[level-membership-for-critical-care-medicine-category]
122 Fluoroquinolones
 Mechanism of Action
Mechanism of Action
DNA gyrase and topoisomerase IV enzymes are thought to be essential for the replication of DNA and partition of replicated chromosomal DNA.1 DNA gyrase, a tetrameric enzyme consisting of two A and two B subunits, is known to be a primary target of fluoroquinolones in gram-negative bacteria and is the only known enzyme capable of introducing negative super-helical twists into bacterial DNA.1,2 The two subunits of gyrase are encoded by gyrA and gyrB, which are also potential sites of mutation and subsequent quinolone resistance.1,3 Topoisomerase IV seems to be a primary target of many fluoroquinolones in gram-positive bacteria such as Staphylococcus aureus and Streptococcus pneumoniae.3,4 Bacterial topoisomerase IV appears to be the principal enzyme that resolves or “decatenates” interlocked daughter DNA circles occurring at the completion of a round of DNA replication, allowing segregation of daughter chromosomes into daughter cells.1,3,4 Topoisomerase IV, like DNA gyrase, is composed of four subunits, two each of the parC and parE gene products.
As part of the topoisomerase reaction mechanism, DNA gyrase and topoisomerase IV transiently break the DNA backbone and pass a double strand of DNA through those breaks, thus introducing a negative supercoil into the DNA strand.1,2 Fluoroquinolone antibiotics have been shown to target DNA gyrase and topoisomerase IV while these enzymes are functionally attached to the DNA strand in the presence of adenosine triphosphate, resulting in a drug/enzyme/DNA complex in which the DNA remains broken.1,2 Cell death apparently results from release of double-stranded DNA breaks from multiple drug/enzyme/DNA complexes throughout the chromosome.1,2 This mechanism of action does not in itself explain why the fluoroquinolones kill bacteria so rapidly, and it has been suggested that additional protein synthesis mechanisms involving unidentified “protein factors,” interference with the “SOS” response involved in the repair of damaged DNA, dissociation of gyrase subunits, and increased oxidative stress may all play a role in the rapidly bactericidal effects of these drugs.1,2 Fluoroquinolones have also been noted to substantially decrease the synthesis of proinflammatory cytokines, although the relevance of this finding to their overall pharmacologic activity is unknown.5
 Antimicrobial Spectrum of Activity
Antimicrobial Spectrum of Activity
Fluoroquinolones have excellent in vitro activity against a wide range of both gram-positive and gram-negative organisms. Representative activities of fluoroquinolones which are currently available and frequently used in critically ill patients are shown in Table 122-1. The entire fluoroquinolone class displays excellent activity against enteric gram-negative aerobic bacteria as well as Haemophilus influenzae, Moraxella catarrhalis, and Neisseria spp. Gastrointestinal (GI) pathogens such as Salmonella spp., Shigella spp., and Campylobacter spp. are also highly susceptible to fluoroquinolones. Although some differences in relative potency exist between individual drugs as determined by the minimum inhibitory concentration (MIC) for these organisms, little difference in clinical efficacy should be expected in the treatment of infections due to susceptible strains. Activity against P. aeruginosa is more variable, however. Ciprofloxacin has traditionally been considered the most active fluoroquinolone against this organism, but data suggest there is little difference between ciprofloxacin and levofloxacin in terms of relative susceptibility of P. aeruginosa strains.6 Ciprofloxacin was active against greater than 95% of P. aeruginosa strains when first released to the market in 1987, but by 2001 both ciprofloxacin and levofloxacin were active against only approximately 65% to 80% of strains. Such high levels of resistance (25%-35%) are still seen among P. aeruginosa strains.6–10 Clinically relevant differences between ciprofloxacin and levofloxacin are further minimized when pharmacokinetic and pharmacodynamic properties are considered.9 Moxifloxacin tends to be the least active of the currently available agents.6,11–13 Nearly all fluoroquinolones adequately inhibit P. aeruginosa at concentrations achieved in the urine.
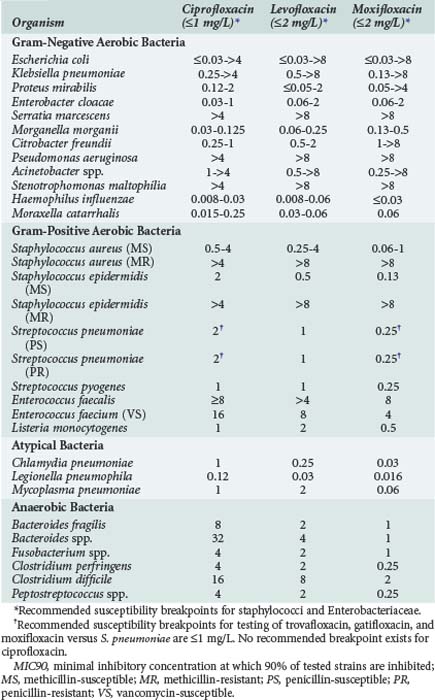
Activity of the fluoroquinolones against other hospital-acquired pathogens is also highly variable. Levofloxacin tends to be slightly more active against Acinetobacter spp., whereas moxifloxacin usually displays the best activity and ciprofloxacin is consistently the least active agent against Stenotrophomonas maltophilia.6,11–13 However, resistance to these latter organisms is quite common, and even agents with the best relative in vitro activity are not reliably clinically effective against many isolates.6,11–13
Several studies have reported that fluoroquinolones produce synergistic activity against gram-negative bacilli when used in combination with β-lactam antibiotics.14,15 These studies primarily evaluated antibiotic synergy against P. aeruginosa due to the frequent use of fluoroquinolones in antipseudomonal treatment regimens; ciprofloxacin and levofloxacin have been shown to achieve synergy against 25% to 75% of tested strains. One previous study also demonstrated synergistic in vitro activity against P. aeruginosa with the combination of moxifloxacin and either ceftazidime or cefepime15; however, additive or synergistic activity with moxifloxacin-containing combinations has not been extensively evaluated against other organisms. The ability of moxifloxacin to produce synergistic activity against P. aeruginosa may often be limited by clinically achievable drug concentrations. The use of ciprofloxacin and levofloxacin in combination regimens is most likely to result in synergistic activity, owing to their more potent activity and higher serum concentrations relative to the bacterial MICs.14,15
Newer fluoroquinolones have improved activity against gram-positive bacteria relative to older agents such as ciprofloxacin. Moxifloxacin has the best overall activity against staphylococci and streptococci, followed by levofloxacin and more distantly by ciprofloxacin.12,13 Levofloxacin and moxifloxacin are reliably active against penicillin-susceptible strains of S. pneumoniae; this activity is also retained against strains of S. pneumoniae resistant to other drug classes including penicillins, macrolides, and sulfonamides. Although ciprofloxacin has only moderate activity against methicillin-susceptible S. aureus (MSSA), newer agents have excellent activity against this organism. None of the fluoroquinolones is reliably active against methicillin-resistant S. aureus (MRSA), and rates of fluoroquinolone resistance among MRSA are quite high. Fluoroquinolones as a class also have only moderate activity against enterococci, with great variability seen among the various agents and specific bacterial strains.11–13 Fluoroquinolones have consistently excellent activity against Listeria monocytogenes.11–13
The activity of various fluoroquinolones against anaerobic bacteria is highly variable. Trovafloxacin was the first commercially available fluoroquinolone with clinically relevant anaerobic activity in vitro, as well as proven clinical efficacy for anaerobic infections including complicated intraabdominal infection. Moxifloxacin has in vitro activity against Bacteroides fragilis, Bacteroides group organisms, Fusobacterium spp., Clostridium spp., and other anaerobes that is generally comparable to trovafloxacin.12,13,16 However, recent data have demonstrated resistance rates in excess of 30% for many clinically important anaerobes, and the appropriateness of moxifloxacin for treatment of serious anaerobic infections is questionable.16 Neither ciprofloxacin nor levofloxacin have clinically relevant activity against anaerobic bacteria.
 Mechanisms of Fluoroquinolone Resistance
Mechanisms of Fluoroquinolone Resistance
Two basic mechanisms of fluoroquinolone resistance have been identified. One involves alteration of DNA gyrase and topoisomerase IV, whereas the other results in reduced drug accumulation within bacterial cells.2,17 Plasmid-mediated resistance, once considered quite rare, appears to be spreading rapidly among enteric gram-negative bacilli in certain geographic regions.18 However, plasmid-mediated resistance is relatively unusual compared with the more typical chromosomally mediated mechanisms of resistance.
Mutations in quinolone-resistance determining regions (QRDR) of topoisomerase enzymes prevent formation of drug/enzyme/DNA complexes, allowing DNA synthesis to occur in the presence of the drugs. Mutations in the genes encoding DNA gyrase (gyrA and gyrB) have been most frequently identified. However, other quinolone-resistant mutations in parC and parE, the genes encoding topoisomerase IV, have also been identified.1,2,17,19 Resistance to fluoroquinolones appears to arise in a stepwise manner. In some species (e.g., gram-negative bacteria), first-step mutations occur in gyrA and occasionally in gyrB, whereas in other species (e.g., S. aureus, S. pneumoniae) first-step mutations occur in parC and less often in parE.17,19 First-step mutations usually result in a low-level resistance (≤fourfold increased MIC), whereas additional mutations in either primary or secondary enzyme targets (second-step mutations) result in high-level resistance to drugs at clinically relevant concentrations. Dual gyrA and parC mutations have been described in clinical isolates of S. pneumoniae; however, it is thought that these strains were selected by fluoroquinolones with less potent antipneumococcal activity (e.g., ciprofloxacin).19–22
The first efflux system for quinolones was identified in Escherichia coli,23 whereas the first evidence for actual efflux-mediated quinolone resistance came from the characterization of S. aureus with overexpression of the norA gene product, a protein that mediates efflux.24 Such efflux may occur in both quinolone-resistant and quinolone-susceptible strains of S. aureus. In some species (e.g., P. aeruginosa), at least two different efflux systems may be present that mediate resistance to multiple other drug classes in addition to fluoroquinolones.25 Although most efflux proteins appear to be relatively nonspecific multidrug transporters whose substrates include hydrophilic fluoroquinolones as well as monocationic organic compounds, relatively substrate-specific efflux pumps have also been described.26
Many other genetic mutations have been described that result in decreased intracellular accumulation of fluoroquinolones and low-level drug resistance. Nearly all these mutations are associated with decreased expression of OmpF, a nonspecific outer membrane porin channel that is a major route of passage of hydrophilic fluoroquinolones through bacterial cellular membranes into the periplasmic space.27 Although decreased membrane permeability is relatively common and easily induced, this is an unusual mechanism for clinically significant resistance. Strains of S. pneumoniae, E. coli, S. aureus, and P. aeruginosa have been identified that possess both altered outer membrane permeability and gyrA mutations, resulting in high-level resistance to all tested fluoroquinolones.28,29 Strains of highly ciprofloxacin-resistant Salmonella with both outer membrane protein alterations and expression of efflux pumps have also been described.30
Fluoroquinolone resistance among pathogens such as S. aureus and P. aeruginosa has been particularly problematic since the introduction of these agents into clinical use. As fluoroquinolones have become more extensively used in the treatment of respiratory tract infections, reports of increasing resistance among S. pneumoniae have focused attention on newly recognized mechanisms of drug action and drug resistance.19–22 Resistance to fluoroquinolones has tended to emerge rapidly in bacteria with lower intrinsic susceptibility (e.g., S. aureus, P. aeruginosa, and Acinetobacter spp.) because potentially fewer mutational steps are required to confer clinically relevant MIC changes.26,31,32 However, fluoroquinolone resistance has also been noted to be an increasing problem among gram-negative bacilli such as Enterobacter spp., Klebsiella pneumoniae, and even E. coli, organisms that were originally considered to be highly susceptible to the drugs.6,8,10 This problem is particularly an issue among isolates from ICUs.10 Development of resistance is also accelerated by the use of drugs with lower in vitro activity, use of inappropriately low doses to treat infections caused by less susceptible organisms, and treatment of infection at sites where quinolone penetration may be decreased.31–34 Development of resistance to one fluoroquinolone usually causes decreased susceptibility to all other agents in the class, although clinically relevant resistance may not necessarily occur. Of note, fluoroquinolone use has also been associated with high rates of cross-resistance among drugs of unrelated antibiotic classes such as the carbapenems, cephalosporins, and aminoglycosides.33–35 Although not well understood, such cross-resistance is probably mediated by up-regulation and/or reduction in multiple efflux pump systems involved in passage of antibiotics through cell membranes and intracellular drug accumulation.33–35 Although the fluoroquinolones remain highly active and clinically effective against a wide variety of important pathogens found in critically ill patients, increasing resistance is clearly an important issue in the clinical use of these drugs.
 Pharmacokinetics
Pharmacokinetics
Pharmacokinetic properties of the currently used fluoroquinolones are shown in Table 122-2. Individual agents in the class exhibit distinct differences in properties such as oral bioavailability, half-lives, extent of metabolism, and routes of excretion. However, fluoroquinolones as a whole are characterized by rapid oral absorption and extensive distribution into many fluids and tissues, resulting in concentrations that are well above the MIC for many gram-negative and gram-positive organisms; serum half-lives are sufficiently long to allow once- or twice-daily dosing. Ciprofloxacin and levofloxacin have been most extensively studied in critically ill patients. Although large interpatient variability and some differences in mean parameters were observed compared with normal volunteers, pharmacokinetics of the drugs were generally similar enough to allow the use of normally recommended doses.36,37
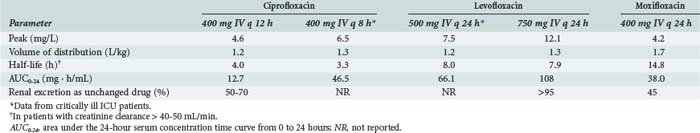
Certain pharmacokinetic features are of particular importance during use of these drugs in critically ill patients. Limited data suggest that fluoroquinolones are well absorbed after oral administration to critically ill patients, although patients must be carefully selected for clinical stability and absence of GI diseases or processes that may affect drug absorption.37
Fluoroquinolones have excellent distribution into many tissues and fluids and often reach concentrations many-fold higher than found in blood. For example, ciprofloxacin achieves tissue-to-serum concentration ratios of approximately 2 in bronchial and lung tissues, 2 in lung tissues, 13 in the kidneys, and up to 30 in the bile.38 Levofloxacin has been shown to achieve pulmonary epithelial lining fluid–to–plasma and alveolar macrophage–to–plasma ratios of 2.1 to 2.3 and 8.9 to 12.0, respectively, 12 hours after multiple-dose administration of levofloxacin, 500 to 750 mg orally.39 Such high tissue and fluid levels have important pharmacodynamic implications (see later) and increase the likelihood of successfully treating infections at these sites. In contrast, penetration of the fluoroquinolones into the cerebrospinal fluid is relatively poor and ranges from 20% to 40% of serum concentrations in the absence of inflamed meninges.
Ciprofloxacin and levofloxacin are excreted to a large degree through the kidneys as unmetabolized drug; doses should therefore be appropriately adjusted in the presence of moderate to severe renal dysfunction to avoid unnecessary drug accumulation. In contrast, elimination of moxifloxacin is relatively insensitive to changes in renal function; this drug is highly metabolized, and even severe renal impairment does not influence dosing requirements. Mild to moderate hepatic impairment does not appear to significantly affect the pharmacokinetics of these agents; however, most drugs have not been well studied in patients with severe or end-stage liver disease, and consideration should be given to empirically decreasing the daily dosage of hepatically eliminated drugs. Ciprofloxacin has been shown to undergo compensatory increases in renal clearance in patients with severe liver disease, and no dosage adjustments are required if renal function is normal40; however, caution is warranted when dosing ciprofloxacin in patients with both hepatic and renal dysfunction.38,40
 Pharmacodynamic Considerations
Pharmacodynamic Considerations
Studies have clearly demonstrated that fluoroquinolones exhibit concentration-dependent bacterial killing.41–48 A number of studies, including a prospectively developed model of the pharmacodynamic response to levofloxacin during treatment of respiratory tract, skin, and urinary tract infections, have provided evidence that achieving a ratio of fluoroquinolone maximum serum concentrations to the bacterial MIC (Cmax/MIC ratio) of greater than 10 to 12 appears to be predictive of clinical drug efficacy and successful bacterial eradication.41–48 The ratio of area under the 24-hour serum concentration time curve to MIC (AUC0-24/MIC) has also been shown in vitro and retrospectively in vivo to be predictive of favorable clinical response and reduced development of resistance.41–48 Although the optimal AUC0-24/MIC ratio breakpoints are still unclear, favorable AUC0-24/MIC ratios appear to be 125 to 250 for gram-negative organisms and 30 to 50 for S. pneumoniae.41–48 Whether either the Cmax/MIC ratio or AUC0-24/MIC ratio is superior to the other parameter and which specific ratios are most predictive of drug efficacy remain somewhat controversial; however, the strong relationships between these pharmacodynamic parameters and clinical and microbiological outcomes during fluoroquinolone therapy have been well established.
 Adverse Effects
Adverse Effects
With some notable exceptions (e.g., trovafloxacin), fluoroquinolones have generally proven to be a safe and well-tolerated class of drugs. The most common adverse effects associated with fluoroquinolones are GI effects such as nausea, vomiting, and diarrhea (∼1% to 5% incidence); rash (<2.5%); and central nervous system (CNS) effects, including headache, dizziness, and sleep disturbances (<1% to 2%). These adverse effects are generally mild and self-limiting and seldom result in discontinuation of fluoroquinolone therapy. With IV preparations, pain and inflammation at the injection site have also been reported.49–52
Adverse GI effects of the fluoroquinolones are thought to be caused by a combination of direct GI irritation and CNS-mediated effects; adverse GI effects may still be seen when these drugs are administered IV. Clostridium difficile–associated colitis has been associated with fluoroquinolone use in several epidemiological studies, but other studies have not confirmed fluoroquinolone use as a significant risk factor for C. difficile infection.53–56 There was initially some concern that newer fluoroquinolones with enhanced anaerobic activity and GI elimination (e.g., moxifloxacin) may perhaps be associated with an increased risk of C. difficile–associated colitis, but whether there is a difference among the various fluoroquinolones, or indeed whether fluoroquinolones in general are truly associated with increased risk of C. difficile infection, is still controversial.53–56
CNS disturbances caused by fluoroquinolones can be broadly divided into two types: those resulting from direct effects of the drugs on the CNS caused by inhibition of γ-aminobutyric acid (GABA) binding, and those resulting from adverse drug-drug interactions (either pharmacokinetic or pharmacodynamic). Of all the fluoroquinolones, levofloxacin is associated with the lowest incidence of adverse CNS events; however, the occurrence of adverse CNS effects with ciprofloxacin and moxifloxacin is similar and only slightly more frequent than with levofloxacin. Seizures have been only rarely reported during fluoroquinolone therapy and usually occurred in the presence of predisposing factors such as seizure disorder, head trauma, anoxia, metabolic disturbances, or concomitant drug therapy with specific interacting agents (i.e., theophylline).49–52
Elevations in serum transaminase, alkaline phosphatase, and/or bilirubin levels have been noted to occur in 2% to 3% of patients receiving fluoroquinolone therapy.49–52 These liver abnormalities are usually mild, are reversible, and do not necessitate discontinuation of therapy. Although trovafloxacin was associated with clinically significant hepatotoxicity and acute hepatic failure, currently used fluoroquinolones have been only rarely associated with liver injury.
Fluoroquinolones as a class have been implicated in causing abnormalities of glucose homeostasis.52 Both hypoglycemia and hyperglycemia have been reported, and patients with preexisting diabetes mellitus or other known glucose abnormalities are apparently at particularly high risk. Glucose abnormalities appeared to occur most commonly with gatifloxacin, and that drug was first restricted in use then removed from the market because of the potentially increased risk. Although other fluoroquinolones have also been associated with glucose abnormalities, the overall risk appears to be quite low. However, glucose levels should be monitored in all acutely ill patients receiving fluoroquinolone therapy, with special care being warranted in elderly patients and those with diabetes mellitus or pre-diabetes.52
The potential of fluoroquinolones to cause cardiac toxicity has also been examined; such toxicity manifests as electrocardiographic prolongation of the corrected QT (QTc) interval and arrhythmias including ventricular tachycardia, ventricular fibrillation, and torsades de pointes.52,57–59 Some controversy exists as to the true risk of cardiac toxicity associated with fluoroquinolones and whether specific agents might be associated with a greater degree of risk. It appears that all currently available fluoroquinolones are capable of causing some degree of QTc prolongation. However, QTc interval prolongation is usually quite minor (mean of <5 to 10 ms, with greatest prolongation usually observed with moxifloxacin), does not predictably occur in all patients exposed to fluoroquinolones, and is of no clinical significance in the vast majority of patients treated with these agents. Patients who may be at particular risk of drug-induced cardiac toxicity and who should be more closely monitored during fluoroquinolone use include those with the following characteristics: advanced age (>60 years), history of significant cardiac disease or previous arrhythmia, presence of electrolyte abnormalities (e.g., potassium, calcium, magnesium), and concomitant use of antiarrhythmic or other drugs known to cause prolongation of the QTc interval.52,57–59
Tendonitis and tendon rupture are unusual complications of fluoroquinolone use. A total of only 33 cases associated with ciprofloxacin or levofloxacin had been reported in the medical literature up until 2003.60 Several studies have subsequently carried out detailed analyses of fluoroquinolone-associated tendonitis and tendon rupture and have confirmed tendinopathies to be uncommon complications of drug use, with an estimated incidence of approximately 1/200,000 treated patients.52,61 The median duration of drug use before the onset of symptoms is approximately 10 to 14 days. Risk factors for the occurrence of tendinopathy appear to include male sex, age older than 60 years, concurrent corticosteroid use, and presence of renal disease.52,61
 Drug-Drug Interactions
Drug-Drug Interactions
Concurrent administration of oral fluoroquinolones with multivalent cation-containing products such as aluminum- or magnesium-containing antacids and products containing calcium, iron, or zinc (including multivitamins with minerals) should be avoided. Concomitant use of these agents with a fluoroquinolone invariably results in a marked reduction of oral absorption of the antimicrobial; bioavailability of fluoroquinolones may be reduced as much as 90% owing to the formation of insoluble chelation complexes in the GI tract that inhibit drug absorption.51,62 Similar changes in oral antibiotic absorption have also been observed with concurrent administration of sucralfate or ferrous sulfate. Effects of enteral feeding formulas on the absorption of fluoroquinolones are variable, but concurrent administration should nevertheless be avoided. Concomitant administration of H2-receptor antagonists and proton-pump inhibitors have no clinically significant effects on the absorption of fluoroquinolones.51,62
Ciprofloxacin has been shown to decrease theophylline clearance by a mean of approximately 25% to 30%.51,62 Levofloxacin and moxifloxacin have no significant effects on theophylline metabolism. Ciprofloxacin was also shown to reduce the clearance of the R-enantiomer of warfarin by 15% to 32%; however, the clearance of the S-enantiomer, which is more potent and is thought to cause the majority of warfarin’s anticoagulant activity, was not affected. Therefore, this interaction was not thought to be clinically significant. No apparent effects on either the R- or S-warfarin concentrations or prothrombin times were noted during concomitant dosing of levofloxacin or moxifloxacin.51,62 Although significant pharmacokinetic or pharmacodynamic interactions between the fluoroquinolones and warfarin have not been documented through studies, several anecdotal case reports have described clinically significant interactions between warfarin and fluoroquinolones. Anticoagulation of any patient receiving concomitant fluoroquinolone and warfarin therapy should therefore be closely monitored.
 Dosing
Dosing
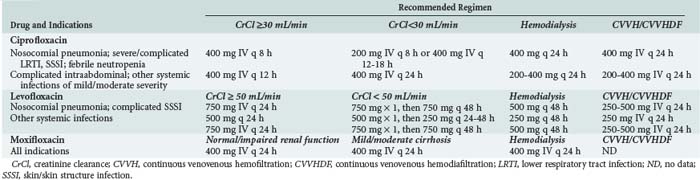
Recommendations for dosing of currently available fluoroquinolones are given in Table 122-3. Recommended regimens in the presence of renal or hepatic dysfunction are also given when appropriate and where data are available. Consideration should always be given to the susceptibility of presumed or documented pathogens, site of infection, severity of infection, presence of organ dysfunction, and pharmacodynamic characteristics of fluoroquinolones when choosing an appropriate dosing regimen for a specific patient.
Key Points
Fish DN. Fluoroquinolone adverse effects and drug interactions. Pharmacotherapy. 2001;21:253S-272S.
Neuhauser MM, Weinstein RA, Rydman R, et al. Antibiotic resistance among gram-negative bacilli in US intensive care units: implications for fluoroquinolone use. JAMA. 2003;289:885-888.
Preston SL, Drusano GL, Berman AL, et al. Pharmacodynamics of levofloxacin: a new paradigm for early clinical trials. JAMA. 1998;279:125-129.
Rebuck JA, Fish DN, Abraham E. Pharmacokinetics of intravenous and oral levofloxacin in critically ill patients in a medical intensive care unit. Pharmacotherapy. 2002;22:1216-1225.
Sahm DF, Critchley IA, Kelly LJ, et al. Evaluation of current activities of fluoroquinolones against gram-negative bacilli using centralized in vitro testing and electronic surveillance. Antimicrob Agents Chemother. 2001;45:267-274.
Hidron AI, Edwards JR, Patel J, et al. NHSN annual update: antimicrobial-resistant pathogens associated with healthcare-associated infections: annual summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2006-2007. Infect Control Hosp Epidemiol. 2008;29:996-1011.
Owens RC, Ambrose PG. Antimicrobial safety: focus on fluoroquinolones. Clin Infect Dis. 2005;41:S144-S157.
1 Hooper DC. Bacterial topoisomerases, anti-topoisomerases, and anti-topoisomerase resistance. Clin Infect Dis. 1998;27(Suppl 1):S54-S63.
2 Hawkey PM. Mechanisms of quinolone action and microbial response. J Antimicrob Chemother. 2003;51(Suppl S1):29-35.
3 Pan XS, Yague G, Fisher LM. Quinolone resistance mutations in Streptococcus pneumoniae GyrA and parC proteins: mechanistic insights into quinolone action from enzymatic analysis, intracellular levels, and phenotypes of wild-type and mutant proteins. Antimicrob Agents Chemother. 2001;45:3140-3147.
4 Drlica K, Zhao X. DNA gyrase, topoisomerase IV, and the 4-quinolones. Microbiol Mol Biol Rev. 1997;61:377-392.
5 Dalhoff A. Immunomodulatory activities of fluoroquinolones. Infection. 2005;33(Suppl 2):55-70.
6 Sahm DF, Critchley IA, Kelly LJ, et al. Evaluation of current activities of fluoroquinolones against gram-negative bacilli using centralized in vitro testing and electronic surveillance. Antimicrob Agents Chemother. 2001;45:267-274.
7 Van Eldere J. Multicentre surveillance of Pseudomonas aeruginosa susceptibility patterns in nosocomial infections. J Antimicrob Chemother. 2003;51:347-352.
8 Neuhauser MM, Weinstein RA, Rydman R, et al. Antibiotic resistance among gram-negative bacilli in US intensive care units: implications for fluoroquinolone use. JAMA. 2003;289:885-888.
9 MacGowan AP, Wootton M, Holt HA. The antibacterial efficacy of levofloxacin and ciprofloxacin against Pseudomonas aeruginosa assessed by combining antibiotic exposure and bacterial susceptibility. J Antimicrob Chemother. 1999;43:345-349.
10 Hidron AI, Edwards JR, Patel J, et al. NHSN annual update: antimicrobial-resistant pathogens associated with healthcare-associated infections: annual summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2006-2007. Infect Control Hosp Epidemiol. 2008;29:996-1011.
11 Van Bambeke F, Michot J-M, Van Eldere J, et al. Quinolones in 2005: an update. Clin Microbiol Infect. 2005;11:256-280.
12 Woodcock JM, Andrews JM, Boswell FJ, et al. In vitro activity of BAY 12-8039, a new fluoroquinolone. Antimicrob Agents Chemother. 1997;41:101-106.
13 Blondeau JM. A review of the comparative in-vitro activities of 12 antimicrobial agents, with a focus on five new “respiratory quinolones.”. J Antimicrob Chemother. 1999;43(Suppl B):1-11.
14 Burgess DS, Nathisuwan S. Cefepime, piperacillin/tazobactam, gentamicin, ciprofloxacin, and levofloxacin alone and in combination against Pseudomonas aeruginosa. Diagn Microbiol Infect Dis. 2002;44:35-41.
15 Fish DN, Choi MK, Jung R. Synergic activity of cephalosporins plus fluoroquinolones against Pseudomonas aeruginosa with resistance to one or both drugs. J Antimicrob Chemother. 2002;50:1045-1049.
16 Snydman DR, Jacobus NV, McDermott LA, et al. Lessons learned from the anaerobe survey: historical perspective and review of the most recent data (2005-2007). Clin Infect Dis. 2010;50(Suppl 1):S26-S33.
17 Jacoby GA. Mechanisms of resistance to quinolones. Clin Infect Dis. 2005;41(Suppl 2):S120-S126.
18 Strahilevitz J, Jacoby GA, Hooper DC, et al. Plasmid-mediated quinolone resistance: a multifaceted threat. Clin Microbiol Rev. 2009;22:664-689.
19 Eliopoulos GM. Quinolone resistance mechanisms in Streptococcus pneumoniae. Clin Infect Dis. 2004;38(Suppl 4):S350-S356.
20 Ho PL, Yung RW, Tsang DN, et al. Increasing resistance of Streptococcus pneumoniae to fluoroquinolones: results of a Hong Kong multicentre study in 2000. J Antimicrob Chemother. 2001;48:659-665.
21 Johnson AP, Sheppard CL, Harnett SJ, et al. Emergence of a fluoroquinolone-resistant strain of Streptococcus pneumoniae in England. J Antimicrob Chemother. 2003;52:953-960.
22 Goldstein EJC, Garabedian-Ruffalo SM. Widespread use of fluoroquinolones versus emerging resistance in pneumococci. Clin Infect Dis. 2002;35:1505-1511.
23 Cohen SP, Hooper DC, Wolfson JS, et al. Endogenous active efflux of norfloxacin in susceptible Escherichia coli. Antimicrob Agents Chemother. 1988;32:1187-1191.
24 Kaatz GW, Seo SM, Ruble CA. Efflux-mediated fluoroquinolone resistance in Staphylococcus aureus. Antimicrob Agents Chemother. 1993;37:1086-1094.
25 Kriengkauykiat J, Porter E, Lomovskaya O, et al. Use of an efflux pump inhibitor to determine the prevalence of efflux-mediated fluoroquinolone resistance and multidrug resistance in Pseudomonas aeruginosa. Antimicrob Agents Chemother. 2005;49:565-570.
26 Kaatz GW, Seo SM, O’Brien L, et al. Evidence for the existence of a multidrug efflux transporter distinct from NorA in Staphylococcus aureus. Antimicrob Agents Chemother. 2000;44:1404-1406.
27 Yang Y-L, Lauderdale T-L, Lo H-J. Molecular mechanisms of fluoroquinolone resistance in Klebsiella. Curr Drug Targets Infect Disorders. 2004;4:295-302.
28 Bast DJ, Low DE, Duncan CL, et al. Fluoroquinolone resistance in clinical isolates of Streptococcus pneumoniae: contributions of type II topoisomerase mutations and efflux to levels of resistance. Antimicrob Agents Chemother. 2000;44:3049-3054.
29 Tanaka M, Zhang YX, Ishida H, et al. Mechanisms of 4-quinolone resistance in quinolone-resistant and methicillin-resistant Staphylococcus aureus isolates from Japan and China. J Med Microbiol. 1995;42:214-219.
30 Giraud E, Cloeckaert A, Kerboeuf D, et al. Evidence for active efflux as the primary mechanism of resistance to ciprofloxacin in Salmonella enterica serovar typhimurium. Antimicrob Agents Chemother. 2000;44:1223-1228.
31 Bakken JS. The fluoroquinolones: how long will their utility last? Scand J Infect Dis. 2004;36:85-92.
32 Le Thomas I, Couetdic G, Clermont O, et al. In vivo selection of a target/efflux double mutant of Pseudomonas aeruginosa by ciprofloxacin therapy. J Antimicrob Chemother. 2001;48:553-555.
33 Lister PD, Wolter DJ, Hanson ND. Antibacterial-resistant Pseudomonas aeruginosa: clinical impact and complex regulation of chromosomally encoded resistance mechanisms. Clin Microbiol Rev. 2009;22:582-610.
34 Poole K, Strikumar R. Multidrug efflux in Pseudomonas aeruginosa: components, mechanisms, and clinical significance. Curr Top Med Chem. 2001;1:59-71.
35 Livermore DM. Multiple mechanisms of antimicrobial resistance in Pseudomonas aeruginosa: our worst nightmare? Clin Infect Dis. 2002:634-640.
36 Conil J-M, Georges B, de Lussy A, Khachman D, Seguin T, Ruiz S, et al. Ciprofloxacin use in critically ill patients: pharmacokinetic and pharmacodynamic approaches. Int J Antimicrob Agents. 2008;32:505-510.
37 Rebuck JA, Fish DN, Abraham E. Pharmacokinetics of intravenous and oral levofloxacin in critically ill patients in a medical intensive care unit. Pharmacotherapy. 2002;22:1216-1225.
38 Aminimanizani A, Beringer P, Jelliffe R. Comparative pharmacokinetics and pharmacodynamics of the newer fluoroquinolone antibacterials. Clin Pharmacokinet. 2001;40:169-187.
39 Gotfried MH, Danziger LH, Rodvold KA. Steady-state plasma and intrapulmonary concentrations of levofloxacin and ciprofloxacin in healthy adult subjects. Chest. 2001;119:1114-1122.
40 Jones EM. The pharmacokinetics of intravenous ciprofloxacin 400 mg 12 hourly in patients with severe sepsis: the effect of renal function and intra-abdominal disease. J Antimicrob Chemother. 1997;40:121-124.
41 Craig WA. Pharmacokinetic/pharmacodynamic parameters: rationale for antibacterial dosing of mice and men. Clin Infect Dis. 1998;26:1-10.
42 DeRyke CA, Lee SY, Kuti JL, Nicolau DP. Optimising dosing strategies of antibacterials utilising pharmacodynamic principles: impact on the development of resistance. Drugs. 2006;66:1-14.
43 Ambrose PG, Bhavnani SM, Rubina CM, et al. Pharmacokinetics-pharmacodynamics of antimicrobial therapy: it’s not just for mice anymore. Clin Infect Dis. 2007;44:79-86.
44 Preston SL, Drusano GL, Berman AL, et al. Pharmacodynamics of levofloxacin: a new paradigm for early clinical trials. JAMA. 1998;279:125-129.
45 Wright DH, Brown GH, Peterson ML, Rotschafer JC. Application of fluoroquinolone pharmacodynamics. J Antimicrob Chemother. 2000;46:669-683.
46 Thomas JK, Forrest A, Bhavnani SM, et al. Pharmacodynamic evaluation of factors associated with the development of bacterial resistance in acutely ill patients during therapy. Antimicrob Agents Chemother. 1998;42:521-527.
47 Lister PD, Sanders CC. Pharmacodynamic comparisons of levofloxacin and ciprofloxacin against Streptococcus pneumoniae. J Antimicrob Chemother. 1999;43:79-86.
48 Ambrose PG, Grasela DM, Grasela TH, et al. Pharmacodynamics of fluoroquinolones against Streptococcus pneumoniae in patients with community-acquired respiratory tract infections. Antimicrob Agents Chemother. 2001;45:2793-2797.
49 Iannini PB. The safety profile of moxifloxacin and other fluoroquinolones in special patient populations. Curr Med Res Opin. 2007;23:1403-1413.
50 Bertino JJr, Fish DN. The safety profile of the fluoroquinolones. Clin Ther. 2000;22:798-817.
51 Fish DN. Fluoroquinolone adverse effects and drug interactions. Pharmacotherapy. 2001;21(Suppl):253S-272S.
52 Owens RC, Ambrose PG. Antimicrobial safety: focus on fluoroquinolones. Clin Infect Dis. 2005;41:S144-S157.
53 Owens RCJr, Donskey CJ, Gaynes RP, et al. Antimicrobial-associated risk factors for Clostridium difficile infection. Clin Infect Dis. 2008;46(Suppl 1):S19-S31.
54 Cadena J, Thompson GRIII, Patterson JE, et al. Clinical predictors and risk factors for relapsing Clostridium difficile infection. Am J Med Sci. 2010;339:350-355.
55 Novell MJ, Morreale CA. The relationship between inpatient fluoroquinolone use and Clostridium difficile-associated diarrhea. Ann Pharmacother. 2010;44:826-831.
56 Weiss K. Clostridium difficile and fluoroquinolones: is there a link? Int J Antimicrobial Agents. 2009;33(Suppl 1):S29-S32.
57 Falagas ME, Rafailidis PI, Rasmarakis ES. Arrhythmias associated with fluoroquinolone therapy. Int J Antimicrob Agents. 2007;29:374-379.
58 Owens RC Jr. Risk assessment for antimicrobial agent-induced QTc interval prolongation and torsades de pointes. Pharmacotherapy. 2001;21:301-319.
59 Frothingham R. Rates of torsades de pointes associated with ciprofloxacin, ofloxacin, levofloxacin, gatifloxacin, and moxifloxacin. Pharmacotherapy. 2001;21:1468-1472.
60 Khaliq Y, Zhanel GG. Fluoroquinolone-associated tendinopathy: a critical review of the literature. Clin Infect Dis. 2003;36:1404-1410.
61 Sendzik J, Lode H, Stahlmann R. Quinolone-induced arthropathy: an update focusing on new mechanistic and clinical data. Int J Antimicrobial Agents. 2009;33:194-200.
62 Pai MP, Momary KM, Rodvold KA. Antibiotic drug interactions. Med Clin North Am. 2006;90:1223-1255.
[/level-membership-for-critical-care-medicine-category][not-level-membership-for-critical-care-medicine-category]
122 Fluoroquinolones
Mechanism of Action
DNA gyrase and topoisomerase IV enzymes are thought to be essential for the replication of DNA and partition of replicated chromosomal DNA.1 DNA gyrase, a tetrameric enzyme consisting of two A and two B subunits, is known to be a primary target of fluoroquinolones in gram-negative bacteria and is the only known enzyme capable of introducing negative super-helical twists into bacterial DNA.1,2 The two subunits of gyrase are encoded by gyrA and gyrB, which are also potential sites of mutation and subsequent quinolone resistance.1,3 Topoisomerase IV seems to be a primary target of many fluoroquinolones in gram-positive bacteria such as Staphylococcus aureus and Streptococcus pneumoniae.3,4 Bacterial topoisomerase IV appears to be the principal enzyme that resolves or “decatenates” interlocked daughter DNA circles occurring at the completion of a round of DNA replication, allowing segregation of daughter chromosomes into daughter cells.1,3,4 Topoisomerase IV, like DNA gyrase, is composed of four subunits, two each of the parC and parE gene products.
As part of the topoisomerase reaction mechanism, DNA gyrase and topoisomerase IV transiently break the DNA backbone and pass a double strand of DNA through those breaks, thus introducing a negative supercoil into the DNA strand.1,2 Fluoroquinolone antibiotics have been shown to target DNA gyrase and topoisomerase IV while these enzymes are functionally attached to the DNA strand in the presence of adenosine triphosphate, resulting in a drug/enzyme/DNA complex in which the DNA remains broken.1,2 Cell death apparently results from release of double-stranded DNA breaks from multiple drug/enzyme/DNA complexes throughout the chromosome.1,2 This mechanism of action does not in itself explain why the fluoroquinolones kill bacteria so rapidly, and it has been suggested that additional protein synthesis mechanisms involving unidentified “protein factors,” interference with the “SOS” response involved in the repair of damaged DNA, dissociation of gyrase subunits, and increased oxidative stress may all play a role in the rapidly bactericidal effects of these drugs.1,2 Fluoroquinolones have also been noted to substantially decrease the synthesis of proinflammatory cytokines, although the relevance of this finding to their overall pharmacologic activity is unknown.5
Antimicrobial Spectrum of Activity
Fluoroquinolones have excellent in vitro activity against a wide range of both gram-positive and gram-negative organisms. Representative activities of fluoroquinolones which are currently available and frequently used in critically ill patients are shown in Table 122-1. The entire fluoroquinolone class displays excellent activity against enteric gram-negative aerobic bacteria as well as Haemophilus influenzae, Moraxella catarrhalis, and Neisseria spp. Gastrointestinal (GI) pathogens such as Salmonella spp., Shigella spp., and Campylobacter spp. are also highly susceptible to fluoroquinolones. Although some differences in relative potency exist between individual drugs as determined by the minimum inhibitory concentration (MIC) for these organisms, little difference in clinical efficacy should be expected in the treatment of infections due to susceptible strains. Activity against P. aeruginosa is more variable, however. Ciprofloxacin has traditionally been considered the most active fluoroquinolone against this organism, but data suggest there is little difference between ciprofloxacin and levofloxacin in terms of relative susceptibility of P. aeruginosa strains.6 Ciprofloxacin was active against greater than 95% of P. aeruginosa strains when first released to the market in 1987, but by 2001 both ciprofloxacin and levofloxacin were active against only approximately 65% to 80% of strains. Such high levels of resistance (25%-35%) are still seen among P. aeruginosa strains.6–10 Clinically relevant differences between ciprofloxacin and levofloxacin are further minimized when pharmacokinetic and pharmacodynamic properties are considered.9 Moxifloxacin tends to be the least active of the currently available agents.6,11–13 Nearly all fluoroquinolones adequately inhibit P. aeruginosa at concentrations achieved in the urine.
Activity of the fluoroquinolones against other hospital-acquired pathogens is also highly variable. Levofloxacin tends to be slightly more active against Acinetobacter spp., whereas moxifloxacin usually displays the best activity and ciprofloxacin is consistently the least active agent against Stenotrophomonas maltophilia.6,11–13 However, resistance to these latter organisms is quite common, and even agents with the best relative in vitro activity are not reliably clinically effective against many isolates.6,11–13
Several studies have reported that fluoroquinolones produce synergistic activity against gram-negative bacilli when used in combination with β-lactam antibiotics.14,15 These studies primarily evaluated antibiotic synergy against P. aeruginosa due to the frequent use of fluoroquinolones in antipseudomonal treatment regimens; ciprofloxacin and levofloxacin have been shown to achieve synergy against 25% to 75% of tested strains. One previous study also demonstrated synergistic in vitro activity against P. aeruginosa with the combination of moxifloxacin and either ceftazidime or cefepime15; however, additive or synergistic activity with moxifloxacin-containing combinations has not been extensively evaluated against other organisms. The ability of moxifloxacin to produce synergistic activity against P. aeruginosa may often be limited by clinically achievable drug concentrations. The use of ciprofloxacin and levofloxacin in combination regimens is most likely to result in synergistic activity, owing to their more potent activity and higher serum concentrations relative to the bacterial MICs.14,15
Newer fluoroquinolones have improved activity against gram-positive bacteria relative to older agents such as ciprofloxacin. Moxifloxacin has the best overall activity against staphylococci and streptococci, followed by levofloxacin and more distantly by ciprofloxacin.12,13 Levofloxacin and moxifloxacin are reliably active against penicillin-susceptible strains of S. pneumoniae; this activity is also retained against strains of S. pneumoniae resistant to other drug classes including penicillins, macrolides, and sulfonamides. Although ciprofloxacin has only moderate activity against methicillin-susceptible S. aureus (MSSA), newer agents have excellent activity against this organism. None of the fluoroquinolones is reliably active against methicillin-resistant S. aureus (MRSA), and rates of fluoroquinolone resistance among MRSA are quite high. Fluoroquinolones as a class also have only moderate activity against enterococci, with great variability seen among the various agents and specific bacterial strains.11–13 Fluoroquinolones have consistently excellent activity against Listeria monocytogenes.11–13
The activity of various fluoroquinolones against anaerobic bacteria is highly variable. Trovafloxacin was the first commercially available fluoroquinolone with clinically relevant anaerobic activity in vitro, as well as proven clinical efficacy for anaerobic infections including complicated intraabdominal infection. Moxifloxacin has in vitro activity against Bacteroides fragilis, Bacteroides group organisms, Fusobacterium spp., Clostridium spp., and other anaerobes that is generally comparable to trovafloxacin.12,13,16 However, recent data have demonstrated resistance rates in excess of 30% for many clinically important anaerobes, and the appropriateness of moxifloxacin for treatment of serious anaerobic infections is questionable.16 Neither ciprofloxacin nor levofloxacin have clinically relevant activity against anaerobic bacteria.
Mechanisms of Fluoroquinolone Resistance
Two basic mechanisms of fluoroquinolone resistance have been identified. One involves alteration of DNA gyrase and topoisomerase IV, whereas the other results in reduced drug accumulation within bacterial cells.2,17 Plasmid-mediated resistance, once considered quite rare, appears to be spreading rapidly among enteric gram-negative bacilli in certain geographic regions.18 However, plasmid-mediated resistance is relatively unusual compared with the more typical chromosomally mediated mechanisms of resistance.
Mutations in quinolone-resistance determining regions (QRDR) of topoisomerase enzymes prevent formation of drug/enzyme/DNA complexes, allowing DNA synthesis to occur in the presence of the drugs. Mutations in the genes encoding DNA gyrase (gyrA and gyrB) have been most frequently identified. However, other quinolone-resistant mutations in parC and parE, the genes encoding topoisomerase IV, have also been identified.1,2,17,19 Resistance to fluoroquinolones appears to arise in a stepwise manner. In some species (e.g., gram-negative bacteria), first-step mutations occur in gyrA and occasionally in gyrB, whereas in other species (e.g., S. aureus, S. pneumoniae) first-step mutations occur in parC and less often in parE.17,19 First-step mutations usually result in a low-level resistance (≤fourfold increased MIC), whereas additional mutations in either primary or secondary enzyme targets (second-step mutations) result in high-level resistance to drugs at clinically relevant concentrations. Dual gyrA and parC mutations have been described in clinical isolates of S. pneumoniae; however, it is thought that these strains were selected by fluoroquinolones with less potent antipneumococcal activity (e.g., ciprofloxacin).19–22
The first efflux system for quinolones was identified in Escherichia coli,23 whereas the first evidence for actual efflux-mediated quinolone resistance came from the characterization of S. aureus with overexpression of the norA gene product, a protein that mediates efflux.24 Such efflux may occur in both quinolone-resistant and quinolone-susceptible strains of S. aureus. In some species (e.g., P. aeruginosa), at least two different efflux systems may be present that mediate resistance to multiple other drug classes in addition to fluoroquinolones.25 Although most efflux proteins appear to be relatively nonspecific multidrug transporters whose substrates include hydrophilic fluoroquinolones as well as monocationic organic compounds, relatively substrate-specific efflux pumps have also been described.26
Many other genetic mutations have been described that result in decreased intracellular accumulation of fluoroquinolones and low-level drug resistance. Nearly all these mutations are associated with decreased expression of OmpF, a nonspecific outer membrane porin channel that is a major route of passage of hydrophilic fluoroquinolones through bacterial cellular membranes into the periplasmic space.27 Although decreased membrane permeability is relatively common and easily induced, this is an unusual mechanism for clinically significant resistance. Strains of S. pneumoniae, E. coli, S. aureus, and P. aeruginosa have been identified that possess both altered outer membrane permeability and gyrA mutations, resulting in high-level resistance to all tested fluoroquinolones.28,29 Strains of highly ciprofloxacin-resistant Salmonella with both outer membrane protein alterations and expression of efflux pumps have also been described.30
Fluoroquinolone resistance among pathogens such as S. aureus and P. aeruginosa has been particularly problematic since the introduction of these agents into clinical use. As fluoroquinolones have become more extensively used in the treatment of respiratory tract infections, reports of increasing resistance among S. pneumoniae have focused attention on newly recognized mechanisms of drug action and drug resistance.19–22 Resistance to fluoroquinolones has tended to emerge rapidly in bacteria with lower intrinsic susceptibility (e.g., S. aureus, P. aeruginosa, and Acinetobacter spp.) because potentially fewer mutational steps are required to confer clinically relevant MIC changes.26,31,32 However, fluoroquinolone resistance has also been noted to be an increasing problem among gram-negative bacilli such as Enterobacter spp., Klebsiella pneumoniae, and even E. coli, organisms that were originally considered to be highly susceptible to the drugs.6,8,10 This problem is particularly an issue among isolates from ICUs.10 Development of resistance is also accelerated by the use of drugs with lower in vitro activity, use of inappropriately low doses to treat infections caused by less susceptible organisms, and treatment of infection at sites where quinolone penetration may be decreased.31–34 Development of resistance to one fluoroquinolone usually causes decreased susceptibility to all other agents in the class, although clinically relevant resistance may not necessarily occur. Of note, fluoroquinolone use has also been associated with high rates of cross-resistance among drugs of unrelated antibiotic classes such as the carbapenems, cephalosporins, and aminoglycosides.33–35 Although not well understood, such cross-resistance is probably mediated by up-regulation and/or reduction in multiple efflux pump systems involved in passage of antibiotics through cell membranes and intracellular drug accumulation.33–35 Although the fluoroquinolones remain highly active and clinically effective against a wide variety of important pathogens found in critically ill patients, increasing resistance is clearly an important issue in the clinical use of these drugs.
