CHAPTER 77 Functional Imaging in Movement Disorders
Functional Imaging of Presynaptic Dopamine Terminals
The pathologic hallmark of PD is degeneration of pigmented dopaminergic neurons in the substantia nigra pars compacta in association with the formation of intraneuronal Lewy bodies. Loss of nigral cells results in profound dopamine depletion in the nigrostriatal projections, with terminals in the posterior part of the putamen being most affected. A loss of dopaminergic nigrostriatal neurons, however, is not exclusive to PD; it can be observed in other neurodegenerative disorders associated with parkinsonism, including multiple system atrophy (MSA), progressive supranuclear palsy (PSP), and corticobasal degeneration. Three different strategies have been developed for imaging the presynaptic function of dopamine terminals: (1) measurement of the binding of presynaptic dopamine transporter (DAT), the plasma membrane protein involved in the reuptake of dopamine from the synaptic cleft, by several PET and SPECT tracers; (2) measurement of dopamine synthesis from exogenous dopa and its vesicular storage, by 18F-dopa PET; and (3) measurement of the binding of type 2 vesicular monoamine transporter, the protein responsible for the transport of dopamine from the cytoplasm into secretory vesicles, with 11C-dihydrotetrabenazine (DTBZ) PET. Figure 77-1 is a schematic representation of a striatal presynaptic dopamine terminal.
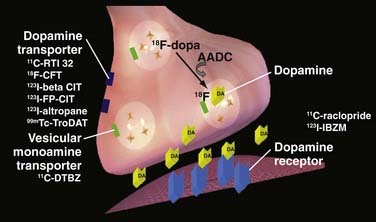
Several PET (11C-CFT, 18F-CFT, 18F-FP-CIT, 11C-RTI-32) and SPECT (123I-β-CIT, 123I-FP-CIT, 123I-altropane, 11C-methylphenidate, 99mTc-TRODAT-1) ligands are now available for the imaging of DAT. Most of these ligands bind to dopamine, noradrenaline, and serotonin transporters. However, because the majority of transporters within the striatum are dopaminergic, reductions in the striatal uptake of these tracers provide a useful marker of DAT functional loss in that region. It must be taken into account that DAT binding may underestimate true terminal density owing to the relative downregulation of DAT in the remaining neurons as a response to reductions in synaptic dopamine. Additionally, there is evidence that DAT binding decreases with age (3.3% to 10% per decade) in healthy subjects.1–3
SPECT is more widely accessible than PET, so more DAT binding data are available with this modality in parkinsonian syndromes. In patients with PD, striatal 123I-β-CIT uptake correlates well with stage of disease and symptom severity, particularly limb bradykinesia and rigidity, but not with rest tremor severity.4–6 Patients with hemiparkinsonism show a pronounced reduction in contralateral striatal 123I-β-CIT binding. Binding in the ipsilateral striatum is also lower than that in healthy controls.7,8 Parallel findings are provided by 123I-FP-CIT SPECT (DaTSCAN), and this is frequently used in routine clinical practice to differentiate idiopathic PD from non–dopamine-deficient syndromes such as secondary nondegenerative parkinsonism and essential tremor.
Measurement of striatal aromatic amino acid decarboxylase activity with 18F-dopa PET is another way to assess the integrity of dopaminergic neurons in vivo. After intravenous administration, 18F-dopa is taken up by the terminals of dopaminergic projections, where it is converted to 18F-dopamine by aromatic amino acid decarboxylase and stored in vesicles. Measurement of 18F-dopa uptake in PD striatum therefore provides a functional measure of the number of remaining dopaminergic terminals. This is supported by pathologic studies, which have demonstrated that levels of striatal 18F-dopa uptake correlate well with nigral cell counts in both humans with various disorders and in nonhuman primates with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced parkinsonism.9,10 Uptake of 18F-dopa may, however, underestimate the degenerative process owing to the compensatory upregulation of aromatic amino acid decarboxylase in the preserved terminals.11
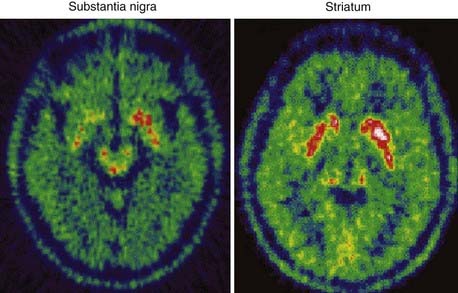
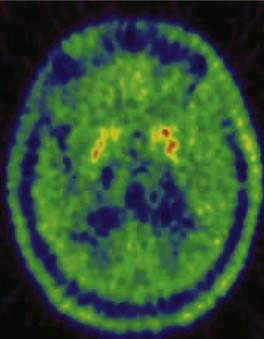
To date, 18F-dopa PET has been the most informative neuroimaging technique. In agreement with the results of neuropathologic studies, PD patients show a gradient of reduced striatal 18F-dopa uptake along a rostrocaudal axis. Patients with hemiparkinsonism have the greatest 18F-dopa uptake reduction in the dorsal posterior putamen contralateral to the symptomatic side,12 whereas patients with more advanced bilateral disease show additional decreases within the ventral and anterior putamen and dorsal caudate (Fig. 77-2). Reductions in 18F-dopa uptake within the ventral head of the caudate are usually observed only in advanced stages of disease. Putaminal uptake of 18F-dopa in PD inversely correlates with the degree of motor disability as measured by the Unified Parkinson’s Disease Rating Scale (UPDRS).13–15 In patients with MSA and PSP, 18F-dopa PET studies reveal reductions in striatal uptake that resemble those observed in PD patients; however, the caudate nucleus is generally more severely affected than in PD, and the reduction of uptake is more homogeneous within the striatal structures.16,17 Figure 77-3 shows striatal 18F-dopa uptake in a patient with MSA.
11C-DTBZ PET is a marker of type 2 vesicular monoamine transporters. It has been suggested that 11C-DTBZ binding provides the most reliable measurement of the density of dopaminergic terminals.18 At present, however, 11C-DTBZ PET is available in only a few centers, and not many studies have been performed with this technique.
Functional Imaging of Dopamine Receptor Availability
In PD patients, striatal 123I-IBZM binding is normal in early disease, and it remains normal or becomes mildly reduced in more advanced disease.19–21 With 11C-raclopride PET, a mild increase in putaminal D2 binding has been observed in untreated PD patients, suggesting either increased D2 receptor availability due to reduced occupancy by endogenous dopamine or receptor upregulation secondary to deafferentation.22–24 As the disease progresses and dopaminergic therapy is started, 11C-raclopride binding normalizes in the putamen and mildly decreases in the caudate,13,24–27 reflecting either disease progression or an effect of treatment.
11C-FLB 457 PET also reveals decreased dopamine receptor availability in the thalamus, anterior cingulate, and dorsolateral prefrontal and temporal cortices in patients with advanced PD.28,29 Scherfler and colleagues30 recently reported that untreated patients with parkinsonism linked to parkin gene mutations show an increase in putaminal 11C-raclopride binding compared with normal subjects. However, at variance with observations in PD patients, levodopa-treated parkin-linked patients show significant reductions in 11C-raclopride binding in both the caudate and the putamen. This could reflect a greater susceptibility to the exposure to dopaminergic medication in parkin-linked patients than in idiopathic PD patients.
In patients with MSA and PSP, both 11C-raclopride binding and 123I-IBZM binding are reduced, suggesting that a degeneration of striatal D2 receptors occurs in these conditions.16,20–22,26,31 Unfortunately, a degree of overlap is seen across the ranges of individual D2 binding data in MSA, PSP, and PD patients and normal subjects. Given this overlap, striatal dopamine binding does not appear to be a sensitive means of discriminating PD from other neurodegenerative causes of parkinsonism.
Finally, reduction of dopamine receptor binding has been proposed as a biomarker of striatal functional integrity in HD. The neurodegenerative process in HD leads to progressive loss of striatal medium-spiny GABA-ergic neurons bearing both dopamine D1 and D2 receptors, along with opioid and benzodiazepine receptors. Decreases of striatal D2 binding in HD patients and asymptomatic HD carriers have been detected with both 11C-raclopride PET and 123I-IBZM SPECT, whereas 123I-epidepride binding was reduced only in patients with advanced HD. Reductions in striatal D1 binding have also been reported with 11C-SCH23390 PET.32–39 Voxel-based statistical parametric mapping of 11C-raclopride PET images has localized a significant reduction of D2 dopamine receptor availability in frontal and temporal areas,38 suggesting that this may play a role in the pathophysiology of cognitive disturbances in early HD.
Functional Imaging of Dopamine Release
Over the past decade, numerous studies have demonstrated the sensitivity of 11C-raclopride PET to fluctuations in synaptic concentrations of dopamine following pharmacologic or behavioral challenges. Rises in synaptic dopamine levels translate into decreases in dopamine D2 receptor availability, which can be detected as reductions in 11C-raclopride binding.40 It has been estimated that a 10% reduction in the availability of D2 receptors for 11C-raclopride binding reflects a fivefold increase in synaptic dopamine levels.41 Using this paradigm, endogenous release of dopamine has been measured after the administration of methamphetamine or levodopa,42–44 during repetitive transcranial magnetic stimulation,45 and during the performance of behaviorally rewarded tasks46 or sequential finger movements47,48 in both normal subjects and PD patients.
In a group of PD patients, the mean reductions in caudate and putamen 11C-raclopride binding following intravenous methamphetamine administration were significantly attenuated compared with normal subjects (8% versus 17% in the caudate; 7% versus 25% in the putamen). The percentage reduction in putamen 11C-raclopride binding after amphetamine administration correlated with both putaminal 18F-dopa uptake and UPDRS scores of individual patients not taking their medication. In the same cohort of patients, statistical parametric mapping localized similar levels of dopamine release in dorsal and ventrolateral prefrontal and orbitofrontal areas in PD patients and normal subjects. These findings indicate that dopamine release in frontal areas after a pharmacologic challenge is preserved even in late stages of PD.42
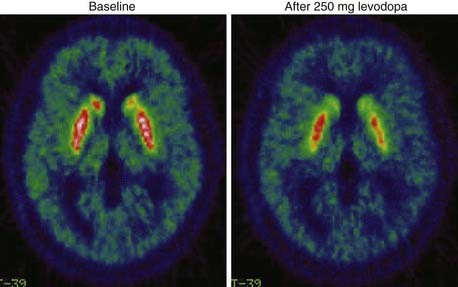
11C-raclopride PET has also been employed to assess the changes in synaptic levels of dopamine that result after exogenous levodopa administration in PD patients (Fig. 77-4).43,44,49,50 All these studies have shown that striatal reductions in 11C-raclopride binding after levodopa become larger as motor disability increases and the disease progresses. The increased synaptic dopamine levels that result from levodopa in more advanced PD probably reflect the reduced dopamine storage capacity of the putamen and fewer dopamine transporters available to clear the transmitter. The relationship between clinical improvement and synaptic dopamine release after a single oral dose of levodopa has been evaluated in a group of patients with advanced PD.43 Although individual improvements in rigidity and bradykinesia correlated well with putaminal dopamine release, this was not the case for tremor or axial symptoms. Interestingly, large putaminal 11C-raclopride binding changes were associated with higher dyskinesia scores. These findings indicate that in advanced PD, the improvement of rigidity and bradykinesia and the presence of dyskinesia after a single dose of oral levodopa are governed by the level of dopamine generated at striatal D2 receptors. They also confirm that relief of parkinsonian tremor and axial symptoms is not related to striatal synaptic dopamine levels and presumably occurs via extrastriatal mechanisms.
Functional Imaging of Cerebral Blood Flow and Glucose Metabolism
Resting Glucose Metabolism
Absolute levels of rCMRGlc are usually normal in PD, but patients show a characteristic profile of relative lentiform nucleus and thalamic hypermetabolism, along with hypometabolism of the lateral and mesial premotor cortex, supplementary motor area (SMA), dorsolateral prefrontal cortex (DLPFC), temporoparietal cortex, and parieto-occipital association regions.51–55 Expression of this PD-related profile can be quantitated and correlates with disease severity as assessed by the UPDRS. The pallidal and putaminal hypermetabolism observed in patients with early PD may represent a compensatory response to deafferentation. Increased 18F-dopa uptake in the globus pallidus interna has also been reported at the onset of PD symptoms.56 Although striatal metabolism is preserved in PD, reduced rCMRGlc is observed in the majority of patients with atypical neurodegenerative parkinsonism such as MSA and PSP.16,57 The patterns of altered regional glucose metabolism in each of these parkinsonian conditions have been used to differentiate them from PD. It has been reported that 18FDG PET has 95% sensitivity and 94% specificity for discriminating patients with PD from healthy volunteers when computer-assisted methodologies are applied.58 However, levels of rCMRGlc are sensitive to coexisting cognitive impairment and depression, and they normalize following treatment with dopaminergic drugs. Using 18FDG PET, it has been possible to detect subclinical cortical dysfunction in about one third of nondemented patients with established PD.53,59,60
The functional effects of HD pathology have also been extensively investigated in vivo with both 18FDG PET and cerebral perfusion SPECT studies. Most studies have reported reduced glucose metabolism and cerebral perfusion in the basal ganglia, particularly the heads of the caudate, and also in association with cortical areas targeting the frontal cortex. Reductions in both cortical and subcortical blood flow and glucose metabolism reportedly correlate with cognitive function in HD. Reduced basal ganglia rCMRGlc is detectable in asymptomatic HD gene mutation carriers. It has been suggested that in many cases this precedes the morphologic changes, such as caudate nucleus atrophy, that are observed on computed tomography and magnetic resonance imaging. In practice, SPECT imaging of caudate blood flow is a less sensitive indicator of caudate dysfunction than is 18FDG PET.61,62
Activation Studies
Several H215O PET studies have compared patterns of regional brain activation during the execution of finger or hand movements by normal subjects, PD patients, and HD patients. Compared with normal subjects, PD patients have reduced regional cerebral blood flow (rCBF) in the rostral SMA and right DLPFC when performing paced movements of a joystick in freely selected directions with the right hand.63 Similar findings were observed in PD patients learning a sequence of finger movements.64 An inability to activate the SMA and DLPFC during learning or selection of movements could explain the difficulty PD patients have acquiring new motor skills and initiating volitional movements. Interestingly, the administration of apomorphine normalized this aberrant activation pattern as soon as the patients switched to an “on” state.65
Other H215O PET and, more recently, functional magnetic resonance imaging studies have shown raised activation in the cerebellum and lateral premotor and parietal areas in PD patients compared with healthy subjects during sequential manual movements.66–69 The hyperactivity in these areas may represent the use of alternative cortical-subcortical circuits to avoid basal ganglia connections, but this is still being debated. It may also explain the beneficial effects of visual and auditory cues when PD patients perform volitional movements. In a recent longitudinal study, 13 patients with recent-onset PD were studied twice with H215O PET. Imaging was performed with the patients off medication at baseline and again after 2 years.70 On both occasions, subjects were asked to perform paced reaching movements toward targets presented in a predictable order. Increasing activation in the pallidum bilaterally and in the left putamen was detected as the disease progressed. In the cortex, motor-related activation increased in the right pre-SMA, anterior cingulate cortex, and left postcentral gyrus. Increases in the right dorsal premotor cortex correlated well with progressive delays in movement initiation, whereas slowing of movement velocity was associated with declining activation in the left DLPFC and dorsal premotor cortex. These findings suggest that with advancing PD, motor performance is associated with the recruitment of brain regions normally involved in the execution of more complex tasks. This may reflect a progressive loss of functional selectivity or inefficient compensatory activation.
There have been only a few activation studies with H215O PET in patients with HD. In one study, changes in rCBF were assessed during the execution of paced joystick movements in freely chosen directions.71 HD patients showed impaired activation of premotor and prefrontal areas in a pattern similar to that observed for PD patients. This finding helps explain the bradykinesia observed in HD patients. Another H215O PET study showed that HD patients have reduced activation of frontostriatal circuitry, along with enhanced activation of parietal areas during a finger opposition task, suggesting compensatory recruitment of normally redundant circuitry.72
Clinical Applications of Functional Neuroimaging in Parkinson’s Disease
Differential Diagnosis
18F-dopa PET and markers of DAT binding such as 123I-β-CIT and 123I-FP-CIT SPECT can differentiate patients with dopamine-deficient parkinsonian syndromes, such as PD, from normal subjects and those with essential tremor with a sensitivity of around 90%.73 It has been reported that 123I-β-CIT SPECT is 100% sensitive and specific for the diagnosis of PD in patients younger than 55 years.74 These approaches can also reveal the presence of dopamine terminal dysfunction in drug-associated parkinsonism74–76 and apparently psychogenic PD.77
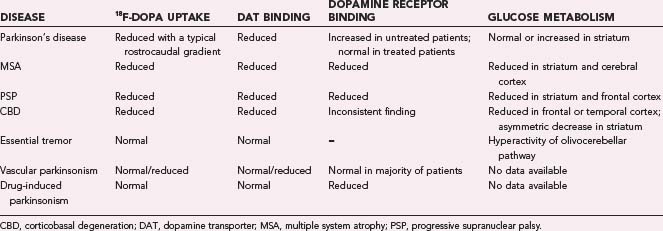
The ability of 18F-dopa PET and markers of DAT binding to differentiate among the different dopamine-deficient parkinsonian syndromes is poor owing to the overlap in individual ranges of putamen tracer uptake in PD, MSA, and PSP patients.78,79 In patients with vascular parkinsonism, 123I-β-CIT and 123I-FP-CIT uptake is heterogeneous; some patients show normal uptake, and others show reduced uptake.74,76 In one study, 99mTc-TRODAT-1 was reported to reliably discriminate between PD and vascular parkinsonism.80 Table 77-1 gives an overview of neuroimaging findings in parkinsonian syndromes.
Detection of Preclinical Disease
18F-dopa PET has demonstrated subclinical dopaminergic dysfunction in one quarter of asymptomatic adult relatives in kindreds with known familial PD.81 One third of those relatives with abnormal imaging went on to develop clinical parkinsonism during a 5-year longitudinal follow-up. 123I-β-CIT SPECT revealed loss of striatal DAT binding in 10% of asymptomatic relatives of PD patients shown to have hyposmia on the University of Pennsylvania Smell Identification Test (UPSIT), another known risk factor for PD.82 Another 18F-dopa PET study reported reduced putaminal 18F-dopa uptake in asymptomatic co-twins of PD patients with apparently sporadic disease. Dopaminergic dysfunction was more common in monozygotic co-twins than in dizygotic co-twins (56% and 18%, respectively). Two of 18 monozygotic co-twins developed clinical parkinsonism over a 4-year follow-up.83 These findings strongly support a role of inheritance in apparently sporadic PD.
Efficacy of Putative Disease-Modifying Agents
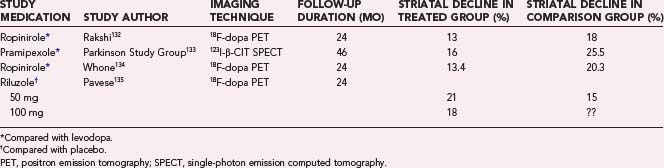
Longitudinal changes in striatal 18F-dopa PET or 123I-β-CIT SPECT have been used as in vivo biomarkers of PD progression in several clinical trials performed to evaluate the efficacy of dopamine agonists, levodopa, and putative neuroprotective agents in modifying the natural course of this condition. Neither the glutamate release inhibitor riluzole nor the mixed lineage kinase inhibitor CEP1347 had a significant effect on disease progression as rated both clinically and with dopaminergic imaging. Use of dopamine agonists slowed the loss of dopamine terminal function in PD relative to levodopa in early PD, but clinically the patients were more improved with the latter medication. In general, functional imaging has either failed to show a neuroprotective effect of the agents on trial or produced findings discordant with clinical outcomes. Table 77-2 gives a brief overview of these studies.
Evaluation of Restorative Approaches
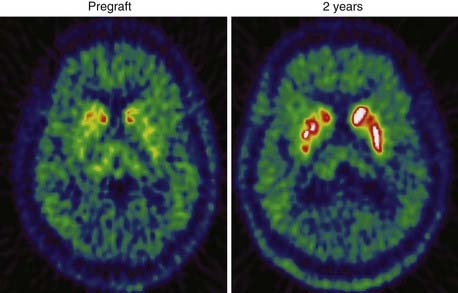
Implantation of human fetal dopamine neurons into the striatum is the most investigated strategy so far. Several open-label uncontrolled studies, albeit with a limited number of patients, have reported significant clinical improvements following striatal transplants. 18F-dopa PET has been used as an in vivo biomarker of graft survival and striatal reinnervation in many of these studies (Fig. 77-5).84–88 Increases from baseline 18F-dopa uptake are thought to reflect the number of graft cells that survive and produce and store dopamine. The DAT marker 123I-IPT SPECT may be a useful alternative to 18F-dopa PET for following implant function in grafted patients. This technique was recently used to demonstrate graft survival over an 8-year follow-up89 in two PD patients who had received transplants. However, Cochen and coworkers were unable to detect increased DAT binding after transplantation in PD patients who demonstrated increased 18F-dopa uptake.90 It is possible that fetal dopamine cells do not always express dopamine transporters.
Using combined 18F-dopa and 11C-raclopride PET, it has been possible to demonstrate in vivo that implanted fetal dopamine cells survive up to 10 years in the striatum and are able to release dopamine following a methamphetamine challenge.86 A significant correlation between 18F-dopa uptake and methamphetamine-induced reductions in 11C-raclopride binding was also found in the putamen containing the graft, suggesting that pharmacologically induced levels of dopamine release by grafts relate to their dopamine storage capacity.42
The effects of fetal grafts on movement-related activation of frontal cortical areas were investigated with H215O PET in four PD patients while they performed joystick tasks. A significant increase in striatal 18F-dopa uptake was already detectable 6.5 months after transplantation. This was associated with a modest clinical improvement on the UPDRS. The impaired mesial premotor and dorsal prefrontal activation seen preoperatively during the performance of freely chosen, paced joystick movements was unchanged at that time point. By 18 months after surgery, there was further significant clinical improvement in the absence of any additional increase in striatal 18F-dopa uptake. Rostral SMA and dorsal prefrontal cortical activation during the performance of joystick movements had significantly improved, however. These findings suggest that the graft is able to form connections in the host brain, restoring the activation of motor cortical areas.88
Based on the encouraging results of these open studies, two prospective, randomized, double-blind controlled trials were performed.91,92 Despite both postmortem and 18F-dopa evidence of some graft function, there was no significant improvement in the primary outcome measures in either of these trials. Younger patients who had received transplants, however, showed a significant improvement in measures of motor severity at 1 year, and all patients were improved by 3 years in the Denver study.91 In the Tampa study, there was a significant clinical improvement at 6 months, but this was subsequently lost—possibly coinciding with the withdrawal of immunosuppression. Troublesome “off-period” dyskinesias occurred in 15% of cases in the Denver series and 56% of those in the Tampa series. It has been suggested that graft-induced dyskinesias may be associated with greater 18F-dopa uptake in the ventral putamen,93 or they may be due to the clumping of transplant cells; however, a recent review came out against the hypothesis that dyskinesias are caused by dopaminergic overgrowth or abnormal dopamine release from the grafts.94,95
We recently used statistical parametric mapping to interrogate 18F-dopa uptake images of individual PD patients before and 1 and 2 years after neural transplantation.95 The patients with the best functional outcome after transplantation exhibited no dopaminergic denervation in areas outside the grafted areas either preoperatively or at 1 or 2 years postoperatively. In contrast, patients with only modest or no clinical benefit showed reductions of 18F-dopa in ventral striatum before or after transplantation. These findings indicate that poor outcome after transplantation is associated with progressive dopaminergic denervation in areas outside the grafts, a process that may have started before surgery.
Recently, 18F-dopa PET was employed to assess the effects of intrastriatal autotransplantation of aggregates of carotid body glomus over a 3-year period. Carotid body cells are dopaminergic and also express glial cell line–derived neurotrophic factor (GDNF). A mild clinical improvement, which was maximal at 6 to 12 months after transplantation, was seen in this study. The clinical improvement reported by the authors ranged from 5% to 74%. 18F-dopa PET showed only a nonsignificant 5% increase in mean putaminal uptake, but there was an inverse relationship between clinical amelioration and annual decline in putaminal 18F-dopa uptake.96
Putaminal infusion of GDNF via an indwelling catheter has been proposed as a restorative approach for PD. GDNF is a potent neurotrophic factor that stimulates embryonic stem cells to differentiate into dopamine cells and protects dopamine neurons against nigral toxins such as MPTP and 6-hydroxydopamine in rodent and primate models of PD. In an early open, uncontrolled human trial, bilateral putaminal infusion of GDNF was continuously administered to five patients with PD at a dose of 14 µg/day. A significant improvement in UPDRS score was seen after 12 months of treatment in all patients. PET studies revealed a postoperative 28% increase in putaminal 18F-dopa uptake.97 A more recent randomized, placebo-controlled study in 34 PD patients confirmed the local increase of 18F-dopa uptake following putaminal infusion of GDNF but failed to show any consistent clinical benefit.98 It seems probable that GDNF is inducing dopamine terminal sprouting in PD striatum, but this may not necessarily communicate with postsynaptic receptors in an effective manner. At present, the future of GDNF infusion as a possible restorative treatment for PD is uncertain.
Evaluation of Ablative Procedures and Deep Brain Stimulation
Surgical therapies have become an effective treatment option in patients with advanced PD who previously experienced good pharmacologic control but now have poor control. Several lines of scientific evidence suggest that loss of striatal dopamine in PD results in an increased inhibitory outflow from the internal globus pallidus (GPi) to the ventral thalamus and cortex; this, in turn, is responsible for the paucity and slowness of movements exhibited by PD patients. Activity of the subthalamic nucleus (STN), which facilitates inhibitory pallidal output, also increases. Surgical strategies therefore aim to decrease internal pallidal and subthalamic outputs to improve movement abnormality. This can be achieved with ablative approaches that produce stereotactic lesions within the basal ganglia structures. Unilateral pallidotomy is the only ablative procedure that is still considered an effective and safe therapy for PD. The procedure leads to moderate improvements in contralateral limb rigidity and bradykinesia but has a dramatic effect on contralateral levodopa-induced dyskinesias, which are abolished or markedly improved in the majority of patients. Unfortunately, bilateral pallidotomy is associated with unacceptable problems with speech and swallowing. Using 18FDG PET, Eidelberg and colleagues99 demonstrated that unilateral pallidotomy reduced preoperative overactivity of the inhibitory pallidothalamic projections. After surgery, PD patients showed significant metabolic increases in primary motor cortex, lateral premotor cortex (LPMC), and DLPFC of the hemisphere that underwent surgery. Improvement in motor performance of the contralateral limb correlated significantly with postoperative reductions in thalamic metabolism and increases in lateral frontal metabolism. Interestingly, preoperative measurements of lentiform glucose metabolism correlated well with postoperative clinical outcome.
Functional effects of pallidotomy on cerebral activation in PD patients have also been reported. After pallidotomy, a group of six PD patients showed significantly increased SMA and LPMC activation when reaching out to grasp lighted targets every 3 seconds while not taking medication.100 Subsequently, Samuel and colleagues101 reported significantly increased activation of the SMA, LPMC, and DLPFC in postpallidotomy PD patients when making paced joystick movements in freely selected directions while off medication. Taken together, the findings of these activation studies lend support to the hypothesis that disruption of inhibitory pallidal input to the ventrolateral thalamus can lead to a normalization of cortical activation in PD and improve movement-related activity in motor association areas.
Ablative procedures, however, have now been largely replaced with implantation of high-frequency stimulating electrodes (deep brain stimulation [DBS]) in the GPi and STN or, more recently, low-frequency stimulation of the pedunculopontine nucleus (PPN). There have been only a few PET reports on the effects of pallidal stimulation on regional brain function in PD. Davis and colleagues102 assessed changes in rCBF induced by subeffective low-frequency and effective high-frequency pallidal stimulation compared with the rest condition when the stimulator was switched off. Only effective GPi stimulation improved contralateral bradykinesia and rigidity in PD patients, and this was associated with increased rCBF in the SMA and in the area of the putamen and globus pallidus externa. Two other H215O PET studies were performed to assess the effects of pallidal stimulation on the execution of a manual motor task. In the first study, clinically effective GPi stimulation in six PD patients did not lead to any significant changes in levels of SMA or DLPFC activation during free-choice joystick movements.103 Conversely, in the second study, clinically effective GPi stimulation led to increased lateral premotor and anterior cingulate activation during reaching movements to touch a visual target.104 Taken together, the findings of these PET studies suggest that pallidotomy and high-frequency pallidal stimulation are both effective in reducing the excess inhibitory drive to premotor and prefrontal areas in PD.
An alternative approach for normalizing thalamocortical activation is to reduce the excitatory input to the GPi from the STN by means of high-frequency electrical stimulation. This procedure dramatically improves bradykinesia and rigidity in PD, and for this reason, the STN has become the preferred target for DBS. STN-DBS, however, is less effective than pallidotomy in relieving levodopa-induced dyskinesias, which is usually achieved by levodopa dose reduction. The exact mechanism by which STN-DBS exerts an antiparkinsonian effect is still unclear, although conduction block probably plays a role. In the stimulator “on” condition, 18FDG PET has shown increased rCMRGlc in the lateral globus pallidus; thalamus; midbrain area; frontal, temporal, and parietal cortices; and anterior cingulate. Decreases have been reported in the cerebellum, orbitofrontal cortex, and parahippocampal gyrus.105–108 Recently, Arai and colleagues108 assessed the effect of unilateral STN stimulation in eight PD patients. Postoperative 18FDG PET in these patients revealed significant metabolic increases in the ipsilateral ventrolateral thalamic areas and decreased metabolism of the contralateral GPi. These results may explain why unilateral STN stimulation often improves parkinsonian symptoms bilaterally.
There have been numerous reports of H215O PET activation findings in PD patients before and after STN stimulation. In most studies, levels of rCBF were measured while PD patients who were not taking medication performed paced joystick movements in freely selected directions. In general, STN stimulation produced relative increases in activation of the rostral SMA, anterior cingulate, DLPFC, globus pallidus, thalamus, and putamen during movement.103,109–112 In most of these studies, STN stimulation was also associated with reduced activation of primary motor cortex, LPMC, and parietal cortex,103,109,110 suggesting a reduction in switching to compensatory pathways.
Based on data from animal models, it has been suggested that STN-DBS may promote dopamine release in the striatum.113,114 However, three independent studies have shown no differences in striatal 11C-raclopride binding in the STN-DBS “on” and “off” conditions. Therefore, these findings do not support the view that STN stimulation works in part by increasing dopaminergic transmission.115–117 Finally, STN-DBS does not seem to have any effect on disease progression. Hilker and colleagues118 assessed PD progression as measured by sequential 18F-dopa PET scans in 30 patients with successful STN-DBS over the first 16 months after surgery. The rates of progression in patients with STN-DBS were similar to previously reported data from longitudinal imaging studies in PD.
Recently, inhibition of STN output has been attempted by the use of gene therapy. The enzyme glutamate decarboxylase (GAD) converts glutamate to GABA, and GAD genes have been unilaterally transfected into the STN of 12 PD patients using an adeno-associated viral vector.119 Significant reductions in thalamic metabolism on the operated side, along with concurrent metabolic increases in ipsilateral motor and premotor cortical regions, were observed. Abnormal elevations in the activity of metabolic networks associated with motor and cognitive functioning in PD patients were evident at baseline. The activity of the motor-related network declined after surgery and persisted at 1 year. These network changes correlated with improved clinical disability ratings.
The PPN is a novel target for DBS. Modulation of PPN activity is believed to be beneficial in the treatment of gait dysfunction and akinesia in PD patients with such symptoms. A recent paper evaluated changes in rCBF at rest during off and on stimulation in an advanced PD patient with unilateral PPN-DBS.120 The authors found that PPN-DBS increased rCBF in the thalamus bilaterally. Double-blind clinical evaluation revealed an improvement in motor function by approximately 20%. If confirmed by further studies, PPN-DBS could represent an important therapeutic option for patients with more complicated forms of movement disorders involving disabling postural instability and deterioration of gait.
Functional Neuroimaging in Huntington’s Disease
Detection of Preclinical Disease
Reduced striatal glucose metabolism and dopamine D1 and D2 receptor binding have been reported in 30% to 50% of asymptomatic adult HD mutation carriers.34,35,37 The marker of microglial activation, 11C-PK11195 PET, shows increased striatal signal in a majority of these subjects.121 These studies clearly indicate that neuroimaging techniques can be used to identify asymptomatic HD gene carriers who are actively progressing toward the symptomatic phase of the disease.
Monitoring of Disease Progression
With the recent advancement in restorative strategies and the advent of putative neuroprotective agents, the use of functional imaging as an in vivo biomarker of disease progression has become highly relevant. The rate of HD progression has been evaluated mostly with 18FDG PET and 11C-raclopride PET. Grafton and colleagues122 reported an annual 3.1% decline in caudate glucose metabolism, whereas a second series reported an annual 2.3% decrease.35 It seems that 11C-raclopride PET is a more sensitive indicator of striatal degeneration. The mean annual rate of striatal 11C-raclopride binding decline reportedly ranges from 4% to 7% in HD patients with early clinical disease and is 4.5% in asymptomatic HD mutation carriers.36–38123 More recently, the annual rate of D2 dopamine receptor loss was assessed in a cohort of HD patients over a 3-year period. Patients were scanned three times with 11C-raclopride PET, and striatal D2 dopamine receptor binding in these patients declined approximately 5% per annum in a linear fashion.38 Taken together, these findings indicate that 11C-raclopride PET is a useful and objective tool for monitoring the effects of neuroprotective drugs and restorative strategies in HD.
Finally, a mean 2% annual loss of striatal D1 receptor measured with 11C-SCH23390 PET has been reported in HD gene carriers with early clinical or subclinical disease.37
Evaluation of Restorative Approaches
Cell replacement therapy in HD is based on the concept that implanted fetal striatal neuroblasts can replace the lost or dysfunctional striatal neurons and establish new striatal-pallidal projections. This approach has been successful in both rodent and nonhuman primate models of HD.124–126 Interestingly, PET was able to demonstrate the regeneration of dopamine receptors after the implantation of fetal striatal tissue in a rodent model of HD.124
To date, only a few studies have been carried out in humans. In the first of these studies (conducted in an open-label fashion in France), five HD patients received grafts of human fetal neuroblasts and were followed over a 12-month period with both clinical evaluations and 18FDG PET to assess metabolic activity. Three of the five patients showed clinical improvement, along with increases in striatal metabolic activity.127 These three patients remained clinically stable over an additional 12-months of follow-up.128 Their final 18FDG PET scans showed improved striatal and cortical metabolism, suggesting restoration of the impaired striatothalamocortical circuitry in these patients. The two patients who did not improve after striatal transplantation showed deterioration in both clinical symptoms and PET findings over 2 years.128
At variance with these findings, another pilot trial carried out in the United States failed to demonstrate any clinical or PET improvement in seven patients who received transplants,129 despite postmortem evidence of graft survival with striatal tissue in one patient.130 In particular, D1 receptor binding did not change significantly after surgery; D2 binding was reduced, suggesting disease progression. Additionally, the patients showed widespread glucose hypometabolism, which remained unaltered over 2 years. The HD patients in this cohort had more advanced disease than those in the French study, and complications from the surgical procedure were relatively common. A different tissue preparation method was also employed. It is not clear whether these factors influenced the final outcome in this study.
Our group recently reported longitudinal clinical and PET assessments in two HD patients who had received fetal striatal allografts 5 years earlier. One patient showed sustained clinical improvement accompanied by increased striatal D2 receptor binding measured with 11C-raclopride PET, suggesting the long-term survival and efficacy of the graft.131 It should be noted that increased dopamine receptor binding is a more selective marker of cell regeneration than are measurements of cerebral metabolic activity. Increased glucose metabolism may, in fact, reflect a glial reaction to grafting and does not specifically indicate graft survival. Taken together, the results of these studies indicate that fetal striatal allografts may be beneficial to some HD patients, although it is not possible to predict which patients might benefit from this procedure. At present, therefore, the role of fetal striatal allografts in HD remains experimental. It is hoped that the results of new clinical trials, which are currently under way, will provide a more definitive answer on the efficacy of this approach in HD.
Bachoud-Levi AC, Rémy P, Nguyen JP, et al. Motor and cognitive improvements in patients with Huntington’s disease after neural transplantation. Lancet. 2000;356:1975-1979.
Brooks DJ, Salmon EP, Mathias CJ, et al. The relationship between locomotor disability, autonomic dysfunction, and the integrity of the striatal dopaminergic system in patients with multiple system atrophy, pure autonomic failure and Parkinson’s disease studied by PET. Brain. 1990;113:1539-1552.
de la Fuente-Fernandez R, Sossi V, et al. Levodopa-induced changes in synaptic dopamine levels increase with progression of Parkinson’s disease: implications for dyskinesias. Brain. 2004;127:2747-2754.
Eckert T, Barnes A, Dhawan V, et al. FDG PET in the differential diagnosis of parkinsonian disorders. Neuroimage. 2005;26:912-921.
Feigin A, Kaplitt MG, Tang C, et al. Modulation of metabolic brain networks after subthalamic gene therapy for Parkinson’s disease. Proc Natl Acad Sci U S A. 2007;104:19559-19564.
Freed CR, Greene PE, Breeze RE, et al. Transplantation of embryonic dopamine neurons for severe Parkinson’s disease. N Engl J Med. 2001;344:710-719.
Freeman TB, Cicchetti F, Hauser RA, et al. Transplanted fetal striatum in Huntington’s disease: phenotypic development and lack of pathology. Proc Natl Acad Sci U S A. 2000;97:13877-13882.
Kaasinen V, Nagren K, Hietala J, et al. Extrastriatal dopamine D2 and D3 receptors in early and advanced Parkinson’s disease. Neurology. 2000;54:1482-1487.
Lang AE, Gill S, Patel NK, et al. Randomized controlled trial of intraputamenal glial cell line-derived neurotrophic factor infusion in Parkinson disease. Ann Neurol. 2006;59:459-466.
Laruelle M. Imaging synaptic neurotransmission with in vivo binding competition techniques: a critical review. J Cereb Blood Flow Metab. 2000;20:423-451.
Morrish PK, Sawle GV, Brooks DJ. Clinical and 18F-dopa PET findings in early Parkinson’s disease. J Neurol Neurosurg Psychiatry. 1995;59:597-600.
Olanow CW, Goetz CG, Kordower JH, et al. A double blind controlled trial of bilateral fetal nigral transplantation in Parkinson’s disease. Ann Neurol. 2003;54:403-414.
Pate BD, Kawamata T, Yamada T, et al. Correlation of striatal fluorodopa uptake in the MPTP monkey with dopaminergic indices. Ann Neurol. 1993;34:331-338.
Pavese N, Evans AH, Tai YF, et al. Clinical correlates of levodopa-induced dopamine release in Parkinson disease: a PET study. Neurology. 2006;67:1612-1617.
Piccini P, Brooks DJ, Bjorklund A, et al. Dopamine release from nigral transplants visualized in vivo in a Parkinson’s patient. Nat Neurosci. 1999;2:1137-1140.
Piccini P, Lindvall O, Bjorklund A, et al. Delayed recovery of movement-related cortical function in Parkinson’s disease after striatal dopaminergic grafts. Ann Neurol. 2000;48:689-695.
Piccini P, Pavese N, Hagell P, et al. Factors affecting the clinical outcome after neural transplantation in Parkinson’s disease. Brain. 2005;128:2977-2986.
Reuter I, Tai YF, Pavese N, et al. Long term clinical and PET outcome of foetal striatal transplantation in Huntington’s disease. J Neurol Neurosurg Psychiatry. 2008;79:948-951.
Sawle GV, Playford ED, Brooks DJ, et al. Asymmetrical pre-synaptic and post-synpatic changes in the striatal dopamine projection in dopa naive parkinsonism. Diagnostic implications of the D2 receptor status. Brain. 1993;116:853-867.
Snow BJ, Tooyama I, McGeer EG, et al. Human positron emission tomographic 18F-dopa studies correlate with dopamine cell counts and levels. Ann Neurol. 1993;34:324-330.
Thobois S, Dominey P, Fraix V, et al. Effects of subthalamic nucleus stimulation on actual and imagined movement in Parkinson’s disease: a PET study. J Neurol. 2002;249:1689-1698.
1 Pirker W, Asenbaum S, Hauk M, et al. Imaging serotonin and dopamine transporters with 123I-beta-CIT SPECT: binding kinetics and effects of normal aging. J Nucl Med. 2000;41:36-44.
2 Tissingh G, Bergmans P, Booij J, et al. 123I-beta-CIT single-photon emission tomography in Parkinson’s disease reveals a smaller decline in dopamine transporters with age than in controls. Eur J Nucl Med. 1997;24:1171-1174.
3 van Dyck CH, Seibyl JP, Malison RT, et al. Age-related decline in striatal dopamine transporter binding with iodine-123-beta-CITSPECT. J Nucl Med. 1995;36:1175-1181.
4 Seibyl JP, Marek KL, Quinlan D, et al. Decreased single-photon emission computed tomographic 123I-beta-CIT striatal uptake correlates with symptom severity in Parkinson’s disease. Ann Neurol. 1995;38:589-598.
5 Brucke T, Asenbaum S, Pirker W, et al. Measurement of dopaminergic degeneration in Parkinson’s disease with 123I-beta-CIT and SPECT. Correlation with clinical findings and comparison with multiple system atrophy and progressive supranuclear palsy. J Neural Transm. 1997;50(Suppl):9-24.
6 Pirker W. Correlation of dopamine transporter imaging with parkinsonian motor handicap: how close is it? Mov Disord. 2003;18(Suppl 7):S43-S51.
7 Tissingh G, Bergmans P, Booij J, et al. Drug-naive patients with Parkinson’s disease in Hoehn and Yahr stages I and II show a bilateral decrease in striatal dopamine transporters as revealed by 123I-beta-CIT SPECT. J Neurol. 1998;245:14-20.
8 Marek KL, Seibyl JP, Zoghbi SS, et al. 123I-beta-CIT/SPECT imaging demonstrates bilateral loss of dopamine transporters in hemi-Parkinson’s disease. Neurology. 1996;46:231-237.
9 Snow BJ, Tooyama I, McGeer EG, et al. Human positron emission tomographic 18F-dopa studies correlate with dopamine cell counts and levels. Ann Neurol. 1993;34:324-330.
10 Pate BD, Kawamata T, Yamada T, et al. Correlation of striatal fluorodopa uptake in the MPTP monkey with dopaminergic indices. Ann Neurol. 1993;34:331-338.
11 Ribeiro MJ, Vidailhet M, Loch C, et al. Dopaminergic function and dopamine transporter binding assessed with positron emission tomography in Parkinson disease. Arch Neurol. 2002;59:580-586.
12 Morrish PK, Sawle GV, Brooks DJ. Clinical and [18F] dopa PET findings in early Parkinson’s disease. J Neurol Neurosurg Psychiatry. 1995;59:597-600.
13 Brooks DJ, Salmon EP, Mathias CJ, et al. The relationship between locomotor disability, autonomic dysfunction, and the integrity of the striatal dopaminergic system in patients with multiple system atrophy, pure autonomic failure and Parkinson’s disease studied by PET. Brain. 1990;113:1539-1552.
14 Broussolle E, Dentresangle C, Landais P, et al. The relation of putamen and caudate nucleus 18F-dopa uptake to motor and cognitive performances in Parkinson’s disease. J Neurol Sci. 1999;166:141-151.
15 Vingerhoets FJG, Schulzer M, Calne D, et al. Which clinical sign of Parkinson’s disease best reflects the nigrostriatal lesion? Ann Neurol. 1997;41:58-64.
16 Antonini A, Leenders KL, Vontobel P, et al. Complementary PET studies of striatal neuronal function in the differential diagnosis between multiple system atrophy and Parkinson’s disease. Brain. 1997;120:2187-2195.
17 Brooks DJ, Ibanez V, Sawle GV, et al. Differing patterns of striatal 18F-dopa uptake in Parkinson’s disease, multiple system atrophy and progressive supranuclear palsy. Ann Neurol. 1990;28:547-555.
18 Lee CS, Samii A, Sossi V, et al. In vivo positron emission tomographic evidence for compensatory changes in presynaptic dopamine nerve terminals in Parkinson’s disease. Ann Neurol. 2000;47:493-503.
19 Playford ED, Brooks DJ. In vivo and in vitro studies of the dopaminergic system in movement disorders. Cerebrovasc Brain Metab Rev. 1992;4:144-171.
20 Schwarz J, Tatsch K, Arnold G, et al. 123I-iodobenzamide-SPECT in 83 patients with de novo parkinsonism. Neurology. 1993;43(Suppl 6):S17-S20.
21 Schulz JB, Klockgether T, Petersen D, et al. Multiple system atrophy: natural history, MRI morphology, and dopamine receptor imaging with 123IBZM-SPECT. J Neurol Neurosurg Psychiatry. 1994;57:1047-1056.
22 Rinne JO, Laihinen A, Rinne UK, et al. PET study on striatal dopamine D2 receptor changes during the progression of early Parkinson’s disease. Mov Disord. 1993;8:134-138.
23 Sawle GV, Playford ED, Brooks DJ, et al. Asymmetrical pre-synaptic and post-synpatic changes in the striatal dopamine projection in dopa naive parkinsonism. Diagnostic implications of the D2 receptor status. Brain. 1993;116:853-867.
24 Turjanski N, Lees AJ, Brooks DJ. In vivo studies on striatal dopamine D1 and D2 site binding in dopa-treated Parkinson’s disease patients with and without dyskinesias. Neurology. 1997;49:717-723.
25 Antonini A, Schwarz J, Oertel WH, et al. 11C-raclopride and positron emission tomography in previously untreated patients with Parkinson’s disease: influence of dopa and lisuride therapy on striatal dopamine D2-receptors. Neurology. 1994;44:1325-1329.
26 Brooks DJ, Ibanez V, Sawle GV, et al. Striatal D2 receptor status in Parkinson’s disease, striatonigral degeneration, and progressive supranuclear palsy measured with 11C-raclopride and PET. Ann Neurol. 1992;31:184-192.
27 Dentresangle C, Veyre L, Le Bars D, et al. Striatal D2 dopamine receptor status in Parkinson’s disease: an 18F-dopa and 11C-raclopride PET study. Mov Disord. 1999;14:1025-1030.
28 Kaasinen V, Nagren K, Hietala J, et al. Extrastriatal dopamine D2 and D3 receptors in early and advanced Parkinson’s disease. Neurology. 2000;54:1482-1487.
29 Kaasinen V, Aalto S, Nagren K, et al. Extrastriatal dopamine D2 receptors in Parkinson’s disease: a longitudinal study. J Neural Transm. 2003;110:591-601.
30 Scherfler C, Khan NL, Pavese N, et al. Upregulation of dopamine D2 receptors in dopaminergic drug-naive patients with parkin gene mutations. Mov Disord. 2006;21:783-788.
31 Plotkin M, Amthauer H, Klaffke S, et al. Combined 123I-FP-CIT and 123I-IBZM SPECT for the diagnosis of parkinsonian syndromes: study on 72 patients. J Neural Transm. 2005;112:677-692.
32 Turjanski N, Weeks R, Dolan R, et al. Striatal D1 and D2 receptor binding in patients with Huntington’s disease and other choreas. A PET study. Brain. 1995;118:689-696.
33 Ichise M, Toyama H, Fornazzari L, et al. Iodine-123-IBZM dopamine D2 receptor and technetium-99m-HMPAO brain perfusion SPECT in the evaluation of patients with and subjects at risk for Huntington’s disease. J Nucl Med. 1993;34:1274-1281.
34 Weeks RA, Piccini P, Harding AE, et al. Striatal D1 and D2 dopamine receptor loss in asymptomatic mutation carriers of Huntington’s disease. Ann Neurol. 1996;40:49-54.
35 Antonini A, Leenders KL, Spiegel R, et al. Striatal glucose metabolism and dopamine D2 receptor binding in asymptomatic gene carriers and patients with Huntington’s disease. Brain. 1996;119:2085-2095.
36 Antonini A, Leenders KL, Eidelberg D. 11C-raclopride-PET studies of the Huntington’s disease rate of progression: relevance of the trinucleotide repeat length. Ann Neurol. 1998;43:253-255.
37 Andrews TC, Weeks RA, Turjanski N, et al. Huntington’s disease progression. PET and clinical observations. Brain. 1999;122:2353-2363.
38 Pavese N, Andrews TC, Brooks DJ, et al. Progressive striatal and cortical dopamine receptor dysfunction in Huntington’s disease: a PET study. Brain. 2003;126:1127-1135.
39 Leslie WD, Greenberg CR, Abrams DN, et al. Clinical deficits in Huntington disease correlate with reduced striatal uptake on iodine-123 epidepride single-photon emission tomography. Eur J Nucl Med. 1999;26:1458-1464.
40 Laruelle M. Imaging synaptic neurotransmission with in vivo binding competition techniques: a critical review. J Cereb Blood Flow Metab. 2000;20:423-451.
41 Breier A, Su TP, Saunders R, et al. Schizophrenia is associated with elevated amphetamine-induced synaptic dopamine concentrations: evidence from a novel positron emission tomography method. Proc Natl Acad Sci U S A. 1997;94:2569-2574.
42 Piccini P, Pavese N, Brooks DJ. Endogenous dopamine release after pharmacological challenges in Parkinson’s disease. Ann Neurol. 2003;53:647-653.
43 Pavese N, Evans AH, Tai YF, et al. Clinical correlates of levodopa-induced dopamine release in Parkinson disease: a PET study. Neurology. 2006;67:1612-1617.
44 de la Fuente-Fernandez R, Sossi V, et al. Levodopa-induced changes in synaptic dopamine levels increase with progression of Parkinson’s disease: implications for dyskinesias. Brain. 2004;127:2747-2754.
45 Strafella AP, Paus T, Fraraccio M, Dagher A. Striatal dopamine release induced by repetitive transcranial magnetic stimulation of the human motor cortex. Brain. 2003;126:2609-2615.
46 Koepp MJ, Gunn RN, Lawrence AD, et al. Evidence for striatal dopamine release during a video game. Nature. 1998;393:266-268.
47 Lawrence AD, Brooks DJ. Neural correlates of reward processing in the human brain: a PET study. Neurology. 1999;52(Suppl 2):A307.
48 Goerendt IK, Messa C, Lawrence AD, et al. Dopamine release during sequential finger movements in Parkinson’s disease: a PET study. Brain. 2003;126:312-325.
49 Tedroff J, Pedersen M, Aquilonius SM, et al. Levodopa-induced changes in synaptic dopamine in patients with Parkinson’s disease as measured by 11C-raclopride displacement and PET. Neurology. 1996;46:1430-1436.
50 de la Fuente-Fernandez R, Lu JQ, Sossi V, et al. Biochemical variations in the synaptic level of dopamine precede motor fluctuations in Parkinson’s disease: PET evidence of increased dopamine turnover. Ann Neurol. 2001;49:298-303.
51 Eidelberg D, Moeller JR, Dhawan V, et al. The metabolic anatomy of Parkinson’s disease: complementary 18F-fluorodeoxyglucose and 18F-fluorodopa positron emission tomographic studies. Mov Disord. 1990;5:203-213.
52 Antonini A, Moeller JR, Nakamura T, et al. The metabolic anatomy of tremor in Parkinson’s disease. Neurology. 1998;51:803-810.
53 Hu MT, Taylor-Robinson SD, Chaudhuri KR, et al. Cortical dysfunction in non-demented Parkinson’s disease patients: a combined 31P-MRS and 18FDG-PET study. Brain. 2000;123:340-352.
54 Fukuda M, Mentis M, Ghilardi MF, et al. Functional correlates of pallidal stimulation for Parkinson’s disease. Ann Neurol. 2001;49:116-155.
55 Lozza C, Marie RM, Baron JC. The metabolic substrates of bradykinesia and tremor in uncomplicated Parkinson’s disease. Neuroimage. 2002;17:688-699.
56 Whone AL, Moore RY, Piccini P, et al. Plasticity of the nigropallidal pathway in Parkinson’s disease. Ann Neurol. 2003;53:206-213.
57 Juh R, Kim J, Moon D, et al. Different metabolic patterns analysis of parkinsonism on the 18F-FDG PET. Eur J Radiol. 2004;51:223-233.
58 Eckert T, Barnes A, Dhawan V, et al. FDG PET in the differential diagnosis of parkinsonian disorders. Neuroimage. 2005;26:912-921.
59 Berding G, Odin P, Brooks DJ, et al. Resting regional cerebral glucose metabolism in advanced Parkinson’s disease studied in the off and on conditions with 18F-FDG PET. Mov Disord. 2001;16:1014-1022.
60 Mentis MJ, McIntosh AR, Perrine K, et al. Relationship among the metabolic patterns that correlate with mnemonic, visuospatial and mood symptoms in Parkinson’s disease. Am J Psychiatry. 2002;159:746-754.
61 Boecker H, Kuwert T, Langen KJ, et al. SPECT with HMPAO compared to PET with FDG in Huntington disease. J Comput Assist Tomogr. 1994;18:542-548.
62 Martin WR, Hoskinson M, Kremer B, et al. Functional caudate imaging in symptomatic Huntington’s disease: positron emission tomography versus single-photon emission computed tomography. J Neuroimaging. 1995;5:227-232.
63 Playford ED, Jenkins IH, Passingham RE, et al. Impaired mesial frontal and putamen activation in Parkinson’s disease: a PET study. Ann Neurol. 1992;32:151-161.
64 Jahanshahi M, Jenkins IH, Brown RG, et al. Self-initiated versus externally triggered movements. An investigation using measurement of regional cerebral blood flow with PET and movement-related potentials in normal and Parkinson’s disease subjects. Brain. 1995;118:913-933.
65 Jenkins IH, Fernandez W, Playford ED, et al. Impaired activation of the supplementary motor area in Parkinson’s disease is reversed when akinesia is treated with apomorphine. Ann Neurol. 1992;32:749-757.
66 Samuel M, Ceballos-Baumann AO, Blin J, et al. Evidence for lateral premotor and parietal overactivity in Parkinson’s disease during sequential and bimanual movements: a PET study. Brain. 1997;120:963-976.
67 Catalan MJ, Ishii K, Honda M, et al. A PET study of sequential finger movements of varying length in patients with Parkinson’s disease. Brain. 1999;122:483-495.
68 Sabatini U, Boulanouar K, Fabre N, et al. Cortical motor reorganization in akinetic patients with Parkinson’s disease. A functional MRI study. Brain. 2000;123:394-403.
69 Haslinger B, Erhard P, Kampfe N, et al. Event-related functional magnetic resonance imaging in Parkinson’s disease before and after levodopa. Brain. 2001;124:558-570.
70 Carbon M, Felice Ghilardi M, Dhawan V, et al. Correlates of movement initiation and velocity in Parkinson’s disease: A longitudinal PET study. Neuroimage. 2007;34:361-370.
71 Weeks RA, Ceballos-Baumann A, Piccini P, et al. Cortical control of movement in Huntington’s disease. A PET activation study. Brain. 1997;120:1569-1578.
72 Bartenstein P, Weindl A, Spiegel S, et al. Central motor processing in Huntington’s disease. A PET study. Brain. 1997;120:1553-1567.
73 Benamer TS, Patterson J, Grosset DG, et al. Accurate differentiation of parkinsonism and essential tremor using visual assessment of 123I-FP-CIT SPECT imaging: the 123I-FP-CIT study group. Mov Disord. 2000;15:503-510.
74 Eerola J, Tienari PJ, Kaakkola S, et al. How useful is 123I-beta-CIT SPECT in clinical practice? J Neurol Neurosurg Psychiatry. 2005;76:1211-1216.
75 Burn DJ, Brooks D. Nigral dysfunction in drug-induced parkinsonism: an 18F-Dopa PET study. Neurology. 1993;43:552-556.
76 Lorberboym M, Djaldetti R, Melamed E, et al. 123I-FP-CIT SPECT imaging of dopamine transporters in patients with cerebrovascular disease and clinical diagnosis of vascular parkinsonism. J Nucl Med. 2004;45:1688-1693.
77 Benaderette S, Zanotti Fregonara P, Apartis E, et al. Psychogenic parkinsonism: a combination of clinical, electrophysiological, and 123I-FP-CIT SPECT scan explorations improves diagnostic accuracy. Mov Disord. 2006;21:310-317.
78 Burn DJ, Sawle GV, Brooks DJ. Differential diagnosis of Parkinson’s disease, multiple system atrophy, and Steele-Richardson-Olszewski syndrome: discriminant analysis of striatal 18F-dopa PET data. J Neurol Neurosurg Psychiatry. 1994;57:278-284.
79 Pirker W, Asenbaum S, Bencsits G, et al. 123I-beta-CIT SPECT in multiple system atrophy, progressive supranuclear palsy, and corticobasal degeneration. Mov Disord. 2000;15:1158-1167.
80 Tzen KY, Lu CS, Yen TC, et al. Differential diagnosis of Parkinson’s disease and vascular parkinsonism by 99mTc-TRODAT-1. J Nucl Med. 2001;42:408-413.
81 Piccini P, Morrish PK, Turjanski N, et al. Dopaminergic function in familial Parkinson’s disease: a clinical and 18F-dopa PET study. Ann Neurol. 1997;41:222-229.
82 Ponsen MM, Stoffers D, Booij J, et al. Idiopathic hyposmia as a preclinical sign of Parkinson’s disease. Ann Neurol. 2004;56:173-181.
83 Piccini P, Burn DJ, Ceravolo R, et al. The role of inheritance in sporadic Parkinson’s disease: evidence from a longitudinal study of dopaminergic function in twins. Ann Neurol. 1999;45:577-582.
84 Remy P, Samson Y, Hantraye P, et al. Clinical correlates of 18F-dopa uptake in five grafted parkinsonian patients. Ann Neurol. 1995;38:580-588.
85 Wenning GK, Odin P, Morrish P, et al. Short- and long-term survival and function of unilateral intrastriatal dopaminergic grafts in Parkinson’s disease. Ann Neurol. 1997;42:95-107.
86 Piccini P, Brooks DJ, Bjorklund A, et al. Dopamine release from nigral transplants visualized in vivo in a Parkinson’s patient. Nat Neurosci. 1999;2:1137-1140.
87 Hauser RA, Freeman TB, Snow BJ, et al. Long-term evaluation of bilateral fetal nigral transplantation in Parkinson disease. Arch Neurol. 1999;56:179-187.
88 Piccini P, Lindvall O, Bjorklund A, et al. Delayed recovery of movement-related cortical function in Parkinson’s disease after striatal dopaminergic grafts. Ann Neurol. 2000;48:689-695.
89 Pogarell O, Koch W, Gildehaus FJ, et al. Long-term assessment of striatal dopamine transporters in Parkinsonian patients with intrastriatal embryonic mesencephalic grafts. Eur J Nucl Med Mol Imaging. 2006;33:407-411.
90 Cochen V, Ribeiro MJ, Nguyen JP, et al. Transplantation in Parkinson’s disease: PET changes correlate with the amount of grafted tissue. Mov Disord. 2003;18:928-932.
91 Freed CR, Greene PE, Breeze RE, et al. Transplantation of embryonic dopamine neurons for severe Parkinson’s disease. N Engl J Med. 2001;344:710-719.
92 Olanow CW, Goetz CG, Kordower JH, et al. A double blind controlled trial of bilateral fetal nigral transplantation in Parkinson’s disease. Ann Neurol. 2003;54:403-414.
93 Ma Y, Feigin A, Dhawan V, et al. Dyskinesia after fetal cell transplantation for parkinsonism: a PET study. Ann Neurol. 2002;52:628-634.
94 Hagell P, Piccini P, Bjorklund A, et al. Dyskinesias following neural transplantation in Parkinson’s disease. Nat Neurosci. 2002;5:627-628.
95 Piccini P, Pavese N, Hagell P, et al. Factors affecting the clinical outcome after neural transplantation in Parkinson’s disease. Brain. 2005;128:2977-2986.
96 Mínguez-Castellanos A, Escamilla-Sevilla F, Hotton GR, et al. Carotid body autotransplantation in Parkinson disease: a clinical and positron emission tomography study. J Neurol Neurosurg Psychiatry. 2007;78:825-831.
97 Gill SS, Patel NK, Hotton GR, et al. Direct brain infusion of glial cell line-derived neurotrophic factor in Parkinson disease. Nat Med. 2003;9:589-595.
98 Lang AE, Gill S, Patel NK, et al. Randomized controlled trial of intraputamenal glial cell line-derived neurotrophic factor infusion in Parkinson disease. Ann Neurol. 2006;59:459-466.
99 Eidelberg D, Moeller JR, Ishikawa T, et al. Regional metabolic correlates of surgical outcome following unilateral pallidotomy for Parkinson’s disease. Ann Neurol. 1996;39:450-459.
100 Grafton ST, Waters C, Sutton J, et al. Pallidotomy increases activity of motor association cortex in Parkinson’s disease—a positron emission tomographic study. Ann Neurol. 1995;37:776-783.
101 Samuel M, Ceballos-Baumann AO, Turjanski N, et al. Pallidotomy in Parkinson’s disease increases SMA and prefrontal activation during performance of volitional movements: An H215O PET study. Brain. 1997;120:1301-1313.
102 Davis KD, Taub E, Houle S, et al. Globus pallidus stimulation activates the cortical motor system during alleviation of parkinsonian symptoms. Nat Med. 1997;3:671-674.
103 Limousin P, Greene J, Pollak P, et al. Changes in cerebral activity pattern due to subthalamic nucleus or internal pallidum stimulation in Parkinson’s disease. Ann Neurol. 1997;42:283-291.
104 Eidelberg D, Nakamura T, Mentis M, et al. Brain activation responses with internal pallidal stimulation in Parkinson’s disease. Neurology. 1999;52(Suppl 2):A176.
105 Hilker R, Voges J, Weisenbach S, et al. Subthalamic nucleus stimulation restores glucose metabolism in associative and limbic cortices and in cerebellum: evidence from a FDG-PET study in advanced Parkinson’s disease. J Cereb Blood Flow Metab. 2004;24:7-16.
106 Zhao YB, Sun BM, Li DY, et al. Effects of bilateral subthalamic nucleus stimulation on resting-state cerebral glucose metabolism in advanced Parkinson’s disease. Chin Med J. 2004;117:1304-1308.
107 Li D, Zuo C, Guan Y, Zhao Y, et al. FDG-PET study of the bilateral subthalamic nucleus stimulation effects on the regional cerebral metabolism in advanced Parkinson disease. Acta Neurochir Suppl. 2006;99:51-54.
108 Arai N, Yokochi F, Ohnishi T, et al. Mechanisms of unilateral STN-DBS in patients with Parkinson’s disease: A PET study. J Neurol. 2008;255:1236-1243.
109 Ceballos-Baumann AO, Boecker H, Bartenstein P, et al. A positron emission tomographic study of subthalamic nucleus stimulation in Parkinson disease: enhanced movement-related activity of motor-association cortex and decreased motor cortex resting activity. Arch Neurol. 1999;56:997-1003.
110 Thobois S, Dominey P, Fraix V, et al. Effects of subthalamic nucleus stimulation on actual and imagined movement in Parkinson’s disease: a PET study. J Neurol. 2002;249:1689-1698.
111 Strafella AP, Dagher A, Sadiko AF. Cerebral blood flow changes induced by subthalamic stimulation in Parkinson’s disease. Neurology. 2003;60:1039-1042.
112 Grafton ST, Turner RS, Desmurget M, et al. Normalizing motor-related brain activity: subthalamic nucleus stimulation in Parkinson disease. Neurology. 2006;66:1192-1199.
113 Benazzouz A, Gao D, Ni Z, Benabid AL. High frequency stimulation of the STN influences the activity of dopamine neurons in the rat. Neuroreport. 2000;11:1593-1596.
114 Bruet N, Windels F, Bertrand A, et al. High frequency stimulation of the subthalamic nucleus increases the extracellular contents of striatal dopamine in normal and partially dopaminergic denervated rats. J Neuropathol Exp Neurol. 2001;60:15-24.
115 Thobois S, Fraix V, Savasta M, et al. Chronic subthalamic nucleus stimulation and striatal D2 dopamine receptors in Parkinson’s disease—a 11C-raclopride PET study. J Neurol. 2003;250:1219-1223.
116 Strafella AP, Sadikot AF, Dagher A. Subthalamic deep brain stimulation does not induce striatal dopamine release in Parkinson’s disease. Neuroreport. 2003;14:1287-1289.
117 Hilker R, Voges J, Ghaemi M, et al. Deep brain stimulation of the subthalamic nucleus does not increase the striatal dopamine concentration in parkinsonian humans. Mov Disord. 2003;18:41-48.
118 Hilker R, Portman AT, Voges J, et al. Disease progression continues in patients with advanced Parkinson’s disease and effective subthalamic nucleus stimulation. J Neurol Neurosurg Psychiatry. 2005;76:1217-1221.
119 Feigin A, Kaplitt MG, Tang C, et al. Modulation of metabolic brain networks after subthalamic gene therapy for Parkinson’s disease. Proc Natl Acad Sci U S A. 2007;104:19559-19564.
120 Strafella AP, Lozano AM, Ballanger B, et al. rCBF changes associated with PPN stimulation in a patient with Parkinson’s disease: a PET study. Mov Disord. 2008;23:1051-1054.
121 Tai YF, Pavese N, Gerhard A, et al. Microglial activation in presymptomatic Huntington’s disease gene carriers. Brain. 2007;130:1759-1766.
122 Grafton ST, Mazziotta JC, Pahl JJ, et al. Serial changes of cerebral glucose metabolism and caudate size in persons at risk for Huntington’s disease. Arch Neurol. 1992;49:1161-1167.
123 Hussey D, Stewart D, Houle S, et al. C-11Raclopride striatal binding potential as a measure of Huntington’s disease progression: implications for prospective neuroprotective studies [abstract]. J Nucl Med. 1998;39:209P.
124 Torres EM, Fricker RA, Hume SP, et al. Assessment of striatal graft viability in the rat in vivo using a small diameter PET scanner. Neuroreport. 1995;6:2017-2021.
125 Kendall AL, Rayment FD, Torres EM, et al. Functional integration of striatal allografts in a primate model of Huntington’s disease. Nat Med. 1998;4:727-729.
126 Palfi S, Condé F, Riche D, et al. Fetal striatal allografts reverse cognitive deficits in a primate model of Huntington disease. Nat Med. 1998;4:963-966.
127 Bachoud-Levi AC, Rémy P, Nguyen JP, et al. Motor and cognitive improvements in patients with Huntington’s disease after neural transplantation. Lancet. 2000;356:1975-1979.
128 Gaura V, Bachoud-Levi AC, Ribeiro MJ, et al. Striatal neural grafting improves cortical metabolism in Huntington’s disease patients. Brain. 2004;127:65-72.
129 Furtado S, Sossi V, Hauser RA, et al. Positron emission tomography after fetal transplantation in Huntington’s disease. Ann Neurol. 2005;58:331-337.
130 Freeman TB, Cicchetti F, Hauser RA, et al. Transplanted fetal striatum in Huntington’s disease: phenotypic development and lack of pathology. Proc Natl Acad Sci U S A. 2000;97:13877-13882.
131 Reuter I, Tai YF, Pavese N, et al. Long term clinical and PET outcome of foetal striatal transplantation in Huntington’s disease. J Neurol Neurosurg Psychiatry. 2008;79:948-951.
132 Rakshi JS, Pavese N, Uema T, et al. A comparison of the progression of early Parkinson’s disease in patients started on ropinirole or L-dopa: an 18F-dopa PET study. J Neural Transm. 2002;109:1433-1443.
133 Parkinson Study Group. Dopamine transporter brain imaging to assess the effects of pramipexole vs levodopa on Parkinson disease progression. JAMA. 2002;287:1653-1661.
134 Whone AL, Watts RL, Stoessl AJ, et al. Slower progression of Parkinson’s disease with ropinirole versus levodopa: The REAL-PET study. Ann Neurol. 2003;54:93-101.
135 Pavese N, Rascol O, Olanow WC, et al. The effects of riluzole on progression of early Parkinson’s disease: An 18F-fluorodopa PET study. Parkinsonism Relat Disord. 2005;11(Suppl 2):229.






