CHAPTER 29 Gastrointestinal Lymphomas
Lymphomas are solid malignancies of the lymphoid system and are subdivided into Hodgkin’s and non-Hodgkin’s lymphomas (NHLs). It was estimated that in 2008, there would be 8,220 and 66,120 new diagnoses of Hodgkin’s and NHL, respectively, in the United States.1 The gastrointestinal tract is very rarely involved with Hodgkin’s lymphoma and will not be discussed in this chapter. There are 0.8 to 1.2 new cases of primary gastrointestinal NHL/100,000 persons/year.2 This accounts for 30% to 50% of all extranodal NHLs, making the gastrointestinal (GI) tract the most common site of extranodal NHL. Lymphomas that involve the GI tract but have the bulk of the disease in nodal areas are managed in a similar fashion to those that do not involve the GI tract. This chapter deals with primary gastrointestinal lymphoma, which is usually defined as “a lymphoma that has presented with the main bulk of disease in the GI tract, with or without involvement of the contiguous lymph nodes, necessitating direction of treatment to that site.”3
BACKGROUND
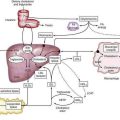
Lymphomas are malignancies of the immune system. In broad terms, the immune system can be thought of as a highly structured and tightly regulated interaction between lymphoid and nonlymphoid tissues aimed at protecting the host from harmful agents.4 Lymphoid cells are produced in the bone marrow and thymus and then arrayed in the lymphoid tissues, which include the lymph nodes, spleen, Waldeyer’s ring, and mucosa-associated lymphoid tissue (MALT; see Chapter 2). The GI tract lymphoid tissue is MALT, typified by the Peyer patches of the terminal ileum. MALT contains B cells at various stages of differentiation, organized into different zones (Fig. 29-1A). B cells that have encountered antigen diffusing across the mucosa enter the germinal center of MALT and undergo repeated immunoglobulin gene mutations (somatic mutations)5; the resultant B cell subclones whose immunoglobulins are highly specific for antigen have a survival advantage over B cells whose immunoglobulins are less specific. These more specific B cells then leave the germinal center, enter the circulation, differentiate into memory B cells or antibody-producing plasma cells, and return to the intestinal mucosa. Memory B cells reside in the marginal zone of MALT. Some marginal zone B cells occupy the epithelial tissue that covers the Peyer patches; these cells are called intraepithelial marginal zone B cells. B cells that have not encountered antigen make up the mantle zone of MALT. T cells play a role in the coordination and delivery of the immune system and thus are also found in MALT (see Fig. 29-1A). Therefore, MALT is composed of B and T cells at various stages of differentiation; immune cells at a given stage of differentiation have characteristic histologic, immunophenotypic, and genetic features. Malignant transformation may occur in a cell at any one of these particular stages of differentiation, leading to a malignancy with distinct clinical pathologic features (see Fig. 29-1B). This way of understanding lymphomas has led to the World Health Organization (WHO) lymphoma system, which recognizes at least 28 different clinical pathologic entities.6
Most lymphomas of the GI tract are B cell lymphomas, with most of these resulting from transformation of marginal zone B cells, classified by the WHO system as extranodal marginal zone B cell lymphomas. However, B cell lymphomas can also arise from other cells of MALT, such as centrocytes of the germinal center (follicular lymphomas) or cells of the mantle zone (mantle cell lymphoma). The precise histogenesis of large B cell lymphomas likely varies from case to case. T cell lymphomas of the GI tract are less common and usually involve malignant transformation of intraepithelial T cells in patients with celiac disease (see Chapter 104).
GI lymphomas most commonly involve the stomach or small intestine, although the oral pharynx, esophagus, colon, or rectum may be involved uncommonly. In developed countries, the stomach is the most common site of involvement (approximately 60% of cases), but in the Middle East, the small intestine is the most common site of GI involvement. Table 29-1 lists the GI lymphomas discussed in this chapter. Clinicians dealing with GI lymphoma are faced with a specific pathologic diagnosis of a lymphoma occurring in a specific site and, in some cases, modified by important patient characteristics, such as human immunodeficiency virus (HIV) infection. This chapter discusses the main clinicopathologic entities that a clinician may encounter. Certain of these GI lymphomas have particular features that warrant more extensive discussion; these include gastric extranodal marginal zone B cell lymphoma of MALT type, gastric diffuse large B cell lymphoma, immunoproliferative small intestinal disease, and enteropathy-type intestinal T cell lymphoma. The other lymphomas that may occur in the GI tract are covered in less detail. Before proceeding to discussion of each entity, we briefly review certain general principles that apply to all subtypes.7
| Gastric Lymphomas |
HIV, human immunodeficiency virus; IPSID, immunoproliferative small intestinal disease; MALT, mucosa-associated lymphoid tissue.
GENERAL PRINCIPLES OF LYMPHOMA MANAGEMENT
DIAGNOSIS
Because of the many subtypes of NHL, lymphoma should be diagnosed and categorized by a hematopathologist. Sufficient tissue is required for an accurate diagnosis. In the GI tract, this often means multiple endoscopic biopsies. Fine-needle aspiration biopsy is not considered sufficient for diagnosis because it only permits analysis of the morphology of individual cells and not an in-depth examination of the background milieu in which those cells reside. The minimal pathologic workup should include light microscopy and immunophenotypic analysis, either by flow cytometry or immunohistochemistry. Staining for immunoglobulin light chains assists in the documentation of monoclonality, with a clear-cut light chain restriction (κ/γ ratio or γ/κ ratio of 10 : 1 or more) strongly suggesting B cell lymphoma. Occasionally, molecular genetic analysis by Southern blot testing or polymerase chain reaction (PCR) assay is indicated to document monoclonal immunoglobulin or T cell receptor gene rearrangements, or to assess characteristic oncogene rearrangements. Microarray analysis of gene expression profiles may yield important prognostic information, but such analyses are not commonly performed in commercial laboratories and thus are not part of the routine pathologic workup.8
STAGING AND PROGNOSTIC ASSESSMENT
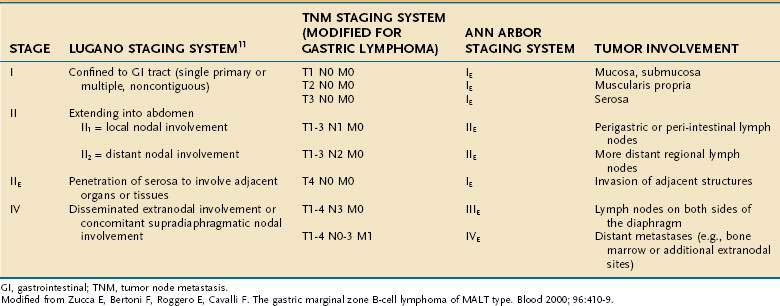
The extent of involvement by NHL is assessed by careful history and physical examination; computed tomography (CT) of the neck, chest, abdomen, and pelvis; positron emission tomography (PET) in cases of high-grade NHL; bone marrow examination; and endoscopic ultrasonography for GI lymphomas.9 Waldeyer’s ring is often involved in gastrointestinal lymphomas, and examination of the upper airway is therefore indicated. The Ann Arbor staging system (Table 29-2),10 which was originally developed for Hodgkin’s lymphoma but is also used for NHL, is deemed by many to be inadequate for staging of GI lymphomas, and several alternative systems have been proposed, two of which are also shown in Table 29-2.11
Prognosis is assessed by defining the distinct lymphoma subtype and evaluating clinical features, including tumor stage, age of the patient, performance status, and serum lactate dehydrogenase (LDH) level. The International Prognostic Index, a model used to predict outcome in patients with aggressive NHL,12 can also be used to assess the prognosis of patients with diffuse large B cell lymphoma of the stomach.13
GASTRIC LYMPHOMAS
Primary gastric lymphomas account for 5% of gastric neoplasms, with an increasing worldwide trend.14 The stomach is the most common extranodal site of lymphoma in developed countries.15 Most of these lymphomas are classified as marginal zone B cell lymphoma of the MALT type16 or as diffuse large B cell lymphoma.
GASTRIC MARGINAL ZONE B CELL LYMPHOMA OF MUCOSA-ASSOCIATED LYMPHOID TISSUE
Extranodal marginal zone B cell lymphoma of MALT, also known as MALT lymphoma, comprises about 8% of all NHLs.17 These lymphomas arise from malignant transformation of B cells from the marginal zone of MALT.18 They may arise from MALT that exists under normal physiologic circumstances (e.g., in Peyer’s patches of the gut) or from MALT that has been acquired in sites of inflammation associated with infection or an autoimmune process. For example, gastric tissue normally does not contain MALT but may acquire it in response to chronic Helicobacter pylori infection (see Chapters 50 and 51).19 Malignant transformation occurs in a small percentage of patients with acquired gastric MALT and results in a lymphoma with generally indolent behavior. The malignant process appears to be driven to a large degree by chronic H. pylori infection, because eradication of the infection leads to regression of the lymphoma in most cases.20
Epidemiology
Gastric marginal zone B cell lymphoma of MALT represents approximately 40% of gastric lymphomas.15 The incidence varies according to the incidence of H. pylori in the population being assessed; the incidence in northeastern Italy, where the rate of H. pylori infection is very high, is roughly 13 times the incidence in the United Kingdom.21 The incidence in H. pylori–infected individuals is from 1 in 30,000 to 1 in 80,000.22 The median age at diagnosis is approximately 60 years, with a wide age range. The male-to-female ratio is equal.
Cause and Pathogenesis
Helicobacter pylori Infection
Several lines of evidence support the key role of H. pylori in the development of gastric MALT lymphoma (see Chapter 50). Infection by H. pylori is present in approximately 90% of cases of gastric MALT lymphoma examined histologically23 and in 98% of cases studied by serology.24,25 The epidemiologic studies cited earlier have shown a close correlation between the prevalence of H. pylori infection and gastric lymphoma in a given population,22 and case-control studies have shown an association between previous H. pylori infection and subsequent development of gastric lymphoma.26 In vitro studies have shown that gastric MALT lymphoma tissue contains T cells that are specifically reactive to H. pylori.27 These H. pylori–reactive T cells support the proliferation of neoplastic B cells.28,29 Many groups have documented the regression of gastric MALT lymphoma after eradication of H. pylori.20,30–33 Of interest, responses of small intestinal and rectal lymphoma to H. pylori eradication have been reported,34,35 although a consistent role of the organism at these nongastric sites is not clear. Lymphomas have also been reported in patients with Helicobacter heilmannii infections, with resolution after eradication of the infection.36
Evidence for Antigen-Driven B Cell Proliferation
As noted, the B cell immunoglobulin variable region (V) genes undergo somatic hypermutation during the T cell–dependent B cell response to antigen5; this leads to the production of new antigen receptors with altered antigen-binding affinity. Resultant B cell clones that express higher affinity antigen receptors have a survival advantage over B cell clones containing receptors with lower affinity. Thus, somatic mutation is a marker for antigen-driven selection of B cell clones. Sequence analysis of malignant B cells from gastric MALT lymphoma shows that the immunoglobulin genes have undergone somatic mutation.37–39
As discussed in Chapter 2, helper T cells support the proliferation of various B cells through cytokines and cell-to-cell interactions.40 These B cells may be specific for the same antigen as the T cells or may be reactive with other antigens. As noted, CD4+ T cells within gastric MALT lymphoma tissue are reactive with H. pylori antigens. The malignant B cells in gastric MALT lymphoma may be reactive with non–H. pylori antigens and, in fact, may be self-reactive. One study has shown that idiotypic immunoglobulins isolated from gastric MALT lymphoma react with a variety of autoantigens.27 Another study has shown that the immunoglobulin genes from gastric MALT lymphoma are derived from germline genes commonly used in the formation of autoantibodies.38 One study has suggested the presence of selecting antigens common to different patients. Analysis of the DNA and amino acid sequences of the antigen-binding region of tumor-derived immunoglobulin from two separate patients showed that although the DNA sequences differed, the resultant amino acid sequences were almost identical.39
Genetic Studies
There are four main chromosomal translocations in extranodal marginal zone lymphomas: t(11;18)(q21;q21), t(14;18)(q32;q21), t(1;14)(p22;q32), and t(3;14)(p14.1;q32). The most common translocation is t(11;18)(q21;q21). Overall, it is found in 30% of cases, but its incidence varies with disease site: it is more common in cases involving the lung and stomach, but rare in other sites.41 The t(11;18) translocation results in the reciprocal fusion of the API-2 and MALT-1 genes. API-2 is an apoptosis inhibitor, and MALT-1 is involved in nuclear factor κB (NF-κB) activation. MALT lymphomas with this translocation do not respond as well to antibiotic therapy aimed at eradicating H. pylori infection as lymphomas without this infection.42 However, they are also less likely to have other chromosomal translocations or transform to more aggressive large cell lymphomas.43,44
The t(14;18)(q32;q21) variant results in the translocation of the MALT-1 gene on chromosome 18q21 to the immunoglobulin gene heavy chain enhancer region, leading to its overexpression, thus differing from the t(14;18) translocation of follicular lymphoma, which involves the bcl-2 gene. This occurs in about 20% of MALT lymphomas overall, although the incidence varies according to the disease site; it is more common in lymphomas occurring in the salivary glands and ocular adnexa, but rare in the GI tract.45,46
Approximately 5% of gastric MALT lymphomas have a t(1;14)(p22;q32) translocation.47 In this translocation, the bcl-10 gene is brought under the control of the immunoglobulin heavy-chain gene enhancer, deregulating its expression. This translocation has been detected only in patients with MALT lymphomas, but those with it often have concurrent trisomies of chromosomes 3, 12, and 18. It is more commonly found in advanced-stage cases, which are less likely to respond to H. pylori eradication.41
The t(3;14)(p14.1;q32) is the latest translocation to be described and results in the juxtaposition of the transcription factor FOXP1 on 3p14.1, next to the immunoglobulin gene heavy (IGH) chain enhancer region.46 The oncogenic role of the IGH-FOXP1 fusion protein is not yet known.
Common Molecular Pathway for MALT Lymphoma Chromosomal Translocations
The first three translocations listed earlier all activate nuclear factor-κB (NF-κB), a transcription factor that increases cell activation, proliferation, and survival.47 In unstimulated B and T lymphocytes, NF-κB is sequestered in the cytoplasm because it is bound to IκB, an inhibitory protein. Phosphorylation of IκB targets it for ubiquitination and degradation, thus releasing NF-κB, which then translocates to the nucleus to function as a transcription factor. The pathways through which IκB is phosphorylated are tightly regulated and involve BCL-10 and MALT-1. Excessive BCL-10 or MALT-1 activity occurring as a consequence of t(11;18), t(14;18), or t(1;14) leads to constitutive NF-κB activation.47,48
Model for Pathogenesis of Gastric MALT Lymphoma
A model for the pathogenesis of gastric MALT lymphoma suggests that the evolution of the disease is a multistage process, comprising the sequential development of H. pylori gastritis, low-grade B cell lymphoma, and then high-grade B cell lymphoma.47,49 This model is supported by gastric biopsies obtained from patients with chronic gastritis taken years before the onset of lymphoma showing B-lymphocytic clones that later gave rise to a clinically evident lymphoma. In this model, H. pylori infection elicits an immune response in which T and B cells are recruited to the gastric mucosa, where MALT is then formed. H. pylori–specific T cells provide growth help to abnormal B cell clones. The abnormal B cells may not be H. pylori–specific and may even be autoreactive. However, their continued proliferation, initially, depends on T cell help. The pivotal role of H. pylori–reactive T cells in driving B cell proliferation may explain why tumor cells tend to remain localized and why the tumor regresses after eradication of H. pylori. However, continued B cell proliferation eventually leads to accumulation of additional genetic abnormalities, resulting in autonomous growth and more aggressive clinical behavior.
Because only a small percentage of H. pylori–infected individuals develops lymphoma, additional currently unknown environmental, microbial, or genetic factors must play a contributory role. H. pylori strains expressing certain proteins such as CagA have been suggested to play a role in the development of gastric lymphoma, but studies have yielded conflicting results.50,51
Pathology
Histology
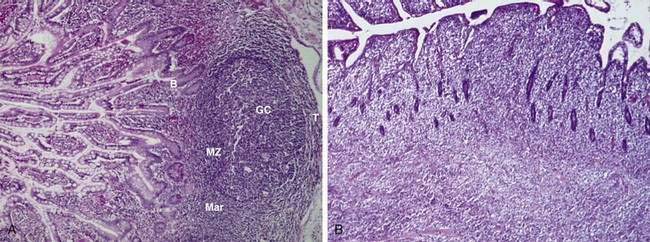
The key histologic feature of low-grade MALT lymphoma is the presence of lymphoepithelial lesions (Fig. 29-2).52,53 These lesions are defined as the unequivocal invasion and partial destruction of gastric glands or crypts by tumor cell aggregates. It should be noted, however, that these lesions can sometimes be seen in cases of florid chronic gastritis. Tumor cells are small to medium-sized lymphocytes, with irregularly shaped nuclei and moderately abundant cytoplasm. The morphology of these cells can vary from small lymphoplasmacytoid cells to monocytoid cells that have abundant pale cytoplasm and well-defined borders. Scattered larger cells or transformed blasts may also be seen. The lymphoma cells infiltrate the lamina propria diffusely and grow around reactive follicles; the germinal centers may be invaded, a phenomenon termed follicular colonization. Because there is a continuous spectrum from the transition of gastritis to lymphoma, diagnosis of borderline cases can be difficult. Various parameters may assist in the distinction, such as the prominence of lymphoepithelial lesions, degree of cytologic atypia, and presence of plasma cells with Dutcher bodies (periodic acid–Schiff [PAS]–positive intranuclear pseudoinclusions).
The presence of large cells can add further complexity to the diagnosis.14 The low-grade MALT lymphoma may have scattered large cells, but the tumor is composed predominantly of small cells. At the other end of the spectrum, gastric lymphomas that contain only large cells or only small areas of small cell MALT-like lymphoma should be classified as diffuse large B cell lymphomas (see later).6 In between the ends of this spectrum are low-grade lymphomas in the process of evolving into more aggressive lymphoma, with increasing numbers of large cells being observed with transformation. Some investigators have proposed histologic grading systems to take this into account; these systems appear to be able to assess prognosis on the basis of large cell percentages and clusters, but it is unclear how reproducible the systems are.20,54
Immunophenotype
Gastric MALT lymphoma cells have the typical immunophenotype of marginal zone B cells. They express pan-B antigens (CD19, CD20, and CD79a) and lack expression of CD5, CD10, CD23, and cyclin D1.55 Further immunostaining by experienced pathologists can aid in identifying lymphoepithelial lesions (see Fig. 29-2) and in distinguishing follicular colonization from follicular lymphoma (a rare occurrence in the stomach; see later).
Molecular Tests of Monoclonality
Southern blotting or PCR assay of immunoglobulin heavy chain rearrangement can assist in the documentation of monoclonality. It should be noted that B cell monoclonality may be detected in H. pylori–associated gastritis (see Chapter 50). Although monoclonality may predict for later development of lymphoma, monoclonality alone does not allow a diagnosis of lymphoma; thus, molecular tests should always be considered in the context of histologic findings.56
Clinical Features
Symptoms, Signs, and Laboratory Tests
The most common symptoms are dyspepsia and epigastric pain. Other less common symptoms include anorexia, weight loss, nausea and/or vomiting, and early satiety.15 Gastric bleeding and B symptoms (fevers, night sweats, weight loss) are rare. Serum levels of serum LDH and β2-microglobulin are usually normal.57
Diagnosis and Staging
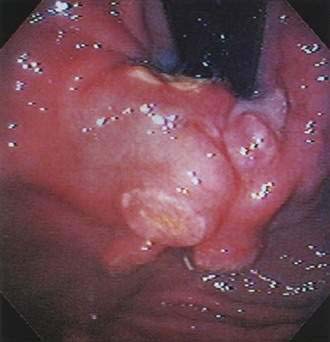
Patients are evaluated by esophagogastroduodenoscopy (EGD). Endoscopic findings include erythema, erosions, and/or ulcers. Diffuse superficial infiltration is typical for MALT lymphoma, whereas masses are more commonly seen in diffuse large B cell lymphoma (Fig. 29-3), an aggressive NHL.58 The most common sites of involvement in the stomach are the pyloric antrum, corpus, and cardia, but biopsies should be taken from all abnormal areas and randomly from each area of the stomach, as well as the duodenum and gastroesophageal junction, because disease is often multifocal.59 Because some lymphomas infiltrate the submucosa without involving the mucosal membrane, biopsies need to be sufficiently deep and large for histopathologic and immunohistochemical analyses. H. pylori infection should be established by histologic studies, breath test, or fecal antigen testing (see Chapter 50).60 Endoscopic ultrasound (EUS) can determine the depth of infiltration and assess for the presence of enlarged perigastric lymph nodes.61–63 Additional staging consists of upper airway examination, CT scans of the chest, abdomen, and pelvis, bone marrow aspiration and biopsy, and measurement of the serum LDH level. PET is not usually helpful in gastric MALT lymphoma because of low uptake of fluorodeoxyglucose (FDG).64,65
Staging System and Prognostic Assessment
In 1994, an international workshop on the staging of GI tract lymphomas proposed the Lugano staging system,11 a modification of the Blackledge system (see Table 29-2). Approximately 75% of gastric MALT lymphomas are confined to the stomach (stage I) at diagnosis66,67 and behave in a clinically indolent fashion; thus, prognosis is good for most patients, with overall survival rates of 80% to 95% at five years. Prognosis is poor in the rare patient with more advanced disease. Additional features associated with a worse prognosis are deep infiltration of the stomach wall, which is associated with a higher likelihood of regional lymph node involvement,68 and high percentages of large cells on histologic evaluation.54
Treatment
Large, randomized clinical trials have not been performed in MALT lymphoma because of the rarity of the disorder. Therefore, treatment recommendations are based on case series and expert opinion. Wotherspoon and colleagues20 first reported that gastric MALT lymphoma could completely regress by endoscopic, histologic, and molecular criteria after eradication of H. pylori. Numerous studies have confirmed these observations,30,33,69–71 and antibiotics aimed at eradicating H. pylori (see Chapter 50) have become the mainstay of therapy for low-grade gastric MALT lymphoma. Even patients with advanced stages of disease can regress with eradication of H. pylori.67 However, it is important to recognize that the current literature in this field is less than optimal in several respects: older studies are limited by insufficient staging procedures and outdated classification systems, none of the reports in the literature is a controlled or randomized trial, and longer follow-up is necessary. Nevertheless, the current literature is sufficient to suggest to most experts in the field that early-stage disease is best managed with a trial of antibiotics, reserving more toxic therapies such as radiation, chemotherapy, or surgery for cases without concomitant H. pylori infection or those that do not respond to antibiotics.57,72 Table 29-3 summarizes treatment according to stage, according to the Lugano staging system.
Table 29-3 Treatment of Gastric Marginal Zone B Cell Lymphoma of MALT Type*
| LUGANO STAGE | TREATMENT†‡ |
|---|---|
| I, with disease limited to mucosa and submucosa | Antibiotics |
| I, with involvement of muscularis propria or serosa; IIE, II | Best treatment unknown at this time. Radiation or chemotherapy is probably a better option than surgery (see text). |
| IV | Chemotherapy for symptomatic disease. Local management with radiation or surgery may be indicated in selected cases. |
Patients with a high percentage of large cells and more advanced-stage disease should be treated as in Table 29-4 for diffuse large B cell lymphoma.
MALT, mucosa-associated lymphoid tissue.
* According to Lugano staging system.
† Patients with Helicobacter pylori infection should be treated with antibiotics to clear the infection, regardless of stage (see Chapter 50).
‡ Patients with a high percentage of large cells and disease limited to the mucosa may respond to antibiotics alone, although further study of this issue is necessary.
Stage I Disease
Most patients fall into this category and can be treated with antibiotic therapy aimed at eradication of H. pylori. Any one of the treatment regimens discussed in Chapter 50 may be used. Follow-up endoscopy with multiple biopsies should be done six to eight weeks after completion of therapy to document clearance of infection and to assess disease regression. Regression of lymphoma, but not necessarily complete regression, is usually evident at this examination. Patients with persistence of infection should be treated with a second-line antibiotic regimen (see Chapter 50).73 Histopathology at this examination can predict ultimate response, with biopsies showing only small foci of lymphoma being predictive of subsequent complete regression and biopsies showing diffuse persistent disease predicting a low likelihood of subsequent complete regression.74 Patients are then followed with endoscopy approximately every six months for two years and then yearly. Overall, approximately 75% of patients with stage I disease confined to the mucosa and submucosa will achieve complete remission. The median time to remission is five months, with remission usually occurring within 12 months; however, time to remission has been reported to be as long as 45 months.30,72,75 Of patients in clinical remission, 50% have tumor clones detected by PCR.76 With continued follow-up of these patients, the malignant clone decreases; current studies have suggested that a positive PCR at histologic remission does not predict for subsequent relapse, but longer follow-up of this issue is necessary.71 Approximately 90% of patients who had a complete clinical remission to H. pylori eradication remain in remission, with a median follow-up of three years; however late relapses can occur. Relapse may occur in association with H. pylori reinfection and can be cured by eradicating the organism again.77 In the absence of H. pylori reinfection, relapse is frequently transient.78
Approximately 25% of patients do not respond to H. pylori eradication. Lack of response is more common in patients with t(11;18) translocation; in one study, 67% of nonresponders harbored this abnormality, whereas only 4% of responders did.42 Lack of response to H. pylori eradication is also seen in patients with translocations t(1;14) and t(1;2)72; lymphomas bearing these translocations can be detected by immunohistochemical staining for nuclear BCL-10.79 Lack of response to H. pylori eradication may also indicate higher grade lesions or more extensive disease (i.e., involving the gastric wall and regional lymph nodes).
The management of patients with localized disease but a significant percentage of large cells is also uncertain. More recent studies have documented remission to H. pylori eradication,80,81 in contrast to earlier studies. For example, in one study of 16 patients with stage IE disease with high-grade histology, 10 of the patients had disease regression with H. pylori eradication and were free of lymphoma with a median follow-up of 43 months.80 Five of the 6 remaining patients responded to multiagent chemotherapy. If this approach is taken, the patient should be followed closely and, if the response is suboptimal, treated with one of the approaches discussed in the following section.
Occasional cases of gastric MALT lymphoma are H. pylori–negative. As would be expected, these patients are much less likely to respond to antibiotic treatment30; however, optimal management remains undefined.
Locally Advanced Disease—Stage I with Involvement of Muscularis or Serosa or Poor Response to H. pylori Eradication (Stage IIE).
Patients with more advanced-stage disease who are H. pylori–positive should also receive antibiotic therapy against H. pylori, but antibiotic therapy alone is usually not sufficient to eradicate the lymphoma. There is currently no consensus regarding the optimal management of this group of patients. Total gastrectomy can cure more than 80% of patients with stage IIE disease but diminishes patients’ quality of life and has not been shown to achieve superior results when compared with more conservative approaches.82,83 Involved field radiation therapy (30 to 40 Gy delivered in four weeks to the stomach and perigastric nodes) produces excellent results with a complete remission rate of 90% to 100% and a five-year disease-free survival of approximately 80%.84–86 Radiation therapy is usually well tolerated and preserves gastric function. Thus, it has become the preferred therapy for patients with advanced-stage disease, those who are negative for H. pylori, and those with persistent disease despite H. pylori treatment.72 Other treatment options in this group include chemotherapy, immunotherapy, or combined chemoimmunotherapy. Single-agent oral chemotherapy using cyclophosphamide87 or chlorambucil has activity,88 as does treatment with purine analogs.89 Immunotherapy with rituximab, a monoclonal antibody against CD20, is also highly efficacious,90,91 although rituximab given with chemotherapy, a common combination in the treatment of aggressive lymphomas, has not been widely tested in low-grade gastric NHLs. The International Extranodal Lymphoma Study Group is currently evaluating the efficacy of this combination in marginal zone lymphomas.
DIFFUSE LARGE B CELL LYMPHOMA OF THE STOMACH
Epidemiology
Approximately 50% of gastric lymphomas are diffuse large B cell lymphomas (DLBCLs). The incidence may be higher in developing than in developed nations, but clinical features appear to be similar.92,93 The median age is approximately 60 years, with a slight male predominance.15,94
Cause and Pathogenesis
The pathogenesis of gastric diffuse large B cell lymphoma is poorly understood.95 Many large cell tumors have components of low-grade MALT tissue and are assumed to have evolved through transformation of low-grade lesions. Frequently, these bear identical rearranged immunoglobulin genes. According to the WHO classification, this is now referred to as diffuse large B cell lymphoma with areas of marginal zone–MALT-type lymphoma.96 However, other DLBCLs have no evidence of associated low-grade MALT tissue. It is unclear whether de novo gastric DLBCL has a worse prognosis than DLBCL with areas of marginal zone–MALT-type lymphoma.68,97 Most studies have suggested this to be the case, but it is unclear what percentage of large cells confers a poorer prognosis.
If the large cell lesions commonly arise from progression of low-grade lesions, then conceivably H. pylori may have a role in the initial pathogenesis. One study has suggested that H. pylori infection is more common in patients whose large cell lesions had a low-grade component.97 As outlined earlier in the discussion of tentative models for H. pylori–induced lymphoma, large cell transformation resulting from genetic events, including loss of p53 and p16, may lead to loss of tumor cells losing their dependence on H. pylori for growth.98 A high incidence of somatic mutations in rearranged immunoglobulin heavy-chain variable genes in one study of diffuse large B cell lymphoma of the stomach has implicated antigen selection in the genesis of the lymphoma. Finally, observation of a response of early-stage large cell lymphomas to H. pylori eradication has suggested a role for the organism, at least in some cases.80,81
Pathology
Diffuse large B cell lymphoma may appear grossly as large ulcers, protruded tumors, or multiple shallow ulcers.99 The most common sites of involvement are the body and antrum of the stomach. Tumors with a low-grade component are more likely to be multifocal than tumors with no low-grade component. Large cell lymphomas typically invade the muscularis propria layer or even more deeply.
Microscopic examination reveals compact clusters, confluent aggregates, or sheets of large cells that resemble immunoblasts or centroblasts, most often with a mixture of the two.99 From 25% to 40% of cases show evidence of derivation from MALT, including dense infiltration of centrocyte-like cells in the lamina propria and typical lymphoepithelial lesions.97
Immunophenotypic analysis shows expression of one or more B cell antigens (CD19, CD20, CD22, CD79a) and CD45.100 Lesions with evidence of low-grade MALT tissue do not express CD10, consistent with their having evolved from the CD10-negative marginal zone low-grade lesions. Lesions without evidence of MALT may or may not express CD10. Genetic analysis reveals monoclonal immunoglobulin gene rearrangements. Bcl-6 is frequently mutated or rearranged.101
It is worth discussing the evolution in terminology regarding diffuse large B cell lymphomas of the stomach. Many pathologists have referred to lymphomas arising in MALT with high-grade features (with or without a component of low-grade disease) as high-grade gastric MALT lymphomas. However, those involved in the development of the WHO classification were concerned that many clinicians had come to regard the term gastric MALT lymphoma as synonymous with a lesion that responds to antibiotics. This is usually not the case with high-grade lesions arising in MALT. Therefore, those involved in formulating the WHO classification96 agreed to use the term extranodal marginal zone B cell lymphoma of MALT type for low-grade lesions, and the term diffuse large B cell lymphoma for high-grade lesions, leaving out the term MALT. Low-grade lesions involving MALT often contain varying proportions of large cells, with a worse prognosis in relation to increased percentage of large cells. However, at this point, a precise grading system for this situation has not been devised and remains a goal of ongoing research.
Clinical Features
Patients present with epigastric pain (70%) or dyspepsia (30%), symptoms similar to those patients with gastric adenocarcinoma.102,103 Large tumors may cause obstruction. Ulcerating lesions may be associated with GI bleeding. B symptoms (fevers, night sweats, weight loss) and elevated serum LDH concentrations are uncommon.
Staging consists of EGD, upper airway examination, CT scans of the chest, abdomen, and pelvis or PET scan, bone marrow aspiration and biopsy, and measurement of the serum LDH level. In addition, endoscopic ultrasonography plays an important role in assessing depth of stomach wall involvement. Lastly, H. pylori infection should be assessed. It is detected in 35% of patients with DLBCL of the stomach and is more common in those with concomitant gastric MALT.104 Most patients have stage I or II disease by the Ann Arbor Staging System (see Table 29-2).105 However, other staging systems have been developed and the use of various systems has made it difficult to compare results of different series. In 1994, a Workshop at the International Conference on Malignant Lymphoma proposed a modification to Blackledge’s system, known as Lugano staging (see Table 29-2).11
Treatment
The optimal management of DLBCL of the stomach is controversial, but the current consensus recommends chemoimmunotherapy with or without radiotherapy as a replacement for surgery (Table 29-4).103 Traditionally, localized disease was approached with surgery alone or surgery followed by radiation and/or chemotherapy for patients with poor prognostic features.106 This approach had the advantage of providing diagnostic and staging information and avoided the risk of perforation or bleeding that was believed to result from treatment with chemotherapy or radiation. Approximately 70% of patients with stage I disease are disease-free five years after surgery. However, several investigators have questioned the role of surgery in the management of localized gastric DLBCL. They noted that with the availability of endoscopy, surgery was no longer necessary for diagnosis and, with the availability of CT and EUS, surgery was no longer necessary for staging. In addition, the risk of bleeding or perforation during chemotherapy is lower than 5% and only a few of those who bleed require urgent gastrectomy.107 Surgery, however, carries a 5% to 10% risk of mortality and is associated with significant morbidity.
Table 29-4 Treatment of Diffuse Large B Cell Lymphoma of the Stomach*
| LUGANO STAGE | TREATMENT |
|---|---|
| I | CHOP† × 3-4 cycles + XRT‡ + rituximab§ |
| II, II1, II2, IIE | CHOP × 3-4 cycles + XRT + rituximab |
| IV | CHOP × 6-8 cycles + XRT + rituximab |
* According to the Lugano staging system, optimal management of this entity is controversial. However, the developing consensus seems to favor combined chemotherapy and radiation and avoidance of surgery (see text).
† Cyclophosphamide, doxorubicin [hydroxydaunorubicin], vincristine [Oncovin], prednisone).
‡ XRT (radiotherapy); usually, 30-40 Gy in 20-30 fractions.
§ The suggestion for the addition of rituximab in this setting involves extrapolation of randomized data from nodal diffuse large B cell lymphoma.
Thus, chemotherapy and radiation were investigated as alternatives to surgery. Retrospective studies have shown similar outcomes in patients treated with surgery alone versus chemotherapy alone.94 A prospective randomized study of patients with DLBCL of the stomach who were randomized to surgery, surgery plus radiotherapy, surgery plus chemotherapy, and chemotherapy alone showed improved complete response rates and overall survival for patients who received surgery plus chemotherapy and chemotherapy alone when compared with those who received surgery alone or surgery plus radiotherapy.108 The German Multicenter Study GIT NHL 01/92 was a prospective nonrandomized study of surgery in conjunction with chemotherapy and radiation versus chemotherapy and radiation alone for primary gastric lymphoma in localized stages. Whether the treatment included surgery was left to the discretion of each participating center. There was no difference in survival rate between those who received surgery followed by chemoradiotherapy and those who received chemoradiotherapy alone.109 These results were confirmed in a second, larger, prospective nonrandomized trial, GIT NHL 02/96.110
For patients with advanced-stage nodal DLBCL, the addition of rituximab, a monoclonal antibody against CD20, to CHOP (cyclophosphamide, doxorubicin [hydroxydaunorubicin], vincristine [Oncovin], prednisone) chemotherapy has been shown to improve overall survival when compared with CHOP alone.111–113 This combination has also been administered to patients with gastric DLBCL and found to be safe and effective.114
The necessity of radiation therapy in the management of gastric DLBCL is also controversial. A small retrospective study of patients with stage I or II primary gastric high-grade DLBCL treated with chemotherapy with or without radiotherapy has shown decreased relapse rates in patients who received consolidative radiotherapy.107 However, this study included only 21 patients, of whom 3 relapsed, and it is thought that a prospective randomized trial is needed.
Thus, standard management of gastric large B cell lymphoma follows standard management of nodal large B cell lymphomas. The treatment of localized (stage I or II) nodal large B cell lymphoma consists of three to six cycles of combination chemotherapy (typically the CHOP regimen) given with rituximab, with or without consolidative radiotherapy.115
DLBCL patients with evidence of H. pylori infection should be treated. Response of large cell lymphoma has been reported after eradication of H. pylori.81,116 However, these studies must be considered preliminary, and most patients treated with antibiotics alone have had disease limited to the mucosa; most patients with DLBCL of the stomach have more advanced disease, and antibiotics alone are considered inadequate treatment.
SMALL INTESTINAL LYMPHOMAS
Small intestinal lymphomas may be divided into B cell tumors and T cell tumors. The B cell tumors include immunoproliferative small intestinal disease (IPSID) and various non-IPSID subtypes, including marginal zone B cell lymphoma of MALT, DLBCL, mantle cell lymphoma, follicular lymphoma, and Burkitt’s lymphoma. Relatively few reports have described the various non-IPSID small intestinal lymphomas, and large series have tended to group together all the lymphoma subtypes when cataloguing manifestations and treatment outcomes.119–121 Given the lack of information about these diseases with regard to their behavior in the intestine, it is probably best to consider them in light of the well-described features of their nodal counterparts. Thus, marginal zone and follicular lymphomas are regarded as indolent processes, incurable but controllable by chemotherapy, and often associated with a relatively long survival. Diffuse large B cell lymphomas, mantle cell lymphomas, and Burkitt’s lymphomas are more aggressive processes, which generally require chemotherapy as part of their management. T cell lymphomas of the small intestine are usually enteropathy-type intestinal T cell lymphomas; other forms of T cell lymphoma have been rarely reported. Recent reports have suggested the existence of a rare natural killer (NK) cell or NK-type T cell intestinal lymphoma.122–125
MARGINAL ZONE B CELL LYMPHOMA OF MALT TYPE
Lymphoma arising in the small intestine may have the characteristics of marginal zone B cell lymphoma, with the same histologic and immunophenotypic features described earlier for gastric marginal zone B cell lymphoma.124 However, an association with H. pylori infection has not been documented, although rare responses to antibiotics have been reported. Most cases occur in older patients who present with melena. The disease is usually present as a single annular or exophytic tumor,126 which may be present anywhere in the small intestine; disease is usually confined to the intestine or to local nodes. Treatment is generally surgical. Some patients have received chemotherapy, but few data are available regarding regimens and outcome. It should be noted that in nodal marginal zone lymphoma, chemotherapy is usually reserved for patients with symptoms, because the disease is understood to be slow-growing and sensitive to chemotherapy, but not curable by it. The five-year survival rate is approximately 75%. As in gastric marginal zone B cell lymphoma, the small intestinal variety may have varying components of large cell transformation. This feature probably confers a worse prognosis, but data are scanty.
DIFFUSE LARGE B CELL LYMPHOMA
DLBCL of the small intestine is similar to its gastric counterpart in histology and clinical behavior. Patients may present with abdominal pain, weight loss, obstruction, abdominal mass, bleeding, and/or perforation. The tumor is usually an exophytic or annular lesion. Histologic findings are similar to those described earlier for gastric diffuse large B cell lymphoma, with some patients having a low-grade component and others having only a large-cell component. Approximately half of patients have localized disease, and half have disease spread to regional or distant nodes. Surgery is usually required, and additional therapy includes anthracycline-containing chemotherapy and the anti-CD20 monoclonal antibody, rituximab.119 In addition, radiotherapy is sometimes indicated. Prognosis depends on disease stage and patient factors, such as age and performance status.
MANTLE CELL LYMPHOMA
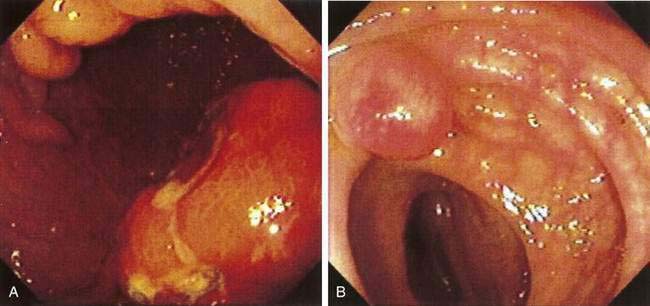
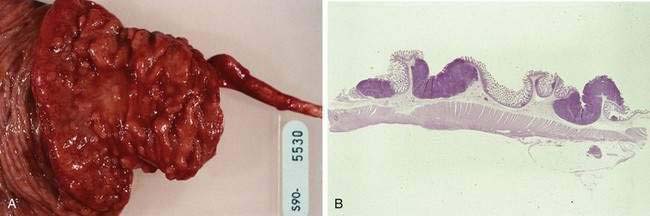
Mantle cell lymphoma is a relatively recently described subtype of B cell NHL.127 Patients typically present with widespread adenopathy and frequently have bone marrow and extranodal involvement. The GI tract is involved in more than 80% of patients, although not all patients with GI involvement are symptomatic.128 The most common manifestation of GI disease is multiple lymphomatous polyposis, in which multiple lymphoid polyps are present in the GI tract (Fig. 29-4).129,130 The most common site of involvement is the ileocecal region, but any other area may be involved from the stomach to the rectum; occasional patients have involvement of all these regions (Fig. 29-5). Involvement of the GI tract may also occur without the appearance of multiple polyps, and the GI tract as the only site of involvement has been reported. When patients have symptoms related to GI involvement, they usually include pain, obstruction, diarrhea, or hematochezia. It should be noted that multiple lymphomatous polyposis can also be seen with other lymphomas, especially marginal zone B cell lymphomas of MALT and follicular lymphomas. Microscopically, mantle cell lymphoma involves the mucosa and submucosa and the malignant cells have the appearance of small atypical lymphocytes, which may surround benign-appearing germinal centers or may efface the lymphoid tissue. The tumor cells express pan-B markers and the T cell marker CD5. The disease is characterized by t(11;14), a translocation that results in rearrangement and overexpression of the bcl-1 gene encoding cyclin D1.131 Patients with obstructive tumor masses require surgical therapy, but the mainstay of treatment is chemotherapy. Although mantle cell lymphoma is initially responsive to chemotherapy, it eventually becomes refractory; median survival is three to five years.
FOLLICULAR LYMPHOMA
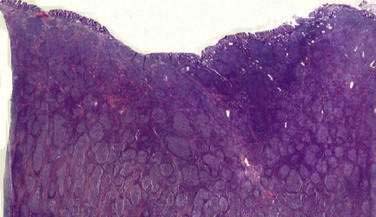
Follicular B cell lymphomas of the GI tract are rare.132 The most common presentation is as an obstructing lesion in the terminal ileum. As noted, patients with this diagnosis may also present with the gross appearance of multiple lymphomatous polyposis. Microscopically, most follicular lymphomas are composed of small cleaved lymphocytes, or centrocytes (Fig. 29-6), with a varying admixture of large cells. The disease is characterized by t(14;18), a translocation that results in overexpression of the bcl-2 gene.133 Obstructing lesions require surgical management. Chemotherapy and radiation are sometimes indicated for the management of this indolent but incurable disorder.
BURKITT’S LYMPHOMA
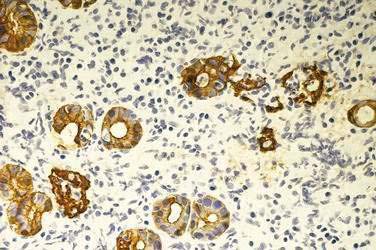
Burkitt’s lymphoma is a highly aggressive malignancy that in patients who are HIV-negative, presents as an endemic form, observed in Africa, or a sporadic form.134 In the sporadic form, patients usually present with disease in the abdomen, with involvement of the distal ileum, cecum, mesentery, or cecum and mesentery. Burkitt’s tumor cells are monomorphic, medium-sized cells with round nuclei, multiple nucleoli, and basophilic cytoplasm (Fig. 29-7). The involved lymphoid tissue microscopically has a starry sky appearance caused by numerous benign macrophages that have ingested apoptotic tumor cells.135 The tumor cells express B cell–associated antigens and surface immunoglobulin. Most cases have a translocation of the c-myc gene, either to the immunoglobulin heavy-chain region on chromosome 14 or to one of the immunoglobulin light-chain regions on chromosomes 2 or 22, resulting in a t(8;14), t(2;8), or t(8;22) translocation.136 Burkitt’s lymphoma is rapidly fatal without treatment but responds immediately to institution of aggressive chemotherapy. Treatment carries a high risk of tumor lysis syndrome. Cure rates are 50% to 90%, depending on the extent of the disease.137,138
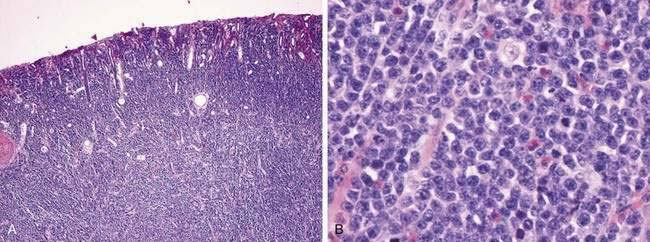
IMMUNOPROLIFERATIVE SMALL INTESTINAL DISEASE
Epidemiology
Immunoproliferative small intestinal disease (also known as α heavy-chain disease and as Mediterranean lymphoma) is confined to certain regions of the world, especially North Africa, Israel, and surrounding Middle Eastern and Mediterranean countries.139 IPSID is seen less often in other areas, including Central and South Africa, India and East Asia, and South and Central America. A diagnosis in North America or Europe should be questioned, unless the patient has previously lived in an endemic area. The disease occurs in individuals with lower socioeconomic status who live in conditions of poor hygiene and sanitation.140 The disease generally occurs in the second or third decade of life, although it has been observed in older individuals. The incidence in males and females is equal.
Cause and Pathogenesis
Several observations have led to the belief that IPSID may be initiated by an infectious agent or agents: (1) an association of the disease with lower socioeconomic status and poor sanitation; (2) a high prevalence of intestinal bacterial overgrowth and parasitosis; (3) a decrease in incidence when living conditions have improved in endemic areas; and (4) a response of early lesions to antibiotic therapy. In addition, it is known that bowel flora stimulate IgA-producing cells, and intestinal biopsies from apparently normal individuals from endemic regions have shown an increase in lamina propria lymphocytes and plasma cells, reminiscent of findings in patients with IPSID. One study detected the presence of Campylobacter jejuni in five of seven patients with IPSID, suggesting a role for this organism in the disease.141
As discussed later, IPSID is associated with the production of an unusual IgA heavy-chain protein, called α heavy chain, which is secreted by plasma cells and is detectable in various body fluids.142,143 The plasma cells, which are the predominant histologic feature in the superficial mucosa, possess surface and cytoplasmic α chain protein. Centrocyte-like cells proliferating deeper in the mucosa have mainly cytoplasmic α chain protein. It is likely that these centrocyte-like cells, stimulated by microbial antigens, differentiate into the plasma cells that secrete the α chain protein characteristic of the disease. Genetic analyses have revealed that cellular proliferations are monoclonal, even in early lesions.144,145
Thus, it can be proposed that in a way somewhat analogous to H. pylori–associated gastric MALT, lymphocytes in intestinal MALT may be stimulated by infectious agents, in particular Campylobacter jejuni,141 and proliferate in response. The lymphocytic response becomes monoclonal and initially depends on the presence of antigen. However, with time, the malignant cells acquire additional genetic changes, causing them to lose their dependence on antigen persistence. This loss of antigen dependence is associated with the development of more aggressive clinical features.
Pathology
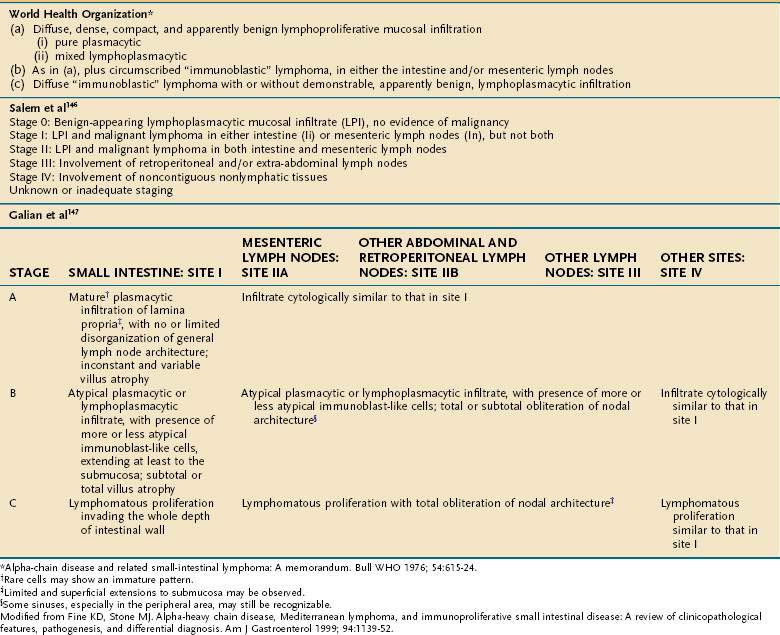
Gross lesions are generally confined to the proximal small intestine, with adenopathy of adjacent mesenteric nodes.146 Although some patients have thickening of mucosal folds only, others have a generalized thickening of the bowel wall, discrete masses, nodules, or polypoid lesions. Although grossly only the proximal bowel wall is involved, histologically the disease is characterized by a dense mucosal and submucosal cellular infiltrate that extends continuously throughout the length of the small intestine. Various pathologic staging systems have been proposed (Table 29-5).146,147 In early-stage disease, the cellular infiltrate is composed of benign-appearing plasma cells or lymphoplasmacytic cells. However, as noted, various studies assessing immunoglobulin gene rearrangements or light chain restriction have suggested that even the earliest infiltrate is monoclonal. This early infiltrate broadens villi and shortens and separates crypts, but epithelial cells remain intact. A histologic variant, the follicular lymphoid type, has been described in some patients (see Fig. 29-7). This variant features a diffuse involvement of the mucosa, with lymphoid follicle-like structures. As the disease progresses to intermediate and late stages, the villi are further broadened and may become completely effaced, crypts are fewer, and the immunoproliferation extends more deeply. Atypical lymphoid cells infiltrate the benign-appearing plasma cells and lymphoplasmacytic cells. With time, the process evolves into overt lymphoma. Mesenteric lymph nodes are enlarged in early lesions, with preserved architecture, although follicles may be shortened by a histologically benign-appearing lymphocytic or plasmacytic infiltrate. As the disease progresses, the lymph node may acquire a more dysplastic appearance.
Clinical Features
Patients usually present with diarrhea, colicky abdominal pain, anorexia, and significant weight loss, with a duration of symptoms from months to years. The diarrhea initially may be intermittent but becomes voluminous and foul-smelling as malabsorption develops. About half of patients have fever. Physical examination reveals evidence of malnutrition, digital clubbing, and peripheral edema. Late physical manifestations are ascites, hepatosplenomegaly, an abdominal mass, and peripheral lymphadenopathy. Endoscopy may reveal thickened mucosal folds, nodules, ulcers, or evidence of submucosal infiltration, rendering the intestine immobile, tender, and indistensible. Small bowel barium radiographs show diffuse dilation of the duodenum, jejunum, and proximal ileum, with thickened mucosal folds. Patients are frequently anemic because of vitamin deficiencies, and the erythrocyte sedimentation rate is elevated in one third of cases. The circulating lymphocyte count is low, and measures of humoral and cellular immunity are impaired. Stool examination frequently reveals Giardia lamblia infestation. As noted, C. jejuni has been implicated in a high percentage of patients by PCR assay, DNA sequencing, fluorescence in situ hybridization, and immunohistochemical studies on intestinal biopsy specimens.141 Serum IgG and IgM levels may be high or low; IgA levels are usually low or undetectable.
The characteristic and unique laboratory abnormality is the presence of the α chain protein.148 This 29- to 34-kd protein is a free α1 heavy chain with an internal deletion of the variable (VH) and CH1 regions. It is devoid of light chains and thus corresponds to the Fc portion of the α1 subunit of IgA. The α chain protein amino terminal contains sequences that are not homologous to any known immunoglobulin sequence. These changes are often the result of insertions or deletions, usually involving the VH-JH and CH2 regions,149 but the source of inserted genetic material is unknown.
The α chain production migrates as a broad band within the α2 and β regions on serum protein electrophoresis. In addition to electrophoresis, the protein can be detected by immunoelectrophoresis or immunoselection (the most sensitive and specific methods)149 in serum, urine, saliva, or intestinal secretions. Detection of α chain protein from these sources is more likely in patients with early disease than in patients with more advanced disease, but, regardless of stage, α chain protein can be detected in tissue sections in most cases of IPSID by immunofluorescence or immunoperoxidase staining of plasma or lymphoma cells.148
It has been postulated that chronic antigenic stimulation of the intestinal IgA secretory apparatus results in expansion of several plasma cell clones. Eventually, a structural mutation occurs in a particular clone, resulting in an internal deletion of part of the α heavy chain. This leads to an inability to make light chains and results in secretion of α chain protein rather than intact IgA.149,150
Diagnosis and Staging
Because the more malignant-appearing histology may be present only in deeper layers of the intestine, endoscopic biopsy alone is often considered an inadequate evaluation; staging laparotomy is therefore strongly recommended by some authors to allow full-thickness intestinal biopsy and biopsy of mesenteric lymph nodes.151 However, it should be noted that some investigators do not routinely perform laparotomies; instead, upper and lower endoscopy, small bowel series, bone marrow biopsies, and fine-needle aspiration of enlarged lymph nodes are performed.152 One of the staging systems may then be applied (see Table 29-5). More advanced disease, poor performance status, and comorbid illnesses portend a worse prognosis.
Treatment
Because of the relative rarity of this lymphoma, no large trials investigating therapy have been carried out.152,153 Patients often require intensive nutritional support.154 Patients with early disease (e.g., Salem stage 0 disease; see Table 29-5) are generally treated with antibiotics for six months or more. The two most commonly used regimens are tetracycline alone and a combination of metronidazole and ampicillin. Response rates have ranged from 33% to 71%139; in one study, the complete response rate was 71%, with disease-free survival of 43% at five years.152 In patients who do not significantly improve by six months or who do not achieve complete remission by 12 months, or who have advanced disease at presentation, chemotherapy should be given. Most investigators recommend anthracycline-containing regimens such as CHOP.155,156 For example, one investigator has reported a complete response of 67% and a survival of 58% at 3.5 years in patients treated with antibiotics, total parenteral nutrition, and anthracycline-based combination chemotherapy.156 However, good results have been reported with non–anthracycline-containing regimens as well; in one report, 56% of patients with advanced disease were free of disease at five years.152 Finally, total abdominal radiotherapy has been used in a small number of patients but, on the basis of current data, it is difficult to assess its proper role.157
ENTEROPATHY-TYPE INTESTINAL T CELL LYMPHOMA
Enteropathy-type T cell lymphoma occurs as a complication of celiac disease (see Chapter 115).158 Malignant transformation of intraepithelial T cells leads to an aggressive malignancy, causing most patients to die within a few months of diagnosis.159,160 Treatment of celiac disease with a gluten-free diet may decrease the risk of this malignancy.161
Epidemiology
Enteropathy-type T cell lymphoma is a rare malignancy and comprises only 0.5% of all NHL cases.162–164 Celiac disease has a prevalence of 0.5% to 1% in the United States and Europe158,165,166 and is more common in whites compared with African Americans and Asians. In a cohort of more than 1000 patients with celiac disease, the most common cause of death was NHL.167 The diagnosis of lymphoma is usually made concomitantly with or shortly after the diagnosis of celiac disease,168 although the two conditions are commonly diagnosed simultaneously, especially in patients who have a long history of malabsorption. Adherence to a gluten-free diet for more than five years appears to reduce the risk of developing lymphoma.161 Males are affected twice as often as females.160 The peak incidence occurs in the sixth or seventh decade of life.169
Cause and Pathogenesis
Enteropathy-type intestinal T cell lymphoma occurs in patients with adult celiac disease.164 As discussed in Chapter 104, celiac disease is characterized by a hereditary sensitivity to gluten.170 Gluten peptides are presented by celiac disease–specific HLA-DQ2 and HLA-DQ8 positive antigen-presenting cells and thus elicit an immune response in which gluten-specific intraepithelial lymphocytes damage intestinal epithelium. Intraepithelial T cells in celiac disease have a normal immunophenotype (CD3+/CD8+) and are polyclonal.171,172 Malignant transformation of intraepithelial T cells results in a monoclonal population of intraepithelial T cells that have an abnormal phenotype.173–176 Monoclonal populations of intraepithelial T cells in celiac mucosa may result in any one of several interrelated processes.176,177 The first condition is refractory celiac disease, a condition in which patients lose responsiveness to a gluten-free diet.178 The second condition, ulcerative jejunitis, is characterized by inflammatory jejunal ulcers and unresponsiveness to a gluten-free diet.179 The third condition is enteropathy-type intestinal T cell lymphoma, an aggressive malignancy of the small intestine.174,175 In patients with any of these three conditions, uninvolved mucosa adjacent to the lesions can contain monoclonal T cells containing the same rearranged T cell receptor genes.180 In addition, patients with ulcerative jejunitis can subsequently develop enteropathy-type intestinal T cell lymphoma, in which the same clone is isolated in the jejunitis and the subsequent lymphoma. Thus, these three conditions have come to be considered to represent a spectrum of disorders mediated by monoclonal intraepithelial T cells.
Comparative genomic hybridization studies have shown recurrent chromosomal gains in enteropathy-type intestinal T cell lymphoma at chromosomes 9q, 7q, 5q, and 1q and recurrent losses at 8p, 13q, and 9p. A gain at 9q is the most common, seen in 58% of cases examined.181 Another study has shown that loss of heterozygosity at chromosome 9q21 is a frequent finding.182 In addition, one study has suggested that gain of chromosome 1q may be an early event in the genesis of enteropathy-type intestinal T cell lymphoma lymphomagenesis.183
Pathology
Tumors typically occur in the jejunum but may occur in other sites of the small intestine. Lymphoma may occur in single or multiple sites. Grossly, the lymphomas commonly appear as ulcerating lesions, with circumferential involvement of the small bowel.160 Lesions may also appear as nodules, plaques, or strictures, but large masses are uncommon. Mesenteric lymph nodes are often enlarged, either because of tumor involvement or of edema and reactive changes. Distant sites, especially the bone marrow or the liver, are sometimes involved.
Histologically, the lymphoma is generally characterized by large, highly pleomorphic cells with numerous, bizarre, multinucleated forms.184,185 However, cells sometimes have a more immunoblastic, anaplasti, or, less commonly, small cell appearance (Fig. 29-8). Eosinophils and other inflammatory cells are often present. Histologic features often vary within the same tumor and between multiple lesions of the same patient. Uninvolved mucosa usually has the typical appearance of celiac disease, with villous atrophy, crypt hyperplasia, plasmacytosis in the lamina propria, and an increase in intraepithelial lymphocytes. However, the enteropathy may be subtle in some cases, with only an increase in the intraepithelial lymphocytes.
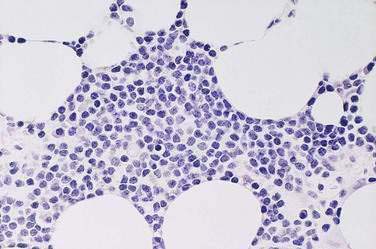
Immunophenotyping typically shows that the malignant cells are CD3+, CD7+, CD4−, CD5−, and contain cytotoxic granules recognized by the antibody TIA-1.173 Surface CD3 may be negative but cytoplasmic CD3e chains are usually detectable.174 Cells are CD8+ in a minority of cases and a small percentage of cases coexpress CD8 and CD56.186 CD103 is commonly positive. Cases with anaplastic morphology are CD30+. Genotypic studies have shown monoclonal T cell receptor gene rearrangements.180,187,188 As noted, monoclonal T cell populations can also be detected in mucosa not involved by lymphoma.
Clinical Features
Patients may have a history of documented celiac disease, with the time to development of lymphoma varying widely. However, at least half of patients have celiac sprue diagnosed at the same time as the lymphoma. The most common symptoms at presentation are abdominal pain, weight loss, diarrhea, or vomiting. Less common symptoms may include fever, night sweats, and small bowel obstruction or perforation. It is rare for patients to have palpable abdominal masses or peripheral lymphadenopathy, but extraintestinal sites of involvement may include the liver, spleen, thyroid, skin, nasal sinus, and brain.158 In one series, 68% of patients had anemia and 25% of patients had elevations of the serum lactate dehydrogenase level.160 Hypoalbuminemia is a common finding at presentation.
Diagnosis is usually made by endoscopic biopsies or full-thickness, laparoscopic small bowel biopsies, but approximately 20% of patients have been diagnosed with barium studies and small bowel biopsies. Traditionally, patients were staged with CT and bone marrow biopsies, but 18F-FDG PET appears to be more sensitive and specific than CT in differentiating enteropathy-associated intestinal T cell lymphoma from refractory celiac disease.189 In the two largest recent clinical studies, the Ann Arbor staging system was used to stage patients (see Table 29-2). In these two combined studies, 9% of the patients had Ann Arbor stage IE disease, 62% had stage IIE disease, and 29% had stage IV disease.160
Treatment
No large controlled trials of therapy for enteropathy-type intestinal T cell lymphoma have been reported. Thus, standard treatment is not well defined. Typically, patients are treated with a combination of surgery and chemotherapy.160 Surgery involves removal of as much tumor as is feasible. Intensive chemotherapy is then administered, with the most common regimens being ones that contain anthracyclines such as CHOP. There is no evidence for superiority of any particular chemotherapy regimen. In one study, in which 24 patients received chemotherapy, small bowel perforation developed in 4 patients and in 3 of these the perforation occurred soon after receiving chemotherapy and was fatal.160 Nutritional status is commonly poor, requiring parenteral nutrition. Because of poor nutritional and performance status, less than 50% of patients are able to complete the prescribed treatment regimen. In one study, the overall response rate to chemotherapy was 58%, with a complete remission rate of 42%.160 However, relapse occurs at a median of six months from the time of diagnosis in approximately 80% of patients, usually in small bowel sites. Various salvage regimens have been tried for patients with relapsed disease, but few relapsed patients have survived. The actuarial one- and five-year survival rates in one study were 39% and 20%, respectively, with one- and five-year failure-free survival rates of only 19% and 3%, respectively. Another series from the United Kingdom found that survival at 2.5 years was only 13%.168 Thus, the prognosis for this lymphoma is poor. There are a few reports of patients treated with high-dose chemotherapy followed by autologous stem cell transplantation, usually with poor outcomes.190–192 Therapy with novel agents are needed. Alemtuzumab (Campath), an anti-CD52 monoclonal antibody, has been used to treat refractory celiac disease.193 Conceivably, earlier diagnosis may improve the outcome. The diagnosis should be considered for patients who present in midlife with celiac disease and for those who have clinical deterioration after having been stable on a gluten-free diet.
UNCOMMON SMALL INTESTINAL LYMPHOMAS
Natural Killer Type T Cell Intestinal Lymphoma
Extranodal NK T cell lymphoma, nasal type, is a distinct pathologic entity in the World Health Organization classification of hematolymphoid malignancies.96 Very rare cases of intestinal NK cell lymphomas have been described.125 Most of the cases reported have not involved patients with celiac sprue or sensitivity to gluten.123,124 Optimal management of this very rare disorder has not been determined. Most patients undergo resection, with some receiving adjuvant chemotherapy.122
OTHER SITES
Waldeyer’s ring lymphomas are usually diffuse large cell lymphomas, but other histologies may be present instead.194,195 Endoscopy and imaging of the remainder of the GI tract should be included in the staging workup, because lymphomatous involvement in other sites may accompany Waldeyer’s ring involvement. Ann Arbor stage I or II diffuse large cell lymphoma is managed with combined anthracycline-based chemotherapy and/or local radiotherapy.115,196
Primary hepatic lymphoma (PHL; see Chapter 35 and 94) is more common in men and has a median age of approximately 50 years.197,198 PHL can present as a single, large, multilobulated mass or as single or multiple nodules. The histology is usually diffuse large B cell, but MALT lymphoma (extranodal marginal B cell lymphoma) has been reported as well (see Table 35-2). Rare cases of T cell hepatic lymphoma have been reported. Diagnosis is usually by needle biopsy. Because of the rarity of the disease, optimal therapy is uncertain. Long-term disease-free survival has been reported after resection, but multiagent chemotherapy is probably most appropriate for diffuse large B cell lymphoma. Less aggressive chemotherapy may be appropriate for lymphomas with marginal zone histology. The anti-CD20 monoclonal antibody, rituximab, has activity in B cell lymphomas and thus may have a role in the treatment of these diseases. An association of hepatitis C and hepatic and splenic marginal zone lymphoma has been established, and response of the lymphoma to hepatitis C treatment has been documented199,200; whether there may be an association of other hepatitis viruses and hepatic lymphomas is unknown.
As discussed in Chapter 60, the pancreas is a rare site of lymphoma.201–203 Patients have a clinical presentation similar to that of pancreatic adenocarcinoma, with abdominal pain and obstructive jaundice; chylous ascites has also been reported. Histology is usually diffuse large B cell, and therapy consists of anthracycline-containing combination chemotherapy (e.g., the CHOP regimen) with rituximab. Patients with biliary obstruction may require a biliary drainage procedure before being treated with chemotherapy to avoid excessive chemotherapy-related toxicity.
Primary colorectal lymphomas (see Chapter 123) most commonly involve the cecum,204,205 with high- or intermediate-grade histology. Most colorectal lymphomas are Ann Arbor stage IE or IIE. Again, therapy is dictated by histology and stage. Resection is the standard therapy, with adjuvant chemotherapy given for patients with aggressive histology.
IMMUNODEFICIENCY-RELATED LYMPHOMAS
POST-TRANSPLANTATION LYMPHOPROLIFERATIVE DISORDERS
The post-transplantation lymphoproliferative disorders (PTLDs)206–208 complicate 0.8% to 20% of cases of those with solid organ transplants (see Chapter 34), with the incidence being highest in heart-lung transplant recipients. PTLDs are also seen in bone marrow transplant recipients, particularly in patients receiving T cell–depleted allografts. PTLD results from proliferation of Epstein-Barr virus (EBV)–transformed B cell clones that have developed in part because of immunosuppression. The histologic appearance of PTLD is highly variable, with lesions being polymorphic or monomorphic; the histology may represent infectious mononucleosis, aggressive NHL, or plasmacytoma.209 Lesions may be polyclonal, oligoclonal, or monoclonal. The clinical presentation also varies greatly, with some patients having a syndrome resembling infectious mononucleosis and some having a more lymphoma-like presentation, with nodal or extranodal disease. Involvement of extranodal areas is common, with the GI tract being a common site. The literature regarding the treatment of PTLD suffers from a lack of prospective trials and lack of standardized histologic classification.
The treatment approach varies, but usually consists initially of withdrawal of immunosuppression210; anthracycline-based chemotherapy is then reserved for patients who fail to respond to this first maneuver. Surgical or radiation therapy may cure patients with localized disease. Other treatments have included acyclovir or ganciclovir directed at EBV and interferon-α. Monoclonal antibodies to B cells such as rituximab have activity in this disorder, and donor leukocyte infusions are frequently used for patients with PTLD that develops after allogeneic bone marrow transplantation.211
HUMAN IMMUNODEFICIENCY VIRUS–ASSOCIATED NON-HODGKIN’S LYMPHOMA
The risk of developing NHL is markedly increased in patients with HIV (see Chapter 33), and development of lymphoma is considered an AIDS-defining condition. These malignancies are B cell neoplasms,212,213 with most cases having small noncleaved cell or diffuse large cell histology. EBV is implicated in about half of non–central nervous system HIV-related lymphomas. HIV-associated NHL typically has an aggressive presentation, with rapidly growing disease and prominent B symptoms. The GI tact is a common site, including unusual sites such as the anus and rectum. Historically, chemotherapy has been poorly tolerated and lower dose chemotherapy regimens have been used.214,215 However, patients with higher CD4+ T cell counts (as is more commonly seen in view of the standard current usage of highly active antiretroviral therapy) may be more able to tolerate full-dose chemotherapy regimens and may have a better prognosis than has been seen in previous studies.216
Primary effusion lymphoma (PEL) is a rather recently described clinicopathologic entity associated with the herpesvirus HHV-8 (Kaposi’s sarcoma–associated virus).217,218 Histology shows a distinctive morphology that bridges large cell immunoblastic lymphoma and anaplastic large cell lymphoma.219 Tumor cells show monoclonal immunoglobulin gene rearrangements but typically lack B cell–associated antigens. HHV-8 is detectable by PCR assay. Patients are usually HIV-positive, but the syndrome has been reported in HIV-negative patients. Patients present with malignant effusions in the pleural or peritoneal cavity, which remain localized to the body cavity of origin. Disease progression is rapid, with survival of only a few weeks to months. Optimal therapy has not been defined.
Bertoni F, Zucca E. State-of-the-art therapeutics: Marginal-zone lymphoma. J Clin Oncol. 2005;23:6415-20. (Ref 57.)
Ferrucci PF, Zucca E. Primary gastric lymphoma pathogenesis and treatment: What has changed over the past 10 years? Br J Haematol. 2007;136:521-38. (Ref 14.)
Gale J, Simmonds PD, Mead GM, et al. Enteropathy-type intestinal T-cell lymphoma: Clinical features and treatment of 31 patients in a single center. J Clin Oncol. 2000;18:795-803. (Ref 160.)
Koch P, del Valle F, Berdel WE, et al. Primary gastrointestinal non-Hodgkin’s lymphoma: I. Anatomic and histologic distribution, clinical features, and survival data of 371 patients registered in the German Multicenter Study GIT NHL 01/92. J Clin Oncol. 2001;19:3861-73. (Ref 15.)
Koch P, del Valle F, Berdel WE, et al. Primary gastrointestinal non-Hodgkin’s lymphoma: II. Combined surgical and conservative or conservative management only in localized gastric lymphoma—results of the prospective German Multicenter Study GIT NHL 01/92. J Clin Oncol. 2001;19:3874-83. (Ref 109.)
Koch P, Probst A, Berdel WE, et al. Treatment results in localized primary gastric lymphoma: Data of patients registered within the German multicenter study (GIT NHL 02/96). J Clin Oncol. 2005;23:7050-9. (Ref 110.)
Lecuit M, Abachin E, Martin A, et al. Immunoproliferative small intestinal disease associated with Campylobacter jejuni. N Engl J Med. 2004;350:239-48. (Ref 141.)
Liu H, Ye H, Ruskone-Fourmestraux A, et al. T(11; 18) is a marker for all stage gastric MALT lymphomas that will not respond to H. pylori eradication. Gastroenterology. 2002;122:1286-94. (Ref 42.)
Nagai S, Mimuro H, Yamada T, et al. Role of Peyer’s patches in the induction of Helicobacter pylori–induced gastritis. Proc Natl Acad Sci U S A. 2007;104:8971-6. (Ref 19.)
Parsonnet J, Hansen S, Rodriguez L, et al. Helicobacter pylori infection and gastric lymphoma. N Engl J Med. 1994;330:1267-71. (Ref 26.)
Romaguera JE, Medeiros LJ, Hagemeister FB, et al. Frequency of gastrointestinal involvement and its clinical significance in mantle cell lymphoma. Cancer. 2003;97:586-91. (Ref 128.)
Wotherspoon AC, Doglioni C, Diss TC, et al. Regression of primary low-grade B-cell gastric lymphoma of mucosa-associated lymphoid tissue type after eradication of Helicobacter pylori. Lancet. 1993;342:575-7. (Ref 20.)
Wundisch T, Thiede C, Morgner A, et al. Long-term follow-up of gastric MALT lymphoma after Helicobacter pylori eradication. J Clin Oncol. 2005;23:8018-24. (Ref 32.)
Zucca E, Bertoni F, Roggero E, Cavalli F. The gastric marginal zone B-cell lymphoma of MALT type. Blood. 2000;96:410-19. (Ref 18.)
Zucca E, Bertoni F, Roggero E, et al. Molecular analysis of the progression from Helicobacter pylori–associated chronic gastritis to mucosa-associated lymphoid-tissue lymphoma of the stomach. N Engl J Med. 1998;338:804-10. (Ref 24.)
1. Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2008. CA Cancer J Clin. 2008;58:71-96.
2. Ducreux M, Boutron MC, Piard F, et al. A 15-year series of gastrointestinal non-Hodgkin’s lymphomas: A population-based study. Br J Cancer. 1998;77:511-14.
3. Isaacson PG. Gastrointestinal lymphomas of T- and B-cell types. Mod Pathol. 1999;12:151-8.
4. Picker L, Siegelman M. Lymphoid tissues and organs. In: Paul WE, editor. Fundamental immunology. 4th ed. Philadelphia: Lippincott-Raven; 1999:479.
5. Jacobs H, Bross L. Towards an understanding of somatic hypermutation. Curr Opin Immunol. 2001;13:208-18.
6. Jaffe ES, Harris NL, Stein H, Vardiman JW, editors. Pathology and genetics of tumours of haematopoietic and lymphoid tissues. Lyon: IARC Press, 2001.
7. van Besien K, Cabanillas F. Clinical manifestations, staging and treatment of non-Hodgkin’s lymphoma. In: Huffman R, Benz EJ, Shattil SJ, editors. Hematology: Basic principles and practice. New York: Churchill Livingstone; 2000:1263.
8. Rosenwald A, Wright G, Chan WC, et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N Engl J Med. 2002;346:1937-47.
9. Sackmann M, Morgner A, Rudolph B, et al. Regression of gastric MALT lymphoma after eradication of Helicobacter pylori is predicted by endosonographic staging. MALT Lymphoma Study Group. Gastroenterology. 1997;113:1087-90.
10. Carbone PP, Kaplan HS, Musshoff K, et al. Report of the Committee on Hodgkin’s Disease Staging Classification. Cancer Res. 1971;31:1860-1.
11. Rohatiner A, d’Amore F, Coiffier B, et al. Report on a workshop convened to discuss the pathological and staging classifications of gastrointestinal tract lymphoma. Ann Oncol. 1994;5:397-400.
12. A predictive model for aggressive non-Hodgkin’s lymphoma. The International Non-Hodgkin’s Lymphoma Prognostic Factors Project. N Engl J Med. 2008;329:987-94.
13. Cortelazzo S, Rossi A, Roggero F, et al. Stage-modified international prognostic index effectively predicts clinical outcome of localized primary gastric diffuse large B-cell lymphoma. International Extranodal Lymphoma Study Group (IELSG). Ann Oncol. 1999;10:1433-40.
14. Ferrucci PF, Zucca E. Primary gastric lymphoma pathogenesis and treatment: What has changed over the past 10 years? Br J Haematol. 2007;136:521-38.
15. Koch P, del Valle F, Berdel WE, et al. Primary gastrointestinal non-Hodgkin’s lymphoma: I. Anatomic and histologic distribution, clinical features, and survival data of 371 patients registered in the German Multicenter Study GIT NHL 01/92. J Clin Oncol. 2001;19:3861-73.
16. Isaacson PG, Spencer J, Wright DH. Classifying primary gut lymphomas. Lancet. 1988;2:1148-9.
17. A clinical evaluation of the International Lymphoma Study Group classification of non-Hodgkin’s lymphoma. The Non-Hodgkin’s Lymphoma Classification Project. Blood. 1997;89:3909-18.
18. Zucca E, Bertoni F, Roggero E, Cavalli F. The gastric marginal zone B-cell lymphoma of MALT type. Blood. 2000;96:410-19.
19. Nagai S, Mimuro H, Yamada T, et al. Role of Peyer’s patches in the induction of Helicobacter pylori–induced gastritis. Proc Natl Acad Sci U S A. 2007;104:8971-6.
20. Wotherspoon AC, Doglioni C, Diss TC, et al. Regression of primary low-grade B-cell gastric lymphoma of mucosa-associated lymphoid tissue type after eradication of Helicobacter pylori. Lancet. 1993;342:575-7.
21. Doglioni C, Wotherspoon AC, Moschini A, et al. High incidence of primary gastric lymphoma in northeastern Italy. Lancet. 1992;339:834-5.
22. Zaki M, Schubert ML. Helicobacter pylori and gastric lymphoma. Gastroenterology. 1995;108:610-12.
23. Wotherspoon AC, Ortiz-Hidalgo C, Falzon MR, Isaacson PG. Helicobacter pylori–associated gastritis and primary B-cell gastric lymphoma. Lancet. 1991;338:1175-6.
24. Zucca E, Bertoni F, Roggero E, et al. Molecular analysis of the progression from Helicobacter pylori–associated chronic gastritis to mucosa-associated lymphoid-tissue lymphoma of the stomach. N Engl J Med. 1998;338:804-10.
25. Eck M, Greiner A, Schmausser B, et al. Evaluation of Helicobacter pylori in gastric MALT-type lymphoma: Differences between histologic and serologic diagnosis. Mod Pathol. 1999;12:1148-51.
26. Parsonnet J, Hansen S, Rodriguez L, et al. Helicobacter pylori infection and gastric lymphoma. N Engl J Med. 1994;330:1267-71.
27. Hussell T, Isaacson PG, Crabtree JE, et al. Immunoglobulin specificity of low grade B cell gastrointestinal lymphoma of mucosa-associated lymphoid tissue (MALT) type. Am J Pathol. 1993;142:285-92.
28. Hussell T, Isaacson PG, Spencer J. Proliferation and differentiation of tumour cells from B-cell lymphoma of mucosa-associated lymphoid tissue in vitro. J Pathol. 1993;169:221-7.
29. Hussell T, Isaacson PG, Crabtree JE, Spencer J. Helicobacter pylori–specific tumour-infiltrating T cells provide contact dependent help for the growth of malignant B cells in low-grade gastric lymphoma of mucosa-associated lymphoid tissue. J Pathol. 1996;178:122-7.
30. Steinbach G, Ford R, Glober G, et al. Antibiotic treatment of gastric lymphoma of mucosa-associated lymphoid tissue. An uncontrolled trial. Ann Intern Med. 1999;131:88-95.
31. Bayerdorffer E, Neubauer A, Rudolph B, et al. Regression of primary gastric lymphoma of mucosa-associated lymphoid tissue type after cure of Helicobacter pylori infection. MALT Lymphoma Study Group. Lancet. 1995;345:1591-4.
32. Wundisch T, Thiede C, Morgner A, et al. Long-term follow-up of gastric MALT lymphoma after Helicobacter pylori eradication. J Clin Oncol. 2005;23:8018-24.
33. Montalban C, Santon A, Boixeda D, et al. Treatment of low-grade gastric mucosa–associated lymphoid tissue lymphoma in stage I with Helicobacter pylori eradication. Long-term results after sequential histologic and molecular follow-up. Haematologica. 2001;86:609-17.
34. Matsumoto T, Iida M, Shimizu M. Regression of mucosa-associated lymphoid-tissue lymphoma of rectum after eradication of Helicobacter pylori. Lancet. 1997;350:115-16.
35. Nagashima R, Takeda H, Maeda K, et al. Regression of duodenal mucosa-associated lymphoid tissue lymphoma after eradication of Helicobacter pylori. Gastroenterology. 1996;111:1674-8.
36. Morgner A, Lehn N, Andersen LP, et al. Helicobacter heilmannii-associated primary gastric low-grade MALT lymphoma: Complete remission after curing the infection. Gastroenterology. 2000;118:821-8.
37. Qin Y, Greiner A, Trunk MJ, et al. Somatic hypermutation in low-grade mucosa-associated lymphoid tissue-type B-cell lymphoma. Blood. 1995;86:3528-34.
38. Du M, Diss TC, Xu C, et al. Ongoing mutation in MALT lymphoma immunoglobulin gene suggests that antigen stimulation plays a role in the clonal expansion. Leukemia. 1996;10:1190-7.
39. Bertoni F, Cazzaniga G, Bosshard G, et al. Immunoglobulin heavy chain diversity genes rearrangement pattern indicates that MALT-type gastric lymphoma B cells have undergone an antigen selection process. Br J Haematol. 1997;97:830-6.
40. Greiner A, Knorr C, Qin Y, et al. Low-grade B cell lymphomas of mucosa-associated lymphoid tissue (MALT-type) require CD40-mediated signaling and Th2-type cytokines for in vitro growth and differentiation. Am J Pathol. 1997;150:1583-93.
41. Ye H, Liu H, Attygalle A, et al. Variable frequencies of t(11; 18)(q21; q21) in MALT lymphomas of different sites: Significant association with CagA strains of H pylori in gastric MALT lymphoma. Blood. 2003;102:1012-18.
42. Liu H, Ye H, Ruskone-Fourmestraux A, et al. T(11; 18) is a marker for all stage gastric MALT lymphomas that will not respond to H. pylori eradication. Gastroenterology. 2002;122:1286-94.
43. Cavalli F, Isaacson PG, Gascoyne RD, Zucca E. MALT lymphomas. Hematology Am Soc Hematol Educ Program. 2001:241-58.
44. Starostik P, Greiner A, Schultz A, et al. Genetic aberrations common in gastric high-grade large B-cell lymphoma. Blood. 2000;95:1180-7.
45. Sanchez-Izquierdo D, Buchonnet G, Siebert R, et al. MALT1 is deregulated by both chromosomal translocation and amplification in B-cell non-Hodgkin lymphoma. Blood. 2003;101:4539-46.
46. Streubel B, Lamprecht A, Dierlamm J, et al. T(14; 18)(q32; q21) involving IGH and MALT1 is a frequent chromosomal aberration in MALT lymphoma. Blood. 2003;101:2335-9.
47. Isaacson PG, Du MQ. MALT lymphoma: From morphology to molecules. Nat Rev Cancer. 2004;4:644-53.
48. Bertoni F, Zucca E. Delving deeper into MALT lymphoma biology. J Clin Invest. 2006;116:22-6.
49. Isaacson PG. Gastric MALT lymphoma: From concept to cure. Ann Oncol. 1999;10:637-45.
50. De JD, van der Hulst RW, Pals G, et al. Gastric non-Hodgkin lymphomas of mucosa-associated lymphoid tissue are not associated with more aggressive Helicobacter pylori strains as identified by CagA. Am J Clin Pathol. 1996;106:670-5.
51. Peng H, Ranaldi R, Diss TC, et al. High frequency of CagA+ Helicobacter pylori infection in high-grade gastric MALT B-cell lymphomas. J Pathol. 1998;185:409-12.
52. Harris NL, Jaffe ES, Stein H, et al. A revised European-American classification of lymphoid neoplasms: a proposal from the International Lymphoma Study Group. Blood. 1994;84:1361-92.
53. Isaacson PG, Spencer J. Malignant lymphoma of mucosa-associated lymphoid tissue. Histopathology. 1987;11:445-62.
54. De JD, Boot H, van HP, Hart GA, Taal BG. Histological grading in gastric lymphoma: pretreatment criteria and clinical relevance. Gastroenterology. 1997;112:1466-74.
55. Isaacson PG, Müller-Hermelink HK, Piris MA, et al. Extranodal marginal zone B-cell lymphoma of mucosa-associated lymphoid tissue (MALT lymphoma). In: Jaffe ES, Harris NL, Stein H, Vardiman JW, editors. Pathology and genetics of tumours of haematopoietic and lymphoid tissues. Lyon: IARC Press; 2001:157.
56. Aiello A, Giardini R, Tondini C, et al. PCR-based clonality analysis: A reliable method for the diagnosis and follow-up monitoring of conservatively treated gastric B-cell MALT lymphomas? Histopathology. 1999;34:326-30.
57. Bertoni F, Zucca E. State-of-the-art therapeutics: Marginal-zone lymphoma. J Clin Oncol. 2005;23:6415-20.
58. Taal BG, Boot H, van Heerde P, et al. Primary non-Hodgkin lymphoma of the stomach: Endoscopic pattern and prognosis in low versus high grade malignancy in relation to the MALT concept. Gut. 1996;39:556-61.
59. Wotherspoon AC, Doglioni C, Isaacson PG. Low-grade gastric B-cell lymphoma of mucosa-associated lymphoid tissue (MALT): A multifocal disease. Histopathology. 1992;20:29-34.
60. Chey WD, Wong BC. American College of Gastroenterology guideline on the management of Helicobacter pylori infection. Am J Gastroenterol. 2007;102:1808-25.
61. Fischbach W, Goebeler-Kolve ME, Greiner A. Diagnostic accuracy of EUS in the local staging of primary gastric lymphoma: Results of a prospective, multicenter study comparing EUS with histopathologic stage. Gastrointest Endosc. 2002;56:696-700.
62. Caletti G, Zinzani PL, Fusaroli P, et al. The importance of endoscopic ultrasonography in the management of low-grade gastric mucosa-associated lymphoid tissue lymphoma. Aliment Pharmacol Ther. 2002;16:1715-22.
63. Nakamura S, Matsumoto T, Suekane H, et al. Predictive value of endoscopic ultrasonography for regression of gastric low-grade and high-grade MALT lymphomas after eradication of Helicobacter pylori. Gut. 2001;48:454-60.
64. Elstrom R, Guan L, Baker G, et al. Utility of FDG-PET scanning in lymphoma by WHO classification. Blood. 2003;101:3875-6.
65. Cheson BD, Pfistner B, Juweid ME, et al. Revised response criteria for malignant lymphoma. J Clin Oncol. 2007;25:579-86.
66. Cogliatti SB, Schmid U, Schumacher U, et al. Primary B-cell gastric lymphoma: A clinicopathological study of 145 patients. Gastroenterology. 1991;101:1159-70.
67. Radaszkiewicz T, Dragosics B, Bauer P. Gastrointestinal malignant lymphomas of the mucosa-associated lymphoid tissue: Factors relevant to prognosis. Gastroenterology. 1992;102:1628-38.
68. Ruskone-Fourmestraux A, Lavergne A, Aegerter PH, et al. Predictive factors for regression of gastric MALT lymphoma after anti-Helicobacter pylori treatment. Gut. 2001;48:297-303.
69. Neubauer A, Thiede C, Morgner A, et al. Cure of Helicobacter pylori infection and duration of remission of low-grade gastric mucosa–associated lymphoid tissue lymphoma. J Natl Cancer Inst. 1997;89:1350-5.
70. Fischbach W, Goebeler-Kolve ME, Dragosics B, et al. Long-term outcome of patients with gastric marginal zone B cell lymphoma of mucosa associated lymphoid tissue (MALT) following exclusive Helicobacter pylori eradication therapy: Experience from a large prospective series. Gut. 2004;53:34-7.
71. Bertoni F, Conconi A, Capella C, et al. Molecular follow-up in gastric mucosa-associated lymphoid tissue lymphomas: Early analysis of the LY03 cooperative trial. Blood. 2002;99:2541-4.
72. Du MQ, Isaccson PG. Gastric MALT lymphoma: From aetiology to treatment. Lancet Oncol. 2002;3:97-104.
73. Hunt RH. Peptic ulcer disease: Defining the treatment strategies in the era of Helicobacter pylori. Am J Gastroenterol. 1997;92:36S-40S.
74. Yamashita H, Watanabe H, Ajioka Y, et al. When can complete regression of low-grade gastric lymphoma of mucosa-associated lymphoid tissue be predicted after helicobacter pylori eradication? Histopathology. 2000;37:131-40.
75. Pinotti G, Zucca E, Roggero E, et al. Clinical features, treatment and outcome in a series of 93 patients with low-grade gastric MALT lymphoma. Leuk Lymphoma. 1997;26:527-37.
76. Thiede C, Wundisch T, Neubauer B, et al. Eradication of Helicobacter pylori and stability of remissions in low-grade gastric B-cell lymphomas of the mucosa-associated lymphoid tissue: Results of an ongoing multicenter trial. Recent Results Cancer Res. 2000;156:125-33.
77. Horstmann M, Erttmann R, Winkler K. Relapse of MALT lymphoma associated with Helicobacter pylori after antibiotic treatment. Lancet. 1994;343:1098-9.
78. Savio A, Zamboni G, Capelli P, et al. Relapse of low-grade gastric MALT lymphoma after Helicobacter pylori eradication: True relapse or persistence? Long-term post-treatment follow-up of a multicenter trial in the north-east of Italy and evaluation of the diagnostic protocol’s adequacy. Recent Results Cancer Res. 2000;156:116-24.
79. Liu H, Ye H, Dogan A, et al. T(11; 18)(q21; q21) is associated with advanced mucosa-associated lymphoid tissue lymphoma that expresses nuclear BCL10. Blood. 2001;98:1182-7.
80. Chen LT, Lin JT, Shyu RY, et al. Prospective study of Helicobacter pylori eradication therapy in stage I(E) high-grade mucosa-associated lymphoid tissue lymphoma of the stomach. J Clin Oncol. 2001;19:4245-51.
81. Morgner A, Miehlke S, Fischbach W, et al. Complete remission of primary high-grade B-cell gastric lymphoma after cure of Helicobacter pylori infection. J Clin Oncol. 2001;19:2041-8.
82. Kuo SH, Chen LT, Wu MS, et al. Long-term follow-up of gastrectomized patients with mucosa-associated lymphoid tissue lymphoma: Need for a revisit of surgical treatment. Ann Surg. 2008;247:265-9.
83. Yoon SS, Coit DG, Portlock CS, Karpeh MS. The diminishing role of surgery in the treatment of gastric lymphoma. Ann Surg. 2004;240:28-37.
84. Schechter NR, Yahalom J. Low-grade MALT lymphoma of the stomach: A review of treatment options. Int J Radiat Oncol Biol Phys. 2000;46:1093-103.
85. Tsang RW, Gospodarowicz MK, Pintilie M, et al. Localized mucosa-associated lymphoid tissue lymphoma treated with radiation therapy has excellent clinical outcome. J Clin Oncol. 2003;21:4157-64.
86. Yahalom J. MALT lymphomas: A radiation oncology viewpoint. Ann Hematol. 2001;80(Suppl 3):B100-5.
87. Nakamura S, Matsumoto T, Suekane H, et al. Long-term clinical outcome of Helicobacter pylori eradication for gastric mucosa-associated lymphoid tissue lymphoma with a reference to second-line treatment. Cancer. 2005;104:532-40.
88. Hammel P, Haioun C, Chaumette MT, et al. Efficacy of single-agent chemotherapy in low-grade B-cell mucosa-associated lymphoid tissue lymphoma with prominent gastric expression. J Clin Oncol. 1995;13:2524-9.
89. Jager G, Neumeister P, Quehenberger F, et al. Prolonged clinical remission in patients with extranodal marginal zone B-cell lymphoma of the mucosa-associated lymphoid tissue type treated with cladribine: 6-year follow-up of a phase II trial. Ann Oncol. 2006;17:1722-3.
90. Conconi A, Martinelli G, Thieblemont C, et al. Clinical activity of rituximab in extranodal marginal zone B-cell lymphoma of MALT type. Blood. 2003;102:2741-5.
91. Martinelli G, Laszlo D, Ferreri AJ, et al. Clinical activity of rituximab in gastric marginal zone non-Hodgkin’s lymphoma resistant to or not eligible for anti-Helicobacter pylori therapy. J Clin Oncol. 2005;23:1979-83.
92. Almasri NM, al-Abbadi M, Rewaily E, et al. Primary gastrointestinal lymphomas in Jordan are similar to those in Western countries. Mod Pathol. 1997;10:137-41.
93. Ibrahim EM, Ezzat AA, Raja MA, et al. Primary gastric non-Hodgkin’s lymphoma: Clinical features, management, and prognosis of 185 patients with diffuse large B-cell lymphoma. Ann Oncol. 1999;10:1441-9.
94. Papaxoinis G, Papageorgiou S, Rontogianni D, et al. Primary gastrointestinal non-Hodgkin’s lymphoma: A clinicopathologic study of 128 cases in Greece. A Hellenic Cooperative Oncology Group study (HeCOG). Leuk Lymphoma. 2006;47:2140-6.
95. Nakamura S, Ye H, Bacon CM, et al. Translocations involving the immunoglobulin heavy chain gene locus predict better survival in gastric diffuse large B-cell lymphoma. Clin Cancer Res. 2008;14:3002-10.
96. Harris NL, Jaffe ES, Diebold J, et al. World Health Organization classification of neoplastic diseases of the hematopoietic and lymphoid tissues: Report of the Clinical Advisory Committee meeting—Airlie House, Virginia, November 1997. J Clin Oncol. 1999;17:3835-49.
97. Hsu C, Chen CL, Chen LT, et al. Comparison of MALT and non-MALT primary large cell lymphoma of the stomach: Does histologic evidence of MALT affect chemotherapy response? Cancer. 2001;91:49-56.
98. Driessen A, Tierens A, Ectors N, et al. Primary diffuse large B cell lymphoma of the stomach: Analysis of somatic mutations in the rearranged immunoglobulin heavy chain variable genes indicates antigen selection. Leukemia. 1999;13:1085-92.
99. Yoshino T, Omonishi K, Kobayashi K, et al. Clinicopathological features of gastric mucosa associated lymphoid tissue (MALT) lymphomas: High-grade transformation and comparison with diffuse large B cell lymphomas without MALT lymphoma features. J Clin Pathol. 2000;53:187-90.
100. Takeshita M, Iwashita A, Kurihara K, et al. Histologic and immunohistologic findings and prognosis of 40 cases of gastric large B-cell lymphoma. Am J Surg Pathol. 2000;24:1641-9.
101. Chen YW, Hu XT, Liang AC, et al. High BCL6 expression predicts better prognosis, independent of BCL6 translocation status, translocation partner, or BCL6-deregulating mutations, in gastric lymphoma. Blood. 2006;108:2373-83.
102. Psyrri A, Papageorgiou S, Economopoulos T. Primary extranodal lymphomas of stomach: Clinical presentation, diagnostic pitfalls and management. Ann Oncol. 2008;19:1992-9.
103. Ferreri AJ, Montalban C. Primary diffuse large B-cell lymphoma of the stomach. Crit Rev Oncol Hematol. 2007;63:65-71.
104. Ferreri AJ, Freschi M, Dell’Oro S, et al. Prognostic significance of the histopathologic recognition of low- and high-grade components in stage I-II B-cell gastric lymphomas. Am J Surg Pathol. 2001;25:95-102.
105. Lister TA, Crowther D, Sutcliffe SB, et al. Report of a committee convened to discuss the evaluation and staging of patients with Hodgkin’s disease: Cotswolds meeting. J Clin Oncol. 1989;7:1630-6.
106. Fischbach W, Dragosics B, Kolve-Goebeler ME, et al. Primary gastric B-cell lymphoma: results of a prospective multicenter study. The German-Austrian Gastrointestinal Lymphoma Study Group. Gastroenterology. 2000;119:1191-02.
107. Ferreri AJ, Cordio S, Ponzoni M, Villa E. Non-surgical treatment with primary chemotherapy, with or without radiation therapy, of stage I-II high-grade gastric lymphoma. Leuk Lymphoma. 1999;33:531-41.
108. Aviles A, Nambo MJ, Neri N, et al. The role of surgery in primary gastric lymphoma: Results of a controlled clinical trial. Ann Surg. 2004;240:44-50.
109. Koch P, del Valle F, Berdel WE, et al. Primary gastrointestinal non-Hodgkin’s lymphoma: II. Combined surgical and conservative or conservative management only in localized gastric lymphoma—results of the prospective German Multicenter Study GIT NHL 01/92. J Clin Oncol. 2001;19:3874-83.
110. Koch P, Probst A, Berdel WE, et al. Treatment results in localized primary gastric lymphoma: Data of patients registered within the German multicenter study (GIT NHL 02/96). J Clin Oncol. 2005;23:7050-9.
111. Feugier P, Van HA, Sebban C, et al. Long-term results of the R-CHOP study in the treatment of elderly patients with diffuse large B-cell lymphoma: A study by the Groupe d’Etude des Lymphomes de l’Adulte. J Clin Oncol. 2005;23:4117-26.
112. Pfreundschuh M, Trumper L, Osterborg A, et al. CHOP-like chemotherapy plus rituximab versus CHOP-like chemotherapy alone in young patients with good-prognosis diffuse large-B-cell lymphoma: A randomised controlled trial by the MabThera International Trial (MInT) Group. Lancet Oncol. 2006;7:379-91.
113. Sehn LH, Donaldson J, Chhanabhai M, et al. Introduction of combined CHOP plus rituximab therapy dramatically improved outcome of diffuse large B-cell lymphoma in British Columbia. J Clin Oncol. 2005;23:5027-33.
114. Wohrer S, Puspok A, Drach J, et al. Rituximab, cyclophosphamide, doxorubicin, vincristine and prednisone (R-CHOP) for treatment of early-stage gastric diffuse large B-cell lymphoma. Ann Oncol. 2004;15:1086-90.
115. Miller TP. The limits of limited stage lymphoma. J Clin Oncol. 2004;22:2982-4.
116. Chen LT, Lin JT, Tai JJ, et al. Long-term results of anti-Helicobacter pylori therapy in early-stage gastric high-grade transformed MALT lymphoma. J Natl Cancer Inst. 2005;97:1345-53.
117. Itatsu T, Miwa H, Ohkura R, et al. Primary gastric T-cell lymphoma accompanied by HTLV-I, HBV and H. pylori infection. Dig Dis Sci. 1999;44:1823-36.
118. Niitsu N, Nakamine H, Kohri M, et al. Primary gastric T-cell lymphoma not associated with human T-lymphotropic virus type I: A case report and review of the literature. Ann Hematol. 2003;82:197-202.
119. Chim CS, Au WY, Shek TW, et al. Primary CD56 positive lymphomas of the gastrointestinal tract. Cancer. 2001;91:525-33.
120. Chuang SS, Jung YC. Natural killer cell lymphoma of small intestine with features of enteropathy but lack of association with celiac disease. Hum Pathol. 2004;35:639-42.
121. Yuan CM, Stein S, Glick JH, Wasik MA. Natural killer-like T-cell lymphoma of the small intestine with a distinct immunophenotype and lack of association with gluten-sensitive enteropathy. Arch Pathol Lab Med. 2003;127:e142-6.
122. Tung CL, Hsieh PP, Chang JH, et al. Intestinal T-cell and natural killer-cell lymphomas in Taiwan with special emphasis on 2 distinct cellular types: natural killer-like cytotoxic T cell and true natural killer cell. Hum Pathol. 2008;39:1018-25.
123. Li B, Shi YK, He XH, et al. Primary non-Hodgkin lymphomas in the small and large intestine: Clinicopathological characteristics and management of 40 patients. Int J Hematol. 2008;87:375-81.
124. Nakamura S, Matsumoto T, Takeshita M, et al. A clinicopathologic study of primary small intestine lymphoma: Prognostic significance of mucosa-associated lymphoid tissue-derived lymphoma. Cancer. 2000;88:286-94.
125. Daum S, Ullrich R, Heise W, et al. Intestinal non-Hodgkin’s lymphoma: A multicenter prospective clinical study from the German Study Group on Intestinal non-Hodgkin’s Lymphoma. J Clin Oncol. 2003;21:2740-6.
126. Saito T, Toyoda H, Yamaguchi M, et al. Ileocolonic lymphomas: A series of 16 cases. Endoscopy. 2005;37:466-9.
127. Bertoni F, Zucca E, Cotter FE. Molecular basis of mantle cell lymphoma. Br J Haematol. 2004;124:130-40.
128. Romaguera JE, Medeiros LJ, Hagemeister FB, et al. Frequency of gastrointestinal involvement and its clinical significance in mantle cell lymphoma. Cancer. 2003;97:586-91.
129. Michopoulos S, Petraki K, Matsouka C, et al. Mantle-cell lymphoma (multiple lymphomatous polyposis) of the entire GI tract. J Clin Oncol. 2008;26:1555-7.
130. Mar N, Khaled S, Kencana F, et al. Multiple lymphomatous polyposis as a sole presentation of mantle cell lymphoma. J Clin Gastroenterol. 2006;40:653-4.
131. Witzig TE. Current treatment approaches for mantle-cell lymphoma. J Clin Oncol. 2005;23:6409-14.
132. Damaj G, Verkarre V, Delmer A, et al. Primary follicular lymphoma of the gastrointestinal tract: A study of 25 cases and a literature review. Ann Oncol. 2003;14:623-9.
133. Salles GA. Clinical features, prognosis and treatment of follicular lymphoma. Hematology Am Soc Hematol Educ Program. 2007:216-25.
134. Blum KA, Lozanski G, Byrd JC. Adult Burkitt leukemia and lymphoma. Blood. 2004;104:3009-20.
135. Harris NL, Horning SJ. Burkitt’s lymphoma—the message from microarrays. N Engl J Med. 2006;354:2495-8.
136. Hecht JL, Aster JC. Molecular biology of Burkitt’s lymphoma. J Clin Oncol. 2000;18:3707-21.
137. Lacasce A, Howard O, Lib S, et al. Modified magrath regimens for adults with Burkitt and Burkitt-like lymphomas: Preserved efficacy with decreased toxicity. Leuk Lymphoma. 2004;45:761-7.
138. Thomas DA, Faderl S, O’Brien S, et al. Chemoimmunotherapy with hyper-CVAD plus rituximab for the treatment of adult Burkitt and Burkitt-type lymphoma or acute lymphoblastic leukemia. Cancer. 2006;106:1569-80.
139. Fine KD, Stone MJ. Alpha-heavy chain disease, Mediterranean lymphoma, and immunoproliferative small intestinal disease: A review of clinicopathological features, pathogenesis, and differential diagnosis. Am J Gastroenterol. 1999;94:1139-52.
140. Khojasteh A, Haghighi P. Immunoproliferative small intestinal disease: Portrait of a potentially preventable cancer from the Third World. Am J Med. 1990;89:483-90.
141. Lecuit M, Abachin E, Martin A, et al. Immunoproliferative small intestinal disease associated with Campylobacter jejuni. N Engl J Med. 2004;350:239-48.
142. Seligmann M, Danon F, Hurez D, et al. Alpha-chain disease: A new immunoglobulin abnormality. Science. 1968;162:1396-7.
143. Isaacson PG, Dogan A, Price SK, Spencer J. Immunoproliferative small-intestinal disease. An immunohistochemical study. Am J Surg Pathol. 1989;13:1023-33.
144. Smith WJ, Price SK, Isaacson PG. Immunoglobulin gene rearrangement in immunoproliferative small intestinal disease (IPSID). J Clin Pathol. 1987;40:1291-7.
145. Isaacson PG, Price SK. Light chains in Mediterranean lymphoma. J Clin Pathol. 1985;38:601-7.
146. Salem P, el-Hashimi L, Anaissie E, et al. Primary small intestinal lymphoma in adults. A comparative study of IPSID versus non-IPSID in the Middle East. Cancer. 1987;59:1670-6.
147. Galian A, Lecestre MJ, Scotto J, et al. Pathological study of alpha-chain disease, with special emphasis on evolution. Cancer. 1977;39:2081-2101.
148. Matuchansky C, Cogne M, Lemaire M, et al. Nonsecretory alpha-chain disease with immunoproliferative small-intestinal disease. N Engl J Med. 1989;320:1534-9.
149. Al-Saleem T, Al-Mondhiry H. Immunoproliferative small intestinal disease (IPSID): A model for mature B-cell neoplasms. Blood. 2005;105:2274-80.
150. Salem PA, Estephan FF. Immunoproliferative small intestinal disease: Current concepts. Cancer J. 2005;11:374-82.
151. Martin IG, Aldoori MI. Immunoproliferative small intestinal disease: Mediterranean lymphoma and alpha heavy chain disease. Br J Surg. 1994;81:20-4.
152. Akbulut H, Soykan I, Yakaryilmaz F, et al. Five-year results of the treatment of 23 patients with immunoproliferative small intestinal disease: A Turkish experience. Cancer. 1997;80:8-14.
153. Ben-Ayed F, Halphen M, Najjar T, et al. Treatment of alpha chain disease. Results of a prospective study in 21 Tunisian patients by the Tunisian-French intestinal Lymphoma Study Group. Cancer. 1989;63:1251-6.
154. O’Keefe SJ, Winter TA, Newton KA, et al. Severe malnutrition associated with alpha-heavy chain disease: Response to tetracycline and intensive nutritional support. Am J Gastroenterol. 1988;83:995-1001.
155. Salimi M, Spinelli JJ. Chemotherapy of Mediterranean abdominal lymphoma. Retrospective comparison of chemotherapy protocols in Iranian patients. Am J Clin Oncol. 1996;19:18-22.
156. el Saghir NS. Combination chemotherapy with tetracycline and aggressive supportive care for immunoproliferative small-intestinal disease lymphoma. J Clin Oncol. 1995;13:794-5.
157. Jones JD. Intestinal lymphomas, including immunoproliferative small intestinal disease. In: Feldman M, Scharschmidt BF, Sleisenger MH, Fordtran JS, editors. Sleisenger and Fordtran’s gastrointestinal and liver disease: Pathophysiology/diagnosis/management. 6th ed. Philadelphia: WB Saunders; 1998:1844.
158. Catassi C, Bearzi I, Holmes GK. Association of celiac disease and intestinal lymphomas and other cancers. Gastroenterology. 2005;128:S79-86.
159. Al-Toma A, Verbeek WH, Hadithi M, et al. Survival in refractory coeliac disease and enteropathy-associated T-cell lymphoma: Retrospective evaluation of single-centre experience. Gut. 2007;56:1373-8.
160. Gale J, Simmonds PD, Mead GM, et al. Enteropathy-type intestinal T-cell lymphoma: Clinical features and treatment of 31 patients in a single center. J Clin Oncol. 2000;18:795-803.
161. Holmes GK, Prior P, Lane MR, et al. Malignancy in coeliac disease—effect of a gluten-free diet. Gut. 1989;30:333-8.
162. Domizio P, Owen RA, Shepherd NA, et al. Primary lymphoma of the small intestine. A clinicopathological study of 119 cases. Am J Surg Pathol. 1993;17:429-42.
163. Otter R, Bieger R, Kluin PM, et al. Primary gastrointestinal non-Hodgkin’s lymphoma in a population-based registry. Br J Cancer. 1989;60:745-50.
164. Catassi C, Fabiani E, Corrao G, et al. Risk of non-Hodgkin lymphoma in celiac disease. JAMA. 2002;287:1413-19.
165. Fasano A, Berti I, Gerarduzzi T, et al. Prevalence of celiac disease in at-risk and not-at-risk groups in the United States: A large multicenter study. Arch Intern Med. 2003;163:286-92.
166. Rostom A, Murray JA, Kagnoff MF. American Gastroenterological Association (AGA) Institute technical review on the diagnosis and management of celiac disease. Gastroenterology. 2006;131:1981-2002.
167. Corrao G, Corazza GR, Bagnardi V, et al. Mortality in patients with coeliac disease and their relatives: A cohort study. Lancet. 2001;358:356-61.
168. Howdle PD, Jalal PK, Holmes GK, Houlston RS. Primary small-bowel malignancy in the UK and its association with coeliac disease. QJM. 2003;96:345-53.
169. O’Farrelly C, Feighery C, O’Briain DS, et al. Humoral response to wheat protein in patients with coeliac disease and enteropathy-associated T cell lymphoma. Br Med J (Clin Res Ed). 1986;293:908-10.
170. National Institutes of Health Consensus Development Conference Statement on Celiac Disease, June 28-30, 2004. Gastroenterology. 2005;128:S1-9.
171. Lundqvist C, Baranov V, Hammarstrom S, et al. Intra-epithelial lymphocytes. Evidence for regional specialization and extrathymic T cell maturation in the human gut epithelium. Int Immunol. 1995;7:1473-87.
172. Russell GJ, Winter HS, Fox VL, Bhan AK. Lymphocytes bearing the gamma delta T-cell receptor in normal human intestine and celiac disease. Hum Pathol. 1991;22:690-4.
173. Bagdi E, Diss TC, Munson P, Isaacson PG. Mucosal intra-epithelial lymphocytes in enteropathy-associated T-cell lymphoma, ulcerative jejunitis, and refractory celiac disease constitute a neoplastic population. Blood. 1999;94:260-4.
174. Cellier C, Patey N, Mauvieux L, et al. Abnormal intestinal intraepithelial lymphocytes in refractory sprue. Gastroenterology. 1998;114:471-81.
175. Cellier C, Delabesse E, Helmer C, et al. Refractory sprue, coeliac disease, and enteropathy-associated T-cell lymphoma. French Coeliac Disease Study Group. Lancet. 2000;356:203-8.
176. Daum S, Weiss D, Hummel M, et al. Frequency of clonal intraepithelial T lymphocyte proliferations in enteropathy-type intestinal T cell lymphoma, coeliac disease, and refractory sprue. Gut. 2001;49:804-12.
177. Isaacson PG. Relation between cryptic intestinal lymphoma and refractory sprue. Lancet. 2000;356:178-9.
178. Ryan BM, Kelleher D. Refractory celiac disease. Gastroenterology. 2000;119:243-51.
179. Cellier C, Cuillerier E, Patey-Mariaud de SN, et al. Push enteroscopy in celiac sprue and refractory sprue. Gastrointest Endosc. 1999;50:613-17.
180. Murray A, Cuevas EC, Jones DB, Wright DH. Study of the immunohistochemistry and T cell clonality of enteropathy-associated T cell lymphoma. Am J Pathol. 1995;146:509-19.
181. Zettl A, Ott G, Makulik A, et al. Chromosomal gains at 9q characterize enteropathy-type T-cell lymphoma. Am J Pathol. 2002;161:1635-45.
182. Obermann EC, Diss TC, Hamoudi RA, et al. Loss of heterozygosity at chromosome 9p21 is a frequent finding in enteropathy-type T-cell lymphoma. J Pathol. 2004;202:252-2.
183. Verkarre V, Romana SP, Cellier C, et al. Recurrent partial trisomy 1q22-q44 in clonal intraepithelial lymphocytes in refractory celiac sprue. Gastroenterology. 2003;125:40-6.
184. Chott A, Vesely M, Simonitsch I, et al. Classification of intestinal T-cell neoplasms and their differential diagnosis. Am J Clin Pathol. 1999;111:S68-74.
185. Isaacson PG, Norton AJ. Malignant lymphoma of the gastrointestinal tract. Extranodal lymphomas. Edinburgh: Churchill Livingston; 1994.
186. Chott A, Haedicke W, Mosberger I, et al. Most CD56+ intestinal lymphomas are CD8+CD5-T-cell lymphomas of monomorphic small to medium size histology. Am J Pathol. 1998;153:1483-90.
187. Diss TC, Watts M, Pan LX, et al. The polymerase chain reaction in the demonstration of monoclonality in T cell lymphomas. J Clin Pathol. 1995;48:1045-50.
188. Isaacson PG, O’Connor NT, Spencer J, et al. Malignant histiocytosis of the intestine: A T-cell lymphoma. Lancet. 1985;2:688-91.
189. Hadithi M, Mallant M, Oudejans J, et al. 18F-FDG PET versus CT for the detection of enteropathy-associated T-cell lymphoma in refractory celiac disease. J Nucl Med. 2006;47:1622-7.
190. Rongey C, Micallef I, Smyrk T, Murray J. Successful treatment of enteropathy-associated T cell lymphoma with autologous stem cell transplant. Dig Dis Sci. 2006;51:1082-6.
191. Bishton MJ, Haynes AP. Combination chemotherapy followed by autologous stem cell transplant for enteropathy-associated T cell lymphoma. Br J Haematol. 2007;136:111-13.
192. Al-Toma A, Verbeek WH, Visser OJ, et al. Disappointing outcome of autologous stem cell transplantation for enteropathy-associated T-cell lymphoma. Dig Liver Dis. 2007;39:634-41.
193. Vivas S, Ruiz de Morales JM, Ramos F, Suarez-Vilela D. Alemtuzumab for refractory celiac disease in a patient at risk for enteropathy-associated T-cell lymphoma. N Engl J Med. 2006;354:2514-15.
194. Li YX, Liu QF, Fang H, et al. Variable clinical presentations of nasal and Waldeyer ring natural killer/T-cell lymphoma. Clin Cancer Res. 2009;15:2905-12.
195. Gurkaynak M, Cengiz M, Akyurek S, et al. Waldeyer’s ring lymphomas: Treatment results and prognostic factors. Am J Clin Oncol. 2003;26:437-40.
196. Horning SJ, Weller E, Kim K, et al. Chemotherapy with or without radiotherapy in limited-stage diffuse aggressive non-Hodgkin’s lymphoma: Eastern Cooperative Oncology Group study 1484. J Clin Oncol. 2004;22:3032-8.
197. Noronha V, Shafi NQ, Obando JA, Kummar S. Primary non-Hodgkin’s lymphoma of the liver. Crit Rev Oncol Hematol. 2005;53:199-207.
198. Bronowicki JP, Bineau C, Feugier P, et al. Primary lymphoma of the liver: Clinical-pathological features and relationship with HCV infection in French patients. Hepatology. 2003;37:781-7.
199. De Renzo A, Perna F, Persico M, et al. Excellent prognosis and prevalence of HCV infection of primary hepatic and splenic non-Hodgkin’s lymphoma. Eur J Haematol. 2008;81:51-7.
200. Salmon JS, Thompson MA, Arildsen RC, Greer JP. Non-Hodgkin’s lymphoma involving the liver: Clinical and therapeutic considerations. Clin Lymphoma Myeloma. 2006;6:273-80.
201. Nayer H, Weir EG, Sheth S, Ali SZ. Primary pancreatic lymphomas: A cytopathologic analysis of a rare malignancy. Cancer. 2004;102:315-21.
202. Nishimura R, Takakuwa T, Hoshida Y, et al. Primary pancreatic lymphoma: Clinicopathological analysis of 19 cases from Japan and review of the literature. Oncology. 2001;60:322-9.
203. Grimison PS, Chin MT, Harrison ML, Goldstein D. Primary pancreatic lymphoma—pancreatic tumours that are potentially curable without resection, a retrospective review of four cases. BMC Cancer. 2006;6:117.
204. Gonzalez QH, Heslin MJ, vila-Cervantes A, et al. Primary colonic lymphoma. Am Surg. 2008;74:214-16.
205. Dionigi G, Annoni M, Rovera F, et al. Primary colorectal lymphomas: Review of the literature. Surg Oncol. 2007;16(Suppl 1):S169-71.
206. Bakker NA, van Imhoff GW. Post-transplant lymphoproliferative disorders: From treatment to early detection and prevention? Haematologica. 2007;92:1447-50.
207. LaCasce AS. Post-transplant lymphoproliferative disorders. Oncologist. 2006;11:674-80.
208. Aucejo F, Rofaiel G, Miller C. Who is at risk for post-transplant lymphoproliferative disorders (PTLD) after liver transplantation? J Hepatol. 2006;44:19-23.
209. Harris NL, Ferry JA, Swerdlow SH. Posttransplant lymphoproliferative disorders: Summary of Society for Hematopathology Workshop. Semin Diagn Pathol. 1997;14:8-14.
210. Tsai DE, Hardy CL, Tomaszewski JE, et al. Reduction in immunosuppression as initial therapy for posttransplant lymphoproliferative disorder: Analysis of prognostic variables and long-term follow-up of 42 adult patients. Transplantation. 2001;71:1076-88.
211. Papadopoulos EB, Ladanyi M, Emanuel D, et al. Infusions of donor leukocytes to treat Epstein-Barr virus–associated lymphoproliferative disorders after allogeneic bone marrow transplantation. N Engl J Med. 1994;330:1185-91.
212. Noy A. Update on HIV lymphoma. Curr Oncol Rep. 2007;9:384-90.
213. Parekh S, Ratech H, Sparano JA. Human immunodeficiency virus-associated lymphoma. Clin Adv Hematol Oncol. 2003;1:295-301.
214. Levine AM, Wernz JC, Kaplan L, et al. Low-dose chemotherapy with central nervous system prophylaxis and zidovudine maintenance in AIDS-related lymphoma. A prospective multi-institutional trial. JAMA. 1991;266:84-8.
215. Kaplan LD, Straus DJ, Testa MA, et al. Low-dose compared with standard-dose m-BACOD chemotherapy for non-Hodgkin’s lymphoma associated with human immunodeficiency virus infection. National Institute of Allergy and Infectious Diseases AIDS Clinical Trials Group. N Engl J Med. 1997;336:1641-8.
216. Little RF, Pittaluga S, Grant N, et al. Highly effective treatment of acquired immunodeficiency syndrome-related lymphoma with dose-adjusted EPOCH: Impact of antiretroviral therapy suspension and tumor biology. Blood. 2003;101:4653-9.
217. Du MQ, Bacon CM, Isaacson PG. Kaposi sarcoma-associated herpesvirus/human herpesvirus 8 and lymphoproliferative disorders. J Clin Pathol. 2007;60:1350-7.
218. Carbone A, Gloghini A. KSHV/HHV8-associated lymphomas. Br J Haematol. 2008;140:13-24.
219. Brimo F, Michel RP, Khetani K, Auger M. Primary effusion lymphoma: a series of 4 cases and review of the literature with emphasis on cytomorphologic and immunocytochemical differential diagnosis. Cancer. 2007;111:224-33.