] Yazaki T., Manz H.J., Rabkin S.D., Martuza R.L. Treatment of human malignant meningiomas by G207, a replication-competent multimutated herpes simplex virus 1. Cancer Res. 1995;55:4752-4756.
[30] Harrow S., Papanastassiou V., Harland J., et al. HSV1716 injection into the brain adjacent to tumour following surgical resection of high-grade glioma: safety data and long-term survival. Gene Ther. 2004;11:1648-1658.
[31] Markert J.M., Medlock M.D., Rabkin S.D., et al. Conditionally replicating herpes simplex virus mutant, G207 for the treatment of malignant glioma: results of a phase I trial. Gene Ther. 2000;7:867-874.
[32] Rampling R., Cruickshank G., Papanastassiou V., et al. Toxicity evaluation of replication-competent herpes simplex virus (ICP 34.5 null mutant 1716) in patients with recurrent malignant glioma. Gene Ther. 2000;7:859-866.
[33] Markert J.M., Coen D.M., Malick A., et al. Expanded spectrum of viral therapy in the treatment of nervous system tumors. J Neurosurg. 1992;77:590-594.
[34] Liu T.C., Zhang T., Fukuhara H., et al. Dominant-negative fibroblast growth factor receptor expression enhances antitumoral potency of oncolytic herpes simplex virus in neural tumors. Clin Cancer Res. 2006;12:6791-6799.
[35] Dirven C.M.F., Grill J., Lamfers M.L., et al. Gene therapy for meningioma: improved gene delivery with targeted adenoviruses. J Neurosurg. 2002;97:441-449.
[36] van Beusechem V.W., Grill J., Mastenbroek D.C., et al. Efficient and selective gene transfer into primary human brain tumors by using single-chain antibody-targeted adenoviral vectors with native tropism abolished. J Virol. 2002;76:2753-2762.
[37] Shu J., Lee J.H., Harwalkar J.A., et al. Adenovirus-mediated gene transfer of dominant negative Ha-Ras inhibits proliferation of primary meningioma cells. Neurosurgery. 1999;44:579-587. discussion 587–8
[38] Chauvet A.E., Kesava P.P., Goh C.S., Badie B. Selective intraarterial gene delivery into a canine meningioma. J Neurosurg. 1998;88:870-873.
[39] Viola J.J., Ram Z., Walbridge S., et al. Adenovirally mediated gene transfer into experimental solid brain tumors and leptomeningeal cancer cells. J Neurosurg. 1995;82:70-76.
[40] Yang W.Q., Senger D., Muzik H., et al. Reovirus prolongs survival and reduces the frequency of spinal and leptomeningeal metastases from medulloblastoma. Cancer Res. 2003;63:3162-3172.
[41] Lun X.Q., Zhou H., Alain T., et al. Targeting human medulloblastoma: oncolytic virotherapy with myxoma virus is enhanced by rapamycin. Cancer Res. 2007;67:8818-8827.
[42] Louis D.N., Scheithauer B.W., Budka H., et al. Kleihues P., editor. Meningiomas, The WHO classification of tumors of the nervous system. Cavenee WK, Lyon, 2002;176-184.
[43] Perry A., Gutmann D.H., Reifenberger G. Molecular pathogenesis of meningiomas. J Neurooncol. 2004;70:183-202.
[44] Hanahan D., Weinberg R.A. The hallmarks of cancer. Cell. 2000;100:57-70.
[45] Carroll R.S., Black P.M., Zhang J., et al. Expression and activation of epidermal growth factor receptors in meningiomas. J Neurosurg. 1997;87:315-323.
[46] Shamah S.M., Alberta J.A., Giannobile W.V., et al. Detection of activated platelet-derived growth factor receptors in human meningioma. Cancer Res. 1997;57:4141-4147.
[47] Todo T., Adams E.F., Fahlbusch R., et al. Autocrine growth stimulation of human meningioma cells by platelet-derived growth factor. J Neurosurg. 1996;84:852-858. discussion 858–9
[48] Evans D.G., Birch J.M., Ramsden R.T. Paediatric presentation of type 2 neurofibromatosis. Arch Dis Child. 1999;81:496-499.
[49] Lorence R.M., Roberts M.S., Groene W.S., Rabin H. Replication-competent, oncolytic Newcastle disease virus for cancer therapy, replication-competent viruses for cancer therapy. In: Virology-Doerr H.W., Driever P.H., Rabkin S.D., editors. Monographs. Basel: Karger; 2001:160-182.
[50] Lee W.H. Characterization of a newly established malignant meningioma cell line of the human brain: IOMM-Lee. Neurosurgery. 1990;27:389-395. discussion 396
[51] Ishiwata I., Ishiwata C., Ishiwata E., et al. In vitro culture of various typed meningiomas and characterization of a human malignant meningioma cell line (HKBMM). Hum Cell. 2004;17:211-217.
[52] van Furth W.R., Laughlin S., Taylor M.D., et al. Imaging of murine brain tumors using a 1.5 Tesla clinical MRI system. Can J Neurol Sci. 2003;30:326-332.
[53] Ragel B.T., Elam I.L., Gillespie D.L., et al. A novel model of intracranial meningioma in mice using luciferase-expressing meningioma cells. Laboratory investigation. J Neurosurg. 2008;108:304-310.
[54] Akat K., Bleck C.K., Lee Y.M., et al. Characterization of a novel type of adherens junction in meningiomas and the derived cell line HBL-52. Cell Tissue Res. 2008;331:401-412.
[55] Ragel B.T., Couldwell W.T., Gillespie D.L., et al. A comparison of the cell lines used in meningioma research. Surg Neurol. 2008;70:295-307.
[56] Puttmann S., Senner V., Braune S., et al. Establishment of a benign meningioma cell line by hTERT-mediated immortalization. Lab Invest. 2005;85:1163-1171.
[57] Cargioli T.G., Ugur H.C., Ramakrishna N., et al. Establishment of an in vivo meningioma model with human telomerase reverse transcriptase. Neurosurgery. 2007;60:750-759. discussion 759–60
[58] Greene H.S., Arnold H. The homologous and heterologous transplantation of brain and brain tumors. J Neurosurg. 1945;2:315-329.
[59] Medhkour A., Van Roey M., Sobel R.A., et al. Implantation of human meningiomas into the subrenal capsule of the nude mouse. A model for studies of tumor growth. J Neurosurg. 1989;71:545-550.
[60] Jensen R.L., Leppla D., Rokosz N., Wurster R.D. Matrigel augments xenograft transplantation of meningioma cells into athymic mice. Neurosurgery. 1998;42:130-135. discussion 135–6
[61] Baia G.S., Dinca E.B., Ozawa T., et al. An orthotopic skull base model of malignant meningioma. Brain Pathol. 2008;18:172-179.
[62] McCutcheon I.E., Friend K.E., Gerdes T.M., et al. Intracranial injection of human meningioma cells in athymic mice: an orthotopic model for meningioma growth. J Neurosurg. 2000;92:306-314.
[63] Hahn W.C., Weinberg R.A. Modelling the molecular circuitry of cancer. Nat Rev Cancer. 2002;2:331-341.
[64] Kalamarides M., Niwa-Kawakita M., Leblois H., et al. Nf2 gene inactivation in arachnoidal cells is rate-limiting for meningioma development in the mouse. Genes Dev. 2002;16:1060-1065.
[65] Phuong L.K., Allen C., Peng K.W., et al. Use of a vaccine strain of measles virus genetically engineered to produce carcinoembryonic antigen as a novel therapeutic agent against glioblastoma multiforme. Cancer Res. 2003;63:2462-2469.
[66] Forsyth P., Roldan G., George D., et al. A phase I trial of intratumoral administration of reovirus in patients with histologically confirmed recurrent malignant gliomas. Mol Ther. 2008;16:627-632.
[67] Lun X., Senger D.L., Alain T., et al. Effects of intravenously administered recombinant vesicular stomatitis virus (VSV(deltaM51)) on multifocal and invasive gliomas. J Natl Cancer Inst. 2006;98:1546-1547.
[68] Ozduman K., Wollmann G., Piepmeier J.M., et al. Systemic vesicular stomatitis virus selectively destroys multifocal glioma and metastatic carcinoma in brain. J Neurosci. 2008;28:1882-1893.
[69] Freeman A.I., Zakay-Rones Z., Gomori J.M., et al. Phase I/II trial of intravenous NDV-HUJ oncolytic virus in recurrent glioblastoma multiforme. Mol Ther. 2006;13:221-228.
[70] Csatary L.K., Gosztonyi G., Szeberenyi J., et al. MTH-68/H oncolytic viral treatment in human high-grade gliomas. J Neurooncol. 2004;67:83-93.
[/level-membership-for-neurosurgery-category][not-level-membership-for-neurosurgery-category]
CHAPTER 57 Gene Therapy for Meningiomas
RATIONALE FOR ALTERNATIVE TREATMENT STRATEGIES IN MENINGIOMAS
Surgery is the mainstay of treatment in meningiomas, and most such tumors can be easily and safely resected surgically with an excellent outcome. Despite an overall favorable outcome, treatment attempts fail in some patients due to intrinsic biology of meningiomas or to difficult localization. Most meningiomas recur after resection if followed long enough.1 Adjuvant treatment modalities such as radiosurgery and radiotherapy are effective, however, with limitations and side effects. Chemotherapy is marginally effective and used as a salvage therapy in cases that have failed other available treatment options. Gene therapy is an appealing new strategy for meningioma treatment. Areas of potential use are (1) recurrent meningiomas, (2) meningiomas in difficult to treat localizations, (3) multiple meningiomas, (4) meningiomas in high-risk patients, (5) metastatic cases, and (6) atypical or malignant meningiomas that have not responded to conventional treatment modalities.
INTRODUCTION TO GENE THERAPY
Gene therapy is defined as an experimental treatment that involves introducing genetic material (DNA or RNA) into a person’s cells for therapeutic purposes. This field initially started with efforts to replace or substitute missing or altered genes that caused disease. Advances in vector design and therapeutic virology and an increased understanding of molecular tumor biology have extended this initial concept to a multitude of promising strategies. When broadly classified, current gene therapy applications to treat brain malignancies fall into three categories (Fig. 57-1):
In Vivo Gene Delivery Methods
A wide range of methods are available for delivery of DNA or RNA into target cells. In vitro techniques such as direct microinjection and chemical (calcium phosphate) or physical (electroporation) transfection methods are not practical for in vivo application. This leaves “naked DNA” administration, liposomal gene delivery, and virally mediated gene transfer as the methods of choice for in vivo gene therapy application. Injection of “naked DNA” plasmids has been shown to result in intracranial gene expression.2 However, with current technology, expression efficiency is very low and the practical value of this technique for brain tumor gene therapy remains to be seen. In contrast, liposomal gene transfer has been established as a promising therapeutic modality for brain tumors.3 Good transfer efficiency, safety, simple design, and their unique capability to carry, not only genetic material, but also proteins, chemicals, or physical particles into a cell make these artificial lipid bilayered vesicles useful and exciting therapeutic tools. However, viral-based vectors and therapeutic viruses are the absolute mainstay in the field of gene therapy.
Viral Therapies
Viruses have evolved as nature’s way to deliver genomic material into cells with unsurpassed efficiency, displaying an overwhelming repertoire of mechanisms. Since the early 1990s advances in the field of molecular biology have provided the tools necessary to manipulate viral genomes. The vast majority of viral vectors are composed of recombinant viruses, often engineered to exhibit attenuated phenotypes and harbor nonviral genes of interest. The search for and the design of appropriate vectors are directed by certain paradigms that depend on the clinical context. Simplified, in gene therapy applications for genetic or degenerative diseases the duration of the expression of the therapeutic gene is a foremost priority. In contrast, antitumor gene therapy generally seeks to destroy the target cell, making transduction efficiency much more important, and a short term gene expression appears to be sufficient. For tumor treatment, viruses have both been used as gene delivery vehicles (viral vectors) or as biological agents that attack and kill tumor cells directly (oncolytic viruses). Gene therapy strategies using adenoviral or retroviral vectors to deliver suicide genes to tumor cells have been the forerunners in the field and both have been used in clinical trials against human high-grade gliomas. In vivo transduction efficiency has been the main problem with retroviruses,4 which can deliver genes only to cells that are actively dividing.5 Insertional mutagenesis is a rare but feared side effect and can lead to the formation of secondary tumors resulting from oncogene activation or tumor-suppressor gene silencing due to the uncontrollable random insertion of the viral transgene into the human genome. Low transfection efficiency and lack of any therapeutic effect in phase III trials of RV-HSV-tk in newly diagnosed glioblastoma led to abandonment of retroviral strategies.6 First-generation adenoviruses (AdV) had been deprived of their replicative capabilities by deletion of variable portions (E1 or E3) of the viral genome.7 As these AdV vectors retain large portions of their viral genome, vectors and transduced cells are highly immunogenic once administered into the immunocompetent host. Consequently, therapeutic effects are short lived, though the enhanced immune response may noticeably contribute synergistically in the setting of anticancer therapy. Newer adenovirus strategies use either highly stripped down “gutless” AdV, requiring helper systems for production8 with minimized immune response and prolonged gene expression or conditional replication competent variants with inherent tumor selective affinity.9 An important aspect of antitumor gene therapy is the spatial distribution of the viral therapeutic. The therapeutic activity increases significantly from cell-limited gene expression over the so-called bystander effect of released therapeutic products to the active spread of replication-competent viruses. Replication-incompetent viral vectors have been proven ineffective in vivo, as only a small percentage of tumor cells can be infected under the most optimal circumstances with the delivered viral load.10,11 Whether the viral therapeutic will replicate in the tissue is an important issue: Nonreplicating viruses are most commonly used to deliver toxic or therapeutic genes. Conditionally replicating viruses are based on replicating viruses but one or more of their viral genes are deleted to make them depend on cellular mechanisms, which are present only in tumor cells. Although this is a very attractive strategy, replicative health of most of these viruses is also impaired.6,12 Replicating viruses are used to infect tumor tissues and viral progeny produced inside the target tissue infects surrounding cells to cause “local self-amplification.” A considerably small amount of virus can therefore be delivered to the target tissue to yield a much heavier therapeutic dose inside the tumor. Such a phenomenon does not exist in any other therapeutic modality. Newer, recombinant adenoviruses were engineered to be conditionally replicative in tumor cells or carried transgenes that are expressed only in specific cell types.13 Recombinant herpes viruses have also been commonly used in gene therapy protocols for brain tumors.6 Recombinant viral vectors based on vaccinia have also been used. In contrast to strategies that use viral vectors to transfer genes to the target cell, other researchers have experimented with oncolytic viruses, which preferentially replicate inside the tumor tissue and kill the tumor cells as a direct result of this infection.14 Other viruses including reovirus; Newcastle disease virus; and measles, vesicular stomatitis, and myxoma virus, have shown merit for infecting some types of brain tumor cells.15
In animal experiments viral infection can easily be monitored inside the target tissue or elsewhere using virally expressed fluorescent reporter genes. Fluorescent reporter genes can be engineered into most viruses, and when combined with tumor models that express other fluorescent proteins, temporal and spatial monitoring of both the oncolytic activity and/or side effects is greatly simplified.16
Immunotherapy/Immunogene Therapy Strategies
Cancer immunotherapy has been intensely studied in the field of malignant melanoma therapy, and the promise in that field among others led in October 2000 to the formation of the Immunotherapy Task Force for malignant brain tumors.17 As with gene therapy and oncolytic virotherapy, the focus of current immunotherapy studies of brain tumors is on tumors of neuroectodermal lineage, that is, glioblastoma and anaplastic astrocytoma. Though the transfer of this strategy to meningioma remains hypothetical at this point, a brief discussion of the current studies is merited.
Besides detection of infectious or foreign organisms the immune system has the capability to recognize and even eliminate cancerous cells. For that response to be effective, certain preconditions need to be met. Tumor-associated antigens (TAAs) need to be expressed and the tumor environment needs to be “immune-compatible,” as certain tumors can generate a local and systemic state of immune suppression. Evidence suggests both membrane and intracellular elements can serve as TAAs.18 However; despite its remarkably high selectivity, the natural immune response falls short in terms of efficiency to control advanced, sizable cancers. The limit of a patient’s immune response seems to be in the range of less than a million tumor cells, which is a negligible fraction compared to the billions of cells of an average sized brain tumor.19 In that regard, immunotherapy can be defined as strategies aiming to amplify the natural immune response to cancerous cells. Both arms of the immune response, the initiating cascade and the executing effector phase, can be targeted.
Earlier attempts of immunotherapy included the use of tumor lysates and microbe preparations such as nonspecific immune activators with little success.20 With increasing knowledge of mechanisms underlying antigen presentation and immune modulation, the direction shifted toward highly specific stimulation of the antitumor response. The main strategies comprise the use of cytokines,21 active vaccination with tumor antigen-pulsed dendritic cells,22 and adoptive immunotherapy using cytotoxic T cells raised and expanded ex vivo.23 Among others, cytokines interleuking-2 (IL-2), IL-4, IL-12, IL-18, interferon (IFN), granulocyte-macrophage colony-stimulating factor (GM-CSF), and B7-2 have been used with promising results alone, in combinations, or as adjuvants to cell-based immunotherapy.24 Applications range from direct peripheral infusion and convection-enhanced delivery to in vivo cytokine gene therapy and ex vivo gene transfer to antitumor dendritic or cytotoxic T cells. Active antitumor vaccination involves the collection of blood mononuclear cells and the in vitro isolation and priming of dendritic cells. Considered the most potent antigen presenting cells, dendritic cells pulsed with tumor cells or tumor lysate can effectively prime T-helper cells once reinjected into the patient’s body, leading to an efficient mounting of an antitumor response by cytotoxic T lymphocytes. In contrast, adoptive immunotransfer focuses on ex vivo expansion and reinjection of tumor-specific effector cells. Several cell types have been described with potential for adoptive immunotherapy: lymphokine-activated killer cells, tumor infiltrating lymphocytes, cytotoxic T lymphocytes, and others.
CURRENT GENE THERAPY STRATEGIES FOR MENINGIOMAS
What Has Been Achieved So Far?
Herpes simplex virus and adenovirus are among the first recombinant virus species used for gene therapy applications. Both viruses have been extensively used both in preclinical and clinical studies against various types of tumors and have shown promise against meningioma. Similarly, poliovirus25 and herpesvirus26 replicons have been found to effectively transduce meningioma cells. Other viruses such as reovirus,27 myxoma virus,28 and retroviruses26 have been reported to be ineffective against meningioma cultures, despite their potential against other kinds of brain tumor.
Herpes simplex virus and adenovirus have been the most common recombinant virus species used for gene therapy attempts. Both viruses have been extensively used both in preclinical and clinical studies against various types of tumors. Herpes simplex was the first oncolytic virus tested against meningiomas.29 Herpes simplex virus is a large, enveloped virus that belongs to the alpha-herpesvirus family and has a 152-kb double-stranded linear DNA genome. The wild-type virus has 80 densely packed genes, approximately 50% of which are necessary for viral replication. Almost 30 kilobases can be replaced with transgenes without affecting viral replication. The virus can infect both replicating and nonreplicating cells. Lytic infection of herpesviruses has three phases (α, β, and γ) and genes taking part in these three phases are responsible for regulating transcription, promoting DNA synthesis and creating a favorable environment for protein synthesis, respectively. Earlier genes in the genome have a suppressor role on late genes. Recombinant HSVs have been designed by making changes in or deleting α-genes (e.g., G47Δ, NV1020), β-genes (DLSPTK, G207), or γ-genes (R3616, G207). Herpes simplex is a large virus with a large transgene capacity, is stable, infects many cell types, and has animal models available for toxicity. Absence of proinflammatory effects and bone marrow toxicity, the availability of antiherpetic chemotherapy, and the absence of genomic integration are advantages of the HSV for gene therapy. In contrast, the large size of the genome, presence of preexisting HSV immunity in humans, and risk of encephalitis from primary or latent infection are the basic concerns associated with HSV. G207, which is the only HSV that was used in experimental studies against meningioma, had been tested in a phase 1 clinical trial against human glioblastoma.30–32 Magnetic resonance imaging (MRI) responses and few long-term survivors have been reported in these glioblastoma trials, with no chronic toxicity observed in survivors.
[/not-level-membership-for-neurosurgery-category]
