[level-membership-for-gastroenterology-and-hepatology-category]
CHAPTER 30 Gastrointestinal Stromal Tumors (GISTs)
INCIDENCE
It is difficult to obtain accurate data regarding the true incidence of GISTs. This is because of referral bias, which concentrates GIST cases with a worse prognosis and a more malignant behavior in academic cancer centers, and also the lack of definitive diagnostic techniques before the molecular definitions of GISTs in 1998 and later. Before 2000, the number of new GIST cases in the United States had been underestimated and underreported. However, it is now recognized that the true incidence of GISTs is much higher than previously believed. Much of this is to the result of an increased recognition of GISTs; new molecularly targeted drugs, such as imatinib mesylate (Gleevec), have become available. A population-based study to assess the incidence of GISTs using the most up-to-date criteria has estimated an annual incidence of approximately 15 cases/million.1 This has increased the estimated incidence of GISTs in the United States alone to approximately 5000 new cases/year.2,3 However, even this figure may underestimate the actual incidence because many smaller lesions, including those detected incidentally by many gastroenterologists on endoscopy performed for other reasons, are almost certainly not revealed in these incidence figures. Not all GIST cases will prove to be life-threatening, because many patients who present with GISTs have limited disease with lesions that are small (smaller than 1 cm) and fully curable with optimal surgery as first-line therapy. It remains a question as to whether there are any true GIST lesions that might be able to be observed without resection.
GISTs occur predominantly in adults, with a median incidence in the fifth decade of life, but these tumors can occur across the age spectrum, from infants to older adults. The incidence has been reported to be slightly higher in men than women. Recent studies of surgical or autopsy specimens of stomach samples resected for non-neoplastic disease have documented a remarkably high incidence of occult microscopic GIST lesions, in the range of 20% to 35%.4,5
LOCATION
Most GISTs (60% to 70%) arise in the stomach, 20% to 30% originate in the small intestine, and less than 10% in the esophagus, colon, and rectum (see later). GISTs can also occur in extraintestinal sites in the abdomen or pelvis such as the omentum, mesentery, or retroperitoneum.6–8 However, data on sites of GIST origin are somewhat limited. Large studies before 1999 may have included other subtypes of mesenchymal neoplasms because these series were not selected by immunohistochemical or molecular genetic markers specific for true GISTs.
CLINICAL FEATURES
The vast majority of GIST metastases at presentation are intra-abdominal, with metastases to the liver, omentum, or peritoneal cavity.6 Metastatic spread to lymph nodes and to other regions via lymphatics is rare; most lesions thought to be nodal metastases by imaging studies simply represent metastatic deposits of tumor nodules in the omentum or peritoneum rather than true lymphatic spread of the disease.
PATHOLOGY AND MOLECULAR PATHOBIOLOGY
UNIQUE CLINICOPATHOLOGIC SUBTYPE OF SARCOMAS (MESENCHYMAL NEOPLASIA)
Although relatively rare compared with epithelial cancers, GISTs represent a form of sarcoma that comprises approximately 1% to 3% of all malignant GI tumors. As noted, the diagnostic criteria for GISTs before 1999 were somewhat subjective, controversial, and perhaps even a bit confusing, with a great deal of room for interobserver variation. The term GIST was initially a purely descriptive term applied by Mazur and Clark in 1983 to define intra-abdominal tumors that were not carcinomas (i.e., nonepithelial) and that did not exhibit features of smooth muscle or nerve cells.9 The morphology of the cells was the dominant feature driving the diagnostic scheme. However, pathologists subsequently recognized that there was no completely clear differential expression of muscle or nerve antigenic markers when careful immunohistochemical analyses were performed on samples identified as GIST lesions by cellular morphology. The expression of differentiation antigens used as markers for muscle cells (e.g., smooth muscle actin) and nerve cells (e.g., S100) was noted to vary widely in GI mesenchymal lesions, leading to interesting hypotheses about whether GIST lesions from different patients were attempting to recapitulate distinct myogenic or neural programs of differentiation.
To accommodate these empirical observations, it was proposed that approximately one third of GIST lesions differentiated along smooth muscle lineages, another third were neurogenic in origin, and the final third lacked any detectable lineage-specific markers (null phenotype) by immunohistochemical analysis.10–12
Nomenclature for GISTs had also been confusingly complex before the availability of molecular- and mechanism-based diagnostic tests. Because of limited understanding of the disease before 1999, GISTs were most commonly diagnosed as leiomyomas or leiomyosarcomas because of the histologic resemblance to these smooth muscle neoplasms. Other terms that had often been applied to a GIST included benign leiomyoblastoma and, recognizing some of the neural characteristics, plexosarcomas13 or gastrointestinal autonomic nerve tumors (GANTs).14 All these terms are now recognized as referring to GISTs. Insightful studies by several pathology groups have noted that the panoply of tumors lumped together as smooth muscle tumors of the GI tract were likely not simply leiomyosarcomas nor benign leiomyomas; a subset of these tumors originating in the bowel wall had several unique histologic features, probably representing a totally different diagnostic group altogether.15,16 Additionally, clinical oncologists had noted that putative leiomyosarcomas of the GI tract had a completely different prognosis than true leiomyosarcomas arising in other parts of the body (e.g., the uterus). The leiomyosarcomas of the GI tract were extraordinarily resistant to standard chemotherapy regimens, further supporting the notion that these lesions represent a different form of cancer entirely.
Immunohistochemical analysis of GISTs in the early 1990s attempted to find specific markers that might distinguish GISTs from other spindle cell tumors of the GI tract, such as schwannomas and sarcomatoid carcinomas. There was some initial enthusiasm for the CD34 antigen as such a marker; however, this antigen is also expressed by hematopoietic stem cells and by vascular and myofibroblastic cells. Also, the sensitivity and specificity of CD34 are low because only approximately half of GIST cases express CD34, and other smooth muscle, myofibroblastic (e.g., desmoid), or Schwann cell tumors can also express CD34. Therefore, CD34 is not a reliable marker to distinguish true GISTs from other neoplasms.17,18
UNCONTROLLED KINASE ACTIVATION: PRIMARY MOLECULAR PATHOGENESIS
A critical advance in the understanding of GISTs at a molecular level occurred in the late 1990s, with the recognition that these tumors exhibited some histopathologic similarities with the pacemaker cells of the gut known as the interstitial cells of Cajal (ICCs).19 ICCs are normally present in the myenteric plexus and serve to coordinate gut peristalsis by assisting the linkage of smooth muscle cells of the bowel wall with the autonomic nervous system (see Chapters 96 to 98). GIST cells and ICCs have certain ultrastructural features in common, such as the combination of neural and muscle phenotypes.
The molecular pathogenesis of GISTs was advanced further by a key observation made by Hirota and colleagues20 in 1998. This group was studying the role played by the KIT receptor tyrosine kinase (RTK) in cell growth and development. Expression of the KIT RTK can be detected by immunohistochemical staining for the CD117 antigen as a marker of the KIT protein. In normal cell signaling, KIT binds its ligand, known as stem cell factor (SCF) or Steel factor; ligand binding brings together two molecules of KIT with subsequent activation of a signaling cascade. A homodimeric complex is formed with these two KIT receptors, leading to cross-phosphorylation of critical tyrosine residues in the intracellular domains of KIT, which activate signal transduction pathways downstream of KIT. The net physiologic effect of normal ligand-induced KIT activation is the controlled stimulation of cell proliferation and enhanced cell survival; therefore, uncontrolled activation could theoretically lead to neoplastic growth and transformation of cells.
Hirota and colleagues20 recognized this potential mechanism and provided the critical confirmation of this theory at the cellular and molecular levels. This same team, in a project led by Nishida, also expanded these observations to familial GISTs, with germline carriage of an activating mutation in the KIT gene encoding the RTK.21 This elegant work supported some of the key biologic similarities between GIST and ICC cells22–25 because both cell types had been shown to express the KIT RTK. It is likely that the cells of GISTs and normal ICCs share a common precursor cell.26,27 The KIT RTK and SCF play essential roles in the development and maintenance of normal ICCs, as well as other cells, including melanocytes, erythrocytes, germ cells, and mast cells. KIT expression is noted in the vast majority (>95%) of GISTs, but KIT is not expressed by true smooth muscle tumors of the GI tract nor by stromal tumors at other anatomic locations, such as endometrial stromal tumors. Although the origin of the neoplastic cells of GISTs remains a matter of active investigation, some data suggest that GISTs originate from CD34-positive stem cells residing within the wall of the gut, which can then differentiate incompletely toward the ICC phenotype.27–29
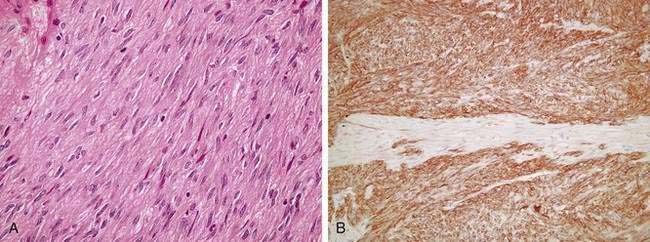
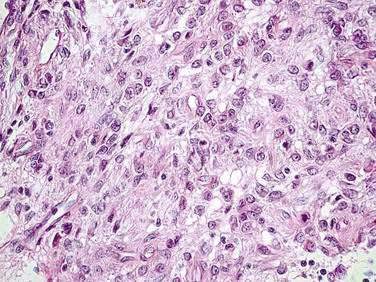
GISTs characteristically exhibit expression of CD117 by immunohistochemical assays (>95% of lesions but, importantly, not 100%, because there are true KIT-negative GIST cases).30 Levels of expression of CD117 (KIT) are generally diffuse and strong in the spindle cell GIST subtype (Fig. 30-1). In contrast, in the epithelioid subtype, CD117 expression is typically focal and weakly positive in a dot-like pattern (Fig. 30-2). As noted, CD34 expression is neither sensitive nor specific for GIST because this antigen can also be noted in desmoid tumors, and approximately 30% to 40% of GIST lesions are negative for CD34.21,26 True leiomyosarcomas express two smooth muscle markers, smooth muscle actin (SMA) and desmin, but fail to express CD117. Schwannomas are usually positive for the neural antigen S100 but are also negative for CD117. Normal mast cells and ICCs in the surrounding stromal tissues serve as ideal positive internal controls because these normal cells strongly express CD117. Activating mutations in the KIT gene were identified in five of six cases of human GISTs originally analyzed by Hirota and colleagues,20 with evidence that the mutations resulted in uncontrolled, ligand-independent activation of the KIT kinase. Genetically engineered cells harboring the mutant overactive KIT proteins were tumorigenic in nude mice, serving as proof of concept that the malignant phenotype was directly induced by the aberrant signaling pathways associated with uncontrolled KIT kinase activation.
The oncogenic potential of mutant, uncontrollably active KIT in the pathogenesis of GISTs in humans has also been supported by the identification of familial syndromes with an autosomal dominant inheritance pattern and an abnormally high incidence of GISTs, usually occurring as multiple foci within any affected individual (see later).21,31,32 Genetic analysis of such kindreds reveals that they harbor germline-activating KIT mutations, similar to the mutations that were first described in sporadic cases of GISTs.
With analyses using optimal tumor specimens and sophisticated technology, it has become clear that KIT mutations can be detected in more than 90% of GIST cells.33–35 Constitutive activation of the KIT kinase enzymatic function has been reported to characterize every GIST sample analyzed by immunoblotting technique, even in cases in which there are no detectable mutations in the KIT gene. The mechanisms whereby nonmutated (wild-type) KIT is maintained in an uncontrollably phosphorylated state are poorly understood and are likely to be fertile ground for future research, with therapeutic potential.
Importantly, the vast majority of GIST cells at initial presentation demonstrate only a single site of mutation in the KIT gene; complex genetic changes in untreated GISTs at initial presentation are vanishingly rare. Gain of function mutations have been identified most commonly in exon 11 of KIT (up to 70% of cases), an exon that encodes the intracellular juxtamembrane domain of the KIT protein. Mutations in the KIT gene locus have also been described in other regions including (in decreasing order of prevalence) exon 9 (the KIT extracellular domain), exon 13 (kinase domain), and exon 17 (kinase domain).33–35 Structural biology studies have revealed the mechanism whereby normal (wild-type) KIT is kept in an autoinhibited conformation until ligand binds; mutational changes in conformation interfere with this autoinhibition and lead to a structural basis for the aberrant activation of the KIT kinase function.36,37
Another key advance in the understanding of GISTs has been the recognition that signaling through other uncontrolled kinases in addition to KIT could drive the neoplastic phenotype of GIST cells. Specifically, it is now recognized that approximately 5% of GIST cells are not through activation and aberrant signaling of the KIT receptor, but rather through mutational activation of the structurally related kinase known as the platelet-derived growth factor receptor-alpha (PDGFRA).38,39
The definitive diagnostic criteria of uncommon CD117-negative lesions that are nevertheless truly GISTs are currently somewhat obscure. GIST lesions can be heterogeneous in the expression of CD117, even within a single mass. It is possible that a needle biopsy could yield cells consistent with a GIST yet be CD117-negative simply by sampling bias alone. However, expert pathologists can also define a rare subset of GISTs (<5% of cases overall) that have no CD117 expression; these are most likely dependent on an alternative kinase such as PDGFRA.38 Molecular analyses of the KIT and PDGFRA genotypes may be useful to define with certainty the group of rare patients with CD117-negative GISTs in the future. Now, it is important to note that the diagnosis of a GIST should be made on the grounds of morphologic, clinicopathologic, and immunohistochemical data, as well as possibly molecular analysis if there is any ambiguity from the other pathologic assessments.
KIT mutations have also been documented in small GISTs (<1 cm in greatest dimension)40; such lesions are most often detected incidentally (e.g., during upper endoscopy for reflux symptoms) and may appear morphologically benign. These findings support the hypothesis that activating mutations in the KIT proto-oncogene represent an early event in the transformation from a normal precursor cell into a GIST lesion. Because lesions in familial GISTs (see later) may not present clinically until the second or third decade of life, or even much later, it is likely that second hits are necessary to attain a more aggressive malignant phenotype. The other key signaling steps that confer a more malignant phenotype to GIST cells remain obscure. However, the unique aspects of the signaling cascades in GISTs are being actively elucidated, and these appear to differ from KIT signaling in hematologic cancers. For example, the STAT5 pathway of leukemic cells is not typically activated in GISTs, whereas STAT1 and STAT3 are activated at a high level.33
BENIGN GASTROINTESTINAL STROMAL TUMORS
The literature before 2000 was somewhat confusing about whether mutational status of KIT could distinguish between so-called benign and malignant GISTs. With the recognition that KIT mutations can be found in even the smallest GIST,40 there is now consensus that KIT genotype alone cannot account for differences between GISTs that may behave in an indolent manner (and which, when small, may be curable by optimal resection alone) versus those that are clearly aggressive and malignant by all functional definitions. It is important to note that a well-differentiated benign cell morphology alone should not provide any reassurance that an individual GIST lesion will pursue a benign clinical course.
Consensus was reached at a meeting held at the National Cancer Institute (NCI) among pathologists with expertise in GISTs. This consensus defined the two most reliable prognostic factors for behavior of a primary GIST as the size of the primary tumor and the number of mitoses, reflecting the proliferative activity of the cells.2 Other factors, such as the specific histologic subtype (epithelioid vs. spindle cell; see Figs. 30-1A and 30-2), the degree of cellular pleomorphism, and patient age may have some contribution to prognosis but are most likely to play a minor role in determining the clinical outcome. Recurrence and survival rates have also been reported to correlate with the location of the primary GIST lesion, with small bowel tumors showing a somewhat worse prognosis. Most important, the consensus panel emphasized that no GIST lesion should ever be considered completely benign. This is because morphologically benign lesions have a finite capacity to recur and metastasize, occasionally several years after initial presentation of the primary disease. Nonetheless, further research is necessary to define the true risks of tiny GIST lesions (<1 cm) because these are far more common than previously appreciated.
DIAGNOSIS
The revolutionary changes in the scientific understanding, diagnostic evaluation, and therapeutic management of GIST patients (discussed later) has led several professional organizations to develop consensus-based (and, whenever possible, evidence-based) clinical practice guidelines. The National Comprehensive Cancer Network (NCCN) has developed extensive publically accessible guidelines to assist clinicians in the management of GIST patients (see the GIST and Soft Tissue Sarcoma Guidelines on www.nccn.org). Also, the European Society of Medical Oncology (ESMO) has published expert-driven clinical practice guidelines.41 Given the rapid progress in this field, these guidelines are reviewed at least annually to ensure that current information is available to assist clinicians in providing the most up-to-date care and accurate information to patients.
DIAGNOSTIC APPROACH TO STROMAL TUMORS BY SITE
Esophageal Tumors
GIST lesions may arise within the esophagus, although this is a rare presentation for larger lesions. Most esophageal GIST lesions are noted incidentally during upper endoscopy performed for some other unrelated symptom or disorder, such as reflux esophagitis. Esophageal GIST lesions may be small (only a few millimeters in size) in this location and may be resected using endoscopic techniques.42 Margins may be involved if a lesion, unsuspected as a GIST and thought to be benign, is simply popped out using an endoscopic procedure. It remains unclear whether watchful waiting with serial endoscopic follow-up is appropriate for any patient with small GIST lesions (<1 cm in maximal dimension).
As noted, histopathology showing putatively benign GIST cells cannot be viewed reassuringly because histology does not perfectly predict the malignant behavior of GISTs. A careful risk-based assessment that takes into account other aspects of the tumor, as well as patient-specific factors (e.g., age, comorbidities, patient preferences), must be performed. It will be important for prospective research to accumulate a larger objective database regarding the outcomes of such patients so that medical decision making can be based on solid evidence. Although a small esophageal GIST lesion is probably a single primary, it is appropriate to take a careful family history and perform computed tomography (CT) scanning of the abdomen and pelvis to ensure that no other lesions are present in the patient’s family, especially if the disease is first detected in a young person. Some series have attempted to distinguish between clinical outcomes of patients with GISTs and those with leiomyosarcomas or other mesenchymal neoplasms, such as leiomyomas.43
Gastric Tumors
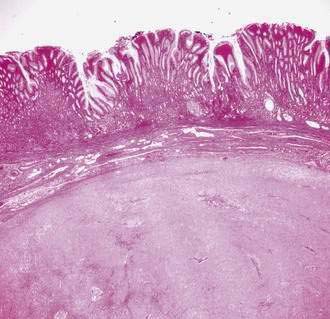
The most common primary site for GISTs is the stomach. Most GIST lesions are submucosal, rather than mucosal, without overlying ulceration (Fig. 30-3). This explains why many GIST masses may only be visualized on endoscopy as a subtle, smooth protrusion with overly normal mucosa.44 Also, this submucosal localization can make diagnostic biopsy through an endoscope difficult. It is not uncommon for superficial biopsies to reveal only normal mucosa, whereas deeper biopsies or histopathology from a definitive resection would show the true underlying GIST cells.
Small Intestinal Tumors
Because complete surgical resection is the treatment of choice for localized GISTs, there is some controversy over whether any preoperative biopsy is necessary or whether that simply represents an extra risk for the patient. This is a challenging subject because other disease entities enter into the differential diagnosis of a large abdominal mass involving the small intestine and mesentery. In general, clinical practice guidelines have suggested that resection may be performed without antecedent biopsy if a GIST is strongly suspected and if surgery can be accomplished without significant risk of morbidity to the patient. If only radical surgery leading to significant functional impairment could remove the lesion, it may be in the patient’s best interest to consider a preoperative biopsy to establish the diagnosis of a GIST. The prognosis of GISTs involving the small intestine is related to the adequacy of resection.6,45 In one series of 50 GIST patients involving the small intestine, 70% could be completely resected, with median overall survival longer than five years for patients with localized or locally advanced disease. Patients who underwent complete resections exhibited a five-year overall survival rate of 42%, while those whose lesions could not be completely resected had a five-year overall survival rate of only 8%.28
IMAGING STUDIES
Endoscopic Ultrasonography
Endoscopic ultrasonography (EUS) is a useful technology for evaluating possible GIST lesions because of their submucosal localization. The paradigm of a GIST lesion visualized by EUS is that of a hypoechoic mass contiguous with the fourth (muscularis propria) or second (muscularis mucosae) layers of the normal gut wall. In one study,46 the features most predictive of so-called benign GI tumors were regular margins, tumor size 3 cm or smaller, and a homogeneous echogenicity pattern. Multivariant analysis identified the presence of cystic spaces and irregular margins as independent predictors of malignant potential. A second study identified tumor size larger than 4 cm, irregular extraluminal borders, echogenic foci larger than 3 mm, and cystic spaces larger than 4 mm as factors that correlated with malignant behavior in GIST.47
Computed Tomography and Magnetic Resonance Imaging
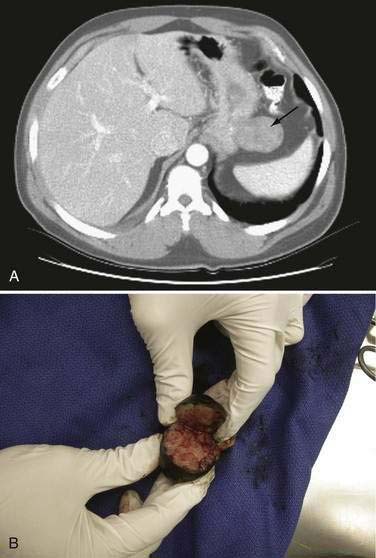
CT is the most effective way to image primary lesions in the stomach because the oral contrast will outline masses and gastric thickening (Fig. 30-4). Differential diagnosis with inadequate gastric distention can be a challenge, especially when monitoring for recurrence following surgery. CT is also essential to stage the extent of disease completely and accurately. For measurable GIST lesions, it is particularly useful to perform CT with noncontrast image acquisition, as well as assess early and late images following the administration of intravenous contrast.
Baseline imaging is critical for GIST, because endoscopic imaging alone may only reveal a small fraction of the underlying tumor. Additionally, imaging patterns (particularly tumor density on CT imaging) can be interpreted qualitatively to assess the impact of targeted therapy using metrics other than tumor size. Tumors that become more hypodense on CT imaging have a more favorable antineoplastic response to targeted therapies, such as the kinase inhibitors imatinib or sunitinib.48
Positron Emission Tomography
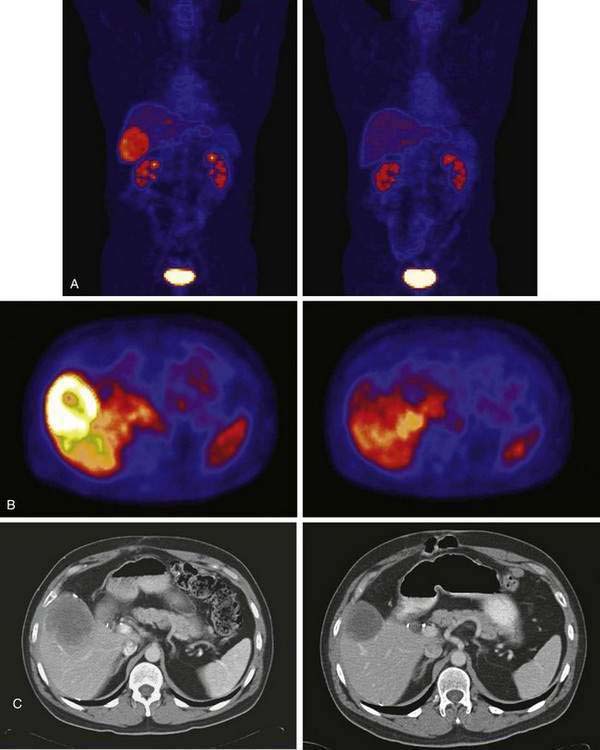
One of the most impressive aspects of GIST diagnostic imaging is the use of 18F-fluorodeoxyglucose (18F-FDG)–positron emission tomography (PET) scans to add complementary information to that obtained by conventional anatomic imaging (Fig. 30-5). Although CT or MRI scanning can assess the size of GIST lesions accurately, the functional imaging of GIST with 18F-FDG–PET can provide useful additional information that can assist clinicians in the management of GIST patients. The actual mechanisms responsible for the high-level avidity of GISTs for the 18F-FDG tracer used most commonly in PET imaging are not yet known; however, it is likely that there is a direct connection between signaling through the overactive KIT RTK and glucose transport proteins. In this way, one could explain the rapid changes in PET imaging associated with inhibition of KIT signaling by pharmacologic means.49,50 Large GIST lesions can demonstrate centers with predominantly cystic or low attenuation characteristics noted on CT or MRI scans. It is clear by 18F-FDG–PET scans that the internal mass of large GIST lesions can often be viewed as metabolically quiescent, likely because of the endogenous necrosis of large lesions in their central portions. Even though GIST lesions can be vascular, the internal portion can nonetheless represent a confluent mass of necrotic material, with the more viable aspects of the GIST pushing out toward the edges of the lesion. Much of the added value of 18F-FDG–PET imaging in serial imaging can also be obtained by qualitative assessment of tumor density obtained via CT imaging.48 However, occasionally, metastatic GIST lesions in the omentum can be subtle and easy to overlook on CT scans because small lesions could blend into the folds of the bowel walls and be difficult for even the most experienced radiologist to detect. 18F-FDG–PET imaging can detect lesions approximately 1 cm or larger in size without difficulty because neither the normal bowel nor omentum will take up the 18F-FDG tracer with excess avidity.
DIFFERENTIAL DIAGNOSIS
GIST was originally described as a monomorphic spindle cell neoplasm. However, it is now clear that GIST can exhibit a wide variety of histologic appearances, ranging from an epithelioid form with large round cells (see Fig. 30-2) to the spindle cell form (see Fig. 30-1), and even a subset of GIST lesions with mixed histology. The spindle cell GIST variant is far more common, representing approximately 70% of cases. The epithelioid, or round cell, pattern represents most of the remaining 30% and may contain an admixture of spindle cells. The epithelioid subset generally was previously diagnosed as leiomyoblastomas, although some may have been mistaken for poorly differentiated carcinomas. The differential diagnosis of GI tract neoplasms that appear to be mesenchymal in origin includes GISTs (80% of the time), but there are definitely true smooth muscle neoplasms of the GI tract, including true leiomyomas and leiomyosarcomas (approximately 15%). Schwannomas account for the remaining 5%. Therefore, the differential diagnosis is complex and requires expert pathology review, as well as adequate and appropriately processed and fixed diagnostic tissues.
As noted, expression of KIT is not limited to GIST cells. Normal ICCs and mast cells express CD117 and depend on KIT for normal growth and development. A relatively limited number of other tumors may also express immunohistochemically detectable CD117. These include certain subsets of soft tissue sarcomas, including Ewing’s sarcoma and angiosarcoma, as well as other neoplasms, such as occasional small cell lung cancers, melanomas, desmoid tumors, seminomas, ovarian carcinomas, mastocytomas, neuroblastomas, adenoid cystic carcinomas, and rare subsets of lymphoma and acute myeloid leukemia.22,23,51,52 It is also relevant to note that expression of the CD117 antigen does not imply the activation of the KIT target, nor does it correlate necessarily with any KIT gene mutation. The same CD117 antigen is expressed by cells harboring normal (wild-type) KIT as those that have activating KIT mutations. Also, expression of KIT protein does not necessarily mean that the protein is involved in the pathogenesis of that specific cancer. In all these regards, GIST was a special example of a disease in which expression correlates universally with kinase activation, and this activation is now validated as being etiologically relevant to the neoplastic behavior of GIST cells.
TREATMENT
ADVANCED GASTROINTESTINAL STROMAL TUMORS: METASTATIC, SURGICALLY UNRESECTABLE, OR RECURRENT DISEASE
Ineffectiveness of Systemic and Locoregional Conventional Chemotherapy
There was universal consensus that treatment of advanced disease represented a pressing unmet medical need before the advent of molecularly targeted therapy. Efforts of medical oncologists to treat GISTs with conventional cytotoxic chemotherapy were universally futile. The rates of benefit or objective antitumor response to various chemotherapy agents for patients with GIST or abdominal leiomyosarcomas were routinely reported to be 0% to 4%.49,53 Some investigators attempted to improve on the dismal results with chemotherapy by administering the drugs via an intraperitoneal route.54 However, because GISTs rarely remain confined to the peritoneal surfaces with the hematogenous dissemination of metastases to the liver and other intra-abdominal locations, and because most of the life-threatening complications of GIST arise from hepatic involvement or from bulk disease affecting the omentum, this intraperitoneal approach was not particularly promising. On the basis of these disappointing results, conventional cytotoxic chemotherapy has generally been regarded as useless for the treatment of patients with GIST.
The mechanisms that might explain the high levels of resistance to chemotherapy exhibited by GIST may result, in part, by the expression of increased levels of P-glycoprotein (the product of the MDR-1 gene) and the multidrug resistance protein in GISTs and other intra-abdominal sarcomas. In one study evaluating the differences in outcome between GISTs and leiomyosarcomas, significantly higher levels of expression of P-glycoprotein (38% vs. 13%) and multidrug resistance protein-1 (35% vs. 13%) were demonstrated in the GIST cells.55 It has been postulated that these cellular efflux pumps may prevent chemotherapy from reaching effective intracellular concentrations in the target GIST cells.
There are some uncontrolled data regarding the potential to control metastatic GIST for a limited time by locoregional techniques, such as hepatic resection, hepatic artery embolization, or chemoembolization. Although a subset of patients with metastatic GISTs involving the liver have demonstrated antitumor responses and a somewhat limited progression-free survival following chemoembolization, the benefits are generally measured in months rather than years; this has not been viewed as a particularly promising strategy for the management of most GIST patients.56,57
Clearly, for patients with metastatic or unresectable GISTs, the prognosis was dismal before the advent of molecularly targeted therapy. For patients with metastatic or recurrent GISTs or GI sarcomas (most of which were likely to have been a true GIST), most studies prior to the introduction of targeted therapy with kinase inhibition had documented poor survival rates, with fatal outcomes from disease progression generally occurring within two years from the date of first recurrence or metastasis.6,53,58
Imatinib Mesylate: First-Generation Selective Tyrosine Kinase Inhibitor Targeting KIT
The initial concept for this molecularly targeted approach came from the collaborative studies of researchers at Ciba-Geigy (now known as Novartis). These studies linked academia and the biopharmaceutical industry in efforts to develop small molecules for inhibition of tyrosine kinases such as the PDGF receptor (useful potentially to block restenosis of coronary stents) and the constitutively active kinase function of the BCR-ABL oncoprotein, which is a target critical to the pathogenesis of chronic myeloid leukemia (CML). A small molecule in the 2-phenylaminopyrimidine class was identified by Druker and colleagues at Ciba-Geigy, with potent inhibitory activity for ABL and the dysregulated BCR-ABL in vitro.59 Additional screening studies from the laboratories of Druker and Buchdunger and associates59–61 demonstrated that this agent, signal transduction inhibitor-571 (STI-571), subsequently called imatinib mesylate (now known by the commercial name of Gleevec in the United States and Glivec elsewhere), could also potently inhibit the tyrosine kinase activity of KIT and platelet-derived growth factor receptor (PDGFR). Subsequent studies performed in a human mast cell leukemia cell line that harbored a KIT mutation similar to the mutations noted in GISTs documented that imatinib could inhibit both mutant and wild-type KIT protein.62 Laboratory experiments testing imatinib in human GIST cell lines with defined activating mutations of KIT have revealed dramatic evidence of anti-GIST activity from this agent. The addition of imatinib to cultured human GIST cells rapidly blocks the constitutive activation of KIT, arrests cell proliferation, and induces apoptosis in the tumor cells.63 By all criteria, therefore, the clinical development of imatinib has been promising as a treatment of GIST to target the fundamental molecular pathogenesis of this disease.
The collaborative worldwide clinical development of imatinib as a molecularly targeted therapy of GIST proceeded at a dramatic pace since the earliest demonstration of the unprecedented activity of this agent (Table 30-1). The first clinical experience with imatinib in the treatment of GIST began in March 2000, with a single-patient pilot study of a woman in Helsinki, Finland, who had far advanced, heavily pretreated, and widely metastatic GIST. The case history of this patient documents the rapid response and sustained clinical benefit from imatinib dosing that this patient enjoyed for approximately three years.64 However, resistance to imatinib (see later) ultimately developed and the patient succumbed to metastatic GIST refractory to selective kinase inhibition.
Table 30-1 Clinical Studies of Imatinib Mesylate in Patients with Metastatic or Unresectable Gastrointestinal Stromal Tumors (GISTs)
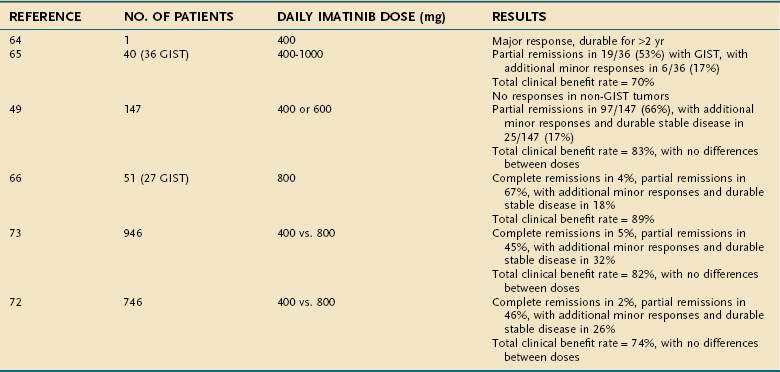
Nonetheless, on the basis of the dramatic and durable benefits in this patient, as well as the striking scientific rationale and strong preclinical data, other studies testing imatinib for GISTs were started. A multicenter United States–Finland collaborative study randomized patients with metastatic GISTs between two dose levels of the drug (400 or 600 mg of imatinib administered orally each day continuously, as long as the disease was stable or responding to therapy).49 The United States–Finland trial rapidly accrued 147 patients with metastatic GIST. Almost concurrently, a dose-finding study was also begun in Europe under the auspices of the European Organization for the Research and Treatment of Cancer (EORTC) Sarcoma Group to test imatinib for GISTs, as well as other forms of sarcomas.65 The maximal tolerated dose of imatinib identified in the EORTC dose-ranging phase I trial was judged to be 800 mg/day (given as 400 mg twice daily); at the higher dose level of 1000 mg daily (given as 500 mg twice daily), unacceptably severe, dose-limiting toxicities, such as nausea, vomiting, and severe edema, were reported.
The results from these two trials in the United States and Europe confirmed the exceptional activity of imatinib in controlling metastatic GIST, inducing objective responses in most patients, providing control of symptoms, and prolonging the survival of these patients in comparison with historical series. The results from these wholly independent trials were remarkably concordant (see Table 30-1), further confirming the dramatic effectiveness of imatinib in the treatment of metastatic GISTs. Most patients with advanced GISTs treated with imatinib exhibited objective responses (almost all partial responses), and an additional subset of patients experienced objectively stable disease associated with imatinib dosing. The median time to objective response was more than three months, although some patients experienced dramatic disease regressions within one week after starting imatinib oral dosing. There were no significant differences in response rates or duration of disease control between the 400 and 600 mg daily dose levels of imatinib in the United States–Finland trial, although the study was relatively underpowered to detect any differences.49 Imatinib was well tolerated overall in both studies. On the basis of these trials, the U.S. Food and Drug Administration (FDA) approved the use of imatinib for the treatment of metastatic or unresectable GIST in early 2002. Approval in Europe and the rest of the world followed quickly thereafter.
In a subsequent trial, the EORTC group went on to expand its exploration of imatinib in GISTs and other forms of sarcomas. In this trial, the high levels of antitumor activity against GISTs were again confirmed, whereas there was no demonstrable benefit for patients with other forms of soft tissue sarcomas.66 This work supports the hypothesis that specific molecular targeting of a signaling pathway crucial to GIST cells can explain the extraordinary activity of imatinib in this disease. Without such a target to inhibit (as in sarcomas other than GISTs), imatinib treatment does not have major anticancer activity. In particular, it is relevant to note that the activation of the PDGFR signaling pathway in dermatofibrosarcoma protuberans (DFSP) has been targeted successfully using imatinib as well, with evidence of clinical benefit for DFSP patients.67–69
With reference to the molecular targeting of kinase inhibition in GISTs, correlative molecular studies performed in conjunction with the United States–Finland trial have documented differences in the activity of imatinib on the basis of the genotype of the GIST lesions treated. Specifically, patients whose GISTs harbored KIT mutations in exon 11 (the most common molecular subtype) had higher rates of objective response and more durable disease control over time with continuous dosing of imatinib than patients whose disease had exon 9 KIT mutations or no detectable KIT mutations at all.70 Imatinib-sensitive PDGFRA mutations can explain the clinical benefit seen in certain GIST patients whose disease does not harbor any KIT mutations.70
The most worrisome adverse events observed in the imatinib treatment of patients with advanced GIST have included hemorrhages from abdominal or GI sites in approximately 5% of GIST patients. These hemorrhagic events were likely related to bleeding from bulky tumor masses, which may have been induced by the potent and rapid antitumor effects of imatinib. There were no deaths on the initial trial directly attributable to the study drug.49
Another fascinating aspect of this work was the finding that imatinib could rapidly and dramatically affect the 18F-FDG tracer uptake by tumors as imaged by functional imaging with PET scans (see Fig. 30-5). The decreases in tumor avidity for 18F-FDG on PET scanning could be detected as early as 24 hours following a single dose of imatinib. PET imaging could detect the biological activity of imatinib far earlier than changes in anatomic measures on CT scanning. The findings of PET scans were also highly reliable, correlating both with beneficial response to imatinib and documenting progressive disease in the small subset of patients with primary resistance to imatinib. These data indicate that functional imaging of GIST with 18F-FDG–PET scanning represents a useful diagnostic modality for early-response assessment with imatinib therapy.50,71 This paradigm demonstrates that PET imaging may be a useful tool for future drug development efforts because the signal of drug activity can be detected rapidly and reliably following effective target inhibition.
To explore more definitively whether there is a clinically significant dose response above the lowest recommended daily dose of 400 mg, two large phase III randomized studies have been conducted. In both studies, patients with advanced metastatic and/or unresectable GISTs were randomized to receive imatinib at 400 mg or 800 mg daily. Patients were allowed to cross over from the lower dose to the higher dose if progression of disease occurred at the lower dose. These studies were considered to be adequately powered to determine whether this twofold difference in imatinib dose would translate into significant clinical benefits as measured by improved response rates, duration of disease control, or survival for patients with advanced unresectable or metastatic GISTs. The most recent updates on these studies have been published,72,73 and together these studies have treated and studied almost 1700 GIST patients worldwide. Importantly, because these trials shared the same fundamental designs at inception, a meta-analysis has now been conducted based on a pooled analysis of all primary data. Although there was no survival difference documented in either trial between these two dose levels, there was an interesting, although subtle, difference observed in terms of duration of disease control (see Table 30-1). Although the North American Sarcoma Intergroup trial demonstrated only a favorable trend in the duration of disease control associated with the higher dose of imatinib, the European-led study noted a modest but statistically significant benefit in favor of the higher dose arm for progression-free survival; however, neither showed any difference in overall survival. In the combined meta-analysis, it is clear that the benefit of the higher dose imatinib was limited solely to the subset of GIST patients whose tumors harbored mutations in KIT exon 9, encoding a mutation in the extracellular domain that promotes dimerization of the kinase.74 This has led to the recommendation by some experts to use the higher dose of imatinib as first-line therapy for advanced GIST in patients whose tumors have documented exon 9 KIT mutations. Such modest benefit must, however, be balanced by the additional toxicities, because the higher dose of imatinib was associated with a greater incidence of adverse effects and led to a greater number of dose reductions for toxicity in these large studies.
The optimal duration of imatinib for patients with metastatic GIST has been defined as lifelong therapy on the basis of current evidence.75 A randomized study in France that discontinued imatinib therapy in GIST patients once a maximal response was reached has found that the disease rapidly recurs following cessation of imatinib dosing.76
For optimal management of metastatic disease, medical oncologists, surgeons, radiologists, and nuclear medicine imaging experts must all collaborate closely to determine the best course of action for any given patient. This important message has been emphasized in the Task Force Report on GIST Clinical Practice Guidelines of the NCCN.75 For example, disease that is initially judged as unresectable may become amenable to surgical excision following a major response induced by imatinib therapy. Most centers recommend surgical resection of such patients because it is feared that residual GIST lesions likely harbor cells with kinase inhibitor resistance mutations, which evolve into clinical appearance of GIST resistance to imatinib and subsequent progression of treatment-refractory disease.
Resistance to imatinib may be primary and manifest as rapid progression of disease despite initial imatinib dosing, although this appears in far less than 20% of patients (see Table 30-1). Alternatively, clonal evolution of GIST may occur over time, with the emergence of resistant disease after more than one or two years of durable response and disease control by imatinib. Several mechanisms of resistance to imatinib in GIST have been described,77 and these are overall similar to the resistance mechanisms that have been described in imatinib-resistant CML.78 It is unclear what role should ideally be played by other modalities (e.g., surgical resection, radiofrequency ablation [RFA], chemoembolization, or other locoregional approaches) for managing metastatic GIST once imatinib has achieved the optimal effect or following the development of resistance to imatinib with oligoclonal or widespread systemic progression. Certainly, many metastatic GIST lesions may remain controlled on imatinib, although limited clonal progression appears as the first sign of resistance to imatinib.79,80 It may be feasible in such patients to resect the resistant clonal growth while maintaining control over most of the disease by continuation of imatinib dosing. In fact, several nonrandomized surgical series have suggested benefit to resection of resistant clones for patients whose GISTs have not developed widespread systemic resistance to kinase inhibitor therapy.81,82 The value of such surgical strategies will be tested in future randomized trials to assess whether early surgical intervention will improve disease control compared with kinase inhibitor therapy alone.
Gastrointestinal Stromal Tumors Resistant to Imatinib: Development of Sunitinib
To address resistance, other kinase inhibitors with varying target specificities have been developed to control GISTs that have become refractory to imatinib. Sunitinib (Sutent) is the first of these that has been FDA-approved for GIST, as well as by worldwide regulatory authorities. Sunitinib inhibits multiple receptor tyrosine kinases including KIT; PDGFRs (alpha and beta); vascular endothelial growth factor receptors (VEGFRs)-1, -2, and -3; FMS-like tyrosine kinase-3 receptor (FLT3); macrophage colony-stimulating factor receptor (CSF-1R); and glial cell–line derived neurotrophic factor receptor (RET, rearranged during transfection). Sunitinib demonstrated important antineoplastic activity in the first phase I clinical trial,83 and subsequently this drug was proven to have potent activity in a definitive international prospective randomized, placebo-controlled clinical trial.84 In molecular analysis of GIST samples from the initial phase I and II trial, primary and secondary mutations in KIT and PDGFRA were shown to affect the treatment outcomes with sunitinib in patients with imatinib-resistant GISTs.85 Sunitinib was effective for treatment of GISTs of all KIT and PDGFRA genotypes prior to imatinib exposure, and sunitinib showed particular efficacy for those with a wild-type genotype, a primary KIT exon 9 mutation, or secondary KIT mutations in exon 13 or 14. Structural biology analyses of these mutated kinases have provided insight as to how sunitinib can inhibit the kinase function when mutations encode certain amino acid changes that induce steric hindrance to the binding and inhibition of imatinib.86 Importantly, the incidence of secondary mutations (especially exon 17 KIT mutations, which confer resistance to imatinib and sunitinib) was higher in patients whose GIST was initially sensitive to imatinib.
In the phase III trial, which led to the regulatory approval of sunitinib, 312 patients with metastatic or surgically unresectable GIST following failure of imatinib caused by resistance or intolerance were randomized to receive sunitinib, 50 mg/day (n = 207), or placebo (n = 105) on a dosing schedule with four weeks of drug dosing followed by a two-week period off drug.84 The primary endpoint of the study was disease control as assessed by time to progression (TTP). The trial was unblinded early when a planned interim efficacy analysis showed that sunitinib was associated with a significant improvement in median TTP of more than fourfold compared with placebo. The median progression-free survival (PFS) for patients receiving sunitinib was significantly greater than for those receiving placebo (sunitinib, 24.1 weeks. vs. placebo, 6.0 weeks). In the initial analysis, sunitinib also significantly improved overall survival (OS; hazard ratio, 0.49; 95% confidence interval [CI], 0.29 to 0.83); at the time of the interim analysis, the median OS had not been reached in the group receiving sunitinib. Although improved disease control was demonstrated with sunitinib, the objective rates of response were very low, even though significantly more patients treated with sunitinib had an objective response compared with placebo (6.8% vs. 0%). Sunitinib was reasonably well tolerated, with the most common adverse events being fatigue, diarrhea, abdominal pain, and nausea. Patients on sunitinib also experienced a greater incidence of skin abnormalities, including palmar-plantar erythrodysesthesia (hand-foot syndrome), oral cavity mucosal irritation, and, with longer exposure, a relatively high incidence of hypothyroidism.87 Certain patients also exhibited cardiac dysfunction, which was in general reversible with temporary discontinuation of sunitinib dosing.88
Thus, sunitinib appears to have unique activity for the management of imatinib-resistant GIST as second-line therapy, but the powerful inhibition of a number of other kinase signaling pathways can induce other unpleasant or medically relevant adverse effects that require close monitoring and possibly adjustment of dosing. Additional studies are evaluating a lower continuous daily dose regimen of sunitinib, which appears to have similar activity with perhaps somewhat improved tolerability.89 Clinical trials are also evaluating several other kinase inhibitors, including the second-generation selective tyrosine kinase inhibitor nilotinib, the multitargeted kinase inhibitor sorafenib, and the combination of imatinib plus RAD001, an inhibitor of the serine-threonine kinase mTOR. In each case, a certain level of clinical efficacy has been demonstrated in limited phase II trials, and larger trials are warranted and necessary to define more precisely whether specific molecular subtypes of GIST might benefit from these treatment strategies. Given the evolution of several different kinase mutations in individual patients with imatinib- and sunitinib-resistant GISTs,90,91 new strategies are needed to control the polyclonal resistance that emerges following failure of first-line therapy. Because mutated kinase proteins appear to maintain dependence on the protective chaperone function of the heat shock protein 90 (Hsp90), inhibitors of Hsp90 such as IPI-504 are being tested in clinical trials as a consequence of promising data from the laboratory.92 The early phase I-II trial experience with the Hsp90 inhibitor IPI-504 has been encouraging,93 and these data have now been translated into other clinical trials to test the value of this novel molecular target for GISTs.
PRIMARY LOCALIZED DISEASE
Definitive expert surgery remains the mainstay of treatment for patients with primary localized GISTs (early-stage GISTs). However, with the advent of highly effective drug therapy, current guidelines recommend that surgical resection of GISTs be undertaken as the first intervention only if there is an acceptably low risk of functional deficit or morbidity from the surgery. If a large GIST were to be detected, it might be judicious to consider such a lesion unresectable without causing unacceptable risk for morbidity. In this case, preoperative administration of imatinib should be considered, because trials have shown that such neoadjuvant administration can be effective at diminishing the size of tumors and thereby facilitating effective surgical intervention.94 This is the specific clinical situation in which early assessment of therapeutic response by 18F-FDG–PET scanning could prove valuable to confirm that the patient’s disease is exhibiting the desired response to imatinib. This should minimize the risk of disease progression that otherwise might put the patient at risk for further growth and invasion into surrounding vital structures. Following maximal response (usually occurring within three to six months), definitive surgery could be performed.
The surgical approach to GIST resection of primary disease must take into account the specific growth and behavior characteristics of this disease. GISTs rarely involve the locoregional lymph nodes, and so extensive lymph node exploration or resection is rarely indicated. GIST lesions are highly vascularized and often exhibit a fragile pseudocapsule; therefore, surgeons should be careful to minimize the risk of tumor rupture, which might subsequently increase the risk of peritoneal dissemination.95 The margins of resection from the tumor specimen should be carefully oriented and examined, and biopsy samples from several different areas of the tumor should be evaluated by the surgical pathologist.
The natural history of early-stage primary GIST has been examined in studies from single-institution referral centers. These are certainly prone to selection bias, and it is clear in this evolving field that many early-stage GIST patients have likely been managed by physicians of multiple specialties, including gastroenterology and general surgery. However, one of the larger GIST series from a referral cancer center evaluated 200 patients followed prospectively at the Memorial Sloan-Kettering Cancer Center6; 80 of these patients (40%) had primary disease managed with complete surgical resection. This latter group, with primary resected GISTs, demonstrated five-year disease-specific survival rates of only 54%, supporting the fact that GISTs, as seen at such an academic referral center, can exhibit a high risk for recurrence and ultimately prove to be a life-threatening disease. On multivariate analysis, large tumor size (>10 cm) was the only factor that reduced disease-specific survival. In an earlier study of 191 so-called GI leiomyosarcomas (of which a sizable proportion were likely to have been true GIST), investigators at the M.D. Anderson Cancer Center reported that smaller tumor size (<5 cm), complete surgical resection without tumor rupture, and low histologic grade of tumor were significant favorable prognostic factors. The propensity of GISTs to recur was also confirmed by these data because only 10% of these patients were disease-free on long-term follow-up.95
Anatomic location of the primary tumor also appears to be an important prognostic factor for primary localized GIST, in addition to tumor size and the proliferation rate of the tumor (as measured by the number of tumor cells in mitosis).96 Assessing the risk of recurrence will be exceedingly important to advise patients and to make reasonable judgments about the potential value of adjuvant systemic therapy with a kinase inhibitor.
Adjuvant Therapy for Patients with Early-Stage Gastrointestinal Stromal Tumors
It is now clear that the administration of imatinib in the postresection (adjuvant) setting has the potential to delay tumor recurrence, especially for patients who present with very large tumors and who are likely at very high risk of disease recurrence and metastatic spread. The activity of adjuvant imatinib in GIST patients who are at moderate to high risk of recurrence has been investigated in two large multicenter trials conducted by several groups, including the American College of Surgeons Oncology Group (ACOSOG) and several European cooperative oncology groups. In the initial adjuvant trials of ACOSOG, imatinib (or placebo) was administered for an arbitrary one year’s duration. In the randomized trial Z9001, administration of imatinib following resection of primary limited GIST significantly prolonged recurrence-free survival compared with placebo (98% vs. 83% free of recurrence at one year; hazard ratio 0.35), although no overall survival benefit has yet been noted because of the very short follow-up period.97 It is certainly possible that a longer duration of adjuvant therapy might lead to even better outcomes, and this is being tested in the other European trials. Given the potential toxicities and costs of imatinib, it is important to decide what constitutes sufficient clinical activity to justify the universal administration of imatinib in the adjuvant setting. Although imatinib has powerful activity in prolonging the survival of patients with recurrent and metastatic GISTs, it is possible that earlier administration of this systemic therapy in the adjuvant setting might not change the natural history of this disease sufficiently to affect overall survival. However, it is possible that with longer duration of dosing in patients at highest risk of recurrence, such trials may document important clinical benefits. These trials in progress will generate crucial evidence on which to base optimal medical practice in the future. Nonetheless, for the moment, the adjuvant activity of imatinib seems clear. It merits a thoughtful discussion of possible risks and benefits in all patients with resected GISTs who are at moderate to high risk of disease recurrence by current risk classification systems.
SPECIAL CONSIDERATIONS
FAMILIAL DISEASE
GIST rarely can be associated with familial inheritance patterns in which several members of a kindred have the disease.20,98–100 In several of these families, KIT mutations have been reported. Additional characteristics of affected family members include cutaneous lesions such as hyperpigmentation or skin lesions that resemble the clinical appearance of urticaria pigmentosa. These skin pigmentation abnormalities are no doubt caused by the effect of the mutationally activated KIT kinase function on melanocyte growth and development. The mechanisms whereby such pigmentation disorders remain focal, rather than disseminated, may provide clues as to why GIST lesions may take decades to appear in these rare familial cases. These cases have tended to be autosomal dominant germline mutations, and the GIST lesions seen in affected members tend to be multifocal. Familial GIST has also been associated with germline mutations in certain subunits of the succinate dehydrogenase genes SDHB, SDHC, and SDHD.101
PEDIATRIC PATIENTS
Although very rare, GIST can affect pediatric patients. The disease seems to have a very different molecular profile, however, because KIT mutations are rarely, if ever, demonstrable in GIST patients younger than 18 years.102–104 The presence of constitutive KIT signaling is nonetheless a common feature of pediatric GIST, although the mechanism of KIT activation in the absence of mutations remains obscure. Wild-type GIST, especially in pediatric patients, appears to exhibit high levels of the insulin-like growth factor-1 receptor, which may provide another therapeutic target for intervention using investigational new inhibitory monoclonal antibodies or small molecules.105
RELATIONSHIP TO OTHER GENETIC SYNDROMES PREDISPOSING TO NEOPLASMS
Several other syndromes that predispose to the development of neoplasms have been described in association with GISTs. One of the more widely known is the Carney triad, which includes GISTs (often multifocal) in addition to pulmonary chondromas and extra-adrenal paragangliomas.106 A variant of this syndrome, known eponymously as the Carney-Stratakis syndrome, has been described, with only GISTs and familial paragangliomas being present.107 Additionally, a linkage between neurofibromatosis type I (NF1) and an increased incidence of GISTs has been widely noted.108,109 Molecular analysis of GIST lesions arising from patients with NF1 disease has documented that these GISTs do not harbor detectable mutations in the KIT gene. It is unclear whether GISTs that arise in the setting of a genetic predisposition syndrome have the same response to imatinib as sporadically occurring GISTs.
Blanke CD, Rankin C, Demetri GD, et al. Phase III randomized intergroup trial assessing imatinib mesylate at two dose levels in patients with unresectable or metastatic gastrointestinal stromal tumor expressing the KIT receptor tyrosine kinase: S0033. J Clin Oncol. 2008;26:626-32. (Ref 72.)
Blay JY, Le Cesne A, Ray-Coquard I, et al. Prospective multicentric randomized phase III study of imatinib in patients with advanced gastrointestinal stromal tumors comparing interruption versus continuation of treatment beyond 1 year: The French Sarcoma Group. J Clin Oncol. 2007;25:1107-13. (Ref 76.)
Choi H, Charnsangavej C, Faria SC, et al. Correlation of computed tomography and positron emission tomography in patients with metastatic gastrointestinal stromal tumor treated at a single institution with imatinib mesylate: proposal of new computed tomography response criteria. J Clin Oncol. 2007;25:1753-9. (Ref 48.)
Debiec-Rychter M, Sciot R, LeCesne A, et al. KIT mutations and dose selection for imatinib in patients with advanced gastrointestinal stromal tumours. Eur J Cancer. 2006;42:1093-103. (Ref 74.)
DeMatteo RP, Maki RG, Singer S, et al. Results of tyrosine kinase inhibitor therapy followed by surgical resection for metastatic gastrointestinal stromal tumor. Ann Surg. 2007;245:347-52. (Ref 82.)
Demetri GD, Benjamin RS, Blanke CD, et al. NCCN Task Force report: Management of patients with gastrointestinal stromal tumor (GIST)—update of the NCCN clinical practice guidelines. J Natl Compr Canc Netw. 2007;5(Suppl 2):S1-29. (Ref 75.)
Eisenberg BL, Harris J, Blanke CD, et al. Phase II trial of neoadjuvant/adjuvant imatinib mesylate (IM) for advanced primary and metastatic/recurrent operable gastrointestinal stromal tumor (GIST): Early results of RTOG 0132/ACRIN 6665. J Surg Oncol. 2009;99:42-7. (Ref 94.)
ESMO Guidelines Working GroupBlay JY, Le Cesne A. Gastrointestinal stromal tumors: ESMO clinical recommendations for diagnosis, treatment and follow-up. Ann Oncol. 2007;18(Suppl 2):ii27-9. (Ref 41.)
Heinrich MC, Maki RG, Corless CL, et al. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumor. J Clin Oncol. 2008;26:5352-9. (Ref 85.)
Liegl B, Kepten I, Le C, et al. Heterogeneity of kinase inhibitor resistance mechanisms in GIST. J Pathol. 2008;216:64-74. (Ref 91.)
McWhinney SR, Pasini B, Stratakis CA, International Carney Triad and Carney-Stratakis Syndrome Consortium. Familial gastrointestinal stromal tumors and germ-line mutations. N Engl J Med. 2007;357:1054-6. (Ref 101.)
Miettinen M, Lasota J. Gastrointestinal stromal tumors: pathology and prognosis at different sites. Semin Diagn Pathol. 2006;23:70-83. (Ref 96.)
Tarn C, Rink L, Merkel E, et al. Insulin-like growth factor 1 receptor is a potential therapeutic target for gastrointestinal stromal tumors. Proc Natl Acad Sci U S A. 2008;105:8387-92. (Ref 105.)
Verweij J, Casali PG, Zalcberg J, et al. Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: Randomised trial. Lancet. 2004;364:1127-34. (Ref 73.)
Wardelmann E, Merkelbach-Bruse S, Pauls K, et al. Polyclonal evolution of multiple secondary KIT mutations in gastrointestinal stromal tumors under treatment with imatinib mesylate. Clin Cancer Res. 2006;12:1743-9. (Ref 90.)
1. Kindblom L-G, Meis-Kindblom J, Bümming P, et al. Incidence, prevalence, phenotype and biologic spectrum of gastrointestinal stromal cell tumors (GIST)—a population-based study of 600 cases [abstract]. Ann Oncol. 2002;13(Suppl 5):157.
2. Fletcher CD, Berman JJ, Corless C, et al. Diagnosis of gastrointestinal stromal tumors: A consensus approach. Hum Pathol. 2002;33:459-65.
3. Miettinen M, El-Rifai W, Sobin LH, Lasota J. Evaluation of malignancy and prognosis of gastrointestinal stromal tumors: A review. Hum Pathol. 2002;33:478-83.
4. Kawanowa K, Sakuma Y, Sakurai S, et al. High incidence of microscopic gastrointestinal stromal tumors in the stomach. Hum Pathol. 2006;37:1527-35.
5. Agaimy A, Wünsch PH, Hofstaedter F, et al. Minute gastric sclerosing stromal tumors (GIST tumorlets) are common in adults and frequently show c-KIT mutations. Am J Surg Pathol. 2007;31:113-20.
6. DeMatteo RP, Lewis JJ, Leung D, et al. Two hundred gastrointestinal stromal tumors: Recurrence patterns and prognostic factors for survival. Ann Surg. 2000;231:51-8.
7. Miettinen M, Lasota J. Gastrointestinal stromal tumors—definition, clinical, histological, immunohistochemical, and molecular genetic features and differential diagnosis. Virchows Arch. 2001;438:1-12.
8. Emory TS, Sobin LH, Lukes L, et al. Prognosis of gastrointestinal smooth-muscle (stromal) tumors: Dependence on anatomic site. Am J Surg Pathol. 1999;23:82-7.
9. Mazur MT, Clark HB. Gastric stromal tumors. Reappraisal of histogenesis. Am J Surg Pathol. 1983;7:507-19.
10. Newman PL, Wadden C, Fletcher CD. Gastrointestinal stromal tumours: Correlation of immunophenotype with clinicopathological features. J Pathol. 1991;164:107-17.
11. Hurlimann J, Gardiol D. Gastrointestinal stromal tumours: An immunohistochemical study of 165 cases. Histopathology. 1991;19:311-20.
12. Pike AM, Lloyd RV, Appelman HD. Cell markers in gastrointestinal stromal tumors. Hum Pathol. 1988;19:830-4.
13. Herrera GA, Pinto de Moraes H, Grizzle WE, et al. Malignant small bowel neoplasm of enteric plexus derivation (plexosarcoma). Light and electron microscopic study confirming the origin of the neoplasm. Dig Dis Sci. 1984;29:275-84.
14. Walker P, Dvorak AM. Gastrointestinal autonomic nerve (GAN) tumor. Ultrastructural evidence for a newly recognized entity. Arch Pathol Lab Med. 1986;110:309-16.
15. Golden T, Stout AP. Smooth muscle tumors of the gastrointestinal tract and retroperitoneal tissues. Gynecol Obstet. 1941;73:784.
16. Stout AP. Bizarre smooth muscle tumors of the stomach. Cancer. 1962;15:400-9.
17. Miettinen M, Virolainen M, Maarit Sarlomo R. Gastrointestinal stromal tumors—value of CD34 antigen in their identification and separation from true leiomyomas and schwannomas. Am J Surg Pathol. 1995;19:207-16.
18. Miettinen M, Monihan JM, Sarlomo-Rikala M, et al. Gastrointestinal stromal tumors/smooth muscle tumors (GISTs) primary in the omentum and mesentery: Clinicopathologic and immunohistochemical study of 26 cases. Am J Surg Pathol. 1999;23:1109-18.
19. Perez-Atayde AR, Shamberger RC, Kozakewich HW. Neuroectodermal differentiation of the gastrointestinal tumors in the Carney triad. An ultrastructural and immunohistochemical study. Am J Surg Pathol. 1993;17:706-14.
20. Hirota S, Isozaki K, Moriyama Y, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998;279:577-80.
21. Nishida T, Hirota S, Taniguchi M, et al. Familial gastrointestinal stromal tumours with germline mutation of the KIT gene. Nat Genet. 1998;19:323-4.
22. Furitsu T, Tsujimura T, Tono T, et al. Identification of mutations in the coding sequence of the proto-oncogene c-kit in a human mast cell leukemia cell line causing ligand-independent activation of c-kit product. J Clin Invest. 1993;92:1736-44.
23. Longley BJ, Tyrrell L, Lu SZ, et al. Somatic c-KIT activating mutation in urticaria pigmentosa and aggressive mastocytosis: Establishment of clonality in a human mast cell neoplasm. Nat Genet. 1996;12:312-14.
24. Nagata H, Worobec AS, Oh CK, et al. Identification of a point mutation in the catalytic domain of the protooncogene c-kit in peripheral blood mononuclear cells of patients who have mastocytosis with an associated hematologic disorder. Proc Natl Acad Sci U S A. 1995;92:10560-4.
25. Tsujimura T, Furitsu T, Morimoto M, et al. Ligand-independent activation of c-kit receptor tyrosine kinase in a murine mastocytoma cell line P-815 generated by a point mutation. Blood. 1994;83:2619-26.
26. Kindblom LG, Remotti HE, Aldenborg F, et al. Gastrointestinal pacemaker cell tumor (GIPACT): Gastrointestinal stromal tumors show phenotypic characteristics of the interstitial cells of Cajal. Am J Pathol. 1998;152:1259-69.
27. Sircar K, Hewlett BR, Huizinga JD, et al. Interstitial cells of Cajal as precursors of gastrointestinal stromal tumors. Am J Surg Pathol. 1999;23:377-89.
28. Sakurai S, Fukasawa T, Chong JM, et al. Embryonic form of smooth muscle myosin heavy chain (SMemb/MHC-B) in gastrointestinal stromal tumor and interstitial cells of Cajal. Am J Pathol. 1999;154:23-8.
29. Wang L, Vargas H, French SW. Cellular origin of gastrointestinal stromal tumors: A study of 27 cases. Arch Pathol Lab Med. 2000;124:1471-15.
30. Lasota J, Wozniak A, Sarlomo-Rikala M, et al. Mutations in exons 9 and 13 of KIT gene are rare events in gastrointestinal stromal tumors. A study of 200 cases. Am J Pathol. 2000;157:1091-5.
31. Maeyama H, Hidaka E, Ota H, et al. Familial gastrointestinal stromal tumor with hyperpigmentation: Association with a germline mutation of the c-kit gene. Gastroenterology. 2001;120:210-15.
32. Isozaki K, Terris B, Belghiti J, et al. Germline-activating mutation in the kinase domain of KIT gene in familial gastrointestinal stromal tumors. Am J Pathol. 2000;157:1581-5.
33. Duensing A, Medeiros F, McConarty B, et al. Mechanisms of oncogenic KIT signal transduction in primary gastrointestinal stromal tumors (GISTs). Oncogene. 2004;23:3999-4006.
34. Rubin BP, Singer S, Tsao C, et al. KIT activation is a ubiquitous feature of gastrointestinal stromal tumors. Cancer Res. 2001;61:8118-21.
35. Lux ML, Rubin BP, Biase TL, et al. KIT extracellular and kinase domain mutations in gastrointestinal stromal tumors. Am J Pathol. 2000;156:791-5.
36. Mol CD, Dougan DR, Schneider TR, et al. Structural basis for the autoinhibition and STI-571 inhibition of c-Kit tyrosine kinase. J Biol Chem. 2004;279:31655-63.
37. Mol CD, Lim KB, Sridhar V, et al. Structure of a c-kit product complex reveals the basis for kinase transactivation. J Biol Chem. 2003;278:31461-4.
38. Heinrich MC, Corless CL, Duensing A, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science. 2003;299:708-10.
39. Hirota S, Ohashi A, Nishida T, et al. Gain-of-function mutations of platelet-derived growth factor receptor alpha gene in gastrointestinal stromal tumors. Gastroenterology. 2003;125:660-7.
40. Corless CL, McGreevey L, Haley A, et al. KIT mutations are common in incidental gastrointestinal stromal tumors one centimeter or less in size. Am J Pathol. 2002;160:1567-72.
41. ESMO Guidelines Working GroupBlay JY, Le Cesne A. Gastrointestinal stromal tumors: ESMO clinical recommendations for diagnosis, treatment and follow-up. Ann Oncol. 2007;18(Suppl 2):ii27-9.
42. Hyun JH, Jeen YT, Chun HJ, et al. Endoscopic resection of submucosal tumors of the esophagus: Results in 62 patients. Endoscopy. 1997;29:165-70.
43. Miettinen M, Sarlomo-Rikala M, Sobin LH, Lasota J. Esophageal stromal tumors: A clinicopathologic, immunohistochemical, and molecular genetic study of 17 cases and comparison with esophageal leiomyomas and leiomyosarcomas. Am J Surg Pathol. 2000;24:211-22.
44. Pidhorecky I, Cheney RT, Kraybill WG, et al. Gastrointestinal stromal tumors: Current diagnosis, biologic behavior, and management. Ann Surg Oncol. 2000;7:705-12.
45. Crosby JA, Catton CN, Davis A, et al. Malignant gastrointestinal stromal tumors of the small intestine: A review of 50 cases from a prospective database. Ann Surg Oncol. 2001;8:50-9.
46. Palazzo L, Landi B, Cellier C, et al. Endosonographic features predictive of benign and malignant gastrointestinal stromal cell tumours. Gut. 2000;46:88-92.
47. Chak A, Canto MI, Rösch T, et al. Endosonographic differentiation of benign and malignant stromal cell tumours. Gastrointest Endosc. 1997;45:468-73.
48. Choi H, Charnsangavej C, Faria SC, et al. Correlation of computed tomography and positron emission tomography in patients with metastatic gastrointestinal stromal tumor treated at a single institution with imatinib mesylate: Proposal of new computed tomography response criteria. J Clin Oncol. 2007;25:1753-9.
49. Demetri GD, von Mehren M, Blanke CD, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002;347:472-80.
50. Stroobants S, Goeminne J, Seegers M, et al. 18FDG-positron emission tomography for the early prediction of response in advanced soft tissue sarcoma treated with imatinib mesylate (Glivec). Eur J Cancer. 2003;39:2012-20.
51. Coffin CM, Dehner LP, Meis-Kindblom JM. Inflammatory myofibroblastic tumor, inflammatory fibrosarcoma, and related lesions: An historical review with differential diagnostic considerations. Semin Diagn Pathol. 1998;15:102-10.
52. Hornick JL, Fletcher CD. Immunohistochemical staining for KIT (CD117) in soft tissue sarcomas is very limited in distribution. Am J Clin Pathol. 2002;117:188-93.
53. Goss GA, Merriam P, Manola J, et al. Clinical and pathological characteristics of gastrointestinal stromal tumors (GIST). Proc Am Soc Clin Oncol. 19, 2000.
54. Eilber FC, Rosen G, Forscher C, et al. Recurrent gastrointestinal stromal sarcomas. Surg Oncol. 2000;9:71-5.
55. Plaat BE, Hollema H, Molenaar WM, et al. Soft tissue leiomyosarcomas and malignant gastrointestinal stromal tumors: Differences in clinical outcome and expression of multidrug resistance proteins. J Clin Oncol. 2000;18:3211-20.
56. Rajan DK, Soulen MC, Clark TW, et al. Sarcomas metastatic to the liver: Response and survival after cisplatin, doxorubicin, mitomycin-C, ethiodol, and polyvinyl alcohol chemoembolization. J Vasc Interv Radiol. 2001;12:187-93.
57. Mavligit GM, Zukwiski AA, Ellis LM, et al. Gastrointestinal leiomyosarcoma metastatic to the liver. Durable tumor regression by hepatic chemoembolization infusion with cisplatin and vinblastine. Cancer. 1995;75:2083-8.
58. Mudan SS, Conlon KC, Woodruff J, et al. Salvage surgery for recurrent gastrointestinal sarcoma: Prognostic factors to guide patient selection. Cancer. 1999;88:66-74.
59. Druker BJ, Tamura S, Buchdunger E, et al. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med. 1996;2:561-6.
60. Buchdunger E, Zimmermann J, Mett H, et al. Inhibition of the Abl protein-tyrosine kinase in vitro and in vivo by a 2-phenylaminopyrimidine derivative. Cancer Res. 1996;56:100-4.
61. Buchdunger E, Cioffi CL, Law N, et al. Abl protein-tyrosine kinase inhibitor STI571 inhibits in vitro signal transduction mediated by c-kit and platelet-derived growth factor receptors. J Pharmacol Exp Ther. 2000;295:139-45.
62. Heinrich MC, Griffith DJ, Druker BJ, et al. Inhibition of c-kit receptor tyrosine kinase activity by STI 571, a selective tyrosine kinase inhibitor. Blood. 2000;96:925-32.
63. Tuveson DA, Willis NA, Jacks T, et al. STI571 inactivation of the gastrointestinal stromal tumor c-KIT oncoprotein: Biological and clinical implications. Oncogene. 2001;20:5054-8.
64. Joensuu H, Roberts PJ, Sarlomo-Rikala M, et al. Effect of the tyrosine kinase inhibitor STI571 in a patient with a metastatic gastrointestinal stromal tumor. N Engl J Med. 2001;344:1052-6.
65. van Oosterom AT, Judson I, Verweij J, et al. European Organisation for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group. Safety and efficacy of imatinib (STI571) in metastatic gastrointestinal stromal tumours: A phase I study. Lancet. 2001;358:1421-3.
66. Verweij J, van Oosterom A, Blay JY, et al. Imatinib mesylate is an active agent for gastrointestinal stromal tumors but does not yield responses in other soft-tissue sarcomas that are unselected for a molecular target. Eur J Cancer. 2003;39:2006-11.
67. Maki RG, Awan RA, Dixon RH, et al. Differential sensitivity to imatinib of 2 patients with metastatic sarcoma arising from dermatofibrosarcoma protuberans. Int J Cancer. 2002;100:623-6.
68. Rubin BP, Schuetze SM, Eary JF, et al. Molecular targeting of platelet-derived growth factor B by imatinib mesylate in a patient with metastatic dermatofibrosarcoma protuberans. J Clin Oncol. 2002;20:3586-91.
69. McArthur GA, Demetri GD, Heinrich M, et al. Imatinib Target Exploration Study. Molecular and clinical analysis of response to imatinib for locally advanced dermatofibrosarcoma protuberans [abstract 781]. Proc Am Soc Clin Oncol. 2003;22:195.
70. Heinrich MC, Corless CL, Demetri GD, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol. 2003;21:4342-9.
71. Van den Abbeele A, Badawi RD, Cliché JP, et al. 18F-FDG-PET predicts response to imatinib mesylate (Gleevec) in patients with advanced gastrointestinal stromal tumors (GIST) [abstract]. Proc Am Soc Clin Oncol. 21, 2002.
72. Blanke CD, Rankin C, Demetri GD, et al. Phase III randomized intergroup trial assessing imatinib mesylate at two dose levels in patients with unresectable or metastatic gastrointestinal stromal tumor expressing the KIT receptor tyrosine kinase: S0033. J Clin Oncol. 2008;26:626-32.
73. Verweij J, Casali PG, Zalcberg J, et al. Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: Randomised trial. Lancet. 2004;364:1127-34.
74. Debiec-Rychter M, Sciot R, LeCesne A, et al. KIT mutations and dose selection for imatinib in patients with advanced gastrointestinal stromal tumours. Eur J Cancer. 2006;42:1093-103.
75. Demetri GD, Benjamin RS, Blanke CD, et al. NCCN Task Force report: Management of patients with gastrointestinal stromal tumor (GIST)—update of the NCCN clinical practice guidelines. J Natl Compr Cancer Netw. 2007;5(Suppl 2):S1-29.
76. Blay JY, Le Cesne A, Ray-Coquard I, et al. Prospective multicentric randomized phase III study of imatinib in patients with advanced gastrointestinal stromal tumors comparing interruption versus continuation of treatment beyond 1 year: The French Sarcoma Group. J Clin Oncol. 2007;25:1107-13.
77. Fletcher JA, Corless CL, Dimitrijevic S, et al. Mechanisms of resistance to imatinib mesylate (IM) in advanced gastrointestinal stromal tumor (GIST) [abstract]. Proc Am Soc Clin Oncol. 2003;22:815.
78. Shah NP, Sawyers CL. Mechanisms of resistance to STI571 in Philadelphia chromosome-associated leukemias. Oncogene. 2003;22:7389-95.
79. Shankar S, van Sonnenberg E, Desai J, et al. Gastrointestinal stromal tumor: New nodule-within-a-mass pattern of recurrence after partial response to imatinib mesylate. Radiology. 2005;235:892-8.
80. Desai J, Shankar S, Heinrich MC, et al. Clonal evolution of resistance to imatinib in patients with metastatic gastrointestinal stromal tumors. Clin Cancer Res. 2007;13:5398-405.
81. Raut CP, Posner M, Desai J, et al. Surgical management of advanced gastrointestinal stromal tumors after treatment with targeted systemic therapy using kinase inhibitors. J Clin Oncol. 2006;24:2325-31.
82. DeMatteo RP, Maki RG, Singer S, et al. Results of tyrosine kinase inhibitor therapy followed by surgical resection for metastatic gastrointestinal stromal tumor. Ann Surg. 2007;245:347-52.
83. Demetri GD, Heinrich MC, Fletcher JA, et al. Molecular target modulation, imaging, and clinical evaluation of gastrointestinal stromal tumor patients treated with sunitinib malate after imatinib failure. Clin Cancer Res. 2009;15:5902-9.
84. Demetri GD, van Oosterom AT, Garrett CR, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: A randomised controlled trial. Lancet. 2006;368:1329-38.
85. Heinrich MC, Maki RG, Corless CL, et al. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumor. J Clin Oncol. 2008;26:5352-9.
86. Gajiwala KS, Wu JC, Christensen J, et al. KIT kinase mutants show unique mechanisms of drug resistance to imatinib and sunitinib in gastrointestinal stromal tumor patients. Proc Natl Acad Sci U S A. 2009;106:1542-7.
87. Desai J, Yassa L, Marqusee E, et al. Hypothyroidism after sunitinib treatment for patients with gastrointestinal stromal tumors. Ann Intern Med. 2006;145:660-4.
88. Chu T, Rupnick MA, Kerkela R, et al. Cardiotoxicity associated with the tyrosine kinase inhibitor sunitinib. Lancet. 2007;370:2011-19.
89. George S, Blay JY, Casal PG, et al. Clinical evaluation of continuous daily dosing of sunitinib malate in patients with advanced gastrointestinal stromal tumor after imatinib failure. Eur J Cancer. 2009;45:1959-68.
90. Wardelmann E, Merkelbach-Bruse S, Pauls K, et al. Polyclonal evolution of multiple secondary KIT mutations in gastrointestinal stromal tumors under treatment with imatinib mesylate. Clin Cancer Res. 2006;12:1743-9.
91. Liegl B, Kepten I, Le C, et al. Heterogeneity of kinase inhibitor resistance mechanisms in GIST. J Pathol. 2008;216:64-74.
92. Bauer S, Yu LK, Demetri GD, Fletcher JA. Heat shock protein 90 inhibition in imatinib-resistant gastrointestinal stromal tumor. Cancer Res. 2006;66:9153-61.
93. Wagner AJ, Morgan JA, Chugh R, et al. Inhibition of heat shock protein 90 (Hsp90) with the novel agent IPI-504 in metastatic GIST following failure of tyrosine kinase inhibitors (TKIs) or other sarcomas: Clinical results from phase I trial [abstract]. J Clin Oncol. 26(Suppl), 2008.
94. Eisenberg BL, Harris J, Blanke CD, et al. Phase II trial of neoadjuvant/adjuvant imatinib mesylate (IM) for advanced primary and metastatic/recurrent operable gastrointestinal stromal tumor (GIST): Early results of RTOG 0132/ACRIN 6665. J Surg Oncol. 2009;99:42-7.
95. Ng EH, Pollock RE, Munsell MF, et al. Prognostic factors influencing survival in gastrointestinal leiomyosarcomas. Implications for surgical management and staging. Ann Surg. 1992;215:68-77.
96. Miettinen M, Lasota J. Gastrointestinal stromal tumors: Pathology and prognosis at different sites. Semin Diagn Pathol. 2006;23:70-83.
97. Dematteo RP, Ballman KV, Antonescu CR, et al. Adjuvant imatinib mesylate after resection of localised, primary gastrointestinal stromal tumour: A radomised, double-blind, placebo-controlled trial. Lancet. 2009;373:1097-104.
98. Isozaki K, Terris B, Belghiti J, et al. Germline-activating mutation in the kinase domain of KIT gene in familial gastrointestinal stromal tumors. Am J Pathol. 2000;157:1581-5.
99. Maeyama H, Hidaka E, Ota H, et al. Familial gastrointestinal stromal tumor with hyperpigmentation: Association with a germline mutation of the c-kit gene. Gastroenterology. 2001;120:210-15.
100. Li F, Fletcher JA, Heinrich MC, et al. Familial gastrointestinal stromal tumors (GISTs) and associated manifestations in a kindred. J Clin Oncol. 2005;23:2735-43.
101. McWhinney SR, Pasini B, Stratakis CA, International Carney Triad and Carney-Stratakis Syndrome Consortium. Familial gastrointestinal stromal tumors and germ-line mutations. N Engl J Med. 2007;357:1054-6.
102. Price VE, Zielenska M, Chilton-MacNeill S, et al. Clinical and molecular characteristics of pediatric gastrointestinal stromal tumors (GISTs). Pediatr Blood Cancer. 2005;45:20-4.
103. Janeway KA, Liegl B, Harlow A, et al. Pediatric KIT wild-type and platelet-derived growth factor receptor alpha–wild-type gastrointestinal stromal tumors share KIT activation but not mechanisms of genetic progression with adult gastrointestinal stromal tumors. Cancer Res. 2007;67:9084-8.
104. Agaram NP, Laquaglia MP, Ustun B, et al. Molecular characterization of pediatric gastrointestinal stromal tumors. Clin Cancer Res. 2008;14:3204-15.
105. Tarn C, Rink L, Merkel E, et al. Insulin-like growth factor 1 receptor is a potential therapeutic target for gastrointestinal stromal tumors. Proc Natl Acad Sci U S A. 2008;105:8387-92.
106. Carney JA. Gastric stromal sarcoma, pulmonary chondroma, and extra-adrenal paraganglioma (Carney triad): Natural history, adrenocortical component, and possible familial occurrence. Mayo Clin Proc. 1999;74:543-52.
107. Carney JA, Stratakis CA. Familial paraganglioma and gastric stromal sarcoma: A new syndrome distinct from the Carney triad. Am J Med Genet. 2002;108:132-9.
108. Zoller ME, Rembeck B, Oden A, et al. Malignant and benign tumors in patients with neurofibromatosis type 1 in a defined Swedish population. Cancer. 1997;79:2125-31.
109. Kinoshita K, Hirota S, Isozaki K, et al. Absence of c-kit gene mutations in gastrointestinal stromal tumours from neurofibromatosis type 1 patients. J Pathol. 2004;202:80-5.
[/level-membership-for-gastroenterology-and-hepatology-category][not-level-membership-for-gastroenterology-and-hepatology-category]
CHAPTER 30 Gastrointestinal Stromal Tumors (GISTs)
INCIDENCE
It is difficult to obtain accurate data regarding the true incidence of GISTs. This is because of referral bias, which concentrates GIST cases with a worse prognosis and a more malignant behavior in academic cancer centers, and also the lack of definitive diagnostic techniques before the molecular definitions of GISTs in 1998 and later. Before 2000, the number of new GIST cases in the United States had been underestimated and underreported. However, it is now recognized that the true incidence of GISTs is much higher than previously believed. Much of this is to the result of an increased recognition of GISTs; new molecularly targeted drugs, such as imatinib mesylate (Gleevec), have become available. A population-based study to assess the incidence of GISTs using the most up-to-date criteria has estimated an annual incidence of approximately 15 cases/million.1 This has increased the estimated incidence of GISTs in the United States alone to approximately 5000 new cases/year.2,3 However, even this figure may underestimate the actual incidence because many smaller lesions, including those detected incidentally by many gastroenterologists on endoscopy performed for other reasons, are almost certainly not revealed in these incidence figures. Not all GIST cases will prove to be life-threatening, because many patients who present with GISTs have limited disease with lesions that are small (smaller than 1 cm) and fully curable with optimal surgery as first-line therapy. It remains a question as to whether there are any true GIST lesions that might be able to be observed without resection.
GISTs occur predominantly in adults, with a median incidence in the fifth decade of life, but these tumors can occur across the age spectrum, from infants to older adults. The incidence has been reported to be slightly higher in men than women. Recent studies of surgical or autopsy specimens of stomach samples resected for non-neoplastic disease have documented a remarkably high incidence of occult microscopic GIST lesions, in the range of 20% to 35%.4,5
LOCATION
Most GISTs (60% to 70%) arise in the stomach, 20% to 30% originate in the small intestine, and less than 10% in the esophagus, colon, and rectum (see later). GISTs can also occur in extraintestinal sites in the abdomen or pelvis such as the omentum, mesentery, or retroperitoneum.6–8 However, data on sites of GIST origin are somewhat limited. Large studies before 1999 may have included other subtypes of mesenchymal neoplasms because these series were not selected by immunohistochemical or molecular genetic markers specific for true GISTs.
CLINICAL FEATURES
The vast majority of GIST metastases at presentation are intra-abdominal, with metastases to the liver, omentum, or peritoneal cavity.6 Metastatic spread to lymph nodes and to other regions via lymphatics is rare; most lesions thought to be nodal metastases by imaging studies simply represent metastatic deposits of tumor nodules in the omentum or peritoneum rather than true lymphatic spread of the disease.
PATHOLOGY AND MOLECULAR PATHOBIOLOGY
UNIQUE CLINICOPATHOLOGIC SUBTYPE OF SARCOMAS (MESENCHYMAL NEOPLASIA)
Although relatively rare compared with epithelial cancers, GISTs represent a form of sarcoma that comprises approximately 1% to 3% of all malignant GI tumors. As noted, the diagnostic criteria for GISTs before 1999 were somewhat subjective, controversial, and perhaps even a bit confusing, with a great deal of room for interobserver variation. The term GIST was initially a purely descriptive term applied by Mazur and Clark in 1983 to define intra-abdominal tumors that were not carcinomas (i.e., nonepithelial) and that did not exhibit features of smooth muscle or nerve cells.9 The morphology of the cells was the dominant feature driving the diagnostic scheme. However, pathologists subsequently recognized that there was no completely clear differential expression of muscle or nerve antigenic markers when careful immunohistochemical analyses were performed on samples identified as GIST lesions by cellular morphology. The expression of differentiation antigens used as markers for muscle cells (e.g., smooth muscle actin) and nerve cells (e.g., S100) was noted to vary widely in GI mesenchymal lesions, leading to interesting hypotheses about whether GIST lesions from different patients were attempting to recapitulate distinct myogenic or neural programs of differentiation.
To accommodate these empirical observations, it was proposed that approximately one third of GIST lesions differentiated along smooth muscle lineages, another third were neurogenic in origin, and the final third lacked any detectable lineage-specific markers (null phenotype) by immunohistochemical analysis.10–12
Nomenclature for GISTs had also been confusingly complex before the availability of molecular- and mechanism-based diagnostic tests. Because of limited understanding of the disease before 1999, GISTs were most commonly diagnosed as leiomyomas or leiomyosarcomas because of the histologic resemblance to these smooth muscle neoplasms. Other terms that had often been applied to a GIST included benign leiomyoblastoma and, recognizing some of the neural characteristics, plexosarcomas13 or gastrointestinal autonomic nerve tumors (GANTs).14 All these terms are now recognized as referring to GISTs. Insightful studies by several pathology groups have noted that the panoply of tumors lumped together as smooth muscle tumors of the GI tract were likely not simply leiomyosarcomas nor benign leiomyomas; a subset of these tumors originating in the bowel wall had several unique histologic features, probably representing a totally different diagnostic group altogether.15,16 Additionally, clinical oncologists had noted that putative leiomyosarcomas of the GI tract had a completely different prognosis than true leiomyosarcomas arising in other parts of the body (e.g., the uterus). The leiomyosarcomas of the GI tract were extraordinarily resistant to standard chemotherapy regimens, further supporting the notion that these lesions represent a different form of cancer entirely.
Immunohistochemical analysis of GISTs in the early 1990s attempted to find specific markers that might distinguish GISTs from other spindle cell tumors of the GI tract, such as schwannomas and sarcomatoid carcinomas. There was some initial enthusiasm for the CD34 antigen as such a marker; however, this antigen is also expressed by hematopoietic stem cells and by vascular and myofibroblastic cells. Also, the sensitivity and specificity of CD34 are low because only approximately half of GIST cases express CD34, and other smooth muscle, myofibroblastic (e.g., desmoid), or Schwann cell tumors can also express CD34. Therefore, CD34 is not a reliable marker to distinguish true GISTs from other neoplasms.17,18
UNCONTROLLED KINASE ACTIVATION: PRIMARY MOLECULAR PATHOGENESIS
A critical advance in the understanding of GISTs at a molecular level occurred in the late 1990s, with the recognition that these tumors exhibited some histopathologic similarities with the pacemaker cells of the gut known as the interstitial cells of Cajal (ICCs).19 ICCs are normally present in the myenteric plexus and serve to coordinate gut peristalsis by assisting the linkage of smooth muscle cells of the bowel wall with the autonomic nervous system (see Chapters 96 to 98). GIST cells and ICCs have certain ultrastructural features in common, such as the combination of neural and muscle phenotypes.
The molecular pathogenesis of GISTs was advanced further by a key observation made by Hirota and colleagues20 in 1998. This group was studying the role played by the KIT receptor tyrosine kinase (RTK) in cell growth and development. Expression of the KIT RTK can be detected by immunohistochemical staining for the CD117 antigen as a marker of the KIT protein. In normal cell signaling, KIT binds its ligand, known as stem cell factor (SCF) or Steel factor; ligand binding brings together two molecules of KIT with subsequent activation of a signaling cascade. A homodimeric complex is formed with these two KIT receptors, leading to cross-phosphorylation of critical tyrosine residues in the intracellular domains of KIT, which activate signal transduction pathways downstream of KIT. The net physiologic effect of normal ligand-induced KIT activation is the controlled stimulation of cell proliferation and enhanced cell survival; therefore, uncontrolled activation could theoretically lead to neoplastic growth and transformation of cells.
Hirota and colleagues20 recognized this potential mechanism and provided the critical confirmation of this theory at the cellular and molecular levels. This same team, in a project led by Nishida, also expanded these observations to familial GISTs, with germline carriage of an activating mutation in the KIT gene encoding the RTK.21 This elegant work supported some of the key biologic similarities between GIST and ICC cells22–25 because both cell types had been shown to express the KIT RTK. It is likely that the cells of GISTs and normal ICCs share a common precursor cell.26,27 The KIT RTK and SCF play essential roles in the development and maintenance of normal ICCs, as well as other cells, including melanocytes, erythrocytes, germ cells, and mast cells. KIT expression is noted in the vast majority (>95%) of GISTs, but KIT is not expressed by true smooth muscle tumors of the GI tract nor by stromal tumors at other anatomic locations, such as endometrial stromal tumors. Although the origin of the neoplastic cells of GISTs remains a matter of active investigation, some data suggest that GISTs originate from CD34-positive stem cells residing within the wall of the gut, which can then differentiate incompletely toward the ICC phenotype.27–29
GISTs characteristically exhibit expression of CD117 by immunohistochemical assays (>95% of lesions but, importantly, not 100%, because there are true KIT-negative GIST cases).30 Levels of expression of CD117 (KIT) are generally diffuse and strong in the spindle cell GIST subtype (Fig. 30-1). In contrast, in the epithelioid subtype, CD117 expression is typically focal and weakly positive in a dot-like pattern (Fig. 30-2). As noted, CD34 expression is neither sensitive nor specific for GIST because this antigen can also be noted in desmoid tumors, and approximately 30% to 40% of GIST lesions are negative for CD34.21,26 True leiomyosarcomas express two smooth muscle markers, smooth muscle actin (SMA) and desmin, but fail to express CD117. Schwannomas are usually positive for the neural antigen S100 but are also negative for CD117. Normal mast cells and ICCs in the surrounding stromal tissues serve as ideal positive internal controls because these normal cells strongly express CD117. Activating mutations in the KIT gene were identified in five of six cases of human GISTs originally analyzed by Hirota and colleagues,20 with evidence that the mutations resulted in uncontrolled, ligand-independent activation of the KIT kinase. Genetically engineered cells harboring the mutant overactive KIT proteins were tumorigenic in nude mice, serving as proof of concept that the malignant phenotype was directly induced by the aberrant signaling pathways associated with uncontrolled KIT kinase activation.
The oncogenic potential of mutant, uncontrollably active KIT in the pathogenesis of GISTs in humans has also been supported by the identification of familial syndromes with an autosomal dominant inheritance pattern and an abnormally high incidence of GISTs, usually occurring as multiple foci within any affected individual (see later).21,31,32 Genetic analysis of such kindreds reveals that they harbor germline-activating KIT mutations, similar to the mutations that were first described in sporadic cases of GISTs.
With analyses using optimal tumor specimens and sophisticated technology, it has become clear that KIT mutations can be detected in more than 90% of GIST cells.33–35 Constitutive activation of the KIT kinase enzymatic function has been reported to characterize every GIST sample analyzed by immunoblotting technique, even in cases in which there are no detectable mutations in the KIT gene. The mechanisms whereby nonmutated (wild-type) KIT is maintained in an uncontrollably phosphorylated state are poorly understood and are likely to be fertile ground for future research, with therapeutic potential.
Importantly, the vast majority of GIST cells at initial presentation demonstrate only a single site of mutation in the KIT gene; complex genetic changes in untreated GISTs at initial presentation are vanishingly rare. Gain of function mutations have been identified most commonly in exon 11 of KIT (up to 70% of cases), an exon that encodes the intracellular juxtamembrane domain of the KIT protein. Mutations in the KIT gene locus have also been described in other regions including (in decreasing order of prevalence) exon 9 (the KIT extracellular domain), exon 13 (kinase domain), and exon 17 (kinase domain).33–35 Structural biology studies have revealed the mechanism whereby normal (wild-type) KIT is kept in an autoinhibited conformation until ligand binds; mutational changes in conformation interfere with this autoinhibition and lead to a structural basis for the aberrant activation of the KIT kinase function.36,37
Another key advance in the understanding of GISTs has been the recognition that signaling through other uncontrolled kinases in addition to KIT could drive the neoplastic phenotype of GIST cells. Specifically, it is now recognized that approximately 5% of GIST cells are not through activation and aberrant signaling of the KIT receptor, but rather through mutational activation of the structurally related kinase known as the platelet-derived growth factor receptor-alpha (PDGFRA).38,39
The definitive diagnostic criteria of uncommon CD117-negative lesions that are nevertheless truly GISTs are currently somewhat obscure. GIST lesions can be heterogeneous in the expression of CD117, even within a single mass. It is possible that a needle biopsy could yield cells consistent with a GIST yet be CD117-negative simply by sampling bias alone. However, expert pathologists can also define a rare subset of GISTs (<5% of cases overall) that have no CD117 expression; these are most likely dependent on an alternative kinase such as PDGFRA.38 Molecular analyses of the KIT and PDGFRA genotypes may be useful to define with certainty the group of rare patients with CD117-negative GISTs in the future. Now, it is important to note that the diagnosis of a GIST should be made on the grounds of morphologic, clinicopathologic, and immunohistochemical data, as well as possibly molecular analysis if there is any ambiguity from the other pathologic assessments.
KIT mutations have also been documented in small GISTs (<1 cm in greatest dimension)40; such lesions are most often detected incidentally (e.g., during upper endoscopy for reflux symptoms) and may appear morphologically benign. These findings support the hypothesis that activating mutations in the KIT proto-oncogene represent an early event in the transformation from a normal precursor cell into a GIST lesion. Because lesions in familial GISTs (see later) may not present clinically until the second or third decade of life, or even much later, it is likely that second hits are necessary to attain a more aggressive malignant phenotype. The other key signaling steps that confer a more malignant phenotype to GIST cells remain obscure. However, the unique aspects of the signaling cascades in GISTs are being actively elucidated, and these appear to differ from KIT signaling in hematologic cancers. For example, the STAT5 pathway of leukemic cells is not typically activated in GISTs, whereas STAT1 and STAT3 are activated at a high level.33
BENIGN GASTROINTESTINAL STROMAL TUMORS
The literature before 2000 was somewhat confusing about whether mutational status of KIT could distinguish between so-called benign and malignant GISTs. With the recognition that KIT mutations can be found in even the smallest GIST,40 there is now consensus that KIT genotype alone cannot account for differences between GISTs that may behave in an indolent manner (and which, when small, may be curable by optimal resection alone) versus those that are clearly aggressive and malignant by all functional definitions. It is important to note that a well-differentiated benign cell morphology alone should not provide any reassurance that an individual GIST lesion will pursue a benign clinical course.
Consensus was reached at a meeting held at the National Cancer Institute (NCI) among pathologists with expertise in GISTs. This consensus defined the two most reliable prognostic factors for behavior of a primary GIST as the size of the primary tumor and the number of mitoses, reflecting the proliferative activity of the cells.2 Other factors, such as the specific histologic subtype (epithelioid vs. spindle cell; see Figs. 30-1A and 30-2), the degree of cellular pleomorphism, and patient age may have some contribution to prognosis but are most likely to play a minor role in determining the clinical outcome. Recurrence and survival rates have also been reported to correlate with the location of the primary GIST lesion, with small bowel tumors showing a somewhat worse prognosis. Most important, the consensus panel emphasized that no GIST lesion should ever be considered completely benign. This is because morphologically benign lesions have a finite capacity to recur and metastasize, occasionally several years after initial presentation of the primary disease. Nonetheless, further research is necessary to define the true risks of tiny GIST lesions (<1 cm) because these are far more common than previously appreciated.
DIAGNOSIS
The revolutionary changes in the scientific understanding, diagnostic evaluation, and therapeutic management of GIST patients (discussed later) has led several professional organizations to develop consensus-based (and, whenever possible, evidence-based) clinical practice guidelines. The National Comprehensive Cancer Network (NCCN) has developed extensive publically accessible guidelines to assist clinicians in the management of GIST patients (see the GIST and Soft Tissue Sarcoma Guidelines on www.nccn.org). Also, the European Society of Medical Oncology (ESMO) has published expert-driven clinical practice guidelines.41 Given the rapid progress in this field, these guidelines are reviewed at least annually to ensure that current information is available to assist clinicians in providing the most up-to-date care and accurate information to patients.
DIAGNOSTIC APPROACH TO STROMAL TUMORS BY SITE
Esophageal Tumors
GIST lesions may arise within the esophagus, although this is a rare presentation for larger lesions. Most esophageal GIST lesions are noted incidentally during upper endoscopy performed for some other unrelated symptom or disorder, such as reflux esophagitis. Esophageal GIST lesions may be small (only a few millimeters in size) in this location and may be resected using endoscopic techniques.42 Margins may be involved if a lesion, unsuspected as a GIST and thought to be benign, is simply popped out using an endoscopic procedure. It remains unclear whether watchful waiting with serial endoscopic follow-up is appropriate for any patient with small GIST lesions (<1 cm in maximal dimension).
As noted, histopathology showing putatively benign GIST cells cannot be viewed reassuringly because histology does not perfectly predict the malignant behavior of GISTs. A careful risk-based assessment that takes into account other aspects of the tumor, as well as patient-specific factors (e.g., age, comorbidities, patient preferences), must be performed. It will be important for prospective research to accumulate a larger objective database regarding the outcomes of such patients so that medical decision making can be based on solid evidence. Although a small esophageal GIST lesion is probably a single primary, it is appropriate to take a careful family history and perform computed tomography (CT) scanning of the abdomen and pelvis to ensure that no other lesions are present in the patient’s family, especially if the disease is first detected in a young person. Some series have attempted to distinguish between clinical outcomes of patients with GISTs and those with leiomyosarcomas or other mesenchymal neoplasms, such as leiomyomas.43
Gastric Tumors
The most common primary site for GISTs is the stomach. Most GIST lesions are submucosal, rather than mucosal, without overlying ulceration (Fig. 30-3). This explains why many GIST masses may only be visualized on endoscopy as a subtle, smooth protrusion with overly normal mucosa.44 Also, this submucosal localization can make diagnostic biopsy through an endoscope difficult. It is not uncommon for superficial biopsies to reveal only normal mucosa, whereas deeper biopsies or histopathology from a definitive resection would show the true underlying GIST cells.
Small Intestinal Tumors
Because complete surgical resection is the treatment of choice for localized GISTs, there is some controversy over whether any preoperative biopsy is necessary or whether that simply represents an extra risk for the patient. This is a challenging subject because other disease entities enter into the differential diagnosis of a large abdominal mass involving the small intestine and mesentery. In general, clinical practice guidelines have suggested that resection may be performed without antecedent biopsy if a GIST is strongly suspected and if surgery can be accomplished without significant risk of morbidity to the patient. If only radical surgery leading to significant functional impairment could remove the lesion, it may be in the patient’s best interest to consider a preoperative biopsy to establish the diagnosis of a GIST. The prognosis of GISTs involving the small intestine is related to the adequacy of resection.6,45 In one series of 50 GIST patients involving the small intestine, 70% could be completely resected, with median overall survival longer than five years for patients with localized or locally advanced disease. Patients who underwent complete resections exhibited a five-year overall survival rate of 42%, while those whose lesions could not be completely resected had a five-year overall survival rate of only 8%.28
IMAGING STUDIES
Endoscopic Ultrasonography
Endoscopic ultrasonography (EUS) is a useful technology for evaluating possible GIST lesions because of their submucosal localization. The paradigm of a GIST lesion visualized by EUS is that of a hypoechoic mass contiguous with the fourth (muscularis propria) or second (muscularis mucosae) layers of the normal gut wall. In one study,46 the features most predictive of so-called benign GI tumors were regular margins, tumor size 3 cm or smaller, and a homogeneous echogenicity pattern. Multivariant analysis identified the presence of cystic spaces and irregular margins as independent predictors of malignant potential. A second study identified tumor size larger than 4 cm, irregular extraluminal borders, echogenic foci larger than 3 mm, and cystic spaces larger than 4 mm as factors that correlated with malignant behavior in GIST.47
Computed Tomography and Magnetic Resonance Imaging
CT is the most effective way to image primary lesions in the stomach because the oral contrast will outline masses and gastric thickening (Fig. 30-4). Differential diagnosis with inadequate gastric distention can be a challenge, especially when monitoring for recurrence following surgery. CT is also essential to stage the extent of disease completely and accurately. For measurable GIST lesions, it is particularly useful to perform CT with noncontrast image acquisition, as well as assess early and late images following the administration of intravenous contrast.
Baseline imaging is critical for GIST, because endoscopic imaging alone may only reveal a small fraction of the underlying tumor. Additionally, imaging patterns (particularly tumor density on CT imaging) can be interpreted qualitatively to assess the impact of targeted therapy using metrics other than tumor size. Tumors that become more hypodense on CT imaging have a more favorable antineoplastic response to targeted therapies, such as the kinase inhibitors imatinib or sunitinib.48
Positron Emission Tomography
One of the most impressive aspects of GIST diagnostic imaging is the use of 18F-fluorodeoxyglucose (18F-FDG)–positron emission tomography (PET) scans to add complementary information to that obtained by conventional anatomic imaging (Fig. 30-5
[/not-level-membership-for-gastroenterology-and-hepatology-category]
