12 Fluid, Electrolyte, and Endocrine Problems
 Be sure to check out the supplementary content available at
Be sure to check out the supplementary content available at Pearls
• The term osmolality refers to the concentration of solute (electrolytes and proteins) per liter of fluid. Serum osmolality reflects extracellular fluid osmolality. It can be estimated with the following formula:
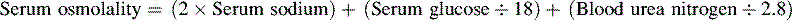
• Acute changes in serum sodium and osmolality can cause acute water shifts between the intracellular and extracellular spaces. An acute fall in serum sodium concentration and osmolality and the resulting intracellular water shift can cause cerebral edema. If neurologic symptoms develop, urgent treatment is needed. In general, significant water shifts into and out of the vascular space are poorly tolerated.
• Critical care practitioners use the child’s estimated maintenance fluid requirement as a baseline and individualize administered fluid and electrolytes to meet patient needs.
• With the development of acidosis or alkalosis, the serum potassium concentration will change in a direction opposite the change in serum pH, in response to reciprocal potassium and hydrogen ion shifts into and out of the cell.
• If the level of consciousness of the child with diabetic ketoacidosis (DKA) deteriorates during treatment, cerebral edema may be present and urgent intervention is needed. If clinical signs of cerebral edema develop, immediate treatment with intravenous (IV) mannitol or hypertonic saline is needed. If the child’s ability to protect the airway or spontaneous ventilation deteriorates, intubation and mechanical ventilation are indicated.
Anatomy and physiology
Fluid Compartments
Water accounts for 65% to 80% of total body weight. The total body water (TBW) volume and distribution are influenced by factors such as age, gender, adipose content, and skeletal muscle mass (Table 12-1). TBW is divided into two compartments (see Evolve Table 12-1 and Evolve Fig. 12-1 in the Chapter 12 Supplement on the Evolve Website for more information): intracellular fluid (ICF) and extracellular fluid (ECF) compartments (Fig. 12-1).
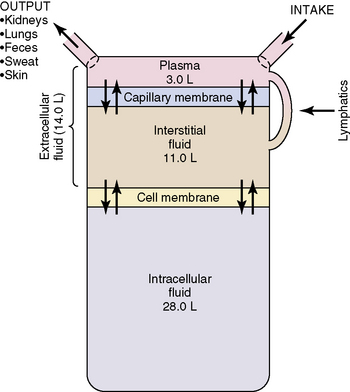
Fig. 12-1 Body fluid compartments showing values for an average 70-kg person.
(From Guyton AC, Hall JE, editors: Textbook of medical physiology, ed 11. Philadelphia, 2006, WB Saunders, p. 292, Fig. 29-1.)
Developmental Considerations
Energy requirements (per kilogram body weight) and metabolic rate are higher in infancy and childhood (for further information, see Chapter 14). The ratio of BSA to volume is significantly higher in infants and children than in adults. As a result, pediatric evaporative fluid losses and fluid requirements per kilogram body weight are higher than those of adults.
Insensible water loss (IWL) is fluid lost through the skin, via evaporation and sweat, and through the respiratory tract. Normal IWL is approximately 300 to 400 mL/m2 BSA per day (for more detailed information, see Evolve Table 12-2 in the Chapter 12 Supplement on the Evolve Website). Fever increases IWL by approximately 0.42 mL/kg per hour for each degree Celsius increase above 37° C. Increased IWL can occur with increased air movement across the skin, and with tachypnea, unless inspired air is humidified. IWL decreases when ambient and inspired air are humidified.
Table 12-2 Formulas for Estimating Daily Maintenance Fluid and Electrolyte Requirements for Children
| Daily Requirements | Hourly Requirements | |
| Fluid Requirements Estimated from Weight* | ||
| Newborn (up to 72 hr after birth) | 60-100 mL/kg (newborns are born with excess body water) | – |
| Up to 10 kg | 100 mL/kg (can increase up to 150 mL/kg to provide caloric requirements if renal and cardiac function are adequate) | 4 mL/kg |
| 11-20 kg | 1000 mL for the first 10 kg + 50 mL/kg for each kg over 10 kg | 40 mL for first 10 kg + 2 mL/kg for each kg over 10 kg |
| 21-30 kg | 1500 mL for the first 20 kg + 25 mL/kg for each kg over 20 kg | 60 mL for first 20 kg + 1 mL/kg for each kg over 20 kg |
| Fluid Requirements Estimated from Body Surface Area (BSA) | ||
| Maintenance | 1500 mL/m2 BSA | – |
| Insensible losses | 300-400 mL/m2 BSA | – |
| Electrolytes | ||
| Sodium (Na) | 2-4 mEq/kg | – |
| Potassium (K) | 1-2 mEq/kg | – |
| Chloride (Cl) | 2-3 mEq/kg | – |
| Calcium (Ca) | 0.5-3 mEq/kg | – |
| Phosphorous (Phos) | 0.5-2 mmol/kg | – |
| Magnesium (Mg) | 0.4-0.9 mEq/kg | – |
* The “maintenance” fluids calculated by these formulas must only be used as a starting point to determine the fluid requirements of an individual patient. If intravascular volume is adequate, children with cardiac, pulmonary, or renal failure or increased intracranial pressure should generally receive less than these calculated “maintenance” fluids. The formula utilizing body weight generally results in a generous “maintenance” fluid total.
Fluid and electrolyte requirements will vary with age and clinical condition. Normal baseline fluid and electrolyte requirements are listed in Table 12-2. Critical care practitioners use estimated maintenance fluid requirements as a baseline and individualize administered fluid and electrolytes to meet patient needs.
Fluid, Electrolyte, and Glucose Balance
Role of Osmolality
The term osmolality refers to the concentration of solute (electrolytes and proteins) per liter of fluid. Serum osmolality reflects ECF osmolality. It can be estimated with the following formula*:
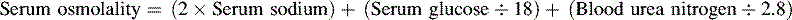
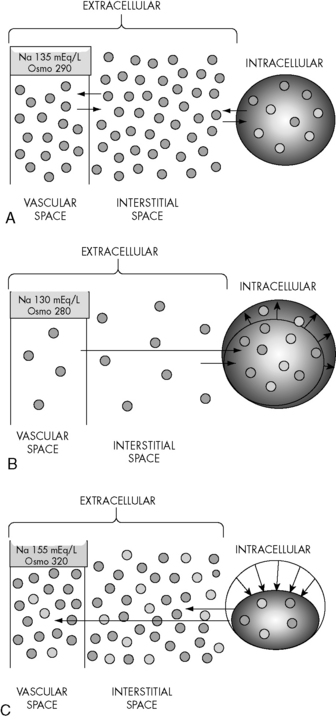
Changes in the osmolality of one body fluid compartment will affect all other compartments. Water shifts between the ICF and the ECF compartments in response to changes in the osmolality of either compartment, moving from the compartment of lower osmolality to the compartment of higher osmolality until osmolality equilibrates. When the osmolality of the extracellular compartment (including the vascular space) decreases, water will shift from the extracellular compartment into cells (Fig. 12-2). Conversely, when the osmolality of the extracellular compartment (including the vascular space) increases, water will shift from the intracellular to the extracellular compartment (including into the vascular space). The volume and acuity of the water shift, as well as likely clinical significance, is determined by the magnitude and acuity of the osmolality gradient between compartments. Significant water shifts can cause neurologic complications.
Renal Influences
The kidneys help maintain fluid balance through filtration and selective reabsorption. Changes in the glomerular filtration rate (GFR) alter the amount of water and sodium excreted or reabsorbed by the kidneys. Expansion of intravascular volume normally increases the GFR, increasing sodium and water excretion. When intravascular volume is depleted, the GFR falls and sodium and water excretion decrease (i.e., more sodium and water are reabsorbed into plasma from the renal filtrate). As noted previously, the kidney is less able to concentrate urine during the first months of life. (See Chapter 13 for more detailed information.)
Endocrine Influences
Several hormones contribute to regulation of fluid and electrolyte balance.
Antidiuretic Hormone (ADH, Vasopressin)
Negative feedback mechanisms normally regulate ADH secretion to maintain a serum osmolality of 275 to 295 mOsm/L. Osmolality receptors located in the brain are stimulated by a rise in serum osmolality (e.g., above 285 mOsm/L or a rise of at least 2%). Volume-sensitive receptors (located in the left atrium and thoracic vessels) and baroreceptors (stretch receptors located in the ascending aorta, pulmonary arteries, and carotid sinus) are stimulated by volume depletion and hypotension. Additional causes of ADH secretion include stress, trauma and severe pain (through activation of cholinergic neurotransmitters in the hypothalamus), angiotensin II, and some medications.3 A normal or low serum osmolality, hypertension, and an increase in left atrial stretch should inhibit ADH secretion.
Although endogenous ADH does not affect vascular tone, exogenous (administered) vasopressin can cause vasoconstriction and increase blood pressure. For further information regarding use vasopressin, see Chapters 6 and 14.
Serum Glucose in Critically Ill or Injured Children
Although a serum glucose concentration of 60 to 180 mg/dL is normally maintained over a wide range of conditions, critically ill or injured children often develop hypoglycemia or hyperglycemia. Infants have high glucose needs and low glycogen stores, so they can rapidly become hypoglycemic during critical illness or injury.24 Providers should monitor serum glucose concentration with point-of-care testing, if possible, and treat hypoglycemia as needed. Treatment of hypoglycemia should avoid frequent, intermittent bolus administration of large quantities of glucose; provision of a continuous source of glucose is preferable.
Hyperglycemia can result from steroid administration, stress response, relative hypoinsulinemia, or insulin resistance and has been associated with increased mortality in critically ill children in some studies. A prospective, randomized study of tight control of serum glucose concentration in critically ill children (targeted to age-adjusted normal fasting glucose concentration) reduced critical care unit mortality,37 but was associated with episodes of hypoglycemia. In general, an insulin infusion (0.5-1 unit regular insulin/kg per hour) is often titrated during the first 18 to 24 hours of critical care therapy to maintain the serum glucose concentration less than 150 mg/dL (range will vary; use your unit protocol). Careful monitoring is required to avoid and treat episodes of hypoglycemia. The ultimate value versus risk of this approach is still under investigation.
Electrolyte homeostasis and common imbalances
Table 12-3 presents a summary of electrolyte imbalances and associated clinical manifestations in critically ill infants and children. In the following sections, approximate ranges for normal and abnormal serum electrolyte concentrations are listed, but providers should use normal ranges referenced by the clinical laboratory in their practice settings.
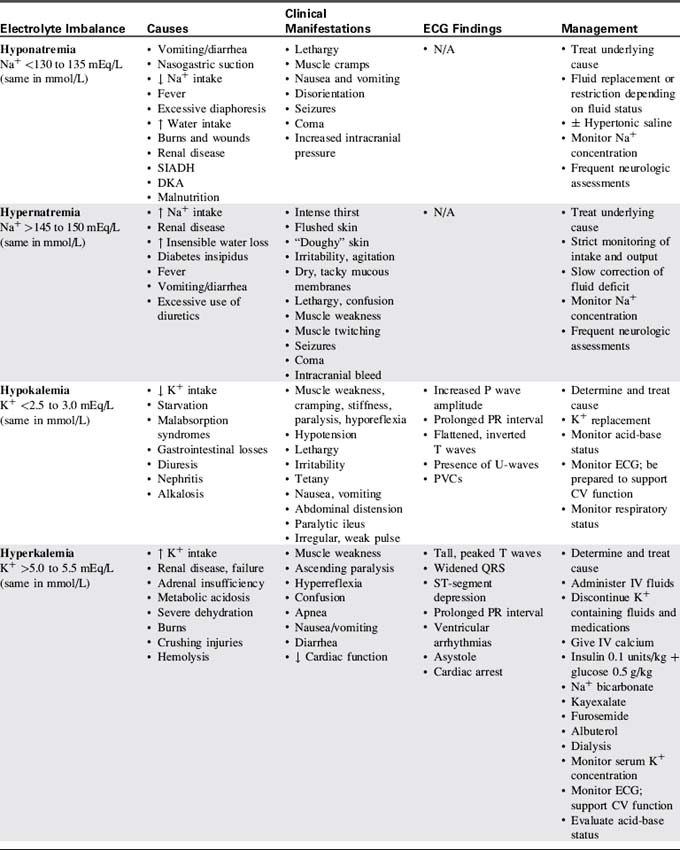
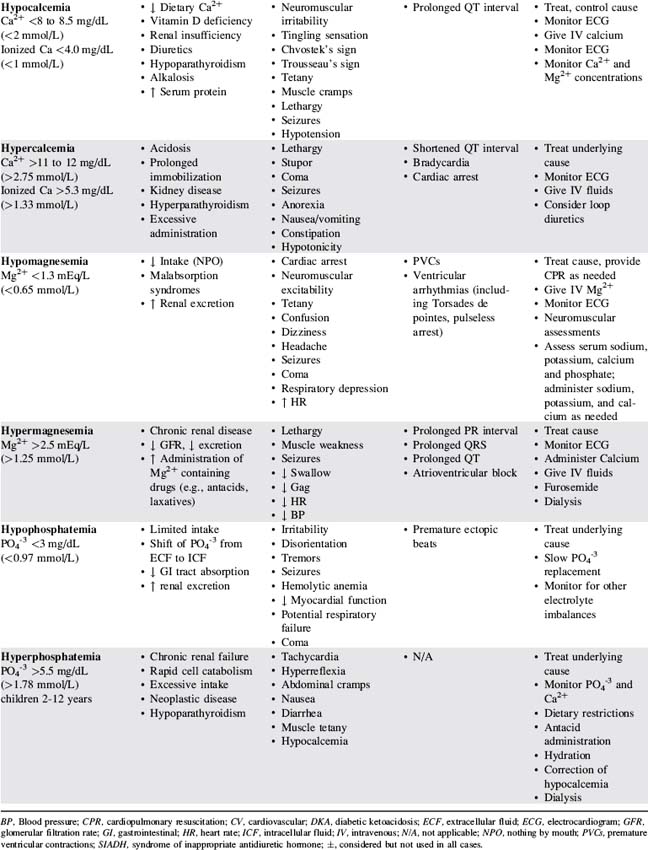
Sodium Imbalance: Hyponatremia
Etiology
Hyponatremia is a low serum sodium concentration, typically less than 130 to 135 mEq/L. It often develops as a complication of disease or therapy. The critically ill infant or child can develop hyponatremia from excessive water intake relative to sodium, excess water retention, increased sodium loss, or a combination of these factors.10
Hyponatremia can occur in conjunction with hypervolemia, euvolemia, or hypovolemia. Hypervolemic hyponatremia is associated with water intoxication, nephrotic syndrome, cardiac failure, renal failure, and the syndrome of inappropriate antidiuretic hormone (SIADH). Hypovolemic hyponatremia can occur with renal losses (e.g., osmotic diuresis, renal tubular acidosis) or extrarenal losses (e.g., diarrhea, vomiting, burns).10 Other potential causes include adrenal insufficiency, excessive use of diuretics, and cerebral salt-wasting syndrome.
Pathophysiology and Clinical Signs and Symptoms
The volume of water shift and the severity of clinical manifestations with hyponatremia are directly related to the acuity and the magnitude of the fall in serum sodium and osmolality. Infants and children who develop hyponatremia that gradually worsens over several days or weeks (e.g., with adrenocortical insufficiency) typically have milder clinical manifestations and may be asymptomatic until the serum sodium is very low.26
Acute hyponatremia that develops within hours or days (i.e., <48 hours) is more likely to produce cerebral edema.26 Seizures and coma are associated with a serum sodium concentration less than 120 mEq/L.
Management
Once the child’s neurologic status is stable and perfusion is adequate, plans are made to replace the sodium deficit. The following formula36 is used to calculate the sodium deficit:
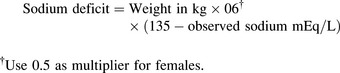
During treatment of hyponatremia, providers must closely monitor the serum sodium concentration and the rate of rise in the concentration. If the serum sodium and osmolality are raised too rapidly, the resulting water shifts from the cellular to the extracellular (including intravascular) compartments can produce neurologic complications including intracranial bleeding (Box 12-1). In general, the serum sodium should be raised no faster than approximately 0.5-1.0 mEq/L per hour.35 Additional important assessments include strict monitoring of intake and output, urine specific gravity, serum electrolytes, serum osmolality, and daily weights.
Rapid correction of hyponatremia has been shown to result in cerebral dysfunction, linked with damage to the myelin sheath of neurons. This myelinolysis is called central pontine myelinolysis if it occurs in the pons (brainstem) and osmotic demyelinization syndrome if it occurs elsewhere in the brain. Signs of brain dysfunction can include decreased level of consciousness, lack of coordination, paralysis, and dysphagia. Because there is no known treatment for such cerebral dysfunction, and it can cause permanent disability, prevention is critical. In general, unless neurologic symptoms indicate the need for more aggressive treatment, providers should aim to correct hyponatremia or hypernatremia no faster than approximately 0.5 mEq/L per hour (or 10-12 mEq/L per day). For more information, consult the National Institutes of Health Web site: http://www.ninds.nih.gov/disorders/central_pontine/central_pontine_myelinolysis.htm.
Sodium Imbalances: Hypernatremia
Pathophysiology and Clinical Signs and Symptoms
Hypernatremia typically increases the serum osmolality. The rise in osmolality stimulates the posterior pituitary to release ADH, which increases renal water reabsorption until the osmolality returns to normal. The increased osmolality associated with hypernatremia also leads to a shift in water from the intracellular to the extracellular compartment, including into the vascular space. This water movement from the cells can cause cellular dehydration and central nervous system dysfunction. Complications including subdural, subarachnoid, and intracerebral bleeding and sinus vein thrombosis can develop with acute and severe increases in serum sodium concentration and the resulting water shift (see Box 12-1). Permanent central nervous system dysfunction can result when the serum sodium concentration is extremely high (e.g., >165-170 mEq/L).12 When serum osmolality is chronically elevated, brain cells will generate idiogenic osmoles to maintain cell volume (Box 12-2).
Management
If the patient with hypernatremia has signs of inadequate tissue perfusion (i.e., shock), administer isotonic crystalloid by bolus (20 mL/kg) until perfusion is adequate. Avoid excessive bolus fluid administration (i.e., beyond that needed to treat shock), because it can contribute to a rapid fall in serum sodium and osmolality resulting in cerebral edema and other neurologic complications (see Box 12-2).
Potassium Homeostasis
The normal serum potassium (K+) concentration is 3.0 to 5.0 mEq/L. The intracellular potassium concentration is much higher, approximately 150 mEq/L.28 Small alterations in serum potassium concentration can significantly affect the transmembrane gradient and therefore neuromuscular and cardiac function. The serum K+ concentration is affected by potassium intake and excretion, renal regulation, and serum pH. Because the intravascular (serum) potassium concentration represents only a small proportion of the total body potassium, accurate interpretation of the child’s potassium balance requires consideration of the child’s clinical status and acid-base balance.
Potassium ions normally shift between the intra- and extracellular compartments with changes in the serum pH (see Evolve Fig. 12-2 in the Chapter 12 Supplement on the Evolve Website). When the serum hydrogen ion concentration increases (i.e., with acidosis or a fall in pH), hydrogen moves from the extracellular to the intracellular compartment, where it is buffered. To maintain a balance of cation movement across cell membranes, the intracellular movement of hydrogen ions is associated with an extracellular shift of potassium. Thus, acidosis or a fall in pH (e.g., with correction of alkalosis) is associated with a rise in serum potassium concentration. In contrast, alkalosis or a rise in serum pH (e.g., with treatment of acidosis) is associated with a fall in serum potassium, because hydrogen ion shifts out of cells and is replaced by potassium.
The kidneys play a critical role in potassium homeostasis. Renal failure limits the kidney’s ability to excrete potassium and may result in hyperkalemia (see Chapter 13).
Potassium Imbalances: Hypokalemia
Etiology
Causes of hypokalemia (serum K+ concentration <2.5 to 3.0 mEq/L) can be classified into three general categories: inadequate potassium intake (rare, but may be iatrogenic), shifts of potassium from the extracellular to the intracellular compartment (e.g., with alkalosis or a rise in serum pH), or excessive losses of potassium (see Evolve Fig. 12-3 in the Chapter 12 Supplement on the Evolve Website for more information). Critically ill patients often develop true potassium deficit following the use of diuretics, especially loop and thiazide diuretics. Some antimicrobial agents (e.g., amphotericin B or carbenicillin) can increase renal potassium losses. Hypokalemia also is associated with severe hypochloremia and the potassium-wasting Bartter’s syndrome.
Pathophysiology and Clinical Signs and Symptoms
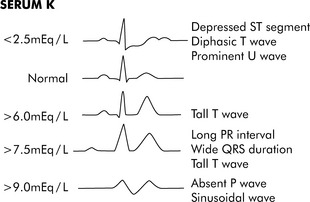
Hypokalemia can cause hyperpolarization of nerve and muscle cells, leading to muscle weakness, slowed nerve impulse conduction, and decreased muscle contraction. Hypokalemia can cause characteristic electrocardiogram (ECG) changes, including development of a U-wave (Fig. 12-3).
Management
The appropriate speed and method of potassium replacement is based on the severity of the hypokalemia and the child’s clinical condition (including acid-base balance, renal function and urine output). Replacement is typically indicated if the serum potassium concentration is below 3 mEq/L or if the child becomes symptomatic. Enteral replacement in a dose of 0.5-1 mEq/kg will usually correct mild, asymptomatic hypokalemia.17 The dose may be repeated every 4 to 8 hours.
IV potassium administration is indicated when the serum potassium concentration is less than 2.5 mEq/L or when milder levels of hypokalemia are present in patients who cannot receive enteral replacement (Box 12-3). A typical IV supplement dose is 0.5 to 1.0 mEq/kg, infused over approximately 1 to 3 hours.6,17 Dilute the potassium adequately and administer it over several hours to prevent bolus or rapid delivery that can produce lethal arrhythmias. Closely monitor the child’s serum potassium concentration and ECG.
Box 12-3 Recommended Intravenous Potassium Dose and Administration Guidelines*
• Daily: 2-5 mEq/kg per day in parenteral nutrition fluid or in divided doses
• Supplement: 0.5-1.0 mEq/kg per dose administered over 1-3 hours via syringe or infusion pump
• Typical maximum concentration


Information consistent with Lacy CF, et al: Lexicomp’s drug information handbook, ed 18. Hudson, OH, 2009, Lexicomp; and Custer JW, Rau RE: The Harriet Lane handbook, ed 18, Philadelphia, 2009, Mosby-Elsevier.
Potassium Imbalances: Hyperkalemia
Etiology
Pseudohyperkalemia is a high reported serum potassium concentration caused by release of intracellular potassium at the time of phlebotomy or in the specimen after collection.28 Intracellular potassium is released during fingerstick or other traumatic blood sampling and can be released in a blood specimen after collection in patients with severe leukocytosis or thrombocytosis. Pseudohyperkalemia should be considered when an unexpected high serum potassium concentration is reported by the laboratory in patients who are asymptomatic and have an otherwise normal electrolyte and/or acid-base status. If there is a question about the accuracy of a high reported serum potassium concentration in an asymptomatic patient, the concentration should be rechecked before therapy is initiated to reduce the serum potassium.
Pathophysiology and Clinical Signs and Symptoms
Additional causes of extracellular (including intravascular) shifts in potassium are: insulin deficiency, rapid cell breakdown, burns, trauma, hyperosmolality (e.g., hyperglycemia, mannitol administration), rhabdomyolysis, and the administration of succinylcholine.23 Unless the amount of cellular potassium release is excessive, such as in crushing injuries, tumor necrosis syndrome, or administration of succinylcholine, such an extracellular potassium shift alone rarely causes clinically significant hyperkalemia.23
Decreased potassium elimination can result from renal dysfunction or the administration of medications such as potassium-sparing diuretics. Although both acute and chronic renal failure can cause hyperkalemia, patients with acute renal failure are at higher risk for life-threatening hyperkalemia because the potassium concentration often rises quite rapidly, before the body is able to mount a compensatory response.28 Acidosis or cell injury in these patients will likely worsen the severity of the hyperkalemia.
Hyperkalemia can produce characteristic changes in the ECG such as a tall, peaked T wave, followed by widening of the QRS complex, S-T segment depression, and decreased R-wave amplitude (see Fig. 12-3). If the serum potassium concentration continues to rise, the P-R interval is prolonged, the amplitude of the P wave decreases, and finally the P wave disappears. Ultimately the patient’s rhythm deteriorates into a classic sine wave. Ventricular arrhythmias or fibrillation can occur at any point during this progression.
Management
Critical care management of hyperkalemia includes recognition of at-risk patients, and rapid identification and correction of the hyperkalemia. Priorities of management are determined by the child’s clinical presentation (for more information regarding treatment of hyperkalemia in the patient with renal failure, see Chapter 13).
Immediate intervention is required when the serum potassium concentration is greater than 6.5 to 7.0 mEq/L or is associated with ECG changes (see Fig. 12-3). Although the evidence supporting emergent therapies for hyperkalemia is largely anecdotal, widely accepted goals are to stabilize the myocardial cellular membrane, expand the ECF volume, shift potassium into the cells, and remove potassium from the body (Table 12-4).16,19,20,24
| Management Goal | Intervention | Comments |
| Stabilize myocardium |
Calcium salts |
Monitor children with respiratory failure carefully, because buffering of sodium bicarbonate increases CO2 and may worsen respiratory acidosis if the lungs cannot clear CO2.
Hypernatremia may develop as complication of this therapy.
ECF, Extracellular fluid; ICF, intracellular fluid; IV, intravenous.
* Ahee P, Crowe AV: The management of hyperkalemia in the emergency department. J Accid Emerg Med 17:188-191, 2000.
† Jones RE, Brashers VL, Huether SE: Alterations of hormonal regulation. In: McCance KL, Huether SE, editors: Pathophysiology: the biologic basis for disease in adults and children, ed 6. Philadelphia, 2009, Mosby-Elsevier.
‡ Chmielewski CM: Hyperkalemic emergencies: mechanisms, manifestations and management. Crit Care Nursing Clin North Am 10:449-458, 1998.
Calcium Imbalances: Hypocalcemia
Etiology
Hypocalcemia (total Ca2+ <8.0 to 8.5 mg/dL [<2.0 mmol/L]; ionized Ca2+ <4.0 mg/dL [<1.0 mmol/L]) is associated with significant morbidity and mortality in critically ill infants and children. Hypocalcemia can result from inadequate calcium intake, reduced calcium absorption, or excessive calcium excretion (Box 12-4).
Management
The healthcare team should immediately treat any acute episode of hypocalcemia in children at risk for impending cardiovascular or neurologic failure. Calcium chloride is the calcium supplement of choice for infants and children, because it provides greater bioavailability of calcium.11,16a An IV dose of 20 mg/kg of 10% calcium chloride provides 5.4 mg/kg of elemental calcium. Calcium gluconate can be used in neonates (IV dose of 60-100 mg/kg of 10% calcium gluconate provides approximately 5.4-9 mg/kg). Administer IV calcium by slow push or infusion and monitor for arrhythmias. Rapid administration of calcium salts (faster than 100 mg/min) is not recommended because it may produce bradycardia and asystole.24,32
Calcium Imbalances: Hypercalcemia
Management
With life-threatening hypercalcemia, aggressive therapy is needed to reduce the serum calcium. Drugs are typically administered to reduce the serum calcium concentration over several days: biphosphonate inhibits bone reabsorption, calcitonin inhibits calcium reabsorption from bone, mithracin reduces osteoclast activity, and gallium nitrate interferes with osteoclast function (for more information, see Table 15-2).4 Administration of a biphosphate such as pamidronate (1 mg/kg) may be indicated in severe hypercalcemia. It can cause mild to moderate hypophosphatemia, hypomagnesemia, and transient hypocalcemia at 2 to 10 days after administration9; therefore, it will be important to monitor these electrolytes.
Magnesium Imbalances: Hypomagnesemia
Pathophysiology and Clinical Signs and Symptoms
Decreased intestinal absorption of magnesium may be intrinsic or induced by medications (e.g., laxatives) or disease.30 Medical conditions associated with decreased absorption include pancreatitis, severe or chronic diarrhea, prolonged vomiting, malabsorption syndromes, and intestinal and biliary fistulas. Prolonged gastric suctioning can reduce serum magnesium.
Management
If hypomagnesemia produces ventricular arrhythmias, cardiopulmonary resuscitation is needed (see Chapter 6) with bolus magnesium administration.24 Acute hypomagnesemia in children at risk for impending cardiovascular or neurologic failure is treated immediately with IV administration of magnesium sulfate (25-50 mg/kg infusion over 10-20 minutes or longer).24 Monitor for arrhythmias and hemodynamic instability during administration of magnesium supplements.30
Magnesium Imbalance: Hypermagnesemia
Management
Patients at risk for hypermagnesemia should be monitored closely, because severe hypermagnesemia can lead to coma or cardiac arrest.27 Management of the pediatric patient with hypermagnesemia includes identification and correction of the cause and administration of calcium salts and IV fluids. Calcium salts antagonize magnesium and can reverse cardiac complications.
Dehydration and hypovolemia
Etiology
In the critically ill child, “third spacing” is a common cause of intravascular volume loss.31 “Third spacing” is a shift of fluid from the vascular space to a space that is neither intravascular nor intracellular. The third space may be interstitial (e.g., with septic shock), on the surface of a burn, or even into the bowel lumen. With third spacing of fluid, a significant volume of fluid is unavailable to the circulation to support cardiac output and systemic perfusion. Third spacing of fluid can develop in conditions such as ascites, pancreatitis, burns, peritonitis, sepsis, and intestinal obstruction. Such third-spacing results in inadequate intravascular volume that can compromise systemic perfusion.
Pathophysiology
Dehydration is classified by severity and by its effect on serum sodium concentration. Hyponatremic dehydration (serum Na+ <130 mEq/L) occurs when there is a relatively greater loss of sodium than water, so the serum sodium concentration falls. This effect is often seen when gastrointestinal fluid losses are replaced with water or other hypotonic solution (e.g., 5% dextrose and water).22 The resulting hyponatremia creates an osmolality gradient between the extracellular (including vascular) and intracellular compartments, so that water shifts from the vascular and interstitial spaces to the intracellular compartment. This water shift increases the severity of the clinical signs of (intravascular) volume depletion at any given volume deficit.
Clinical Signs and Symptoms
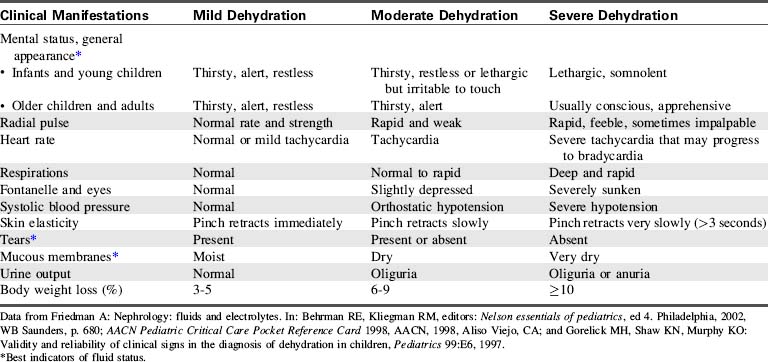
The clinical manifestations of dehydration will vary based on the severity of the fluid deficit and the serum sodium concentration. Table 12-5 shows a comparison of the clinical manifestations of mild, moderate and severe dehydration.
Management
If signs of shock are present (i.e., signs of poor peripheral perfusion or hypotension), initial management focuses on restoring intravascular volume to treat shock. Typically, 20 mL/kg IV boluses of isotonic crystalloid are administered until signs of shock are corrected. Infants or children with a severe fluid volume deficit may require resuscitation with 60 mL/kg or more of isotonic crystalloid.11
During volume resuscitation, providers should closely monitor signs of systemic perfusion (including mental status, skin perfusion and urine output), heart rate and blood pressure, to evaluate response to therapy. The fluid bolus administration is repeated, as needed. If volume resuscitation is adequate, the blood pressure and signs of systemic perfusion and blood pressure should improve and the heart rate should decrease toward normal; if the child fails to improve, additional boluses are needed or there may be other causes of the child’s shock. See Chapter 6 for further details of the management of hypovolemic shock.
Once initial fluid resuscitation is complete, ongoing management includes estimation and replacement of water and electrolyte deficits. In isotonatremic or hyponatremic dehydration, the fluid and electrolyte deficit is typically replaced over a period of 24 to 36 hours to restore intravascular volume within a short period of time while preventing overexpansion of the ECF.10 In the presence of hyponatremic dehydration, the need for sodium replacement should be carefully evaluated.
In hypernatremic dehydration, replacement of fluid and electrolyte deficit is typically accomplished more slowly, over a period of 48 to 72 hours. The goal is to prevent rapid decreases in the serum sodium and osmolality during rehydration because such decreases can produce an intracellular water shift and neurologic complications. Replacement fluids to treat the fluid deficit are administered in addition to routine maintenance fluids (see Table 12-2) and replacement of any ongoing fluid losses (e.g., emesis or diarrhea).
Guidelines for parenteral fluid therapy
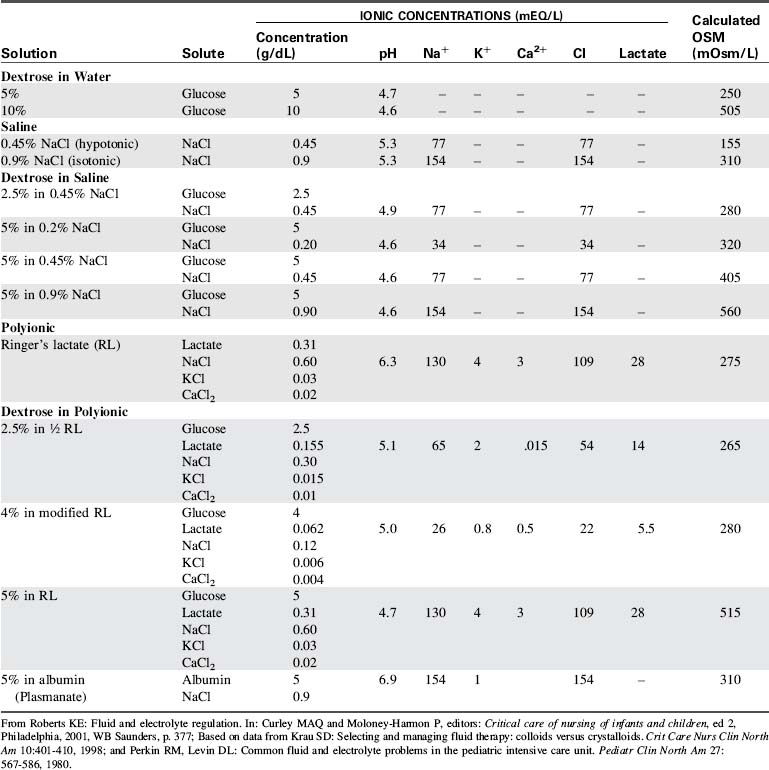
Parenteral fluid therapy is ubiquitous in pediatric critical care. See Table 12-6 for a comparison of common parenteral fluid solutions.
IV maintenance fluids are administered to provide daily free water and electrolyte requirements in a fasting patient. Fluid administration rate for each patient begins with an estimation of maintenance fluid requirements based on the formula developed by Holliday and Segar in 1957.13 These estimated requirements are then tailored to meet the needs of the patient.
For many years, hypotonic crystalloids (e.g., 5% dextrose with 0.2% sodium chloride) were routinely used for maintenance and replacement fluids for critically ill infants and children, based on the assumption that critically ill patients were likely to have a tendency to retain sodium and water. However, after reports of fatal hyponatremia in hospitalized children,1,2,21 a systematic review of the literature by Choong et al.5 estimated that the odds of developing hyponatremia after administration of hypotonic solutions were 17-fold higher than with administration of isotonic solutions. In addition, the administration of isotonic solutions was not associated with hypernatremia; in fact, some patients receiving isotonic fluids still developed hyponatremia.5
Currently, there is insufficient evidence to identify use of a single IV fluid (e.g., hypotonic or isotonic) as the standard of care for pediatric maintenance therapy. Until clinical trials compare the safety and efficacy of different IV fluid regimens, practitioners must continue to individualize IV fluid selection and administration rate for each patient.3,5 Providers should monitor serum electrolytes and clinical status during parenteral fluid therapy to prevent or rapidly detect and treat any imbalances that develop.
Specific diseases
Syndrome of Inappropriate Antidiuretic Hormone
Etiology
SIADH is characterized by ADH secretion in the absence of a physiologic stimulus. Patients with SIADH demonstrate low serum osmolality, hyponatremia, and hypervolemia. Causes of SIADH include central nervous system injury (e.g., surgery, tumor, traumatic brain injury) or infection, disease or damage to the pituitary stalk or hypothalamus, holoprosencephaly, spinal cord injury or surgery, chemotherapy, pulmonary disease, and liver disease.34 The critically ill patient with any of these conditions requires close monitoring for the development of SIADH.
Management
Management of SIADH is targeted at the prevention of complications associated with hyponatremia. Early detection of neurologic deterioration is critical; therefore, frequent neurologic assessments are needed. If neurologic complications develop, including a change in mental status or seizure activity, immediate treatment includes administration of hypertonic saline (e.g., 2-4 mL/kg of 3% saline should raise serum sodium approximately 1 to 2 mEq/L) and loop diuretics (e.g., furosemide).34 These therapies should raise the serum sodium sufficiently to slow the intracellular water shift.
Cerebral Salt Wasting
Etiology
Cerebral salt wasting (CSW) is caused by increased circulating levels of ANP with resulting natriuresis and contraction of intravascular volume, including potential hypovolemia. Causes of CSW are the same as those causing SIADH (see sections, Specific Diseases and Syndrome of Inappropriate Antidiuretic Hormone). When CSW is associated with a head injury, it typically develops 2 to 7 days after the injury. CSW can also complicate congestive heart failure, Cushing’s syndrome, DKA, and hyperaldosteronism.33
Pathophysiology
Intracranial disease or injury and complications of other disease are thought to alter neurologic secretion of ANP and other natriuretic peptides.25 Increased circulating levels of ANP lead to increased excretion of a large amount of sodium in a high volume of urine. Like SIADH, CSW produces hyponatremia with a low serum osmolality and a high urine sodium and urine osmolality. However, because ANP and other natriuretic peptides produce a high urine volume, the patient also develops mild to moderate dehydration.34
Clinical Signs and Symptoms
Clinical manifestations of CSW include the signs and symptoms of hyponatremia (see “Hyponatremia” earlier in chapter) and those of mild to moderate dehydration (see Table 12-5). Laboratory studies confirm hyponatremia (serum Na+ <130 mEq/L) and increased urinary sodium (urine Na+ >80 mEq/L). Although not routinely monitored, serum levels of ANP are increased.33
Management
Because the priorities of management differ, it is important to distinguish CSW from SIADH (Table 12-7).14,33 Treatment of CSW includes sodium replacement with oral salts or hypertonic saline, volume replacement with isotonic crystalloids, and vigilant monitoring for and prompt treatment of adverse effects of hyponatremia and hypovolemia.25 Mineralocorticoid therapy (fludrocortisone) can enhance sodium reabsorption and expand intravascular fluid volume in patients with CSW.29
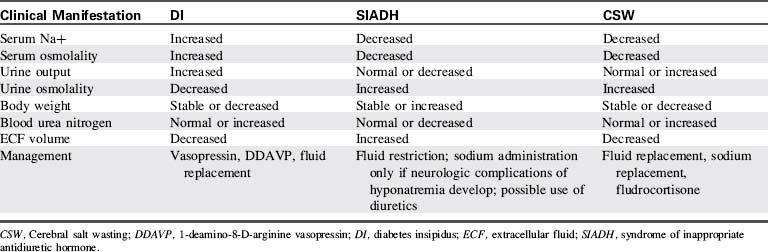
Diabetes Insipidus
Clinical Signs and Symptoms
Patients at risk should be monitored closely for the clinical manifestations of hypovolemia and hypernatremia. Laboratory studies consistent with DI (see Table 12-7) include elevated serum sodium (Na+ >150 mEq/L) and osmolality (>295 mOsm/L) and low urine osmolality (<100-200 mOsm/L).33
Management
Critical care management of DI includes administration of exogenous vasopressin (aqueous vasopressin [5-10 units subcutaneously] or lysine vasopressin [2-4 units IV]) or the vasopressin analog, 1-Deamino-8-d-Arginine Vasopressin (DDAVP[5-20 mcg intranasally every 12-24 hours]), careful fluid replacement, close monitoring of fluid intake and output, and close monitoring of serum sodium and other electrolytes.33,34 While urine volume is high, it is also important to monitor for the signs and symptoms of hypovolemic shock and support electrolyte balance.
Diabetic Ketoacidosis
Etiology
DKA is a life-threatening complication of diabetes mellitus that includes the combination of severe hyperglycemia and ketoacidosis. Approximately 40% of children with diabetes mellitus present with DKA as their first clinical manifestation of diabetes mellitus.25
Type I diabetes mellitus in children results from a loss of insulin-secreting pancreatic beta cells. The most common form of type I diabetes is triggered by a combination of genetic susceptibility and environmental factors, including some viruses, that produce autoimmune destruction of beta cells.15
Pathophysiology
Ketoacids—acetoacetic acid and beta-hydroxybutyric acid—are released from adipocytes in instances of prolonged insulin deficiency. The presence of high levels of these circulating ketoacids results in ketoacidosis.34 Children with DKA always experience acidosis and hyperosmolar dehydration with glucosuria and osmotic diuresis.
Finally, dehydration, acidosis, and osmotic water shifts into brain cells (see Box 12-1) can contribute to life-threatening cerebral edema or intracranial thrombotic events.18 A major difference between children and adults with DKA is the susceptibility of the young to potentially fatal episodes of cerebral edema. Proposed mechanisms of cerebral edema include: accumulation of intracellular idiogenic osmoles (see Box 12-2); vasopressin and atrial natriuretic factor producing water retention, natriuresis, and hyponatremia; disruption in cell membrane ion exchange processes, causing intracellular ion movement and cell swelling; hypoxia and ischemia; ketones and acidosis contributing to the inflammatory cascade; and abnormalities in water (aquaporin) channels in the glial cells of the brain.18
Symptomatic cerebral edema in children with DKA can cause death or neurologic devastation.20 Additional intracranial pathologic conditions—including bleeding, thrombosis, infarction or emboli—can cause altered mental status in children with DKA.18
Clinical Signs and Symptoms
Children with DKA have severe hyperglycemia (serum glucose ≥180 mg/dL or >10 mmol/L) and ketoacidosis. Additional signs and symptoms are related to the severity of the resulting hyperosmolality, volume depletion, and acidosis. Children with DKA generally present with a 5% to 10% fluid deficit7 caused by the massive water losses secondary to glucosuria.
Dehydration, acidosis and osmotic fluid shifts in the brain can contribute to life-threatening cerebral edema or intracranial thrombotic events. These central nervous system complications can produce decreased responsiveness, posturing, abnormal respiratory patterns, and signs of increased intracranial pressure and cerebral herniation.18 Table 12-8 shows a summary of the clinical manifestations of DKA and their underlying mechanisms.
| Signs and Symptoms | Underlying Mechanisms |
| Hyperglycemia, dehydration, cardiovascular collapse | Osmotic diuresis secondary to hyperglycemia |
| Acidosis | Accumulation of ketoacids and lactic acid in serum from lipolysis, resulting in increased H+ concentration in serum |
| Electrolyte imbalance: sodium | Total body hyponatremia because of passive loss of electrolytes as a result of osmotic diuresis; however, serum sodium may be high as a result of hemoconcentration |
| Electrolyte imbalance: potassium | In acidosis, potassium shifts from ICF to ECF; therefore, initially serum potassium may be normal or even high, but will decrease as a result of osmotic diuresis and correction of acidosis |
| Cardiac arrhythmia | Hypokalemia or hyperkalemia |
| Ketonuria | Serum ketone levels rise above the renal threshold and are spilled in the urine |
| Hyperpnea | Compensatory mechanism; effort to blow off excess CO2 and thereby correct the degree of acidosis |
| Mental status changes, cerebral edema | Possibly related to rate of correction of hyperglycemia, level of acidosis, fluid shifts |
ECF, Extracellular fluid; ICF, intracellular fluid.
From Trimarchi T: Endocrine critical care problems. In: Curley MAQ, Moloney-Harmon PA, editors: Critical care nursing of infants and children, ed 2, Philadelphia, 2001, WB Saunders, p. 817.
Management
Priorities of management of DKA in order of importance are: assessment and support of neurologic function (including support of patent airway and effective ventilation), assessment and support of circulation (including volume resuscitation), insulin therapy, and restoration and maintenance of electrolyte, glucose and acid-base balance (Box 12-5). Consideration is also given to confounding factors, such as infection, injury or other patient problems.
Box 12-5 Priorities of Management of Diabetic Ketoacidosis in Order of Importance*
1. Assess and support neurologic function, including support of patent airway and effective ventilation
2. Assess and stabilize circulation (including fluid resuscitation)
4. Restore and maintain electrolyte, glucose and acid-base balance
5. Identify confounding factors (infection, drugs, injury, other diagnoses)
Although very few children present with neurologic compromise (primarily cerebral edema) this can be a very poor prognostic indicator. Most who present with this finding perish and those who survive are often severely neurologically compromised with little or no independent function.20
If the child’s level of consciousness deteriorates, cerebral edema may be present, and urgent intervention is needed (see diagnostic criteria for cerebral edema21a in Box 12-6). If clinical signs of cerebral edema develop, immediate treatment with IV mannitol (0.2-1.0 g/kg over 30-60 minutes) is indicated. Hypertonic saline (e.g., 3% sodium chloride, 10 mL/kg, administered IV over 30 minutes) may be used as an alternative to mannitol. If the child’s ability to protect the airway or spontaneous ventilation deteriorates, intubation and mechanical ventilation are indicated.
Box 12-6 Bedside Neurologic Evaluation of Children with Diabetic Ketoacidosis
From Muir AB, et al: Cerebral edema in childhood diabetic ketoacidosis. Diabetes Care 24:1541-1546, 2004.
The child’s volume deficit is initially treated with the administration of normal saline or lactated Ringer’s. The quantity of fluid administered is determined by the patient’s clinical severity; if shock is present, bolus administration of isotonic crystalloids is needed. For less significant volume deficit, typically 10 to 20 mL/kg is administered over 1 to 2 hours and repeated as needed.18 Replacement of the total fluid deficit is planned over approximately 48 hours or longer. Isotonic crystalloids are used for initial fluid replacement; the sodium content may then be reduced to 0.45% sodium chloride with potassium added as the child’s condition stabilizes.
Replacement of insulin is generally accomplished by continuous IV infusion. An infusion of regular insulin (0.05 to 0.1 unit/kg per hour) is titrated to gradually reduce the serum glucose at the rate of approximately 50 to 100 mg/dL per hour. Simultaneous infusion of 10% glucose IV can be titrated to control the maximum rate of decline in the serum glucose to no more than 100 mg/dL per hour. Glucose infusion is preferable to slowing the insulin infusion, because reducing the insulin dose can worsen the patient’s acidosis.18
As noted previously, patients with DKA typically have deficits of water, sodium, potassium, and phosphate. Although serum electrolyte concentrations may be normal at the time of initial clinical presentation, with treatment hyponatremia, hypokalemia, and hypophosphatemia are likely to be present. Replacement fluids are selected to address these deficits. Some landmark studies suggest that phosphate replacement is of no clinical value.8,38 There is no evidence that the addition of sodium bicarbonate to correct acidosis is of value, and it may contribute to hypokalemia and cerebral edema.18 To prevent a fall in serum osmolality when the serum glucose is reduced, the serum sodium concentration should rise approximately 1.6 mEq/L for every 100 mg/dL fall in serum glucose concentration.
Box 12-7 contains a typical management plan for children with DKA. As a general rule, patients in DKA recover from acidosis within 24 hours after presentation.
Box 12-7 Management Plan for Children with Diabetic Ketoacidosis
1. First Priority: Perform repeat assessments of neurologic function, airway and ventilation
2. Second priority: Assess and stabilize circulation (including acute management of severe hyperkalemia associated with acidosis).












4. Assess and maintain serum sodium concentration.
5. Treat potassium and phosphate imbalance.


 Treat severe hyperkalemia (>7 mEq/L and ECG changes) with calcium and consider sodium bicarbonate administration (see Table 12-4).
Treat severe hyperkalemia (>7 mEq/L and ECG changes) with calcium and consider sodium bicarbonate administration (see Table 12-4).
 The absence of ketones and serum glucose <200 mg/dL indicates readiness for transition to subcutaneous closing.
The absence of ketones and serum glucose <200 mg/dL indicates readiness for transition to subcutaneous closing.7. Assess for infection (most frequent cause of DKA)
Modified from Trimarchi T: Endocrine problems in critically ill children: an overview. AACN Clin Issues 17:66-78, 2006.
Adrenal Insufficiency
Etiology
Secondary adrenocortical deficiency occurs as the result of interrupted communication in the hypothalamic-pituitary-adrenal axis. Secondary adrenal insufficiency is due to deficiencies in corticotrophin releasing hormone (CRH) from the hypothalamus or adrenocorticotropic hormone from the anterior pituitary.15 The most common cause of adrenal insufficiency in children is the abrupt cessation of administration of exogenous corticosteroids.34 Adrenal insufficiency should be considered in any critically ill child with persistent shock despite fluid resuscitation and vasopressor therapy (e.g., unresponsive or fluid-refractory septic shock).
Pathophysiology
• Aldosterone deficiency leads to decreased renal reabsorption of sodium and water and increased reabsorption of potassium.
• Isolated glucocorticoid deficiency results in hypoglycemia, elevated levels of ADH (as in SIADH, producing hypervolemia and dilutional hyponatremia), and decreased secretion of and responsiveness to catecholamines. This condition in turn results in the development of myocardial depression, hypotension, and diminished arterial tone.
Clinical Signs and Symptoms
The initial presentation of acute adrenal insufficiency is often that of profound circulatory failure with the clinical manifestations of hypovolemic, distributive, and cardiogenic shock (see Chapter 6.) Children may also exhibit hyponatremia, hyperkalemia, and hypoglycemia and their corresponding clinical manifestations, as presented earlier in this chapter.
Management
Treatment for aldosterone deficiency and volume depletion includes judicious fluid resuscitation with an isotonic crystalloid (typically normal saline, possibly with 5% or 10% dextrose) to expand intravascular volume. Excessive fluid replacement can be harmful in instances of isolated glucocorticoid deficiency when SIADH is already present. If the child exhibits the clinical manifestations of hyperkalemia, urgent treatment with calcium, glucose plus insulin, and other strategies listed in Table 12-4 are needed.
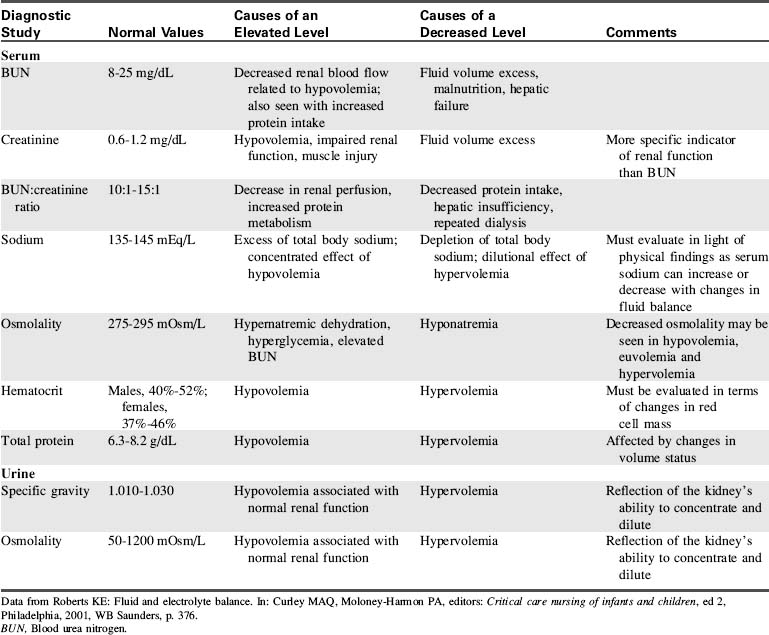
Diagnostic tests
Diagnostic studies are a key component in identifying disruptions in fluid or electrolyte balance. Table 12-9 provides an overview of the most common diagnostic tests used to evaluate fluid balance. However, these diagnostic studies are just one piece of the puzzle when evaluating a child with a disruption in fluid or electrolyte balance. The laboratory results should be evaluated in light of the child’s history and clinical presentation.
1 Arieff A.I., Ayus J.C., Fraser C.L. Hyponatraemia and death or permanent brain damage in healthy children. BMJ. 1992;304:1218-1222.
2 Armon K., et al. Hyponatremia and hypokalemia during intravenous fluid administration. Arch Dis Child. 2008;93:285-287.
3 Beck C.E. Hypotonic versus isotonic maintenance intravenous fluid therapy in hospitalized children: a systematic review. Clin Pediatr. 2007;46:764-770.
4 Brashers V.L., Jones R.E. Mechanisms of hormonal regulation. In McCance K.L., Huether S.E., editors: Pathophysiology: the biologic basis for disease in adults and children, ed 6, Philadelphia: Mosby-Elsevier, 2009.
5 Choong K., et al. Hypotonic versus isotonic saline in hospitalized children: a systematic review. Arch Dis Child. 2006;91:828-835.
6 Custer J.W., Rau R.E. The Harriet Lane handbook, ed 18. Philadelphia: Mosby-Elsevier; 2009.
7 Dunger D.B., et al. European Society for Paediatric Endocrinology/Lawson Wilkins Pediatric Endocrine Society consensus statement on diabetic ketoacidosis in children and adolescents. Pediatrics. 2004;113:e133-e140.
8 Fisher J.N., Kitabchi A.E. A randomized study of phosphate therapy in the treatment of diabetic ketoacidosis. J Clin Endocrinol Metab. 1983;57:177-180.
9 Elisaf M., Kalaitzidis R., Siamopoulos K.C. Multiple electrolyte abnormalities after pamidronate administration. Nephron. 1998;79:337-339.
10 Friedman A.L. Nephrology: fluid and electrolytes. In: Behrman R.E., Kliegman R.M., editors. Nelson essentials of pediatrics. ed 4. Philadelphia: WB Saunders; 2002:671-709.
11 Hazinski M.F., et al. Fluid therapy and medications for shock and cardiac arrest. In: PALS provider manual. Dallas: American Heart Association; 2002.
12 Hellerstein S. Fluids and electrolytes: clinical aspects. Pediatr Rev. 1993;14:103-115.
13 Holliday M.A., Segar W.E. The maintenance need for water in parenteral fluid therapy. Pediatrics. 1957;19:823-832.
14 Jimenez R., et al. Cerebral salt wasting syndrome in children with acute central nervous system injury. Ped Neuro. 2006;35:261-263.
15 Jones R.E., Brashers V.L., Huether S.E. Alterations of hormonal regulation. In McCance K.L., Huether S.E., editors: Pathophysiology: the biologic basis for disease in adults and children, ed 6, Philadelphia: Mosby-Elsevier, 2009.
16 Kamel K.S., Wei C. Controversial issues in the treatment of hyperkalemia. Nephrol Dial Transplant. 2003;18:2215-2218.
17 Lacy C.F., et al. Lexicomp’s drug information handbook, ed 18. Hudson, OH: Lexicomp; 2009.
17a Kleinman ME, Chameides L, Schexnayder SM, et al. Part 14: pediatric advanced life support: 2010 American Heart Association Guidelines for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care. Circulation. 2010;122(18 Suppl 3):S876-908.
18 Levin D.L. Cerebral edema in diabetic ketoacidosis. Pediatr Crit Care Med. 2008;9:320-329.
19 Mahoney B.A., et al. Emergency interventions for hyperkalaemia. Cochrane Database Syst Rev. 2005. April 18 (2) CD003235
20 Marcin J.P., et alAmerican Academy of Pediatrics. The Pediatric Emergency Medicine Collaborative Research Committee: factors associated with adverse outcomes in children with diabetic ketoacidosis-related cerebral edema. J Pediatr. 2002;141:793-797.
21 Moritz M.L., Ayus J.C. Prevention of hospital-acquired hyponatremia: a case for using isotonic saline. Pediatrics. 2003;111:227-230.
21a Muir A.B., et al. Cerebral edema in childhood diabetic ketoacidosis. Diabetes Care. 2004;24:1541-1546.
22 Neville K.A., et al. Isotonic is better than hypotonic saline for intravenous rehydration of children with gastroenteritis: a prospective randomized study. Arch Dis Child. 2006;91:226-232.
23 Nyirenda MJ, et al: Clinical review: hyperkalemia, Br Med J 339:4114
24 Ralston M., et al. PALS provider manual. Dallas: American Heart Association; 2006.
25 Relvas M.S. Endocrine emergencies. In: Conway E.E., editor. Pediatric multiprofessional critical care review. Des Plaines, IL: Society of Critical Care Medicine; 2006:365-376.
26 Reynolds M., Padfield P.L., Seckl J.R. Disorders of sodium balance. Br Med J. 2006;332:702-705.
27 Roberts K.E. Fluid and electrolyte regulation. In Curley M.A.Q., Moloney-Harmon P., editors: Critical care nursing of infants and children, ed 2, Philadelphia: WB Saunders, 2001.
28 Schaefer T.J., Wolford R.W. Disorders of potassium. Emerg Med Clin North Am. 2005;23:723-747.
29 Springate J. Cerebral salt wasting. Available at: http://www.emedicine.com/ped/topic354.htm, 2008. Accessed July 4
30 Springate J.E., Kala G.K. Hypomagnesemia. Available at: http://emedicine.medscape.com/article/922142-overview, 2010. Accessed January 4
31 Stark J. A comprehensive analysis of the fluid and electrolytes system. Crit Care Nursing Clin North Am. 1998;10:471-475.
32 Styne D.M., Glaser N.S. Endocrinology. In: Behrman R.E., Kliegman R.M., editors. Nelson essentials of pediatrics. ed 4. Philadelphia: WB Saunders; 2002:711-766.
33 Trimarchi T. Endocrine critical care problems. In: Curley M.A.Q., Moloney-Harmon P.A., editors. Critical care nursing of infants and children. ed 2. Philadelphia: WB Saunders; 2001:805-819.
34 Trimarchi T. Endocrine problems in critically ill children: an overview. AACN Clin Issues. 2006;17:66-78.
35 Vellaichamy M. Hypernatremia. Available at: http://www.emedicine.com/ped/topic1082.htm, 2008. Accessed July 4
36 Vellaichamy M. Hyponatremia. Available at: http://www.emedicine.com/ped/topic1124.htm, 2008. Accessed July 4
37 Vlasselaers D., et al. Intensive insulin therapy for patients in paediatric intensive care: a prospective, randomised controlled study. Lancet. 2009;373:547-556.
38 Wilson H.K., et al. Phosphate therapy in diabetic ketoacidosis. Arch Intern Med. 1982;142:517-520.