19 Gastrointestinal, Liver and Nutritional Alterations
After reading this chapter, you should be able to:
• describe the changes in normal gastrointestinal physiology and metabolism associated with critical illness
• integrate theoretical knowledge of the nutritional requirements, assessment of and potential for malnutrition in the critically ill with clinical practice and rationalise selected nutritional support strategies for specific patients
• identify patients at risk for the development of stress ulcers and rationalise therapeutic interventions for their prevention
• discuss the effects of critical illness on hepatic function and evaluate the consequences of liver dysfunction
• describe the treatment of liver failure, including liver support therapies and transplantation
• critically analyse the role of glycaemic control in the context of critical illness
• describe the physiological changes that occur during diabetic ketoacidosis and rationalise assessment and treatment strategies.
Gastrointenstinal Physiology
Digestion and absorption of nutrients such as carbohydrates, amino acids, minerals and water are key functions of the gastrointestinal system. Digestive enzymes are responsible for breaking down food into smaller substances that can be absorbed by the gastrointestinal tract. While some digestion begins in the oral cavity (for example, the breakdown of starch into sugar by salivary amylase), the stomach, pancreas, and small intestine secrete the most enzymes responsible for digestion (Table 19.1). The small bowel plays an important part in the digestion and absorption of these nutrients, where the processes of diffusion, facilitated diffusion, osmosis and active transport are responsible for absorption of 90% of all nutrients.1 The remaining 10% of nutrients are absorbed in the large intestine.
| Location | Enzymes | Target substance |
|---|---|---|
| Oral cavity | Salivary amylase (ptyalin) | Starch and glycogen |
| Bromelain | Protein | |
| Stomach | Pepsin | Proteins |
| Gelatinase | Proteoglycans in meat (gelatine and collagen) | |
| Gastric amylase | Starch | |
| Gastric lipase | Triglyceride | |
| Chymosin | Milk | |
| Pancreas | Trypsin, chymotrypsin, carboxypeptidase, elasatases | Proteins |
| Pancreatic lipase | Triglycerides | |
| Pancreatic amylase | Carbohydrates | |
| Small intestine | Sucrase | Sucrose |
| Lactase | Lactose | |
| Maltase | Maltose (into 2 molecules of glucose) | |
| Isomaltase | Maltose into isomaltose | |
| Intestinal lipase | Fatty acid |
The gastrointestinal tract also plays a role in immunity. It has a variety of mechanisms in place that prevent the movement of substances (other than nutrients, water and electrolytes) into the systemic circulation (see Table 19.2). In the setting of critical illness, where gastrointestinal hypoperfusion may be present, these protective functions may be diminished, so it is essential to understand the alterations in normal gastrointestinal physiology that occur during critical illness.
TABLE 19.2 Protective mechanisms of the gastrointestinal system and impact of critical illness1,3–12
| Mechanism | Action |
|---|---|
| Motility | Propels bacteria through the GI tract. In critical illness, motility may be altered because of enteric nerve impairment and altered smooth muscle function, inflammation (mediated by cytokines and nitric oxide), gut injury, hypoperfusion, medications (opioids, dopamine), electrolyte disturbances, hyperglycaemia, sepsis and increased intracranial pressure.3 |
| Hydrochloric acid secretion | Reduces gastric acidity and destroys bacteria. Parietal cells in the stomach produce hydrochloric acid and keep the intragastric environment relatively acidic (pH approx 4.0). An acidic pH has bactericidal and bacteriostatic properties,4 thus limiting overgrowth in the stomach. |
| Bicarbonate | Bicarbonate ions bind with hydrogen ions to form water and carbon dioxide, preventing the hydrogen ions (acid) from damaging the duodenal wall.5 |
| Bile salts | Bile salts provide protection against bacteria by breaking down the liposaccharide portion of endotoxins,6 thereby detoxifying gram-negative bacteria in the gastrointestinal tract. The deconjugation of bile salts into secondary bile acids inhibits the proliferation of pathogens and may destroy their cell walls.7 |
| Mucin production | Prevents the adhesion of bacteria to the wall of the GI tract. Mucous cells secrete large quantities of very thick, alkaline mucus (approximately 1 mm thick in the stomach). Glycoproteins present in the mucus prevent bacteria from adhering to and colonising the mucosal wall.8 |
| Epithelial cell shedding | Limits bacterial adhesion. The mucosal lining of the entire gastrointestinal tract is composed of epithelial cells that create a physical barrier to bacterial invasion. These cells are replaced approximately every 3–5 days9 limiting bacterial colonisation. |
| Zonea occludulns (tight junctions surrounding each cell in the epithelial sheet) | The junctions between epithelial cells provide a barrier to microorganisms. Intermediate junctions (zonula adherens) function primarily in cell–cell adhesion, while the tight junctions (zonula occludens) limit the movement of bacteria and toxins across the gut wall.10 |
| Gut-associated lymphoid tissue | Protection against bacterial invasion is provided by gut-associated lymphoid tissue,11 capable of cell-mediated and humoral-mediated immune responses.12 |
| Kupffer cells | Kupffer cells in the liver and spleen provide a back-up defence against pathogens that cross the barrier of the gastrointestinal wall and enter the systemic circulation.1 |
Alterations to Normal Gastrointestinal Physiology in Critical Illness
During critical illness, the digestion and absorption of nutrients may be altered. Gastric acid production is commonly thought to increase in critical illness, although evidence suggests that many critically ill patients do not hypersecrete gastric acid13 with increased gastric pH being observed in some critically ill patients, even in the absence of pharmacological inhibition of gastric acid secretion.14,15 The ability of the small intestine to absorb nutrients can be impaired during critical illness,16 although most critically ill patients appear to be able to tolerate enteral nutrition, making the clinical significance of impaired absorption unclear.
Some alterations to normal gastrointestinal physiology in critical illness relate to hypoperfusion and decreased oxygenation in this area and have high metabolic demands. Historically, gastrointestinal dysfunction in critical illness was described in relation to symptoms, such as gastrointestinal bleeding, mechanical obstruction, and pancreatitis17 resulting from ischaemia.18 However, the presence of covert ischaemia has resulted in a heightened interest in the prevention and early detection of gastrointestinal ischaemia in the critically ill, in an attempt to minimise ischaemia-related dysfunction.
Gastrointestinal Mucosal Hypoperfusion
The gastrointestinal system is particularly susceptible to alterations in regional blood flow and oxygen delivery because it has a higher critical oxygen delivery (DO2) than the rest of the body. Splanchnic vasoconstriction is also proportionally greater than other vascular beds and the countercurrent O2 exchange between vessels within the villi further contribute to decreased regional oxygen delivery.5
During shock states, decreased blood flow from vasoconstriction occurs in this region first. It is the last place to be restored following successful resuscitation.19 In shock states, the gastrointestinal system attempts to maintain adequate cellular oxygenation by increasing the amount of oxygen extracted from the blood. This increase in oxygen extraction may prevent serious compromise of tissue oxygenation even in the presence of reduced oxygen delivery.20
During periods of ischaemia and hypoxia, oxygen free-radicals are generated as byproducts of anaerobic metabolism. With successful resuscitation of the gastrointestinal tract, blood flow and oxygen delivery are restored but the oxygen free-radicals are liberated, contributing to the microvascular and mucosal changes characteristic of ischaemia and reperfusion of the gut mucosa.21
Consequences of Gastrointestinal Hypoperfusion
The consequences of gastrointestinal hypoperfusion are significant, and include disruption of the physical barrier to pathogens; disruption of chemical control of bacterial overgrowth; decreased peristalsis; and reduced immunological activities of gastrointestinal-associated lymphoid tissue. In health, all of these mechanisms work efficiently to contain bacteria within the gastrointestinal tract. During critical illness, however, reduced oxygenation contributes to decreased cellular function and failure of the protective mechanisms described in Table 19.2. Consequently, bacterial proliferation and translocation from the gastrointestinal tract to the systemic circulation may occur.22
Changes in gastrointestinal perfusion also has the capacity to affect hepatic perfusion, oxygenation and function. In approximately 50% of critically ill patients, ischaemic hepatitis or ‘shock liver’ occurs, which is evidenced by jaundice, elevation of liver function tests or overt hepatic dysfunction.23 Ischaemic hepatitis can vary from a mild elevation of serum aminotransferase and bilirubin levels in septic patients, to an acute elevation following haemodynamic shock. Ischaemic hepatic injury influences morbidity and mortality but remains underdiagnosed, probably because the clinical signs become apparent long after hypoperfusion has occurred. Physiological changes contributing to ischaemic hepatitis include changes to the portal and arterial blood supply as well as hepatic microcirculation. The degree to which the liver is damaged is directly related to the severity and duration of hypoperfusion, and both anoxic and reperfusion injury can damage hepatocytes and the vascular endothelium.23
Alterations to Normal Metabolism in Critical Illness
There is little information describing the changes to the exocrine function in the gastrointestinal system during critical illness, and it is uncertain how critical illness influences the metabolism of nutrients. While there is data to demonstrate that the secretion of hydrochloric acid by the parietal cells in the stomach is decreased, it is not certain whether the exocrine failure also extends to a decreased pepsin secretion. It is also possible that secretion of digestive enzymes might also be influenced by critical illness-induced pancreatitis, although clear data demonstrating this level of dysfunction are unavailable.16
Nutrition
Optimal nutritional support in the critically ill aims to prevent, detect and correct malnutrition, optimise the patient’s metabolic state, reduce morbidity and improve recovery.24 The metabolic response of stress or injury is hypermetabolism. There is an increased release of cytokines (e.g. interleukin-1, interleukin-6, tumor necrosis factor-α) and production of counter-regulatory hormones (e.g. catecholamines, cortisol, glucagon and growth hormone) that induce catabolism and oppose the anabolic effects of insulin.25 Hypercatabolism occurs with the imbalance between anabolism (i.e. the chemical process by which complex molecules, such as peptides, proteins, polysaccharides, lipids and nucleic acids, are synthesised from simpler molecules) and catabolism (i.e. the convergent process, in which many different types of molecules are broken down into relatively few types of end products). To compensate for the altered metabolic regulation, neuroendocrine stimulation increases the mobilisation and consumption of nutrients, such as glycogen and protein, from existing body stores. As the metabolic rate rises, nutritional requirements in critical illness are increased, characterised by a rise in resting energy expenditure and oxygen consumption, which in some critically ill patients can be increased by over 50%.26 Depletion of body energy stores result from alterations in protein, carbohydrate and fat metabolism.27 In addition to the rise in metabolic demands, patients who are critically ill often experience a concomitant fall in nutritional intake. The metabolic and nutrition alterations vary with the stress level, severity of illness, type of injury, organ dysfunction and nutrition status.25
To maintain normal cellular function, body cells require adequate amounts of the six basic nutrients: carbohydrates, fats and proteins to provide energy, vitamins, minerals and water to catalyse metabolic processes. Unlike normal metabolism, which preferentially uses carbohydrates and fats for energy, the hypermetabolic state associated with critical illness consumes proportionally more fats and proteins than carbohydrates to generate energy.28 As a consequence of the gluconeogenesis and the synthesis of acute-phase proteins, there is a decrease in lean body mass and negative nitrogen balance.
Consequences of Malnutrition
When adequate and timely nutrition support is not provided, body energy and protein depletion can occur with negative consequences on patient outcome.29 Critically ill patients require adequate nutrition to limit muscle wasting, respiratory and gastrointestinal dysfunction and alterations in immunity, all of which are associated with malnutrition.30 Respiratory support is often necessary during critical illness, and changes in respiratory muscle function and ventilatory drive may contribute to an increase in the number of ventilator days. Furthermore, infection rates may be increased in malnourished critically ill patients. The decrease in lean body mass and negative nitrogen balance is associated with delayed wound healing and a higher risk of infection.28
These complications contribute to increased length of stay, cost, morbidity and mortality.31 The degree of critical illness and hypercatabolism varies between patients and is often difficult to accurately determine. For this reason it is necessary to assess, as accurately as possible, the nutritional requirements of each individual patient.
Nutritional Assessment
The majority of studies report cumulative energy deficit or caloric debt is associated with worse clinical outcomes.32–35 Krishnan and colleagues,36 however, describe better clinical outcomes for patients fed fewer than the target nutrition goals when compared to those who received near target goals. Nutritional assessment includes patient history, physical examination and assessment of nutritional indices (see Table 19.3), but is often unreliable in the critically ill patient.37,38 Clinical judgement remains the most common way of assessing a patient’s nutritional status, and is shown to be as reliable as biochemical tests.39–41 Clinical judgement takes into consideration recent weight loss, reduced dietary intake, anorexia, vomiting, diarrhoea, muscle wasting and signs of nutritional deficiency.42 Appreciation of the importance of nutritional assessment and the impact of malnutrition in the critically ill informs management and is likely to improve outcomes.30
| Assessment | Limitations in critical illness |
|---|---|
| Subjective global assessment | Not validated in the critically ill40,43 |
| Biochemical markers: | |
Decreased sensitivity because of 20-day half-life; influenced by fluid balance/shifts42
Half-life of 8 days but lacks the sensitivity and specificity for determining nitrogen balance;43 influenced by fluid balance/shifts
Most sensitive with a half-life of 2 days,44 but changes may result from the metabolic response to illness rather than change in nutritional status; influenced by fluid balance/shifts
Determining Nutritional Requirements
Determining caloric requirements is largely dependent on energy expenditure, influenced by patient activity, stage of illness, type of injury and previous nutritional status.42 Indirect calorimetry is the ‘gold standard’ and most precise way of determining the nutritional requirements in critical illness.45 Energy expenditure is measured using the oxygen consumption obtained from carbon dioxide levels (PaCO2), or using a metabolic monitor. It is infrequently used in critical care settings, possibly because of the high equipment costs and unreliability in the critically ill.46
Calculating basal energy expenditure using the Harris-Benedict equation is a common, but less precise, method of determining nutritional requirements.42,47,48 The Harris-Benedict equation, and others, takes into account the age, height, weight and gender of the patient, with adjustments made for treatment, disease process and metabolic state. Importantly, these equations fail to find any significant benefits in outcomes, most likely because they do not measure energy requirement.49
The Prognostic Inflammatory Nutrition Index (PINI) uses the elevations in acute phase proteins (alpha-1-acid glycoprotein and C-reactive protein [CRP]) that occur with simultaneous reductions in transport proteins (albumin and pre-albumin) in a simple formula to stratify critically ill patients by risk of complications or death.50
Nutrition Support
For patients in ICU who are unable to take oral nutrition, enteral nutrition (EN), parenteral nutrition (PN) or combined EN and PN is available. The best method of providing nutrition to the critically ill who cannot have oral feeding is controversial. Infectious complications have been associated with PN when used alone,51 but no differences in infectious complications were seen with concurrent use of EN and PN.52 In a meta-analysis, PN was associated with reduced mortality when comparing PN with delayed EN despite the increased risk for infectious complications associated with PN.51 Meta-analyses are limited by the quality of the studies included in the analyses.53,54 Recent guidelines advocate early enteral nutrition53,55–59 but better evidence is needed.60
Enteral Nutrition
EN has benefits beyond the supply of nutrients to the body,61 including:
• gut-derived mucosal immunity62 and decrease in septic complications63–65
• preservation of gastrointestinal mucosal integrity66,67
• improved gastrointestinal mucosal cell growth and replacement68
Absence of enteral nutrients (despite the provision of PN) has been linked to atrophy of the intestinal villi, a reduction in the number of epithelial cells produced, reduced gastrointestinal mucosal thickness, and ineffective functioning of the intestinal brush border enzymes of the gastrointestinal mucosa.59,70–73 Stimulating and improving gastrointestinal immune function is an important goal of early EN.59 Early enteral feeding (within 48 hours) is recommended.55,56
Hypocaloric Intake in the Critically Ill
A significant number of hospitalised patients receiving EN do not have their nutritional needs met.70,71 Hypocaloric feeding in the first few days of critical illness may be beneficial,36,74–77 but results are conflicting.34,78–80 The belief that early enteral feeding prevents gut dysfunction independently of calorie intake81 perpetuates the acceptance of administration of EN below the nutrition target.33,70,82,83 In most cases, hypocaloric feeding is unnecessary and avoidable.84,85 Severe underfeeding over a short time particularly during the initial week of ICU stay is associated with the formation of an energy debt that leads to increased infections, complications and longer ICU stays.34 Factors that contribute to unintentional hypocaloric feeding include staffing shortages, unavailability of feeds/equipment, low priorities for feeding, fasting for clinical investigations, blockages in feeding tubes and variations in feed prescriptions.86 Delivery issues, such as elective interruption for investigative procedures or operations, contributed to hypocaloric feeding with only 76% of prescribed feeds delivered to critically ill patients.87 Similar results were observed in mechanically-ventilated patients,88 where more than 36% of patients received less than 90% of their caloric requirements.
Enteral Feeding Protocols
Enteral feeding protocols improve the delivery of enteral feeds87,89,90 and have been shown to improve clinical outcomes.83,91,92 But protocols vary widely between units and institutions,24,58,93–95 primarily as a consequence of the shortage of reliable and valid research into the effective delivery of enteral nutrition. In the absence of strong research evidence, rituals are embraced and rarely challenged.86 Furthermore, the implementation and sustainability of guidelines is influenced by multiple factors, e.g. clinicians, patients, context and contents of guidelines.96
Management of Enteral Feeding
Routes of enteral feeding
The insertion of enteral feeding tubes into the correct place in the critically ill can be difficult because of reduced cough reflex, altered sensorium and use of sedative and narcotic medications.97 Wide-bore nasogastric tubes (sump tubes) are most commonly used in the critically ill in the early stages of enteral feeding. Because long-term use of wide-bore tubes can contribute to sinusitis, a fine-bore feeding tube is often introduced if enteral feeding is expected to continue beyond a few days. Should prolonged enteral feeding be anticipated (longer than 1 month), gastrostomy, duodenostomy or jejunostomy tubes may also be used.98 Postpyloric feeding has not been shown to be beneficial over gastric feeding,99,100 but is useful for later enteral feeding in patients if gastric atony is present and the patient has persistent high gastric residual volumes.101
For some critically ill patients, gastric secretions may increase when small bowel feeding is initiated.102 A double-lumen tube is available, one lumen for gastric aspiration and decompression and the second for simultaneous jejunal feeding, but these tubes are not widely used in the clinical setting.103
Assessment of enteral feeding tube placement
Correct placement of enteral feeding tubes in the critically ill can be difficult.104,105 Misplacement of the feeding tube into the tracheobronchial tree are important complications of tube insertion.106 Additional complications such as infusion of tube feedings, pneumothorax, pneumonitis, hydropneumothorax, bronchopleural fistula, empyema and pulmonary hemorrhage have been reported.107–112 While confirmation of tube placement is routinely done with radiography, this approach does not prevent incorrect placement occurring during insertion; less reliable methods of confirming tube placement include the use of auscultation and aspiration, and other novel methods such as capnography.105,113
• Aspirate from critically ill patients who receive continuous feedings may have the appearance of unchanged formula, regardless of the site of the feeding tube, therefore this method should not be used.114
• Analysis of the pH of gastric secretions is not reliable. A pH of 0–5 may be used to indicate gastric placement of enteral feeding tubes, although this technique may be problematic for patients receiving histamine-2-receptor antagonists or proton pump inhibitors. If the aspirated fluid has a low pH, it may be assumed that the fluid originated in the stomach but the pH of fluid from an infected pleural space can also be acidic,115 therefore pH testing as a sole method to determine tube placement is not recommended.116
• End-tidal CO2 (ETCO2) detectors: capnometry and capnography. Capnometry and capnography use ETCO2 CO2 detectors to evaluate enteral tube placement but they are not used in routine clinical practice.107,117–122 Differentiating between oesophageal, stomach or intestinal placement is not possible.116
• Pepsin/trypsin. Measuring the concentrations of pepsin and trypsin in feeding tube aspirates can be used as a method of predicting tube placement however methods to measure pepsin and trypsin at the bedside are currently unavailable.123
In the absence of X-ray, several approaches should be used in combination to verify tube position. Metheny and colleagues114 found measuring: (a) length of tubing extending from the insertion site, (b) volume of aspirate from the feeding tube, (c) appearance of the aspirate, and (d) pH of the aspirate were able to correctly differentiate between gastric and bowel tube placement during continuous feedings in 81% of the predictions. Ongoing assessment of feeding tube placement is also essential, as feeding tubes may migrate after initial placement.
Feeding regimens
Once the enteral feeding tube is successfully placed, administration of the feeding solution can begin using a variety of methods, including bolus, intermittent and continuous enteral feeding (see Table 19.4). Bolus enteral feeding is rarely used in the critically ill, but it is less clear whether intermittent or continuous feeding is more beneficial.124–126 Because of inconclusive evidence regarding feeding regimens, decisions are best based on individual patient assessment and the clinician’s clinical judgement.
Commencing enteral feeding
The starting rate for enteral feeding is controversial, with suggestions in the range of 10–100 mL/h,86 and the commonest starting rate being 30 mL/h, despite there being no empirical data on which to base this recommendation. Increasing the rate of enteral feeding is equally variable, but strategies to progress patients towards meeting their daily caloric requirements should be employed. When a patient has experienced a prolonged period of starvation or total parenteral nutrition, the approach to enteral feeding is somewhat more reserved, as the risk of refeeding syndrome is increased.130–132 Although not common, this syndrome is associated with severe derangement in fluid and electrolyte levels (particularly hypophosphataemia, hypomagnesaemia and hypokalaemia), and may result in significant morbidity and mortality.
Managing complications of enteral feeding
Once enteral feeding is established, it is important to assess for such complications as:
This intolerance to enteral feeding can result in gastric distension, diarrhoea and increased GRV.87,133,134
Critically ill patients exhibit elevated gastric residual volume for a variety of reasons including feeding intolerance135–139 and reduced gastric motility.135,136,140 Monitoring tolerance to enteral feeding through the measurement of gastric residual volume has always been viewed as an important aspect of nursing management, although consensus on what constitutes a high gastric residual and any recommendations for interventions remain controversial. Ceasing feeds in response to gastric residual volume is questionable,141 particularly as a balanced enteral diet in itself has a prokinetic effect.142
Practice tip
In determining feeding intolerance, a single high gastric residual volume in the absence of physical examination or radiographic findings should not result in the cessation of enteral feeding. Persisting with enteral feeding has demonstrated benefits. It is thought that a balanced enteral diet, in itself, has a prokinetic effect.143
Development of diarrhoea is another complication for enterally fed patients, and is a common reason why enteral feeding is often reduced or ceased. Diarrhoea may contribute to fluid and electrolyte disorders, patient (and nursing) distress, and a higher cost of patient care.144 Unfortunately, defining diarrhoea is problematic, as it is a subjective assessment that relies on nursing interpretation rather than on quantifiable assessment of stool weight.145 There are various aetiologies for diarrhoea in the enterally fed, critically ill patient, including:
Probiotic administration may limit the development of diarrhoea,149 although its efficacy is yet to be established.150,151
Enteral feeding solutions present an excellent medium for the growth of microorganisms,152 and bacterial contamination of enteral feeds is common.153–155 Strategies to limit bacterial contamination of enteral feeding solutions include:
• meticulous preparation of feeding solutions and equipment156
• commercially prepared formula used in preference to decanted feeds157–159
• use of closed feeding systems93,160,161
• limiting the time feeding solution is kept at room temperature once opened and hang times93,148,157,162–168
• meticulous attention to hand washing and limiting manipulation of the enteral nutrition bags and delivery system at the bedside153,155
Prevention of pulmonary aspiration
An important complication of enteral feeding is the development of pulmonary aspiration and nosocomial pneumonia. Determining whether aspiration has occurred is difficult, even for experienced clinicians. High gastric residual volumes have been linked to the potential for pulmonary aspiration, although this has not been shown in research.141 Oropharyngeal secretions can contribute to nosocomial pneumonia and subglottic aspiration has improved outcomes.169 Nursing strategies to improve gastric emptying includes elevation of the head of the bed 30–45 degrees (unless otherwise contraindicated),170 reducing the likelihood of gastro-oesophageal reflux, which is present in up to 30% of patients in the supine position.
Prokinetic agents can improve gastric emptying and feeding tolerance, and avoid gastro-oesophageal reflux and pulmonary aspiration. Cisapride, erythromycin and metoclopramide have all been used clinically to improve gastrointestinal motility. A systematic review noted that, as a class of drugs, promotility agents have a beneficial effect on gastrointestinal motility in the critically ill patient.171 These prokinetic agents do, however, have undesirable effects. Use of erythromycin is associated with the development of bacterial resistance, and metoclopramide is associated with numerous systemic side effects. Erythromycin is more effective than metoclopramide in treating gastric intolerance among patients receiving enteral nutrition.172 However, combination therapy with erythromycin and metoclopramide is more effective than erythromycin alone in improving the delivery of enteral nutrition.173
Assessment of pulmonary aspiration
• The dye method involves the addition of blue food colouring to the enteral feeding formula, theoretically making it possible to visualise gastric contents if they have been inhaled into the tracheobronchial tree. However, the use of blue dye is poorly standardised and has a low sensitivity in detecting microaspiration.174 The use of methylene blue is not recommended because of associated side effects and high costs.175 There have been case reports of blue dye absorption describing discolouration of the skin, urine, serum and organs,176 and refractory hypotension and severe acidosis, suggesting poisoning by a mitochondrial toxin.177,178 These safety concerns, coupled with minimal benefits, have resulted in the recommendation that the practice of using blue food colouring in enteral feeding solutions be abandoned.179
• Measurement of glucose in tracheobronchial secretions is another method to detect pulmonary aspiration.180 As these secretions normally contain <5 mg/dL glucose, higher amounts of glucose may indicate the aspiration of glucose-rich enteral feeding formula.68However, differences in enteral feeding solutions affect the sensitivity of this method, with low glucose solutions being more difficult to detect. Also, patients not receiving enteral feeding can have detectable glucose in aspirates.181 This is further confounded by the presence of blood, which is closely associated with glucose values >20 mg/dL; consequently, any blood in the respiratory tract could contribute to a false-positive result.181 These findings led to the consensus that glucose monitoring in respiratory secretions should also be abandoned.179
• Measurement of pepsin in tracheobronchial secretions has been used in an animal study suggested that the detection of pepsin, a component of gastric secretions, may be useful in determining pulmonary aspiration.182 however, further investigation in acutely ill patients receiving enteral feeding is necessary.
Parenteral Nutrition
The appropriate use of PN in the context of critical illness continues to be debated.183–185 EN is the preferred method of nutritional support because it is less expensive and is associated with fewer infectious and metabolic complications than PN. However, it is not uncommon for critically ill patients to have difficulty in meeting daily caloric intake34,71 and this may necessitate supplementation of enteral nutrition with PN or the sole provision of nutritional support through parenteral means (as TPN). For patients who are unable to be fed by the enteral route and who were healthy prior to ICU admission, with no evidence of protein-calorie malnutrition, then it is recommended that PN be initiated after 3–7 days186 of hospitalisation.187 The lack of agreement on the efficacy of PN means that the use of this therapy varies both within and between countries.58,186,187
PN solutions contain carbohydrates, lipids, proteins, electrolytes, vitamins and trace elements. PN, whether supplementary or complete, provides daily allowances of nutrients and minerals. The components of PN are listed in Table 19.5. The addition of vitamins and trace elements to PN solutions is necessary, particularly as water-soluble vitamins and trace elements are rapidly depleted (see Table 19.6). Glucose is the primary energy source in PN solutions. Concentrations of 10–70% glucose may be used in PN solutions although the final concentration of the solution should be no more than 35%. The high concentration of PN solutions can cause thrombosis so PN is normally infused via a central venous catheter (CVC). Peripheral administration can be considered when the final solution concentration is 10–12%,188 but is not usually used in the context of critical illness because high volumes of PN would be required to meet caloric requirements.189
• Content varies in amino acid solutions, so content in relation to patient requirements needs to be considered.
• Monitoring of electrolyte status is essential, particularly serum phosphate levels if the amino acid solution used is phosphate-free.
• The balance between chloride and acetate is monitored, as administration of additional sodium or potassium may result in acid–base imbalances.
TABLE 19.6 Trace elements in TPN192
| Trace element | Action |
|---|---|
| Zinc | Wound healing |
| Iron | Haemoglobin synthesis |
| Copper | Erythrocyte maturation and lipid metabolism |
| Manganese | Calcium and phosphorus metabolism |
| Cobalt | Essential constituent of vitamin B12 |
| Iodine | Thyroxine synthesis |
| Chromium | Glucose utilisation |
Catheter insertion, ongoing care and replacement are similar to that with any other CVC. A dedicated CVC, or lumen of a multilumen CVC, should be used for PN.191,193 Manipulation of the CVC and tubing should be avoided to minimise infection of the catheter.
Routine monitoring of the patient’s fluid balance, glucose, biochemical profile, full blood count, triglycerides, trace elements and vitamins is necessary. The patient is also assessed for signs of complications associated with the administration of PN (see Table 19.7).
TABLE 19.7 Short-term metabolic complications associated with total parenteral nutrition
| Complication | Cause | Detection and treatment |
|---|---|---|
| Hyperosmolar coma | Occurs acutely if a rapid infusion of hypertonic fluid is administered. Infusion can cause severe osmotic diuresis, resulting in electrolyte abnormalities, dehydration and malfunction of the central nervous system. | Daily blood samples, accurate measurements of fluid balance, routine blood samples. Reduce infusion rate, correct electrolyte imbalances. |
| Electrolyte imbalance | Disturbances in serum electrolytes, particularly sodium potassium, urea and creatinine, may occur early in the treatment of TPN. Electrolyte imbalances can be caused by the patient’s underlying medical condition; requirements vary with individual patients’ needs. Can be caused by inadequate or excessive administration of intravenous fluids. | Daily blood samples taken early in treatment to detect abnormalities. Replacement fluid as required, extra intravenous fluids may be required during the stabilisation period. |
| Hyperglycaemia | Critically ill patients may be resistant to insulin because of the secretion of ACTH and adrenaline. This promotes the secretion of glycogen, which inhibits the insulin response to hyperglycaemia. | Monitor the patient’s blood sugar 4-hourly after commencement of treatment or as required. Monitor daily urinalysis for glucose and ketones. An insulin infusion may be required to keep blood sugar levels within prescribed limits. |
| Rebound hypoglycaemia | May occur on discontinuation of TPN because hyperinsulinism may occur after prolonged intravenous nutrition. A rise in serum insulin occurs with infusion, and thus sudden cessation of infusion can result in hypoglycaemia. | Glucose infusion rate should be gradually reduced over the final hour of infusion before discontinuing. Some patients may receive a 10% glucose solution after cessation of TPN. |
| Hypophosphataemia | Glucose infusion results in the continuous release of insulin, stimulating anabolism and resulting in rapid influx of phosphorus into muscle cells. The greatest risk is to malnourished patients with overzealous administration of feeding. Patients who are hyperglycaemic, who require insulin therapy during TPN or who have a history of alcoholism or chronic weight loss may require extra phosphate in the early stages of treatment. | Monitor phosphate levels daily. Hypophosphataemia will usually appear after 24–48 hours of feeding. Reduce the carbohydrate load and give phosphate supplementation. |
| Lipid clearance | Lipids are broken down in the bloodstream with the aid of lipoprotein lipase found in the epithelium of capillaries in many tissues. A syndrome known as fat overload syndrome can occur when infusion of lipid is administered that is beyond the patient’s clearing capacity, resulting in lipid deposits in the capillaries. | Blood samples should be taken after the first infusion commences (within 6 hours) to observe for lipid in the blood. |
| Side effects of lipid infusion | Some patients suffer symptoms either during or after an infusion of lipid mix parenteral nutrition. The exact cause is unknown. The patient may complain of headache, nausea or vomiting, and generally feels unwell. | Treat mild symptoms. If tolerated, the TPN solution of non-protein calories can be given in the form of glucose. However, it is essential that the regimen includes some fat to prevent the development of fatty acid deficiency. |
| Anaphylactic shock | This is a rare complication but may occur as a reaction to the administration of a lipid. | It may be necessary to administer adrenaline and/or steroids, and to provide supportive therapy as required. |
| Glucose intolerance | TPN using glucose as the main source of calories is associated with a rise in oxygen consumption and CO2 production. The workload imposed by the high CO2 production may precipitate respiratory distress in susceptible patients, particularly those requiring mechanical ventilation. | Observe patients for signs of respiratory distress. Provide non-protein calories in the form of glucose lipid mix. Slow initial rate of infusion. |
| Liver function | Abnormalities with liver function can be associated with TPN. May be attributable to hepatic stenosis with moderate hepatomegaly; patient may also develop jaundice. Liver function tests often return to normal after cessation of therapy; however, TPN can lead to severe hepatic dysfunction in neonates. | Monitor liver function tests twice weekly. There are several factors that may contribute to development of abnormal liver function tests. These most often occur after a period of time and appear to be more of a problem when there is an excess calorie intake or in glucose-based regimens. |
ACTH = adrenocorticotrophic hormone.
Stress-Related Mucosal Disease
The reported incidence of stress-related mucosal damage is variable194 and complicated by definitions of end points, difficulty in measuring the end points, and the heterogeneity of the patient populations.195 With occult bleeding (drop in haemoglobin level or positive stool occult blood test) as an endpoint, it is estimated that 15–50% of critically ill patients would be reported to have stress-related mucosal damage.196,197 Reported incidence is reduced to 25% or less when haematemesis or nasogastric lavage positive for bright red blood is used as an endpoint to describe clinically overt bleeding.198,199 The incidence of clinically significant bleeding, that is bleeding associated with hypotension, tachycardia, and a drop in haemoglobin level necessitating transfusion, is estimated to be 3–4%.194
Factors influencing the development of stress-related mucosal disease include splanchnic hypoperfusion200 which may influence mucosal ischaemia and reperfusion injury,201 maintenance of the gastric mucosa by sufficient microcirculation and the mucus-bicarbonate gel layer,202 decreased prostaglandin levels which impairs mucus replenishment and increased nitric oxide synthase which contributes to reperfusion injury and cell death.203 The protective mechanisms and factors which promote injury are detailed in Table 19.8.
TABLE 19.8 Factors contributing to stress-related mucosal disease204
| Factors | Mechanism | Action |
|---|---|---|
| Protective mechanisms | Mucosal prostaglandins | Protect the mucosa by stimulating blood flow, mucus and bicarbonate production205 Stimulate epithelial cell growth and repair |
| Mucosal bicarbonate barrier | Forms a physical barrier to acid and pepsin, preventing injury to the epithelium206 | |
| Epithelial restitution and regeneration | Epithelial cells rapidly regenerate but the process is highly metabolic and may be impaired by physiological stress206 | |
| Mucosal blood flow | Mucosal blood flow helps remove acid from the mucosa, supplies bicarbonate and oxygen to the mucosal epithelial cells207 | |
| Cell membrane and tight junctions | Tight junctions between mucosal epithelial cells prevents the back diffusion of hydrogen ions208 | |
| Factors promoting injury | Acid | Acid is a key issue in the pathogenesis of stress-related mucosal injury however not all critically ill patients hypersecrete acid.14,208 However small amounts of acid may still cause injury and the prevention of acid secretion has led to a reduction in injury209 |
| Pepsin | May cause direct injury to the mucosa210 Facilitates the lysis of clots211 |
|
| Mucosal hypoperfusion | Reduced mucosal blood flow results in reduced oxygen and nutrient delivery, making epithelial cells susceptible to injury.208 Contributes to mucosal acid-base imbalances Results in the formation of free radicals |
|
| Reperfusion injury | Nitric oxide, which causes vasodilation and hyperaemia, is released during hypoperfusion and results in an increase in cell-damaging cytokines | |
| Intramucosal acid–base balance | The mucus layer protects the epithelium and traps bicarbonate ions that neutralise acid thus a decrease in bicarbonate secretion results in intramucosal acidosis and local injury206 | |
| Systemic acidosis | Results in increased intramucosal acidity207 | |
| Free oxygen radicals | Generated as a result of tissue hypoxia, free oxygen radicals cause oxidative injury to the mucosa212 | |
| Bile salts | Bile salts reflux from the duodenum into the stomach and may have a role in stress-related damage although the exact mechanism is uncertain213 | |
| Heliobacter pylori | Conflicting results about the role of H. pylori as a cause of stress-induced mucosal disease in the critically ill214 |
Risk Factors for Stress-Related Mucosal Disease
A number of risk factors are associated with the development of stress-related mucosal disease, including respiratory failure requiring at least 48 hours of mechanical ventilation and coagulopathy,215 acute hepatic failure, hypotension, chronic renal failure, prolonged nasogastric tube placement, alcohol abuse, sepsis and an increased serum concentration of anti-Helicobacter pylori (H. pylori) immunoglobulin A.216
Mortality rates for critically ill patients who develop stress-related mucosal disease approximate 50–77% and higher than for those who do not develop this complication.200 Consequently, there is a strong imperative to implement stress-ulcer prophylaxis, particularly in those patients who are considered at risk.
Preventing Stress-Related Mucosal Disease
Prophylaxis for stress-related mucosal disease is often part of the care of the critically ill although evidence demonstrating an added benefit when this therapy is applied to those patients who are not identified as at risk for developing stress-related mucosal disease, is limited.201 Nevertheless, it is common for the majority of critically ill patients to receive some form of stress-ulcer prophylaxis during their episode of critical illness. There are a variety of pharmacological strategies that can be used to prevent stress ulcers from developing. These include antacids, sucralfate, histamine-2-receptor antagonists and proton pump inhibitors (PPIs).201
Antacids
Antacids directly neutralise gastric acid and have been shown to be effective in reducing significant stress-related bleeding.217 One of the disadvantages of this therapy is the time-intensive nature of administering antacids every 1–2 hours. Furthermore, antacids can contribute to further complications (e.g. aluminium toxicity, hypophosphataemia, diarrhoea or hypermagnesaemia). These factors have led to their infrequent use within the critical care setting.200,218,219
Histamine-2-Receptor Antagonists
Histamine-2-receptor antagonists (H2RAs) are commonly used in the critically ill to inhibit the production of gastric acid, which is achieved by the drug binding to the histamine-2 receptor on the basement membrane of the parietal cell.196 However, gastric acid secretion may also occur through stimulation of the acetylcholine or gastrin receptors present in parietal cells;220 therefore complete blocking of gastric acid production does not occur when H2RAs are used. A further limitation of H2RA is the development of tolerance that may occur within 72 hours of administration.221 Nevertheless, this pharmacological strategy to prevent stress-related mucosal disease remains commonplace in critical care.222
The decrease in gastric acidity as a result of H2RA use may be beneficial from the perspective of preventing stress-related mucosal disease, but changes in gastric pH could lead to bacterial overgrowth in the stomach, microaspiration, and consequently an increase in the incidence of nosocomial pneumonia,223 although there is some research that does not support this notion.209
Proton Pump Inhibitors
Proton pump inhibitors (PPIs) have a greater ability to maintain an increased intragastric pH than H2RAs.224 These drugs work by irreversibly binding to the proton pump, effectively blocking all three receptors responsible for gastric acid secretion by the parietal cell.196,201 PPIs are also able to limit vagally-mediated gastric acid secretion.200
Clinical evaluation of the efficacy of PPIs is somewhat limited; few studies have specifically studied the prophylactic use of PPIs for stress-related mucosal diseases12,225–227 and many are limited by small sample sizes. Although PPIs are similar to H2RA in the ability to raise the gastric pH above 4, a level considered adequate to prevent stress ulceration, PPIs are more likely to maintain the pH at greater than 6, which may be necessary to maintain clotting in those patients at risk of rebleeding from peptic ulcer.201
PPIs that may be administered intravenously include omeprazole, esomeprazole and pantoprazole. Omeprazole has the highest potential for drug interation and interferes with the metabolism of some drugs commonly used in intensive care, including cyclosporine, diazepam, phenytoin and warfarin.203 Pantoprazole has the lowest potential for drug interactions.200
Sucralfate
Sucralfate provides protection against stress-related mucosal disease through a number of mechanisms. Sucralfate provides a protective barrier on the surface gastric epithelium, stimulates mucus and bicarbonate secretion, stimulates epithelial renewal, improves mucosal blood flow and enhances prostaglandin release.196 Given orally or via a nasogastric tube, sucralfate is well tolerated but appears to be less effective than H2RAs in decreasing clinically significant bleeding.228 Earlier reports comparing sucralfate with ranitidine showed a decrease in the development of pneumonia in those patients receiving sucralfate; however, these findings were not supported in a subsequent Level I randomised controlled trial.228
Enteral Nutrition
It is thought that the presence of enteral feeding solution results in an increase in intragastric pH, thereby minimising acid injury. Several studies have demonstrated a lower incidence of stress-related bleeding in mechanically-ventilated229 and burn patients,230 while others were unable to show a significant effect on increasing gastric pH.231 A lack of well-designed prospective studies examining the role of enteral nutrition in stress-ulcer prophylaxsis prevents the use of this therapy as a sole therapeutic agent for this purpose.201
Liver Dysfunction
Related Anatomy and Physiology
The liver is the largest internal organ, weighing approximately 1200–1600 g in the adult. It receives approximately 25% of total cardiac output through a dual vascular supply consisting of the hepatic artery and portal vein.232 About 75% of the hepatic blood flow arises from the portal vein with the remaining 25% from the hepatic artery. Anatomically, the liver consists of 4 lobes: the major left and right lobes, and the minor caudate and quadrate lobes. The right lobe is considerably larger than the left lobe. Functionally, the liver is divided into eight segments each with their own inflow and outflow blood supply and biliary drainage. Hepatic lobules, or liver acini, are small units consisting of a single or double layer of hepatocytes arranged in plates interspersed with capillaries (sinusoids) that receive inflowing blood from the portal vein and hepatic artery. To safeguard the body from the entrance of toxins absorbed from the intestines, the sinusoids are lined by macrophages known as Kupffer cells. The hepatic vein then drains effluent blood from the liver into the general circulation.1
The arrangement of the circulation to the liver with its rich vascular architecture enables it to perform the vital functions of carbohydrate, fat and protein metabolism; production of bile to aid in digestion; the production, conjugation and elimination of bilirubin; immunological and inflammatory responses; glycogen storage; and detoxification of toxins and drugs.1
Mechanisms of Liver Cell Injury
Liver cell injury and death can occur either as a direct result of injury to the cell, resulting in cell necrosis, or as a result of ‘cellular stress’ and the triggering of apoptotic pathways, leading to ‘programmed cell death’.233 Major factors for the triggering of the apoptotic pathway are hypoxia with resulting ischaemia and reperfusion; reactive oxygen metabolites resulting from alcohol or drug ingestion; accumulation of bile acids resulting from cholestasis; and inflammatory cytokines such as tumour necrosis factor alpha (TNF-α).233 The apoptotic pathway results in the deconstruction of the cellular structure from the inside out, while necrosis results in cell rupture and release of cellular contents. Although these processes may overlap, it is thought that the apoptotic pathway is a way of preventing the inflammatory response that is triggered with cell necrosis. The activation of the inflammatory response results in secondary liver cell injury and contributes to the multiple organ dysfunction seen in liver failure.233,234
The degree and time course of liver cell damage from viral hepatitis depends on the immune response. Immune recognition and destruction of infected cells may result in either clearance of the virus or ongoing inflammation, cell death and fibrosis if the virus is not cleared. This process may progress over 20–40 years to cirrhosis and hepatocellular carcinoma.235 Chronic excessive alcohol intake may also result in a slower chronic course of liver injury that eventually results in cirrhosis, liver failure or hepatocellular carcinoma.236
Liver cells may also be injured by the toxic effects of drugs or their metabolites, as in paracetamol overdose, or by drugs at therapeutic doses (e.g. non-steroidal anti-inflammatory drugs, phenytoin, antimalarial agents). Other poisoning from the ingestion of mushrooms (e.g. Amanita phalloides), and from recreational drug use (e.g. ecstasy and amphetamines), may result in liver cell death and liver failure.237 Diseases of the biliary system such as primary biliary cirrhosis and primary sclerosing cholangitis also result in liver dysfunction and failure.238
The liver has a remarkable regenerative capacity. After injury and necrosis, liver cells rapidly regenerate around areas of surviving cells to restore the lost tissue whilst maintaining homeostasis during hepatic regeneration.234,239,240 However, with chronic injury, fibrosis or scarring occurs, resulting in the loss of the functional architecture and cell mass and ultimately in cirrhosis. Cirrhosis results in destruction of the normal liver vasculature, increased resistance to blood flow, and back pressure into the portal circulation. Dilation of the venous system leading into the liver results in the formation of varices.241
Liver cell injury may occur to such a degree that a critical amount of hepatic necrosis results in the failure of the liver to maintain metabolic, synthetic and clearance functions leading to death. Liver cell injury may also occur more slowly, giving rise to chronic liver injury.236
Epidemiology of Viral Hepatitis
In developed countries such as Australia and New Zealand, viral infection, primarily from hepatitis B and hepatitis C viruses, is the major cause of liver cell injury leading to liver failure.242,243 Although viral hepatitis can result in acute liver failure, it more often results in chronic disease that may lead to cirrhosis and hepatocellular carcinoma.243 While the prevalence of hepatitis B in Australia and New Zealand is generally low, infection rates among social subgroups, such as the socially disadvantaged, migrants from Asian countries, injecting drug users, homosexual males, and those with a history of incarceration, are high.244,245 Hepatitis C is blood-borne, with intravenous drug use the cause of about 80% of hepatitis C infections. Blood screening has greatly reduced the incidence of hepatitis C infections.246 In Australia in 2009, approximately 217,000 people were living with chronic hepatitis C infection, with 46,000 in the moderate to severe liver disease category.247 However, about 25% of people with exposure to hepatitis C virus have cleared the virus and are not chronically infected. It is estimated that the number of people with hepatitis C will increase in Australia and New Zealand due to lack of access to antiviral therapy.247–249 Vertical transmission (transmission from the mother to the child during the perinatal period) at birth is a major cause of such infections in children.243
Liver Dysfunction/Failure
Liver dysfunction can be acute or chronic. Chronic liver disease is usually associated with cirrhosis and can develop from viral (hepatitis B and C), drug (alcohol), metabolic (Wilson’s disease), or autoimmune (primary biliary cirrhosis) conditions. Acute liver failure (ALF) is an uncommon condition associated with rapid liver dysfunction leading to jaundice, hepatic encephalopathy and coagulopathy.250 The term ‘fulminant hepatic failure’ is often used synonymously; however, it has been proposed that ‘hyperacute’, ‘acute’ and ‘sub-acute’ liver failure should be used instead.251 In this classification, hyperacute refers to patients who develop encephalopathy within 7 days of the onset of jaundice, acute liver failure should be used in patients between 8–28 days from jaundice to encephalopathy and sub-acute liver failure when encephalopathy occurs within 5–12 weeks of the onset of jaundice.251 This has not received universal acceptance with the terms fulminant and sub-fulminant hepatic failure still used in clinical practice.
ALF without preexisting liver disease can result from drug reactions, toxins or viral infection, or from the effect on inflammatory mediators released in response to tissue injury. Liver failure can also occur as an acute decompensation of chronic liver disease (acute-on-chronic liver failure: AoCLF) or as an end-stage decompensation in chronic liver failure. AoCLF can be precipitated by bacterial or viral infection, bleeding or intoxication, and results in the same clinical syndrome as seen in ALF.234
End-stage decompensation of chronic liver failure represents irreversible deterioration with inadequate residual function to maintain homeostasis, and liver transplantation is the only viable treatment (see later in the chapter). However, in AoCLF, the function of the residual liver cell mass may be adequate to maintain hepatic homeostasis if the precipitating event can be treated.234,236
Liver dysfunction is also a common consequence of critical illness,252,253 and may be caused by inadequate perfusion leading to ischaemic injury or as a result of the inflammatory response in sepsis.234 Given the number of drugs that critically ill patients receive, the possibility of liver injury as a result of drug reactions and toxicity should always be considered.
Consequences of Liver Failure
The consequences of liver failure manifest as a syndrome of hepatic encephalopathy (HE), hepatorenal syndrome (HRS), oesophageal and gastric varices, ascites, respiratory compromise, haemodynamic instability, susceptibility to infection, coagulopathy and metabolic derangement.234,236,237,254
Hepatic Encephalopathy
Hepatic encephalopathy is a reversible neuropsychiatric complication due to metabolic dysfunction associated with liver disease.255 The cerebral effects of liver failure may manifest as an altered sleep–wake cycle, mild confusion/disorientation, asterixis (i.e. abnormal tremor, especially in the hands) and coma. Patients with AoCLF may develop a mild degree of cerebral oedema, while a differential feature of ALF is the risk of death from cerebral oedema and raised intracranial pressure.256
The exact mechanisms responsible for the development of hepatic encephalopathy are unknown, although raised ammonia levels resulting from the failure of the liver urea cycle are thought to be central to the pathogenesis. The raised ammonia levels disrupt the blood–brain barrier, which leads to the development of cerebral oedema. Ammonia levels also seem to be related to the disruption of neurotransmission, resulting in decreased cerebral function.234,236,256 In addition, reactive oxidative species causing oxidative stress and inflammatory cytokine release have been suggested, and the exact pathophysio-logy is yet to be fully elucidated.257
Previously, hepatic encephalopathy has been classified using the West Haven criteria,258 a four-stage scale according to the severity of clinical signs and symptoms (Table 19.9). However, the West Haven system has poor sensitivity and no inherent metric component. For instance, for patients with grades III–IV encephalopathy, the Glasgow Coma Scale (GCS) is probably a more sensitive tool for neurological assessment.256 Accordingly, other grading criteria have been proposed259,260 but are yet to be validated in large clinical trials.
TABLE 19.9 West Haven grading of hepatic encephalopathy258,261
| Grade | Characteristics |
|---|---|
| I |
Hepatorenal Syndrome
Hepatorenal syndrome (HRS) is the development of renal failure in patients with severe liver disease (acute or chronic), in the absence of any other identifiable cause of renal dysfunction.262 HRS that develops rapidly in the setting of ALF or AoCLF is classified as type 1 HRS, while type 2 HRS is slowly progressing and is usually associated with diuretic-resistant ascites.262,263
The pathophysiological features of HRS appear to be caused by an inflammatory response from the injured liver, resulting in upregulation of nitric oxide production (a vasodilator) and splanchnic vasodilation.234,236,262,263 Splanchnic vasodilation results in redistribution of circulating blood volume and a lowered mean arterial pressure. The reduction in perfusion pressure results in an enhanced sympathetic nervous system response and local renal autoregulatory responses. The net result of these effects is a reduction in renal blood flow and increased activity of the renin–angiotensin–aldosterone system, resulting in sodium (aldosterone) and water retention (arginine vasopressin; see Chapter 18).
Varices and Variceal Bleeding
The development of varices and variceal bleeding arises from portal hypertension. This manifests when blood flowing from an area of high pressure (i.e. the cirrhotic liver) to areas of lower pressure (i.e. the collateral circulation, involving veins of the oesophagus, spleen, intestines and stomach), causes the tiny, thin-walled vessels to become engorged and dilated, forming varices that are vulnerable to gastric secretions, resulting in rupture and haemorrhage.241,264 Variceal haemorrhage is a major cause of acute decompensation and a reason for admission to the ICU. It is an acute clinical event characterised by severe gastrointestinal haemorrhage presenting as haematemesis, with or without melaena, and haemodynamic instability (tachycardia and hypotension).241,264
Respiratory Compromise
Patients with liver failure may have poor oxygen exchange, fluctuating GCS that requires intubation for airway protection and hepatopulmonary syndrome (HPS). HPS is found in 15–20% of patients with cirrhosis.265 It is defined as pulmonary microvascular dilation resulting in impaired oxygenation, and it is generally assumed that vascular production of vasodilators, specifically nitric oxide, underlies the vasodilation in HPS. It has also been hypothesised that the mechanisms that trigger HPS are the same as those that result in the hyperdynamic circulation (low systemic vascular resistance and high cardiac output) seen in liver failure.265 Other factors, such as pleural effusions or severe ascites, may impinge on ventilation.
Haemodynamic Instability, Susceptibility to Infection, Coagulopathy and Metabolic Derangement
The hyperdynamic, low vascular resistance picture, similar to that associated with sepsis, is seen in liver dysfunction. This probably results from the production of vasodilator substances (nitric oxide) from the inflammatory response of the injured liver cells.234 Sepsis may also be a complication of liver dysfunction because of the failure of the liver to produce acute-phase proteins and the impaired function of Kupffer cells.237
Hepatocyte damage leads to a decreased production of the majority of clotting factors and, therefore, haemostasis. Therefore, the risk of bleeding is elevated.266 Disordered metabolic function and failure of synthetic function can manifest as unstable blood glucose levels.
Independent Practice
Early signs of the patient presenting with ALF are malaise, loss of appetite, fatigue, nausea, jaundice, bruising, bleeding, inflamed/enlarged liver, possibly epigastric and right-upper-quadrant pain, high or low blood glucose levels (which require monitoring, at least every 4 hours; patients may require insulin infusion or 10–50% dextrose infusion), deranged liver function tests (LFTs) and fluctuating GCS due to cerebral oedema.237 If acute liver failure is suspected, admission to an ICU is recommended to monitor for further deterioration, and provide supportive management and airway protection. The patient presenting with AoCLF will have similar symptoms but will present with other unique characteristics. Cirrhosis and portal hypertension will often lead to oesophageal and gastric varices, ascites, hepatorenal and hepatopulmonary syndrome, malnutrition, bone disease, sepsis, palmar erythema, spider naevi and feminisation in males.267
Neurological Considerations
Cerebral oedema is present in 80% of patients with grade IV encephalopathy and is the leading cause of death due to brain herniation.268 Patients with cerebral oedema and raised intracranial pressure due to ALF are managed primarily as patients with acute head injury (see Chapter 17).
Collaborative Practice
Assessment of Liver Function
Patients presenting with ALF require a careful history to establish the cause of liver injury. The well-known signs of chronic liver disease (e.g. palmar erythema, spider naevi and ascites) may not be present. Biochemical and haematological tests determine whether liver cell injury is occurring, with liver synthesis and clearance functions assessed by albumin level and prothrombin time, and bilirubin level respectively.269 These measures have been incorporated into a scoring system to determine liver dysfunction and prognostic information for liver transplantation suitability (model for end-stage liver disease [MELD], see later in this chapter under Transplantation).270,271 These tests have been summarised in Table 19.10.253,272
| Blood test | Normal value | Description |
|---|---|---|
| Alanine aminotransferase (ALT) and aspartate aminotransferase (AST) | ALT: <35 U/L AST: <40 U/L |
• ALT and AST are enzymes that indicate liver cell damage; they are produced within the liver cells (hepatocytes) and leak out into the general circulation when the liver cells are damaged. • ALT is a more specific indication of liver inflammation. • In acute liver injury, ALT and AST may be elevated to the high 100s, even 1000s, U/L. • In chronic liver damage such as hepatitis or cirrhosis, there may be mild to moderate elevation (100–300 U/L). • ALT and AST are commonly used to measure the course of chronic hepatitis and the response to treatments. |
GGT: Males <50 U/L
Females <30 U/L
• These are enzymes that indicate obstruction to the biliary system.
• They are produced in the liver, or within the larger bile channels outside the liver.
• The GGT is used as the supplementary test to be sure that a rise in ALP is indeed coming from the liver or biliary tree.
• A rise in GGT but normal ALP may indicate liver enzyme changes induced by alcohol or medications, causing no injury to the liver.
• ALP and GGT are commonly used to measure bile flow obstructions due to disorders such as gallstones, a tumour blocking the common bile duct, biliary tree damage, alcoholic liver disease or drug-induced hepatitis.
• Liver injury or cholestasis results in an elevated bilirubin level.
• Raised unconjugated bilirubin without an accompanying rise in conjugated bilirubin is consistent with red blood cell destruction (haemolysis).
• Raised bilirubin levels result in jaundice.
• In cases of chronic liver disease, bilirubin levels usually remain normal until significant damage occurs and cirrhosis develops.
• In cases of ALF, bilirubin levels will rise rapidly and result in marked jaundice; the degree of rise is indicative of the severity of illness.
Treatment
ALF or AoCLF therapy often involves the support and treatment of the consequences of liver failure, such as sepsis, encephalopathy, renal failure and coagulopathy (see Table 19.11). One specific support therapy that may be used to prevent further liver cell injury is administration of N-acetylcysteine (NAC), a glutathione donor that acts to replenish liver cellular stores of this scavenger of toxic oxygen free-radicals. Inflammation, the accumulation of bile acids, and ischaemia/reperfusion results in the build-up of oxygen free-radicals, which can induce hepatic necrosis if not controlled.237
• Paracentesis is very effective at reducing ascites and is a simple procedure to remove fluid and an aid in diagnosis.
• Correction of coagulopathy or thrombocytopenia should be considered when the INR is greater than 2.5 or the platelet count markedly reduced.
• Paracentesis may aid in determining the cause of ascites (ascites-serum albumin gradient, ascitic cytology, microscopy and culture for acid-fast bacilli, chylous ascites) and in establishing or excluding primary or secondary peritonitis in patients with ascites (ascitic WCC and neutrophil count, culture).
• Litres of ascites are normally removed, and the volume is replaced with IV concentrated albumin to prevent fluid shifts and hypotension.
• Mean arterial pressures, central venous pressures, heart rate and urine output are carefully monitored during the procedure. For every litre of ascites removed, 6–8 g albumin is infused.275
Oesophageal balloon tamponade and transjugular intrahepatic portosystemic stent/shunt
There are two types of balloon tamponade devices available on the market: the Sengstaken-Blakemore tube (see Figure 19.1) and the Linton tube. The Sengstaken-Blakemore is a four-lumen tube with oesophageal and gastric balloons, and oesophageal and gastric aspiration ports. The benefit of this tube is that direct pressure can be applied on gastric and oesophageal varices by balloon inflation and traction.276 The Linton tube has one lumen for inflation of the pear-shaped gastric balloon and two additional lumens for oesophageal and gastric aspiration.
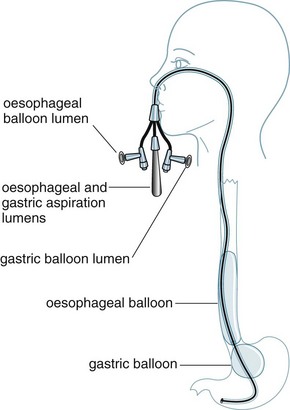
Prior to insertion (oral or nasal), balloons are lubricated, checked for leakage, and the distance to the cardio-oesophageal junction is estimated (nose to ear, then to xiphisternum). Once inserted, the gastric balloon is inflated with 50 mL air and pulled back until resistance is felt. Position (lying compressed against the cardio-oesophageal junction) is confirmed by X-ray. Then the gastric balloon is inflated according to the manufacturer’s instructions and traction is applied using a weight (500 or 1000 mL IV fluid bag) attached to rope; traction is applied via a pulley and IV pole at the foot of the bed. Nursing care276 of patients involves:
• head of the bed raised at least 30 degrees to facilitate gastric emptying and prevent aspiration
• ensuring that gastric/oesophageal ports are on free drainage, with regular monitoring of type and amount of drainage
• ensuring that correct traction is maintained, with regular checking of tube migration and checking position at nares/lips at regular intervals (4/24 hours).
Tamponade is generally maintained for 24–48 hours, then traction is removed and the balloon deflated to assess for further bleeding. If the patient is stabilised, endoscopy can be performed. If bleeding persists, the balloon(s) is/are reinflated and traction reapplied.264,276
Once the patient has been stabilised, a transjugular intrahepatic portosystemic stent/shunt (TIPS) may be con-sidered to control variceal haemorrhage. TIPS is a metal expandable stent inserted to decompress the portal venous system.277
Extracorporeal liver support
The aim of extracorporeal liver support therapy is to allow time for liver recovery or to provide support until a liver transplant is possible.234 Either biological or non-biological systems are available for liver support. Biological systems utilise pig hepatocytes or hepatoma cells to achieve removal of toxins,234 but this requires complex technical support in specialist centres. Non-biological systems are similar to renal replacement circuits, and use albumin as a dialysis medium or dialyse against an activated charcoal medium as a mechanism for toxin removal and liver support.278
Liver Transplantation
Liver transplantation is the definitive treatment for patients suffering acute and chronic end-stage liver failure when other supportive critical care therapies have been exhausted.250,279
In Australia, the first liver transplant was undertaken in Brisbane in 1985.280 Liver transplantation commenced in 1998 at Auckland Hospital in New Zealand.281 Between 1985 and December 2009, 3533 orthotopic liver transplants were performed in Australia and New Zealand on 3277 patients.282 There are six liver transplant units in Australia and New Zealand: the Royal Prince Alfred and Children’s Hospitals, Sydney; the Austin and Royal Children’s Hospitals, Melbourne; the Princess Alexandra and Royal Children’s Hospitals, Brisbane; Flinders Medical Centre, Adelaide; Sir Charles Gairdner Hospital, Perth; and Auckland Hospital, New Zealand.282
Surgical refinement and postoperative management of liver transplantation has reduced time in critical care and reduced overall hospital length of stay. Survival rates of all patients who have undergone liver transplantation exceed 80% at 5 years,283 with children having superior survival rates to adults.282
Indications for Transplantation
Indications for liver transplantation are patients with severe liver disease in whom alternative treatments have been exhausted. Categories consist of acute liver failure, end-stage liver disease, metabolic liver disease and primary liver cancer.284 Timing and patient selection is of critical importance, as this has contributed to the success of transplantation. Re-transplantation for any disorder is considered only in patients with acceptable predicted survival.283
Contraindications for Transplantation
Generally, Australian centres set upper age limits of around 65 years for liver transplantation, but occasionally older patients are considered. Patients with extra-hepatic malignancy and uncontrolled systemic infection (contraindication to high-dose immunosuppressive therapy) are unsuitable for transplantation. In addition, patients with alcoholic liver disease with social instability and patients with inadequate or absent social support are relative contradictions due to increased risk of non-adherence to immunosuppressive therapy.283
Recipient Selection
The model for end-stage liver disease (MELD) and paediatric end-stage liver disease (PELD) scoring systems are used for liver transplantation eligibility in Australia and New Zealand.283,285 The MELD score is a mathematical model that includes bilirubin, creatinine and INR which was originally devised to predict survival after TIPS.286 The MELD score is the best predictor of pre-transplant mortality, and eliminates the subjectiveness of the CTP score regarding the presence and degree of ascites and hepatic encephalopathy.285,287,288
Once the need for transplantation is established, the decision to allocate a donor liver to a patient is based on donor and recipient blood group; donor size and size of recipient; suitability of donor liver for splitting; severity of disease; matching of functional status of donor with severity of liver disease; and hepatitis B and C status of donor and recipient.283
Surgical Techniques
Orthotopic Liver Transplantation
Two main techniques are used for OLTx: portal bypass or the piggyback technique. Portal bypass occurs where an internal temporary portocaval shunt or external veno-venous bypass is used.289–291 In the piggyback technique, the recipient’s inferior vena cava (IVC) is left and the donor IVC is piggybacked onto the recipient’s IVC. The advantages of this technique include haemodynamic stability during the anhepatic phase, reduced operating times and reduced use of blood products, enabling a shorter length of hospital stay.292 The use of T tubes, to monitor bile outflow, leaks, stenosis, and to provide direct access to the biliary system to perform controlled cholangiograms and interventional radiographic procedures293,294 are now not common. It has been shown that there were fewer biliary complications and costs were reduced (there were fewer radiographic interventions) without insertion of a T tube.295
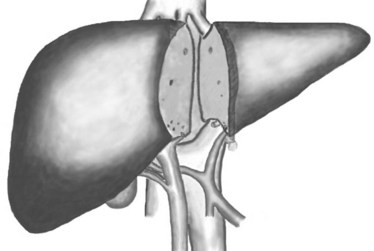
Split-liver Transplantation
The disparity between the increasing number of people on transplant waiting lists and the shortage of donor livers available has led to several innovative strategies. Split-liver transplantation occurs when the cadaver organ is divided for two recipients, with the larger right segment going to an adult and the smaller left lobe to a child (see Figure 19.2).273,296 The complication rate is higher in split-liver than whole-liver transplants due to biliary leaks and anastomosis strictures. The risk of complications and the potential for small-sized grafts are taken into consideration when selecting a recipient patient for transplant. Furthermore, not all donor livers are suitable for splitting. Within the first few months, a split liver will regenerate until it is a full-sized liver; in children it also grows and develops at the same rate as the children. This technique has significantly reduced the number of children waiting for liver transplantation, although little impact has been made on adult waiting lists.273,296
Adult Living Donor Liver Transplantation
Living donor liver transplantation (LDLT) is an established option for paediatric patients with end-stage liver disease.297 This technique involves removal of the left lobe from the live donor, usually the recipient’s parent, which is then transplanted into the child. It is a relatively straightforward procedure, with little risk to the donor.297,298
Adult-to-adult LDLT involves transplantation of the right lobe of the liver from a donor to an adult recipient, offering hope to patients with end stage liver disease (ESLD). The operation has been performed with some success, although there are significant risks to the donor, including death and morbidity.298
Postoperative Management
Initial Nursing Considerations
The initial postoperative care is similar for all liver transplant patients. However, progress, stability and discharge from critical care can be affected by the patient’s preoperative condition and severity of liver failure. The unique pathophysiology inherent in the end-stage liver failure patient will predispose to varying effects on coagulopathy and cardiopulmonary, neurological, haemodynamic and metabolic functions.299,300 These issues are discussed below.
Blood loss and coagulopathy
The major risk during and post-surgery is massive blood loss, due to a combination of factors. The surgical process itself involves anastomosis of major arteries and veins, usually in the setting of significant portal hypertension, predisposing the patient to bleeding and hypovolaemia during surgery and anastomotic leaks post-surgery.291 Patients with ESLD will also be coagulopathic from hepatic synthetic dysfunction, leading to failure of synthetic clotting factors. Correction of coagulopathy with blood products such as FFP, platelets, cryoprecipitate and factor VIIa may control minor postoperative bleeding, but if bleeding continues an exploratory laparotomy may be required. Conversely, it is not desirable to overcorrect coagulopathy, due to the potential for vascular complications such as hepatic artery thrombosis. Careful monitoring is required to identify and manage hypotension, tachycardia, excessive blood loss from drains, falling haemoglobin, abdominal swelling and oozing from insertion sites. Thrombocytopenia is a common postoperative problem, with platelet counts often falling in the first week post-transplant. If platelet counts are low, a platelet transfusion may be necessary, especially prior to removal of drains, lines, cannulae and sheaths.
Cardiovascular
Haemodynamic instability in the early postoperative period may be due to hypovolaemia or haemorrhage. Treatment includes fluid boluses to increase preload and the initiation of inotropes may be necessary.301 The patient with ESLD may present with a hyperdynamic profile: high cardiac output, low systemic vascular resistance, and low mean arterial pressure,302 although this usually reverses one week after transplantation.300
Neurological
The most frequent neurological complications relate to patients with preexisting encephalopathy and seizures. In ALF patients, cerebral oedema with raised intracranial pressure (ICP) is common, and after liver transplantation, cerebral oedema may take up to 48 hours to subside. Therefore, continuation of preoperative measures to reduce ICP are necessary. These include the head of the bed at 30 degrees, head, neck and body alignment, ensuring that endotracheal tapes are not constrictive, aiding venous return and preventing cerebral congestion, reducing neurological stimuli and timing activities to prevent spikes in ICP (see Chapter 17).302,303
Respiratory
Preexisting pulmonary complications associated with liver disease can affect postoperative recovery and need to be considered when weaning ventilation and maintaining adequate oxygenation. Patients posttransplant often experience bibasal collapse and consolidation, and are prone to infection. Incentive spirometry, chest physiotherapy, early mobilisation and adequate pain relief are recommended.300
Gastrointestinal
Patients with end-stage liver disease often have malnutrition and bone disease, which may influence post-operative management. Fluid overload and ascites can quite often mask signs of malnutrition. Early nutrition is imperative in the postoperative period. If caloric intake is inadequate, consultation with a dietitian will assist with enteral supplementation. Total parenteral nutrition is rarely required.304
Renal
Renal dysfunction is a significant posttransplantation problem. Risk factors include preexisting renal disease or hepatorenal syndrome, intraoperative hypotension, extensive transfusion of blood products, nephrotoxic drugs such as cyclosporin and tacrolimus, sepsis, and graft dysfunction.305 Hepatorenal syndrome is reversible post-transplantation. Patients who are receiving renal support such as CRRT usually require continuation of renal support postoperatively for a period of time until recovery of kidney function is evident (see Chapter 18).
Graft dysfunction and rejection
Acute graft rejection was the most challenging obstacle in the early years of transplantation, but with the development of current immunosuppressive therapy, acute rejection can be avoided, resulting in improved success rates of transplantation.306 Immunosuppressive therapy is commenced intraoperatively with a high-dose steroid such as methylprednisolone and antibiotic (ticarcillin). Patients are then placed on a triple-therapy regimen consisting of steroids such as methylprednisolone and, later, prednisone, azathioprine and either tacrolimus or cyclosporin.306,307
Allograft dysfunction occurs within 48 hours of transplantation, and is characterised by varying degrees of coma, renal failure, worsening coagulopathy, poor bile production and marked elevation in the liver enzymes (AST, ALT) and worsening acidosis. The cause of allograft dysfunction is not always known; possible causes are injury to the liver, either before or during the donor operation procedure, ischaemic-reperfusion injury or graft stenosis. Acute rejection is generally evident in the second week posttransplant, and is generally suspected with a rise in liver enzymes, a decline in bile quality (accessible only if a T tube is present), occasional fever and tachycardia.308
Primary graft non-function is defined as failure of the graft to function in the first postoperative week. It is manifested by failure to regain consciousness, sustained elevated transaminases, increasing coagulopathy, acidosis and poor bile production. Causes include massive haemorrhagic necrosis, ischaemia-reperfusion injury and hepatic artery thrombosis. It may be difficult to distinguish allograft dysfunction, which may recover, from primary graft non-function, which will not recover, and the only solution is retransplant.308
Confirmation of rejection is by liver biopsy but this is not always possible if the patient is coagulopathic; if the diagnosis is positive, rejection is treated with high-dose steroid pulse therapy, followed by reducing doses of oral prednisone. The majority of rejection episodes respond well to pulse steroid therapy. Previously, treatment with a course of antilymphocyte (e.g. monoclonal antilymphocyte globulin, OKT3)306,307 was recommended, but has now been shown to increase the risk of hepatitis development in patients with transplants for hepatitis C infections.309
Late Complications
Readmission to critical care after liver transplantation is not uncommon, with 1 in 5 patients returning due to complications. The most common factors include cardiopulmonary dysfunction from infection or fluid overload, respiratory failure from collapse and consolidation, tachypnoea, recipient age, preoperative liver function, bilirubin, the amount of blood products administered intraoperatively, graft dysfunction, severe sepsis and postoperative surgical complications such as bleeding and biliary anastomotic leaks.310 Outcomes are affected by intraoperative and postoperative complications, renal failure, advanced liver disease and malnutrition.311
Glycaemic Control in Critical Illness
Hyperglycaemia and increased insulin resistance are characteristics of the stress response and activation of the sympathetic nervous system: adrenal and hypothalamic–pituitary–adrenal (HPA) axis responses to critical illness.312 Hyperglycaemia has been considered a beneficial adaptive response to stress to provide energy substrate to the organs involved in the ‘fight or flight’ response.313 However, there is some, although inconsistent, evidence of the association of hyperglycaemia with high mortality and morbidity.313–317 Hyperglycaemia has been associated with: poor wound healing and higher rates of infection after surgery in diabetic patients; higher risk of death after myocardial infarction in diabetic and non-diabetic patients; and poor outcomes after stroke.313,318,319
The complexity of the physiological processes associated with hyperglycaemia in critical illness and the sophisticated research required to generate valid information renders clinical decision-making related to glycaemic control challenging. Since the first landmark study of glycaemic control in the critically ill,320 there have been at least 26 randomised controlled trials investigating tight glycaemic control.321 Contradictory results, even from those studies that appear to have used similar methods, has continued to fuel the debate on tight glycaemic control with some experts urging caution and others seeing tight glycaemic control as a marker of quality practice.322 The discrepancies in these studies have been attributed to many factors including the variability in target ranges for blood glucose, methods of blood glucose measurement, difficulty for some studies to achieve separation of the treatment and control groups, compliance with the therapy, and employment of different nutritional strategies.323
Of particular importance when implementing tight glycaemic control, is monitoring for hypoglycaemia. Two large clinical trials of tight glycaemic control – NICE-SUGAR314 and the COIITSS Study324 – reported reasonably high rates of hypoglycaemia (6.8% and 16.4% respectively), highlighting the need for vigilance in assessing blood sugar levels. The time and frequency of blood glucose measurement that may be required for some patients may impact on the provision of patient care, and the inability to perform the testing as often as required may potentially contribute to underdetection of hypoglycaemia. Another potentially important factor that may contribute to underdetection of hypoglycaemia is fatigue in nurses caring for the critically ill. Louie and colleagues.325 reported the results of a single-centre study that found the increased number of antecedent shifts worked by bedside nurses was associated with an increased incidence of hypoglycaemia.
The validity of blood glucose measurement is also an important consideration. Many of the studies to date have measured blood glucose sampled from arterial, venous and capillary blood. The use of capillary blood in testing blood glucose may be problematic, particularly in those patients for whom hypoperfusion is an issue.326–329 Techniques to measure blood glucose include point-of-care testing meters, blood gas analysers and formal laboratory testing. Formal laboratory testing is considered ‘gold standard’ for blood glucose measurement although the delay in receiving has resulted in point-of-care testing meters being common in clinical practice.330 An important consideration with formal blood testing is the potential drop in glucose concentration of up to 6% within the first hour after the blood is taken,331 highlighting the importance of ensuring blood specimens are delivered to the laboratory in a timely fashion and priority is given to sample testing.
Point-of-care testing of blood glucose is common in critical care setting. Research examining the measurement of blood glucose using these devices is conflicting with some studies reporting good performance of devices while others report that the devices are unsatisfactory. A problematic aspect of evaluating point-of-care devices is the failure of many of these to conform332 with quality guidelines for conducting and reporting glucose monitor evaluation studies.333,334
It is clear that hyperglycaemia should be avoided, however, the inconsistencies in published studies mean that an agreed specified target for blood glucose in the critically ill patient population is difficult to achieve.323 The optimal target blood glucose level is unknown and may differ depending on the patient’s clinical presentation.335 Recommendations for patients with sepsis suggest that blood glucose levels be kept lower than 180 mg/dL with a goal blood glucose approximating 150 mg/dL.336
Incidence of Diabetes in Australasia
Diabetes is known to cause substantial morbidity and mortality in Australia. The prevalence of diabetes in Australia is rising and follows a global trend.337 Reasons for this include an increase in the rates of obesity, physical inactivity, the ageing population, better detection of diabetes and longer survival of affected individuals.338,339 In 2004–05, the prevalence of diagnosed diabetes among Australians was 3.6%.337 The rate of diabetes generally increased with age for both males and females, although declining slightly for both sexes at age 75 years and over. Males had higher rates of diabetes than females at ages 45–54 years, 65–74 years and 75 years and over, and ranged from 0.8% vs 0.7% (0–44 years) to 16.3% vs 11.7% (65–74 years). New Zealand is experiencing a diabetes epidemic that has the biggest impact in the Māori and Pacific ethnic groups. The incidence of diabetes was forecast to nearly double by 2011, and to be accompanied by a rise in mortality.340
Diabetic Ketoacidosis
Diabetic ketoacidosis (DKA) is a metabolic derangement resulting from a relative or absolute insulin deficiency, characterised by hyperglycaemia (>11.1 mmol/L), metabolic acidosis (pH <7.3) and ketosis (raised blood ketone bodies or ketonuria). It is usually precipitated, in insulin- and non-insulin-dependent diabetics, by infection or the omission (or inadequate dosing) of insulin.341 It may also be the cause of the first presentation in new-onset diabetes. Hyperglycaemic hyperosmolar non-ketotic state (HHNS) is seen more often in older patients with type 2 diabetes, and is characterised by hyperglycaemia and the pathological consequences of extreme dehydration. Unlike DKA, where there is insufficient insulin, in HHNS insulin excretion is maintained, so lipolysis and ketoacidosis do not feature. Although DKA and HHNS are considered separate entities, DKA and HHNS may coexist in about a third of cases, especially among older patients.342 Additionally, DKA is increasingly being identified in patients with type 2 diabetes.343
Pathophysiology
The metabolic profile seen in DKA is similar to that seen in the fasting state, with substrate utilisation shifting from glucose to fat in insulin-sensitive tissues (fat, liver, muscles). The brain is insulin-insensitive, and requires a continuous supply of glucose to support metabolism even in a fasting state or DKA.344
Inadequate production (or administration) of insulin to meet metabolic need (or a rise in metabolic demand resulting from the stress of infection, trauma or surgery, for instance) is associated with a rise in the secretion of the counterregulatory hormones glucagon, the catecholamines and cortisol.344 The effects of the counterregulatory hormones are presented in Box 19.1.
Box 19.1
Hyperglycaemia results from increased gluconeogenesis (glucose production from precursors other than carbo-hydrates, e.g. amino acids), the conversion of glycogen stores to glucose (glycogenolysis) and the reduced uptake of glucose resulting from insulin deficiency.344 Free fatty acids (FFAs) and glycerol are produced by the breakdown of triglycerides that results from increased catecholamine secretion.344 Metabolism of FFA results in accumulation of ketone bodies or ketoacids (acetone, beta-hydroxybutyrate, acetoacetate).344 These compensatory mechanisms are ultimately responsible for the patho-logical effects seen in DKA (see Table 19.12). The pathophysiology of DKA is illustrated in Figure 19.3.
TABLE 19.12 Pathological effects of diabetic ketoacidosis (DKA)
| Mechanism | Action |
|---|---|
| Cellular dehydration and intravascular volume depletion |
• Ketoacids are fully dissociated at physiological pH (strong acids). Because of the complete dissociation, acetoacetate and beta-hydroxybutyrate are strong ions (anions).345
• The metabolic acidosis is explained by extracellular (and intracellular) buffering of the dissociated H+, resulting in a decrease in bicarbonate.166 Alternatively, the acidosis can be explained by accumulation of strong anions (acetoacetate and beta-hydroxybutyrate) with resulting reduction of the strong ion difference, causing an increased H+ dissociation from plasma water and thus a metabolic acidosis.347,348
• The presence of ketone bodies widens the anion gap, strong ion gap and base excess gap. These ‘gaps’ can be used to assess the degree of ketonaemia. As ketosis resolves, an acidosis caused by high chloride relative to sodium levels is often seen and probably results from administration of normal saline in the initial resuscitation, especially in the setting of decreased renal function where the ability to excrete chloride is reduced.
• The osmotic diuresis results in potassium, phosphate and magnesium loss.
• Total body potassium losses are particularly significant, as potassium shifts from the intracellular to the extracellular space in concert with the osmotically driven water shift. Acidosis and lack of insulin exacerbates the potassium shift. The final pathway for potassium loss is via the urine.345
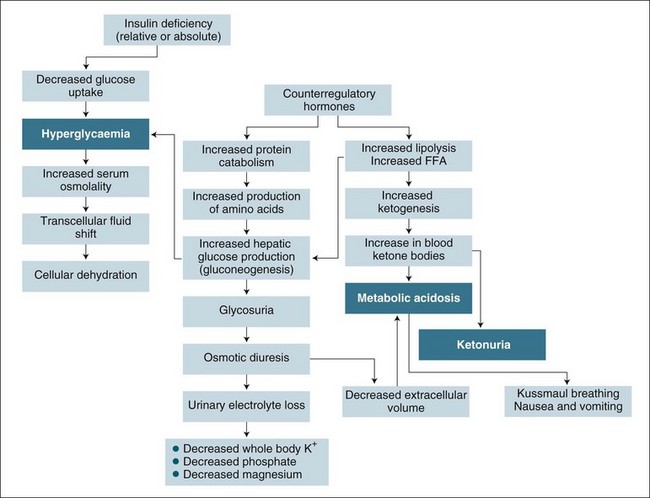
Nursing Practice
Management of HHNS is similar to that for DKA, and includes respiratory support, fluid replacement, insulin treatment to turn off ketogenesis and the accompanying metabolic derangement, electrolyte replacement, correction of acidosis (in DKA), monitoring for and prevention of complications hypoglycaemia, hypokalaemia, hyperglycaemia, and fluid volume overload, and patient teaching and support.341,350,351 Assessment of blood glucose levels is essential. Effectiveness of treatment is usually assessed by resolution of the acidosis and the control of hyperglycaemia. Regular testing of arterial blood gases, blood sugar and electrolytes (especially potassium) is vital until the blood sugar has stabilised and the ketosis and acidosis resolves.344 Considering that fewer patients are now admitted to ICU with DKA and HHNS, understanding the management of these patients is vital and protocols have been developed to guide practice.350,351
Blood ketones (beta-hydroxybutyrate) can now easily be measured using blood from a fingerprick with a bedside handheld monitor. It has been suggested that blood ketone monitoring allows for insulin titration with reference to ketones in addition to usual blood sugar monitoring.352 An outline of the collaborative treatment of DKA and HHNS is presented in Table 19.13.
• A soluble insulin is usually administered via continuous infusion to allow rapid titration of dose.
• Blood glucose levels and blood chemistry should be regularly monitored.
• Care is taken to prevent too rapid a change in blood sugar level, as this will cause a rapid reduction in the extracellular fluid osmolarity. This rapid reduction would result in fluid shift from the extracellular space to the intracellular space, which may result in cerebral oedema.
• There is a risk of hypoglycaemia resulting from insulin therapy. Sympathetic activation accompanies a low blood glucose level and results in sweating, tremor, tachycardia and anxiety. Reduced blood glucose levels also cause global CNS depression and result in depression of the level of consciousness and possibly fitting. Severe hypoglycaemia with a blood glucose level <2 mmol/L is a medical emergency and is treated with administration of 50 mL 50% glucose.
• Intravenous potassium replacement will be required.
• Plasma potassium levels will fall rapidly as a result of commencement of insulin therapy and to a lesser extent with rehydration. Insulin causes the lowering of plasma potassium by mediating the re-entry of potassium into the intracellular compartment.
DKA = diabetic ketoacidosis; HHNS = hyperglycaemic hyperosmolar non-ketotic state.
Summary
Research vignette
Abstract
Objectives
The study aimed to determine the effect of returning or discarding the gastric residual volume (GRV) on gastric emptying delays and feeding, electrolyte balance and patient outcomes among critically ill patients. Gastric emptying delay (GED) was defined as the difficulty in maintaining GRV within safe limits, i.e. below 5 ml/kg. The GED was categorised as light GED (151–250 mL/6 hours), moderate GED (251–350 mL/6 hours) or severe GED (>350 mL/6 hours).353
Methods
The prospective, randomised clinical trial was conducted in a single medical-surgical intensive care unit (ICU) of a public university hospital. Patients admitted to the ICU for longer than 48 hours, aged 18 or older, who had haemodynamic monitoring and were fed enterally or parenterally were recruited to the study over one year. Participants were excluded if connected to an intermittent gastric aspiration system. Computer-generated randomisation was used to randomise participants to the return (intervention) or discard (control) group. The estimated sample size (59 participants in each group) was informed by sample size calculations. The study finished for a participant if: (1) there was no need for further GRV monitoring, (2) occurrence of adverse event associated with the procedure (pulmonary aspiration or cardio-respiratory arrest during or immediately after the procedure), (3) faecal aspirates, (4) major protocol error or (5) death. Gastric residual volumes were checked every 6 hours and an algorithm was used to guide management of GRV. Data were collected by the investigators or by the trained registered nurses from the ICU and included the incidence of (1) blocked NGT; (2) pulmonary aspiration episodes; (3) intolerance episodes (nausea, vomiting, diarrhoea and abdominal distension); (4) enteral feeding delays; (5) hyperkalaemia episodes; (6) hyperglycaemia episodes and (7) discomfort episodes, identified by significant changes in vital signs and also from the Ramsay sedation score.354
Critique
The value for maintaining GRV within safe limits, i.e. below 5 mL/kg was cited by Horn and colleagues355 in their secondary analysis of intermittent versus continuous feeding in a paediatric ICU. Horn and colleagues355 used the value recommended by Taylor and Baker356 (the primary reference) in their paper published on paediatric enteral nutrition. There was no information on how this value was derived and it may not be appropriate for adults. The GED was categorised into three groups but the rationale for using these categories was not provided. Other important outcomes of interest are not well defined. For example testing glucose values in pulmonary secretions is not an acceptable method to define pulmonary aspiration. The report does not explicitly define the ‘discard’ group.
There is a wide variation in the management of GRV and little available evidence to guide practice.357 The volume of GRV considered excessive and the ideal frequency of checking GRV have not been established. Similarly, whether to return or discard gastric aspirate is controversial. The argument to support return of aspirates to maintain electrolyte and fluid balance was not shown in this study. Discarding aspirates minimises handling of feed delivery systems and risk of contamination but exposes staff to splash injury; these events were not measured. While this study provides some information about GED, high quality research is needed to answer some of these difficult questions. There is not enough evidence from this study to guide or change practice.
Learning activities
1. How do changes to the gastrointestinal system in critical illness influence your patient’s ability to achieve their energy–protein goals?
2. With reference to the case study, what factors may contribute to malnutrition and how might you address these in your clinical practice?
3. After reviewing the case study, what interruptions to enteral feeding were necessary and what could have been avoided? What impact might repeated interruptions have on patient outcomes?
4. Review your patients’ notes and calculate what their total daily caloric intake should be. Once you have obtained this figure, compare the prescribed intake to the actual intake. If patients have not received their total daily caloric intake, consider what factors may have contributed to this and how these might be overcome in future.
5. Identify what types of stress ulcer prophylaxis are used in your clinical area. Discuss with your colleagues the advantage of these strategies over other pharmacological agents to prevent the development of stress-related mucosal disease.
6. Consider why acute liver dysfunction/failure causes serious systemic sequelae, such as coagulopathy and hepatic encephalopathy, and why liver function can be restored following the insult.
7. Identify the current practice for glycaemia control in the unit you work in or have access to. If tight glycaemia control is used, identify the practices that have been instituted to minimise the incidence and severity of hypoglycaemia. If tight glycaemia control is not used, identify what protocol is used and what BSL threshold is used. Describe the rationale that supports the practice you identify.
8. Compare and contrast the physiological changes that occur in DKA and HHNS. How do these differences influence the management strategy for restoring normoglycaemia?
American Society for Parenteral and Enteral Nutrition. http://www.nutritioncare.org/.
Australasian Society for Parenteral and Enteral Nutrition. http://www.auspen.org.au/.
The Australia and New Zealand Liver Transplant Registry. http://www.anzltr.org/.
Critical Care Nutrition. http://www.criticalcarenutrition.com/.
European Association for the Study of the Liver. http://www.easl.eu/.
The European Society for Clinical Nutrition and Metabolism. http://www.espen.org/.
Online MELD Calculator. http://optn.transplant.hrsa.gov/resources/professionalResources.asp?index=8.
The Transplantation Society of Australia and New Zealand. http://www.tsanz.com.au/.
1 McCance KL, Huether S. Pathophysiology: the biologic basis for disease in adults and children, 6th edn. St Louis: Mosby Elsevier; 2010.
2 Huether S. Structure and function of the digestive system. McCance KL, Huether S. Pathophysiology: the biologic basis for disease in adults and children, 6th edn, St Louis: Mosby Elsevier, 2010.
3 Ukleja A. Altered GI motility in critically ill patients: Current understanding of pathophysiology, impact, and diagnostic approach. Nutr Clin Pract. 2010;25(1):16–25.
4 Husebye E. The pathogenesis of gastrointestinal bacterial overgrowth. Chemotherapy. 2005;51(Suppl 1):1–22.
5 Lash O’Neill P. Gastrointestinal system: Target organ and source of multiple organ dysfunction syndrome. In: Huddleston Secor V, ed. Multiple organ dysfunction and failure: pathophysiology and clinical implications. 2nd edn. St Louis: Mosby; 1996:215–237.
6 Lord LM, Sax HC. The role of the gut in critical illness. AACN Clin Issues. 1994;5(4):450–458.
7 Flock MH, Binder JJ, Filburn B, Gershengoren W. The effect of bile acids on intestinal microflora. Am J Clin Nutr. 1972;25:1814–1826.
8 Dark DS, Pingleton SK. Nutrition and nutritional support in critically ill patients. J Intensive Care Med. 1993;8(1):16–33.
9 Phillips MC, Olson LR. The immunologic role of the gastrointestinal tract. Crit Care Nurs Clin North Am. 1993;5(1):107–120.
10 Wells CL, Erlandsen SL. Bacterial translocation: Intestinal epithelial permeability. In: Rombeau JL, Takala J. Gut dysfunction in critical illness. 26th edn. Berlin: Springer; 1996:131–149.
11 McVay LD. Immunology of the gut. In: Rombeau JL, Takala J. Gut dysfunction in critical illness. 26th edn. Berlin: Springer; 1996:76–101.
12 Phillips JO, Metzler MH, Palmieri MT, Huckfeldt RE, Dahl NG. A prospective study of simplified omeprazole suspension for the prophylaxis of stress-related mucosal damage. Crit Care Med. 1996;24(11):1793–1800.
13 Fennerty MB. Rationale for the therapeutic benefits of acid-supression therapy in the critically ill patient. Medscape Gastroenterology. 6(3), 2004.
14 Stannard VA, Hutchinson A, Morris DL, Byrne A. Gastric exocrine “failure” in critically ill patients: Incidence and associated features. BMJ. 1988;296(6616):155–156.
15 Higgins D, Mythen MG, Webb AR. Low intramucosal pH is associated with failure to acidify the gastric lumen in response to pentagastrin. Intensive Care Med. 1994;20(2):105–108.
16 Groeneveld ABJ. Gastrointestinal exocrine failure in critical illness. In: Rombeau JL, Takala J. Gut dysfunction in critical illness. 26th edn. Berlin: Springer; 1996:297–306.
17 Marshall JC. Clinical markers of gastrointestinal dysfunction. In: Rombeau JL, Takala J. Gut dysfunction in critical illness. 26th edn. Berlin: Springer; 1996:114–130.
18 Haglund U. Gut ischaemia. Gut. 1994;35(Suppl1):S73–S76.
19 Vallet B, Neviere R, Chagnon J-L. Gastrointestinal mucosal ischaemia. In: Rombeau JL, Takala J. Gut dysfunction in critical illness. 26th edn. Berlin: Springer; 1996:233–245.
20 Antonsson JB, Engstrom L, Rasmussen I, Wollert S, Haglund UH. Changes in gut intramucosal pH and gut oxygen extraction ratio in a porcine model of peritonitis and hemorrhage. Crit Care Med. 1995;23(11):1872–1881.
21 Haglund U. Intestinal mucosal blood flow and oxygenation in sepsis. Brit J Intens Care. 1993 February:49–54.
22 Johnson D, Mayers I. Multiple organ dysfunction syndrome: a narrative review. Can J Anaesth. 2001;48(5):502–509.
23 Strassburg CP. Gastrointestinal disorders of the critically ill. Shock liver. Best Prac Res Clin Gastroenterol. 2003;17(3):369–381.
24 Royal College of Surgeons, England. Nutrition support for adults oral nutrition support: Enteral tube feeding and parenteral nutrition – methods, evidence and guidance. London: England: Royal College of Surgeons; 2006. [Cited October 2010]. Available from http://www.rcseng.ac.uk/new_rcseng/content/publications/docs/nutrition_support_guidelines.html
25 Btaiche IF, Chan LN, Pleva M, Kraft MD. Critical illness, gastrointestinal complications, and medication therapy during enteral feeding in critically ill adult patients. Nutr Clin Pract. 2010;25(1):32–49.
26 Harrington L. Nutrition in critically ill adults: key processes and outcomes. Crit Care Nurs Clin North Am. 2004;16(4):459–465.
27 Dabrowski GP, Steinberg SM, Ferrara JJ, Flint LM. A critical assessment of endpoints of shock resuscitation. Surg Clin North Am. 2000;80(3):825–844.
28 Cartwright MM. The metabolic response to stress: a case of complex nutrition support management. Crit Care Nurs Clin North Am. 2004;16(4):467–487.
29 Biolo G, Grimble G, Preiser JC, Leverve X, Jolliet P, et al. Position paper of the ESICM Working Group on Nutrition and Metabolism. Metabolic basis of nutrition in intensive care unit patients: ten critical questions. Intensive Care Med. 2002;28(11):1512–1520.
30 Rodriguez L. Nutritional status: assessing and understanding its value in the critical care setting. Crit Care Nurs Clin North Am. 2004;15(4):509–514.
31 Giner M, Laviano A, Meguid MM, Gleason JR. In 1995 a correlation between malnutrition and poor outcome in critically ill patients still exists. Nutrition. 1996;12(1):23–29.
32 Alberda C, Gramlich L, Jones N, Jeejeebhoy K, Day AG, et al. The relationship between nutritional intake and clinical outcomes in critically ill patients: results of an international multicenter observational study. Intensive Care Med. 2009;35(10):1728–1737.
33 Petros S, Engelmann L. Enteral nutrition delivery and energy expenditure in medical intensive care patients. Clin Nutr. 2006;25(1):51–59.
34 Villet S, Chiolero RL, Bollmann MD, Revelly JP, Cayeux RNM, et al. Negative impact of hypocaloric feeding and energy balance on clinical outcome in ICU patients. Clin Nutr. 2005;24(4):502–509.
35 Rubinson L, Diette GB, Song X, Brower RG, Krishnan JA. Low caloric intake is associated with nosocomial bloodstream infections in patients in the medical intensive care unit. Crit Care Med. 2004;32(2):350–357.
36 Krishnan JA, Parce PB, Martinez A, Diette GB, Brower RG. Caloric intake in medical ICU patients: consistency of care with guidelines and relationship to clinical outcomes. Chest. 2003;124(1):297–305.
37 Sabol VK. Nutrition assessment of the critically ill adult. AACN Clin Issues. 2004;15(4):595–606.
38 Raguso CA, Dupertuis YM, Pichard C. The role of visceral proteins in the nutritional assessment of intensive care unit patients. Curr Opin Clin Nutr Metab Care. 2003;6(2):211–216.
39 Sungurtekin H, Sungurtekin U, Oner O, Okke D. Nutrition assessment in critically ill patients. Nutr Clin Pract. 2008-9;23(6):635–641.
40 Detsky DS, Baker JP, Mendelson RA, Wolman SL, Wesson DE, Jeejeebhoy KN. Evaluating the accuracy of nutritional assessment techniques applied to hospitalized patients: Methodology and comparison. J Parenter Enteral Nutr. 1984;8(2):153.
41 Jeejeebhoy KN, Detsky AS, Baker JP. Assessment of nutritional status. J Parenter Enteral Nutr. 1990;14(Suppl):S193–S196.
42 Atkinson M, Worthley LIG. Nutrition in the critically ill patient: Part 1. Essential physiology and pathophysiology. Crit Care Resusc. 2003;5(2):109–120.
43 Manning EMC, Shenkin A. Nutritional assessment in the critically ill. Crit Care Clinics. 1995;11(3):603–634.
44 Fitzsimmons L, Hadley SA. Nutritional management of the metabolically stressed patients. Crit Care Nurs Q. 1994;17(3):1–13.
45 American Dietetic Association. Critical illness nutrition practice recommendations: Determining resting metabolic rate. Chicago, IL: American Dietetic Association; 2006. [Cited October 2010]; Available from http://www.adaevidencelibrary.com/topic.cfm?cat=3016
46 Dobb GJ. Enteral nutrition. Oh TE, ed. Intensive Care, 4th edn, Oxford: Butterworth Heinemann, 1997.
47 Miles JM. Energy expenditure in hospitalized patients: implications for nutritional support. Mayo Clin Proc. 2006;81(6):809–816.
48 Elamin EM, Camporesi E. Evidence-based nutritional support in the intensive care unit. Int Anesthesiol Clin. 2009;47(1):121–138.
49 Caba D, Ochoa JB. How many calories are necessary during critical illness? Gastrointest Endosc Clin N Am. 2007;17(4):703–710.
50 Ingenbleek Y, Carpentier YA. A prognostic inflammatory and nutritional index scoring critically ill patients. Int J Vitam Nutr Res. 1985;55(1):91–101.
51 Simpson F, Doig G. Parenteral vs. enteral nutrition in the critically ill patient: a meta-analysis of trials using the intention to treat principle. Intensive Care Med. 2005;31(1):12–23.
52 Dhaliwal R, Jurewitsch B, Harrietha D, Heyland DK. Combination enteral and parenteral nutrition in critically ill patients: harmful or beneficial? A systematic review of the evidence. Intensive Care Med. 2004;30(8):1666–1671.
53 Doig GS, Simpson F, Sweetman EA. Evidence-based nutrition support in the intensive care unit: an update on reported trial quality. Curr Opin Clin Nutr Metab Care. 2009;12(2):201–206.
54 Doig GS, Simpson F, Delaney A. A review of the true methodological quality of nutritional support trials conducted in the critically ill: time for improvement. Anesth Analg. 2005;100(2):527–533.
55 McClave SA, Martindale RG, Vanek VW, McCarthy M, Roberts P, et al. Guidelines for the provision and assessment of nutrition support therapy in the adult critically ill patient: Society of Critical Care Medicine (SCCM) and American Society for Parenteral and Enteral Nutrition (A.S.P.E.N. J Parenter Enteral Nutr. 2009;33(3):277–316.
56 Kreymann KG. Early nutrition support in critical care: a European perspective. Curr Opin Clin Nutr Metab Care. 2008;11(2):156–159.
57 Artinian V, Krayem H, DiGiovine B. Effects of early enteral feeding on the outcome of critically ill mechanically ventilated medical patients. Chest. 2006;129(4):960–967.
58 Heyland DK, Dhaliwal R, Drover JW, Gramlich L, Dodek P. Canadian clinical practice guidelines for nutrition support in mechanically ventilated, critically ill adult patients. J Parenter Enteral Nutr. 2003;27(5):355–373.
59 Kudsk KA. Beneficial effect of enteral feeding. Gastrointest Endosc Clin N Am. 2007;17(4):647–662.
60 Heighes P, Doig GS, Sweetman EA, Simpson F. An overview of evidence from systematic reviews evaluating early enteral nutrition in critically ill patients: more convincing evidence is needed. Anaesth Intensive Care. 2010;38(1):167–174.
61 McClave SA, Heyland DK. The physiologic response and associated clinical benefits from provision of early enteral nutrition. Nutr Clin Pract. 2009;24(3):305–315.
62 Hermsen JL, Sano Y, Kudsk KA. Food fight! Parenteral nutrition, enteral stimulation and gut-derived mucosal immunity. Langenbecks Arch Surg. 2009;394(1):17–30.
63 Sano Y, Gomez FE, Kang W, Lan J, Maeshima Y, et al. Intestinal polymeric immunoglobulin receptor is affected by type and route of nutrition. J Parenter Enteral Nutr. 2007;31(5):351–356.
64 King BK, Kudsk KA. Can an enteral diet decrease sepsis after trauma? Adv Surg. 1997;31:53–78.
65 Bower RH, Cerra FB, Bershadsky B, Licari JJ, Hoyt DB, et al. Early enteral administration of a formula (Impact) supplemented with arginine, nucleotides, and fish oil in intensive care unit patients: Results of a multicenter, prospective, randomized, clinical trial. Crit Care Med. 1995;23(3):436–449.
66 Sax HC. Early nutritional support in critical illness is important. Crit Care Clinics. 1996;12(3):661.
67 Heyland DK, Cook DJ, Guyatt GH. Enteral nutrition in the critically ill patient: a critical review of the evidence. Intens Care Med. 1993;19(8):435–442.
68 Romito RA. Early administration of enteral nutrients in critically ill patients. AACN Clin Issues. 1995;6(2):242–256.
69 Marik PE. Death by total parenteral nutrition: part deux. Crit Care Med. 2006;34(12):3062.
70 Engel JM, Muhling J, Junger A, Menges T, Karcher B, Hempelmann G. Enteral nutrition practice in a surgical intensive care unit: what proporation of energy expenditure is delivered enterally? Clin Nutr. 2003;22(2):187–192.
71 De Jonghe B, Appere-De-Vechi C, Fournier M, Tran B, Merrer J, et al. A prospective survey of nutritional support practices in intensive care unit patients: what is prescribed? What is delivered? Crit Care Med. 2001;29(1):8–12.
72 King BK, Kudsk KA, Li J, Wu Y, Renegar KB. Route and type of nutrition influence mucosal immunity to bacterial pneumonia. Ann Surg. 1999;229(2):272–278.
73 Lin M-T, Saito H, Fukushima R, Inaba T, Fukatsu K, Inoue T, et al. Route of nutritional supply influences local, systemic, and remote organ responses to intraperitoneal bacterial challenge. Ann Surg. 1996;223(1):84–93.
74 Malone AM. Permissive underfeeding: its appropriateness in patients with obesity, patients on parenteral nutrition, and non-obese patients receiving enteral nutrition. Curr Gastroenterol Rep. 2007;9(4):317–322.
75 Ibrahim EH, Mehringer L, Prentice D, Sherman G, Schaiff R, et al. Early versus late enteral feeding of mechanically ventilated patients: results of a clinical trial. J Parenter Enteral Nutr. 2002;26(3):174–181.
76 Dickerson RN. Hypocaloric feeding of obese patients in the intensive care unit. Curr Opin Clin Nutr Metab Care. 2005;8(2):189–196.
77 Zaloga GP, Roberts P. Permissive underfeeding. New Horiz. 1994;2(2):257–263.
78 Berger MM, Chiolero RL. Hypocaloric feeding: pros and cons. Curr Opin Crit Care. 2007;13(2):180–186.
79 Casadei E, Scolletta S, Franchi F, Mongelli P, Gimomarelli P. Effects of hypocaloric feeding on clincal outcome in ICU patient. Crit Care. 2006;10(Suppl 1):217.
80 Dvir D, Cohen J, Singer P. Computerized energy balance and complications in critically ill patients: an observational study. Clin Nutr. 2006;25(1):37–44.
81 Hadfield RJ, Sinclair DG, Houldsworth PE, Evans TW. Effects of enteral and parenteral nutrition on gut mucosal permeability in the critically ill. Am J Respir Crit Care Med. 1995;152(5 Pt 1):1545–1548.
82 Singer P, Pichard C, Heidegger CP, Wernerman J. Considering energy deficit in the intensive care unit. Curr Opin Clin Nutr Metab Care. 2010;13(2):170–176.
83 Doig GS, Simpson F, Finfer S, Delaney A, Davies AR, et al. Effect of evidence-based feeding guidelines on mortality of critically ill adults: A cluster randomized controlled trial. JAMA. 2008;300(23):2731–2741.
84 Briggs D. Nasogastric feeding practice in intensive care units: a study. Nurs Stand. 1996;10(49):42–45.
85 O’Meara D, Mireles-Cabodevila E, Frame F, Hummell AC, Hammel J, et al. Evaluation of delivery of enteral nutrition in critically ill patients receiving mechanical ventilation. Am J Crit Care. 2008;17(1):53–61.
86 Marshall A, West S. Nutritional intake in the critically ill: Improving practice through research. Australian Crit Care. 2004;17(1):6–15.
87 Adam S, Batson S. A study of problems associated with the delivery of enteral feed in critically ill patients in five ICUs in the UK. Intens Care Med. 1997;23:261–266.
88 McClave SA, Lowen CC, Kleber JJ, Nicholson JF, Jimmerson SC, et al. Are patients fed appropriately according to their caloric requirements? J Parenter Enteral Nutr. 1998;22(6):375–381.
89 Main BJ, Morrison DL. Development of a clinical pathway for enteral nutrition. Nutr Clin Pract. 1998;13(1):20–24.
90 Birner V, Ratheiser K, Kranz A, Zauner C, Kramer L, et al. Nutritional protocols improve energy supply and reduce nitrogen loss in critical illness. Crit Care. 1998;2(1):75.
91 Barr J, Hecht M, Flavin KE, Khorana A, Gould MK. Outcomes in critically ill patients before and after the implementation of an evidence-based nutritional management protocol. Chest. 2004;125(4):1446–1457.
92 Heyland DK, Dhaliwal R, Day A, Jain M, Drover J. Validation of the Canadian clinical practice guidelines for nutrition support in mechanically ventilated, critically ill adult patients: results of a prospective observational study. Crit Care Med. 2004;32(11):2260–2266.
93 Bankhead R, Boullata J, Brantley S, Corkins M, Guenter P, et al. Enteral nutrition practice recommendations. J Parenter Enteral Nutr. 2009;33(2):122–167.
94 Kreymann KG, Berger MM, Deutz NE, Hiesmayr M, Jolliet P, et al. ESPEN Guidelines on Enteral Nutrition: Intensive care. Clin Nutr. 2006;25(2):210–223.
95 Doig GS. Evidence-based guidelines for nutritional support of the critically ill: results of a bi-national guidelines development conference. Carlton, Victoria: Australian and New Zealand Intensive Care Society; 2005. [Cited October 2010]. Available from http://www.guideline.gov/content.aspx?id=8012
96 Jones NE, Suurdt J, Ouelette-Kuntz H, Heyland DK. Implementation of the Canadian Clinical Practice Guidelines for Nutrition Support: a multiple case study of barriers and enablers. Nutr Clin Pract. 2007;22(4):449–457.
97 Booker KJ, Niedringhaus L, Eden B, Arnold JS. Comparison of 2 methods of managing gastric residual volumes from feeding tubes. Am J Crit Care. 2000;9(5):318–324.
98 Levy H. Nasogastric and nasoenteric feeding tubes. Gastrointest Endosc Clin N Am. 1998;8(3):529–549.
99 White H, Sosnowski K, Tran K, Reeves A, Jones M. A randomised controlled comparison of early post-pyloric versus early gastric feeding to meet nutritional targets in ventilated intensive care patients. Crit Care. 2009;13(6):R187.
100 Drover JW. Gastric versus postpyloric feeding. Gastrointest Endosc Clin N Am. 2007;17(4):765–775.
101 Berger MM, Soguel L. Feed the ICU patient ‘gastric’ first, and go post-pyloric only in case of failure. Crit Care. 2010;14(1):123.
102 Chendrasekhar A. Jejunal feeding in the absence of reflux increases nasogastric output in critically ill trauma patients. Am Surg. 1996;62(11):887–888.
103 Gentilello LM, Cortes V, Castro M, Byers PM. Enteral nutriton with simultaneous gastric decompression in critically ill patients. Crit Care Med. 1993;21:392–395.
104 de Aguilar-Nascimento JE, Kudsk KA. Clinical costs of feeding tube placement. J Parenter Enteral Nutr. 2007;31(4):269–273.
105 Metheny NA, Meert KL, Clouse RE. Complications related to feeding tube placement. Curr Opin Gastroenterol. 2007;23(2):178–182.
106 Metheny NA, Clouse RE, Chang YH, Stewart BJ, Oliver DA, Kollef MH. Tracheobronchial aspiration of gastric contents in critically ill tube-fed patients: frequency, outcomes, and risk factors. Crit Care Med. 2006;34(4):1007–1015.
107 Burns SM, Carpenter R, Blevins C, Bragg S, Marshall M, et al. Detection of inadvertent airway intubation during gastric tube insertion: Capnography versus a colorimetric carbon dioxide detector. Am J Crit Care. 2006;15(2):188–195.
108 Rassias AJ, Ball PA, Corwin HL. A prospective study of tracheopulmonary complications associated with the placement of narrow-bore enteral feeding tubes. Crit Care. 1997;2:25–28.
109 Bankier AA, Wiesmayr MN, Henk C, Turetschek K, Winkelbauer F, et al. Radiographic detection of intrabronchial malpositions of nasogastric tubes and subsequent complications in intensive care unit patients. Intensive Care Med. 1997;23(4):406–410.
110 Wendell GD, Lenchner GS, Promisloff RA. Pneumothorax complicating small-bore feeding tube placement. Arch Intern Med. 1991;151(3):599–602.
111 Dorsey JS, Cogordan J. Nasotracheal intubation and pulmonary parenchymal perforation. An unusual complication of naso-enteral feeding with small-diameter feeding tubes. Chest. 1985;87(1):131–132.
112 Hendry PJ, Akyurekli Y, McIntyre R, Quarrington A, Keon WJ. Bronchopleural complications of nasogastric feeding tubes. Crit Care Med. 1986;14(10):892–894.
113 Metheny NA, Davis-Jackson J, Stewart BJ. Effectiveness of an aspiration risk-reduction protocol. Nurs Res. 2010;59(1):18–25.
114 Metheny NA, Schnelker R, McGinnis J, Zimmerman G, Duke C, Merritt B, et al. Indicators of tubesite during feedings. J Neurosci Nurs. 2005;37(6):320–325.
115 Gharib AM, Stern EJ, Sherbin VL, Rohrmann CA. Nasogastric and feeding tubes: The importance of proper placement. Postgrad Med. 1996;99(5):165–176.
116 Elpern EH, Killeen K, Talla E, Perez G, Gurka D. Capnometry and air insufflation for assessing initial placement of gastric tubes. Am J Crit Care. 2007;16(6):544–549.
117 Kindopp AS, Drover JW, Heyland DK. Capnography confirms correct feeding tube placement in intensive care unit patients. Can J Anaesth. 2001;48(7):705–710.
118 Araujo-Preza CE, Melhado ME, Gutierrez FJ, Maniatis T, Castellano MA. Use of capnometry to verify feeding tube placement. Crit Care Med. 2002;30(10):2255–2259.
119 Asai T, Stacey M. Confirmation of feeding tube position; how about capnography? Anaesthesia. 1994;49(5):451.
120 Thomas BW, Falcone RE. Confirmation of nasogastric tube placement by colorimetric indicator detection of carbon dioxide: a preliminary report. J Am Coll Nutr. 1998;17(2):195–197.
121 Burns SM, Carpenter R, Truwit JD. Report on the development of a procedure to prevent placement of feeding tubes into the lungs using end-tidal CO2 measurements. Crit Care Med. 2001;29(5):936–939.
122 D’Souza DR, Kilam SA, D’Souza U, Janzen EP, Sipos RA. Pulmonary complications of feeding tubes: a new technique of insertion and monitoring malposition. J Crit Care. 1994;37(5):404–408.
123 Metheny NA, Stewart BJ, Smith L, Yan H, Diebold M, Clouse RE. pH and concentration of pepsin and trypsin in feeding tube aspirates as predictors of tube placement. J Parenter Enteral Nutr. 1997;21(5):279–285.
124 Ciocon JO, Galindo-Ciocon DJ, Tiessen C, Galindo D. Continuous compared with intermittent tube feeding in the elderly. J Parenter Enteral Nutr. 1992;16(6):525–528.
125 Westfall UE, Heitkemper MM. Systemic responses to different enteral feeding schedules in rats. Nurs Res. 1992;41(3):144–150.
126 Trocki O, Mochizuki H, Dominioni L, Heyd T, Alexander JW. Comparison of continuous and intermittent tube feedings in burned animals. J Burn Care Rehabil. 1986;7(2):130–137.
127 Horbal Shuster M. Enteral feeding of the critically ill. AACN Clin Issues. 1994;5(4):459–475.
128 Grodner M, Long Anderson S, de Young S. Foundations and clinical applications of nutrition: a nursing approach. St Louis: Mosby; 1996.
129 Elpern EH. Advantages of enteral feeding. Perspectives in Respiratory Nursing. 1998;May:5–6.
130 Marik PE, Bedigian MK. Refeeding hypophosphatemia in critically ill patients in an intensive care unit. A prospective study. Arch Surg. 1996;131(10):1043–1047.
131 Hearing SD. Refeeding syndrome. BMJ. 2004;328(7445):908–909.
132 Kraft MD, Btaiche IF, Sacks GS. Review of the refeeding syndrome. Nutr Clin Pract. 2005;20(6):625–633.
133 Reintam A, Parm P, Kitus R, Kern H, Starkopf J. Gastrointestinal symptoms in intensive care patients. Acta Anaesthesiol Scand. 2009;53(3):318–324.
134 Pancorbo-Hidalgo PL, Garcia-Fernandex FP, Ramirez-Perez C. Complications associated with enteral nutrition by nasogastric tube in an internal medicine unit. J Clin Nurs. 2001;10(4):482–490.
135 Chapman MJ, Nguyen NQ, Fraser RJ. Gastrointestinal motility and prokinetics in the critically ill. Curr Opin Crit Care. 2007;13(2):187–194.
136 Weekes E, Elia M. Observations on the patterns of 24-hour energy expenditure changes in body composition and gastric emptying in head-injured patients receiving nasogastric tube feeding. J Parenter Enteral Nutr. 1996;20(1):31–37.
137 Spapen HD, Duinslaeger L, Diltoer M, Gillet R, Bossuyt A, Huyghens LP. Gastric emptying in critically ill patients is accelerated by adding cisapride to a standard enteral feeding protocol: results of a prospective, randomized, controlled trial. Crit Care Med. 1995;23(3):481–485.
138 Inglis TJ, Sproat LJ, Sherratt MJ, Hawkey PM, Gibson JS, Shah MV. Gastroduodenal dysfunction as a cause of gastric bacterial overgrowth in patients undergoing mechanical ventilation of the lungs. Br J Anaesth. 1992;68(5):499–502.
139 Kao CH, ChangLai SP, Chieng PU, Yen TC. Gastric emptying in head-injured patients. Am J Gastroenterol. 1998;93(7):1108–1112.
140 Mentec H, Dupont H, Bocchetti M, Cani P, Ponche F, Bleichner G. Upper digestive intolerance during enteral nutrition in critically ill patients: Frequency, risk factors and complications. Crit Care Med. 2001;29(10):1955–1961.
141 McClave SA, Lukan JK, Stefater JA, Lowen CC, Looney SW, et al. Poor validity of residual volumes as a marker for risk of aspiration in critically ill patients. Crit Care Med. 2005;33(2):324–330.
142 Metheny NA, Schallom ME, Edwards SJ. Effect of gastrointestinal motility and feeding tube site on aspiration risk in critically ill patients: A review. Heart Lung. 2004;33(3):131–145.
143 Kesek DR, Akerlind L, Karlsson T. Early enteral nutrition in the cardiothoracic intensive care unit. Clin Nutr. 2002;21(4):303–307.
144 Bowling TE, Silk DBA. Enteral feeding – problems and solutions. Euro J Clin Nutr. 1994;48:379–385.
145 Guenter PA, Sweed MR. A valid and reliable tool to quantify stool output in tube-fed patients. J Parenter Enteral Nutr. 1998;22(3):147–151.
146 Caines C, Gill MV, Cunha BA. Non-clostridium difficile nosocomial diarrhea in the intensive care unit. Heart Lung. 1997;26(1):83–84.
147 O’Keefe GE, Gentilello LM, Maier RV. Incidence of infectious complications associated with the use of histamine 2 -receptor antagonists in critically ill trauma patients. Ann Surg. 1998;227(1):120–125.
148 Bliss DZ, Johnson S, Savik K, Clabots CR, Willard K, Gerding DN. Acquisition of Clostridium difficile and Clostridium difficile-associated diarrhea in hospitalized patients receiving tube feeding. Ann Intern Med. 1998;129(12):1012–1019.
149 Bleichner G, Blehaut H, Mentec H, Moyse D. Saccharomyces boulardii prevents diarrhea in critically ill tube fed patients: a multi-centre, randomised, double-blind placebo controlled trial. Intens Care Med. 1997;23:517–523.
150 Bengmark S. Gut microbial ecology in critical illness: is there a role for prebiotics, probiotics and synbiotics? Curr Opin Crit Care. 2002;8(2):145–151.
151 Frohmander TJ, Chaboyer WP, Robertson IK, Gowardman J. Decrease in frequency of liquid stool in enterally fed critically ill patients given the multispecies probiotic VSL#3: a pilot trial. Am J Crit Care. 2010;19(3):e1–11.
152 Anderton A. Enteral tube feeds as a source of infection: Can we reduce the risk? Nutrition. 1999;15(1):55–57.
153 Roy S, Rigal M, Doit C, Fontan JE, Machinot S, et al. Bacterial contamination of enteral nutrition in a paediatric hospital. J Hosp Infect. 2005;59(4):311–316.
154 Mehall JR, Kite CA, Saltzman DA, Wallett T, Jackson RJ, Smith SD. Prospective study of the incidence and complications of bacterial contamination of enteral feeding in neonates. J Pediatr Surg. 2002;37(8):1177–1182.
155 Anderton A. Reducing bacterial contamination in enteral tube feeds. Br J Nurs. 1995;4(7):368–376.
156 Patchell CJ, Anderton A, Holden C, MacDonald A, George RH, Booth IW. Reducing bacterial contamination of enteral feeds. Arch Dis Child. 1998;78(2):166–168.
157 Mathus-Vliegen EM, Bredius MW, Binnekade JM. Analysis of sites of bacterial contamination in an enteral feeding system. J Parenter Enteral Nutr. 2006;30(6):519–525.
158 Beattie TK, Anderton A. Decanting versus sterile pre-filled nutrient containers – the microbiological risks in enteral feeding. Int J Environ Health Res. 2001;11:81–93.
159 McKinlay J, Anderton A, Wood W, Gould IM. Endogenous bacterial contamination of enteral feeding systems during administration of feeds to hospital patients. J Hum Nutr Diet. 1995;8(1):308.
160 Vanek VW. Closed versus open enteral delivery systems: A quality improvement study. Nutr Clin Pract. 2000;15(5):234–243.
161 Beattie TK, Anderton A. Microbiological evaluation of four enteral feeding systems which have been deliberately subjected to faulty handling procedures. J Hosp Infect. 1999;42(1):11–20.
162 Vonberg RP, Gastmeier P. Hospital-acquired infections related to contaminated substances. J Hosp Infect. 2007;65(1):15–23.
163 Neely AN, Mayes T, Gardner J, Kagan RJ, Gottschlich MM. A microbiologic study of enteral feeding hang time in a burn hospital: can feeding costs be reduced without compromising patient safety? Nutr Clin Pract. 2006;21(6):610–616.
164 Matlow A, Jacobson M, Wray R, Goldman C, Streitenberger L, et al. Enteral tube hub as a reservoir for transmissible enteric bacteria. Am J Infect Control. 2006;34(3):131–133.
165 Okuma T, Nakamura M, Totake H, Fukunaga Y. Microbial contamination of enteral feeding formulas and diarrhea. Nutrition. 2000;16(9):719–722.
166 Payne-James JJ, Rana SK, Bray MJ, McSwiggan DA, Silk DB. Retrograde (ascending) bacterial contamination of enteral diet administration systems. J Parenter Enteral Nutr. 1992;16(4):369–373.
167 Moffitt SK, Gohman SM, Sass KM, Faucher KJ. Clinical and laboratory evaluation of a closed enteral feeding system under cyclic feeding conditions: A microbial and cost evaluation. Nutrition. 1997;13(7–8):622–628.
168 Kohn CL. The relationship between enteral formula contamination and length of enteral delivery set usage. J Parenter Enteral Nutr. 1991;15(5):567–571.
169 Valles J, Artigas A, Rello J, Bonsoms N, Fontanals D, Blanch L. Continuous aspiration of subglottic secretions in preventing ventilator-associated pneumonia. Ann Inter Med. 1995;122(3):179–186.
170 Torres A, Serra-Batlles J, Ros E, Piera C, Puig de la Bellacasa J, et al. Pulmonary aspiration of gastric contents in patients receiving mechanical ventilation: the effect of body position. Ann Intern Med. 1992;116(7):540–543.
171 Booth CM, Heyland DK, Paterson WG. Gastrointesintal promotility drugs in the critical care setting: a systematic review of the evidence. Crit Care Med. 2002;30(7):1429–1435.
172 Nguyen NQ, Chapman MJ, Fraser RJ, Bryant LK, Holloway RH. Erythromycin is more effective than metoclopramide in the treatment of feed intolerance in critical illness. Crit Care Med. 2007;35(2):483–489.
173 Nguyen NQ, Chapman M, Fraser RJ, Bryant LK, Burgstad C, Holloway RH. Prokinetic therapy for feed intolerance in critical illness: one drug or two? Crit Care Med. 2007;35(11):2561–2567.
174 Metheny NA, Dahms TE, Stewart BJ, Stone K, Edwards SJ, et al. Efficacy of dye-stained enteral formula in detecting pulmonary aspiration. Chest. 2002;122(1):276–281.
175 Maloney JP, Ryan TA. Detection of aspiration in enterally fed patients: a requiem for bedside monitors of aspiration. J Parenter Enteral Nutr. 2002;26(6):S34–S42.
176 Maloney JP, Ryan TA, Brasel KJ, Binion DG, Johnson DR, et al. Food dye use in enteral feedings: A review and a call for a moratorium. Nutr Clin Pract. 2002;17(3):169–181.
177 Clay AS, Behnia M, Brown KK. Mitochondiral disease: A pulmonary and critical care medicine perspecitive. Chest. 2001;30:634–648.
178 Grap MJ, Cantley M, Munro CL, Corley MC. Use of backrest elevation in critical care: A pilot study. Am J Crit Care. 1999;8(1):475–480.
179 McClave SA, DeMeo MT, DeLegge MH, DiSario JA, Heyland DK, et al. North American Summit on Aspiration in the Critically Ill Patient: consensus statement. J Parenter Enteral Nutr. 2002;26(6 Suppl):S80–S85.
180 Metheny NA, St.John RE, Clouse RE. Measurement of glucose in tracheobronchial secretions to detect aspiration of enteral feedings. Heart Lung. 1998;27(5):285–292.
181 Metheny NA, Clouse RE. Bedside methods for detecting aspiration in tube-fed patients. Chest. 1997;111(3):724–731.
182 Metheny NA, Dahms TE, Chang YH, Stewart BJ, Frank PA, Clouse RE. Detection of pepsin in tracheal secretions after forced small-volume aspirations of gastric juice. J Parenter Enteral Nutr. 2004;28(2):79–84.
183 Koretz RL, Lipman TO, Klein S. AGA technical review on parenteral nutrition. Gastroenterology. 2001;121(4):970–1001.
184 Braunschweig CL, Levy P, Sheean PM, Wang X-H. Enteral compared with parenteral nutrition: a meta-analysis. Am J Clin Nutr. 2001;74(4):534–542.
185 Sena MJ, Utter GH, Cuschieri J, Maier RV, Tompkins RG, et al. Early supplemental parenteral nutrition is associated with increased infectious complications in critically ill trauma patients. J Am Coll Surg. 2008;207(4):459–467.
186 Singer P, Berger MM, Van den Berghe G, Biolo G, Calder P, et al. ESPEN Guidelines on Parenteral Nutrition: intensive care. Clin Nutr. 2009;28(4):387–400.
187 Martindale RG, McClave SA, Vanek VW, McCarthy M, Roberts P, et al. Guidelines for the provision and assessment of nutrition support therapy in the adult critically ill patient: Society of Critical Care Medicine and American Society for Parenteral and Enteral Nutrition: Executive Summary. Crit Care Med. 2009;37(5):1757–1761.
188 Hagle M. Parenteral nutrition. Weinstein SM, ed. Plumer’s principles & practice of intravenous therapy, 8th edn, Philadelphia: Lippincott Williams & Wilkins, 2006.
189 Ziegler TR. Parenteral nutrition in the critically ill patient. N Engl J Med. 2009;361(11):1088–1097.
190 Pennington C. What is parenteral nutrition? In: Hamilton H, ed. Total parenteral nutrition: a practical guide for nurses. London: Churchill Livingstone, 2000.
191 Mirtallo J, Canada T, Johnson D, Kumpf V, Petersen C, et al. Safe practices for parenteral nutrition. J Parenter Enteral Nutr. 2004;28(6):S39–S70.
192 Koss W, Joshi N. Prescribing total parenteral nutrition. Nutrition. 2003;19:819–820.
193 Hamilton H. Choosing the appropriate catheter for patients requiring parenteral nutrition. In: Hamilton H, ed. Total parenteral nutrition: a practical guide for nurses. London: Churchill Livingstone, 2000.
194 Choung RS, Talley NJ. Epidemiology and clinical presentation of stress-related peptic damage and chronic peptic ulcer. Curr Mol Med. 2008;8(4):253–257.
195 ASHP Therapeutic Guidelines on Stress Ulcer Prophylaxis. ASHP Commission on Therapeutics and approved by the ASHP Board of Directors on November 14, 1998. Am J Health Syst Pharm. 1999;56(4):347–379.
196 Duerksen DR. Stress-related mucosal disease in critically ill patients. Best Prac Res Clin Gastroenterol. 2003;17(3):327–344.
197 Shuman RB, Schuster DP, Zuckerman GR. Prophylactic therapy for stress ulcer bleeding: a reappraisal. Ann Intern Med. 1987;106(4):562–567.
198 Fennerty MB. Pathophysiology of the upper gastrointestinal tract in the critically ill patient: rationale for the therapeutic benefits of acid suppression. Crit Care Med. 2002;30(6 Suppl):S351–S355.
199 Mutlu GM, Mutlu EA, Factor P. GI complications in patients receiving mechanical ventilation. Chest. 2001;119(4):1222–1241.
200 Sesler JM. Stress-related mucosal disease in the intensive care unit: an update on prophylaxis. AACN Adv Crit Care. 2007;18(2):119–126. quiz 27–8
201 Ali T, Harty RF. Stress-induced ulcer bleeding in critically ill patients. Gastroenterol Clin North Am. 2009;38(2):245–265.
202 Laine L, Takeuchi K, Tarnawski A. Gastric mucosal defense and cytoprotection: bench to bedside. Gastroenterology. 2008;135(1):41–60.
203 Spirt MJ, Stanley S. Update on stress ulcer prophylaxis in critically ill patients. Crit Care Nurse. 2006;26(1):18–20.
204 Marshall AP. The gut in critical illness. In: Carlson K, ed. AACN Advanced Critical Care Nursing. Philadelphia: Elsevier, 2009.
205 Hawkey CJ, Rampton DS. Prostaglandins and the gastrointestinal mucosa: are they important in its function, disease, or treatment? Gastroenterology. 1985;89(5):1162–1188.
206 Beejay U, Wolfe MM. Acute gastrointestinal bleeding in the intensive care unit: a gastroenterologist’s perspective. Gastroenterol Clin North Am. 2000;29:309–336.
207 Durham RM, Shapiro MJ. Stress gastritis revisited. Surg Clin North Am. 1991;71:791–810.
208 Goldin GF, Peura DA. Stress-related mucosal damage. What to do or not to do. Gastrointestinal Endoscopy Clin N Am. 1996;6:505–526.
209 Cook D, Guyatt G, Marshall J, Leasa D, Fuller H, et al. A comparison of sucralfate and randitine for the prevention of upper gastrointestinal bleeding in patients requiring mechanical ventilation. N Engl J Med. 1998;338:791–797.
210 Schiessel R, Feil W, Wenzl E. Mechanisms of stress ulceration and implications for treatment. Gastroenterol Clin North Am. 1990;19(1):101–120.
211 Fennerty MB. Pathophysiology of the upper gastrointestinal tract in the critically lll patient: Rationale of the therapeutic benefits of acid suppression. Crit Care Med. 2002;30(6Suppl):S351–S355.
212 Perry MA, Wadhwa S, Parks DA, Pickard W, Granger DN. Role of oxygen radicals in ischemia-induced lesions in the cat stomach. Gastroenterology. 1986;90(2):362–367.
213 Ritchie WP, Jr., Mercer D. Mediators of bile acid-induced alterations in gastric mucosal blood flow. Am J Surg. 1991;161(1):126–130.
214 Robertson MS, Cade JF, Clancy RL. Heliobacter pylori infection in intensive care: increased prevalence and a new nosocomial infection. Crit Care Med. 1999;27:1276–1280.
215 Cook DJ, Fuller HD, Guyatt GH, Marshall JC, Leasa D, et al. Risk factors for gastrointestinal bleeding in critically ill patients. Canadian Critical Care Trials Group. N Engl J Med. 1994;330(6):377–381.
216 Ellison RT, Perez-Perez G, Welsh CH, Blaser MJ, Riester KA, et al. Risk factors for upper gastrointestinal bleeding in intensive care unit patients: role of Helicobacter pylori. Federal Hyperimmune Immunoglobulin Therapy Study Group. Crit Care Med. 1996;24(12):1974–1981.
217 Beejay U, Wolfe MM. Acute gastrointestinal bleeding in the intensive care unit. The gastroenterologist’s perspective. Gastroenterol Clin North Am. 2000;29(2):309–336.
218 Martindale RG. Contemporary strategies for the prevention of stress-related mucosal bleeding. Am J Health Syst Pharm. 2005;62(10 Suppl 2):S11–S17.
219 Daley RJ, Rebuck JA, Welage LS, Rogers FB. Prevention of stress ulceration: current trends in critical care. Crit Care Med. 2004;32(10):2008–2013.
220 Pisegna JR. Pharmacology of acid suppression in the hospital setting: Focus on proton pump inhibition. Crit Care Med. 2002;30(6Suppl):S356–S361.
221 Merki HS, Wilder-Smith CH. Do continuous infusions of omeprazole and ranitidine retain their effect with prolonged dosing? Gastroenterology. 1994;106(1):60–64.
222 Spirt MJ. Acid suppression in critically ill patients: What does the evidence support? Pharmacotherapy. 2003;23(10 Part 2):S87–S93.
223 Metz DC. Preventing the gastrointestinal consequences of stress-related mucosal disease. Curr Med Res Opin. 2005;21(1):11–18.
224 Steinberg KP. Stress-related mucosal disease in the critically ill patient: Risk factors and strategies to prevent stress-related bleeding in the intensive care unit. Crit Care Med. 2002;30(6Suppl):S362–S364.
225 Brett S. Science review: The use of proton pump inhibitors for gastric acid suppression in critical illness. Crit Care. 2005;19(1):45–50.
226 Cash BD. Evidence-based medicine as it applies to acid suppression in the hospitalized patient. Crit Care Med. 2002;30(6Suppl):S373–S378.
227 Levy MJ, Seelig CB, Robinson NJ, Ranney JE. Comparison of omeprazole and ranitidine for stress ulcer prophylaxis. Dig Dis Sci. 1997;42(6):1255–1259.
228 Cook D, Guyatt G, Marshall J, Leasa D, Fuller H, et al. A comparison of sucralfate and ranitidine for the prevention of upper gastrointestinal bleeding in patients requiring mechanical ventilation. Canadian Critical Care Trials Group. N Engl J Med. 1998;338(12):791–797.
229 Pingleton SK. Treating the patient with upper gastrointestinal hemorrhage. Geriatrics. 1983;38(8):61–64. 69, 72
230 Moscona R, Kaufman T, Jacobs R, Hirshowitz B. Prevention of gastrointestinal bleeding in burns: the effects of cimetidine or antacids combined with early enteral feeding. Burns Including Thermal Injury. 1985;12(1):65–67.
231 Bonten MJ, Gaillard CA, van der Hulst R, de Leeuw PW, van der Geest S, et al. Intermittent enteral feeding: the influence on respiratory and digestive tract colonization in mechanically ventilated intensive-care-unit patients. Am J Respir Crit Care Med. 1996;154(2 Pt 1):394–399.
232 Mushlin P, Gelman S. Hepatic physiology and pathophysiology. Miller R, Eriksson LI, Fleisher LA, Wiener-Kronish JP, Young WL. Miller’s Anesthesia, 7th edn, Philadelphia: Churchill Livingstone, 2009.
233 Kaplowitz N. Mechanisms of liver cell injury. J Hepatol. 2000;32(Suppl 1):39–47.
234 Riordan SM, Williams R. Acute liver failure: targeted artificial and hepatocyte-based support of liver regeneration and reversal of multiorgan failure. J Hepatol. 2000;32(Suppl 1):63–76.
235 Australian Government Department of Health and Ageing. Hepatitis overview. Canberra, ACT: Australian Government Department of Health and Ageing; 2005. [cited April 2005]. Available from http://www.health.gov.au/internet/wcms/publishing.nsf/Content/health-pubhlth-strateg-hiv_hepc-hepc-index.htm
236 Sen S, Williams R, Jalan R. The pathophysiological basis of acute-on-chronic liver failure. Liver. 2002;22(Suppl. 2):5–13.
237 Rahman T, Hodgson H. Clinical management of acute hepatic failure. Intens Care Med. 2001;27:467–476.
238 Ryder SD, Beckingham IJ. ABC of diseases of liver, pancreas, and biliary system: Other causes of parenchymal liver disease. BMJ. 2001;322(7281):290–292.
239 Court FG, Wemyss-Holden SA, Dennison AR, Maddern GJ. The mystery of liver regeneration. Br J Surg. 2002;89(9):1089–1095.
240 Michalopoulos GK. Liver regeneration. J Cell Physiol. 2007;213(2):286–300.
241 Lubel JS, Angus PW. Modern management of portal hypertension. Intern Med J. 2005;35(1):45–49.
242 Ryder SD, Beckingham IJ. ABC of diseases of liver, pancreas, and biliary system: Acute hepatitis. BMJ. 2001;322(7279):151–153.
243 Ryder SD, Beckingham IJ. ABC of diseases of liver, pancreas, and biliary systems: Chronic viral hepatitis. BMJ. 2001;322(7280):219–221.
244 Kaldor JM, Plant AJ, Thompson SC, Longbottom H, Rowbottom J. The incidence of hepatitis B infection in Australia: an epidemiological review. Med J Aust. 1996;165:322–326.
245 New Zealand Ministry of Health. The feasibility of screening for and surveillance of hepatitis B in a single geographic area. Wellington, New Zealand: New Zealand Ministry of Health, 1996. [cited June 2005]. Available from http://www.moh.govt.nz/moh.nsf/49b6bf07a4b7346dcc256fb300005a51/e795f5b0cddb25274c256671000c194c/$FILE/hepb.pdf
246 Law MG, Dore GJ, Bath N, Thompson S, Crofts N, et al. Modelling hepatitis C virus incidence, prevalence and long-term sequelae in Australia. Int J Epidemiol. 2001;32:717–724.
247 National Centre in HIV Epidemiology and Clinical Research. HIV/AIDS, viral hepatitis and sexually transmissible infections in Australia: Annual Surveillance Report 2009. Sydney, Australia: NCHECR, The University of NSW; 2009.
248 Hepatitis C Virus Projections Working Group. Estimates and projections of the hepatitis C Virus epidemic in Australia 2006. Sydney, NSW, Australia: National Centre in HIV Epidemiology and Clinical Research, The University of New South Wales; 2006.
249 New Zealand Government Ministry of Health. Action on hepatic C prevention. Wellington, NZ: New Zealand Government; 2002.
250 Foston TP, Carpentar D. Acute liver failure. Crit Care Nurs Clin North Am. 2010;22(3):395–402.
251 O’Grady JG, Schalm SW, Williams R. Acute liver failure: redefining the syndromes. Lancet. 1993;342(8866):273–275.
252 Maynard ND, Bihari DJ, Dalton N, Beale R, Smithies MN, Mason RC. Liver function and splanchnic ischemia in critically ill patients. Chest. 1997;111:180–187.
253 Sakka SG, Reinhart K, Meier-Hellmann A. Prognostic value of the indocyanine green plasma dissappearance rate in critically ill patients. Chest. 2002;122(55):1715–1720.
254 Riordan SM, Williams R. Fulminant hepatic failure. Clin Liver Dis. 2000;4(1):25–45.
255 Butterworth RF. Pathogenesis of hepatic encephalopathy: update on molecular mechanisms. Indian J Gastroenterol. 2003;22(Suppl 2):S11–S16.
256 Vaquero J, Chung C, Cahill ME, Blei AT. Pathogenesis of hepatic encephalopathy in acute liver failure. Semin Liver Dis. 2003;23(3):259–269.
257 Haussinger D, Schliess F. Pathogenetic mechanisms of hepatic encephalopathy. Gut. 2008;57(8):1156–1165.
258 Conn HO, Leevy CM, Vlahcevic ZR, Rodgers JB, Maddrey WC, et al. Comparison of lactulose and neomycin in the treatment of chronic portal-systemic encephalopathy. A double blind controlled trial. Gastroenterology. 1977;72(4 Pt 1):573–583.
259 Ortiz M, Cordoba J, Doval E, Jacas C, Pujadas F, et al. Development of a clinical hepatic encephalopathy staging scale. Aliment Pharmacol Ther. 2007;26(6):859–867.
260 Hassanein TI, Hilsabeck RC, Perry W. Introduction to the Hepatic Encephalopathy Scoring Algorithm (HESA). Dig Dis Sci. 2008;53(2):529–538.
261 Ferenci P, Lockwood A, Mullen K, Tarter R, Weissenborn K, Blei AT. Hepatic encephalopathy – definition, nomenclature, diagnosis, and quantification: final report of the working party at the 11th World Congresses of Gastroenterology, Vienna, 1998. Hepatology. 2002;35(3):716–721.
262 Dagher L, Moore K. The hepatorenal syndrome. Gut. 2001;49:729–737.
263 Gines P, Guevara M, Arroyo V, Rodes J. Hepatorenal syndrome. Lancet. 2003;362:1819–1827.
264 Sharara AI, Rockey DC. Medical prognosis. Gastroesophageal variceal hemorrhage. New Engl J Med. 2001;345(9):669–681.
265 Fallon MB. Mechanisms of pulmonary vascular complications of liver disease: hepatopulmonary syndrome. J Clin Gastroenterol. 2005;39(4 Suppl 2):S138–S142.
266 Cook DJ, Fuller HD, Guyatt GH. Risk factors for gastrointestinal bleeding in critically ill patients. New Engl J Med. 1994;330(6):377–381.
267 Ryder SD. Clinical assessment of liver disease. Medicine. 2007;35(1):1–4.
268 Sizer E, Wendon J. Acute liver failure. Bersten AD, Soni N. Oh’s intensive care manual, 6th edn, Oxford: Butterworth Heinemann, 2009.
269 Sherlock S, Dooley J. Diseases of the liver and biliary system, 9th edn. London: Blackwell; 1995.
270 Botta F, Ciannini E, Romagnoli P, Fasoli A, Malfatti F, et al. MELD scoring system is useful for predicting prognosis in patients with liver cirrohosis and is correlated with residual liver function: a European study. Gut. 2003;52(1):134–139.
271 Schneider PD. Preoperative assessment of liver function. Surg Clin North Am. 2004;84(2):355–373.
272 Kimura S, Yoshioka R, Shibuya M, Sakano T, Tanaka R, Matsuyama S. Indocyanine green elminination rate detects hepatocellular dysfunction early in septic shock and correlates with survival. Crit Care Med. 2001;29(6):1159–1163.
273 Renz JF, Emond JC, Yersiz H, Ascher NL, Busuttil RW. Split-liver transplantation in the United States: outcomes of a national survey. Ann Surg. 2004;239(2):172–181.
274 Menon KV, Kamath PS. Managing the complications of cirrhosis. Mayo Clin Proc. 2000;75(5):501–509.
275 Marrero J, Martinez FJ, Hyzy R. Advances in critical care hepatology. Am J Resp Crit Care Med. 2003;168(12):1421–1426.
276 Christensen T. The treatment of oesophageal varices using a Sengstaken-Blakemore tube: considerations for nursing practice. Nurs Crit Care. 2004;9(2):58–63.
277 Rose JDG. Transjugular intrahepatic portosystemic stent shunt. Care of the Critically Ill. 1995;11(2):50–52.
278 Davenport A. Extracorporeal support for patients with hepatic failure. Hemodial Int. 2003;7(3):256–263.
279 Liou IW, Larson AM. Role of liver transplantation in acute liver failure. Semin Liver Dis. 2008;28(2):201–209.
280 Lynch S, Kerlin P, Wall D, Ong TH, Shepherd RW, et al. The Queensland Liver Transplant Programme: the first two years. Med J Aust. 1987;147(8):380–385.
281 Gane E, McCall J, Streat S, Gunn K, Yeong ML, et al. Liver transplantation in New Zealand: the first four years. N Z Med J. 2002;115(1159):U120.
282 Lynch S, Balderson G. ANZLT Registry Report. Brisbane, QLD: Australian and New Zealand Liver Transplant Registry; 2010.
283 The Transplant Society of Australia and New Zealand (TSANZ). Organ transplanation from deceased donors: Consensus statement on eligibility criteria and allocation protocols. Sydney: TSANZ; 2010.
284 O’Leary JG, Lepe R, Davis GL. Indications for liver transplantation. Gastroenterology. 2008;134(6):1764–1776.
285 Trotter JF, Osgood MJ. MELD scores of liver transplant recipients according to size of waiting list: impact of organ allocation and patient outcomes. JAMA. 2004;291(15):1871–1874. 21
286 Malinchoc M, Kamath PS, Gordon FD, Peine CJ, Rank J, ter Borg PC. A model to predict poor survival in patients undergoing transjugular intrahepatic portosystemic shunts. Hepatology. 2000;31(4):864–871.
287 Freeman RB, Jr., Wiesner RH, Roberts JP, McDiarmid S, Dykstra DM, Merion RM. Improving liver allocation: MELD and PELD. Am J Transplant. 2004;4(Suppl 9):114–131.
288 Kamath PS, Kim WR. The model for end-stage liver disease (MELD). Hepatology. 2007;45(3):797–805.
289 Sizer E, Wendon J. Liver transplantation. Bersten AD, Soni N. Oh’s intensive care manual, 6th edn, Oxford: Butterworth Heinemann, 2009.
290 Maletic-Staschak S. Orthotopic liver transplantation. The surgical procedure. AORN J. 1984;39(1):35–39.
291 Smith SE, Brumm J, Crim BJ. Liver transplantation. Semin Perioper Nurs. 1992;1(1):10–19.
292 Reddy KS, Johnston TD, Putnam LA, Isley M, Ranjan D. Piggyback technique and selective use of veno-venous bypass in adult orthotopic liver transplantation. Clin Transplant. 2000;14(4 Pt 2):370–374.
293 Urbani L, Campatelli A, Romagnoli J, Catalano G, Sartoni G, et al. T-tube removal after liver transplantation: a new technique that reduces biliary complications. Transplantation. 2002;74(3):410–413.
294 Randall HB, Wachs ME, Somberg KA, Lake JR, Emond JC, et al. The use of the T tube after orthotopic liver transplantation. Transplantation. 1996;61(2):258–261.
295 Shimoda M, Saab S, Morrisey M, Ghobrial RM, Farmer DG, et al. A cost-effectiveness analysis of biliary anastomosis with or without T-tube after orthotopic liver transplantation. Am J Transplant. 2001;1(2):157–161.
296 Renz JF, Yersiz H, Reichert PR, Hisatake GM, Farmer DG, et al. Split-liver transplantation: a review. Am J Transplant. 2003;3(11):1323–1335.
297 Crawford M, Shaked A. The liver transplant operation. Graft. 2003;6(2):98–109.
298 Russo MW, Brown RS, Jr. Adult living donor liver transplantation. Am J Transplant. 2004;4(4):458–465.
299 McGilvray ID, Greig PD. Critical care of the liver transplant patient: an update. Curr Opin Crit Care. 2002;8(2):178–182.
300 Saner F, Kavuk I, Lang H, Fruhauf NR, Paul A, et al. Postoperative ICU management in liver transplant patients. Eur J Med Res. 2003;8(11):511–516.
301 Gruppi LA, Killen AR, Rodriguez W. Liver transplantation: key nursing diagnoses. Dimen Crit Care Nurs. 1990;9(5):272–279.
302 Larsen FS, Strauss G, Knudsen GM, Herzog TM, Hansen BA, Secher NH. Cerebral perfusion, cardiac output, and arterial pressure in patients with fulminant hepatic failure. Crit Care Med. 2000;28(4):996–1000.
303 Bronster DJ, Emre S, Boccagni P, Sheiner PA, Schwartz ME, Miller CM. Central nervous system complications in liver transplant recipients – incidence, timing, and long-term follow-up. Clin Transplant. 2000;14(1):1–7.
304 Donaghy A. Issues of malnutrition and bone disease in patients with cirrhosis. J Gastroenterol Hepatol. 2002;17(4):462–466.
305 Lynn M, Abreo K, Zibari G, McDonald J. End-stage renal disease in liver transplants. Clin Transplant. 2001;15(Suppl 6):66–69.
306 Tippner C, Nashan B, Hoshino K, Schmidt-Sandte E, Akimaru K, et al. Clinical and subclinical acute rejection early after liver transplantation: contributing factors and relevance for the long-term course. Transplantation. 2001;72(6):1122–1128.
307 Cohen SM. Current immunosuppression in liver transplantation. Am J Ther. 2002;9(2):119–125.
308 Lucey MR, Neuberger J, Shaked A. Liver transplantation. Washington: Landes Bioscience; 2003.
309 Rosen HR, Shackleton CR, Higa L, Gralnek IM, Farmer DA, et al. Use of OKT3 is associated with early and severe recurrence of hepatitis C after liver transplantation. Am J Gastroenterol. 1997;92(9):1453–1457.
310 Levy MF, Greene L, Ramsay MA, Jennings LW, Ramsay KJ, et al. Readmission to the intensive care unit after liver transplantation. Crit Care Med. 2001;29(1):18–24.
311 Bilbao I, Armadans L, Lazaro JL, Hidalgo E, Castells L, Margarit C. Predictive factors for early mortality following liver transplantation. Clin Transplant. 2003;17(5):401–411.
312 Van Cromphaut SJ. Hyperglycaemia as part of the stress response: the underlying mechanisms. Best Pract Res Clin Anaesthesiol. 2009;23(4):375–386.
313 Robinson LE, van Soeren MH. Insulin resistance and hyperglycemia in critical illness: role of insulin in glycemic control. AACN Clin Issues. 2004;15(1):45–62.
314 Finfer S, Chittock DR, Su SY, Blair D, Foster D, et al. Intensive versus conventional glucose control in critically ill patients. N Engl J Med. 2009;360(13):1283–1297.
315 Arabi YM, Dabbagh OC, Tamim HM, Al-Shimemeri AA, Memish ZA, et al. Intensive versus conventional insulin therapy: a randomized controlled trial in medical and surgical critically ill patients. Crit Care Med. 2008;36(12):3190–3197.
316 De La Rosa Gdel C, Donado JH, Restrepo AH, Quintero AM, Gonzalez LG, et al. Strict glycaemic control in patients hospitalised in a mixed medical and surgical intensive care unit: a randomised clinical trial. Crit Care. 2008;12(5):R120.
317 Devos P, Preiser JC. Impact of tight glucose control by intensive insulin therapy on ICU mortality and the rate of hypoglycaemia: final results of the Glucontrol study. Intens Care Med. 2007;33(Supp2):S189.
318 Weir CJ, Murray GD, Dyker AG, Lees KR. Is hyperglycaemia an independant predictor of poor outcome after acute stroke? Results of a long term follow up study. BMJ. 1997;314:1303–1306.
319 Capes SE, Hunt D, Malmberg K, Gerstein HC. Stress hyperglycaemia and increased risk of death after myocardial infarction in patients with and without diabetes: a systematic review. Lancet. 2000;355(9206):773–778.
320 Van den Berghe G, Wouters P, Bouilon R, Weekers F, Verwaest C, et al. Outcome benefit of intensive insulin therapy in the critically ill: insulin dose versus glycemic control. Crit Care Med. 2003;31(2):359–366.
321 Griesdale DE, de Souza RJ, van Dam RM, Heyland DK, Cook DJ, et al. Intensive insulin therapy and mortality among critically ill patients: a meta-analysis including NICE-SUGAR study data. CMAJ. 2009;180(8):821–827.
322 Padkin A. How to weigh the current evidence for clinical practice. Best Pract Res Clin Anaesthesiol. 2009;23(4):487–496.
323 Mesotten D, Van den Berghe G. Clinical benefits of tight glycaemic control: focus on the intensive care unit. Best Pract Res Clin Anaesthesiol. 2009;23(4):421–429.
324 Annane D, Cariou A, Maxime V, Azoulay E, D’Honneur G, et al. Corticosteroid treatment and intensive insulin therapy for septic shock in adults: a randomized controlled trial. JAMA. 2010;303(4):341–348.
325 Louie K, Cheema R, Dodek P, Wong H, Wilmer A, et al. Intensive nursing work schedules and the risk of hypoglycaemia in critically ill patients who are receiving intravenous insulin. Qual Saf Health Care. 2010. doi:10.1136/qshc.2009.036020
326 Petersen JR, Graves DF, Tacker DH, Okorodudu AO, Mohammad AA, Cardenas VJ, Jr. Comparison of POCT and central laboratory blood glucose results using arterial, capillary, and venous samples from MICU patients on a tight glycemic protocol. Clin Chim Acta. 2008;396(1–2):10–13.
327 Slater-MacLean L, Cembrowski G, Chin D, Shalapay C, Binette T, et al. Accuracy of glycemic measurements in the critically ill. Diabetes Technol Ther. 2008;10(3):169–177.
328 Desachy A, Vuagnat AC, Ghazali AD, Baudin OT, Longuet OH, et al. Accuracy of bedside glucometry in critically ill patients: influence of clinical characteristics and perfusion index. Mayo Clin Proc. 2008;83(4):400–405.
329 Critchell CD, Savarese V, Callahan A, Aboud C, Jabbour S, Marik P. Accuracy of bedside capillary blood glucose measurements in critically ill patients. Intensive Care Med. 2007;33(12):2079–2084.
330 Wahl HG. How accurately do we measure blood glucose levels in intensive care unit (ICU) patients? Best Pract Res Clin Anaesthesiol. 2009;23(4):387–400.
331 Vesper HW, Archibold E, Myers GL. Assessment of trueness of glucose measurement instruments with different specimen matrices. Clin Chim Acta. 2005;358(1–2):68–74.
332 Mahoney J, Ellison J. Assessing the quality of glucose monitor studies: a critical evaluation of published reports. Clin Chem. 2007;53(6):1122–1128.
333 Bossuyt PM, Reitsma JB, Bruns DE, Gatsonis CA, Glasziou PP, et al. The STARD statement for reporting studies of diagnostic accuracy: explanation and elaboration. The Standards for Reporting of Diagnostic Accuracy Group. Croat Med J. 2003;44(5):639–650.
334 National Committee for Clinical Laboratory Standards (NCCLS). Point-of-care blood glucose testing in acute and chronic care facilities; approved guideline, 2nd edn. Wayne, PA: NCCLS; 2002.
335 Van den Berghe G, Schetz M, Vlasselaers D, Hermans G, Wilmer A, et al. Clinical review: Intensive insulin therapy in critically ill patients: NICE-SUGAR or Leuven blood glucose target? J Clin Endocrinol Metab. 2009;94(9):3163–3170.
336 Dellinger RP, Levy MM, Carlet JM, Bion J, Parker MM, et al. Surviving Sepsis Campaign: international guidelines for management of severe sepsis and septic shock: 2008. Crit Care Med. 2008;36(1):296–327.
337 Australian Institute of Health and Welfare (AIHW). Diabetes prevalance in Australia: an assessment of national data sources. Canberra: AIHW; 2009.
338 Wild S, Roglic G, Green A, Sicree R, King H. Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care. 2004;27(5):1047–1053.
339 Colagiur IS, Borch-Johnsen K, Glumer C, Vistisen D. Is there really an epidemic of type 2 diabetes? Diabetologia. 2005;48:1459–1463.
340 Tobias M, Cheung J. Diabetes in New Zealand, models and forecasts 1996–2011. Wellington, New Zealand: New Zealand Ministry of Health; 2002. [cited June 2005]. Available from http://www.moh.govt.nz/moh.nsf/7004be0c19a98f8a4c25692e007bf833/16a3945b4714f9fccc256b74000f3551?OpenDocument
341 Brenner ZR. Management of hyperglycemic emergencies. AACN Clin Issues. 2006;17(1):56–65. quiz 91–3
342 Yared Z, Chiasson JL. Ketoacidosis and the hyperosmolar hyperglycemic state in adult diabetic patients. Diagnosis and treatment. Minerva Med. 2003;94(6):409–418.
343 Newton CA, Raskin P. Diabetic ketoacidosis in type 1 and type 2 diabetes mellitus: clinical and biochemical differences. Arch Intern Med. 2004;164(17):1925–1931.
344 Schmitz K. Providing the best possible care: an overview of the current understanding of diabetic ketoacidosis. Aust Crit Care. 2000;13(1):22–27.
345 Bardsley JK, Want LL. Overview of diabetes. Crit Care Nurs Q. 2004;27(2):106–112.
346 Dunstan DW, Zimmet PZ, Welborn TA, De Courten MP, Sicree RA, et al. The rising prevalence of diabetes and impaired glucose tolerance: The Australian Diabetes, Obesity and Lifestyle Study. Diabetes Care. 2002;25(2):829–834.
347 Boyle M, Lawrence J. An easy method of mentally estimating the metabolic component of acid/base balance using the Fencl-Stewart approach. Anaesth Intensive Care. 2003;31(5):538–547.
348 Hardern RD, Quinn ND. Emergency management of diabetic ketoacidosis in adults. Emerg Med J. 2003;20(3):210–213.
349 Kitabachi AE, Wall BM. Diabetic ketoacidosis. Med Clin North Am. 1995;79(1):9–37.
350 De Beer K, Michael S, Thacker M, Wynne E, Pattni C, et al. Diabetic ketoacidosis and hyperglycaemic hyperosmolar syndrome – clinical guidelines. Nurs Crit Care. 2008;13(1):5–11.
351 Bull SV, Douglas IS, Foster M, Albert RK. Mandatory protocol for treating adult patients with diabetic ketoacidosis decreases intensive care unit and hospital lengths of stay: results of a nonrandomized trial. Crit Care Med. 2007;35(1):41–46.
352 Wallace TM, Matthews DR. Recent advances in the monitoring and management of diabetic ketoacidosis. Q J M. 2004;97(12):773–780.
353 Juve-Udina ME, Valls-Miro C, Carreno-Granero A, Martinez-Estalella G, Monterde-Prat D, et al. To return or to discard? Randomised trial on gastric residual volume management. Intens Crit Care Nurs. 2009;25(5):258–267.
354 Ramsay MA, Savege TM, Simpson BR, Goodwin R. Controlled sedation with alphaxalone-alphadolone. BMJ. 1974;2(5920):656–659.
355 Horn D, Chaboyer W, Schluter PJ. Gastric residual volumes in critically ill paediatric patients: a comparison of feeding regimens. Aust Crit Care. 2004;17(3):98–100.
356 Taylor R, Baker A. Enteral nutrition in critical illness: Part One. Paediatr Nurs. 1999;11(7):16–20.
357 Williams T, Leslie G. A review of the nursing care of enteral feeding tubes in critically ill adults: part II. Intens Crit Care Nurs. 2004;21(1):5–15.