CHAPTER 9 Food Allergies
BACKGROUND, DEFINITIONS, AND PREVALENCE
The first recorded account of food allergy was provided by Hippocrates, but it was not until 1921 that the classic experiment of Prausnitz initiated investigation on a scientific level and established the immunologic basis of allergic reactions.1 In this experiment, Prausnitz injected serum from his patient, Kustner, who was allergic to fish, into his own skin; the next day he injected fish extract into the same areas and into control sites. Local reactions proved sensitivity could be transferred by a factor in serum from an allergic to a nonallergic person. In 1950, Loveless demonstrated the inaccuracy of diagnosing food allergy by history in her report of the first blinded, placebo-controlled food trials in patients with milk allergy.2 In the following three decades, standardized protocols for the evaluation of food allergy were developed, and the double-blind, placebo-controlled oral food challenge (DBPCFC) emerged as the accepted standard for the diagnosis of food allergy.3
PREVALENCE
About 6% of young children and 3.5% of adults in the United States have food allergies.4 The prevalence of food allergies is greatest in the first few years of life and decreases over the first decade. The most common food allergens in young children include milk (2.5%), egg (1.5%), peanut (0.8%), wheat (∼0.4%), and soy (∼0.4%). Other than peanut, most childhood food allergies are outgrown by the end of the first decade. Almost all infants who develop cow’s milk allergy do so in the first year of life, with about two thirds experiencing IgE-mediated reactions and 35% going on to develop other food allergies.5 Peanut, tree nut, sesame, and seafood allergies tend to be lifelong, but about 20% of young children with peanut allergy develop clinical tolerance.6 Food allergies may persist after childhood into adulthood or develop in adulthood, with the most common food allergies in adults consisting of shellfish (2%), peanut (0.6%), tree nuts (0.4%), and fish (0.4%).7 About 5% of the U.S. population experiences allergic reactions to raw fruits and vegetables. Most of these reactions are caused by cross-reactivity between homologous proteins in pollens, such as ragweed, and certain fruits and vegetables, such as melons and bananas (oral allergy syndrome), in adolescents and adults who have seasonal allergic rhinitis. The prevalence of food allergies appears to be increasing.8 Studies from the United States and United Kingdom have indicated that the prevalence of peanut allergy has doubled in young children during the past decade.9,10 In addition, children with atopic disorders have a higher prevalence of food allergies; for example, 35% to 40% of children with moderate to severe atopic dermatitis have IgE-mediated food allergy.11
PATHOGENESIS
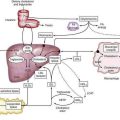
A single-cell layer of columnar intestinal epithelial cells (IECs) separates the external environment from the loosely organized lymphoid tissue of the lamina propria. A highly efficient gastrointestinal mucosal barrier has evolved, however, that provides an enormous surface area for processing and absorbing ingested food and discharging waste products.12 This barrier uses physiologic and immunologic barriers to prevent the penetration of foreign antigens (Table 9-1). The physiologic barrier is composed of the following: epithelial cells, joined by tight junctions and covered with a thick mucus layer that traps particles, bacteria, and viruses; trefoil factors (TFFs, 7 to 12 kd), protease-resistant proteins secreted by mucus-secreting cells of the stomach (TFF1, TFF2) and intestine (TFF3) that help strengthen and promote restoration of the barrier; and luminal and brush border enzymes, bile salts, and extremes of pH, all serving to destroy pathogens and render antigens non-immunogenic. Innate (natural killer [NK] cells, polymorphonuclear leukocytes, macrophages, epithelial cells, and Toll-like receptors) and adaptive immune (intraepithelial and lamina propria lymphocytes, Peyer’s patches, secretory immunoglobulin A [S-IgA] and cytokines) responses provide an active barrier to foreign antigens. Developmental immaturity of various components of the intestinal barrier and immune system reduces the efficiency of the infant mucosal barrier. For example, the activity of various enzymes is suboptimal in the newborn period and the S-IgA system is not fully mature until four years of age. This immature state of the mucosal barrier may play a role in the increased prevalence of gastrointestinal infections and food allergies seen in the first few years of life. In addition, studies have shown that alteration of the physiologic barrier function, such as gastric acidity, can lead to increased IgE sensitization in children and adults.13
Table 9-1 Physiologic and Immunologic Barriers of the Gastrointestinal Tract
SigA, secretory immunoglobulin A; IgG, immunoglobulin G.
Despite the evolution of this complex mucosal barrier, about 2% of ingested food antigens are absorbed and transported throughout the body in an immunologically intact form, even through the normal mature intestine.12 In an elegant series of experiments performed more than 75 years ago, Walzer and colleagues used sera from food-allergic patients to sensitize volunteers passively and demonstrated that immunologically intact antigens cross the mucosal barrier and disseminate rapidly throughout the body.14–16 Increased gastric acidity and the presence of food in the intestine decrease antigen absorption, whereas hypochlorhydria (e.g., H2 blocker– and proton pump inhibitor–induced) and ingestion of alcohol increase antigen absorption.15 The immunologically intact proteins that elude the intestinal barrier usually do not provoke adverse reaction, because most individuals have developed tolerance, but in a sensitized individual, allergic reactions will occur. Although more common in the developing GALT of young children, it is clear that cellular and IgE-mediated allergic responses to foods can develop at any age.
The dominant response in GALT is suppression, or tolerance. The means whereby the immune system is educated to avoid sensitization to ingested food antigens is not well understood, but studies have suggested that antigen-presenting cells, especially IECs and various dendritic cells, and regulatory T cells play a central role.12 High-dose tolerance is caused by lymphocyte anergy, resulting from antigen T cell receptor ligation in the absence of costimulatory signals, whereas low-dose tolerance is mediated by regulatory T cells. Five different regulatory T cells have been identified in conjunction with intestinal immunity: (1) Th3 cells, a population of CD4+ cells that secrete transforming growth factor-β (TGF-β); (2) Th1 cells, CD4+ cells that secrete interleukin-10 (IL-10); (3) CD4+,CD25+ regulatory T cells; (4) CD8+ suppressor T cells; and (5) γδ T cells.17 IECs have been shown to be nonprofessional antigen-presenting cells (APCs) that can process luminal antigen and present it to CD4+ T cells on class II major histocompatibility complex (MHC) molecules. IECs, however, lack a second signal necessary to activate T cells, thus suggesting there is another mechanism whereby these cells can induce tolerance to food antigens. Extracellular proteins that are internalized by professional APCs (e.g., monocytes, macrophages, dendritic cells) into vesicles are processed and displayed by class II MHC molecules to CD4+ T cells, whereas proteins in the cytosol of nucleated cells are processed and displayed by class I MHC molecules to CD8+ T cells. IECs also can present lipid and glycolipid antigens to CD8+ suppressor T cells by a nonclassic—that is, non-MHC, class I molecule (CD1d)—and other novel membrane molecules that interact with CD8+ T cells (see Fig. 9-1). In addition, dendritic cells residing within the lamina propria and noninflammatory environment of Peyer’s patches express IL-10 and IL-4, which favor the generation of tolerance. It has been suggested that T cells primed in the local mucosal environment induce tolerance, whereas T cells primed in the mesenteric lymph nodes, either from antigen reaching the nodes in lymph or carried there by circulating dendritic cells, differentiate and travel to the mucosa, where they induce local immune responses.18 Recently, the unique role of the oral mucosa and its relation to tolerance induction via Langerhans cells has been increasingly appreciated.19
It is likely that the commensal bowel flora also play a role in shaping the mucosal immune response. It is estimated that there are 1012 to 1014 bacteria/g of colonic tissue, which means that there are more bacteria in the colon than cells in the body.12 Bowel flora is largely established in the first 24 hours after birth, is dependent on maternal flora, genetics, and local environment, and is relatively stable throughout life. The importance of bowel flora in the development of oral tolerance induction is suggested by the fact that mice raised in a germ-free environment from birth fail to develop normal tolerance.20 Studies in which lactating mothers and their offspring were fed Lactobacillus suggest that probiotics may be beneficial in preventing some atopic disorders, such as eczema,21 but results from other studies are not consistent.
IECs also may play a central regulatory role in determining the rate and pattern of uptake of ingested antigens. Studies in sensitized rats have indicated that intestinal antigen transport proceeds in two phases.22 In the first phase, transepithelial transport occurs via endosomes, is antigen-specific and mast cell–independent, and occurs 10 times faster in sensitized rats compared with nonsensitized control animals. Antigen-specific IgE antibodies bound to the mucosal surface of IECs via FcεRII are responsible for this accelerated allergen entry.23 In the second phase, paracellular transport predominates. Loosening of the tight junctions occurs as a result of factors released by mast cells activated in the first phase. Whereas the first antigen-specific pathway involves antibody, the second nonspecific pathway most likely involves cytokines. Consistent with this concept, IECs express receptors for a number of cytokines (IL-1, IL-2, IL-6, IL-10, IL-12, IL-15, granulocyte-monocyte colony-stimulating factor [GM-CSF], and interferon-γ [IFN-γ]), and have been shown to be functionally altered by exposure to these cytokines.
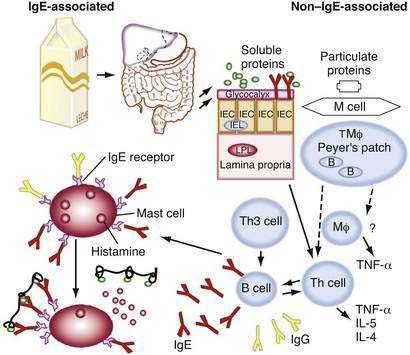
Although the development and mechanistic features of non–IgE-mediated food-allergic responses are poorly understood, the development of IgE-mediated responses has been well characterized. Sensitivity to allergens (generally glycoproteins) is the result of a series of molecular and cellular interactions involving APCs, T cells, and B cells.24 APCs present small peptide fragments (T cell epitopes) in conjunction with MHC class II molecules to T cells. T cells bearing the appropriate complementary T cell receptor (TCR) will bind to the peptide-MHC complex. This interactive first signal leads to T cell proliferation and cytokine generation and the generation of a second signal (e.g., IL-4) that promotes an IgE response (Th2-like cell activation). These cells and their products, in turn, interact with B cells bearing appropriate antigen-specific receptors, leading to isotype switching and the generation of antigen-specific IgE. At all stages, a number of specific cytokines are secreted that modulate the cell interactions. The antigen-specific IgE then binds to surface receptors of mast cells, basophils, macrophages, and other APCs, arming the immune system for an allergic reaction upon the next encounter with the specific antigen. A breakdown in mucosal integrity, caused by infection or other inflammatory processes, leads to increased intestinal permeability, which results in antigens bypassing the normal tolerogenic presentation by IECs and, under some circumstances, leads to allergic sensitization.
Oral tolerance of humoral and cellular immunity has been demonstrated in rodents and humans. Feeding of keyhole limpet hemocyanin to human volunteers resulted in T cell tolerance but priming of B cells at both mucosal and systemic sites.25 The failure of human infants to develop oral tolerance or the breakdown of oral tolerance in older individuals results in the development of food allergy. Young infants are more prone to develop food-allergic reactions because of the immaturity of their immunologic system and, to some extent, the gastrointestinal (GI) tract (see Table 9-1). Exclusive breast-feeding promotes the development of oral tolerance and may prevent some food allergy and atopic dermatitis.26,27 The protective effect of breast milk appears to be the result of several factors, including decreased content of foreign proteins, the presence of S-IgA (which provides passive protection against foreign protein and pathogens), and the presence of soluble factors (e.g., prolactin), which may induce earlier maturation of the intestinal barrier and the infant’s immune response. The antibacterial activity of breast milk is well established, but the ability of breast milk S-IgA to prevent food antigen penetration is less clear. Low concentrations of food-specific IgG, IgM, and IgA antibodies commonly are found in the serum of normal persons. Food protein–specific IgG antibodies tend to rise in the first months following the introduction of a food and then generally decline, even though the food protein continues to be ingested. Persons with various inflammatory bowel disorders (e.g., celiac disease, food allergy) frequently have high levels of food-specific IgG and IgM antibodies, although there is no evidence that these antibodies are pathogenic. Increased lymphocyte proliferation or IL-2 production following food antigen stimulation in vitro is frequently seen in patients with inflammatory bowel disorders, but it also occurs in normal persons.28 Antigen-specific T-cell proliferation in vitro alone does not represent a marker of immunopathogenicity, but simply reflects response to antigen exposure.
In genetically predisposed individuals, as noted, antigen presentation leads to excessive Th2 responsiveness (lymphocytes secreting IL-4, IL-5, IL-10, and IL-13), resulting in increased IgE production and expression of Fc epsilon (FcεI) receptors on a variety of cells.24 These IgE antibodies bind high-affinity FcεI receptors on mast cells, basophils, and dendritic cells, as well as low-affinity FcεII (CD23) receptors on macrophages, monocytes, lymphocytes, eosinophils, and platelets. When food allergens penetrate mucosal barriers and reach IgE antibodies bound to mast cells or basophils, the cells are activated and mediators (e.g., histamine, prostaglandins, and leukotrienes) are released that induce vasodilation, smooth muscle contraction, and mucus secretion, and lead to symptoms of immediate hypersensitivity. These activated mast cells also may release a variety of cytokines (e.g., IL-4, IL-5, IL-6, tumor necrosis factor-α [TNF-α], platelet-activating factor), which may induce the IgE-mediated late-phase inflammation. Various symptoms have been associated with IgE-mediated allergic reactions—generalized: shock; cutaneous: urticaria, angioedema, and a pruritic morbilliform rash; oral and GI: lip, tongue, and palatal pruritus and swelling, vomiting, and diarrhea; and upper and lower respiratory: ocular pruritus and tearing associated with nasal congestion, laryngeal edema, and wheezing. A rise in the plasma histamine level has been associated with the development of these symptoms after blinded food challenges.29 In IgE-mediated GI reactions, endoscopic observation has revealed local vasodilation, edema, mucus secretion, and petechial hemorrhage.30 Cell-mediated hypersensitivity reactions are believed responsible for allergic eosinophilic esophagitis and gastroenteritis (AEE, AEG). It is believed that activated T cells secrete IL-5 and other cytokines, attracting eosinophils and inducing the inflammatory response that causes the delayed onset of symptoms.31 Expansion studies of T cells from biopsy specimens of milk-induced AEG patients have revealed large numbers of CD4+ Th2 cells.32
CLINICAL FEATURES
As depicted in Table 9-2, a number of GI food hypersensitivity disorders have been described. Clinically, these disorders are generally divided into two main categories: IgE-Th2–mediated and non–IgE (cell)-mediated hypersensitivities. There are a number of other disorders, however, that may result in symptoms similar to food-allergic reactions; these must be excluded during the evaluation (Table 9-3).
Table 9-3 Disorders That Must Be Differentiated from Food Hypersensitivities
Long before IgE antibodies were identified, studies of food hypersensitivity focused on radiologic changes associated with immediate hypersensitivity reactions. In one of the first of these reports, hypertonicity of the transverse and pelvic colon and hypotonicity of the cecum and ascending colon were noted following wheat feeding to an allergic patient.33 In a later report, gastric retention, hypermotility of the small intestine, and colonic spasm were observed in 4 patients studied after administering barium containing specific food allergens.34 In a third study, fluoroscopy was used to compare the effect of barium contrast with and without food allergens in 12 food-allergic children35; gastric hypotonia and retention of the allergen test meal, prominent pylorospasm, and increased or decreased peristaltic activity of the intestines were noted.
In the late 1930s, the rigid gastroscope was used to observe reactions in the stomachs of allergic patients. One study evaluated patients with GI food allergy or wheezing exacerbated by food ingestion and control subjects.36 Thirty minutes after a food allergen was placed on the gastric mucosa, patients with GI food allergy had markedly hyperemic and edematous patches of thick gray mucus and scattered petechiae at these sites, similar to those reported earlier by Walzer, in passively sensitized intestinal mucosal sites.15 Only mild hyperemia of the gastric mucosa was noted in patients with wheezing provoked by food ingestion. Subsequent studies confirmed these earlier observations and established an IgE-mediated mechanism for the reactions30; they demonstrated food-specific IgE antibodies and increased numbers of intestinal mast cells prior to challenge in food-allergic patients compared with normal controls, and significant decreases in stainable mast cells and tissue histamine content following a positive food challenge.
IgE-MEDIATED DISORDERS
Pollen-Food Allergy Syndrome
The pollen-food allergy syndrome (oral allergy syndrome) is a form of immediate contact hypersensitivity confined predominantly to the oropharynx and rarely involving other target organs.37 Symptoms include the rapid onset of pruritus and angioedema of the lips, tongue, palate and throat, generally followed by a rapid resolution of symptoms, and most commonly associated with the ingestion of various fresh (uncooked) fruits and vegetables. Symptoms result from local IgE-mediated reactions to conserved homologous proteins (sequences of amino acids in peptide backbones shared by plant pollens and fruit and vegetable proteins that remain unchanged through evolution) that are heat-labile (i.e., readily destroyed by cooking) and shared by certain fruits, vegetables, and some plant pollens.38 Patients with seasonal allergic rhinitis (hay fever) secondary to birch or ragweed pollen sensitivity often are afflicted with this syndrome. For example, in up to 50% of patients with ragweed-induced allergic rhinitis, ingestion of melons (e.g., watermelon, cantaloupe, honeydew) and bananas will provoke oral symptoms,39–41 whereas in birch pollen–allergic patients, symptoms may develop following the ingestion of raw potatoes, carrots, celery, apples, hazelnuts, and kiwi. Diagnosis is based on classic history and positive prick skin tests (e.g., prick and prick—pricking the fresh fruit or vegetable with a needle and then pricking the skin of the patient) with the implicated fresh fruits or vegetables.42
Gastrointestinal Allergy
Gastrointestinal allergy is a relatively common form of IgE-mediated hypersensitivity, which generally accompanies allergic manifestations in other target organs (e.g., skin, airway) and results in a variety of symptoms.4 Symptoms typically develop within minutes to two hours of consuming a food and consist of nausea, abdominal pain, cramps, vomiting, and/or diarrhea. In some infants, frequent ingestion of a food allergen appears to induce partial desensitization of gastrointestinal mast cells resulting in a subclinical reaction, with the only symptom reported being poor appetite and periodic abdominal pain. Diagnosis is established by clinical history, evidence of food-specific IgE antibodies (positive skin prick tests or serum food-specific IgE antibodies), resolution of symptoms following complete elimination of the suspected food, and recurrence of symptoms following oral food challenges. GI allergy is common in IgE-mediated food allergies, with more than 50% of children experiencing abdominal symptoms during double-blind, placebo-controlled food challenges.28
Infantile Colic
Infantile colic is an ill-defined syndrome of paroxysmal fussiness characterized by inconsolable agonized crying, drawing up of the legs, abdominal distention, and excessive gas. It generally develops in the first two to four weeks of life and persists through the third to fourth months of life.43 Various psychosocial and dietary factors have been implicated in the cause of infantile colic, but trials in bottle-fed and breast-fed infants have suggested that IgE-mediated hypersensitivity occasionally may be a pathogenic factor, possibly in 10% to 15% of colicky infants. Diagnosis of food-induced colic is established by the implementation of several brief trials of hypoallergenic formula. In infants with food allergen–induced colic, symptoms are generally short-lived, so prolonged restricted diets are generally unnecessary. Periodic rechallenges should be done every three to four months to determine when eliminated foods can be returned to the infant’s diet.
MIXED IgE- AND NON–IgE-MEDIATED DISORDERS
Allergic eosinophilic esophagitis, gastroenteritis, and proctocolitis (AEE, AEG, AEP) may be caused by IgE- and/or non–IgE-mediated food allergies and are characterized by eosinophilic infiltration of the esophagus, stomach, and/or intestinal walls with peripheral eosinophilia in up to 50% of patients (see Chapter 27 for a more complete discussion).31,39–41 In the esophagus, basal hyperplasia and papillary lengthening are seen. The eosinophilic infiltrate may involve the mucosal, muscular, and/or serosal layers of the stomach or small intestine. Eosinophilic invasion of the muscular layer leads to thickening and rigidity of the stomach and small intestine, which may manifest as obstruction, whereas infiltration of the serosa commonly results in eosinophilic ascites. In most children with AEE-AEG, food-induced IgE- and non–IgE-mediated reactions have been implicated in pathogenesis.44 Patients with IgE-mediated food-induced symptoms generally have atopic disease (atopic dermatitis, allergic rhinitis, and/or asthma), elevated serum IgE concentrations, positive skin prick tests to various foods and inhalants, peripheral blood eosinophilia, iron deficiency anemia, and hypoalbuminemia.
Allergic Eosinophilic Esophagitis
AEE manifests predominantly in young children, especially boys, with reflux or vomiting, irritability, food refusal, early satiety, and failure to thrive,45,46 whereas adults are more likely to present with reflux, epigastric or chest pain, dysphagia, and food impaction.43,47 Food-induced AEE was first demonstrated in a group of 10 children with postprandial abdominal pain, early satiety or food refusal, vomiting or retching, failure to thrive, and refractoriness to standard medical therapy (4 of 10 had undergone Nissen fundoplication).44 Following six to eight weeks of an amino acid-based formula (Neocate) plus corn and apples, symptoms completely resolved in eight patients and were markedly improved in two others. Esophageal biopsies revealed a marked reduction or clearing of the eosinophilic infiltrate and significant improvements in the basal zone hyperplasia and length of the vascular papillae. Symptoms could be reproduced with the introduction of certain foods. Some patients appear to have an association of pulmonary and esophageal inflammation, and some report seasonal esophageal symptoms.31,48 AEE appears to have increased in prevalence over the past decade, an observation some authors believe may be explained by the increased early use of antacids and prokinetic agents in young infants with symptoms of reflux. Because murine models of food-induced anaphylaxis require the use of antacids for sensitization,49,50 it is thought that antireflux medications may further compromise the young infant’s intestinal barrier function. In a cohort study of 152 adults using H2 blockers or proton pump inhibitors for three months, 10% of patients experienced an increase in food-specific IgE and 15% developed de novo IgE to specific foods.51 Untersmayr and colleagues have noted that the use of antacid medications can lead to food sensitivity in children and adults.13
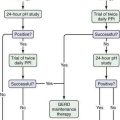
Diagnosis of AEE is based on a suggestive history, the demonstration of an eosinophilic infiltrate in the esophageal mucosa (>20 eosinophils/high-power field [×40]), and the absence of GERD, as evidenced by a normal pH monitoring study of the distal esophagus or lack of response to high-dose proton pump inhibitors.52 Multiple biopsies are necessary because of the potential patchiness of the lesions; a single esophageal biopsy specimen has a sensitivity of 55%, whereas taking five biopsy specimens increases sensitivity to 100%.53 Esophagoscopy may reveal mucosal rings, furrowing, ulcerations, whitish papules (which represent eosinophilic abscesses), or strictures, but endoscopic findings are normal in at least one third of patients with AEE. There is some evidence suggesting that atopy patch testing may be useful in identifying foods responsible for the allergic inflammation, but further studies are necessary to confirm these early reports.54 Elimination of suspect foods for six to ten weeks should lead to resolution and normalization of esophageal histology, although clinical symptoms should improve substantially in three to six weeks.41,55,56 Challenges consist of reintroducing the suspected food allergen and evaluating for recurrence of symptoms and/or eosinophilic infiltrate on biopsy. If food allergens are not identified as provoking agents, oral glucocorticoids generally are required to alleviate symptoms. Although symptoms usually respond to glucocorticoid therapy, recurrence of symptoms is frequent when steroids are discontinued.57 Topical glucocorticoid therapy with swallowed fluticasone spray or viscous budesonide has been shown to induce remission in 50% to 80% of patients, but esophageal candidiasis may occur in up to 20% of patients using this form of treatment.58,59 If exacerbations recur, a daily regimen of low-dose prednisone or prednisolone or prednisone every other day may be successful in suppressing symptoms.60 Recent evidence suggests that anti–IL-5 may be useful in this disorder.61
Allergic Eosinophilic Gastroenteritis
AEG manifests with abdominal pain, nausea, vomiting, diarrhea, and weight loss.62 Generalized edema secondary to hypoalbuminemia may occur in some infants and young children with marked protein-losing enteropathy, often in the presence of minimal gastrointestinal symptoms (e.g., occasional vomiting and diarrhea).63 Rarely AEG may manifest as pyloric stenosis in infants with outlet obstruction and postprandial projectile emesis.64
The immunopathogenesis of AEG is not known, but is believed to involve primarily cell-mediated mechanisms. A subset of patients have exacerbations of symptoms following the ingestion of food to which they have specific IgE antibodies, but most reactions do not appear to involve this mechanism. Peripheral blood T cells from all AEG patients evaluated have been shown to secrete excessive amounts of Th2 cytokines, IL-4, and IL-5 in vitro, compared with normal controls,65 and T cells expanded from duodenal biopsies of AEG patients express Th2 cytokines in vitro following antigen stimulation.32
The diagnosis of AEG is dependent on a suggestive history, gastrointestinal biopsy specimens demonstrating a prominent eosinophilic infiltration, and peripheral eosinophilia, which occurs in about 50% of patients. Lesions are not uniform; therefore, multiple biopsies are often necessary.62 Allergy skin testing may be helpful in some cases to identify causative foods, but often a therapeutic trial of an elemental diet for six to 10 weeks is necessary to determine whether food allergy is provoking the disorder. In a study of children with AEG and protein-losing enteropathy, institution of an amino acid-based formula therapy brought about resolution of symptoms and normalization of intestinal histology.63 As with AEE, if no sensitization is found, a trial of glucocorticoids is recommended, although relapses frequently occur when they are discontinued. The long-term prognosis of this disorder is not well characterized. In one series of children with AEG and protein-losing enteropathy, follow-up for 2.5 to 5.5 years revealed persistence of food-responsive disease.
Allergic Eosinophilic Proctocolitis
AEP generally presents in the first few months of life and is most often secondary to cow’s milk or soy protein hypersensitivity. Over half of reported cases now occur in breast-fed infants because of food antigens that are passed in maternal breast milk.66,67 Affected infants usually appear healthy, often have normally formed stools, and generally are evaluated because of the presence of gross or occult blood in their stools. Blood loss typically is minor but occasionally can produce anemia. Lesions generally are confined to the distal large bowel and consist of mucosal edema, with infiltration of eosinophils in the epithelium and lamina propria. In severe cases with crypt destruction, neutrophils also are prominent. The immunologic mechanism underlying this disorder is not known, but is believed to involve a cell-mediated reaction. There is no evidence that IgE antibodies are involved in this disorder and therefore skin prick testing or evaluation of food-specific IgE antibodies is not helpful. Diagnosis can be established when elimination of the responsible allergen leads to resolution of hematochezia, generally with dramatic improvement within 72 hours of appropriate food allergen elimination. Complete clearing and resolution of mucosal lesions may take up to one month. Reintroduction of the allergen leads to recurrence of symptoms within several hours to days. Sigmoidoscopic findings vary and range from areas of patchy mucosal injection to severe friability, with small aphthoid ulcerations and bleeding. Colonic biopsy reveals a prominent eosinophilic infiltrate in the crypt epithelia and lamina propria. Children with cow’s milk and soy protein–induced proctocolitis usually outgrow their protein sensitivity (i.e., become clinically tolerant within six months to two years of allergen avoidance), but occasionally refractory cases are seen.
NON–IgE-MEDIATED DISORDERS
Some gastrointestinal food-allergic disorders are clearly not IgE-mediated, and are believed to be the result of various cell-mediated mechanisms. Consequently, tests for evidence of food-specific IgE antibodies are of no value to identify the responsible food in these disorders. These non–IgE-mediated hypersensitivities are believed to result from different abnormal antigen processing and/or cell-mediated mechanisms, and may be divided into the following syndromes: dietary protein-induced enterocolitis and dietary protein-induced enteropathy.68
Dietary Protein-Induced Enterocolitis Syndrome
Dietary protein-induced enterocolitis syndrome is a disorder most commonly seen in young infants, presenting between one week and three months of age, with protracted vomiting and diarrhea that not infrequently results in dehydration.69,70 About one third of infants with severe diarrhea develop acidosis and transient methemoglobinemia. Cow’s milk and/or soy protein most often are responsible, but enterocolitis secondary to egg, wheat, rice, oat, peanut, nuts, chicken, turkey, and fish sensitivities has also been reported in older individuals.71 Breast-fed babies almost never develop symptoms while breast-feeding, but may be sensitized through food proteins passed in the breast milk and experience a reaction on the first few feedings of the whole food.72,73 Similar reactions to seafood (e.g., shrimp, crab, lobster), with symptoms developing about two to four hours following ingestion, often are reported in adults.
Stools frequently contain occult blood, polymorphonuclear neutrophils, and eosinophils. Jejunal biopsies reveal flattened villi, edema, and increased numbers of lymphocytes, eosinophils, and mast cells. Food challenges generally result in vomiting and diarrhea within one to three hours, and result in hypotension in about 15% of cases. The immunopathogenesis of this syndrome remains unknown. Some studies suggest that food antigen-induced secretion of TNF-α from local mononuclear cells (e.g., macrophages, dendritic cells) may account for the reaction.74 Other studies indicate that the disorder may be caused by lower expression of type 1 TGF-β receptors than type 2 receptors, suggesting differential contributions of each receptor to the diverse biological activities of TGF-β in the intestinal epithelium.75 Some studies have suggested that atopy patch testing with the suspected food may be useful in distinguishing which children will develop symptoms following ingestion, but most such evidence is not convincing.76 Diagnosis can be established when elimination of the responsible allergen leads to resolution of symptoms within 72 hours and oral challenge provokes symptoms.72 Secondary disaccharidase deficiency may persist longer, however, and may result in ongoing diarrhea for up to two weeks. Oral food challenges consist of administering 0.3 to 0.6 g/kg body weight of the suspected protein allergen while monitoring the peripheral blood white cell count. Vomiting generally develops within one to four hours of administering the challenge food, whereas diarrhea or loose stools often develop after four to eight hours. In conjunction with a positive food challenge, the absolute neutrophil count in the peripheral blood will increase at least 3500 cells/mm3 within four to six hours of developing symptoms, and neutrophils and eosinophils may be found in the stools. About 15% of food antigen challenges lead to profuse vomiting, dehydration, and hypotension, so they must be performed under medical supervision.
Dietary Protein-Induced Enteropathy
Dietary protein-induced enteropathy (excluding celiac disease) frequently manifests in the first several months of life with diarrhea (mild to moderate steatorrhea in about 80%) and poor weight gain.67,77 Symptoms include protracted diarrhea, vomiting in up to two thirds of patients, failure to thrive, and malabsorption, demonstrated by the presence of reducing substances in the stools, increased fecal fat, and abnormal d-xylose absorption. Cow’s milk sensitivity is the most frequent cause of this syndrome, but it also has been associated with sensitivities to soy, egg, wheat, rice, chicken, and fish. The diagnosis is established by identifying and excluding the responsible allergen from the diet, which should result in resolution of symptoms within several days to weeks. On endoscopy, patchy villus atrophy is evident and biopsy reveals a prominent mononuclear round cell infiltrate and a small number of eosinophils, similar to celiac disease, but generally much less extensive. Colitic features such as mucus and gross or microscopic hematochezia usually are absent, but anemia occurs in about 40% of affected infants and protein loss occurs in most. Complete resolution of the intestinal lesions may require 6 to 18 months of allergen avoidance. Unlike celiac disease, loss of protein sensitivity and clinical reactivity frequently occurs, but the natural history of this disorder has not been well studied.
Celiac Disease
Celiac disease (CD) is a more extensive enteropathy leading to malabsorption (see details in Chapter 104). Total villus atrophy and extensive cellular infiltrate are associated with sensitivity to gliadin, the alcohol-soluble portion of gluten found in wheat, rye, and barley. CD is strongly associated with HLA-DQ2 (α1*0501, β1*0201), which is present in more than 90% of CD patients.78 The incidence of CD has been reported as 1 in 250 in the United States. The striking increase in CD in Sweden compared with genetically similar Denmark,79 and the variation in prevalence associated with changes in patterns of gluten feeding in Sweden,80 strongly implicate environmental factors (e.g., feeding practices) in the cause of this disorder.81 The intestinal inflammation in CD is precipitated by exposure to gliadin and is associated with increased mucosal activity of tissue transglutaminase (tTG), which deamidates gliadin in an ordered and specific fashion, creating epitopes that bind efficiently to DQ2 and are recognized by T cells.82
Initial symptoms often include diarrhea or frank steatorrhea, abdominal distention and flatulence, weight loss, and occasionally nausea and vomiting. Oral ulcers and other extraintestinal symptoms secondary to malabsorption are not uncommon. Villus atrophy of the small bowel is a characteristic feature of CD patients who are ingesting gluten. IgA antibodies to gluten are present in more than 80% of adults and children with untreated CD.83 In addition, patients generally have increased IgG antibodies to a variety of foods, presumably the result of increased food antigen absorption. Diagnosis has been dependent on demonstrating biopsy evidence of villus atrophy and an inflammatory infiltrate, resolution of biopsy findings after six to 12 weeks of gluten elimination, and recurrence of biopsy changes following gluten challenge. Revised diagnostic criteria have been proposed, however, that require greater dependency on serologic studies. Quantitation of IgA tTG antibodies may be used for screening in children older than two years. Diagnosis of CD, however, requires an intestinal biopsy showing clear-cut evidence of villus atrophy plus resolution of symptoms on a gluten-free diet, with serologic follow-up showing disappearance of the antibodies to confirm the diagnosis further.84,85 Once the diagnosis of CD is established, lifelong elimination of gluten-containing foods is necessary to control symptoms and possibly to avoid the increased risk of gastrointestinal malignancy.86
Dermatitis Herpetiformis
Dermatitis herpetiformis (DH) is a chronic blistering skin disorder associated with a gluten-sensitive enteropathy. It is characterized by a chronic, intensely pruritic, papulovesicular rash symmetrically distributed over the extensor surfaces and buttocks.87,88 The histology of the intestinal lesion is almost identical to that seen in CD, although villus atrophy and the inflammatory infiltrate are generally milder and T-cell lines isolated from intestinal biopsy specimens of DH patients produce significantly more IL-4 than T cell lines isolated from CD patients.89 Although many patients have minimal or no gastrointestinal complaints, biopsy of the small bowel generally confirms intestinal involvement. Elimination of gluten from the diet generally leads to resolution of skin symptoms and normalization of intestinal findings over several months. Administration of sulfones, the mainstay of therapy, leads to rapid resolution of skin symptoms, but has almost no effect on intestinal symptoms.
Other Gastrointestinal Disorders
Several other disorders have been suggested to be caused by food protein hypersensitivity. Ingestion of pasteurized whole cow’s milk by infants younger than six months may lead to occult GI blood loss and occasionally to iron deficiency anemia.90,91 Substitution of heat-processed infant formula (including cow’s milk–derived formulas) for whole cow’s milk generally leads to resolution of symptoms within three days. Gastroesophageal reflux (GER) in young infants may be the result of food-induced AEE. In a study of 204 infants younger than one year with GER (diagnosed with a 24-hour esophageal pH test and esophageal biopsy),85 42% were diagnosed with cow’s milk–induced reflux by blinded milk challenges. These infants experienced resolution of GER and normalization of pH studies once cow’s milk was eliminated from the diet.92 Constipation also has been reported to be caused by milk allergy,93 although the underlying mechanism is not clear. Circumstantial evidence suggests a possible role of food allergy in inflammatory bowel disease (Crohn’s disease and ulcerative colitis), but convincing evidence of an immunopathogenic role remains to be established.
DIAGNOSIS
The diagnosis of food allergy is a clinical exercise involving a careful history, physical examination, and selective laboratory studies. Various tests are used for the evaluation of food hypersensitivity (see Sicherer and Sampson4 and Sampson94). In some cases, the medical history may be useful in diagnosing food allergy (e.g., acute anaphylaxis after the isolated ingestion of peanuts). Fewer than 50% of reported food-allergic reactions, however, can be verified by a double-blind, placebo-controlled food challenge. Information useful in establishing that a food-allergic reaction has occurred and in constructing an appropriate oral food challenge includes the following: (1) food presumed to have provoked the reaction; (2) quantity of the suspected food ingested; (3) length of time between ingestion and development of symptoms; (4) type of symptoms provoked; and (5) whether similar symptoms developed on other occasions when the food was eaten. Although any food may induce an allergic reaction, a few foods are responsible for the vast majority of reactions (Table 9-4).
Table 9-4 Foods Responsible for Most Food Hypersensitivity Disorders
| IgE-MEDIATED FOOD HYPERSENSITIVITIES* | NON–IgE-MEDIATED FOOD HYPERSENSITIVITIES† |
|---|---|
| Milk | Barley |
| Egg | Beef, lamb |
| Peanuts | Egg |
| Shellfish | Fish |
| Tree nuts | Milk |
| Sesame | Shellfish |
| Fish | Soy |
| Soy | Wheat |
| Wheat | White potato |
* Listed in order of overall prevalence.
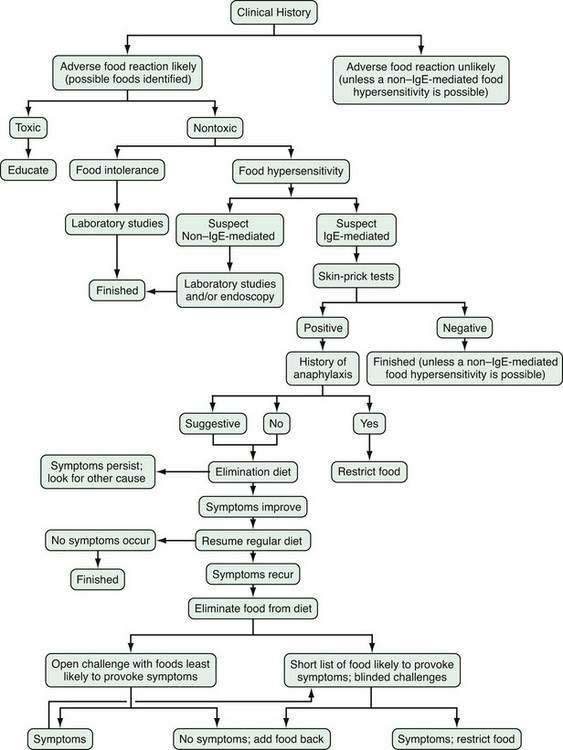
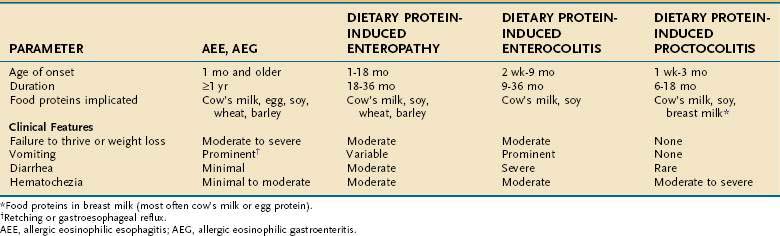
Figure 9-2 depicts a standard approach for evaluating and managing adverse food reactions. If an IgE-mediated disorder is suspected, selected skin prick tests or quantification of food-specific IgE antibodies (e.g., ImmunoCAP) followed by an appropriate exclusion diet and blinded food challenge are warranted. If a non–IgE-mediated GI hypersensitivity disorder is suspected, laboratory and endoscopic studies (with or without oral food challenges) are required to arrive at the correct diagnosis (see earlier). Table 9-5 compares the main features of four non–IgE-mediated food-allergic disorders. An exclusion diet eliminating all foods suspected by history and/or skin testing (for IgE-mediated disorders) should be conducted for one to two weeks in suspected IgE-mediated disorders, food-induced enterocolitis, and benign eosinophilic proctocolitis. Exclusion diets may need to be extended for as long as 12 weeks in other suspected GI hypersensitivity disorders (e.g., food protein-induced enteropathy, AEE, or AEG) and may require the use of elemental diets (e.g., Vivonex, Neocate One+, or EleCare) to exclude all antigens. If no improvement is noted and dietary compliance is ensured, it is unlikely that food allergy is involved. Before undertaking blinded food challenges (single- or double-blind), suspect foods should be eliminated from the diet for 7 to 14 days before challenge and even longer in some disorders when secondary disaccharidase deficiency may have developed, as noted earlier. Prescribing elimination diets, like prescribing medications, may have adverse effects (e.g., malnutrition or eating disorders) and should not be done in the absence of evidence that they are likely to be beneficial.
TREATMENT AND NATURAL HISTORY
Once the diagnosis of food hypersensitivity is established, strict elimination of the offending allergen is the only proven therapy. Patients must be taught to scrutinize food labels to detect potential sources of hidden food allergens.95 Drugs such as H1 and H2 antihistamines and glucocorticoids modify symptoms to food allergens but, overall, have minimal efficacy or unacceptable side effects. Anti–IL-5 antibodies have shown promise in the treatment of eosinophilic disorders.61 The prevalence of food hypersensitivity is greatest in the first few years of life, but most young children outgrow their food hypersensitivity within three to five years, except possibly for IgE-mediated hypersensitivities to peanuts, nuts, and seafood.28 Although younger children are more likely to outgrow food hypersensitivity, older children and adults also may lose their food hypersensitivity (i.e., develop clinical tolerance and be able to ingest the food without symptoms) if the responsible food allergen can be identified and eliminated from the diet for a period of time.96,97
Breiteneder H, Clare Mills EN. Plant food allergens—structural and functional aspects of allergenicity. Biotechnol Adv. 2005;23:395-9. (Ref 38.)
Chehade M, Magid MS, Mofidi S, et al. Allergic eosinophilic gastroenteritis with protein-losing enteropathy: intestinal pathology, clinical course, and long-term follow-up. J Pediatr Gastroenterol Nutr. 2006;42:516-21. (Ref 63.)
Chehade M, Mayer L. Oral tolerance and its relation to food hypersensitivities. J Allergy Clin Immunol. 2005;115:3-12. (Ref 12.)
Furuta GT, Liacouras CA, Collins MH, et al. Eosinophilic esophagitis in children and adults: A systematic review and consensus recommendations for diagnosis and treatment. Gastroenterol. 2007;133:1342-63. (Ref 52.)
Khan S, Orenstein SR. Eosinophilic gastroenteritis. Gastroenterol Clin North Am. 2008;37:333-48. (Ref 62.)
Leffler DA, Kelly CP. Update on the evaluation and diagnosis of celiac disease. Curr Opin Allergy Clin Immunol. 2006;6:191-6. (Ref 78.)
Novak N, Haberstok J, Bieber T, Allam JP. The immune privilege of the oral mucosa. Trends Mol Med. 2008;14:191-8. (Ref 19.)
Nowak-Wegrzyn A, Sampson HA, Wood RA, Sicherer SH. Food protein-induced enterocolitis syndrome caused by solid food proteins. Pediatr. 2003;111:829-35. (Ref 71.)
Rothenberg ME. Eosinophilic gastrointestinal disorders (EGID). J Allergy Clin Immunol. 2004;113:11-28. (Ref 31.)
Sampson HA. Food allergy. Part 2: Diagnosis and management. J Allergy Clin Immunol. 1999;103:981-99. (Ref 94.)
Sampson HA, Sicherer SH, Birnbaum AH. AGA technical review on the evaluation of food allergy in gastrointestinal disorders. American Gastroenterological Association. Gastroenterol. 2001;120:1026-40. (Ref 68.)
Sicherer SH. Food protein-induced enterocolitis syndrome: Clinical perspectives. J Pediatr Gastroenterol Nutr. 2000;30:S45-9. 2000. (Ref 70.)
Sicherer SH, Sampson HA. Food allergy. J Allergy Clin Immunol. 2006;117:S470-5. (Ref 4.)
Vercelli D. Immunoglobulin E and its regulators. Curr Opin Allergy Clin Immunol. 2001;1:61-5. (Ref 24.)
Wickens K, Black PN, Stanley TV, et al. A differential effect of 2 probiotics in the prevention of eczema and atopy: A double-blind, randomized, placebo-controlled trial. J Allergy Clin Immunol. 2008;122:788-94. (Ref 21.)
1. Prausnitz C, Kustner H. Studies on supersensitivity. Centrabl Bakteriol. 1921;86:160-9.
2. Loveless MH. Milk allergy: A survey of its incidence; experiments with a masked ingestion test. J Allergy. 1950;21:489-99.
3. Bock SA, Sampson HA, Atkins FM, et al. Double-blind, placebo-controlled food challenge (DBPCFC) as an office procedure: A manual. J Allergy Clin Immunol. 1988;82:986-97.
4. Sicherer SH, Sampson HA. Food allergy. J Allergy Clin Immunol. 2006;117:S470-5.
5. Host A, Halken S, Jacobsen HP, et al. Clinical course of cow’s milk protein allergy/intolerance and atopic diseases in childhood. Pediatr Allergy Immunol. 2002;13(Suppl 15):23-8.
6. Skolnick HS, Conover-Walker MK, Koerner CB, et al. The natural history of peanut allergy. J Allergy Clin Immunol. 2001;107:367-74.
7. Sicherer SH, Munoz-Furlong A, Sampson HA. Prevalence of seafood allergy in the United States determined by a random telephone survey. J Allergy Clin Immunol. 2004;114:159-65.
8. Branum AM, Lukacs AL. Food allergy among U.S. children: Trends in prevalence and hospitalizations. National Center for Health Statistics Data Brief No. 10. Hyattsville, Md: National Center for Health Statistics; 2008.
9. Grundy J, Matthews S, Bateman B, et al. Rising prevalence of allergy to peanut in children: Data from 2 sequential cohorts. J Allergy Clin Immunol. 2002;110:784-9.
10. Sicherer SH, Munoz-Furlong A, Sampson HA. Prevalence of peanut and tree nut allergy in the United States determined by means of a random digit dial telephone survey: A 5-year follow-up study. J Allergy Clin Immunol. 2003;112:1203-7.
11. Eigenmann PA, Sicherer SH, Borkowski TA, et al. Prevalence of IgE-mediated food allergy among children with atopic dermatitis. Pediatr. 1998;101:E8.
12. Chehade M, Mayer L. Oral tolerance and its relation to food hypersensitivities. J Allergy Clin Immunol. 2005;115:3-12.
13. Untersmayr E, Jensen-Jarolim E. The role of protein digestibility and antacids on food allergy outcomes. J Allergy Clin Immunol. 2008;121:1301-8.
14. Brunner M, Walzer M. Absorption of undigested proteins in human beings: The absorption of unaltered fish protein in adults. Arch Intern Med. 1928;42:173-9.
15. Walzer M. Allergy of the abdominal organs. J Lab Clin Med. 1941;26:1867-77.
16. Walzer M. Absorption of allergens. J Allergy. 1942;13:554-62.
17. Mowat AM, Parker LA, Beacock-Sharp H, et al. Oral tolerance: Overview and historical perspectives. Ann N Y Acad Sci. 2004;1029:1-8.
18. Mowat AM. Anatomical basis of tolerance and immunity to intestinal antigens. Nat Rev Immunol. 2003;3:331-41.
19. Novak N, Haberstok J, Bieber T, Allam JP. The immune privilege of the oral mucosa. Trends Mol Med. 2008;14:191-8.
20. Sudo N, Sawamura S, Tanaka K, et al. The requirement of intestinal bacterial flora for the development of an IgE production system fully susceptible to oral tolerance induction. J Immunol. 1997;159:1739-45.
21. Wickens K, Black PN, Stanley TV, et al. A differential effect of 2 probiotics in the prevention of eczema and atopy: A double-blind, randomized, placebo-controlled trial. J Allergy Clin Immunol. 2008;122:788-94.
22. Berin MC, Kiliaan AJ, Yang PC, et al. Rapid transepithelial antigen transport in rat jejunum: Impact of sensitization and the hypersensitivity reaction. Gastroenterology. 1997;113:856-64.
23. Yang PC, Berin MC, Yu LC, et al. Enhanced intestinal transepithelial antigen transport in allergic rats is mediated by IgE and CD23 (FcepsilonRII). J Clin Invest. 2000;106:879-86.
24. Vercelli D. Immunoglobulin E and its regulators. Curr Opin Allergy Clin Immunol. 2001;1:61-5.
25. Husby S. Normal immune responses to ingested foods. J Pediatr Gastroenterol Nutr. 2000;30:S13-19.
26. Gdalevich M, Mimouni D, David M, Mimouni M. Breast-feeding and the onset of atopic dermatitis in childhood: A systematic review and meta-analysis of prospective studies. J Am Acad Dermatol. 2001;45:520-7.
27. Gdalevich M, Mimouni D, Mimouni M. Breast-feeding and the risk of bronchial asthma in childhood: A systematic review with meta-analysis of prospective studies. J Pediatr. 2001;139:261-6.
28. Sampson HA. Food allergy. Part 1: Immunopathogenesis and clinical disorders. J Allergy Clin Immunol. 1999;103:717-28.
29. Sampson HA, Jolie PL. Increased plasma histamine concentrations after food challenges in children with atopic dermatitis. N Engl J Med. 1984;311:372-6.
30. Reimann HJ, Lewin J. Gastric mucosal reactions in patients with food allergy. Am J Gastroenterol. 1988;83:1212-19.
31. Rothenberg ME. Eosinophilic gastrointestinal disorders (EGID). J Allergy Clin Immunol. 2004;113:11-28.
32. Beyer K, Castro R, Birnbaum A, et al. Human milk-specific mucosal lymphocytes of the gastrointestinal tract display a Th2 cytokine profile. J Allergy Clin Immunol. 2002;109:707-13.
33. Eyermann C. X-ray demonstration of colonic reaction in food allergy. J Missouri Med Assoc. 1927;24:129-32.
34. Fries JH, Zizmor J. Roentgen studies of children with alimentary disturbances due to food allergy. Am J Dis Child. 1937;54:1239-51.
35. Rowe AH. Roentgen studies of patients with gastro-intestinal food allergy. JAMA. 1933;100:394-400.
36. Pollard H, Stuart G. Experimental reproduction of gastric allergy in human beings with controlled observations on the mucosa. J Allergy. 1942;13:467-73.
37. Wang J. Oral allergy syndrome. In: Metcalfe DD, Sampson HA, Simon RA, editors. Food allergy. Oxford: Blackwell Publishing; 2008:133.
38. Breiteneder H, Clare Mills EN. Plant food allergens—structural and functional aspects of allergenicity. Biotechnol Adv. 2005;23:395-99.
39. Liacouras CA, Spergel JM, Ruchelli E, et al. Eosinophilic esophagitis: A 10-year experience in 381 children. Clin Gastroenterol Hepatol. 2005;3:1198-206.
40. Liacouras CA. Eosinophilic esophagitis: Treatment in 2005. Curr Opin Gastroenterol. 2006;22:147-52.
41. Potter JW, Saeian K, Staff D, et al. Eosinophilic esophagitis in adults: An emerging problem with unique esophageal features. Gastrointest Endosc. 2004;59:355-61.
42. Ortolani C, Ispano M, Pastorello EA, et al. Comparison of results of skin prick tests (with fresh foods and commercial food extracts) and RAST in 100 patients with oral allergy syndrome. J Allergy Clin Immunol. 1989;83:683-90.
43. Hill DJ, Hosking CS. Infantile colic and food hypersensitivity. J Pediatr Gastroenterol Nutr. 2000;30(Suppl):S67-76.
44. Kelly KJ, Lazenby AJ, Rowe PC, et al. Eosinophilic esophagitis attributed to gastroesophageal reflux: Improvement with an amino-acid based formula. Gastroenterology. 1995;109:1503-12.
45. Liacouras C. Eosinophilic esophagitis in children and adults. J Pediatr Gastroenterol Nutr. 2003;37:S23-8.
46. Putnam PE. Eosinophilic esophagitis in children: Clinical manifestations. Gastroenterol Clin North Am. 2008;37:369-81.
47. Prasad GA, Talley NJ. Eosinophilic esophagitis in adults. Gastroenterol Clin North Am. 2008;37:349-68.
48. Fogg MI, Ruchelli E, Spergel JM. Pollen and eosinophilic esophagitis. J Allergy Clin Immunol. 2003;112:796-7.
49. Li XM, Serebrisky D, Lee SY, et al. A murine model of peanut anaphylaxis: T- and B-cell responses to a major peanut allergen mimic human responses. J Allergy Clin Immunol. 2000;106(Pt 1):150-8.
50. Untersmayr E, Scholl I, Swoboda I, et al. Antacid medication inhibits digestion of dietary proteins and causes food allergy: A fish allergy model in BALB/c mice. J Allergy Clin Immunol. 2003;112:616-23.
51. Untersmayr E, Bakos N, Scholl I, et al. Anti-ulcer drugs promote IgE formation toward dietary antigens in adult patients. FASEB J. 2005;19:656-8.
52. Furuta GT, Liacouras CA, Collins MH, et al. Eosinophilic esophagitis in children and adults: a systematic review and consensus recommendations for diagnosis and treatment. Gastroenterology. 2007;133:1342-63.
53. Gonsalves N, Policarpio-Nicolas M, Zhang Q. Histopathologic variability and endoscopic correlates in adults with eosinophilic esophagitis. Gastrointest Endosc. 2006;64:313-19.
54. Spergel JM, Brown-Whitehorn T, Beausoleil JL, et al. Predictive values for skin prick test and atopy patch test for eosinophilic esophagitis. J Allergy Clin Immunol. 2007;119:509-11.
55. Markowitz JE, Spergel JM, Ruchelli E, Liacouras C. Elemental diet is an effective treatment for eosinophilic esophagitis in children and adolescents. Am J Gastroenterol. 2003;98:777-82.
56. Kagalwalla AF, Sentongo TA, Ritz S, et al. Effect of six-food elimination diet on clinical and histologic outcomes in eosinophilic esophagitis. Clin Gastroenterol Hepatol. 2006;4:1097-102.
57. Liacouras CA, Wenner WJ, Brown K, Ruchelli E. Primary eosinophilic esophagitis in children: Successful treatment with oral corticosteroids. J Pediatr Gastroenterol Nutr. 1998;26:380-5.
58. Konikoff MR, Noel RJ, Blanchard C, et al. A randomized, double-blind, placebo-controlled trial of fluticasone propionate for pediatric eosinophilic esophagitis. Gastroenterology. 2006;131:1381-91.
59. Aceves SS, Bastian JF, Newbury RO, Dohil R. Oral viscous budesonide: A potential new therapy for eosinophilic esophagitis in children. Am J Gastroenterol. 2007;102:2271-9.
60. Lee C, Changchien C, Chen P, et al. Eosinophilic gastroenteritis: 10 years’ experience. Am J Gastroenterol. 1993;88:70-4.
61. Stein ML, Collins MH, Villanueva JM, et al. Anti–IL-5 (mepolizumab) therapy for eosinophilic esophagitis. J Allergy Clin Immunol. 2006;118:1312-19.
62. Khan S, Orenstein SR. Eosinophilic gastroenteritis. Gastroenterol Clin North Am. 2008;37:333-48.
63. Chehade M, Magid MS, Mofidi S, et al. Allergic eosinophilic gastroenteritis with protein-losing enteropathy: Intestinal pathology, clinical course, and long-term follow-up. J Pediatr Gastroenterol Nutr. 2006;42:516-521.
64. Khan S, Orenstein SR. Eosinophilic gastroenteritis masquerading as pyloric stenosis. Clin Pediatr (Phila). 2000;39:55-7.
65. Jaffe J, James S, Mullins G, et al. Evidence for an abnormal profile of interleukin-4 (IL-4), IL-5, and gamma interferon in peripheral blood T cells from patients with allergic eosinophilic gastroenteritis. J Clin Immunol. 1994;14:299-309.
66. Lake AM. Food-induced eosinophilic proctocolitis. J Pediatr Gastroenterol Nutr. 2000;30:S58-60.
67. Maloney J, Nowak-Wegrzyn A. Educational clinical case series for pediatric allergy and immunology: Allergic proctocolitis, food protein-induced enterocolitis syndrome and allergic eosinophilic gastroenteritis with protein-losing gastroenteropathy as manifestations of non–IgE-mediated cow’s milk allergy. Pediatr Allergy Immunol. 2007;18:360-7.
68. Sampson HA, Sicherer SH, Birnbaum AH. AGA technical review on the evaluation of food allergy in gastrointestinal disorders. American Gastroenterological Association. Gastroenterol. 2001;120:1026-40.
69. Powell G. Food protein-induced enterocolitis of infancy: Differential diagnosis and management. Compr Ther. 1986;12:28-37.
70. Sicherer SH. Food protein-induced enterocolitis syndrome: clinical perspectives. J Pediatr Gastroenterol Nutr. 2000;30:S45-9.
71. Nowak-Wegrzyn A, Sampson HA, Wood RA, Sicherer SH. Food protein-induced enterocolitis syndrome caused by solid food proteins. Pediatrics. 2003;111:829-35.
72. Sicherer SH, Eigenmann PA, Sampson HA. Clinical features of food protein-induced enterocolitis syndrome. J Pediatr. 1998;133:214-19.
73. Hojsak I, Kljaic-Turkalj M, Misak Z, Kolacek S. Rice protein-induced enterocolitis syndrome. Clin Nutr. 2006;25:533-6.
74. Benlounes N, Candalh C, Matarazzo P, et al. The time-course of milk antigen-induced TNF-alpha secretion differs according to the clinical symptoms in children with cow’s milk allergy. J Allergy Clin Immunol. 1999;104(Pt 1):863-9.
75. Chung HL, Hwang JB, Park JJ, Kim SG. Expression of transforming growth factor beta1, transforming growth factor type I and II receptors, and TNF-alpha in the mucosa of the small intestine in infants with food protein-induced enterocolitis syndrome. J Allergy Clin Immunol. 2002;109:150-4.
76. Fogg MI, Brown-Whitehorn TA, Pawlowski NA, Spergel JM. Atopy patch test for the diagnosis of food protein-induced enterocolitis syndrome. Pediatr Allergy Immunol. 2006;17:351-5.
77. Savilahti E. Food-induced malabsorption syndromes. J Pediatr Gastroenterol Nutr. 2000;30:S61-6.
78. Leffler DA, Kelly CP. Update on the evaluation and diagnosis of celiac disease. Curr Opin Allergy Clin Immunol. 2006;6:191-6.
79. Weile B, Cavell B, Nivenius K, Krasilnikoff P. Striking differences in the incidence of childhood celiac disease between Denmark and Sweden: A plausible explanation. J Pediatr Gastroenterol Nutr. 1995;21:64-8.
80. Ivarsson A, Persson LA, Nystrom L, et al. Epidemic of coeliac disease in Swedish children. Acta Paediatr. 2000;89:165-71.
81. Olsson C, Hernell O, Hornell A, et al. Difference in celiac disease risk between Swedish birth cohorts suggests an opportunity for primary prevention. Pediatrics. 2008;122:528-34.
82. Anderson RP, Degano P, Godkin AJ, et al. In vivo antigen challenge in celiac disease identifies a single transglutaminase-modified peptide as the dominant A-gliadin T-cell epitope. Nat Med. 2000;6:337-42.
83. Lurz E, Scheidegger U, Spalinger J, et al. Clinical presentation of celiac disease and the diagnostic accuracy of serologic markers in children. Eur J Pediatr. 2008;168:839-45.
84. Hill ID, Dirks MH, Liptak GS, et al. Guideline for the diagnosis and treatment of celiac disease in children: Recommendations of the North American Society for Pediatric Gastroenterology, Hepatology and Nutrition. J Pediatr Gastroenterol Nutr. 2005;40:1-19.
85. Rostom A, Murray JA, Kagnoff MF. American Gastroenterological Association (AGA) Institute technical review on the diagnosis and management of celiac disease. Gastroenterol. 2006;131:1918-2002.
86. Gao Y, Kristinsson SY, Goldin LR, et al. Increased risk for non-Hodgkin lymphoma in individuals with celiac disease and a potential familial association. Gastroenterology. 2009;136:91-8.
87. Nicolas ME, Krause PK, Gibson LE, Murray JA. Dermatitis herpetiformis. Int J Dermatol. 2003;42:588-600.
88. Alonso-Llamazares J, Gibson LE, Rogers RSIII. Clinical, pathologic, and immunopathologic features of dermatitis herpetiformis: Review of the Mayo Clinic experience. Int J Dermatol. 2007;46:910-19.
89. Hall RP, Smith AD, Streilein RD. Increased production of IL-4 by gut T-cell lines from patients with dermatitis herpetiformis compared to patients with isolated gluten-sensitive enteropathy. Dig Dis Sci. 2000;45:2036-43.
90. Ziegler EE, Fomon SJ, Nelson SE, et al. Cow milk feeding in infancy: Further observations on blood loss from the gastrointestinal tract. J Pediatr. 1990;116:11-18.
91. Fernandes SM, de Morais MB, Amancio OM. Intestinal blood loss as an aggravating factor of iron deficiency in infants aged 9 to 12 months fed whole cow’s milk. J Clin Gastroenterol. 2008;42:152-6.
92. Iacono G, Carroccio A, Cavataio F, et al. Gastroesophageal reflux and cow’s milk allergy in infants: A prospective study. J Allergy Clin Immunol. 1996;97:822-7.
93. Daher S, Tahan S, Sole D, et al. Cow’s milk protein intolerance and chronic constipation in children. Pediatr Allergy Immunol. 2001;12:339-42.
94. Sampson HA. Food allergy. Part 2: Diagnosis and management. J Allergy Clin Immunol. 1999;103:981-99.
95. Groetch M. Diets and nutrition. In: Metcalfe DD, Sampson HA, Simon RA, editors. Food allergy: Adverse reactions to foods and food additives. Oxford: Blackwell Publishing; 2008:482.
96. Sampson HA, Scanlon SM. Natural history of food hypersensitivity in children with atopic dermatitis. J Pediatr. 1989;115:23-7.
97. Pastorello E, Stocchi L, Pravetonni V, et al. Role of the food elimination diet in adults with food allergy. J Allergy Clin Immunol. 1989;84:475-83.