Other classes exhibit antiemetic properties. 5-HT3 antagonists such as ondansetron and granisetron can prevent postoperative vomiting, radiation therapy–induced symptoms, and cancer chemotherapy–induced emesis, but also are used for other causes of emesis with limited evidence for efficacy. Tricyclic antidepressant agents provide symptomatic benefit in patients with chronic idiopathic nausea and functional vomiting as well as in long-standing diabetic patients with nausea and vomiting. Other antidepressants such as mirtazapine and olanzapine also may exhibit antiemetic effects.
GASTROINTESTINAL MOTOR STIMULANTS
Drugs that stimulate gastric emptying are used for gastroparesis (Table 54-2). Metoclopramide, a combined 5-HT4 agonist and D2 antagonist, is effective in gastroparesis, but antidopaminergic side effects, such as dystonias and mood and sleep disturbances, limit use in ~25% of cases. Erythromycin increases gastroduodenal motility by action on receptors for motilin, an endogenous stimulant of fasting motor activity. Intravenous erythromycin is useful for inpatients with refractory gastroparesis, but oral forms have some utility. Domperidone, a D2 antagonist not available in the United States, exhibits prokinetic and antiemetic effects but does not cross into most brain regions; thus, anxiety and dystonic reactions are rare. The main side effects of domperidone relate to induction of hyperprolactinemia via effects on pituitary regions served by a porous blood-brain barrier.
Refractory motility disorders pose significant challenges. Intestinal pseudoobstruction may respond to the somatostatin analogue octreotide, which induces propagative small-intestinal motor complexes. Acetylcholinesterase inhibitors such as pyridostigmine are also observed to benefit some patients with small-bowel dysmotility. Pyloric injections of botulinum toxin are reported in uncontrolled studies to reduce gastroparesis symptoms, but small controlled trials observe benefits no greater than sham treatments. Surgical pyloroplasty has improved symptoms in case series. Placing a feeding jejunostomy reduces hospitalizations and improves overall health in some patients with drug-refractory gastroparesis. Postvagotomy gastroparesis may improve with near-total gastric resection; similar operations are now being tried for other gastroparesis etiologies. Implanted gastric electrical stimulators may reduce symptoms, enhance nutrition, improve quality of life, and decrease health care expenditures in medication-refractory gastroparesis, but small controlled trials do not report convincing benefits.
SAFETY CONSIDERATIONS
Safety concerns about selected antiemetics have been emphasized. Centrally acting antidopaminergics, especially metoclopramide, can cause irreversible movement disorders such as tardive dyskinesia, particularly in older patients. This complication should be carefully explained and documented in the medical record. Some agents with antiemetic properties including domperidone, erythromycin, tricyclics, and 5-HT3 antagonists can induce dangerous cardiac rhythm disturbances, especially in those with QTc interval prolongation on electrocardiography (ECG). Surveillance ECG testing has been advocated for some of these agents.
SELECTED CLINICAL SETTINGS
Some cancer chemotherapies are intensely emetogenic (Chap. 103e). Combining a 5-HT3 antagonist, an NK1 antagonist, and a glucocorticoid provides significant control of both acute and delayed vomiting after highly emetogenic chemotherapy. Unlike other drugs in the same class, the 5-HT3 antagonist palonosetron exhibits efficacy at preventing delayed chemotherapy-induced vomiting. Benzodiazepines such as lorazepam can reduce anticipatory nausea and vomiting. Miscellaneous therapies with benefit in chemotherapy-induced emesis include cannabinoids, olanzapine, and alternative therapies like ginger. Most antiemetic regimens produce greater reductions in vomiting than in nausea.
Clinicians should exercise caution in managing pregnant patients with nausea. Studies of the teratogenic effects of antiemetic agents provide conflicting results. Few controlled trials have been performed in nausea of pregnancy. Antihistamines such as meclizine and doxylamine, antidopaminergics such as prochlorperazine, and antiserotonergics such as ondansetron demonstrate limited efficacy. Some obstetricians offer alternative therapies such as pyridoxine, acupressure, or ginger.
Managing cyclic vomiting syndrome is a challenge. Prophylaxis with tricyclic antidepressants, cyproheptadine, or β-adrenoceptor antagonists can reduce the severity and frequency of attacks. Intravenous 5-HT3 antagonists combined with the sedating effects of a benzodiazepine like lorazepam are a mainstay of treatment of acute flares. Small studies report benefits with antimigraine agents, including the 5-HT1 agonist sumatriptan, as well as selected anticonvulsants such as zonisamide and levetiracetam.
INDIGESTION
MECHANISMS
The most common causes of indigestion are gastroesophageal reflux and functional dyspepsia. Other cases are a consequence of organic illness.
Gastroesophageal Reflux Gastroesophageal reflux results from many physiologic defects. Reduced lower esophageal sphincter (LES) tone contributes to reflux in scleroderma and pregnancy and may be a factor in some patients without systemic illness. Others exhibit frequent transient LES relaxations (TLESRs) that cause bathing of the esophagus by acid or nonacidic fluid. Overeating and aerophagia override the barrier function of the LES, whereas reductions in esophageal body motility or salivary secretion prolong fluid exposure. Increased intragastric pressure promotes gastroesophageal reflux in obesity. The role of hiatal hernias is controversial—most reflux patients have hiatal hernias, but most with hiatal hernias do not have excess heartburn.
Gastric Motor Dysfunction Disturbed gastric motility may contribute to gastroesophageal reflux in up to one-third of cases. Delayed gastric emptying is also found in ~30% of functional dyspeptics. Conversely, some dyspeptics exhibit rapid gastric emptying. The relation of these defects to symptom induction is uncertain; studies show poor correlation between symptom severity and degrees of motor dysfunction. Impaired gastric fundus relaxation after eating (i.e., accommodation) may underlie selected dyspeptic symptoms like bloating, nausea, and early satiety in ~40% of patients.
Visceral Afferent Hypersensitivity Disturbed gastric sensation is another pathogenic factor in functional dyspepsia. Visceral hypersensitivity was first reported in IBS with demonstration of heightened perception of rectal balloon inflation without changes in compliance. Similarly, ~35% of dyspeptic patients note discomfort with fundic distention to lower pressures than healthy controls. Others with dyspepsia exhibit hypersensitivity to chemical stimulation with capsaicin or with acid or lipid exposure in the duodenum. Some individuals with functional heartburn without increased acid or nonacid reflux are believed to have heightened perception of normal esophageal pH and volume.
Other Factors Helicobacter pylori has a clear etiologic role in peptic ulcer disease, but ulcers cause a minority of dyspepsia cases. H. pylori is a minor factor in the genesis of functional dyspepsia. Functional dyspepsia is associated with chronic fatigue, produces reduced physical and mental well-being, and is exacerbated by stress. Anxiety, depression, and somatization may have contributing roles in some cases. Functional MRI studies show increased activation of several brain regions, emphasizing contributions from central nervous system factors. Analgesics cause dyspepsia, whereas nitrates, calcium channel blockers, theophylline, and progesterone promote gastroesophageal reflux. Other stimuli that induce reflux include ethanol, tobacco, and caffeine via LES relaxation. Genetic factors may promote development of reflux and dyspepsia.
DIFFERENTIAL DIAGNOSIS
Gastroesophageal Reflux Disease Gastroesophageal reflux disease (GERD) is prevalent. Heartburn is reported once monthly by 40% of Americans and daily by 7–10%. Most cases of heartburn occur because of excess acid reflux, but reflux of nonacidic fluid produces similar symptoms. Alkaline reflux esophagitis produces GERD-like symptoms most often in patients who have had surgery for peptic ulcer disease. Ten percent of patients with heartburn exhibit normal esophageal acid exposure and no increase in nonacidic reflux (functional heartburn).
Functional Dyspepsia Nearly 25% of the populace has dyspepsia at least six times yearly, but only 10–20% present to clinicians. Functional dyspepsia, the cause of symptoms in >60% of dyspeptic patients, is defined as ≥3 months of bothersome postprandial fullness, early satiety, or epigastric pain or burning with symptom onset at least 6 months before diagnosis in the absence of organic cause. Functional dyspepsia is subdivided into postprandial distress syndrome, characterized by meal-induced fullness, early satiety, and discomfort, and epigastric pain syndrome, which presents with epigastric burning pain unrelated to meals. Most cases follow a benign course, but some with H. pylori infection or on nonsteroidal anti-inflammatory drugs (NSAIDs) develop ulcers. As with idiopathic gastroparesis, some cases of functional dyspepsia result from prior infection.
Ulcer Disease In most GERD patients, there is no destruction of the esophagus. However, 5% develop esophageal ulcers, and some form strictures. Symptoms cannot distinguish nonerosive from erosive or ulcerative esophagitis. A minority of cases of dyspepsia stem from gastric or duodenal ulcers. The most common causes of ulcer disease are H. pylori infection and use of NSAIDs. Other rare causes of gastroduodenal ulcers include Crohn’s disease (Chap. 351) and Zollinger-Ellison syndrome (Chap. 348), resulting from gastrin overproduction by an endocrine tumor.
Malignancy Dyspeptic patients often seek care because of fear of cancer, but few cases result from malignancy. Esophageal squamous cell carcinoma occurs most often with long-standing tobacco or ethanol intake. Other risks include prior caustic ingestion, achalasia, and the hereditary disorder tylosis. Esophageal adenocarcinoma usually complicates prolonged acid reflux. Eight to 20% of GERD patients exhibit esophageal intestinal metaplasia, termed Barrett’s metaplasia, a condition that predisposes to esophageal adenocarcinoma (Chap. 109). Gastric malignancies include adenocarcinoma, which is prevalent in certain Asian societies, and lymphoma.
Other Causes Opportunistic fungal or viral esophageal infections may produce heartburn but more often cause odynophagia. Other causes of esophageal inflammation include eosinophilic esophagitis and pill esophagitis. Biliary colic is in the differential diagnosis of unexplained upper abdominal pain, but most patients with biliary colic report discrete acute episodes of right upper quadrant or epigastric pain rather than the chronic burning, discomfort, and fullness of dyspepsia. Twenty percent of patients with gastroparesis report a predominance of pain or discomfort rather than nausea and vomiting. Intestinal lactase deficiency as a cause of gas, bloating, and discomfort occurs in 15–25% of whites of northern European descent but is more common in blacks and Asians. Intolerance of other carbohydrates (e.g., fructose, sorbitol) produces similar symptoms. Small-intestinal bacterial overgrowth may cause dyspepsia, often associated with bowel dysfunction, distention, and malabsorption. Eosinophilic infiltration of the duodenal mucosa is described in some dyspeptics, particularly with postprandial distress syndrome. Celiac disease, pancreatic disease (chronic pancreatitis, malignancy), hepatocellular carcinoma, Ménétrier’s disease, infiltrative diseases (sarcoidosis, eosinophilic gastroenteritis), mesenteric ischemia, thyroid and parathyroid disease, and abdominal wall strain cause dyspepsia. Gluten sensitivity in the absence of celiac disease is reported to evoke unexplained upper abdominal symptoms. Extraperitoneal etiologies of indigestion include congestive heart failure and tuberculosis.
55 |
Diarrhea and Constipation |
Diarrhea and constipation are exceedingly common and, together, exact an enormous toll in terms of mortality, morbidity, social inconvenience, loss of work productivity, and consumption of medical resources. Worldwide, >1 billion individuals suffer one or more episodes of acute diarrhea each year. Among the 100 million persons affected annually by acute diarrhea in the United States, nearly half must restrict activities, 10% consult physicians, ~250,000 require hospitalization, and ~5000 die (primarily the elderly). The annual economic burden to society may exceed $20 billion. Acute infectious diarrhea remains one of the most common causes of mortality in developing countries, particularly among impoverished infants, accounting for 1.8 million deaths per year. Recurrent, acute diarrhea in children in tropical countries results in environmental enteropathy with long-term impacts on physical and intellectual development.
Constipation, by contrast, is rarely associated with mortality and is exceedingly common in developed countries, leading to frequent self-medication and, in a third of those, to medical consultation. Population statistics on chronic diarrhea and constipation are more uncertain, perhaps due to variable definitions and reporting, but the frequency of these conditions is also high. United States population surveys put prevalence rates for chronic diarrhea at 2–7% and for chronic constipation at 12–19%, with women being affected twice as often as men. Diarrhea and constipation are among the most common patient complaints presenting to internists and primary care physicians, and they account for nearly 50% of referrals to gastroenterologists.
Although diarrhea and constipation may present as mere nuisance symptoms at one extreme, they can be severe or life-threatening at the other. Even mild symptoms may signal a serious underlying gastrointestinal lesion, such as colorectal cancer, or systemic disorder, such as thyroid disease. Given the heterogeneous causes and potential severity of these common complaints, it is imperative for clinicians to appreciate the pathophysiology, etiologic classification, diagnostic strategies, and principles of management of diarrhea and constipation, so that rational and cost-effective care can be delivered.
NORMAL PHYSIOLOGY
While the primary function of the small intestine is the digestion and assimilation of nutrients from food, the small intestine and colon together perform important functions that regulate the secretion and absorption of water and electrolytes, the storage and subsequent transport of intraluminal contents aborally, and the salvage of some nutrients that are not absorbed in the small intestine after bacterial metabolism of carbohydrate allows salvage of short-chain fatty acids. The main motor functions are summarized in Table 55-1. Alterations in fluid and electrolyte handling contribute significantly to diarrhea. Alterations in motor and sensory functions of the colon result in highly prevalent syndromes such as irritable bowel syndrome (IBS), chronic diarrhea, and chronic constipation.
|
NORMAL GASTROINTESTINAL MOTILITY: FUNCTIONS AT DIFFERENT ANATOMIC LEVELS |
Abbreviation: MMC, migrating motor complex.
NEURAL CONTROL
The small intestine and colon have intrinsic and extrinsic innervation. The intrinsic innervation, also called the enteric nervous system, comprises myenteric, submucosal, and mucosal neuronal layers. The function of these layers is modulated by interneurons through the actions of neurotransmitter amines or peptides, including acetylcholine, vasoactive intestinal peptide (VIP), opioids, norepinephrine, serotonin, adenosine triphosphate (ATP), and nitric oxide (NO). The myenteric plexus regulates smooth-muscle function through intermediary pacemaker-like cells called the interstitial cells of Cajal, and the submucosal plexus affects secretion, absorption, and mucosal blood flow. The enteric nervous system receives input from the extrinsic nerves, but it is capable of independent control of these functions.
The extrinsic innervations of the small intestine and colon are part of the autonomic nervous system and also modulate motor and secretory functions. The parasympathetic nerves convey visceral sensory pathways from and excitatory pathways to the small intestine and colon. Parasympathetic fibers via the vagus nerve reach the small intestine and proximal colon along the branches of the superior mesenteric artery. The distal colon is supplied by sacral parasympathetic nerves (S2–4) via the pelvic plexus; these fibers course through the wall of the colon as ascending intracolonic fibers as far as, and in some instances including, the proximal colon. The chief excitatory neurotransmitters controlling motor function are acetylcholine and the tachykinins, such as substance P. The sympathetic nerve supply modulates motor functions and reaches the small intestine and colon alongside their arterial vessels. Sympathetic input to the gut is generally excitatory to sphincters and inhibitory to non-sphincteric muscle. Visceral afferents convey sensation from the gut to the central nervous system (CNS); initially, they course along sympathetic fibers, but as they approach the spinal cord they separate, have cell bodies in the dorsal root ganglion, and enter the dorsal horn of the spinal cord. Afferent signals are conveyed to the brain along the lateral spinothalamic tract and the nociceptive dorsal column pathway and are then projected beyond the thalamus and brainstem to the insula and cerebral cortex to be perceived. Other afferent fibers synapse in the prevertebral ganglia and reflexly modulate intestinal motility, blood flow, and secretion.
INTESTINAL FLUID ABSORPTION AND SECRETION
On an average day, 9 L of fluid enter the gastrointestinal (GI) tract, ~1 L of residual fluid reaches the colon, and the stool excretion of fluid constitutes about 0.2 L/d. The colon has a large capacitance and functional reserve and may recover up to four times its usual volume of 0.8 L/d, provided the rate of flow permits reabsorption to occur. Thus, the colon can partially compensate for excess fluid delivery to the colon that may result from intestinal absorptive or secretory disorders.
In the small intestine and colon, sodium absorption is predominantly electrogenic (i.e., it can be measured as an ionic current across the membrane because there is not an equivalent loss of a cation from the cell), and uptake takes place at the apical membrane; it is compensated for by the export functions of the basolateral sodium pump. There are several active transport proteins at the apical membrane, especially in the small intestine, whereby sodium ion entry is coupled to monosaccharides (e.g., glucose through the transporter SGLT1, or fructose through GLUT-5). Glucose then exits the basal membrane through a specific transport protein, GLUT-5, creating a glucose concentration gradient between the lumen and the intercellular space, drawing water and electrolytes passively from the lumen. A variety of neural and nonneural mediators regulate colonic fluid and electrolyte balance, including cholinergic, adrenergic, and serotonergic mediators. Angiotensin and aldosterone also influence colonic absorption, reflecting the common embryologic development of the distal colonic epithelium and the renal tubules.
SMALL-INTESTINAL MOTILITY
During the fasting period, the motility of the small intestine is characterized by a cyclical event called the migrating motor complex (MMC), which serves to clear nondigestible residue from the small intestine (the intestinal “housekeeper”). This organized, propagated series of contractions lasts, on average, 4 min, occurs every 60–90 min, and usually involves the entire small intestine. After food ingestion, the small intestine produces irregular, mixing contractions of relatively low amplitude, except in the distal ileum where more powerful contractions occur intermittently and empty the ileum by bolus transfers.
ILEOCOLONIC STORAGE AND SALVAGE
The distal ileum acts as a reservoir, emptying intermittently by bolus movements. This action allows time for salvage of fluids, electrolytes, and nutrients. Segmentation by haustra compartmentalizes the colon and facilitates mixing, retention of residue, and formation of solid stools. There is increased appreciation of the intimate interaction between the colonic function and the luminal ecology. The resident microorganisms, predominantly anaerobic bacteria, in the colon are necessary for the digestion of unabsorbed carbohydrates that reach the colon even in health, thereby providing a vital source of nutrients to the mucosa. Normal colonic flora also keeps pathogens at bay by a variety of mechanisms. In health, the ascending and transverse regions of colon function as reservoirs (average transit time, 15 h), and the descending colon acts as a conduit (average transit time, 3 h). The colon is efficient at conserving sodium and water, a function that is particularly important in sodium-depleted patients in whom the small intestine alone is unable to maintain sodium balance. Diarrhea or constipation may result from alteration in the reservoir function of the proximal colon or the propulsive function of the left colon. Constipation may also result from disturbances of the rectal or sigmoid reservoir, typically as a result of dysfunction of the pelvic floor, the anal sphincters, the coordination of defecation, or dehydration.
COLONIC MOTILITY AND TONE
The small intestinal MMC only rarely continues into the colon. However, short duration or phasic contractions mix colonic contents, and high-amplitude (>75 mmHg) propagated contractions (HAPCs) are sometimes associated with mass movements through the colon and normally occur approximately five times per day, usually on awakening in the morning and postprandially. Increased frequency of HAPCs may result in diarrhea or urgency. The predominant phasic contractions in the colon are irregular and non-propagated and serve a “mixing” function.
Colonic tone refers to the background contractility upon which phasic contractile activity (typically contractions lasting <15 s) is superimposed. It is an important cofactor in the colon’s capacitance (volume accommodation) and sensation.
COLONIC MOTILITY AFTER MEAL INGESTION
After meal ingestion, colonic phasic and tonic contractility increase for a period of ~2 h. The initial phase (~10 min) is mediated by the vagus nerve in response to mechanical distention of the stomach. The subsequent response of the colon requires caloric stimulation (e.g., intake of at least 500 kcal) and is mediated, at least in part, by hormones (e.g., gastrin and serotonin).
DEFECATION
Tonic contraction of the puborectalis muscle, which forms a sling around the rectoanal junction, is important to maintain continence; during defecation, sacral parasympathetic nerves relax this muscle, facilitating the straightening of the rectoanal angle (Fig. 55-1). Distention of the rectum results in transient relaxation of the internal anal sphincter via intrinsic and reflex sympathetic innervation. As sigmoid and rectal contractions, as well as straining (Valsalva maneuver), which increases intraabdominal pressure, increase the pressure within the rectum, the rectosigmoid angle opens by >15°. Voluntary relaxation of the external anal sphincter (striated muscle innervated by the pudendal nerve) in response to the sensation produced by distention permits the evacuation of feces. Defecation can also be delayed voluntarily by contraction of the external anal sphincter.
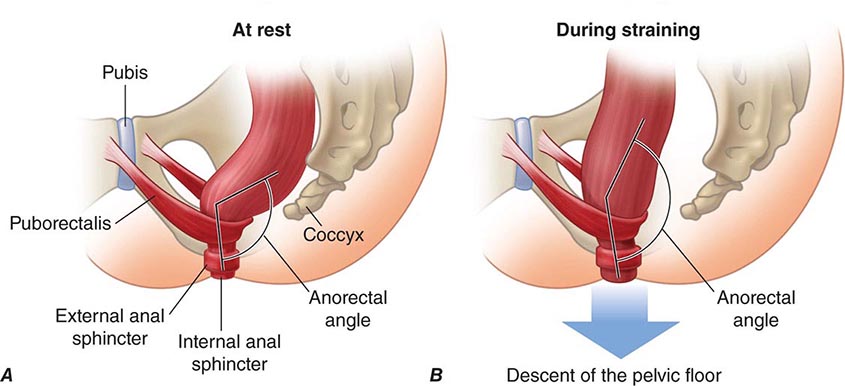
FIGURE 55-1 Sagittal view of the anorectum (A) at rest and (B) during straining to defecate. Continence is maintained by normal rectal sensation and tonic contraction of the internal anal sphincter and the puborectalis muscle, which wraps around the anorectum, maintaining an anorectal angle between 80° and 110°. During defecation, the pelvic floor muscles (including the puborectalis) relax, allowing the anorectal angle to straighten by at least 15°, and the perineum descends by 1–3.5 cm. The external anal sphincter also relaxes and reduces pressure on the anal canal. (Reproduced with permission from A Lembo, M Camilleri: N Engl J Med 349:1360, 2003.)
DIARRHEA
DEFINITION
Diarrhea is loosely defined as passage of abnormally liquid or unformed stools at an increased frequency. For adults on a typical Western diet, stool weight >200 g/d can generally be considered diarrheal. Diarrhea may be further defined as acute if <2 weeks, persistent if 2–4 weeks, and chronic if >4 weeks in duration.
Two common conditions, usually associated with the passage of stool totaling <200 g/d, must be distinguished from diarrhea, because diagnostic and therapeutic algorithms differ. Pseudodiarrhea, or the frequent passage of small volumes of stool, is often associated with rectal urgency, tenesmus, or a feeling of incomplete evacuation, and accompanies IBS or proctitis. Fecal incontinence is the involuntary discharge of rectal contents and is most often caused by neuromuscular disorders or structural anorectal problems. Diarrhea and urgency, especially if severe, may aggravate or cause incontinence. Pseudodiarrhea and fecal incontinence occur at prevalence rates comparable to or higher than that of chronic diarrhea and should always be considered in patients complaining of “diarrhea.” Overflow diarrhea may occur in nursing home patients due to fecal impaction that is readily detectable by rectal examination. A careful history and physical examination generally allow these conditions to be discriminated from true diarrhea.
ACUTE DIARRHEA
More than 90% of cases of acute diarrhea are caused by infectious agents; these cases are often accompanied by vomiting, fever, and abdominal pain. The remaining 10% or so are caused by medications, toxic ingestions, ischemia, food indiscretions, and other conditions.
Infectious Agents Most infectious diarrheas are acquired by fecal-oral transmission or, more commonly, via ingestion of food or water contaminated with pathogens from human or animal feces. In the immunocompetent person, the resident fecal microflora, containing >500 taxonomically distinct species, are rarely the source of diarrhea and may actually play a role in suppressing the growth of ingested pathogens. Disturbances of flora by antibiotics can lead to diarrhea by reducing the digestive function or by allowing the overgrowth of pathogens, such as Clostridium difficile (Chap. 161). Acute infection or injury occurs when the ingested agent overwhelms or bypasses the host’s mucosal immune and nonimmune (gastric acid, digestive enzymes, mucus secretion, peristalsis, and suppressive resident flora) defenses. Established clinical associations with specific enteropathogens may offer diagnostic clues.
In the United States, five high-risk groups are recognized:
1. Travelers. Nearly 40% of tourists to endemic regions of Latin America, Africa, and Asia develop so-called traveler’s diarrhea, most commonly due to enterotoxigenic or enteroaggregative Escherichia coli as well as to Campylobacter, Shigella, Aeromonas, norovirus, Coronavirus, and Salmonella. Visitors to Russia (especially St. Petersburg) may have increased risk of Giardia-associated diarrhea; visitors to Nepal may acquire Cyclospora. Campers, backpackers, and swimmers in wilderness areas may become infected with Giardia. Cruise ships may be affected by outbreaks of gastroenteritis caused by agents such as norovirus.
2. Consumers of certain foods. Diarrhea closely following food consumption at a picnic, banquet, or restaurant may suggest infection with Salmonella, Campylobacter, or Shigella from chicken; enterohemorrhagic E. coli (O157:H7) from undercooked hamburger; Bacillus cereus from fried rice or other reheated food; Staphylococcus aureus or Salmonella from mayonnaise or creams; Salmonella from eggs; Listeria from uncooked foods or soft cheeses; and Vibrio species, Salmonella, or acute hepatitis A from seafood, especially if raw. State departments of public health issue communications regarding food-related illnesses, which may have originated domestically or been imported, but ultimately cause epidemics in the United States (e.g., the Cyclospora epidemic of 2013 in midwestern states that resulted from bagged salads).
3. Immunodeficient persons. Individuals at risk for diarrhea include those with either primary immunodeficiency (e.g., IgA deficiency, common variable hypogammaglobulinemia, chronic granulomatous disease) or the much more common secondary immunodeficiency states (e.g., AIDS, senescence, pharmacologic suppression). Common enteric pathogens often cause a more severe and protracted diarrheal illness, and, particularly in persons with AIDS, opportunistic infections, such as by Mycobacterium species, certain viruses (cytomegalovirus, adenovirus, and herpes simplex), and protozoa (Cryptosporidium, Isospora belli, Microsporida, and Blastocystis hominis) may also play a role (Chap. 226). In patients with AIDS, agents transmitted venereally per rectum (e.g., Neisseria gonorrhoeae, Treponema pallidum, Chlamydia) may contribute to proctocolitis. Persons with hemochromatosis are especially prone to invasive, even fatal, enteric infections with Vibrio species and Yersinia infections and should avoid raw fish.
4. Daycare attendees and their family members. Infections with Shigella, Giardia, Cryptosporidium, rotavirus, and other agents are very common and should be considered.
5. Institutionalized persons. Infectious diarrhea is one of the most frequent categories of nosocomial infections in many hospitals and long-term care facilities; the causes are a variety of microorganisms but most commonly C. difficile. C. difficile can affect those with no history of antibiotic use and may be acquired in the community.
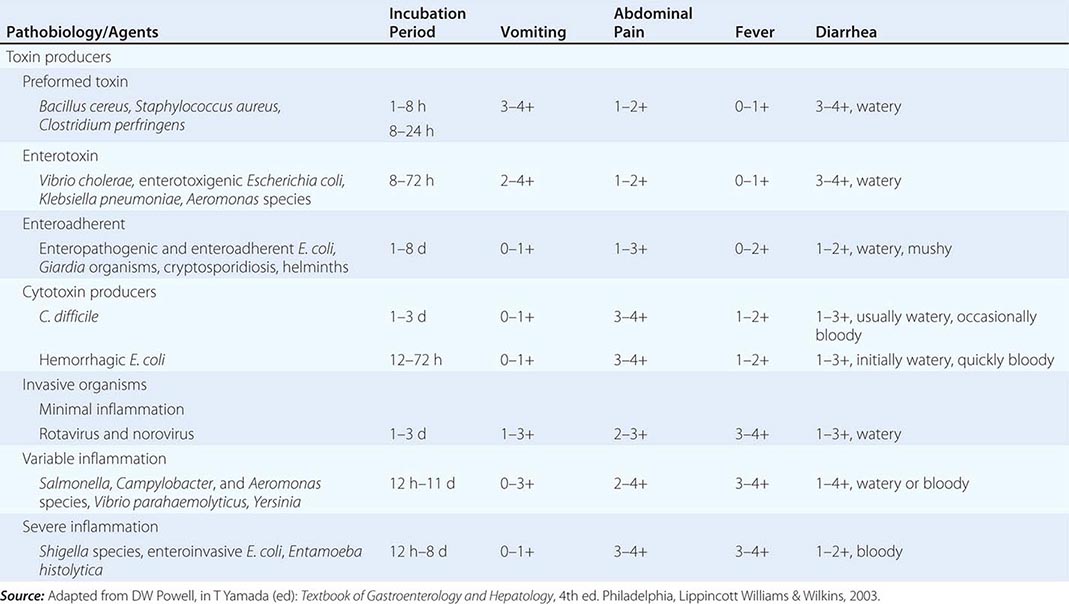
The pathophysiology underlying acute diarrhea by infectious agents produces specific clinical features that may also be helpful in diagnosis (Table 55-2). Profuse, watery diarrhea secondary to small-bowel hypersecretion occurs with ingestion of preformed bacterial toxins, enterotoxin-producing bacteria, and enteroadherent pathogens. Diarrhea associated with marked vomiting and minimal or no fever may occur abruptly within a few hours after ingestion of the former two types; vomiting is usually less, abdominal cramping or bloating is greater, and fever is higher with the latter. Cytotoxin-producing and invasive microorganisms all cause high fever and abdominal pain. Invasive bacteria and Entamoeba histolytica often cause bloody diarrhea (referred to as dysentery). Yersinia invades the terminal ileal and proximal colon mucosa and may cause especially severe abdominal pain with tenderness mimicking acute appendicitis.
|
ASSOCIATION BETWEEN PATHOBIOLOGY OF CAUSATIVE AGENTS AND CLINICAL FEATURES IN ACUTE INFECTIOUS DIARRHEA |
Finally, infectious diarrhea may be associated with systemic manifestations. Reactive arthritis (formerly known as Reiter’s syndrome), arthritis, urethritis, and conjunctivitis may accompany or follow infections by Salmonella, Campylobacter, Shigella, and Yersinia. Yersiniosis may also lead to an autoimmune-type thyroiditis, pericarditis, and glomerulonephritis. Both enterohemorrhagic E. coli (O157:H7) and Shigella can lead to the hemolytic-uremic syndrome with an attendant high mortality rate. The syndrome of postinfectious IBS has now been recognized as a complication of infectious diarrhea. Similarly, acute gastroenteritis may precede the diagnosis of celiac disease or Crohn’s disease. Acute diarrhea can also be a major symptom of several systemic infections including viral hepatitis, listeriosis, legionellosis, and toxic shock syndrome.
Other Causes Side effects from medications are probably the most common noninfectious causes of acute diarrhea, and etiology may be suggested by a temporal association between use and symptom onset. Although innumerable medications may produce diarrhea, some of the more frequently incriminated include antibiotics, cardiac antidysrhythmics, antihypertensives, nonsteroidal anti-inflammatory drugs (NSAIDs), certain antidepressants, chemotherapeutic agents, bronchodilators, antacids, and laxatives. Occlusive or nonocclusive ischemic colitis typically occurs in persons >50 years; often presents as acute lower abdominal pain preceding watery, then bloody diarrhea; and generally results in acute inflammatory changes in the sigmoid or left colon while sparing the rectum. Acute diarrhea may accompany colonic diverticulitis and graft-versus-host disease. Acute diarrhea, often associated with systemic compromise, can follow ingestion of toxins including organophosphate insecticides; amanita and other mushrooms; arsenic; and preformed environmental toxins in seafood, such as ciguatera and scombroid. Acute anaphylaxis to food ingestion can have a similar presentation. Conditions causing chronic diarrhea can also be confused with acute diarrhea early in their course. This confusion may occur with inflammatory bowel disease (IBD) and some of the other inflammatory chronic diarrheas that may have an abrupt rather than insidious onset and exhibit features that mimic infection.
APPROACH TO THE PATIENT:
Acute Diarrhea
The decision to evaluate acute diarrhea depends on its severity and duration and on various host factors (Fig. 55-2). Most episodes of acute diarrhea are mild and self-limited and do not justify the cost and potential morbidity rate of diagnostic or pharmacologic interventions. Indications for evaluation include profuse diarrhea with dehydration, grossly bloody stools, fever ≥38.5°C (≥101°F), duration >48 h without improvement, recent antibiotic use, new community outbreaks, associated severe abdominal pain in patients >50 years, and elderly (≥70 years) or immunocompromised patients. In some cases of moderately severe febrile diarrhea associated with fecal leukocytes (or increased fecal levels of the leukocyte proteins, such as calprotectin) or with gross blood, a diagnostic evaluation might be avoided in favor of an empirical antibiotic trial (see below).
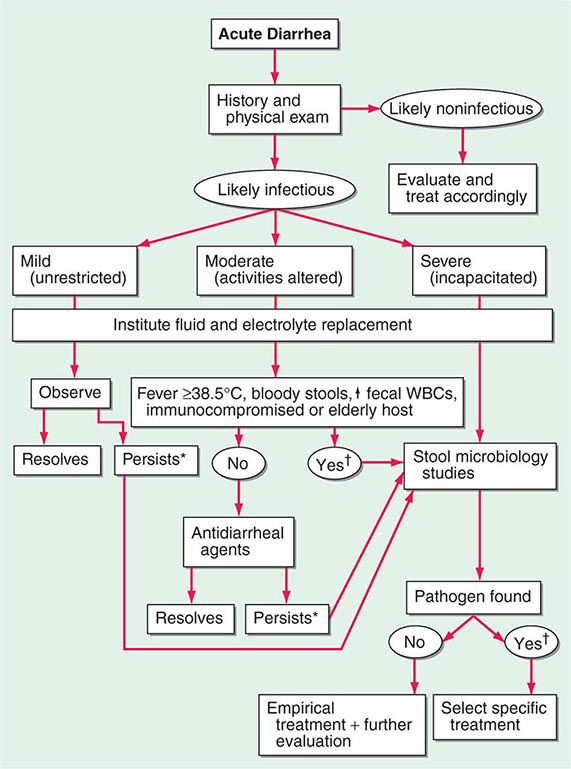
FIGURE 55-2 Algorithm for the management of acute diarrhea. Consider empirical treatment before evaluation with (*) metronidazole and with (†) quinolone. WBCs, white blood cells.
The cornerstone of diagnosis in those suspected of severe acute infectious diarrhea is microbiologic analysis of the stool. Workup includes cultures for bacterial and viral pathogens, direct inspection for ova and parasites, and immunoassays for certain bacterial toxins (C. difficile), viral antigens (rotavirus), and protozoal antigens (Giardia, E. histolytica). The aforementioned clinical and epidemiologic associations may assist in focusing the evaluation. If a particular pathogen or set of possible pathogens is so implicated, then either the whole panel of routine studies may not be necessary or, in some instances, special cultures may be appropriate as for enterohemorrhagic and other types of E. coli, Vibrio species, and Yersinia. Molecular diagnosis of pathogens in stool can be made by identification of unique DNA sequences; and evolving microarray technologies have led to more rapid, sensitive, specific, and cost-effective diagnosis.
Persistent diarrhea is commonly due to Giardia (Chap. 247), but additional causative organisms that should be considered include C. difficile (especially if antibiotics had been administered), E. histolytica, Cryptosporidium, Campylobacter, and others. If stool studies are unrevealing, flexible sigmoidoscopy with biopsies and upper endoscopy with duodenal aspirates and biopsies may be indicated. Brainerd diarrhea is an increasingly recognized entity characterized by an abrupt-onset diarrhea that persists for at least 4 weeks, but may last 1–3 years, and is thought to be of infectious origin. It may be associated with subtle inflammation of the distal small intestine or proximal colon.
Structural examination by sigmoidoscopy, colonoscopy, or abdominal computed tomography (CT) scanning (or other imaging approaches) may be appropriate in patients with uncharacterized persistent diarrhea to exclude IBD or as an initial approach in patients with suspected noninfectious acute diarrhea such as might be caused by ischemic colitis, diverticulitis, or partial bowel obstruction.
CHRONIC DIARRHEA
Diarrhea lasting >4 weeks warrants evaluation to exclude serious underlying pathology. In contrast to acute diarrhea, most of the causes of chronic diarrhea are noninfectious. The classification of chronic diarrhea by pathophysiologic mechanism facilitates a rational approach to management, although many diseases cause diarrhea by more than one mechanism (Table 55-3).
|
MAJOR CAUSES OF CHRONIC DIARRHEA ACCORDING TO PREDOMINANT PATHOPHYSIOLOGIC MECHANISM |
Abbreviation: FODMAP, fermentable oligosaccharides, disaccharides, monosaccharides, and polyols.
Secretory Causes Secretory diarrheas are due to derangements in fluid and electrolyte transport across the enterocolonic mucosa. They are characterized clinically by watery, large-volume fecal outputs that are typically painless and persist with fasting. Because there is no malabsorbed solute, stool osmolality is accounted for by normal endogenous electrolytes with no fecal osmotic gap.
MEDICATIONS Side effects from regular ingestion of drugs and toxins are the most common secretory causes of chronic diarrhea. Hundreds of prescription and over-the-counter medications (see earlier section, “Acute Diarrhea, Other Causes”) may produce diarrhea. Surreptitious or habitual use of stimulant laxatives (e.g., senna, cascara, bisacodyl, ricinoleic acid [castor oil]) must also be considered. Chronic ethanol consumption may cause a secretory-type diarrhea due to enterocyte injury with impaired sodium and water absorption as well as rapid transit and other alterations. Inadvertent ingestion of certain environmental toxins (e.g., arsenic) may lead to chronic rather than acute forms of diarrhea. Certain bacterial infections may occasionally persist and be associated with a secretory-type diarrhea.
BOWEL RESECTION, MUCOSAL DISEASE, OR ENTEROCOLIC FISTULA These conditions may result in a secretory-type diarrhea because of inadequate surface for reabsorption of secreted fluids and electrolytes. Unlike other secretory diarrheas, this subset of conditions tends to worsen with eating. With disease (e.g., Crohn’s ileitis) or resection of <100 cm of terminal ileum, dihydroxy bile acids may escape absorption and stimulate colonic secretion (cholerheic diarrhea). This mechanism may contribute to so-called idiopathic secretory diarrhea or bile acid diarrhea (BAD), in which bile acids are functionally malabsorbed from a normal-appearing terminal ileum. This idiopathic bile acid malabsorption (BAM) may account for an average of 40% of unexplained chronic diarrhea. Reduced negative feedback regulation of bile acid synthesis in hepatocytes by fibroblast growth factor 19 (FGF-19) produced by ileal enterocytes results in a degree of bile-acid synthesis that exceeds the normal capacity for ileal reabsorption, producing BAD. An alternative cause of BAD is a genetic variation in the receptor proteins (β-klotho and fibroblast growth factor 4) on the hepatocyte that normally mediate the effect of FGF-19. Dysfunction of these proteins prevents FGF-19 inhibition of hepatocyte bile acid synthesis.
Partial bowel obstruction, ostomy stricture, or fecal impaction may paradoxically lead to increased fecal output due to fluid hypersecretion.
HORMONES Although uncommon, the classic examples of secretory diarrhea are those mediated by hormones. Metastatic gastrointestinal carcinoid tumors or, rarely, primary bronchial carcinoids may produce watery diarrhea alone or as part of the carcinoid syndrome that comprises episodic flushing, wheezing, dyspnea, and right-sided valvular heart disease. Diarrhea is due to the release into the circulation of potent intestinal secretagogues including serotonin, histamine, prostaglandins, and various kinins. Pellagra-like skin lesions may rarely occur as the result of serotonin overproduction with niacin depletion. Gastrinoma, one of the most common neuroendocrine tumors, most typically presents with refractory peptic ulcers, but diarrhea occurs in up to one-third of cases and may be the only clinical manifestation in 10%. While other secretagogues released with gastrin may play a role, the diarrhea most often results from fat maldigestion owing to pancreatic enzyme inactivation by low intraduodenal pH. The watery diarrhea hypokalemia achlorhydria syndrome, also called pancreatic cholera, is due to a non-β cell pancreatic adenoma, referred to as a VIPoma, that secretes VIP and a host of other peptide hormones including pancreatic polypeptide, secretin, gastrin, gastrin-inhibitory polypeptide (also called glucose-dependent insulinotropic peptide), neurotensin, calcitonin, and prostaglandins. The secretory diarrhea is often massive with stool volumes >3 L/d; daily volumes as high as 20 L have been reported. Life-threatening dehydration; neuromuscular dysfunction from associated hypokalemia, hypomagnesemia, or hypercalcemia; flushing; and hyperglycemia may accompany a VIPoma. Medullary carcinoma of the thyroid may present with watery diarrhea caused by calcitonin, other secretory peptides, or prostaglandins. Prominent diarrhea is often associated with metastatic disease and poor prognosis. Systemic mastocytosis, which may be associated with the skin lesion urticaria pigmentosa, may cause diarrhea that is either secretory and mediated by histamine or inflammatory due to intestinal infiltration by mast cells. Large colorectal villous adenomas may rarely be associated with a secretory diarrhea that may cause hypokalemia, can be inhibited by NSAIDs, and are apparently mediated by prostaglandins.
CONGENITAL DEFECTS IN ION ABSORPTION Rarely, defects in specific carriers associated with ion absorption cause watery diarrhea from birth. These disorders include defective Cl–/HCO3– exchange (congenital chloridorrhea) with alkalosis (which results from a mutated DRA [down-regulated in adenoma] gene) and defective Na+/H+ exchange (congenital sodium diarrhea), which results from a mutation in the NHE3 (sodium-hydrogen exchanger) gene and results in acidosis.
Some hormone deficiencies may be associated with watery diarrhea, such as occurs with adrenocortical insufficiency (Addison’s disease) that may be accompanied by skin hyperpigmentation.
Osmotic Causes Osmotic diarrhea occurs when ingested, poorly absorbable, osmotically active solutes draw enough fluid into the lumen to exceed the reabsorptive capacity of the colon. Fecal water output increases in proportion to such a solute load. Osmotic diarrhea characteristically ceases with fasting or with discontinuation of the causative agent.
OSMOTIC LAXATIVES Ingestion of magnesium-containing antacids, health supplements, or laxatives may induce osmotic diarrhea typified by a stool osmotic gap (>50 mosmol/L): serum osmolarity (typically 290 mosmol/kg) – (2 × [fecal sodium + potassium concentration]). Measurement of fecal osmolarity is no longer recommended because, even when measured immediately after evacuation, it may be erroneous because carbohydrates are metabolized by colonic bacteria, causing an increase in osmolarity.
CARBOHYDRATE MALABSORPTION Carbohydrate malabsorption due to acquired or congenital defects in brush-border disaccharidases and other enzymes leads to osmotic diarrhea with a low pH. One of the most common causes of chronic diarrhea in adults is lactase deficiency, which affects three-fourths of nonwhites worldwide and 5–30% of persons in the United States; the total lactose load at any one time influences the symptoms experienced. Most patients learn to avoid milk products without requiring treatment with enzyme supplements. Some sugars, such as sorbitol, lactulose, or fructose, are frequently malabsorbed, and diarrhea ensues with ingestion of medications, gum, or candies sweetened with these poorly or incompletely absorbed sugars.
WHEAT AND FODMAP INTOLERANCE Chronic diarrhea, bloating, and abdominal pain are recognized as symptoms of nonceliac gluten intolerance (which is associated with impaired intestinal or colonic barrier function) and intolerance of fermentable oligosaccharides, disaccharides, monosaccharides, and polyols (FODMAPs). The latter’s effects represent the interaction between the GI microbiome and the nutrients.
Steatorrheal Causes Fat malabsorption may lead to greasy, foul-smelling, difficult-to-flush diarrhea often associated with weight loss and nutritional deficiencies due to concomitant malabsorption of amino acids and vitamins. Increased fecal output is caused by the osmotic effects of fatty acids, especially after bacterial hydroxylation, and, to a lesser extent, by the neutral fat. Quantitatively, steatorrhea is defined as stool fat exceeding the normal 7 g/d; rapid-transit diarrhea may result in fecal fat up to 14 g/d; daily fecal fat averages 15–25 g with small-intestinal diseases and is often >32 g with pancreatic exocrine insufficiency. Intraluminal maldigestion, mucosal malabsorption, or lymphatic obstruction may produce steatorrhea.
INTRALUMINAL MALDIGESTION This condition most commonly results from pancreatic exocrine insufficiency, which occurs when >90% of pancreatic secretory function is lost. Chronic pancreatitis, usually a sequel of ethanol abuse, most frequently causes pancreatic insufficiency. Other causes include cystic fibrosis; pancreatic duct obstruction; and, rarely, somatostatinoma. Bacterial overgrowth in the small intestine may deconjugate bile acids and alter micelle formation, impairing fat digestion; it occurs with stasis from a blind-loop, small-bowel diverticulum or dysmotility and is especially likely in the elderly. Finally, cirrhosis or biliary obstruction may lead to mild steatorrhea due to deficient intraluminal bile acid concentration.
MUCOSAL MALABSORPTION Mucosal malabsorption occurs from a variety of enteropathies, but it most commonly occurs from celiac disease. This gluten-sensitive enteropathy affects all ages and is characterized by villous atrophy and crypt hyperplasia in the proximal small bowel and can present with fatty diarrhea associated with multiple nutritional deficiencies of varying severity. Celiac disease is much more frequent than previously thought; it affects ~1% of the population, frequently presents without steatorrhea, can mimic IBS, and has many other GI and extraintestinal manifestations. Tropical sprue may produce a similar histologic and clinical syndrome but occurs in residents of or travelers to tropical climates; abrupt onset and response to antibiotics suggest an infectious etiology. Whipple’s disease, due to the bacillus Tropheryma whipplei and histiocytic infiltration of the small-bowel mucosa, is a less common cause of steatorrhea that most typically occurs in young or middle-aged men; it is frequently associated with arthralgias, fever, lymphadenopathy, and extreme fatigue, and it may affect the CNS and endocardium. A similar clinical and histologic picture results from Mycobacterium avium-intracellulare infection in patients with AIDS. Abetalipoproteinemia is a rare defect of chylomicron formation and fat malabsorption in children, associated with acanthocytic erythrocytes, ataxia, and retinitis pigmentosa. Several other conditions may cause mucosal malabsorption including infections, especially with protozoa such as Giardia; numerous medications (e.g., olmesartan, mycophenolate mofetil, colchicine, cholestyramine, neomycin); amyloidosis; and chronic ischemia.
POSTMUCOSAL LYMPHATIC OBSTRUCTION The pathophysiology of this condition, which is due to the rare congenital intestinal lymphangiectasia or to acquired lymphatic obstruction secondary to trauma, tumor, cardiac disease or infection, leads to the unique constellation of fat malabsorption with enteric losses of protein (often causing edema) and lymphocytopenia. Carbohydrate and amino acid absorption are preserved.
Inflammatory Causes Inflammatory diarrheas are generally accompanied by pain, fever, bleeding, or other manifestations of inflammation. The mechanism of diarrhea may not only be exudation but, depending on lesion site, may include fat malabsorption, disrupted fluid/electrolyte absorption, and hypersecretion or hypermotility from release of cytokines and other inflammatory mediators. The unifying feature on stool analysis is the presence of leukocytes or leukocyte-derived proteins such as calprotectin. With severe inflammation, exudative protein loss can lead to anasarca (generalized edema). Any middle-aged or older person with chronic inflammatory-type diarrhea, especially with blood, should be carefully evaluated to exclude a colorectal tumor.
IDIOPATHIC INFLAMMATORY BOWEL DISEASE The illnesses in this category, which include Crohn’s disease and chronic ulcerative colitis, are among the most common organic causes of chronic diarrhea in adults and range in severity from mild to fulminant and life-threatening. They may be associated with uveitis, polyarthralgias, cholestatic liver disease (primary sclerosing cholangitis), and skin lesions (erythema nodosum, pyoderma gangrenosum). Microscopic colitis, including both lymphocytic and collagenous colitis, is an increasingly recognized cause of chronic watery diarrhea, especially in middle-aged women and those on NSAIDs, statins, proton pump inhibitors (PPIs), and selective serotonin reuptake inhibitors (SSRIs); biopsy of a normal-appearing colon is required for histologic diagnosis. It may coexist with symptoms suggesting IBS or with celiac sprue or drug-induced enteropathy. It typically responds well to anti-inflammatory drugs (e.g., bismuth), to the opioid agonist loperamide, or to budesonide.
PRIMARY OR SECONDARY FORMS OF IMMUNODEFICIENCY Immunodeficiency may lead to prolonged infectious diarrhea. With selective IgA deficiency or common variable hypogammaglobulinemia, diarrhea is particularly prevalent and often the result of giardiasis, bacterial overgrowth, or sprue.
EOSINOPHILIC GASTROENTERITIS Eosinophil infiltration of the mucosa, muscularis, or serosa at any level of the GI tract may cause diarrhea, pain, vomiting, or ascites. Affected patients often have an atopic history, Charcot-Leyden crystals due to extruded eosinophil contents may be seen on microscopic inspection of stool, and peripheral eosinophilia is present in 50–75% of patients. While hypersensitivity to certain foods occurs in adults, true food allergy causing chronic diarrhea is rare.
OTHER CAUSES Chronic inflammatory diarrhea may be caused by radiation enterocolitis, chronic graft-versus-host disease, Behçet’s syndrome, and Cronkhite-Canada syndrome, among others.
Dysmotility Causes Rapid transit may accompany many diarrheas as a secondary or contributing phenomenon, but primary dysmotility is an unusual etiology of true diarrhea. Stool features often suggest a secretory diarrhea, but mild steatorrhea of up to 14 g of fat per day can be produced by maldigestion from rapid transit alone. Hyperthyroidism, carcinoid syndrome, and certain drugs (e.g., prostaglandins, prokinetic agents) may produce hypermotility with resultant diarrhea. Primary visceral neuromyopathies or idiopathic acquired intestinal pseudoobstruction may lead to stasis with secondary bacterial overgrowth causing diarrhea. Diabetic diarrhea, often accompanied by peripheral and generalized autonomic neuropathies, may occur in part because of intestinal dysmotility.
The exceedingly common IBS (10% point prevalence, 1–2% per year incidence) is characterized by disturbed intestinal and colonic motor and sensory responses to various stimuli. Symptoms of stool frequency typically cease at night, alternate with periods of constipation, are accompanied by abdominal pain relieved with defecation, and rarely result in weight loss.
Factitial Causes Factitial diarrhea accounts for up to 15% of unexplained diarrheas referred to tertiary care centers. Either as a form of Munchausen syndrome (deception or self-injury for secondary gain) or eating disorders, some patients covertly self-administer laxatives alone or in combination with other medications (e.g., diuretics) or surreptitiously add water or urine to stool sent for analysis. Such patients are typically women, often with histories of psychiatric illness, and disproportionately from careers in health care. Hypotension and hypokalemia are common co-presenting features. The evaluation of such patients may be difficult: contamination of the stool with water or urine is suggested by very low or high stool osmolarity, respectively. Such patients often deny this possibility when confronted, but they do benefit from psychiatric counseling when they acknowledge their behavior.
APPROACH TO THE PATIENT:
Chronic Diarrhea
The laboratory tools available to evaluate the very common problem of chronic diarrhea are extensive, and many are costly and invasive. As such, the diagnostic evaluation must be rationally directed by a careful history, including medications, and physical examination (Fig. 55-3A). When this strategy is unrevealing, simple triage tests are often warranted to direct the choice of more complex investigations (Fig. 55-3B). The history, physical examination (Table 55-4), and routine blood studies should attempt to characterize the mechanism of diarrhea, identify diagnostically helpful associations, and assess the patient’s fluid/electrolyte and nutritional status. Patients should be questioned about the onset, duration, pattern, aggravating (especially diet) and relieving factors, and stool characteristics of their diarrhea. The presence or absence of fecal incontinence, fever, weight loss, pain, certain exposures (travel, medications, contacts with diarrhea), and common extraintestinal manifestations (skin changes, arthralgias, oral aphthous ulcers) should be noted. A family history of IBD or sprue may indicate those possibilities. Physical findings may offer clues such as a thyroid mass, wheezing, heart murmurs, edema, hepatomegaly, abdominal masses, lymphadenopathy, mucocutaneous abnormalities, perianal fistulas, or anal sphincter laxity. Peripheral blood leukocytosis, elevated sedimentation rate, or C-reactive protein suggests inflammation; anemia reflects blood loss or nutritional deficiencies; or eosinophilia may occur with parasitoses, neoplasia, collagen-vascular disease, allergy, or eosinophilic gastroenteritis. Blood chemistries may demonstrate electrolyte, hepatic, or other metabolic disturbances. Measuring IgA tissue transglutaminase antibodies may help detect celiac disease. Bile acid diarrhea is confirmed by a scintigraphic radiolabeled bile acid retention test; however, this is not available in many countries. Alternative approaches are a screening blood test (serum C4 or FGF-19), measurement of fecal bile acids, or a therapeutic trial with a bile acid sequestrant (e.g., cholestyramine or colesevelam).
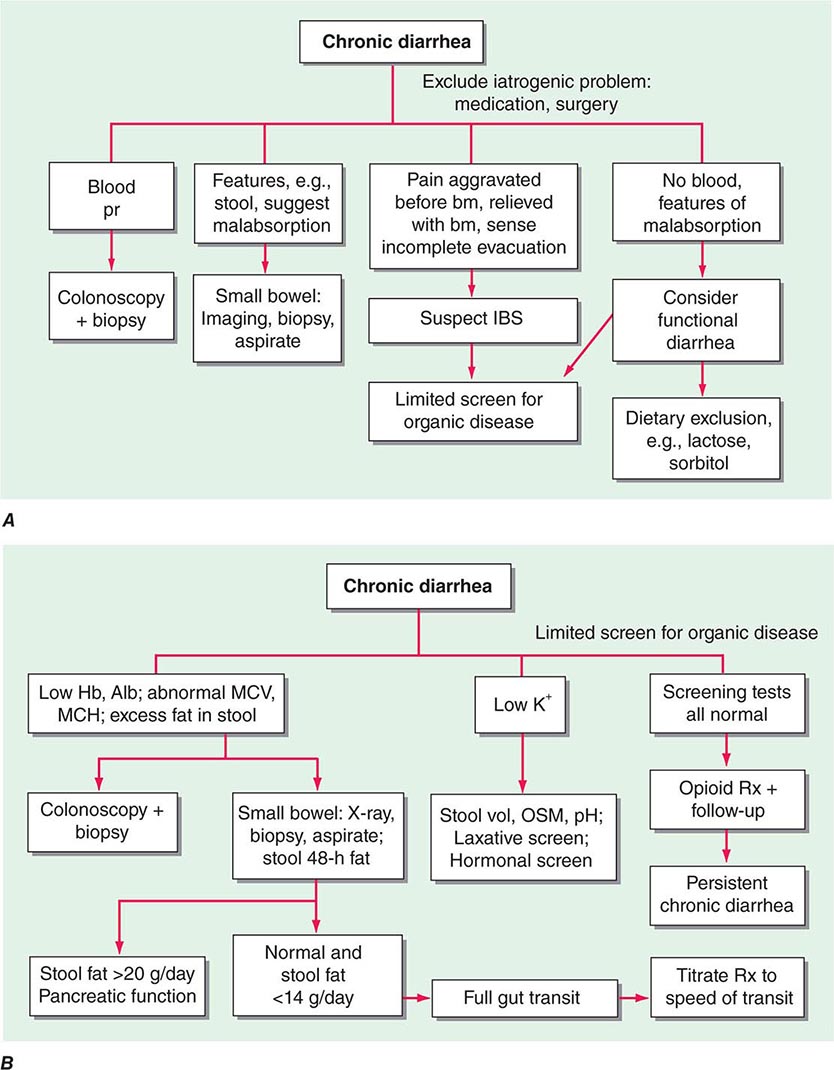
FIGURE 55-3 Chronic diarrhea. A. Initial management based on accompanying symptoms or features. B. Evaluation based on findings from a limited age-appropriate screen for organic disease. Alb, albumin; bm, bowel movement; Hb, hemoglobin; IBS, irritable bowel syndrome; MCH, mean corpuscular hemoglobin; MCV, mean corpuscular volume; OSM, osmolality; pr, per rectum. (Reprinted from M Camilleri: Clin Gastroenterol Hepatol. 2:198, 2004.)
|
PHYSICAL EXAMINATION IN PATIENTS WITH CHRONIC DIARRHEA |
A therapeutic trial is often appropriate, definitive, and highly cost-effective when a specific diagnosis is suggested on the initial physician encounter. For example, chronic watery diarrhea, which ceases with fasting in an otherwise healthy young adult, may justify a trial of a lactose-restricted diet; bloating and diarrhea persisting since a mountain backpacking trip may warrant a trial of metronidazole for likely giardiasis; and postprandial diarrhea persisting following resection of terminal ileum might be due to bile acid malabsorption and be treated with cholestyramine or colesevelam before further evaluation. Persistent symptoms require additional investigation.
Certain diagnoses may be suggested on the initial encounter (e.g., idiopathic IBD); however, additional focused evaluations may be necessary to confirm the diagnosis and characterize the severity or extent of disease so that treatment can be best guided. Patients suspected of having IBS should be initially evaluated with flexible sigmoidoscopy with colorectal biopsies to exclude IBD, or particularly microscopic colitis, which is clinically indistinguishable from IBS with diarrhea; those with normal findings might be reassured and, as indicated, treated empirically with antispasmodics, antidiarrheals, or antidepressants (e.g., tricyclic agents). Any patient who presents with chronic diarrhea and hematochezia should be evaluated with stool microbiologic studies and colonoscopy.
In an estimated two-thirds of cases, the cause for chronic diarrhea remains unclear after the initial encounter, and further testing is required. Quantitative stool collection and analyses can yield important objective data that may establish a diagnosis or characterize the type of diarrhea as a triage for focused additional studies (Fig. 55-3B). If stool weight is >200 g/d, additional stool analyses should be performed that might include electrolyte concentration, pH, occult blood testing, leukocyte inspection (or leukocyte protein assay), fat quantitation, and laxative screens.
For secretory diarrheas (watery, normal osmotic gap), possible medication-related side effects or surreptitious laxative use should be reconsidered. Microbiologic studies should be done including fecal bacterial cultures (including media for Aeromonas and Plesiomonas), inspection for ova and parasites, and Giardia antigen assay (the most sensitive test for giardiasis). Small-bowel bacterial overgrowth can be excluded by intestinal aspirates with quantitative cultures or with glucose or lactulose breath tests involving measurement of breath hydrogen, methane, or other metabolite. However, interpretation of these breath tests may be confounded by disturbances of intestinal transit. Upper endoscopy and colonoscopy with biopsies and small-bowel x-rays (formerly barium, but increasingly CT with enterography or magnetic resonance with enteroclysis) are helpful to rule out structural or occult inflammatory disease. When suggested by history or other findings, screens for peptide hormones should be pursued (e.g., serum gastrin, VIP, calcitonin, and thyroid hormone/thyroid-stimulating hormone, urinary 5-hydroxyindolacetic acid, histamine).
Further evaluation of osmotic diarrhea should include tests for lactose intolerance and magnesium ingestion, the two most common causes. Low fecal pH suggests carbohydrate malabsorption; lactose malabsorption can be confirmed by lactose breath testing or by a therapeutic trial with lactose exclusion and observation of the effect of lactose challenge (e.g., a liter of milk). Lactase determination on small-bowel biopsy is not generally available. If fecal magnesium or laxative levels are elevated, inadvertent or surreptitious ingestion should be considered and psychiatric help should be sought.
For those with proven fatty diarrhea, endoscopy with small-bowel biopsy (including aspiration for Giardia and quantitative cultures) should be performed; if this procedure is unrevealing, a small-bowel radiograph is often an appropriate next step. If small-bowel studies are negative or if pancreatic disease is suspected, pancreatic exocrine insufficiency should be excluded with direct tests, such as the secretin-cholecystokinin stimulation test or a variation that could be performed endoscopically. In general, indirect tests such as assay of fecal elastase or chymotrypsin activity or a bentiromide test have fallen out of favor because of low sensitivity and specificity.
Chronic inflammatory-type diarrheas should be suspected by the presence of blood or leukocytes in the stool. Such findings warrant stool cultures; inspection for ova and parasites; C. difficile toxin assay; colonoscopy with biopsies; and, if indicated, small-bowel contrast studies.
CONSTIPATION
DEFINITION
Constipation is a common complaint in clinical practice and usually refers to persistent, difficult, infrequent, or seemingly incomplete defecation. Because of the wide range of normal bowel habits, constipation is difficult to define precisely. Most persons have at least three bowel movements per week; however, low stool frequency alone is not the sole criterion for the diagnosis of constipation. Many constipated patients have a normal frequency of defecation but complain of excessive straining, hard stools, lower abdominal fullness, or a sense of incomplete evacuation. The individual patient’s symptoms must be analyzed in detail to ascertain what is meant by “constipation” or “difficulty” with defecation.
Stool form and consistency are well correlated with the time elapsed from the preceding defecation. Hard, pellety stools occur with slow transit, whereas loose, watery stools are associated with rapid transit. Both small pellety or very large stools are more difficult to expel than normal stools.
The perception of hard stools or excessive straining is more difficult to assess objectively, and the need for enemas or digital disimpaction is a clinically useful way to corroborate the patient’s perceptions of difficult defecation.
Psychosocial or cultural factors may also be important. A person whose parents attached great importance to daily defecation will become greatly concerned when he or she misses a daily bowel movement; some children withhold stool to gain attention or because of fear of pain from anal irritation; and some adults habitually ignore or delay the call to have a bowel movement.
CAUSES
Pathophysiologically, chronic constipation generally results from inadequate fiber or fluid intake or from disordered colonic transit or anorectal function. These result from neurogastroenterologic disturbance, certain drugs, advancing age, or in association with a large number of systemic diseases that affect the GI tract (Table 55-5). Constipation of recent onset may be a symptom of significant organic disease such as tumor or stricture. In idiopathic constipation, a subset of patients exhibit delayed emptying of the ascending and transverse colon with prolongation of transit (often in the proximal colon) and a reduced frequency of propulsive HAPCs. Outlet obstruction to defecation (also called evacuation disorders) accounts for about a quarter of cases presenting with constipation in tertiary care and may cause delayed colonic transit, which is usually corrected by biofeedback retraining of the disordered defecation. Constipation of any cause may be exacerbated by hospitalization or chronic illnesses that lead to physical or mental impairment and result in inactivity or physical immobility.
|
CAUSES OF CONSTIPATION IN ADULTS |
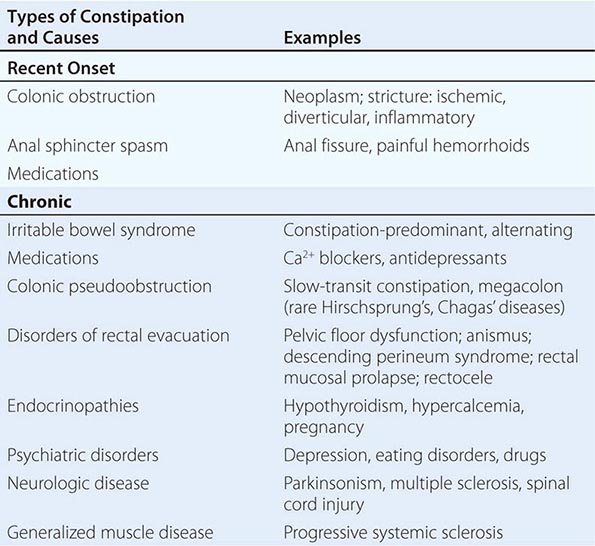
Constipation
A careful history should explore the patient’s symptoms and confirm whether he or she is indeed constipated based on frequency (e.g., fewer than three bowel movements per week), consistency (lumpy/hard), excessive straining, prolonged defecation time, or need to support the perineum or digitate the anorectum to facilitate stool evacuation. In the vast majority of cases (probably >90%), there is no underlying cause (e.g., cancer, depression, or hypothyroidism), and constipation responds to ample hydration, exercise, and supplementation of dietary fiber (15–25 g/d). A good diet and medication history and attention to psychosocial issues are key. Physical examination and, particularly, a rectal examination should exclude fecal impaction and most of the important diseases that present with constipation and possibly indicate features suggesting an evacuation disorder (e.g., high anal sphincter tone, failure of perineal descent, or paradoxical puborectalis contraction during straining to simulate stool evacuation).
The presence of weight loss, rectal bleeding, or anemia with constipation mandates either flexible sigmoidoscopy plus barium enema or colonoscopy alone, particularly in patients >40 years, to exclude structural diseases such as cancer or strictures. Colonoscopy alone is most cost-effective in this setting because it provides an opportunity to biopsy mucosal lesions, perform polypectomy, or dilate strictures. Barium enema has advantages over colonoscopy in the patient with isolated constipation because it is less costly and identifies colonic dilation and all significant mucosal lesions or strictures that are likely to present with constipation. Melanosis coli, or pigmentation of the colon mucosa, indicates the use of anthraquinone laxatives such as cascara or senna; however, this is usually apparent from a careful history. An unexpected disorder such as megacolon or cathartic colon may also be detected by colonic radiographs. Measurement of serum calcium, potassium, and thyroid-stimulating hormone levels will identify rare patients with metabolic disorders.
Patients with more troublesome constipation may not respond to fiber alone and may be helped by a bowel-training regimen, which involves taking an osmotic laxative (e.g., magnesium salts, lactulose, sorbitol, polyethylene glycol) and evacuating with enema or suppository (e.g., glycerine or bisacodyl) as needed. After breakfast, a distraction-free 15–20 min on the toilet without straining is encouraged. Excessive straining may lead to development of hemorrhoids, and, if there is weakness of the pelvic floor or injury to the pudendal nerve, may result in obstructed defecation from descending perineum syndrome several years later. Those few who do not benefit from the simple measures delineated above or require long-term treatment or fail to respond to potent laxatives should undergo further investigation (Fig. 55-4). Novel agents that induce secretion (e.g., lubiprostone, a chloride channel activator, or linaclotide, a guanylate cyclase C agonist that activates chloride secretion) are also available.
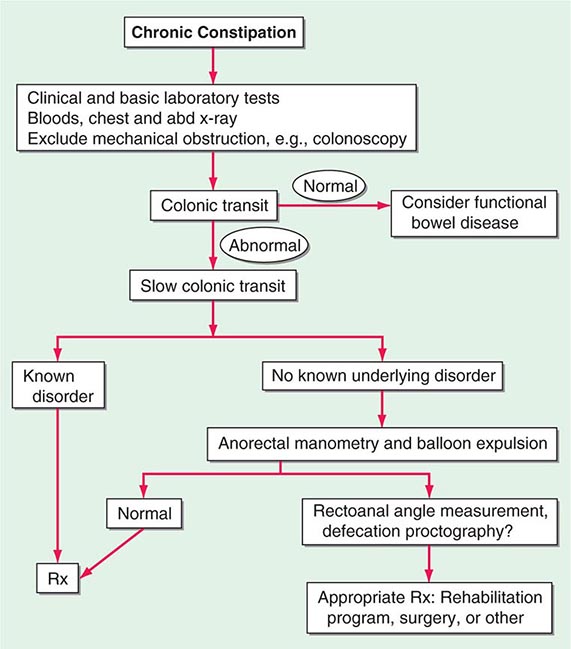
FIGURE 55-4 Algorithm for the management of constipation. abd, abdominal.
INVESTIGATION OF SEVERE CONSTIPATION
A small minority (probably <5%) of patients have severe or “intractable” constipation; about 25% have evacuation disorders. These are the patients most likely to require evaluation by gastroenterologists or in referral centers. Further observation of the patient may occasionally reveal a previously unrecognized cause, such as an evacuation disorder, laxative abuse, malingering, or psychological disorder. In these patients, evaluations of the physiologic function of the colon and pelvic floor and of psychological status aid in the rational choice of treatment. Even among these highly selected patients with severe constipation, a cause can be identified in only about one-third of tertiary referral patients, with the others being diagnosed with normal transit constipation.
Measurement of Colonic Transit Radiopaque marker transit tests are easy, repeatable, generally safe, inexpensive, reliable, and highly applicable in evaluating constipated patients in clinical practice. Several validated methods are very simple. For example, radiopaque markers are ingested; an abdominal flat film taken 5 days later should indicate passage of 80% of the markers out of the colon without the use of laxatives or enemas. This test does not provide useful information about the transit profile of the stomach and small bowel.
Radioscintigraphy with a delayed-release capsule containing radiolabeled particles has been used to noninvasively characterize normal, accelerated, or delayed colonic function over 24–48 h with low radiation exposure. This approach simultaneously assesses gastric, small bowel (which may be important in ~20% of patients with delayed colonic transit because they reflect a more generalized GI motility disorder), and colonic transit. The disadvantages are the greater cost and the need for specific materials prepared in a nuclear medicine laboratory.
Anorectal and Pelvic Floor Tests Pelvic floor dysfunction is suggested by the inability to evacuate the rectum, a feeling of persistent rectal fullness, rectal pain, the need to extract stool from the rectum digitally, application of pressure on the posterior wall of the vagina, support of the perineum during straining, and excessive straining. These significant symptoms should be contrasted with the simple sense of incomplete rectal evacuation, which is common in IBS.
Formal psychological evaluation may identify eating disorders, “control issues,” depression, or post-traumatic stress disorders that may respond to cognitive or other intervention and may be important in restoring quality of life to patients who might present with chronic constipation.
A simple clinical test in the office to document a non-relaxing puborectalis muscle is to have the patient strain to expel the index finger during a digital rectal examination. Motion of the puborectalis posteriorly during straining indicates proper coordination of the pelvic floor muscles. Motion anteriorly with paradoxical contraction during simulated evacuation indicates pelvic floor dysfunction.
Measurement of perineal descent is relatively easy to gauge clinically by placing the patient in the left decubitus position and watching the perineum to detect inadequate descent (<1.5 cm, a sign of pelvic floor dysfunction) or perineal ballooning during straining relative to bony landmarks (>4 cm, suggesting excessive perineal descent).
A useful overall test of evacuation is the balloon expulsion test. A balloon-tipped urinary catheter is placed and inflated with 50 mL of water. Normally, a patient can expel it while seated on a toilet or in the left lateral decubitus position. In the lateral position, the weight needed to facilitate expulsion of the balloon is determined; normally, expulsion occurs with <200 g added or unaided within 2 min.
Anorectal manometry, when used in the evaluation of patients with severe constipation, may find an excessively high resting (>80 mmHg) or squeeze anal sphincter tone, suggesting anismus (anal sphincter spasm). This test also identifies rare syndromes, such as adult Hirschsprung’s disease, by the absence of the rectoanal inhibitory reflex.
Defecography (a dynamic barium enema including lateral views obtained during barium expulsion or a magnetic resonance defecogram) reveals “soft abnormalities” in many patients; the most relevant findings are the measured changes in rectoanal angle, anatomic defects of the rectum such as internal mucosal prolapse, and enteroceles or rectoceles. Surgically remediable conditions are identified in only a few patients. These include severe, whole-thickness intussusception with complete outlet obstruction due to funnel-shaped plugging at the anal canal or an extremely large rectocele that fills preferentially during attempts at defecation instead of expulsion of the barium through the anus. In summary, defecography requires an interested and experienced radiologist, and abnormalities are not pathognomonic for pelvic floor dysfunction. The most common cause of outlet obstruction is failure of the puborectalis muscle to relax; this is not identified by barium defecography, but can be demonstrated by magnetic resonance defecography, which provides more information about the structure and function of the pelvic floor, distal colorectum, and anal sphincters.
Neurologic testing (electromyography) is more helpful in the evaluation of patients with incontinence than of those with symptoms suggesting obstructed defecation. The absence of neurologic signs in the lower extremities suggests that any documented denervation of the puborectalis results from pelvic (e.g., obstetric) injury or from stretching of the pudendal nerve by chronic, long-standing straining. Constipation is common among patients with spinal cord injuries, neurologic diseases such as Parkinson’s disease, multiple sclerosis, and diabetic neuropathy.
Spinal-evoked responses during electrical rectal stimulation or stimulation of external anal sphincter contraction by applying magnetic stimulation over the lumbosacral cord identify patients with limited sacral neuropathies with sufficient residual nerve conduction to attempt biofeedback training.
In summary, a balloon expulsion test is an important screening test for anorectal dysfunction. Rarely, an anatomic evaluation of the rectum or anal sphincters and an assessment of pelvic floor relaxation are the tools for evaluating patients in whom obstructed defecation is suspected and is associated with symptoms of rectal mucosal prolapse, pressure of the posterior wall of the vagina to facilitate defecation (suggestive of anterior rectocele), or prior pelvic surgery that may be complicated by enterocele.
56 |
Involuntary Weight Loss |
Involuntary weight loss (IWL) is frequently insidious and can have important implications, often serving as a harbinger of serious underlying disease. Clinically important weight loss is defined as the loss of 10 pounds (4.5 kg) or >5% of one’s body weight over a period of 6–12 months. IWL is encountered in up to 8% of all adult outpatients and 27% of frail persons age 65 years and older. There is no identifiable cause in up to one-quarter of patients despite extensive investigation. Conversely, up to half of people who claim to have lost weight have no documented evidence of weight loss. People with no known cause of weight loss generally have a better prognosis than do those with known causes, particularly when the source is neoplastic. Weight loss in older persons is associated with a variety of deleterious effects, including hip fracture, pressure ulcers, impaired immune function, and decreased functional status. Not surprisingly, significant weight loss is associated with increased mortality, which can range from 9% to as high as 38% within 1 to 2.5 years in the absence of clinical awareness and attention.
PHYSIOLOGY OF WEIGHT REGULATION WITH AGING
(See also Chaps. 94e and 415e) Among healthy aging people, total body weight peaks in the sixth decade of life and generally remains stable until the ninth decade, after which it gradually falls. In contrast, lean body mass (fat-free mass) begins to decline at a rate of 0.3 kg per year in the third decade, and the rate of decline increases further beginning at age 60 in men and age 65 in women. These changes in lean body mass largely reflect the age-dependent decline in growth hormone secretion and, consequently, circulating levels of insulin-like growth factor type I (IGF-I) that occur with normal aging. Loss of sex steroids, at menopause in women and more gradually with aging in men, also contributes to these changes in body composition. In the healthy elderly, an increase in fat tissue balances the loss in lean body mass until very old age, when loss of both fat and skeletal muscle occurs. Age-dependent changes also occur at the cellular level. Telomeres shorten, and body cell mass—the fat-free portion of cells—declines steadily with aging.
Between ages 20 and 80, mean energy intake is reduced by up to 1200 kcal/d in men and 800 kcal/d in women. Decreased hunger is a reflection of reduced physical activity and loss of lean body mass, producing lower demand for calories and food intake. Several important age-associated physiologic changes also predispose elderly persons to weight loss, such as declining chemosensory function (smell and taste), reduced efficiency of chewing, slowed gastric emptying, and alterations in the neuroendocrine axis, including changes in levels of leptin, cholecystokinin, neuropeptide Y, and other hormones and peptides. These changes are associated with early satiety and a decline in both appetite and the hedonistic appreciation of food. Collectively, they contribute to the “anorexia of aging.”
CAUSES OF INVOLUNTARY WEIGHT LOSS
Most causes of IWL belong to one of four categories: (1) malignant neoplasms, (2) chronic inflammatory or infectious diseases, (3) metabolic disorders (e.g., hyperthyroidism and diabetes), or (4) psychiatric disorders (Table 56-1). Not infrequently, more than one of these causes can be responsible for IWL. In most series, IWL is caused by malignant disease in a quarter of patients and by organic disease in one-third, with the remainder due to psychiatric disease, medications, or uncertain causes.
|
CAUSES OF INVOLUNTARY WEIGHT LOSS |
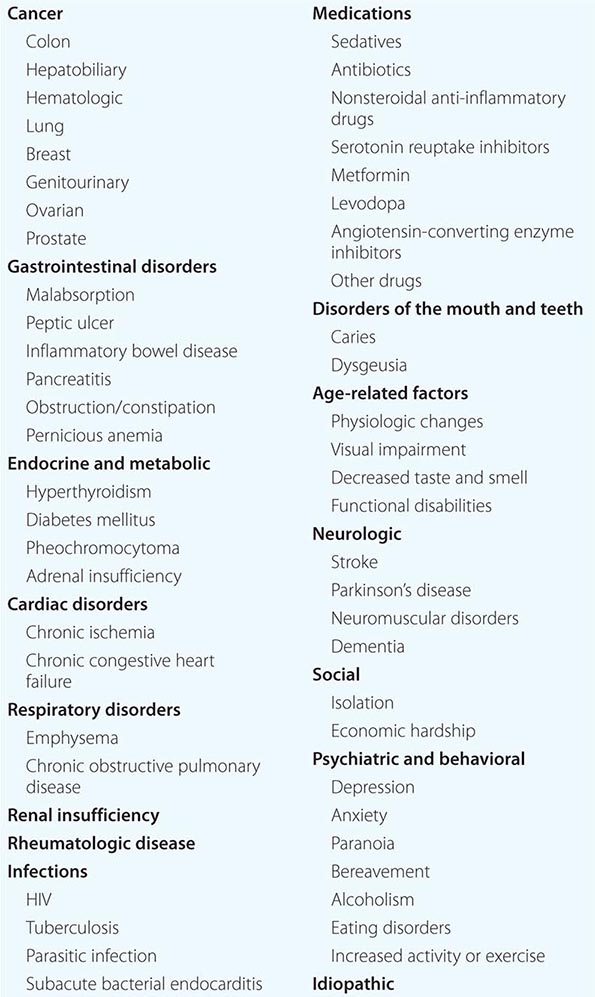
The most common malignant causes of IWL are gastrointestinal, hepatobiliary, hematologic, lung, breast, genitourinary, ovarian, and prostate. Half of all patients with cancer lose some body weight; one-third lose more than 5% of their original body weight, and up to 20% of all cancer deaths are caused directly by cachexia (through immobility and/or cardiac/respiratory failure). The greatest incidence of weight loss is seen among patients with solid tumors. Malignancy that reveals itself through significant weight loss usually has a very poor prognosis.
In addition to malignancies, gastrointestinal causes are among the most prominent causes of IWL. Peptic ulcer disease, inflammatory bowel disease, dysmotility syndromes, chronic pancreatitis, celiac disease, constipation, and atrophic gastritis are some of the more common entities. Oral and dental problems are easily overlooked and may manifest with halitosis, poor oral hygiene, xerostomia, inability to chew, reduced masticatory force, nonocclusion, temporomandibular joint syndrome, edentulousness, and pain due to caries or abscesses.
Tuberculosis, fungal diseases, parasites, subacute bacterial endocarditis, and HIV are well-documented causes of IWL. Cardiovascular and pulmonary diseases cause unintentional weight loss through increased metabolic demand and decreased appetite and caloric intake. Uremia produces nausea, anorexia, and vomiting. Connective tissue diseases may increase metabolic demand and disrupt nutritional balance. As the incidence of diabetes mellitus increases with aging, the associated glucosuria can contribute to weight loss. Hyperthyroidism in the elderly may have less prominent sympathomimetic features and may present as “apathetic hyperthyroidism” or T3 toxicosis (Chap. 405).
Neurologic injuries such as stroke, quadriplegia, and multiple sclerosis may lead to visceral and autonomic dysfunction that can impair caloric intake. Dysphagia from these neurologic insults is a common mechanism. Functional disability that compromises activities of daily living (ADLs) is a common cause of undernutrition in the elderly. Visual impairment from ophthalmic or central nervous system disorders such as a tremor can limit the ability of people to prepare and eat meals. IWL may be one of the earliest manifestations of Alzheimer’s dementia.
Isolation and depression are significant causes of IWL that may manifest as an inability to care for oneself, including nutritional needs. A cytokine-mediated inflammatory metabolic cascade can be both a cause of and a manifestation of depression. Bereavement can be a cause of IWL and, when present, is more pronounced in men. More intense forms of mental illness such as paranoid disorders may lead to delusions about food and cause weight loss. Alcoholism can be a significant source of weight loss and malnutrition.
Elderly persons living in poverty may have to choose whether to purchase food or use the money for other expenses, including medications. Institutionalization is an independent risk factor, as up to 30–50% of nursing home patients have inadequate food intake.
Medications can cause anorexia, nausea, vomiting, gastrointestinal distress, diarrhea, dry mouth, and changes in taste. This is particularly an issue in the elderly, many of whom take five or more medications.
ASSESSMENT
The four major manifestations of IWL are (1) anorexia (loss of appetite), (2) sarcopenia (loss of muscle mass), (3) cachexia (a syndrome that combines weight loss, loss of muscle and adipose tissue, anorexia, and weakness), and (4) dehydration. The current obesity epidemic adds complexity, as excess adipose tissue can mask the development of sarcopenia and delay awareness of the development of cachexia. If it is not possible to measure weight directly, a change in clothing size, corroboration of weight loss by a relative or friend, and a numeric estimate of weight loss provided by the patient are suggestive of true weight loss.
Initial assessment includes a comprehensive history and physical, a complete blood count, tests of liver enzyme levels, C-reactive protein, erythrocyte sedimentation rate, renal function studies, thyroid function tests, chest radiography, and an abdominal ultrasound (Table 56-2). Age, sex, and risk factor–specific cancer screening tests, such as mammography and colonoscopy, should be performed (Chap. 100). Patients at risk should have HIV testing. All elderly patients with weight loss should undergo screening for dementia and depression by using instruments such as the Mini-Mental State Examination and the Geriatric Depression Scale, respectively (Chap. 11). The Mini Nutritional Assessment (www.mna-elderly.com) and the Nutrition Screening Initiative (http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1694757/) are also available for the nutritional assessment of elderly patients. Almost all patients with a malignancy and >90% of those with other organic diseases have at least one laboratory abnormality. In patients presenting with substantial IWL, major organic and malignant diseases are unlikely when a baseline evaluation is completely normal. Careful follow-up rather than undirected testing is advised since the prognosis of weight loss of undetermined cause is generally favorable.
|
ASSESSMENT AND TESTING FOR INVOLUNTARY WEIGHT LOSS |
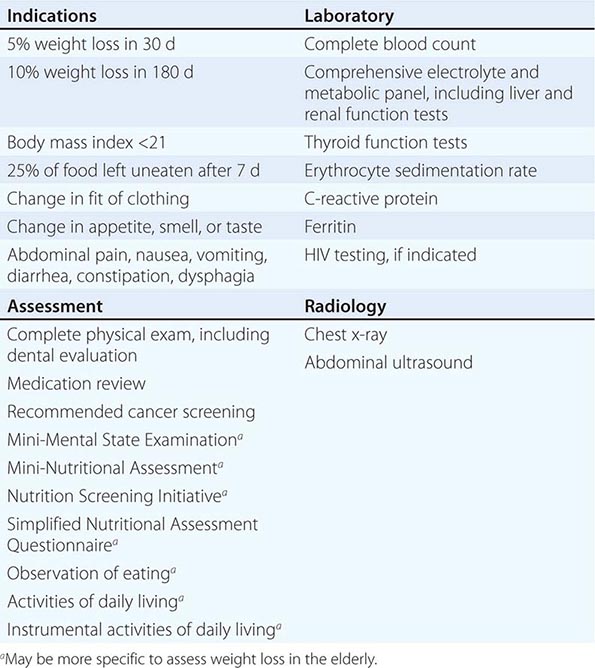
57 |
Gastrointestinal Bleeding |
Gastrointestinal bleeding (GIB) accounts for ~150 hospitalizations per 100,000 population annually in the United States, with upper GIB (UGIB) ~1.5–2 times more common than lower GIB (LGIB) The incidence of GIB has decreased in recent decades, primarily due to a reduction in UGIB, and the mortality has also decreased to <5%. Patients today rarely die from exsanguination, but rather die due to decompensation of other underlying illnesses.
GIB presents as either overt or occult bleeding. Overt GIB is manifested by hematemesis, vomitus of red blood or “coffee-grounds” material; melena, black, tarry, foul-smelling stool; and/or hematochezia, passage of bright red or maroon blood from the rectum. Occult GIB may be identified in the absence of overt bleeding when patients present with symptoms of blood loss or anemia such as lightheadedness, syncope, angina, or dyspnea; or when routine diagnostic evaluation reveals iron deficiency anemia or a positive fecal occult blood test. GIB is also categorized by the site of bleeding as UGIB, LGIB, or obscure GIB if the source is unclear.
SOURCES OF GASTROINTESTINAL BLEEDING
Upper Gastrointestinal Sources of Bleeding (Table 57-1) Peptic ulcers are the most common cause of UGIB, accounting for ~50% of cases. Mallory-Weiss tears account for ~5–10% of cases. The proportion of patients bleeding from varices varies widely from ~5–40%, depending on the population. Hemorrhagic or erosive gastropathy (e.g., due to nonsteroidal anti-inflammatory drugs [NSAIDs] or alcohol) and erosive esophagitis often cause mild UGIB, but major bleeding is rare.
|
SOURCES OF BLEEDING IN PATIENTS HOSPITALIZED FOR UPPER GASTROINTESTINAL BLEEDING |
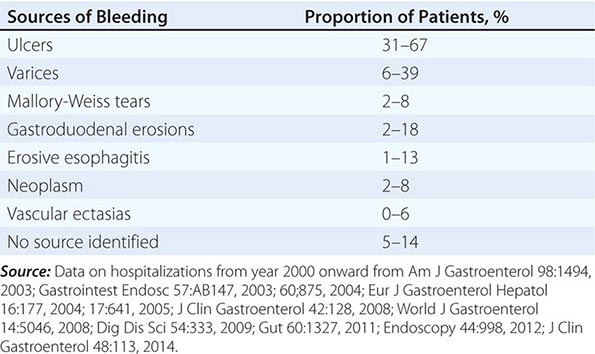
PEPTIC ULCERS Characteristics of an ulcer at endoscopy provide important prognostic information. One-third of patients with active bleeding or a nonbleeding visible vessel have further bleeding that requires urgent surgery if they are treated conservatively. These patients benefit from endoscopic therapy with bipolar electrocoagulation, heater probe, injection therapy (e.g., absolute alcohol, 1:10,000 epinephrine), and/or clips with reductions in bleeding, hospital stay, mortality, and costs. In contrast, patients with clean-based ulcers have rates of recurrent bleeding approaching zero. If stable with no other reason for hospitalization, such patients may be discharged home after endoscopy. Patients without clean-based ulcers usually remain in the hospital for 3 days because most episodes of recurrent bleeding occur within 3 days.
Randomized controlled trials document that high-dose, constant-infusion IV proton pump inhibitor (PPI) (80-mg bolus and 8-mg/h infusion), designed to sustain intragastric pH >6 and enhance clot stability, decreases further bleeding and mortality in patients with high-risk ulcers (active bleeding, nonbleeding visible vessel, adherent clot) when given after endoscopic therapy. Patients with lower-risk findings (flat pigmented spot or clean base) do not require endoscopic therapy and receive standard doses of oral PPI. Approximately one-third of patients with bleeding ulcers will rebleed within the next 1–2 years if no preventive strategies are employed. Prevention of recurrent bleeding focuses on the three main factors in ulcer pathogenesis, Helicobacter pylori, NSAIDs, and acid. Eradication of H. pylori in patients with bleeding ulcers decreases rates of rebleeding to <5%. If a bleeding ulcer develops in a patient taking NSAIDs, the NSAIDs should be discontinued. If NSAIDs must be given, a cyclooxygenase 2 (COX-2) selective inhibitor (coxib) plus a PPI should be used. PPI co-therapy alone or a coxib alone is associated with an annual rebleeding rate of ~10% in patients with a recent bleeding ulcer, whereas the combination of a coxib and PPI provides a further significant decrease in recurrent ulcer bleeding. Patients with established cardiovascular disease who develop bleeding ulcers while taking low-dose aspirin should restart aspirin as soon as possible after their bleeding episode (1–7 days). A randomized trial showed that failure to restart aspirin was associated with no significant difference in rebleeding (5% vs. 10% at 30 days) but a significant increase in mortality at 30 days (9% vs. 1%) and 8 weeks (13% vs. 1%) compared with immediate reinstitution of aspirin. Patients with bleeding ulcers unrelated to H. pylori or NSAIDs should remain on PPI therapy indefinitely. Peptic ulcers are discussed in Chap. 348.
MALLORY-WEISS TEARS The classic history is vomiting, retching, or coughing preceding hematemesis, especially in an alcoholic patient. Bleeding from these tears, which are usually on the gastric side of the gastroesophageal junction, stops spontaneously in 80–90% of patients and recurs in only 0–10%. Endoscopic therapy is indicated for actively bleeding Mallory-Weiss tears. Angiographic therapy with embolization and operative therapy with oversewing of the tear are rarely required. Mallory-Weiss tears are discussed in Chap. 347.
ESOPHAGEAL VARICES Patients with variceal hemorrhage have poorer outcomes than patients with other sources of UGIB. Urgent endoscopy within 12 h is recommended in cirrhotics with UGIB, and if esophageal varices are present, endoscopic ligation is performed and an IV vasoactive medication (e.g., octreotide 50 μg bolus and 50 μg/h infusion) is given for 2–5 days. Combination endoscopic and medical therapy appears to be superior to either therapy alone in decreasing rebleeding. In patients with advanced liver disease (e.g., Child-Pugh class C with score 10–13), a transjugular intrahepatic portosystemic shunt (TIPS) should be strongly considered within the first 1–2 days of hospitalization because randomized trials show significant decreases in rebleeding and mortality compared with standard endoscopic and medical therapy. Over the long term, treatment with nonselective beta blockers plus endoscopic ligation is recommended because the combination of endoscopic and medical therapy is more effective than either alone in reduction of recurrent esophageal variceal bleeding.
In patients who have persistent or recurrent bleeding despite endoscopic and medical therapy, TIPS is recommended. Decompressive surgery (e.g., distal splenorenal shunt) may be considered instead of TIPS in patients with well-compensated cirrhosis.
Portal hypertension is also responsible for bleeding from gastric varices, varices in the small and large intestine, and portal hypertensive gastropathy and enterocolopathy. Bleeding gastric varices due to cirrhosis are treated with endoscopic injection of tissue adhesive (e.g., n-butyl cyanoacrylate), if available; if not, TIPS is performed.
HEMORRHAGIC AND EROSIVE GASTROPATHY (“GASTRITIS”) Hemorrhagic and erosive gastropathy, often labeled gastritis, refers to endoscopically visualized subepithelial hemorrhages and erosions. These are mucosal lesions and do not cause major bleeding due to the absence of arteries and veins in the mucosa. Erosions develop in various clinical settings, the most important of which are NSAID use, alcohol intake, and stress. Half of patients who chronically ingest NSAIDs have erosions, whereas up to 20% of actively drinking alcoholic patients with symptoms of UGIB have evidence of subepithelial hemorrhages or erosions.
Stress-related gastric mucosal injury occurs only in extremely sick patients, such as those who have experienced serious trauma, major surgery, burns covering more than one-third of the body surface area, major intracranial disease, or severe medical illness (i.e., ventilator dependence, coagulopathy). Severe bleeding should not develop unless ulceration occurs. The mortality rate in these patients is quite high because of their serious underlying illnesses.
The incidence of bleeding from stress-related gastric mucosal injury has decreased dramatically in recent years, most likely due to better care of critically ill patients. Pharmacologic prophylaxis for bleeding may be considered in the high-risk patients mentioned above. Meta-analyses of randomized trials indicate that PPIs are more effective than H2 receptor antagonists in reduction of overt and clinically important UGIB without differences in mortality or nosocomial pneumonia.
OTHER CAUSES Other less frequent causes of UGIB include erosive duodenitis, neoplasms, aortoenteric fistulas, vascular lesions (including hereditary hemorrhagic telangiectasias [Osler-Weber-Rendu] and gastric antral vascular ectasia [“watermelon stomach”]), Dieulafoy’s lesion (in which an aberrant vessel in the mucosa bleeds from a pinpoint mucosal defect), prolapse gastropathy (prolapse of proximal stomach into esophagus with retching, especially in alcoholics), and hemobilia or hemosuccus pancreaticus (bleeding from the bile duct or pancreatic duct).
Small-Intestinal Sources of Bleeding Small-intestinal sources of bleeding (bleeding from sites beyond the reach of the standard upper endoscope) are often difficult to diagnose and are responsible for the majority of cases of obscure GIB. Fortunately, small-intestinal bleeding is uncommon. The most common causes in adults are vascular ectasias, tumors (e.g., GI stromal tumor, carcinoid, adenocarcinoma, lymphoma, metastases), and NSAID-induced erosions and ulcers. Other less common causes in adults include Crohn’s disease, infection, ischemia, vasculitis, small-bowel varices, diverticula, Meckel’s diverticulum, duplication cysts, and intussusception.
Meckel’s diverticulum is the most common cause of significant LGIB in children, decreasing in frequency as a cause of bleeding with age. In adults <40–50 years, small-bowel tumors often account for obscure GIB; in patients >50–60 years, vascular ectasias and NSAID-induced lesions are more commonly responsible.
Vascular ectasias should be treated with endoscopic therapy if possible. Although estrogen/progesterone compounds have been used for vascular ectasias, a large double-blind trial found no benefit in prevention of recurrent bleeding. Octreotide is also used, based on case series but no randomized trials. A randomized trial reported significant benefit of thalidomide and awaits further confirmation. Other isolated lesions, such as tumors, are generally treated with surgical resection.
Colonic Sources of Bleeding Hemorrhoids are probably the most common cause of LGIB; anal fissures also cause minor bleeding and pain. If these local anal processes, which rarely require hospitalization, are excluded, the most common causes of LGIB in adults are diverticula, vascular ectasias (especially in the proximal colon of patients >70 years), neoplasms (primarily adenocarcinoma), colitis (ischemic, infectious, idiopathic inflammatory bowel disease), and postpolypectomy bleeding. Less common causes include NSAID-induced ulcers or colitis, radiation proctopathy, solitary rectal ulcer syndrome, trauma, varices (most commonly rectal), lymphoid nodular hyperplasia, vasculitis, and aortocolic fistulas. In children and adolescents, the most common colonic causes of significant GIB are inflammatory bowel disease and juvenile polyps.
Diverticular bleeding is abrupt in onset, usually painless, sometimes massive, and often from the right colon; chronic or occult bleeding is not characteristic. Clinical reports suggest that bleeding colonic diverticula stop bleeding spontaneously in ~80% of patients and, on long-term follow-up, rebleed in ~15–25% of patients. Case series suggest endoscopic therapy may decrease recurrent bleeding in the uncommon case when colonoscopy identifies the specific bleeding diverticulum. When diverticular bleeding is found at angiography, transcatheter arterial embolization by superselective technique stops bleeding in a majority of patients. If bleeding persists or recurs, segmental surgical resection is indicated.
Bleeding from right colonic vascular ectasias in the elderly may be overt or occult; it tends to be chronic and only occasionally is hemodynamically significant. Endoscopic hemostatic therapy may be useful in the treatment of vascular ectasias, as well as discrete bleeding ulcers and postpolypectomy bleeding. Surgical therapy is generally required for major, persistent, or recurrent bleeding from the wide variety of colonic sources of GIB that cannot be treated medically, angiographically, or endoscopically.
APPROACH TO THE PATIENT:
Gastrointestinal Bleeding
INITIAL ASSESSMENT
Measurement of the heart rate and blood pressure is the best way to initially assess a patient with GIB. Clinically significant bleeding leads to postural changes in heart rate or blood pressure, tachycardia, and, finally, recumbent hypotension. In contrast, the hemoglobin does not fall immediately with acute GIB, due to proportionate reductions in plasma and red cell volumes (i.e., “people bleed whole blood”). Thus, hemoglobin may be normal or only minimally decreased at the initial presentation of a severe bleeding episode. As extravascular fluid enters the vascular space to restore volume, the hemoglobin falls, but this process may take up to 72 h. Transfusion is recommended when the hemoglobin drops below 7 g/dL, based on a large randomized trial showing this restrictive transfusion strategy decreases rebleeding and death in acute UGIB compared with a transfusion threshold of 9 g/dL. Patients with slow, chronic GIB may have very low hemoglobin values despite normal blood pressure and heart rate. With the development of iron-deficiency anemia, the mean corpuscular volume will be low and red blood cell distribution width will increase.
DIFFERENTIATION OF UGIB FROM LGIB
Hematemesis indicates an upper GI source of bleeding (above the ligament of Treitz). Melena indicates blood has been present in the GI tract for at least 14 h, and as long as 3–5 days. The more proximal the bleeding site, the more likely melena will occur. Hematochezia usually represents a lower GI source of bleeding, although an upper GI lesion may bleed so briskly that blood transits the bowel before melena develops. When hematochezia is the presenting symptom of UGIB, it is associated with hemodynamic instability and dropping hemoglobin. Bleeding lesions of the small bowel may present as melena or hematochezia. Other clues to UGIB include hyperactive bowel sounds and an elevated blood urea nitrogen (due to volume depletion and blood proteins absorbed in the small intestine).
A nonbloody nasogastric aspirate may be seen in up to ~18% of patients with UGIB, usually from a duodenal source. Even a bile-stained appearance does not exclude a bleeding postpyloric lesion because reports of bile in the aspirate are incorrect in ~50% of cases. Testing of aspirates that are not grossly bloody for occult blood is not useful.
EVALUATION AND MANAGEMENT OF UGIB (FIG. 57-1)
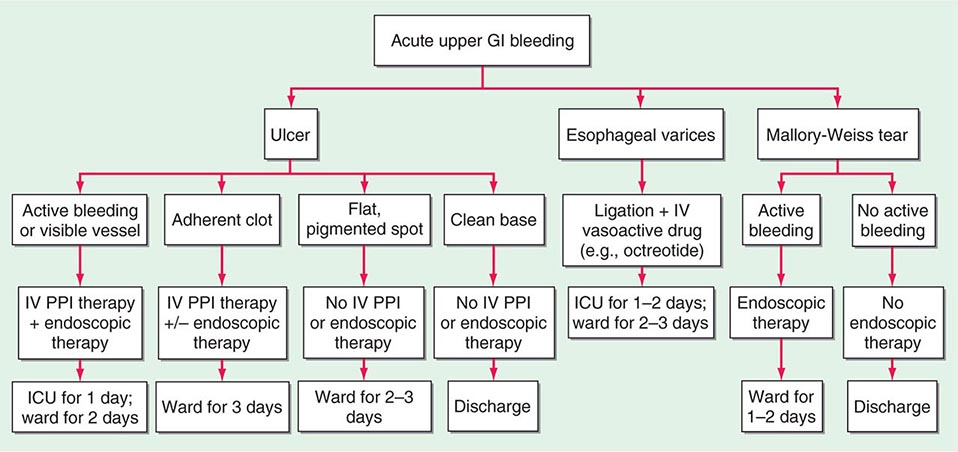
FIGURE 57-1 Suggested algorithm for patients with acute upper gastrointestinal (GI) bleeding. Recommendations on level of care and time of discharge assume patient is stabilized without further bleeding or other concomitant medical problems. ICU, intensive care unit; PPI, proton pump inhibitor.
At presentation, patients are generally stratified as higher or lower risk for further bleeding and death. Baseline characteristics predictive of rebleeding and death include hemodynamic compromise (tachycardia or hypotension), increasing age, and comorbidities. PPI infusion may be considered at presentation: it decreases high-risk ulcer stigmata (e.g., active bleeding) and need for endoscopic therapy but does not improve clinical outcomes such as further bleeding, surgery, or death. Treatment to improve endoscopic visualization with the promotility agent erythromycin, 250 mg intravenously ~30 min before endoscopy, also may be considered: it provides a small but significant increase in diagnostic yield and decrease in second endoscopies but is not documented to decrease further bleeding or death. Cirrhotic patients presenting with UGIB should be placed on antibiotics (e.g., quinolone, ceftriaxone) and started on a vasoactive medication (octreotide, terlipressin, somatostatin, vapreotide) upon presentation, even before endoscopy. Antibiotics decrease bacterial infections, rebleeding, and mortality in this population, and vasoactive medications appear to improve control of bleeding in the first 12 h after presentation.
Upper endoscopy should be performed within 24 h in most patients with UGIB. Patients at higher risk (e.g., hemodynamic instability, cirrhosis) may benefit from more urgent endoscopy within 12 h. Early endoscopy is also beneficial in low-risk patients for management decisions. Patients with major bleeding and high-risk endoscopic findings (e.g., varices, ulcers with active bleeding or a visible vessel) benefit from endoscopic hemostatic therapy, whereas patients with low-risk lesions (e.g., clean-based ulcers, nonbleeding Mallory-Weiss tears, erosive or hemorrhagic gastropathy) who have stable vital signs and hemoglobin and no other medical problems can be discharged home.
EVALUATION AND MANAGEMENT OF LGIB (FIG. 57-2)
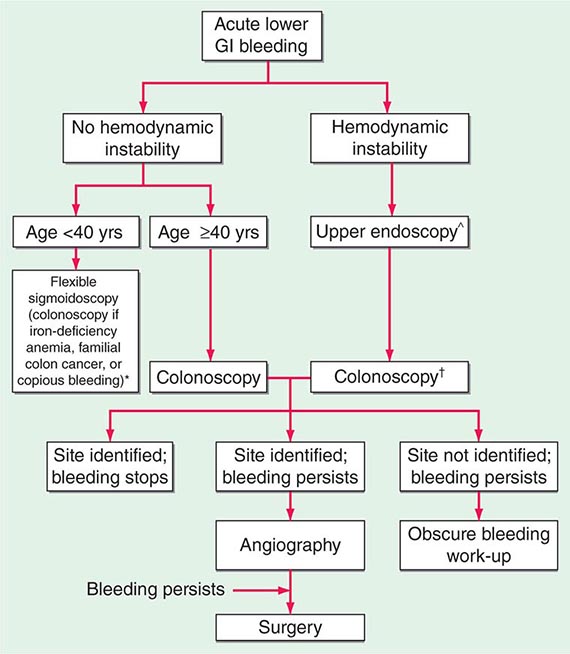
FIGURE 57-2 Suggested algorithm for patients with acute lower gastrointestinal (GI) bleeding. *Some suggest colonoscopy for any degree of rectal bleeding in patients <40 years as well. ^If upper GI endoscopy reveals definite source, no further evaluation is needed. †If massive bleeding does not allow time for colonic lavage, proceed to angiography.
Patients with hematochezia and hemodynamic instability should have upper endoscopy to rule out an upper GI source before evaluation of the lower GI tract.
Colonoscopy after an oral lavage solution is the procedure of choice in most patients admitted with LGIB unless bleeding is too massive, in which case angiography is recommended. Sigmoidoscopy is used primarily in patients <40 years old with minor bleeding. In patients with no source identified on colonoscopy, imaging studies may be employed. 99mTc-labeled red cell scan allows repeated imaging for up to 24 h and may identify the general location of bleeding. However, radionuclide scans should be interpreted with caution because results, especially from later images, are highly variable. Multidector computed tomography (CT) “angiography” is an increasingly used technique that is likely superior to nuclear scintigraphy. In active LGIB, angiography can detect the site of bleeding (extravasation of contrast into the gut) and permits treatment with embolization. Even after bleeding has stopped, angiography may identify lesions with abnormal vasculature, such as vascular ectasias or tumors.
EVALUATION AND MANAGEMENT OF OBSCURE GIB
Obscure GIB is defined as persistent or recurrent bleeding for which no source has been identified by routine endoscopic and contrast x-ray studies; it may be overt (melena, hematochezia) or occult (iron-deficiency anemia). Current guidelines suggest angiography as the initial test for massive obscure bleeding, and video capsule endoscopy, which allows examination of the entire small intestine, for all others. Push enteroscopy, usually performed with a pediatric colonoscope, to inspect the entire duodenum and proximal jejunum also may be considered as an initial evaluation. A systematic review of 14 trials comparing push enteroscopy to capsule revealed “clinically significant findings” in 26% and 56% of patients, respectively. However, in contrast to enteroscopy, lack of control of the capsule prevents its manipulation and full visualization of the intestine; in addition, tissue cannot be sampled and therapy cannot be applied.
If capsule endoscopy is positive, management is dictated by the finding. If capsule endoscopy is negative, current recommendations suggest patients may either be observed or, if their clinical course mandates (e.g., recurrent bleeding, need for transfusions or hospitalization), undergo further testing. “Deep” enteroscopy (e.g., double-balloon, single-balloon, and spiral enteroscopy) is commonly the next test undertaken in patients with clinically important obscure GIB because it allows the endoscopist to examine, obtain specimens from, and provide therapy to much or all of the small intestine. CT and magnetic resonance enterography also are used to examine the small intestine. Other imaging techniques sometimes used in evaluation of obscure GIB include 99mTc-labeled red blood cell scintigraphy, multidetector CT “angiography,” angiography, and 99mTc-pertechnetate scintigraphy for Meckel’s diverticulum (especially in young patients). If all tests are unrevealing, intraoperative endoscopy is indicated in patients with severe recurrent or persistent bleeding requiring repeated transfusions.
POSITIVE FECAL OCCULT BLOOD TEST
Fecal occult blood testing is recommended only for colorectal cancer screening and may be used beginning at age 50 in average-risk adults and beginning at age 40 in adults with a first-degree relative with colorectal neoplasm at ≥60 years or two second-degree relatives with colorectal cancer. A positive test necessitates colonoscopy. If evaluation of the colon is negative, further workup is not recommended unless iron-deficiency anemia or GI symptoms are present.
58 |
Jaundice |
Jaundice, or icterus, is a yellowish discoloration of tissue resulting from the deposition of bilirubin. Tissue deposition of bilirubin occurs only in the presence of serum hyperbilirubinemia and is a sign of either liver disease or, less often, a hemolytic disorder. The degree of serum bilirubin elevation can be estimated by physical examination. Slight increases in serum bilirubin level are best detected by examining the sclerae, which have a particular affinity for bilirubin due to their high elastin content. The presence of scleral icterus indicates a serum bilirubin level of at least 51 μmol/L (3 mg/dL). The ability to detect scleral icterus is made more difficult if the examining room has fluorescent lighting. If the examiner suspects scleral icterus, a second site to examine is underneath the tongue. As serum bilirubin levels rise, the skin will eventually become yellow in light-skinned patients and even green if the process is long-standing; the green color is produced by oxidation of bilirubin to biliverdin.
The differential diagnosis for yellowing of the skin is limited. In addition to jaundice, it includes carotenoderma, the use of the drug quinacrine, and excessive exposure to phenols. Carotenoderma is the yellow color imparted to the skin of healthy individuals who ingest excessive amounts of vegetables and fruits that contain carotene, such as carrots, leafy vegetables, squash, peaches, and oranges. In jaundice the yellow coloration of the skin is uniformly distributed over the body, whereas in carotenoderma the pigment is concentrated on the palms, soles, forehead, and nasolabial folds. Carotenoderma can be distinguished from jaundice by the sparing of the sclerae. Quinacrine causes a yellow discoloration of the skin in 4–37% of patients treated with it.
Another sensitive indicator of increased serum bilirubin is darkening of the urine, which is due to the renal excretion of conjugated bilirubin. Patients often describe their urine as tea- or cola-colored. Bilirubinuria indicates an elevation of the direct serum bilirubin fraction and, therefore, the presence of liver disease.
Serum bilirubin levels increase when an imbalance exists between bilirubin production and clearance. A logical evaluation of the patient who is jaundiced requires an understanding of bilirubin production and metabolism.
PRODUCTION AND METABOLISM OF BILIRUBIN
(See also Chap. 359) Bilirubin, a tetrapyrrole pigment, is a breakdown product of heme (ferroprotoporphyrin IX). About 70–80% of the 250–300 mg of bilirubin produced each day is derived from the breakdown of hemoglobin in senescent red blood cells. The remainder comes from prematurely destroyed erythroid cells in bone marrow and from the turnover of hemoproteins such as myoglobin and cytochromes found in tissues throughout the body.
The formation of bilirubin occurs in reticuloendothelial cells, primarily in the spleen and liver. The first reaction, catalyzed by the microsomal enzyme heme oxygenase, oxidatively cleaves the α bridge of the porphyrin group and opens the heme ring. The end products of this reaction are biliverdin, carbon monoxide, and iron. The second reaction, catalyzed by the cytosolic enzyme biliverdin reductase, reduces the central methylene bridge of biliverdin and converts it to bilirubin. Bilirubin formed in the reticuloendothelial cells is virtually insoluble in water due to tight internal hydrogen bonding between the water-soluble moieties of bilirubin—i.e., the bonding of the proprionic acid carboxyl groups of one dipyrrolic half of the molecule with the imino and lactam groups of the opposite half. This configuration blocks solvent access to the polar residues of bilirubin and places the hydrophobic residues on the outside. To be transported in blood, bilirubin must be solubilized. Solubilization is accomplished by the reversible, noncovalent binding of bilirubin to albumin. Unconjugated bilirubin bound to albumin is transported to the liver. There, the bilirubin—but not the albumin—is taken up by hepatocytes via a process that at least partly involves carrier-mediated membrane transport. No specific bilirubin transporter has yet been identified (Chap. 359, Fig. 359-1).
After entering the hepatocyte, unconjugated bilirubin is bound in the cytosol to a number of proteins including proteins in the glutathione-S-transferase superfamily. These proteins serve both to reduce efflux of bilirubin back into the serum and to present the bilirubin for conjugation. In the endoplasmic reticulum, bilirubin is solubilized by conjugation to glucuronic acid, a process that disrupts the internal hydrogen bonds and yields bilirubin monoglucuronide and diglucuronide. The conjugation of glucuronic acid to bilirubin is catalyzed by bilirubin uridine diphosphate-glucuronosyl transferase (UDPGT). The now-hydrophilic bilirubin conjugates diffuse from the endoplasmic reticulum to the canalicular membrane, where bilirubin monoglucuronide and diglucuronide are actively transported into canalicular bile by an energy-dependent mechanism involving the multidrug resistance–associated protein 2 (MRP2).
The conjugated bilirubin excreted into bile drains into the duodenum and passes unchanged through the proximal small bowel. Conjugated bilirubin is not taken up by the intestinal mucosa. When the conjugated bilirubin reaches the distal ileum and colon, it is hydrolyzed to unconjugated bilirubin by bacterial β-glucuronidases. The unconjugated bilirubin is reduced by normal gut bacteria to form a group of colorless tetrapyrroles called urobilinogens. About 80–90% of these products are excreted in feces, either unchanged or oxidized to orange derivatives called urobilins. The remaining 10–20% of the urobilinogens are passively absorbed, enter the portal venous blood, and are re-excreted by the liver. A small fraction (usually <3 mg/dL) escapes hepatic uptake, filters across the renal glomerulus, and is excreted in urine.
MEASUREMENT OF SERUM BILIRUBIN
The terms direct and indirect bilirubin—i.e., conjugated and unconjugated bilirubin, respectively—are based on the original van den Bergh reaction. This assay, or a variation of it, is still used in most clinical chemistry laboratories to determine the serum bilirubin level. In this assay, bilirubin is exposed to diazotized sulfanilic acid and splits into two relatively stable dipyrrylmethene azopigments that absorb maximally at 540 nm, allowing photometric analysis. The direct fraction is that which reacts with diazotized sulfanilic acid in the absence of an accelerator substance such as alcohol. The direct fraction provides an approximation of the conjugated bilirubin level in serum. The total serum bilirubin is the amount that reacts after the addition of alcohol. The indirect fraction is the difference between the total and the direct bilirubin levels and provides an estimate of the unconjugated bilirubin in serum.
With the van den Bergh method, the normal serum bilirubin concentration usually is 17 μmol/L (<1 mg/dL). Up to 30%, or 5.1 μmol/L (0.3 mg/dL), of the total may be direct-reacting (conjugated) bilirubin. Total serum bilirubin concentrations are between 3.4 and 15.4 μmol/L (0.2 and 0.9 mg/dL) in 95% of a normal population.
Several new techniques, although less convenient to perform, have added considerably to our understanding of bilirubin metabolism. First, studies using these methods demonstrate that, in normal persons or those with Gilbert’s syndrome, almost 100% of the serum bilirubin is unconjugated; <3% is monoconjugated bilirubin. Second, in jaundiced patients with hepatobiliary disease, the total serum bilirubin concentration measured by these new, more accurate methods is lower than the values found with diazo methods. This finding suggests that there are diazo-positive compounds distinct from bilirubin in the serum of patients with hepatobiliary disease. Third, these studies indicate that, in jaundiced patients with hepatobiliary disease, monoglucuronides of bilirubin predominate over diglucuronides. Fourth, part of the direct-reacting bilirubin fraction includes conjugated bilirubin that is covalently linked to albumin. This albumin-linked bilirubin fraction (delta fraction, or biliprotein) represents an important fraction of total serum bilirubin in patients with cholestasis and hepatobiliary disorders. The delta fraction (delta bilirubin) is formed in serum when hepatic excretion of bilirubin glucuronides is impaired and the glucuronides accumulate in serum. By virtue of its tight binding to albumin, the clearance rate of delta bilirubin from serum approximates the half-life of albumin (12–14 days) rather than the short half-life of bilirubin (about 4 h).
The prolonged half-life of albumin-bound conjugated bilirubin accounts for two previously unexplained enigmas in jaundiced patients with liver disease: (1) that some patients with conjugated hyperbilirubinemia do not exhibit bilirubinuria during the recovery phase of their disease because the bilirubin is covalently bound to albumin and therefore not filtered by the renal glomeruli, and (2) that the elevated serum bilirubin level declines more slowly than expected in some patients who otherwise appear to be recovering satisfactorily. Late in the recovery phase of hepatobiliary disorders, all the conjugated bilirubin may be in the albumin-linked form.
MEASUREMENT OF URINE BILIRUBIN
Unconjugated bilirubin is always bound to albumin in the serum, is not filtered by the kidney, and is not found in the urine. Conjugated bilirubin is filtered at the glomerulus, and the majority is reabsorbed by the proximal tubules; a small fraction is excreted in the urine. Any bilirubin found in the urine is conjugated bilirubin. The presence of bilirubinuria implies the presence of liver disease. A urine dipstick test (Ictotest) gives the same information as fractionation of the serum bilirubin and is very accurate. A false-negative result is possible in patients with prolonged cholestasis due to the predominance of delta bilirubin, which is covalently bound to albumin and therefore not filtered by the renal glomeruli.
APPROACH TO THE PATIENT:
Jaundice
The goal of this chapter is not to provide an encyclopedic review of all of the conditions that can cause jaundice. Rather, the chapter is intended to offer a framework that helps a physician to evaluate the patient with jaundice in a logical way (Fig. 58-1).
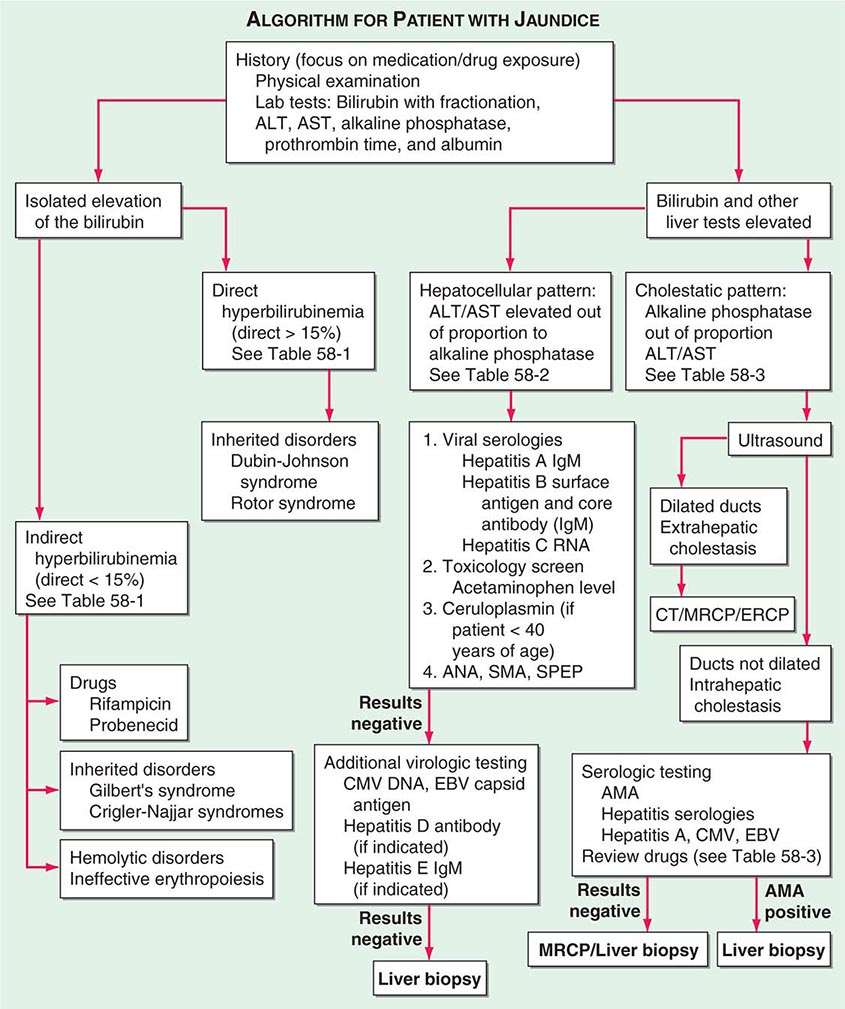
FIGURE 58-1 Evaluation of the patient with jaundice. ALT, alanine aminotransferase; AMA, antimitochondrial antibody; ANA, antinuclear antibody; AST, aspartate aminotransferase; CMV, cytomegalovirus; EBV, Epstein-Barr virus; LKM, liver-kidney microsomal antibody; MRCP, magnetic resonance cholangiopancreatography; SMA, smooth-muscle antibody; SPEP, serum protein electrophoresis.
Simply stated, the initial step is to perform appropriate blood tests in order to determine whether the patient has an isolated elevation of serum bilirubin. If so, is the bilirubin elevation due to an increased unconjugated or conjugated fraction? If the hyperbilirubinemia is accompanied by other liver test abnormalities, is the disorder hepatocellular or cholestatic? If cholestatic, is it intra- or extrahepatic? All of these questions can be answered with a thoughtful history, physical examination, and interpretation of laboratory and radiologic tests and procedures.
The bilirubin present in serum represents a balance between input from the production of bilirubin and hepatic/biliary removal of the pigment. Hyperbilirubinemia may result from (1) overproduction of bilirubin; (2) impaired uptake, conjugation, or excretion of bilirubin; or (3) regurgitation of unconjugated or conjugated bilirubin from damaged hepatocytes or bile ducts. An increase in unconjugated bilirubin in serum results from overproduction, impaired uptake, or conjugation of bilirubin. An increase in conjugated bilirubin is due to decreased excretion into the bile ductules or backward leakage of the pigment. The initial steps in evaluating the patient with jaundice are to determine (1) whether the hyperbilirubinemia is predominantly conjugated or unconjugated in nature and (2) whether other biochemical liver tests are abnormal. The thoughtful interpretation of limited data permits a rational evaluation of the patient (Fig. 58-1). The following discussion will focus solely on the evaluation of the adult patient with jaundice.
ISOLATED ELEVATION OF SERUM BILIRUBIN
Unconjugated Hyperbilirubinemia The differential diagnosis of isolated unconjugated hyperbilirubinemia is limited (Table 58-1). The critical determination is whether the patient is suffering from a hemolytic process resulting in an overproduction of bilirubin (hemolytic disorders and ineffective erythropoiesis) or from impaired hepatic uptake/conjugation of bilirubin (drug effect or genetic disorders).
|
CAUSES OF ISOLATED HYPERBILIRUBINEMIA |
Hemolytic disorders that cause excessive heme production may be either inherited or acquired. Inherited disorders include spherocytosis, sickle cell anemia, thalassemia, and deficiency of red cell enzymes such as pyruvate kinase and glucose-6-phosphate dehydrogenase. In these conditions, the serum bilirubin level rarely exceeds 86 μmol/L (5 mg/dL). Higher levels may occur when there is coexistent renal or hepatocellular dysfunction or in acute hemolysis, such as a sickle cell crisis. In evaluating jaundice in patients with chronic hemolysis, it is important to remember the high incidence of pigmented (calcium bilirubinate) gallstones found in these patients, which increases the likelihood of choledocholithiasis as an alternative explanation for hyperbilirubinemia.
Acquired hemolytic disorders include microangiopathic hemolytic anemia (e.g., hemolytic-uremic syndrome), paroxysmal nocturnal hemoglobinuria, spur cell anemia, immune hemolysis, and parasitic infections (e.g., malaria and babesiosis). Ineffective erythropoiesis occurs in cobalamin, folate, and iron deficiencies. Resorption of hematomas and massive blood transfusions both can result in increased hemoglobin release and overproduction of bilirubin.
In the absence of hemolysis, the physician should consider a problem with the hepatic uptake or conjugation of bilirubin. Certain drugs, including rifampin and probenecid, may cause unconjugated hyperbilirubinemia by diminishing hepatic uptake of bilirubin. Impaired bilirubin conjugation occurs in three genetic conditions: Crigler-Najjar syndrome types I and II and Gilbert’s syndrome. Crigler-Najjar type I is an exceptionally rare condition found in neonates and characterized by severe jaundice (bilirubin >342 μmol/L [>20 mg/dL]) and neurologic impairment due to kernicterus, frequently leading to death in infancy or childhood. These patients have a complete absence of bilirubin UDPGT activity, usually due to mutations in the critical 3′ domain of the UDPGT gene; are totally unable to conjugate bilirubin; and hence cannot excrete it.
Crigler-Najjar type II is somewhat more common than type I. Patients live into adulthood with serum bilirubin levels of 103–428 μmol/L (6–25 mg/dL). In these patients, mutations in the bilirubin UDPGT gene cause the reduction—but not the complete eradication—of the enzyme’s activity. Bilirubin UDPGT activity can be induced by the administration of phenobarbital, which can reduce serum bilirubin levels in these patients. Despite marked jaundice, these patients usually survive into adulthood, although they may be susceptible to kernicterus under the stress of intercurrent illness or surgery.
Gilbert’s syndrome is also marked by the impaired conjugation of bilirubin (to approximately one-third of normal) due to reduced bilirubin UDPGT activity. Patients with Gilbert’s syndrome have mild unconjugated hyperbilirubinemia, with serum levels almost always <103 μmol/L (6 mg/dL). The serum levels may fluctuate, and jaundice is often identified only during periods of fasting. The molecular defect in Gilbert’s syndrome is linked to a reduction in transcription of the bilirubin UDPGT gene due to mutations in the promoter and, rarely, in the coding region. Unlike both Crigler-Najjar syndromes, Gilbert’s syndrome is very common. The reported incidence is 3–7% of the population, with males predominating over females by a ratio of 2–7:1.
Conjugated Hyperbilirubinemia Elevated conjugated hyperbilirubinemia is found in two rare inherited conditions: Dubin-Johnson syndrome and Rotor syndrome (Table 58-1). Patients with either condition present with asymptomatic jaundice. The defect in Dubin-Johnson syndrome is the presence of mutations in the gene for MRP2. These patients have altered excretion of bilirubin into the bile ducts. Rotor syndrome may represent a deficiency of the major hepatic drug uptake transporters OATP1B1 and OATP1B3. Differentiating between these syndromes is possible but is clinically unnecessary due to their benign nature.
ELEVATION OF SERUM BILIRUBIN WITH OTHER LIVER TEST ABNORMALITIES
The remainder of this chapter will focus on the evaluation of patients with conjugated hyperbilirubinemia in the setting of other liver test abnormalities. This group of patients can be divided into those with a primary hepatocellular process and those with intra- or extrahepatic cholestasis. This distinction, which is based on the history and physical examination as well as the pattern of liver test abnormalities, guides the clinician’s evaluation (Fig. 58-1).
History A complete medical history is perhaps the single most important part of the evaluation of the patient with unexplained jaundice. Important considerations include the use of or exposure to any chemical or medication, whether physician-prescribed, over-the-counter, complementary, or alternative medicines (e.g., herbal and vitamin preparations) or other drugs such as anabolic steroids. The patient should be carefully questioned about possible parenteral exposures, including transfusions, intravenous and intranasal drug use, tattooing, and sexual activity. Other important points include recent travel history; exposure to people with jaundice; exposure to possibly contaminated foods; occupational exposure to hepatotoxins; alcohol consumption; the duration of jaundice; and the presence of any accompanying signs and symptoms, such as arthralgias, myalgias, rash, anorexia, weight loss, abdominal pain, fever, pruritus, and changes in the urine and stool. While none of the latter manifestations is specific for any one condition, any of them can suggest a particular diagnosis. A history of arthralgias and myalgias predating jaundice suggests hepatitis, either viral or drug-related. Jaundice associated with the sudden onset of severe right-upper-quadrant pain and shaking chills suggests choledocholithiasis and ascending cholangitis.
Physical Examination The general assessment should include evaluation of the patient’s nutritional status. Temporal and proximal muscle wasting suggests long-standing disease such as pancreatic cancer or cirrhosis. Stigmata of chronic liver disease, including spider nevi, palmar erythema, gynecomastia, caput medusae, Dupuytren’s contractures, parotid gland enlargement, and testicular atrophy, are commonly seen in advanced alcoholic (Laennec’s) cirrhosis and occasionally in other types of cirrhosis. An enlarged left supraclavicular node (Virchow’s node) or a periumbilical nodule (Sister Mary Joseph’s nodule) suggests an abdominal malignancy. Jugular venous distention, a sign of right-sided heart failure, suggests hepatic congestion. Right pleural effusion in the absence of clinically apparent ascites may be seen in advanced cirrhosis.
The abdominal examination should focus on the size and consistency of the liver, on whether the spleen is palpable and hence enlarged, and on whether ascites is present. Patients with cirrhosis may have an enlarged left lobe of the liver, which is felt below the xiphoid, and an enlarged spleen. A grossly enlarged nodular liver or an obvious abdominal mass suggests malignancy. An enlarged tender liver could signify viral or alcoholic hepatitis; an infiltrative process such as amyloidosis; or, less often, an acutely congested liver secondary to right-sided heart failure. Severe right-upper-quadrant tenderness with respiratory arrest on inspiration (Murphy’s sign) suggests cholecystitis. Ascites in the presence of jaundice suggests either cirrhosis or malignancy with peritoneal spread.
Laboratory Tests A battery of tests are helpful in the initial evaluation of a patient with unexplained jaundice. These include total and direct serum bilirubin measurement with fractionation; determination of serum aminotransferase, alkaline phosphatase, and albumin concentrations; and prothrombin time tests. Enzyme tests (alanine aminotransferase [ALT], aspartate aminotransferase [AST], and alkaline phosphatase [ALP]) are helpful in differentiating between a hepatocellular process and a cholestatic process (Table 358-1; Fig. 58-1)—a critical step in determining what additional workup is indicated. Patients with a hepatocellular process generally have a rise in the aminotransferases that is disproportionate to that in ALP, whereas patients with a cholestatic process have a rise in ALP that is disproportionate to that of the aminotransferases. The serum bilirubin can be prominently elevated in both hepatocellular and cholestatic conditions and therefore is not necessarily helpful in differentiating between the two.
In addition to enzyme tests, all jaundiced patients should have additional blood tests—specifically, an albumin level and a prothrombin time—to assess liver function. A low albumin level suggests a chronic process such as cirrhosis or cancer. A normal albumin level is suggestive of a more acute process such as viral hepatitis or choledocholithiasis. An elevated prothrombin time indicates either vitamin K deficiency due to prolonged jaundice and malabsorption of vitamin K or significant hepatocellular dysfunction. The failure of the prothrombin time to correct with parenteral administration of vitamin K indicates severe hepatocellular injury.
The results of the bilirubin, enzyme, albumin, and prothrombin time tests will usually indicate whether a jaundiced patient has a hepatocellular or a cholestatic disease and offer some indication of the duration and severity of the disease. The causes and evaluations of hepatocellular and cholestatic diseases are quite different.
Hepatocellular Conditions Hepatocellular diseases that can cause jaundice include viral hepatitis, drug or environmental toxicity, alcohol, and end-stage cirrhosis from any cause (Table 58-2). Wilson’s disease occurs primarily in young adults. Autoimmune hepatitis is typically seen in young to middle-aged women but may affect men and women of any age. Alcoholic hepatitis can be differentiated from viral and toxin-related hepatitis by the pattern of the aminotransferases: patients with alcoholic hepatitis typically have an AST-to-ALT ratio of at least 2:1, and the AST level rarely exceeds 300 U/L. Patients with acute viral hepatitis and toxin-related injury severe enough to produce jaundice typically have aminotransferase levels >500 U/L, with the ALT greater than or equal to the AST. While ALT and AST values <8 times normal may be seen in either hepatocellular or cholestatic liver disease, values 25 times normal or higher are seen primarily in acute hepatocellular diseases. Patients with jaundice from cirrhosis can have normal or only slightly elevated aminotransferase levels.
|
HEPATOCELLULAR CONDITIONS THAT MAY PRODUCE JAUNDICE |
When the clinician determines that a patient has a hepatocellular disease, appropriate testing for acute viral hepatitis includes a hepatitis A IgM antibody assay, a hepatitis B surface antigen and core IgM antibody assay, a hepatitis C viral RNA test, and, depending on the circumstances, a hepatitis E IgM antibody assay. Because it can take many weeks for hepatitis C antibody to become detectable, its assay is an unreliable test if acute hepatitis C is suspected. Studies for hepatitis D and E viruses, Epstein-Barr virus (EBV), and cytomegalovirus (CMV) may also be indicated. Ceruloplasmin is the initial screening test for Wilson’s disease. Testing for autoimmune hepatitis usually includes an antinuclear antibody assay and measurement of specific immunoglobulins.
Drug-induced hepatocellular injury can be classified as either predictable or unpredictable. Predictable drug reactions are dose-dependent and affect all patients who ingest a toxic dose of the drug in question. The classic example is acetaminophen hepatotoxicity. Unpredictable or idiosyncratic drug reactions are not dose-dependent and occur in a minority of patients. A great number of drugs can cause idiosyncratic hepatic injury. Environmental toxins are also an important cause of hepatocellular injury. Examples include industrial chemicals such as vinyl chloride, herbal preparations containing pyrrolizidine alkaloids (Jamaica bush tea) or Kava Kava, and the mushrooms Amanita phalloides and A. verna, which contain highly hepatotoxic amatoxins.
Cholestatic Conditions When the pattern of the liver tests suggests a cholestatic disorder, the next step is to determine whether it is intra- or extrahepatic cholestasis (Fig. 58-1). Distinguishing intrahepatic from extrahepatic cholestasis may be difficult. History, physical examination, and laboratory tests often are not helpful. The next appropriate test is an ultrasound. The ultrasound is inexpensive, does not expose the patient to ionizing radiation, and can detect dilation of the intra- and extrahepatic biliary tree with a high degree of sensitivity and specificity. The absence of biliary dilation suggests intrahepatic cholestasis, while its presence indicates extrahepatic cholestasis. False-negative results occur in patients with partial obstruction of the common bile duct or in patients with cirrhosis or primary sclerosing cholangitis (PSC), in which scarring prevents the intrahepatic ducts from dilating.
Although ultrasonography may indicate extrahepatic cholestasis, it rarely identifies the site or cause of obstruction. The distal common bile duct is a particularly difficult area to visualize by ultrasound because of overlying bowel gas. Appropriate next tests include CT, magnetic resonance cholangiopancreatography (MRCP), endoscopic retrograde cholangiopancreatography (ERCP), and endoscopic ultrasound (EUS). CT scanning and MRCP are better than ultrasonography for assessing the head of the pancreas and for identifying choledocholithiasis in the distal common bile duct, particularly when the ducts are not dilated. ERCP is the “gold standard” for identifying choledocholithiasis. Beyond its diagnostic capabilities, ERCP allows therapeutic interventions, including the removal of common bile duct stones and the placement of stents. MRCP has replaced ERCP as the initial diagnostic test in cases where the need for intervention is thought to be small. EUS displays sensitivity and specificity comparable to that of MRCP in the detection of bile duct obstruction. EUS also allows biopsy of suspected malignant lesions, but is invasive and requires sedation.
In patients with apparent intrahepatic cholestasis, the diagnosis is often made by serologic testing in combination with percutaneous liver biopsy. The list of possible causes of intrahepatic cholestasis is long and varied (Table 58-3). A number of conditions that typically cause a hepatocellular pattern of injury can also present as a cholestatic variant. Both hepatitis B and C viruses can cause cholestatic hepatitis (fibrosing cholestatic hepatitis). This disease variant has been reported in patients who have undergone solid organ transplantation. Hepatitis A and E, alcoholic hepatitis, and EBV or CMV infections may also present as cholestatic liver disease.
|
CHOLESTATIC CONDITIONS THAT MAY PRODUCE JAUNDICE |
Drugs may cause intrahepatic cholestasis that is usually reversible after discontinuation of the offending agent, although it may take many months for cholestasis to resolve. Drugs most commonly associated with cholestasis are the anabolic and contraceptive steroids. Cholestatic hepatitis has been reported with chlorpromazine, imipramine, tolbutamide, sulindac, cimetidine, and erythromycin estolate. It also occurs in patients taking trimethoprim; sulfamethoxazole; and penicillin-based antibiotics such as ampicillin, dicloxacillin, and clavulanic acid. Rarely, cholestasis may be chronic and associated with progressive fibrosis despite early discontinuation of the offending drug. Chronic cholestasis has been associated with chlorpromazine and prochlorperazine.
Primary biliary cirrhosis is an autoimmune disease predominantly affecting middle-aged women and characterized by progressive destruction of interlobular bile ducts. The diagnosis is made by the detection of antimitochondrial antibody, which is found in 95% of patients. Primary sclerosing cholangitis is characterized by the destruction and fibrosis of larger bile ducts. The diagnosis of PSC is made with cholangiography (either MRCP or ERCP), which demonstrates the pathognomonic segmental strictures. Approximately 75% of patients with PSC have inflammatory bowel disease.
The vanishing bile duct syndrome and adult bile ductopenia are rare conditions in which a decreased number of bile ducts are seen in liver biopsy specimens. The histologic picture is similar to that in primary biliary cirrhosis. This picture is seen in patients who develop chronic rejection after liver transplantation and in those who develop graft-versus-host disease after bone marrow transplantation. Vanishing bile duct syndrome also occurs in rare cases of sarcoidosis, in patients taking certain drugs (including chlorpromazine), and idiopathically.
There are also familial forms of intrahepatic cholestasis. The familial intrahepatic cholestatic syndromes include progressive familial intrahepatic cholestasis (PFIC) types 1–3 and benign recurrent cholestasis (BRC). PFIC1 and BRC are autosomal recessive diseases that result from mutations in the ATP8B1 gene that encodes a protein belonging to the subfamily of P-type ATPases; the exact function of this protein remains poorly defined. While PFIC1 is a progressive condition that manifests in childhood, BRC presents later and is marked by recurrent episodes of jaundice and pruritus; the episodes are self-limited but can be debilitating. PFIC2 is caused by mutations in the ABCB11 gene, which encodes the bile salt export pump, and PFIC3 is caused by mutations in the multidrug-resistant P-glycoprotein 3. Cholestasis of pregnancy occurs in the second and third trimesters and resolves after delivery. Its cause is unknown, but the condition is probably inherited, and cholestasis can be triggered by estrogen administration.
Other causes of intrahepatic cholestasis include total parenteral nutrition (TPN); nonhepatobiliary sepsis; benign postoperative cholestasis; and a paraneoplastic syndrome associated with a number of different malignancies, including Hodgkin’s disease, medullary thyroid cancer, renal cell cancer, renal sarcoma, T cell lymphoma, prostate cancer, and several gastrointestinal malignancies. The term Stauffer’s syndrome has been used for intrahepatic cholestasis specifically associated with renal cell cancer. In patients developing cholestasis in the intensive care unit, the major considerations should be sepsis, ischemic hepatitis (“shock liver”), and TPN jaundice. Jaundice occurring after bone marrow transplantation is most likely due to veno-occlusive disease or graft-versus-host disease. In addition to hemolysis, sickle cell disease may cause intrahepatic and extrahepatic cholestasis. Jaundice is a late finding in heart failure caused by hepatic congestion and hepatocellular hypoxia. Ischemic hepatitis is a distinct entity of acute hypoperfusion characterized by an acute and dramatic elevation in the serum aminotransferases followed by a gradual peak in serum bilirubin.
Jaundice with associated liver dysfunction can be seen in severe cases of Plasmodium falciparum malaria. The jaundice in these cases is due to a combination of indirect hyperbilirubinemia from hemolysis and both cholestatic and hepatocellular jaundice. Weil’s disease, a severe presentation of leptospirosis, is marked by jaundice with renal failure, fever, headache, and muscle pain.
Causes of extrahepatic cholestasis can be split into malignant and benign (Table 58-3). Malignant causes include pancreatic, gallbladder, and ampullary cancers as well as cholangiocarcinoma. This last malignancy is most commonly associated with PSC and is exceptionally difficult to diagnose because its appearance is often identical to that of PSC. Pancreatic and gallbladder tumors as well as cholangiocarcinoma are rarely resectable and have poor prognoses. Ampullary carcinoma has the highest surgical cure rate of all the tumors that present as painless jaundice. Hilar lymphadenopathy due to metastases from other cancers may cause obstruction of the extrahepatic biliary tree.
Choledocholithiasis is the most common cause of extrahepatic cholestasis. The clinical presentation can range from mild right-upper-quadrant discomfort with only minimal elevations of enzyme test values to ascending cholangitis with jaundice, sepsis, and circulatory collapse. PSC may occur with clinically important strictures limited to the extrahepatic biliary tree. IgG4-associated cholangitis is marked by stricturing of the biliary tree. It is critical that the clinician differentiate this condition from PSC as it is responsive to glucocorticoid therapy. In rare instances, chronic pancreatitis causes strictures of the distal common bile duct, where it passes through the head of the pancreas. AIDS cholangiopathy is a condition that is usually due to infection of the bile duct epithelium with CMV or cryptosporidia and has a cholangiographic appearance similar to that of PSC. The affected patients usually present with greatly elevated serum alkaline phosphatase levels (mean, 800 IU/L), but the bilirubin level is often near normal. These patients do not typically present with jaundice.
GLOBAL CONSIDERATIONS
![]() While extrahepatic biliary obstruction and drugs are common causes of new-onset jaundice in developed countries, infections remain the leading cause in developing countries. Liver involvement and jaundice are observed with numerous infections, particularly malaria, babesiosis, severe leptospirosis, infections due to Mycobacterium tuberculosis and the Mycobacterium avium complex, typhoid fever, viral hepatitis secondary to infection with hepatitis viruses A–E, EBV and CMV infections, late phases of yellow fever, dengue hemorrhagic fever, schistosomiasis, fascioliasis, clonorchiasis, opisthorchiasis, ascariasis, echinococcosis, hepatosplenic candidiasis, disseminated histoplasmosis, cryptococcosis, coccidioimycosis, ehrlichiosis, chronic Q fever, yersiniosis, brucellosis, syphilis, and leprosy. Bacterial infections that do not necessarily involve the liver and bile ducts may also lead to jaundice, as in cholestasis of sepsis.
While extrahepatic biliary obstruction and drugs are common causes of new-onset jaundice in developed countries, infections remain the leading cause in developing countries. Liver involvement and jaundice are observed with numerous infections, particularly malaria, babesiosis, severe leptospirosis, infections due to Mycobacterium tuberculosis and the Mycobacterium avium complex, typhoid fever, viral hepatitis secondary to infection with hepatitis viruses A–E, EBV and CMV infections, late phases of yellow fever, dengue hemorrhagic fever, schistosomiasis, fascioliasis, clonorchiasis, opisthorchiasis, ascariasis, echinococcosis, hepatosplenic candidiasis, disseminated histoplasmosis, cryptococcosis, coccidioimycosis, ehrlichiosis, chronic Q fever, yersiniosis, brucellosis, syphilis, and leprosy. Bacterial infections that do not necessarily involve the liver and bile ducts may also lead to jaundice, as in cholestasis of sepsis.
ACKNOWLEDGMENT
This chapter is a revised version of chapters that have appeared in prior editions of Harrison’s in which Marshall M. Kaplan was a co-author together with Daniel Pratt.
59 |
Abdominal Swelling and Ascites |
ABDOMINAL SWELLING
Abdominal swelling is a manifestation of numerous diseases. Patients may complain of bloating or abdominal fullness and may note increasing abdominal girth on the basis of increased clothing or belt size. Abdominal discomfort is often reported, but pain is less frequent. When abdominal pain does accompany swelling, it is frequently the result of an intraabdominal infection, peritonitis, or pancreatitis. Patients with abdominal distention from ascites (fluid in the abdomen) may report the new onset of an inguinal or umbilical hernia. Dyspnea may result from pressure against the diaphragm and the inability to expand the lungs fully.
CAUSES
The causes of abdominal swelling can be remembered conveniently as the six Fs: flatus, fat, fluid, fetus, feces, or a “fatal growth” (often a neoplasm).
Flatus Abdominal swelling may be the result of increased intestinal gas. The normal small intestine contains approximately 200 mL of gas made up of nitrogen, oxygen, carbon dioxide, hydrogen, and methane. Nitrogen and oxygen are consumed (swallowed), whereas carbon dioxide, hydrogen, and methane are produced intraluminally by bacterial fermentation. Increased intestinal gas can occur in a number of conditions. Aerophagia, the swallowing of air, can result in increased amounts of oxygen and nitrogen in the small intestine and lead to abdominal swelling. Aerophagia typically results from gulping food; chewing gum; smoking; or as a response to anxiety, which can lead to repetitive belching. In some cases, increased intestinal gas is the consequence of bacterial metabolism of excess fermentable substances such as lactose and other oligosaccharides, which can lead to production of hydrogen, carbon dioxide, or methane. In many cases, the precise cause of abdominal distention cannot be determined. In some persons, particularly those with irritable bowel syndrome and bloating, the subjective sense of abdominal pressure is attributable to impaired intestinal transit of gas rather than increased gas volume. Abdominal distention—an objective increase in girth—is the result of a lack of coordination between diaphragmatic contraction and anterior abdominal wall relaxation, a response in some cases to an increase in intraabdominal volume loads. Occasionally, increased lumbar lordosis accounts for apparent abdominal distention.
Fat Weight gain with an increase in abdominal fat can result in an increase in abdominal girth and can be perceived as abdominal swelling. Abdominal fat may be caused by an imbalance between caloric intake and energy expenditure associated with a poor diet and sedentary lifestyle; it also can be a manifestation of certain diseases, such as Cushing’s syndrome. Excess abdominal fat has been associated with an increased risk of insulin resistance and cardiovascular disease.
Fluid The accumulation of fluid within the abdominal cavity (ascites) often results in abdominal distention and is discussed in detail below.
Fetus Pregnancy results in increased abdominal girth. Typically, an increase in abdominal size is first noted at 12–14 weeks of gestation, when the uterus moves from the pelvis into the abdomen. Abdominal distention may be seen before this point as a result of fluid retention and relaxation of the abdominal muscles.
Feces In the setting of severe constipation or intestinal obstruction, increased stool in the colon leads to increased abdominal girth. These conditions are often accompanied by abdominal discomfort or pain, nausea, and vomiting and can be diagnosed by imaging studies.
Fatal Growth An abdominal mass can result in abdominal swelling. Enlargement of the intraabdominal organs, specifically the liver (hepatomegaly) or spleen (splenomegaly), or an abdominal aortic aneurysm can result in abdominal distention. Bladder distention also may result in abdominal swelling. In addition, malignancies, abscesses, or cysts can grow to sizes that lead to increased abdominal girth.
IMAGING AND LABORATORY EVALUATION
Abdominal x-rays can be used to detect dilated loops of bowel suggesting intestinal obstruction or ileus. Abdominal ultrasonography can detect as little as 100 mL of ascitic fluid, hepatosplenomegaly, a nodular liver, or a mass. Ultrasonography is often inadequate to detect retroperitoneal lymphadenopathy or a pancreatic lesion because of overlying bowel gas. If malignancy or pancreatic disease is suspected, CT can be performed. CT may also detect changes associated with advanced cirrhosis and portal hypertension (Fig. 59-1).
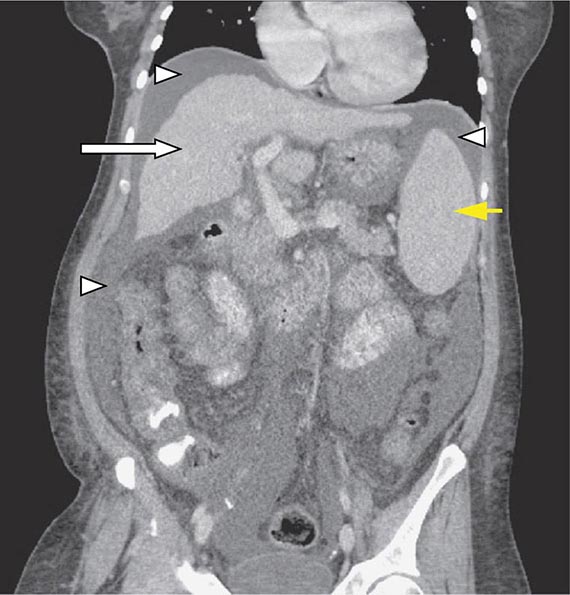
FIGURE 59-1 CT of a patient with a cirrhotic, nodular liver (white arrow), splenomegaly (yellow arrow), and ascites (arrowheads).
Laboratory evaluation should include liver biochemical testing, serum albumin level measurement, and prothrombin time determination (international normalized ratio) to assess hepatic function as well as a complete blood count to evaluate for the presence of cytopenias that may result from portal hypertension or of leukocytosis, anemia, and thrombocytosis that may result from systemic infection. Serum amylase and lipase levels should be checked to evaluate the patient for acute pancreatitis. Urinary protein quantitation is indicated when nephrotic syndrome, which may cause ascites, is suspected.
In selected cases, the hepatic venous pressure gradient (pressure across the liver between the portal and hepatic veins) can be measured via cannulation of the hepatic vein to confirm that ascites is caused by cirrhosis (Chap. 365). In some cases, a liver biopsy may be necessary to confirm cirrhosis.
ASCITES
PATHOGENESIS IN THE PRESENCE OF CIRRHOSIS
Ascites in patients with cirrhosis is the result of portal hypertension and renal salt and water retention. Similar mechanisms contribute to ascites formation in heart failure. Portal hypertension signifies elevation of the pressure within the portal vein. According to Ohm’s law, pressure is the product of resistance and flow. Increased hepatic resistance occurs by several mechanisms. First, the development of hepatic fibrosis, which defines cirrhosis, disrupts the normal architecture of the hepatic sinusoids and impedes normal blood flow through the liver. Second, activation of hepatic stellate cells, which mediate fibrogenesis, leads to smooth-muscle contraction and fibrosis. Finally, cirrhosis is associated with a decrease in endothelial nitric oxide synthetase (eNOS) production, which results in decreased nitric oxide production and increased intrahepatic vasoconstriction.
The development of cirrhosis is also associated with increased systemic circulating levels of nitric oxide (contrary to the decrease seen intrahepatically) as well as increased levels of vascular endothelial growth factor and tumor necrosis factor that result in splanchnic arterial vasodilation. Vasodilation of the splanchnic circulation results in pooling of blood and a decrease in the effective circulating volume, which is perceived by the kidneys as hypovolemia. Compensatory vasoconstriction via release of antidiuretic hormone ensues; the consequences are free water retention and activation of the sympathetic nervous system and the renin angiotensin aldosterone system, which lead in turn to renal sodium and water retention.
PATHOGENESIS IN THE ABSENCE OF CIRRHOSIS
Ascites in the absence of cirrhosis generally results from peritoneal carcinomatosis, peritoneal infection, or pancreatic disease. Peritoneal carcinomatosis can result from primary peritoneal malignancies such as mesothelioma or sarcoma, abdominal malignancies such as gastric or colonic adenocarcinoma, or metastatic disease from breast or lung carcinoma or melanoma (Fig. 59-2). The tumor cells lining the peritoneum produce a protein-rich fluid that contributes to the development of ascites. Fluid from the extracellular space is drawn into the peritoneum, further contributing to the development of ascites. Tuberculous peritonitis causes ascites via a similar mechanism; tubercles deposited on the peritoneum exude a proteinaceous fluid. Pancreatic ascites results from leakage of pancreatic enzymes into the peritoneum.
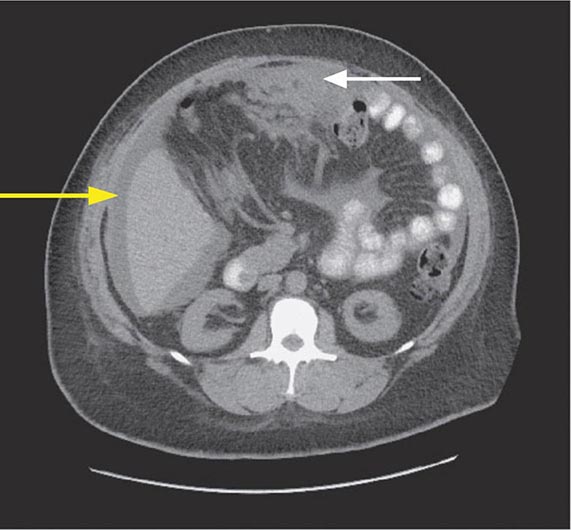
FIGURE 59-2 CT of a patient with peritoneal carcinomatosis (white arrow) and ascites (yellow arrow).
CAUSES
Cirrhosis accounts for 84% of cases of ascites. Cardiac ascites, peritoneal carcinomatosis, and “mixed” ascites resulting from cirrhosis and a second disease account for 10–15% of cases. Less common causes of ascites include massive hepatic metastasis, infection (tuberculosis, Chlamydia infection), pancreatitis, and renal disease (nephrotic syndrome). Rare causes of ascites include hypothyroidism and familial Mediterranean fever.
EVALUATION
Once the presence of ascites has been confirmed, the etiology of the ascites is best determined by paracentesis, a bedside procedure in which a needle or small catheter is passed transcutaneously to extract ascitic fluid from the peritoneum. The lower quadrants are the most frequent sites for paracentesis. The left lower quadrant is preferred because of the greater depth of ascites and the thinner abdominal wall. Paracentesis is a safe procedure even in patients with coagulopathy; complications, including abdominal wall hematomas, hypotension, hepatorenal syndrome, and infection, are infrequent.
Once ascitic fluid has been extracted, its gross appearance should be examined. Turbid fluid can result from the presence of infection or tumor cells. White, milky fluid indicates a triglyceride level >200 mg/dL (and often >1000 mg/dL), which is the hallmark of chylous ascites. Chylous ascites results from lymphatic disruption that may occur with trauma, cirrhosis, tumor, tuberculosis, or certain congenital abnormalities. Dark brown fluid can reflect a high bilirubin concentration and indicates biliary tract perforation. Black fluid may indicate the presence of pancreatic necrosis or metastatic melanoma.
The ascitic fluid should be sent for measurement of albumin and total protein levels, cell and differential counts, and, if infection is suspected, Gram’s stain and culture, with inoculation into blood culture bottles at the patient’s bedside to maximize the yield. A serum albumin level should be measured simultaneously to permit calculation of the serum-ascites albumin gradient (SAAG).
The SAAG is useful for distinguishing ascites caused by portal hypertension from nonportal hypertensive ascites (Fig. 59-3). The SAAG reflects the pressure within the hepatic sinusoids and correlates with the hepatic venous pressure gradient. The SAAG is calculated by subtracting the ascitic albumin concentration from the serum albumin level and does not change with diuresis. A SAAG ≥1.1 g/dL reflects the presence of portal hypertension and indicates that the ascites is due to increased pressure in the hepatic sinusoids. According to Starling’s law, a high SAAG reflects the oncotic pressure that counterbalances the portal pressure. Possible causes include cirrhosis, cardiac ascites, hepatic vein thrombosis (Budd-Chiari syndrome), sinusoidal obstruction syndrome (veno-occlusive disease), or massive liver metastases. A SAAG <1.1 g/dL indicates that the ascites is not related to portal hypertension, as in tuberculous peritonitis, peritoneal carcinomatosis, or pancreatic ascites.
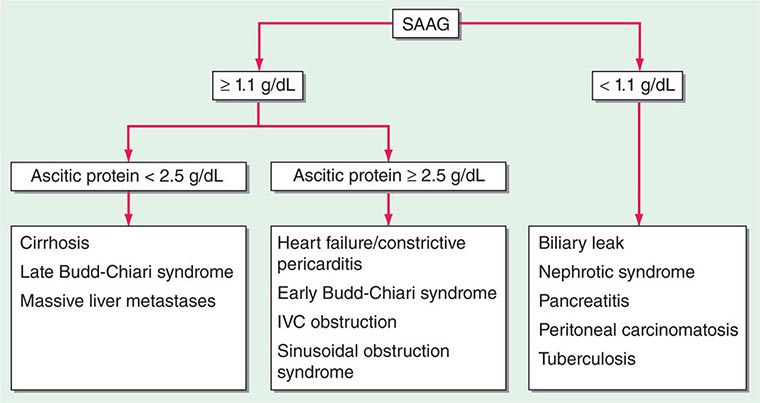
FIGURE 59-3 Algorithm for the diagnosis of ascites according to the serum-ascites albumin gradient (SAAG). IVC, inferior vena cava.
For high-SAAG (≥1.1) ascites, the ascitic protein level can provide further clues to the etiology (Fig. 59-3). An ascitic protein level of ≥2.5 g/dL indicates that the hepatic sinusoids are normal and are allowing passage of protein into the ascites, as occurs in cardiac ascites, early Budd-Chiari syndrome, or sinusoidal obstruction syndrome. An ascitic protein level <2.5 g/dL indicates that the hepatic sinusoids have been damaged and scarred and no longer allow passage of protein, as occurs with cirrhosis, late Budd-Chiari syndrome, or massive liver metastases. Pro-brain-type natriuretic peptide (BNP) is a natriuretic hormone released by the heart as a result of increased volume and ventricular wall stretch. High levels of BNP in serum occur in heart failure and may be useful in identifying heart failure as the cause of high-SAAG ascites.
Further tests are indicated only in specific clinical circumstances. When secondary peritonitis resulting from a perforated hollow viscus is suspected, ascitic glucose and lactate dehydrogenase (LDH) levels can be measured. In contrast to “spontaneous” bacterial peritonitis, which may complicate cirrhotic ascites (see “Complications,” below), secondary peritonitis is suggested by an ascitic glucose level <50 mg/dL, an ascitic LDH level higher than the serum LDH level, and the detection of multiple pathogens on ascitic fluid culture. When pancreatic ascites is suspected, the ascitic amylase level should be measured and is typically >1000 mg/dL. Cytology can be useful in the diagnosis of peritoneal carcinomatosis. At least 50 mL of fluid should be obtained and sent for immediate processing. Tuberculous peritonitis is typically associated with ascitic fluid lymphocytosis but can be difficult to diagnose by paracentesis. A smear for acid-fast bacilli has a diagnostic sensitivity of only 0 to 3%; a culture increases the sensitivity to 35–50%. In patients without cirrhosis, an elevated ascitic adenosine deaminase level has a sensitivity of >90% when a cut-off value of 30–45 U/L is used. When the cause of ascites remains uncertain, laparotomy or laparoscopy with peritoneal biopsies for histology and culture remains the gold standard.
COMPLICATIONS
Spontaneous bacterial peritonitis (SBP; Chap. 159) is a common and potentially lethal complication of cirrhotic ascites. Occasionally, SBP also complicates ascites caused by nephrotic syndrome, heart failure, acute hepatitis, and acute liver failure but is rare in malignant ascites. Patients with SBP generally note an increase in abdominal girth; however, abdominal tenderness is found in only 40% of patients, and rebound tenderness is uncommon. Patients may present with fever, nausea, vomiting, or the new onset of or exacerbation of preexisting hepatic encephalopathy.
SBP is defined by a polymorphonuclear neutrophil (PMN) count of ffi250/ L in the ascitic fluid. Cultures of ascitic fluid typically reveal one bacterial pathogen. The presence of multiple pathogens in the setting of an elevated ascitic PMN count suggests secondary peritonitis from a ruptured viscus or abscess (Chap. 159). The presence of multiple pathogens without an elevated PMN count suggests bowel perforation from the paracentesis needle. SBP is generally the result of enteric bacteria that have translocated across an edematous bowel wall. The most common pathogens are gram-negative rods, including Escherichia coli and Klebsiella, as well as streptococci and enterococci.
Treatment of SBP with an antibiotic such as IV cefotaxime is effective against gram-negative and gram-positive aerobes. A 5-day course of treatment is sufficient if the patient improves clinically. Nosocomial or health care–acquired SBP is frequently caused by multidrug-resistant bacteria, and initial antibiotic therapy should be guided by the local bacterial epidemiology.
Cirrhotic patients with a history of SBP, an ascitic fluid total protein concentration <1 g/dL, or active gastrointestinal bleeding should receive prophylactic antibiotics to prevent SBP; oral daily norfloxacin is commonly used. Diuresis increases the activity of ascitic fluid protein opsonins and may decrease the risk of SBP.
Hepatic hydrothorax occurs when ascites, often caused by cirrhosis, migrates via fenestrae in the diaphragm into the pleural space. This condition can result in shortness of breath, hypoxia, and infection. Treatment is similar to that for cirrhotic ascites and includes sodium restriction, diuretics, and, if needed, thoracentesis or TIPS placement. Chest tube placement should be avoided.
SECTION 7 |
ALTERATIONS IN RENAL AND URINARY TRACT FUNCTION |
60e |
Dysuria, Bladder Pain, and the Interstitial Cystitis/Bladder Pain Syndrome |
Dysuria and bladder pain are two symptoms that commonly call attention to the lower urinary tract.
DYSURIA
Dysuria, or pain that occurs during urination, is commonly perceived as burning or stinging in the urethra and is a symptom of several syndromes. The presence or absence of other symptoms is often helpful in distinguishing among these conditions. Some of these syndromes differ in men and women.
WOMEN
Approximately 50% of women experience dysuria at some time in their lives; ∼20% report having had dysuria within the past year. Most dysuria syndromes in women can be categorized into two broad groups: bacterial cystitis and lower genital tract infections.
Bacterial cystitis is usually caused by Escherichia coli; a few other gram-negative rods and Staphylococcus saprophyticus can also be responsible. Bacterial cystitis is acute in onset and manifests not only as dysuria but also as urinary frequency, urinary urgency, suprapubic pain, and/or hematuria.
The lower genital tract infections include vaginitis, urethritis, and ulcerative lesions; many of these infections are caused by sexually transmitted organisms and should be considered particularly in young women who have new or multiple sexual partners or whose partner(s) do not use condoms. The onset of dysuria associated with these syndromes is more gradual than in bacterial cystitis and is thought (but not proven) to result from the flow of urine over damaged epithelium. Frequency, urgency, suprapubic pain, and hematuria are reported less frequently than in bacterial cystitis. Vaginitis, caused by Candida albicans or Trichomonas vaginalis, presents as vaginal discharge or irritation. Urethritis is a consequence of infection by Chlamydia trachomatis or Neisseria gonorrhoeae. Ulcerative genital lesions may be caused by herpes simplex virus and several other specific organisms.
Among women presenting with dysuria, the probability of bacterial cystitis is ∼50%. This figure rises to >90% if four criteria are fulfilled: dysuria and frequency without vaginal discharge or irritation. Present standards suggest that women meeting these four criteria, if they are otherwise healthy, are not pregnant, and have an apparently normal urinary tract, can be diagnosed with uncomplicated bacterial cystitis and treated empirically with appropriate antibiotics. Other women with dysuria should be further evaluated by urine dipstick, urine culture, and a pelvic examination.
MEN
Dysuria is less common among men. The syndromes presenting as dysuria are similar to those in women but with some important distinctions.
In the majority of men with dysuria, frequency, urgency, and/or suprapubic, penile, and/or perineal pain, the prostate is involved, either as the source of infection or as an obstruction to urine flow. Bacterial prostatitis is usually caused by E. coli or another gram-negative rod, with one of two presentations. Acute bacterial prostatitis presents with fever and chills; prostate examination should be gentle or not performed at all, as massage may result in a wave of bacteremia. Chronic bacterial prostatitis presents as recurrent episodes of bacterial cystitis; prostate examination with massage demonstrates prostatic bacteria and leukocytes. Benign prostatic hyperplasia (BPH) can obstruct urine flow, with consequent symptoms of weak stream, hesitancy, and dribbling. If a bacterial infection develops behind the obstructing prostate, dysuria and other symptoms of cystitis will occur. Men whose symptoms are consistent with bacterial cystitis should be evaluated with urinalysis and urine culture.
Several sexually transmitted infections can manifest as dysuria. Urethritis (usually without urinary frequency) presents as a urethral discharge and can be caused by C. trachomatis, N. gonorrhoeae, Mycoplasma genitalium, Ureaplasma urealyticum, or T. vaginalis. Herpes simplex, chancroid, and other ulcerous lesions may present as dysuria, again without urinary frequency.
For further discussion, see Chaps. 162 and 163.
EITHER WOMEN OR MEN
Other causes of dysuria may be found in patients of either sex. Some cases are acute and include lower urinary tract stones, trauma, and urethral exposure to topical chemicals. Others may be relatively chronic and attributable to lower urinary tract cancers, certain medications, Behçet’s syndrome, reactive arthritis, a poorly understood entity known as chronic urethral syndrome, and interstitial cystitis/bladder pain syndrome (see below).
BLADDER PAIN
Studies indicate that patients perceive pain as coming from the urinary bladder if it is suprapubic in location, alters with bladder filling or emptying, and/or is associated with urinary symptoms such as urgency and frequency. Bladder pain occurring acutely (i.e., over hours or a day or two) is helpful in distinguishing bacterial cystitis from urethritis, vaginitis, and other genital infections. Chronic or recurrent bladder pain may accompany lower urinary tract stones; bladder, uterine, cervical, vaginal, urethral, or prostate cancer; urethral diverticulum; cystitis induced by radiation or certain medications; tuberculous cystitis; bladder neck obstruction; neurogenic bladder; urogenital prolapse; or BPH. In the absence of these conditions, the diagnosis of interstitial cystitis/bladder pain syndrome (IC/BPS) should be considered.
INTERSTITIAL CYSTITIS/BLADDER PAIN SYNDROME
Most clinicians with outpatient practices see undiagnosed cases of IC/BPS. This chronic condition is characterized by pain perceived to be from the urinary bladder, urinary urgency and frequency, and nocturia. The majority of cases are diagnosed in women. Symptoms wax and wane for months or years or possibly even for the rest of the patient’s life. The spectrum of symptom intensity is broad. The pain can be excruciating, urgency can be distressing, frequency can be up to 60 times per 24 h, and nocturia can cause sleep deprivation. These symptoms can disrupt daily activities, work schedules, and personal relationships; patients with IC/BPS report less life satisfaction than do those with end-stage renal disease.
IC/BPS is not a new disease, having first been described in the late nineteenth century in a patient with the symptoms mentioned above and a single ulcer visible on cystoscopy (now called a Hunner’s lesion after the urologist who first reported it). Over the ensuing decades, it became clear that many patients with similar symptoms had no ulcer. It is now appreciated that only up to 10% of patients with IC/BPS have a Hunner’s lesion. The definition of IC/BPS, its diagnostic features, and even its name continue to evolve. The American Urological Association has defined IC/BPS as “an unpleasant sensation (pain, pressure, discomfort) perceived to be related to the urinary bladder, associated with lower urinary tract symptoms of more than six weeks’ duration, in the absence of infection or other identifiable causes.”
Many patients with IC/BPS also have other syndromes, such as fibromyalgia, chronic fatigue syndrome, irritable bowel syndrome, and migraine. These syndromes collectively are known as functional somatic syndromes (FSSs): chronic conditions in which pain and fatigue are prominent features but laboratory tests and histologic findings are normal. Like IC/BPS, the FSSs often are associated with depression and anxiety. The majority of FSSs affect more women than men, and more than one FSS can affect a single patient. Because of its similar features and comorbidity, IC/BPS sometimes is considered an FSS.
EPIDEMIOLOGY
Contemporary population studies of IC/BPS in the United States indicate a prevalence of 3–6% among women and 2–4% among men. For decades, it was thought that IC/BPS occurred mostly in women. These prevalence findings, however, have generated research aimed at determining the proportion of men who have symptoms usually diagnosed as chronic prostatitis (now known as chronic prostatitis/chronic pelvic pain syndrome) but who actually have IC/BPS.
Among women, the average age at onset of IC/BPS symptoms is the early forties, but the range is from childhood through the early sixties. Risk factors (antecedent features that distinguish cases from controls) primarily have been FSSs. Indeed, the odds of IC/BPS increase with the number of such syndromes present. Surgery was long thought to be a risk factor for IC/BPS, but analyses adjusting for FSSs refuted that association. About one-third of patients appear to have bacterial cystitis at the onset of IC/BPS.
The natural history of IC/BPS is not known. Although studies from urology and urogynecology practices have been interpreted as showing that IC/BPS lasts for the lifetime of the patient, population studies suggest that some individuals with IC/BPS do not consult specialists and may not seek medical care at all, and most prevalence studies do not show an upward trend with age—a pattern that would be expected with incident cases throughout adulthood followed by lifetime persistence of a nonfatal disease. It may be reasonable to conclude that patients in a urology practice represent those with the most severe and recalcitrant IC/BPS.
PATHOLOGY
For the ≤10% of IC/BPS patients who have a Hunner’s lesion, the term interstitial cystitis may indeed describe the histopathologic picture. Most of these patients have substantive inflammation, mast cells, and granulation tissue. However, in the 90% of patients without such lesions, the bladder mucosa and interstitium are relatively normal, with scant inflammation.
ETIOLOGY
Numerous hypotheses about the pathogenesis of IC/BPS have been put forward. It is not surprising that most early theories focused on the bladder. For instance, IC/BPS has been investigated as a chronic bladder infection. Sophisticated technologies have not identified a causative organism in urine or in bladder tissue; however, the patients studied by these methods had IC/BPS of long duration, and the results do not preclude the possibility that infection may trigger the syndrome or may be a feature of early IC/BPS. Other inflammatory factors, including a role for mast cells, have been postulated, but (as noted above) the 90% of patients without a Hunner’s ulcer have little bladder inflammation and do not have a prominence of mast cells in bladder tissue. Autoimmunity has been considered, but autoantibodies are low in titer, nonspecific, and thought to be a result rather than a cause of IC/BPS. Increased permeability of the bladder mucosa due to defective epithelium or glycosaminoglycan (the bladder’s mucous coating) has been studied frequently, but the findings have been inconclusive.
Investigations of causes outside the bladder have been prompted by the presence of comorbid FSSs. Many patients with FSSs have abnormal pain sensitivity as evidenced by (1) low pain thresholds in body areas unrelated to the diagnosed syndrome, (2) dysfunctional descending neurologic control of tactile signals, and (3) enhanced brain responses to touch in functional neuroimaging studies. Moreover, in patients with IC/BPS, body surfaces remote from the bladder are more sensitive to pain than is the case in individuals without IC/BPS. All these findings are consistent with upregulation of sensory processing in the brain. Indeed, a prevailing theory is that these concomitantly occurring syndromes have in common an abnormality of brain processing of sensory input. However, antecedence is a critical criterion for causality, and no study has demonstrated that abnormal pain sensitivity precedes either IC/BPS or the FSSs.
CLINICAL PRESENTATION
In some patients, IC/BPS has a gradual onset, and/or the cardinal symptoms of pain, urgency, frequency, and nocturia appear sequentially in no consistent order. Other patients can identify the exact date of onset of IC/BPS symptoms. More than half of the latter patients describe dysuria beginning on that date. As stated, only a minority of IC/BPS patients who obtain medical care soon after symptom onset have uropathogenic bacteria or leukocytes in the urine. These patients—and many others with new-onset IC/BPS—are treated with antibiotics for presumptive bacterial cystitis or, if male, chronic bacterial prostatitis. Persistent or recurring symptoms without bacteriuria eventually prompt a differential diagnosis, and IC/BPS is considered. Traditionally, the diagnosis of IC/BPS has been delayed for years, but recent interest in the disease has shortened this interval.
The pain of IC/BPS includes suprapubic prominence and changes with the voiding cycle. Two-thirds of women with IC/BPS report two or more sites of pain. The most common site (involved in 80% of women) and generally the one with the most severe pain is the suprapubic area. About 35% of female patients have pain in the urethra, 25% in other parts of the vulva, and 30% in nonurogenital areas, mostly the low back and also the anterior or posterior thighs or the buttocks. The pain of IC/BPS is most commonly described as aching, pressing, throbbing, tender, and/or piercing. What may distinguish IC/BPS from other pelvic pain is that, in 95% of patients, bladder filling exacerbates the pain and/or bladder emptying relieves it. Almost as many patients report a puzzling pattern in which certain dietary substances worsen the pain of IC/BPS. Smaller proportions—but still the majority—of patients report that their IC/BPS pain is worsened by menstruation, stress, tight clothing, exercise, and riding in a car as well as during or after vaginal intercourse.
The urethral and vulvar pains of IC/BPS merit special mention. In addition to the descriptive adjectives for IC/BPS mentioned above, these pains commonly are described as burning, stinging, and sharp and as being worsened by touch, tampons, and vaginal intercourse. Patients report that urethral pain increases during urination and generally lessens afterward. These characteristics have commonly resulted in diagnosis of the urethral pain of IC/BPS as chronic urethral syndrome and the vulvar pain as vulvodynia.
In many patients with IC/BPS, there is a link between pain and urinary urgency; that is, two-thirds of patients describe the urge to urinate as a desire to relieve their bladder pain. Only 20% report that the urge stems from a desire to prevent incontinence; indeed, very few patients with IC/BPS are incontinent. As mentioned above, urinary frequency can be severe, with ∼85% of patients voiding more than 10 times per 24 h and some as often as 60 times. Voiding continues through the night, and nocturia is common, frequent, and often associated with sleep deprivation.
Beyond these common symptoms of IC/BPS, additional urinary and other symptoms may be present. Among the urinary symptoms are difficulty in starting urine flow, perceptions of difficulty in emptying the bladder, and bladder spasms. Other symptoms include the manifestations of comorbid FSSs as well as symptoms that do not constitute recognized syndromes, such as numbness, muscle spasms, dizziness, ringing in the ears, and blurred vision.
The pain, urgency, and frequency of IC/BPS can be debilitating. Proximity to a bathroom is a continual focus, and patients report difficulties in the workplace, leisure activities, travel, and simply leaving home. Familial and sexual relationships can be strained.
DIAGNOSIS
Traditionally, IC/BPS has been considered a rare condition that is diagnosed by urologists at cystoscopy. However, this disorder is much more common than once was thought; it is now being considered earlier in its course and is being diagnosed and managed more often by primary care clinicians. Results of physical examination, urinalysis, and urologic procedures are insensitive and/or nonspecific. Thus, diagnosis is based on the presence of appropriate symptoms and the exclusion of diseases with a similar presentation.
Three categories of disorders can be considered in the differential diagnosis of IC/BPS. The first comprises diseases that manifest as bladder pain (see above) or urinary symptoms. Among the latter diseases is overactive bladder, a chronic condition of women and men that presents as urgency and frequency and that can be distinguished from IC/BPS by the patient’s history: pain is not a feature of overactive bladder, and its urgency arises from the need to avoid incontinence. Endometriosis is a special case: it can be asymptomatic or can cause pelvic pain, dysmenorrhea, and dyspareunia—i.e., types of pain that mimic IC/BPS. Endometrial implants on the bladder (although uncommon) can cause urinary symptoms, and the resulting syndrome can mimic IC/BPS. Even if endometriosis is identified, it is difficult in the absence of bladder implants to determine whether it is causative of or incidental to the symptoms of IC/BPS in a specific woman.
The second category of disorders encompasses the FSSs that can accompany IC/BPS. IC/BPS can be misdiagnosed as gynecologic chronic pelvic pain, irritable bowel syndrome, or fibromyalgia. The correct diagnosis may be entertained only when either changes of pain with altered bladder volume or urinary symptoms become more prominent.
The third category involves syndromes that IC/BPS mimics by way of its referred pain, such as vulvodynia and chronic urethral syndrome. Therefore, IC/BPS should be considered in the differential diagnosis of persistent or recurrent “urinary tract infection” (UTI) with sterile urine cultures; overactive bladder with pain; chronic pelvic pain, endometriosis, vulvodynia, or FSSs with urinary symptoms; and “chronic prostatitis.” As mentioned above, important clues to the diagnosis of IC/BPS are pain that changes with bladder volume or with certain foods or drinks. Common among these are chilies, chocolate, citrus fruits, tomatoes, alcohol, caffeinated drinks, and carbonated beverages; full lists of common trigger foods are available at the websites cited in the treatment section below.
Cystoscopy under anesthesia formerly was thought to be necessary for the diagnosis of IC/BPS because of its capacity to reveal a Hunner’s lesion or—in the 90% of patients without an ulcer—petechial hemorrhages after bladder distention. However, because Hunner’s lesions are uncommon in IC/BPS and petechiae are nonspecific, cystoscopy is no longer necessary for diagnosis. Accordingly, the indications for urologic referral have evolved toward the need to rule out other diseases or to administer more advanced treatment.
A typical patient presents to the primary clinician after days, weeks, or months of pain, urgency, frequency, and/or nocturia. The presence of urinary nitrites, leukocytes, or uropathogenic bacteria should prompt treatment for UTI in women and chronic bacterial prostatitis in men. Persistence or recurrence of symptoms in the absence of bacteriuria should prompt a pelvic examination for women, an assay for serum prostate-specific antigen for men, and urine cytology and inclusion of IC/BPS in the differential diagnosis for both sexes.
In the diagnosis of IC/BPS, inquiries about pain, pressure, and discomfort are useful; IC/BPS should be considered if any of these sensations are noted in one or more anterior or posterior sites between the umbilicus and the upper thighs. Nondirective questions about the effect of bladder volume changes include “As your next urination approaches, does this pain get better, get worse, or stay the same?” and “After you urinate, does this pain get better, get worse, or stay the same?” Establishing that the pain is exacerbated by the consumption of certain foods and drinks not only supports the diagnosis of IC/BPS but also serves as the basis for one of the first steps in managing this syndrome. A nondirective way to ask about urgency is to describe it to the patient as a compelling urge to urinate that is difficult to postpone; follow-up questions can determine whether this urge is intended to relieve pain or prevent incontinence. To assess severity and provide quantitative baseline measures, pain and urgency should be estimated by the patient on a scale of 0–10, with 0 being none and 10 the worst imaginable. Frequency per 24-h period should be determined and nocturia assessed as the number of times per night the patient is awakened by the need to urinate.
About half of patients with IC/BPS have intermittent or persistent microscopic hematuria; this manifestation and the need to exclude bladder stones or cancer require urologic or urogynecologic referral. Initiation of therapy for IC/BPS does not hamper subsequent urologic evaluation.
61 |
Azotemia and Urinary Abnormalities |
Normal kidney functions occur through numerous cellular processes to maintain body homeostasis. Disturbances in any of these functions can lead to abnormalities that may be detrimental to survival. Clinical manifestations of these disorders depend on the pathophysiology of renal injury and often are identified as a complex of symptoms, abnormal physical findings, and laboratory changes that constitute specific syndromes. These renal syndromes (Table 61-1) may arise from systemic illness or as primary renal disease. Nephrologic syndromes usually consist of several elements that reflect the underlying pathologic processes, typically including one or more of the following: (1) reduction in glomerular filtration rate (GFR) (azotemia), (2) abnormalities of urine sediment (red blood cells [RBCs], white blood cells [WBCs], casts, and crystals), (3) abnormal excretion of serum proteins (proteinuria), (4) disturbances in urine volume (oliguria, anuria, polyuria), (5) presence of hypertension and/or expanded total body fluid volume (edema), (6) electrolyte abnormalities, and (7) in some syndromes, fever/pain. The specific combination of these findings should permit identification of one of the major nephrologic syndromes (Table 61-1) and allow differential diagnoses to be narrowed so that the appropriate diagnostic and therapeutic course can be determined. All these syndromes and their associated diseases are discussed in more detail in subsequent chapters. This chapter focuses on several aspects of renal abnormalities that are critically important for distinguishing among those processes: (1) reduction in GFR leading to azotemia, (2) alterations of the urinary sediment and/or protein excretion, and (3) abnormalities of urinary volume.
|
INITIAL CLINICAL AND LABORATORY DATA BASE FOR DEFINING MAJOR SYNDROMES IN NEPHROLOGY |
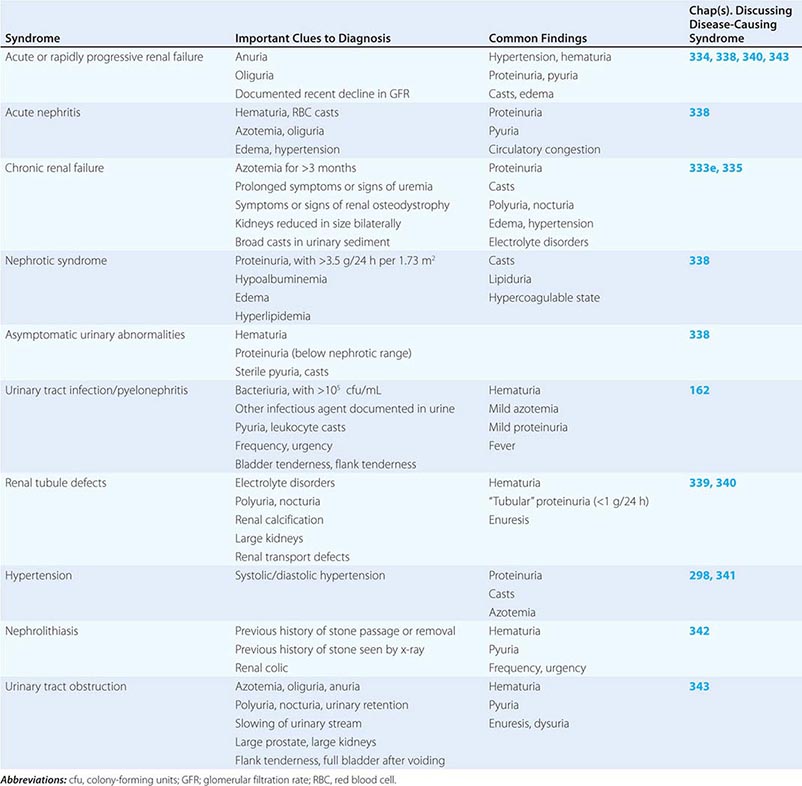
AZOTEMIA
ASSESSMENT OF GFR
Monitoring the GFR is important in both hospital and outpatient settings, and several different methodologies are available. GFR is the primary metric for kidney “function,” and its direct measurement involves administration of a radioactive isotope (such as inulin or iothalamate) that is filtered at the glomerulus into the urinary space but is neither reabsorbed nor secreted throughout the tubule. GFR—i.e., the clearance of inulin or iothalamate in milliliters per minute—is calculated from the rate of appearance of the isotope in the urine over several hours. In most clinical circumstances, direct GFR measurement is not feasible, and the plasma creatinine level is used as a surrogate to estimate GFR. Plasma creatinine (PCr) is the most widely used marker for GFR, which is related directly to urine creatinine (UCr) excretion and inversely to PCr. On the basis of this relationship (with some important caveats, as discussed below), GFR will fall in roughly inverse proportion to the rise in PCr. Failure to account for GFR reductions in drug dosing can lead to significant morbidity and death from drug toxicities (e.g., digoxin, aminoglycosides). In the outpatient setting, PCr serves as an estimate for GFR (although much less accurate; see below). In patients with chronic progressive renal disease, there is an approximately linear relationship between 1/PCr (y axis) and time (x axis). The slope of that line will remain constant for an individual; when values deviate, an investigation for a superimposed acute process (e.g., volume depletion, drug reaction) should be initiated. Signs and symptoms of uremia develop at significantly different levels of PCr, depending on the patient (size, age, and sex), underlying renal disease, existence of concurrent diseases, and true GFR. Generally, patients do not develop symptomatic uremia until renal insufficiency is severe (GFR <15 mL/min).
A significantly reduced GFR (either acute or chronic) is usually reflected in a rise in PCr, leading to retention of nitrogenous waste products (defined as azotemia) such as urea. Azotemia may result from reduced renal perfusion, intrinsic renal disease, or postrenal processes (ureteral obstruction; see below and Fig. 61-1). Precise determination of GFR is problematic, as both commonly measured indices (urea and creatinine) have characteristics that affect their accuracy as markers of clearance. Urea clearance may underestimate GFR significantly because of urea reabsorption by the tubule. In contrast, creatinine is derived from muscle metabolism of creatine, and its generation varies little from day to day.
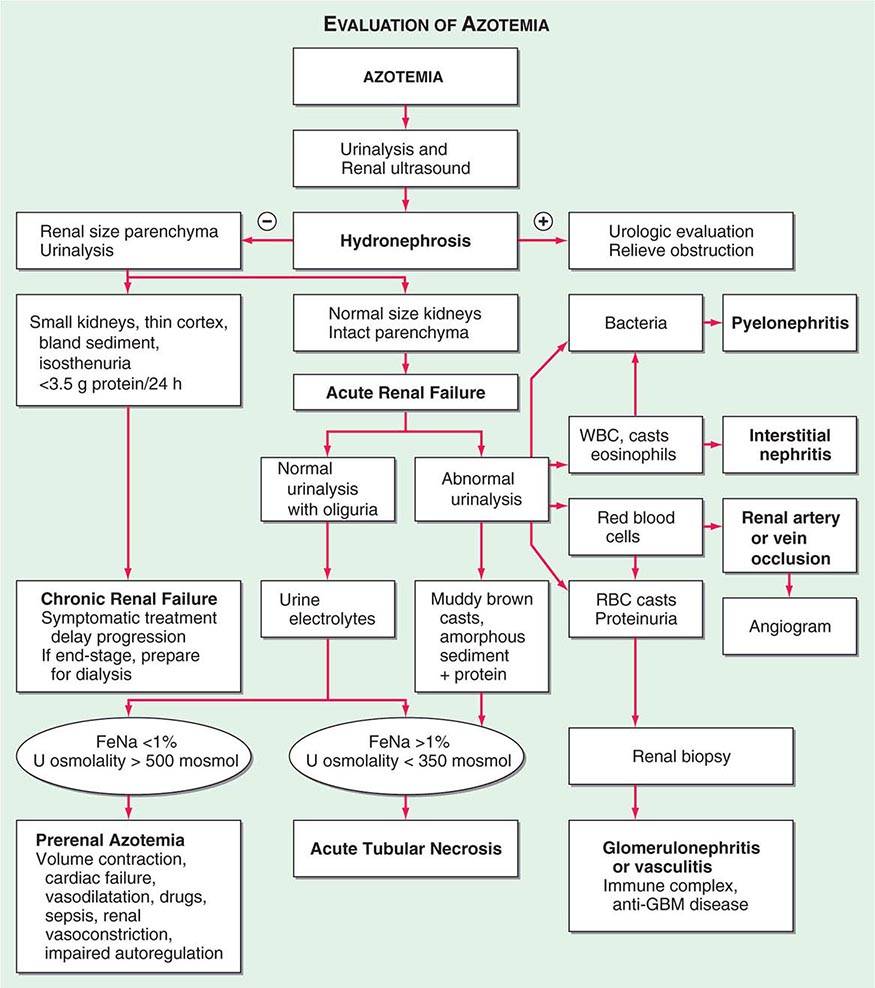
FIGURE 61-1 Approach to the patient with azotemia. FeNa, fractional excretion of sodium; GBM, glomerular basement membrane; RBC, red blood cell; WBC, white blood cell.
Creatinine clearance (CrCl), an approximation of GFR, is measured from plasma and urinary creatinine excretion rates for a defined period (usually 24 h) and is expressed in milliliters per minute: CrCl = (Uvol × UCr)/(PCr × Tmin). Creatinine is useful for estimating GFR because it is a small, freely filtered solute that is not reabsorbed by the tubules. PCr levels can increase acutely from dietary ingestion of cooked meat, however, and creatinine can be secreted into the proximal tubule through an organic cation pathway (especially in advanced progressive chronic kidney disease), leading to overestimation of GFR. When a timed collection for CrCl is not available, decisions about drug dosing must be based on PCr alone. Two formulas are used widely to estimate kidney function from PCr: (1) Cockcroft-Gault and (2) four-variable MDRD (Modification of Diet in Renal Disease).
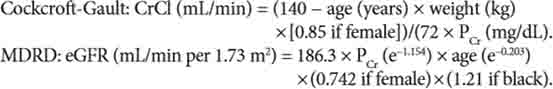
Numerous websites are available to assist with these calculations (www.kidney.org/professionals/kdoqi/gfr_calculator.cfm). A newer CKD-EPI eGFR, which was developed by pooling several cohorts with and without kidney disease who had data on directly measured GFR, appears to be more accurate:
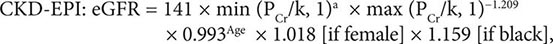
where PCr is plasma creatinine, k is 0.7 for females and 0.9 for males, a is –0.329 for females and –0.411 for males, min indicates the minimum of PCr/k or 1, and max indicates the maximum of PCr/k or 1 (http://www.qxmd.com/renal/Calculate-CKD-EPI-GFR.php).
There are limitations to all creatinine-based estimates of GFR. Each equation, along with 24-h urine collection for measurement of creatinine clearance, is based on the assumption that the patient is in steady state, without daily increases or decreases in PCr as a result of rapidly changing GFR. The MDRD equation is better correlated with true GFR when the GFR is <60 mL/min per 1.73 m2. The gradual loss of muscle from chronic illness, chronic use of glucocorticoids, or malnutrition can mask significant changes in GFR with small or imperceptible changes in PCr. Cystatin C, a member of the cystatin superfamily of cysteine protease inhibitors, is produced at a relatively constant rate from all nucleated cells. Serum cystatin C has been proposed to be a more sensitive marker of early GFR decline than is PCr; however, like serum creatinine, cystatin C is influenced by the patient’s age, race, and sex and also is associated with diabetes, smoking, and markers of inflammation.
APPROACH TO THE PATIENT:
Azotemia
Once GFR reduction has been established, the physician must decide if it represents acute or chronic renal injury. The clinical situation, history, and laboratory data often make this an easy distinction. However, the laboratory abnormalities characteristic of chronic renal failure, including anemia, hypocalcemia, and hyperphosphatemia, often are present as well in patients presenting with acute renal failure. Radiographic evidence of renal osteodystrophy (Chap. 335) can be seen only in chronic renal failure but is a very late finding, and these patients are usually undergoing dialysis. The urinalysis and renal ultrasound can facilitate distinguishing acute from chronic renal failure. An approach to the evaluation of azotemic patients is shown in Fig. 61-1. Patients with advanced chronic renal insufficiency often have some proteinuria, nonconcentrated urine (isosthenuria; isosmotic with plasma), and small kidneys on ultrasound, characterized by increased echogenicity and cortical thinning. Treatment should be directed toward slowing the progression of renal disease and providing symptomatic relief for edema, acidosis, anemia, and hyperphosphatemia, as discussed in Chap. 335. Acute renal failure (Chap. 334) can result from processes that affect renal blood flow (prerenal azotemia), intrinsic renal diseases (affecting small vessels, glomeruli, or tubules), or postrenal processes (obstruction of urine flow in ureters, bladder, or urethra) (Chap. 343).
PRERENAL FAILURE
Decreased renal perfusion accounts for 40–80% of cases of acute renal failure and, if appropriately treated, is readily reversible. The etiologies of prerenal azotemia include any cause of decreased circulating blood volume (gastrointestinal hemorrhage, burns, diarrhea, diuretics), volume sequestration (pancreatitis, peritonitis, rhabdomyolysis), or decreased effective arterial volume (cardiogenic shock, sepsis). Renal perfusion also can be affected by reductions in cardiac output from peripheral vasodilation (sepsis, drugs) or profound renal vasoconstriction (severe heart failure, hepatorenal syndrome, agents such as nonsteroidal anti-inflammatory drugs [NSAIDs]). True or “effective” arterial hypovolemia leads to a fall in mean arterial pressure, which in turn triggers a series of neural and humoral responses, including activation of the sympathetic nervous and renin-angiotensin-aldosterone systems and antidiuretic hormone (ADH) release. GFR is maintained by prostaglandin-mediated relaxation of afferent arterioles and angiotensin II–mediated constriction of efferent arterioles. Once the mean arterial pressure falls below 80 mmHg, GFR declines steeply.
Blockade of prostaglandin production by NSAIDs can result in severe vasoconstriction and acute renal failure. Blocking angiotensin action with angiotensin-converting enzyme (ACE) inhibitors or angiotensin receptor blockers (ARBs) decreases efferent arteriolar tone and in turn decreases glomerular capillary perfusion pressure. Patients taking NSAIDs and/or ACE inhibitors/ARBs are most susceptible to hemodynamically mediated acute renal failure when blood volume is reduced for any reason. Patients with bilateral renal artery stenosis (or stenosis in a solitary kidney) are dependent on efferent arteriolar vasoconstriction for maintenance of glomerular filtration pressure and are particularly susceptible to a precipitous decline in GFR when given ACE inhibitors or ARBs.
Prolonged renal hypoperfusion may lead to acute tubular necrosis (ATN), an intrinsic renal disease that is discussed below. The urinalysis and urinary electrolyte measurements can be useful in distinguishing prerenal azotemia from ATN (Table 61-2). The urine Na and osmolality of patients with prerenal azotemia can be predicted from the stimulatory actions of norepinephrine, angiotensin II, ADH, and low tubule fluid flow rate. In prerenal conditions, the tubules are intact, leading to a concentrated urine (>500 mosmol), avid Na retention (urine Na concentration, <20 mmol/L; fractional excretion of Na, <1%), and UCr/PCr >40 (Table 61-2). The prerenal urine sediment is usually normal or has hyaline and granular casts, whereas the sediment of ATN usually is filled with cellular debris, tubular epithelial casts, and dark (muddy brown) granular casts.
|
LABORATORY FINDINGS IN ACUTE RENAL FAILURE |
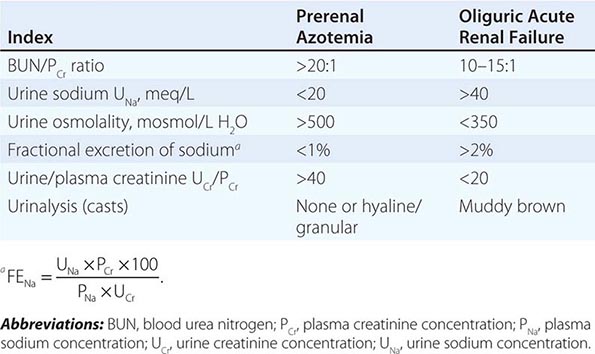
POSTRENAL AZOTEMIA
Urinary tract obstruction accounts for <5% of cases of acute renal failure but is usually reversible and must be ruled out early in the evaluation (Fig. 61-1). Since a single kidney is capable of adequate clearance, obstructive acute renal failure requires obstruction at the urethra or bladder outlet, bilateral ureteral obstruction, or unilateral obstruction in a patient with a single functioning kidney. Obstruction is usually diagnosed by the presence of ureteral and renal pelvic dilation on renal ultrasound. However, early in the course of obstruction or if the ureters are unable to dilate (e.g., encasement by pelvic or periureteral tumors), the ultrasound examination may be negative. The specific urologic conditions that cause obstruction are discussed in Chap. 343.
INTRINSIC RENAL DISEASE
When prerenal and postrenal azotemia have been excluded as etiologies of renal failure, an intrinsic parenchymal renal disease is present. Intrinsic renal disease can arise from processes involving large renal vessels, intrarenal microvasculature and glomeruli, or the tubulointerstitium. Ischemic and toxic ATN account for ~90% of cases of acute intrinsic renal failure. As outlined in Fig. 61-1, the clinical setting and urinalysis are helpful in separating the possible etiologies. Prerenal azotemia and ATN are part of a spectrum of renal hypoperfusion; evidence of structural tubule injury is present in ATN, whereas prompt reversibility occurs with prerenal azotemia upon restoration of adequate renal perfusion. Thus, ATN often can be distinguished from prerenal azotemia by urinalysis and urine electrolyte composition (Table 61-2 and Fig. 61-1). Ischemic ATN is observed most frequently in patients who have undergone major surgery, trauma, severe hypovolemia, overwhelming sepsis, or extensive burns. Nephrotoxic ATN complicates the administration of many common medications, usually by inducing a combination of intrarenal vasoconstriction, direct tubule toxicity, and/or tubule obstruction. The kidney is vulnerable to toxic injury by virtue of its rich blood supply (25% of cardiac output) and its ability to concentrate and metabolize toxins. A diligent search for hypotension and nephrotoxins usually uncovers the specific etiology of ATN. Discontinuation of nephrotoxins and stabilization of blood pressure often suffice without the need for dialysis while the tubules recover. An extensive list of potential drugs and toxins implicated in ATN is found in Chap. 334.
Processes involving the tubules and interstitium can lead to acute kidney injury (AKI), a subtype of acute renal failure. These processes include drug-induced interstitial nephritis (especially by antibiotics, NSAIDs, and diuretics), severe infections (both bacterial and viral), systemic diseases (e.g., systemic lupus erythematosus), and infiltrative disorders (e.g., sarcoidosis, lymphoma, or leukemia). A list of drugs associated with allergic interstitial nephritis is found in Chap. 340. Urinalysis usually shows mild to moderate proteinuria, hematuria, and pyuria (~75% of cases) and occasionally WBC casts. The finding of RBC casts in interstitial nephritis has been reported but should prompt a search for glomerular diseases (Fig. 61-1). Occasionally, renal biopsy will be needed to distinguish among these possibilities. The finding of eosinophils in the urine is suggestive of allergic interstitial nephritis or atheroembolic renal disease and is optimally observed with Hansel staining. The absence of eosinophiluria, however, does not exclude these etiologies.
Occlusion of large renal vessels, including arteries and veins, is an uncommon cause of acute renal failure. A significant reduction in GFR by this mechanism suggests bilateral processes or, in a patient with a single functioning kidney, a unilateral process. Renal arteries can be occluded with atheroemboli, thromboemboli, in situ thrombosis, aortic dissection, or vasculitis. Atheroembolic renal failure can occur spontaneously but most often is associated with recent aortic instrumentation. The emboli are cholesterol-rich and lodge in medium and small renal arteries, with a consequent eosinophil-rich inflammatory reaction. Patients with atheroembolic acute renal failure often have a normal urinalysis, but the urine may contain eosinophils and casts. The diagnosis can be confirmed by renal biopsy, but this procedure is often unnecessary when other stigmata of atheroemboli are present (livedo reticularis, distal peripheral infarcts, eosinophilia). Renal artery thrombosis may lead to mild proteinuria and hematuria, whereas renal vein thrombosis typically induces heavy proteinuria and hematuria. These vascular complications often require angiography for confirmation and are discussed in Chap. 341.
Diseases of the glomeruli (glomerulonephritis and vasculitis) and the renal microvasculature (hemolytic-uremic syndromes, thrombotic thrombocytopenic purpura, and malignant hypertension) usually present with various combinations of glomerular injury: proteinuria, hematuria, reduced GFR, and alterations of sodium excretion that lead to hypertension, edema, and circulatory congestion (acute nephritic syndrome). These findings may occur as primary renal diseases or as renal manifestations of systemic diseases. The clinical setting and other laboratory data help distinguish primary renal diseases from systemic diseases. The finding of RBC casts in the urine is an indication for early renal biopsy (Fig. 61-1), as the pathologic pattern has important implications for diagnosis, prognosis, and treatment. Hematuria without RBC casts can also be an indication of glomerular disease; this evaluation is summarized in Fig. 61-2. A detailed discussion of glomerulonephritis and diseases of the microvasculature is found in Chap. 340.
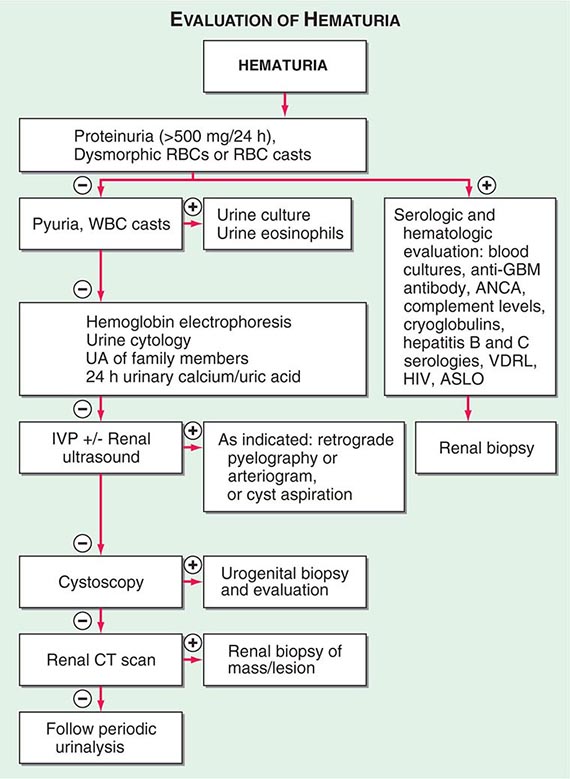
FIGURE 61-2 Approach to the patient with hematuria. ANCA, antineutrophil cytoplasmic antibody; ASLO, antistreptolysin O; CT, computed tomography; GBM, glomerular basement membrane; IVP, intravenous pyelography; RBC, red blood cell; UA, urinalysis; VDRL, Venereal Disease Research Laboratory; WBC, white blood cell.
OLIGURIA AND ANURIA
Oliguria refers to a 24-h urine output <400 mL, and anuria is the complete absence of urine formation (<100 mL). Anuria can be caused by total urinary tract obstruction, total renal artery or vein occlusion, and shock (manifested by severe hypotension and intense renal vasoconstriction). Cortical necrosis, ATN, and rapidly progressive glomerulonephritis occasionally cause anuria. Oliguria can accompany acute renal failure of any etiology and carries a more serious prognosis for renal recovery in all conditions except prerenal azotemia. Nonoliguria refers to urine output >400 mL/d in patients with acute or chronic azotemia. With nonoliguric ATN, disturbances of potassium and hydrogen balance are less severe than in oliguric patients, and recovery to normal renal function is usually more rapid.
ABNORMALITIES OF THE URINE
PROTEINURIA
The evaluation of proteinuria is shown schematically in Fig. 61-3 and typically is initiated after detection of proteinuria by dipstick examination. The dipstick measurement detects only albumin and gives false-positive results at pH >7.0 or when the urine is very concentrated or contaminated with blood. Because the dipstick relies on urinary albumin concentration, a very dilute urine may obscure significant proteinuria on dipstick examination. Quantification of urinary albumin on a spot urine sample (ideally from a first morning void) by measurement of an albumin-to-creatinine ratio (ACR) is helpful in approximating a 24-h albumin excretion rate (AER), where ACR (mg/g) ≈AER (mg/24 h). Furthermore, proteinuria that is not predominantly due to albumin will be missed by dipstick screening. This information is particularly important for the detection of Bence-Jones proteins in the urine of patients with multiple myeloma. Tests to measure total urine protein concentration accurately rely on precipitation with sulfosalicylic or trichloracetic acid (Fig. 61-3).
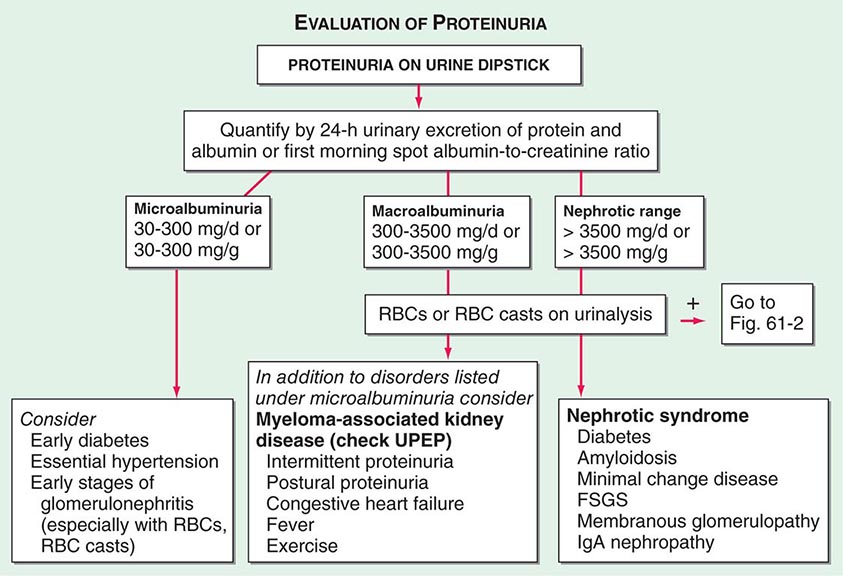
FIGURE 61-3 Approach to the patient with proteinuria. Investigation of proteinuria is often initiated by a positive dipstick on routine urinalysis. Conventional dipsticks detect predominantly albumin and provide a semiquantitative assessment (trace, 1+, 2+, or 3+), which is influenced by urinary concentration as reflected by urine specific gravity (minimum, <1.005; maximum, 1.030). However, more exact determination of proteinuria should employ a spot morning protein/creatinine ratio (mg/g) or a 24-h urine collection (mg/24 h). FSGS, focal segmental glomerulosclerosis; RBC, red blood cell; UPEP, urine protein electrophoresis.
The magnitude of proteinuria and its composition in the urine depend on the mechanism of renal injury that leads to protein losses. Both charge and size selectivity normally prevent virtually all plasma albumin, globulins, and other high-molecular-weight proteins from crossing the glomerular wall; however, if this barrier is disrupted, plasma proteins may leak into the urine (glomerular proteinuria; Fig. 61-3). Smaller proteins (<20 kDa) are freely filtered but are readily reabsorbed by the proximal tubule. Traditionally, healthy individuals excrete <150 mg/d of total protein and <30 mg/d of albumin. However, even at albuminuria levels <30 mg/d, risk for progression to overt nephropathy or subsequent cardiovascular disease is increased. The remainder of the protein in the urine is secreted by the tubules (Tamm-Horsfall, IgA, and urokinase) or represents small amounts of filtered β2-microglobulin, apoproteins, enzymes, and peptide hormones. Another mechanism of proteinuria entails excessive production of an abnormal protein that exceeds the capacity of the tubule for reabsorption. This situation most commonly occurs with plasma cell dyscrasias, such as multiple myeloma, amyloidosis, and lymphomas, that are associated with monoclonal production of immunoglobulin light chains.
The normal glomerular endothelial cell forms a barrier composed of pores of ~100 nm that retain blood cells but offer little impediment to passage of most proteins. The glomerular basement membrane traps most large proteins (>100 kDa), and the foot processes of epithelial cells (podocytes) cover the urinary side of the glomerular basement membrane and produce a series of narrow channels (slit diaphragms) to allow molecular passage of small solutes and water but not proteins. Some glomerular diseases, such as minimal change disease, cause fusion of glomerular epithelial cell foot processes, resulting in predominantly “selective” (Fig. 61-3) loss of albumin. Other glomerular diseases can present with disruption of the basement membrane and slit diaphragms (e.g., by immune complex deposition), resulting in losses of albumin and other plasma proteins. The fusion of foot processes causes increased pressure across the capillary basement membrane, resulting in areas with larger pore sizes (and more severe “nonselective” proteinuria (Fig. 61-3).
When the total daily urinary excretion of protein is >3.5 g, hypoalbuminemia, hyperlipidemia, and edema (nephrotic syndrome; Fig. 61-3) are often present as well. However, total daily urinary protein excretion >3.5 g can occur without the other features of the nephrotic syndrome in a variety of other renal diseases, including diabetes (Fig. 61-3). Plasma cell dyscrasias (multiple myeloma) can be associated with large amounts of excreted light chains in the urine, which may not be detected by dipstick. The light chains are filtered by the glomerulus and overwhelm the reabsorptive capacity of the proximal tubule. Renal failure from these disorders occurs through a variety of mechanisms, including proximal tubule injury, tubule obstruction (cast nephropathy), and light chain deposition (Chap. 340). However, not all excreted light chains are nephrotoxic.
Hypoalbuminemia in nephrotic syndrome occurs through excessive urinary losses and increased proximal tubule catabolism of filtered albumin. Edema forms from renal sodium retention and reduced plasma oncotic pressure, which favors fluid movement from capillaries to interstitium. To compensate for the perceived decrease in effective intravascular volume, activation of the renin-angiotensin system, stimulation of ADH, and activation of the sympathetic nervous system take place, promoting continued renal salt and water reabsorption and progressive edema. Despite these changes, hypertension is uncommon in primary kidney diseases resulting in the nephrotic syndrome (Fig. 61-3 and Chap. 338). The urinary loss of regulatory proteins and changes in hepatic synthesis contribute to the other manifestations of the nephrotic syndrome. A hypercoagulable state may arise from urinary losses of antithrombin III, reduced serum levels of proteins S and C, hyperfibrinogenemia, and enhanced platelet aggregation. Hypercholesterolemia may be severe and results from increased hepatic lipoprotein synthesis. Loss of immunoglobulins contributes to an increased risk of infection. Many diseases (some listed in Fig. 61-3) and drugs can cause the nephrotic syndrome; a complete list is found in Chap. 338.
HEMATURIA, PYURIA, AND CASTS
Isolated hematuria without proteinuria, other cells, or casts is often indicative of bleeding from the urinary tract. Hematuria is defined as two to five RBCs per high-power field (HPF) and can be detected by dipstick. A false-positive dipstick for hematuria (where no RBCs are seen on urine microscopy) may occur when myoglobinuria is present, often in the setting of rhabdomyolysis. Common causes of isolated hematuria include stones, neoplasms, tuberculosis, trauma, and prostatitis. Gross hematuria with blood clots usually is not an intrinsic renal process; rather, it suggests a postrenal source in the urinary collecting system. Evaluation of patients presenting with microscopic hematuria is outlined in Fig. 61-2. A single urinalysis with hematuria is common and can result from menstruation, viral illness, allergy, exercise, or mild trauma. Persistent or significant hematuria (>3 RBCs/HPF on three urinalyses, a single urinalysis with >100 RBCs, or gross hematuria) is associated with significant renal or urologic lesions in 9.1% of cases. The level of suspicion for urogenital neoplasms in patients with isolated painless hematuria and nondysmorphic RBCs increases with age. Neoplasms are rare in the pediatric population, and isolated hematuria is more likely to be “idiopathic” or associated with a congenital anomaly. Hematuria with pyuria and bacteriuria is typical of infection and should be treated with antibiotics after appropriate cultures. Acute cystitis or urethritis in women can cause gross hematuria. Hypercalciuria and hyperuricosuria are also risk factors for unexplained isolated hematuria in both children and adults. In some of these patients (50–60%), reducing calcium and uric acid excretion through dietary interventions can eliminate the microscopic hematuria.





