CHAPTER 73 FRONTOTEMPORAL DEMENTIA
After Alzheimer’s disease, Lewy body disease, and vascular dementia, frontotemporal dementias (FTDs) as a group constitute a significant percentage of the degenerative dementias, accounting for 5% to 7% of some autopsy series. FTD is overrepresented among the early-onset dementias (manifesting before the age of 70), accounting for 8% to 17% of patients in such cases. Arnold Pick provided the first clinical and pathological description of an aphasic dementia in 1892.1 The term Pick’s disease was subsequently used to describe patients with behavioral changes and circumscribed atrophy affecting frontal and temporal lobes of the brain. Mesulam (2001) later described the progressive aphasias in relation to preferential left hemisphere degeneration.2 Advances in the study of dementia have improved genetic and biochemical characterization of this group of disorders and are discussed in this chapter. Case examples are provided in order to familiarize the reader with approaches to evaluation of these disorders. Future directions of research are also discussed.
CLINICAL FEATURES
Varied terminology makes the literature on FTD somewhat confusing. This ultimately resulted in a consensus among experts in the field on preferred terminology (Table 73-1). Neary and associates (1998) outlined the clinical features commonly encountered in frontotemporal lobar degeneration (so-called Neary criteria).3 Pick’s complex of diseases is another term proposed for this group of disorders by Kertesz and colleagues (1998),4 but at present, frontotemporal dementia is the preferred term. The Neary classification describes three major clinical syndromes (Tables 73-2 to 73-4). The progressive aphasias include nonfluent primary progressive aphasia (PPA) and semantic dementia, which involve degeneration affecting the left frontal and temporal lobes respectively. The behavioral manifestation of FTD involves degeneration of both frontal lobes, although asymmetrical degeneration can occur. Although uncommon, cases of primarily right temporal degeneration exist with clinical features of prosopagnosia and/or associative agnosia. Unfortunately, clinical manifestations of FTD that do not strictly conform to the proposed criteria also occur. For example, aphasic dementias that do not meet criteria for PPA or semantic dementia exist, and some patients otherwise meeting criteria for an FTD syndrome may exhibit symptoms implicating parietal lobe involvement. The aphasic and behavioral manifestations of FTD can also overlap.
* A problem with this preferred terminology is the persistent use of overlapping abbreviations for both clinical and pathologic aspects of these disorders, such as “FTD.”
TABLE 73-2 Frontal Lobe Dementia
Adapted from Neary D, Snowden JS, Gustafson L, et al: Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 1998; 51:1546-1554.
TABLE 73-3 Progressive Nonfluent Aphasia
Adapted from Neary D, Snowden JS, Gustafson L, et al: Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 1998; 51: 1546-1554.
TABLE 73-4 Semantic Dementia/Progressive Fluent Aphasia
Adapted from Neary D, Snowden JS, Gustafson L, et al: Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 1998; 51:1546–1554.
Frontotemporal Dementia with Parkinsonism
FTD with parkinsonism linked to chromosome 17 (FTDP-17) is a rare clinical syndrome with autosomal dominant inheritance and characterized by progressive cognitive and behavioral disturbance and parkinsonism. Genetic analysis of affected families localized the mutations to 17q21-22, in the gene coding for the microtubule-associated protein tau.5 Clinical and pathological variabilities exist even among affected family members with the same mutation, presumably in relation to other differing genetic or environmental factors.
Frontotemporal Dementia with Motor Neuron Disease
Clinical features of FTD and motor neuron disease can coexist in the same individual.6 There is no predictable temporal relationship between the cognitive and motor features of the syndrome. Also of interest are cases of clinically diagnosed FTD lacking motor symptoms in which pathological findings of motor neuron disease are encountered. Just as in classic amyotrophic lateral sclerosis, motor symptoms of dysphagia or respiratory failure adversely affect prognosis in patients with FTD.
Corticobasal Degeneration
Corticobasal degeneration is a pathological diagnosis and is clinically characterized by asymmetrical rigidity, apraxia, and alien limb phenomena. However, many patients with corticobasal degeneration do not have asymmetrical rigidity and apraxia.7 Relevant to this discussion is that pathological corticobasal degeneration may manifest clinically as FTD. This furthers the concept that both clinical and pathological findings implicating involvement of regions other than the frontal and temporal lobes should not prevent the diagnosis of FTD.
PATHOLOGY
Nearly a century after Pick first described the pathological features of frontotemporal degeneration, different pathological subtypes were described as well.8 Three major pathological divisions were identified: Pick’s disease type A, with neuronal loss, astrocytic gliosis, Pick bodies, and swollen neurons; Pick’s disease type B, with neuronal loss, astrocytic gliosis, and swollen neurons; and Pick’s disease type C, with neuronal loss and gliosis. Additional advances in the field of pathology have allowed further characterization of the FTDs through immunohistochemical and biochemical techniques.
Further classification of FTD is achieved through the use of microscopy. Affected cortical regions typically show neuronal loss, microvacuolation, astrocytic gliosis centered on cortical layer II, and, in some cases, ballooned neurons. A major biochemical subdivision of degenerative dementias, including FTD, involves the presence or absence of tau protein–related pathology. There are more than 20 recognized “tauopathies,” as shown in Table 73-5,9 with Pick’s disease, corticobasal degeneration, FTDP-17, PSP, and Alzheimer’s disease most commonly identified in cases with a clinical manifestation of FTD. Assays for ubiquitin-positive inclusions are required when no tau, synuclein, or amyloid pathology is identified. Intranuclear or cytoplasmic ubiquitin inclusions, as well as ubiquitinated neurites (axons and dendrites), may be noted. Such inclusions may also be found in motor neurons in amyotrophic lateral sclerosis, which furthers the overlap between motor neuron disease and FTD. Intraneuronal ubiquitin inclusions have been reported in familial cases of FTD, and a comprehensive review of the neuropathology of FTD is presented by Munoz and colleagues (2003).10 Neuronal intranuclear ubiquitin inclusions appear to be the distinction between familial and sporadic cases of FTD with ubiquitin immunoreactivity.11 Presence of neuronal loss, gliosis, and microvacuolation in the absence of any recognizable inclusions defines dementia lacking distinctive histological features.12 This entity less commonly identified as careful immunohistochemical analysis often reveals ubiquitin pathology. In addition to the identification of ubiquitin-immunoreactive inclusions, in many cases initially characterized as dementia lacking distinctive histological features, there also appears to be a high prevalence of hippocampal sclerosis.13
TABLE 73-5 Diseases in Which Filamentous Tau Protein Deposits Have Been Described
It is possible to further classify the tau protein–based forms of FTD. Six isoforms of the tau protein exist, one half referred to as “three-repeat forms” and the remainder as “four-repeat forms.” This terminology refers to the presence of three or four repeated sequences in the tau protein, representing microtubule binding sites. The fourth repeated sequence is coded for by exon 10 of the tau gene; alternate splicing of exon 10 generates three-repeat or four-repeat isoforms. Tauopathies characterized by four-repeat isoforms include corticobasal degeneration, PSP, and argyrophilic grain disease, whereas Pick’s disease is characterized by four-repeat isoforms, shown in Figure 73-1.
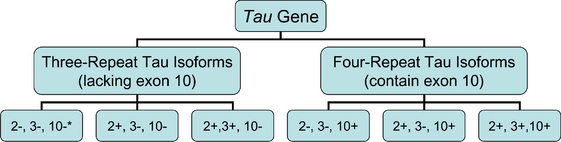
GENETICS
Tau Gene
Tau mutations account for only a small percentage of FTD in general, although they constitute the most recognized genetic cause of FTD. FTDP-17 is inherited in an autosomal dominant manner; multiple kindreds have been recognized in many countries. Three mutations account for more than one half of the known cases of FTDP-17, despite there being more than 30 known mutations. These are the P301L mutation, the mutation of the 5′ splice site of exon 10 at position +16, and the N279K mutation, depicted in Figure 73-2. Each mutation can be associated with a classic FTD phenotype, although the +16 mutation and N279K mutation typically produce features of parkinsonism, the latter with features resembling PSP. Other features encountered in FTDP-17 include paranoid delusions, antisocial behavior, seizures, and, on occasion, amyotrophy. Most mutations affecting the splicing of exon 10 are associated with parkinsonism. Clinical differences among individuals harboring the same mutation suggest that other genetic or environmental factors influence phenotype.
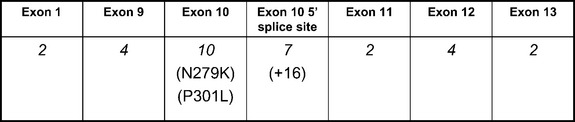
Knowledge of tau protein biochemistry facilitates recognition of the effects of tau gene mutations, with an excellent review by Hutton (2000). The tau protein is soluble in the normal human brain. Microtubule binding is mediated by specific domains in the carboxy-terminal portion of the protein, coded for by exons 9, 10, 11, and 12 of the tau gene. These domains are composed of tandem repeat sequences of 31 amino acids with either three or four microtubule-binding domains present on the protein. Alternative splicing of exon 10 dictates the number of microtubule binding domains, and an approximate 1 : 1 ratio of three-repeat and four-repeat tau isoforms exists in the normal human brain. Disruption of this balance between three-repeat and four-repeat isoforms is associated with the pathological inclusions of FTDP-17. Nearly all mutations that have been identified in families with FTDP-17 are located from exons 9 to 13, or in the 5′ splice site of the intron after exon 10. Mutations in exon 10 and the intronic 5′ splice site after exon 10 (see Fig. 73-2) appear to increase the amount of four-repeat tau isoforms produced. The abnormal ratio of tau isoforms appears to decrease tau microtubule binding and to increase tau aggregation into insoluble filaments. Despite increased knowledge of the biochemical effects of these mutations, clinical heterogeneity remains largely unexplained. For example, phenotypes resembling the behavioral manifestations of FTD, corticobasal degeneration, and PSP have all been reported with the P301L mutation.
Tau Haplotype
There are two extended tau haplotypes (H1 and H2), consisting of a series of polymorphisms throughout the tau gene in linkage disequilibrium. The tau H1 haplotype is associated with PSP and corticobasal degeneration; it accounts for 90% of the haplotypes in affected patients, in comparison with 78% in the normal population.14 The tau H2 haplotype appears more common in the SD subtype of FTD when compared to other FTD subtypes (Short, Graff-Radford, et al, 2002).15
Apolipoprotein E Genotype
Although the apolipoprotein E (apoE) genotype is a known genetic risk factor for Alzheimer’s disease, its effect on FTD remains controversial. This largely stems from the same problem of FTD classification: Different studies involved different clinical and pathological criteria. For example, the apoE4 allele has been reported to be overrepresented in both Pick’s disease with Pick bodies16 and in patients with semantic dementia.15
Other Loci
Familial forms of FTD have also been reported with linkage to chromosomes 3 and 9. The former involves a kindred with clinical FTD and pathology that is consistent with dementia lacking distinctive histology,17 whereas the latter is characterized by affected individuals expressing clinical features of motor neuron disease.18 In several kindreds with familial FTD, the disease has been linked to chromosome 17q21-22 with no mutation in the tau gene identified.19
Genetic Testing
Clinical genetic testing for known tau gene mutations is available (Table 73-6). As with other untreatable genetic disorders such as Huntington’s disease, caution should be used when arranging for genetic testing. Referral for genetic counseling would be appropriate before testing.
TABLE 73-6 Centers Providing Clinical Screening for Tau Gene Mutations
CLINICAL VIGNETTES
Cases 1 through 4 illustrate the clinical features outlined in the Neary criteria (see Table 73-1).
Case 1: Frontal Lobe Dementia
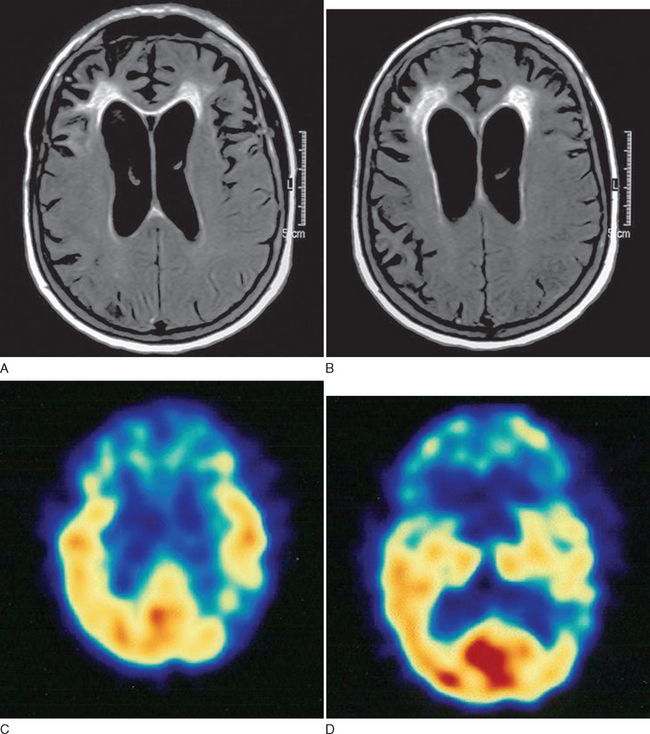
This case demonstrates typical clinical features of frontal lobe dementia in which prominent behavioral changes dominate the initial symptom complex. The family history is suggestive of a hereditary basis for her particular syndrome. Figure 73-3 demonstrates typical imaging findings in frontal lobe dementia.
Case 2: Progressive Nonfluent Aphasia
This case is illustrative of progressive nonfluent aphasia, a subtype of PPA. The preservation of general cognitive function despite the language disturbance is typical, as is the early absence of any significant behavioral disturbance (Fig. 73-4).
Case 3: Semantic Dementia
There was a family history of memory difficulties in the patient’s mother in her 80s but no clear language disturbance.
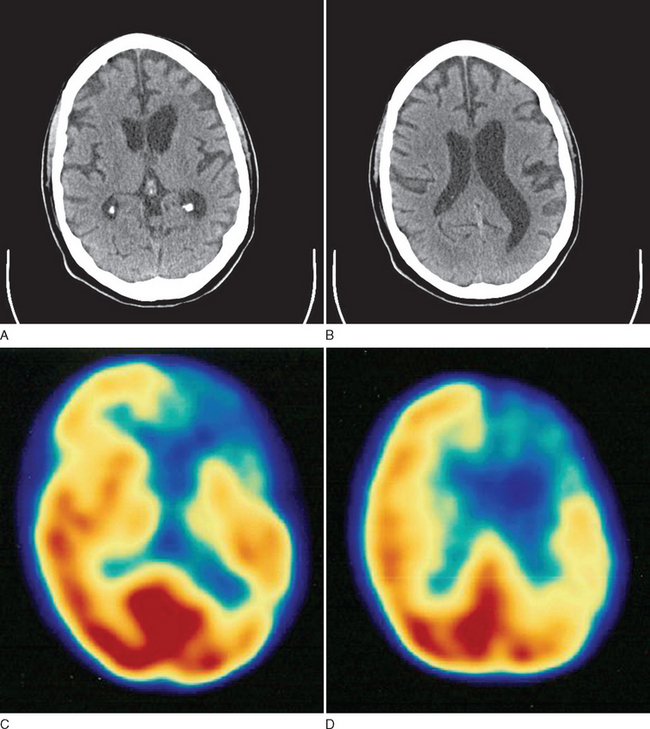
This case demonstrates typical history and examination findings for semantic dementia, a subtype of PPA. Patients generally exhibit prominent anomia and progressively lose the meaning of words, initially less common ones but then even common words as the disease progresses. Figure 73-5 shows typical imaging findings in a case of semantic dementia.
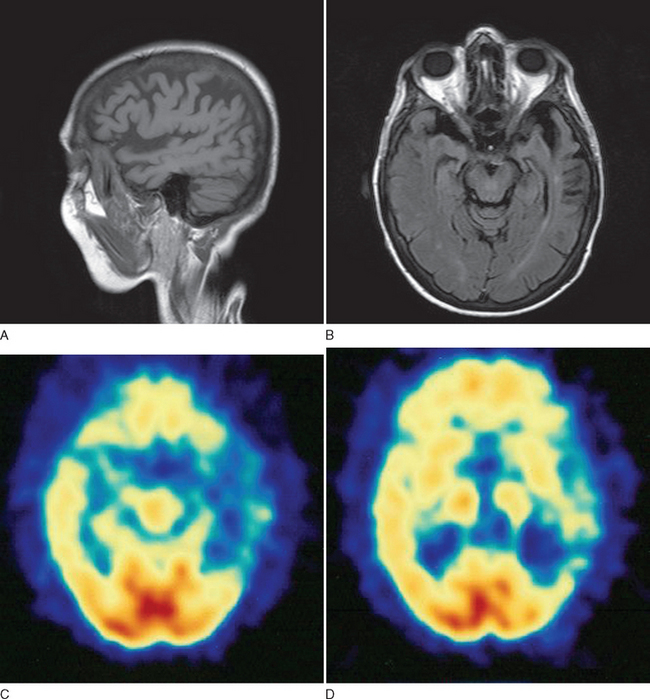
Case 4: Progressive Prosopagnosia
A 73-year-old right-handed mechanic related a 5-year history of progressive difficulties recognizing people. These difficulties first involved acquaintances he encountered while at the store or church, but over time they progressed to include even family members. He often would greet people when addressed and would be able to recognize them only by the sound of their voice. This difficulty extended beyond recognizing people: He also found that he had developed problems recognizing different makes and models of vehicles, which he found quite frustrating in light of his prior occupation.
On examination, he scored 28 of 38 on a Short Test of Mental Status, missing points for digit span, learning, calculation, construction, and delayed recall. He could not recognize pictures of celebrities from a magazine, although he knew they were “famous people.” When shown a picture of a grizzly bear standing in a mountain stream and was asked what was depicted, he stated, “an animal.” When asked to elaborate, he speculated, “It might be a dog.” He exhibited moderate anomia on the Boston Naming Test during neuropsychological assessment. His general neurological examination findings were unremarkable. Laboratory studies were unrevealing. MRI of his brain showed bitemporal atrophy, greater on the right side than on the left.
This case demonstrates a case of progressive prosopagnosia with prominent difficulties recognizing familiar faces and even objects. The ability to recognize categories of items such as “vehicles” or “animals” with inability to subcategorize them can be seen and was evident in this particular patient. Figure 73-6 demonstrates typical imaging findings in a case of prosopagnosia related to prominent right temporal lobe degeneration.
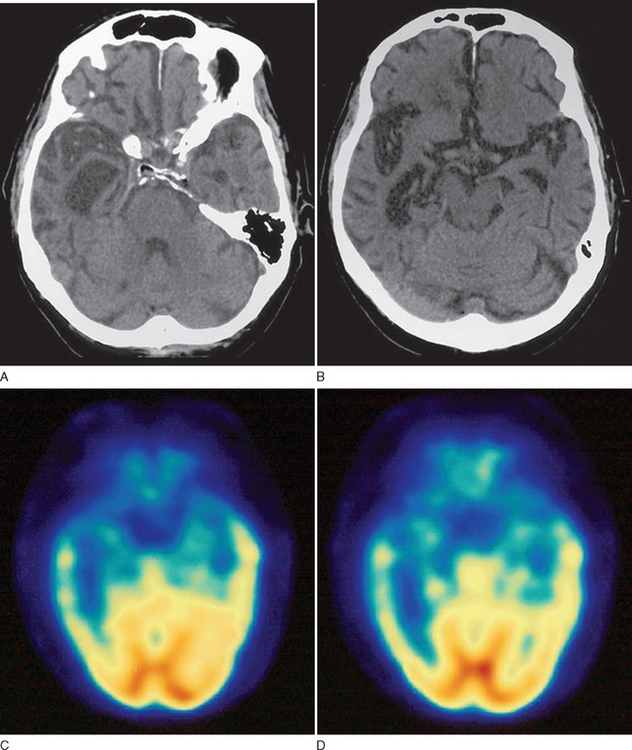
IMAGING
In all patients presenting with progressive impairment of cognitive or behavioral functions, an imaging study of the brain is warranted, to rule out mass lesions or other structural abnormalities. For example, a tumor involving the frontal lobes could produce manifestations mimicking the clinical features of FTD. In FTD, the frontal and temporal lobe atrophy is often evident on standard computed tomography and MRI of the brain. Asymmetrical atrophy may be evident in the setting of PPA and semantic dementia. Longitudinal volumetric MRI demonstrates predictable patterns of regional change among the subtypes of FTD; semantic dementia exhibits the most dramatic longitudinal change in the temporal lobe.20 White matter abnormalities, best appreciated on fluid-attenuated inversion recovery MRI sequences, may also be evident in the underlying affected cortical regions. Functional neuroimaging may be helpful, especially in cases lacking obvious regional atrophy. Regional hypoperfusion on single photon emission computed tomography or hypometabolism on positron emission tomography may be evident in the frontal and temporal lobes, but further clinicopathological data are necessary to determine the diagnostic utility of these two imaging modalities.
THERAPY
There are no approved pharmacological therapies for FTD. Off-label use of cholinesterase inhibitors and memantine for the cognitive symptoms of FTD occurs, but their efficacy is not established. A randomized controlled trial of trazodone also demonstrated promising results for behavioral symptoms.21 Atypical neuroleptic agents and anticonvulsants with mood-stabilizing properties can also be considered.
Patients with PPA or semantic dementia may benefit from formal speech therapy. Caregiver burden is an important issue for all dementing illnesses, but because of the relatively younger age at onset in patients with FTD, this problem can be particularly difficult. Educational material about available community resources such as the Alzheimer’s Association should be made available. Social workers or geriatric counselors can provide invaluable information to patients and their families regarding care options. Internet resources for FTD are listed in Table 73-7.
| The Association for Frontotemporal Dementias | http://www.ftd-picks.org/ |
| Pick’s Disease Support Group | http://www.pdsg.org.uk |
| Northwestern Alzheimer’s Disease Center Primary Progressive Aphasia (PPA) Program | http://www.brain.northwestern.edu/ppa/ppa.html |
| University of California, San Francisco, Memory and Aging Center | http://memory.ucsf.edu/Education/Disease/ftd.html |
| Alzheimer’s Association | http://www.alz.org/ |
| National Institute of Neurological Disorders and Stroke (NINDS) | www.ninds.nih.gov |
| GeneTests Web Site | http://www.geneclinics.org/ |
| Online Mendelian Inheritance in Man (OMIM) | http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=OMIM |
FUTURE DIRECTIONS
In summary, the term frontotemporal dementia encompasses a group of disorders that have diverse clinical, pathological, biochemical, and genetic features. Better understanding of these disorders will be achieved only through collaboration of specialists in both the clinical and basic science disciplines. A multicenter initiative to monitor patients with FTD longitudinally is already under way and is hoped to enable additional therapeutic trials in the future. Powerful research tools such as transgenic animal models will expedite development of therapies specifically targeting the underlying pathophysiology of FTD and other degenerative disorders affecting the nervous system.22 Development of assays for biomarkers such as phosphorylated tau species may allow more accurate differentiation between FTD and other neurodegenerative disorders, especially early in the disease course.23 Although research focusing on FTD may appear daunting in light of the heterogeneity of the condition, any advances made will potentially affect patients with a host of other related disorders and should therefore be avidly pursued.
ACKNOWLEDGMENTS
Hutton M. Molecular genetics of chromosome 17 tauopathies. Ann N Y Acad Sci. 2000;920:63-73.
Kertesz A, Munoz D. Pick’s disease, frontotemporal dementia, and Pick complex: emerging concepts. Arch Neurol. 1998;55:302-304.
Mesulam MM. Primary progressive aphasia. Ann Neurol. 2001;49:425-432.
Munoz DG, Dickson DW, Bergeron C, et al. The neuropathology and biochemistry of frontotemporal dementia. Ann Neurol. 2003;54(Suppl 5):S24-S28.
Neary D, Snowden JS, Gustafson L, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology. 1998;51:1546-1554.
1 Pick A. Uber die Beziehungen der senilen Hirnantropie zur aphasie. Prager Medizinishe Wochenscrift. 1892;17:165-167.
2 Mesulam MM. Primary progressive aphasia. Ann Neurol. 2001;49:425-432.
3 Neary D, Snowden JS, Gustafson L, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology. 1998;51:1546-1554.
4 Kertesz A, Munoz D. Pick’s disease, frontotemporal dementia, and Pick complex: emerging concepts. Arch Neurol. 1998;55:302-304.
5 Hutton M. Molecular genetics of chromosome 17 tauopathies [Review]. Ann N Y Acad Sci. 2000;920:63-73.
6 Strong MJ, Lomen-Hoerth C, Caselli RJ, et al. Cognitive impairment, frontotemporal dementia, and the motor neuron diseases. Ann Neurol. 2003;54(Suppl 5):S20-S23.
7 Boeve BF, Maraganore DM, Parisi JE, et al. Pathologic heterogeneity in clinically diagnosed corticobasal degeneration. Neurology. 1999;53:795-800.
8 Constantinidis J, Richard J, Tissot R. Pick’s disease. Histological and clinical correlations. Eur Neurol. 1974;11:208-217.
9 Goedert M. Introduction to the tauopathies. In: Dickson D, editor. Neurodegeneration: The Molecular Pathology of Dementia and Movement Disorders. Basel, Switzerland: ISN Neuropath Press; 2003:82-85.
10 Munoz DG, Dickson DW, Bergeron C, et al. The neuropathology and biochemistry of frontotemporal dementia. Ann Neurol. 2003;54(Suppl 5):S24-S28.
11 Mackenzie IR, Feldman H. Neuronal intranuclear inclusions distinguish familial FTD-MND type from sporadic cases. Dement Geriatr Cogn Disord. 2004;17:333-336.
12 Knopman DS, Mastri AR, Frey WH2nd, et al. Dementia lacking distinctive histologic features: a common non-Alzheimer degenerative dementia. Neurology. 1990;40:251-256.
13 Josephs KA, Jones AG, Dickson DW. Hippocampal sclerosis and ubiquitin-positive inclusions in dementia lacking distinctive histopathology. Dement Geriatr Cogn Disord. 2004;17:342-345.
14 Baker M, Litvan I, Houlden H, et al. Association of an extended haplotype in the tau gene with progressive supranuclear palsy. Hum Mol Genet. 1999;8:711-715.
15 Short R, Graff-Radford N, Adamson J, et al. Differences in tau and apolipoprotein E polymorphism frequencies in sporadic frontotemporal lobar degeneration syndromes. Arch Neurol. 2002;59:611-615.
16 Kalman J, Juhasz A, Majtenyi K, et al. Apolipoprotein E polymorphism in Pick’s disease and in Huntington’s disease. Neurobiol Aging. 2000;21:555-558.
17 Brown J. Chromosome 3-linked frontotemporal dementia [Review]. Cell Mol Life Sci. 1998;54:925-927.
18 Hosler BA, Siddique T, Sapp PC, et al. Linkage of familial amyotrophic lateral sclerosis with frontotemporal dementia to chromosome 9q21-q22. JAMA. 2000;284:1664-1669.
19 Ghetti B, Hutton M, et al. Frontotemporal dementia and parkinsonism linked to chromosome 17 associated with tau gene mutations (FTDP-17T). Dickson D, editor. Neurodegeneration: The Molecular Pathology of Dementia and Movement Disorders. vol 1. Basel, Switzerland: ISN Neuropath Press; 2003:86-102.
20 Whitwell JL, Anderson VM, Scahill RI, et al. Longitudinal patterns of regional change on volumetric MRI in frontotemporal lobar degeneration. Dement Geriatr Cogn Disord. 2004;17:307-310.
21 Lebert F, Stekke W, Hasenbroekx C, et al. Frontotemporal dementia: a randomised, controlled trial with trazodone. Dement Geriatr Cogn Disord. 2004;17:355-359.
22 Hutton M, Lewis J, Dickson D, et al. Analysis of tauopathies with transgenic mice [Review]. Trends Mol Med. 2001;7:467-470.
23 Hampel H, Teipel SJ. Total and phosphorylated tau proteins: evaluation as core biomarker candidates in frontotemporal dementia. Dement Geriatr Cogn Disord. 2004;17:350-354.






