68 Friedreich’s ataxia
Salient features
History
Examination
• Cerebellar signs, ataxia being a constant sign
• Impaired vibration and joint sense
• Absence of deep tendon reflexes (caused by degeneration of peripheral nerves)
• Distal muscle wasting (in 50% of cases), especially in the hands.
• Check for nystagmus (present in 25% of the cases), scanning speech, intention tremor
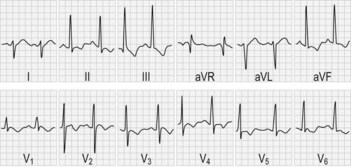
• Examine the heart for hypertrophic cardiomyopathy (Fig. 68.2)
• Check the eyes for optic atrophy (present in 30% of cases)
• Check the spine for kyphoscoliosis
• Check the urine for sugar (10% of patients have diabetes)
• Check IQ, looking for intellectual deterioration
• Tell the examiner that you would like to do a 25 ft (7.5 m) walk test to determine ambulatory capacity.
Advanced-level questions
Why are the deep tendon reflexes absent even though plantars are upgoing?
This is caused by a combination of pyramidal weakness with peripheral neuropathy.
What is the forme fruste of this condition?
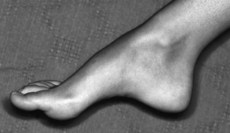
Pes cavus or hammer toes, without any other signs, are seen in family members of such patients.
What are the clinical criteria for diagnosis of Friedreich’s ataxia?
Harding’s criteria (Brain 1981;104:589):
• Essential criteria are onset before the age of 25 years, ataxia of limbs and gait, absent knee and ankle jerks, extensor plantars, autosomal recessive inheritance, motor conduction velocity greater than 40 m/s, small or absent sensory nerve action potentials, dysarthria within 5 years of onset.
• Additional criteria (present in two-thirds) are scoliosis, pyramidal weakness of lower limbs, absent upper limb reflexes, loss of vibration and joint position sense in the legs, abnormal ECG and pes cavus.
• Other features (present in <50%) are nystagmus, optic atrophy, deafness, distal muscle wasting and diabetes.
Name a few syndromes with spinocerebellar degeneration
• Roussy–Lévy disease: hereditary spinocerebellar degeneration with atrophy of lower limb muscles and loss of deep tendon reflexes
• Bassen–Kornzweig syndrome (p. 724): caused by cellular deficiency of vitamin E (α-tocopherol) resulting from a defect in the α-tocopherol-transfer protein, and abetalipoproteinaemia associated with a defect of VLDL
• Olivopontocerebellar degeneration: first described in 1882. This has an autosomal dominant inheritance and has been mapped to the HLA loci on the short arm of chromosome 6 where a highly polymorphic CAG repeat sequence occurs. The CAG repeat sequence is longer than normal and unstable in affected patients
• Machado–Joseph disease: dominant inheritance, first described in families of Portuguese origin. Clinical features include progressive ataxia, ophthalmoparesis, spasticity, dystonia, amyotrophy and parkinsonism. This disorder has been linked to chromosome 14 and is caused by the expansion of unstable CAG repeat sequences
• Dentatorubral pallidoluysian atrophy is similar to Machado–Joseph disease but maps on the short arm of chromosome 12. The abnormally expanded CAG repeat sequences identified in the gene for olivopontocerebellar degeneration, Machado–Joseph disease and dentatorubral pallidoluysian atrophy each result in the expression of a specific ataxin.