CHAPTER 6 GASTROINTESTINAL SYSTEM
GASTROINTESTINAL TRACT IN CRITICAL ILLNESS






Manifestations of gastrointestinal tract failure
Failure of the gastrointestinal tract during critical illness may present in a number of ways. These are listed in Box 6.1.
The principle aim of investigation of gastrointestinal dysfunction in the critically ill patients is to exclude serious, remediable, intra-abdominal pathology. In some cases, the combination of history, clinical examination and blood results in the context of the overall clinical picture will suffice. In many cases however, intra-abdominal imaging will be required. Occasionally laparotomy or laparoscopy may be necessary to exclude serious pathology.
DIARRHOEA
 As a manifestation of multisystem disorder. For example, generalized tissue hypoxia, tissue oedema and vascular endothelial failure affect the function of all tissues. The gut is no exception to this. These pathological abnormalities are associated with a failure in cellular function and metabolic pathways, which may persist for some time. Consequently, gut failure (either diarrhoea or constipation) is often a characteristic feature of the patient in the intensive care unit.
As a manifestation of multisystem disorder. For example, generalized tissue hypoxia, tissue oedema and vascular endothelial failure affect the function of all tissues. The gut is no exception to this. These pathological abnormalities are associated with a failure in cellular function and metabolic pathways, which may persist for some time. Consequently, gut failure (either diarrhoea or constipation) is often a characteristic feature of the patient in the intensive care unit.





GASTROINTESTINAL BLEEDING
Slow bleeding from the gastrointestinal tract may occur, and is a common cause of reduced haemoglobin in the critically ill. Less commonly, but of greater immediate concern is acute massive GIT haemorrhage, which may be life threatening. The source is usually the upper GIT. Common causes are listed in Box 6.2. Bleeding of this sort can result in haematemesis, melaena or even frank rectal blood loss. Lower GIT bleeding is less common, but may still be life threatening.
Box 6.2 Causes of upper gastrointestinal tract haemorrhage
Gastric erosion and ulceration
Management
 Large-bore vascular access is obtained and resuscitation of hypovolaemia commenced. In massive bleeds full haemodynamic monitoring, including arterial line, CVP or some other measure of volume status, are valuable.
Large-bore vascular access is obtained and resuscitation of hypovolaemia commenced. In massive bleeds full haemodynamic monitoring, including arterial line, CVP or some other measure of volume status, are valuable.






ABDOMINAL COMPARTMENT SYNDROME
The presence of blood, free fluid or gas in the abdominal cavity, splanchnic and tissue oedema and organomegally can lead to a progressive increase in the intra-abdominal pressure and to decreased splanchnic perfusion. This may result in gut, renal and liver hypoperfusion with associated clinical features. If suspected, intra-abdominal pressure can be measured via an indwelling urinary catheter (Fig. 6.1).
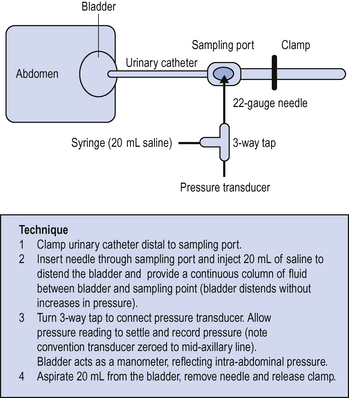
Normal intra-abdominal pressure is less than 15–20 mmHg. Intra-abdominal pressures above 20 mmHg with evidence of end organ dysfunction are suggestive of abdominal compartment syndrome. Decompression of the abdomen may be required. Seek surgical advice. Once opened, it may be impossible to close the abdomen at first laparotomy. The abdomen may be left ‘open’ under sterile plastic coverings for later closure when the swelling has subsided.
HEPATIC FAILURE
Liver failure is defined as hyperacute where the onset of encephalopathy occurs within 7 days of the onset of jaundice, acute where the interval is 7–28 days, and subacute where it is between 28 days and 6 months. Longer intervals represent chronic liver failure. The term fulminant liver failure refers to an earlier classification where encephalopathy occurs within 8 weeks of the onset of jaundice. It thus encompasses acute and hyperacute liver failure.
Clinical features
Hypoglycaemia is common and develops any time in the first few days of the condition. It gradually resolves as the liver failure improves. Acid – base homeostasis is altered and either alkalosis or acidosis may complicate this. A metabolic acidosis is the more sinister. Concurrently with the development of the metabolic derangement, conscious level may become impaired. Encephalopathy is graded as in Table 6.1.
| Grade 0 | Normal |
| Grade 1 | Mild confusion (may not be immediately evident) |
| Grade 2 | Drowsiness |
| Grade 3 | Severe drowsiness, inappropriate words/phrases, grinding teeth |
| Grade 4 | Unrousable |
True‘infective’ sepsis may occur. A rising WCC, falling platelet count, a PT whose recovery becomes arrested or a worsening acidosis should all be regarded as suspicious.
Management
 Acute hepatic failure carries a high mortality and treatment options include transplantation. Early discussion with a specialist centre is therefore mandatory.
Acute hepatic failure carries a high mortality and treatment options include transplantation. Early discussion with a specialist centre is therefore mandatory.






 The prothrombin time (PT) is one of the key prognostic factors determining whether the optimal treatment of acute liver failure is likely to be medical or surgical. Therefore, do not treat coagulopathy (by, for example, giving fresh frozen plasma) except in cases of life threatening aemorrhage.
The prothrombin time (PT) is one of the key prognostic factors determining whether the optimal treatment of acute liver failure is likely to be medical or surgical. Therefore, do not treat coagulopathy (by, for example, giving fresh frozen plasma) except in cases of life threatening aemorrhage.Metabolic issues
 Hypoglycaemia is common. All infusions should be made up in dextrose solutions (5–20%), and dextrose (5–50%) infused to maintain a normal blood sugar.
Hypoglycaemia is common. All infusions should be made up in dextrose solutions (5–20%), and dextrose (5–50%) infused to maintain a normal blood sugar.








Prognosis
Without transplantation, the prognosis is poorest in the subacute group and best in the hyperacute group. Within this group, the prognosis is poorer in those at the extremes of age, those with non-A, non-B hepatitis and drug dyscrasia. The overall mortality in patients with grade IV encephalopathy is 70%. With liver transplantation (i.e. in those patients at the highest risk of death with optimal medical management), the 1-year mortality is between 30 and 50%.
ACUTE PANCREATITIS
Inflammation of the pancreas and autodigestion may be precipitated by a number of triggers, some of which are listed in Box 6.3. The commonest causes are alcohol and biliary obstruction. Frequently no cause can be identified.
Box 6.3 Common causes of pancreatitis
Biliary obstruction (gallstones) / ERCP
Investigations
Localized posterior perforations of duodenum or stomach may be difficult to distinguish. Chest and abdominal plain films should confirm the absence of free gas or pneumonia, and may show calcium deposition. In severe cases the typical radiographic features of ARDS may be present. Plain film and ultrasound may confirm the presence of gallstones or other underlying biliary lesion. Blood glucose and arterial gases should be closely monitored. As the condition progresses, serial CT scanning is valuable to monitor pancreatic viability and to diagnose pancreatic cysts and pseudocysts.