[level-membership-for-pediatrics-category]
Fetal Therapy
The first open fetal surgical procedure was performed at the University of California, San Francisco (UCSF) in 1982.1 Since then, more than 515 fetal interventions have been performed at UCSF over the past 30 years without maternal mortality. Over the past 30 years, a number of advances have been developed to allow a broader application of fetal intervention. These techniques, including maternal hysterotomy, minimal-access fetoscopy, and percutaneous fetal access, were initially tested and validated in animal models. More recently, in utero therapy has shown to be beneficial for non-life-threatening conditions such as myelomeningocele (MMC). In this chapter, we will present an overview of the current state of fetal surgery and will review specific fetal problems outlining current management strategies.
General Principles
The primary morbidity following fetal surgery has been, and remains, preterm labor resulting in premature delivery, usually between 25 and 35 gestational weeks. Preventing preterm labor after fetal intervention remains problematic. Complications can also arise from endotracheal intubation, general anesthesia, epidural and spinal anesthesia, blood transfusion, premature rupture of membranes, chorioamniotic separation, chorioamnionitis, and placental abruption. Long-term morbidity from the hysterotomy includes infertility, uterine rupture with the current and future pregnancies, and mandatory cesarean section with future pregnancies. Notably, in reviewing our experience, subsequent fertility following fetal intervention has been good.2
Fetal Access
Ultrasound-guided percutaneous procedures are performed through small skin incisions on the mother’s abdominal wall. During these operations, real-time ultrasound is needed to visualize the fetal and maternal anatomy.3 Catheters and shunts can be inserted into the fetus to drain cystic masses, ascites, or pleural fluid into the amniotic space. In addition, radio frequency ablation (RFA) probes can be deployed into the amniotic space to treat various twin gestational anomalies. The needles used to place these catheters, as well as the RFA device, are approximately 1.5–2 mm in diameter, minimizing morbidity to the mother and irritation of the uterus.4,5
Open fetal procedures require general anesthesia with a combination of preoperative indomethacin and high mean alveolar concentration of inhalational agents to maintain uterine relaxation.6–8 An epidural is also inserted for postoperative analgesia.
Anomalies Amenable to Fetal Surgery
Congenital Diaphragmatic Hernia
Despite significant advances in neonatal respiratory support, survival for children born with congenital diaphragmatic hernia (CDH) remains only 60–70% throughout the USA. Additionally, survival for prenatally diagnosed CDH may be as low as 25% due to IUFD and stillborns that are not included in conventional postnatal survival data.9–11 This high mortality rate has made CDH a primary area of interest for the development of effective prenatal intervention. In fact, improving outcomes specifically for CDH was a significant driving force in the genesis of fetal surgery at UCSF.
Prognostic Criteria
One of the key elements in developing fetal intervention for CDH has been identifying what factors will identify those fetuses at the greatest risk for a poor outcome. The factors most consistently associated with a poor outcome on prenatal ultrasound are (1) the presence of liver herniation into the chest; and (2) a low lung-to-head ratio (LHR). In our experience, survival has been 100% in fetuses with CDH that do not have liver herniation on prenatal ultrasound and 56% in fetuses with CDH and liver herniation into the chest.12 The LHR is calculated as the area of the contralateral lung at the level of the cardiac atria divided by the head circumference. This LHR value has been shown to statistically correlate with survival: 100% survival with an LHR greater than 1.35, 61% survival with an LHR between 0.6 and 1.35, and 0% survival with an LHR less than 0.6.12
While the LHR has been a reliable predictor of outcomes at our center, other institutions have suggested the LHR does not account for discrepant growth rates between the head and lung during gestation and therefore may not be reliable at certain gestational ages.13,14 To account for this, the observed to expected LHR (OE LHR) has been proposed. The OE LHR is represented as a percentage of what the expected LHR would be in a normal fetus of the same gestational age. For left-sided defects, an OE LHR <25% is associated with an 18% survival whereas an OE LHR >45% correlates with 89% survival.13,15
Magnetic resonance imaging (MRI) for volumetric measurement of the lungs is a promising modality for prognosis with CDH.16 MRI can be used to calculate the percent-predicted lung volume (PPLV). Results for PPLV have varied. In one study, a PPLV >20% was associated with 100% survival whereas survival was only 40% when PPLV was <15%.17 In another study, a PPLV <25% was associated with a 13% survival and a PPLV >35% correlated with 83% survival.18 MRI can also be used to determine the percentage of liver herniation, although the prognostic value of this finding is still being investigated.18
Fetal Interventions
CDH and its effect on fetal lung development has been studied in animal models.19,20 In the fetal lamb model, compression of the lungs, either with an intrathoracic balloon or by creation of a diaphragmatic hernia, results in uniformly fatal pulmonary hypoplasia. However, in utero correction of the compressing lesion leads to sufficient lung growth and development, which improves postnatal survival.20
This concept of early, in utero correction of CDH has been studied and applied in humans.21,22 Fetal surgery for CDH initially involved open repair of the diaphragmatic defect. The first successful case was reported in 1990 which demonstrated the feasibility of open fetal repair using a two-step approach which involved creation of an abdominal silo to accommodate the reduced viscera and prevent compression of the umbilical vessels.23 This initial success was followed by a prospective trial at UCSF comparing open fetal surgery to postnatal repair in severe cases of prenatally diagnosed CDH. However, in this study, there was no difference in survival or in the need for extracorporeal membranous oxygenation (ECMO) between fetal repair and postnatal repair.22,24 Concordant with this effort, investigators at UCSF observed that fetuses with congenital high airway obstruction syndrome (CHAOS) had pulmonary hyperplasia.25 Also, fetal tracheal occlusion had been shown to cause pulmonary hyperplasia.26 In this condition, the lung parenchyma creates fluid that is ‘exhaled’ by the fetus. Occluding the trachea causes a build-up of this fluid and subsequent pulmonary hyperplasia.27,28 The inability to improve outcomes with open fetal repair for severe cases of CDH led to an interest in this physiologic process.29
The first eight patients were treated with open hysterotomy and tracheal occlusion with a metallic clip.30 This approach proved to be problematic for several reasons. First, the open hysterotomy led to significant prematurity due to premature labor. Second, the use of clips was associated with tracheal stenosis and also required a stringent delivery plan—which was later described as the ex utero intrapartum treatment (EXIT) procedure—whereby the fetus was exposed through a hysterotomy and maintained on utero–placental circulation while the clip was removed and a patent airway established prior to delivering the baby.31 However, outcomes with this approach were poor with only a 15% survival rate.30
Ongoing advancements in fetal surgery led to fetoscopic balloon placement for tracheal occlusion (Fig. 10-1). This technique has the advantages of being less invasive, a lower risk of tracheal stenosis, and the balloon being much easier to remove, although still necessitating an EXIT procedure. Results in the first eight cases were favorable with a 75% survival rate compared to a 38% survival rate in historical, case-matched controls managed with postnatal repair.32
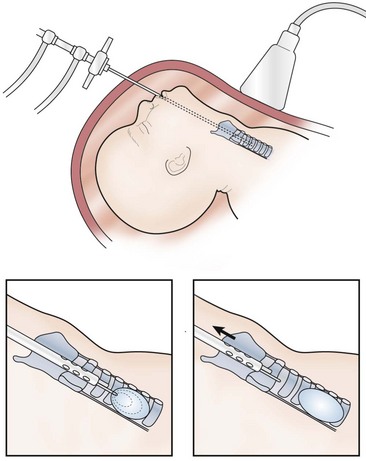
FIGURE 10-1 This schematic diagram shows the method of fetoscopic tracheal occlusion. A fetoscope is placed into the fetal mouth, the airway is identified, and a balloon is inserted into the trachea by using both fetoscopic and ultrasonographic visualization.
These early results led to an National Institutes of Health (NIH) funded, prospective randomized trial comparing in utero fetoscopic tracheal occlusion to standard postnatal care for fetuses diagnosed with severe left-sided CDH (liver up and LHR <1.4) and no other detectable anomalies. However, results of the trial showed no difference in survival between the tracheal occlusion group and the standard postnatal care group (73% vs 77%, respectively).33 Unexpectedly, the survival in the postnatal repair group was considerably greater when compared to historical controls. Although this study did not demonstrate a difference in survival between the prenatal intervention group and the postnatal group, the results of this trial demonstrate the tremendous importance of proper randomized controlled trials for novel fetal surgical procedures.
Further data regarding fetal tracheal occlusion have suggested that temporary, short-term reversible tracheal occlusion may be preferable to a longer duration of occlusion. Animal models of fetal tracheal occlusion have demonstrated that long-term tracheal occlusion can be deleterious to type II pneumocytes (the cells that secrete surfactant) and that this adverse effect is not seen with a shorter duration of tracheal occlusion.34 To test the hypothesis that temporary fetal tracheal occlusion is better, Deprest et al. studied patients undergoing fetal tracheal balloon occlusion who also had the balloon removed prenatally to limit the duration of occlusion.35 In this group of patients, improved lung growth was evident on fetal MRI and was also associated with improved postnatal survival. While reversal of the tracheal occlusion requires a second maternal and fetal intervention for balloon removal, it obviates the need for an EXIT procedure at birth. Early results have been favorable.31,36
These promising findings with temporary tracheal occlusion have led to its current application in Europe. The European FETO consortium has reported a 48% survival rate among 210 cases of severe CDH treated with temporary fetal tracheal occlusion, and the Eurofetus group is currently sponsoring a prospective fetal tracheal occlusion trial that seeks to determine the ideal time and duration for tracheal occlusion.37 Our group at UCSF is currently offering reversible, fetal tracheal balloon occlusion for fetuses with liver herniation in the chest and an LHR of <1.0, as these babies continue to have a very high mortality.38 This study has Food and Drug Administration oversight, and involves percutaneous placement of a fetoscopic tracheal balloon between 26 and 28 weeks gestation, with removal of the balloon via a second percutaneous fetoscopic procedure between 32 and 34 weeks.
Neoplasms
Fortunately, fetal neoplasms are rare. When they do occur, most are benign. However, if they become large enough, they can impede venous return to the heart or cause high-output heart failure via arteriovenous shunting. Such shunting can lead to non-immune fetal hydropic changes such as polyhydramnios, placentomegaly, skin and scalp edema, and pleural, pericardial, and peritoneal fluid accumulation. When only one compartment is involved, this is considered early fetal hydrops; when two or more compartments are affected, then true hydrops is present. If left untreated, hydrops is nearly always fatal.39–40 The two most common prenatally diagnosed neoplasms that cause nonimmune fetal hydrops are congenital pulmonary airway malformations (CPAM) and sacrococcygeal teratomas (SCT).
Congenital Pulmonary Airway Malformations
CPAMs are pulmonary lesions with a broad range of clinical presentations. This new terminology includes congenital cystic adenomatoid malformations (CCAM) and bronchopulmonary sequestrations. CCAMs are much more likely than sequestrations to cause nonimmune fetal hydrops. CCAMs are characterized by an overgrowth of respiratory bronchioles with the formation of cysts of various sizes.41–44 Most fetuses diagnosed with a CCAM develop normally, and can be followed with serial ultrasound studies. These asymptomatic patients then undergo standard, postnatal resection. A small percentage of patients with the prenatal diagnosis of CCAM will develop non-immune hydrops.43,44
Various measurements have been developed to predict which fetuses are at risk for developing hydrops. The most accepted measurement is the CCAM volume ratio (CVR), defined as the product of the three longest measurements of the lesion on ultrasound multiplied by the constant 0.52, and then divided by the head circumference. Crombleholme and colleagues identified a CVR of 1.6 as a cut-off for an increased likelihood of developing hydrops.45 When the CVR is <1.6, there is only a 2% risk of developing hydrops. When the CVR is >1.6, there is an 80% chance of developing hydrops.
CCAMs that are predominantly microcystic have a more predictable course than the macrocystic ones. Microcystic or solid CCAMs undergo steady growth that tends to plateau at 26 to 28 weeks gestation. At this point, fetal growth exceeds that of the CCAM. For this reason, patients with microcystic or solid CCAMs should be followed closely up to 26 to 28 weeks gestation at which point the interval between ultrasound examinations can be lengthened if the pregnancy has been otherwise uncomplicated. In contrast, macrocystic CCAMs undergo abrupt enlargement due to rapid fluid accumulation in a dominant cyst. Therefore, macrocystic CCAMs require close follow-up with serial ultrasound throughout the duration of the pregnancy.42,46
If a fetus develops hydrops at a viable gestational age, early delivery should be considered. Hydropic fetuses who are not yet viable outside the uterus, and have a dominant macrocystic lesion, are appropriate candidates for a thoracoamniotic shunt.47 Needle drainage alone has not been found to be an effective therapy as rapid re-accumulation of fluid in the cyst necessitates repeat intervention. In the largest single-center experience with thoracoamniotic shunts, shunting led to a mean 51% volume reduction in the size of the lesion and a 70% survival rate.48 Other institutions have reported similar survival rates.49 Despite shunting, these babies can still have significant respiratory distress at birth and should be delivered at a tertiary referral center.
Open fetal thoracotomy and CCAM resection is an option in the pre-viable fetus with a microcystic or solid lesion. This is performed through an open hysterotomy. A thoracotomy is made through the fifth intercostal space, and the lobe containing the CCAM is identified and exteriorized through the incision (Fig. 10-2). The pulmonary hilar structures are then mass ligated using an endoloop or endoscopic stapler. The thoracotomy is then closed in layers.50–51
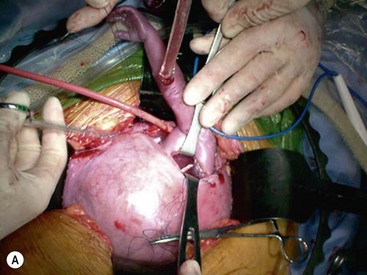
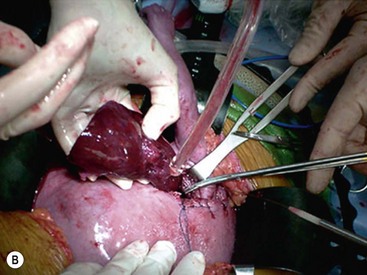
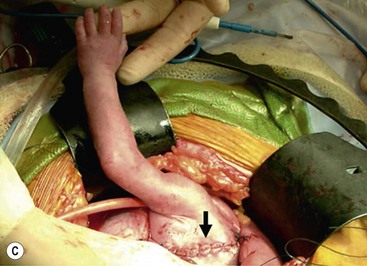
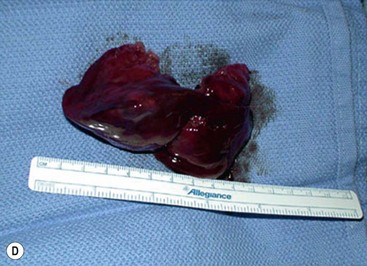
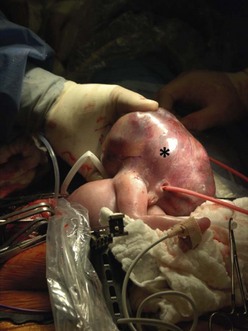
FIGURE 10-2 These photographs depict an infant with a large left upper lobe CCAM undergoing in utero left lobectomy. (A) The infant’s left arm is visualized. Note the maternal hysterotomy and the left fetal thoracotomy (with retractors inserted) through the fifth intercostal space. (B) The left upper lobe containing the CCAM has been identified and exteriorized through the thoracotomy incision. The pulmonary hilar structures were mass ligated using an endoloop. (C) The fetal thoracotomy incision (arrow) has been closed. (D) The left upper lobe specimen containing the CCAM is seen.
In a group of 120 patients with the prenatal diagnosis of CPAM from UCSF and Children’s Hospital of Philadelphia (CHOP), 79 had no evidence of hydrops.51 Of these, 76 were followed expectantly and all survived. Three fetuses without evidence of hydrops and with large dominant cysts underwent thoracoamniotic shunting. All three fetuses survived. Twenty-five hydropic fetuses were followed with no intervention. All mothers delivered prematurely and all fetuses died perinatally. Sixteen fetuses with hydrops underwent intervention: 13 underwent open fetal surgery while three underwent thoracoamniotic shunting. Two of the three survived in the group that underwent shunt insertion and eight of 13 survived in the open fetal surgery group.
Despite positive results with open fetal resection in the hydropic fetus, there has been a shift away from this therapy in the last five years due to the efficacy of maternal steroids. This finding was discovered serendipitously at UCSF during the preparation of several hydropic fetuses for open fetal surgery.52 In these cases, maternal steroids were administered to enhance fetal lung maturity. Preoperative ultrasound studies showed resolution of the hydrops and those fetuses survived to delivery and beyond. Thirteen patients with microcystic CCAMs, nine of which were complicated by hydrops, had an overall survival rate of 85% with resolution of hydrops in seven of nine fetuses.53 CHOP has reported a series of 11 patients, five of which had hydrops, and all survived after receiving steroids.54
Currently, we recommend maternal betamethasone for fetuses with nonimmune hydrops or a CVR >1.6. Steroids can be re-dosed, but repeated administration of maternal steroids beyond three to five courses can result in untoward effects such as reduced birth weight.55 It is widely accepted that steroids are most effective for predominantly microcystic or solid lesions as this is the component of the malformation that responds to steroids. Macrocystic lesions are less likely to respond.
Sacrococcygeal Teratoma
Recent evidence has identified the tumor volume to fetal weight ratio (TFR) as an important prognostic indicator.56 Tumor volume is calculated using the greatest length, width, and height of the tumor as measured by ultrasound or MRI; fetal weight can be calculated by ultrasound as well. In the initial report of ten fetuses with SCT, a TFR >0.12 was associated with an 80% incidence of hydrops and a 60% mortality, whereas a TFR <0.12 was associated with 100% survival.56 UCSF has recently presented our experience in 37 fetuses with SCT and confirmed that a TFR <0.12 was a favorable prognostic finding up to 24 weeks. Between 24–32 weeks, a TFR of <0.11 was associated with better outcomes. In addition, we also found that cystic SCTs had a more favorable prognosis than solid ones.57
The fetus with SCT has a high risk for mortality, especially when associated with non-immune fetal hydrops. The group at CHOP has published their experience with 30 fetuses with SCT.58 There were 14 survivors and four pregnancies were terminated. Fifteen fetuses had solid tumors. Of those, four developed signs of hydrops and underwent fetal debulking operations. Three of the four survived. In the UCSF experience with 65 prenatally diagnosed SCTs, the overall survival was 44%.57,59 Nineteen of these pregnancies were complicated by fetal hydrops of which eight underwent a fetal intervention and had a 38% survival. In the 11 patients with hydrops who did not undergo fetal intervention, there was only one survivor. Overall, 15 patients with SCT have undergone fetal intervention at UCSF (excluding patients who had cyst aspiration to facilitate delivery): six underwent open resection, five underwent RFA, one underwent alcohol ablation, one had therapeutic cyst aspiration to relieve urinary tract obstruction, one had RFA followed by EXIT-to-resection, and one had EXIT-to-resection alone. Overall survival was 33%. Although ten patients survived to delivery, the mean gestational age was 28.1 weeks and there was a 50% neonatal mortality rate.57
The most common approach for fetal SCT resection is a maternal hysterotomy with resection or debulking of the tumor (Fig. 10-3). A predominantly cystic lesion may be amenable to percutaneous drainage or placement of a shunt which may not be necessary given the favorable prognosis for cystic SCTs. However, immediate decompression of an SCT may be needed just prior to delivery to prevent dystocia or to facilitate cesarean delivery. Tumor debulking using percutaneous coagulation techniques, such as with RFA or laser coagulation, to decrease the vascular shunt are minimally invasive alternatives to open resection that may warrant further investigation.40,60
Abnormalities of Twin Gestations
Twin–Twin Transfusion Syndrome
Twin–twin transfusion syndrome (TTTS) is the most common complication of monochorionic twin pregnancies.61 In such twin pregnancies, the two fetuses share a single placenta with normal vascular connections (arterial-to-venous, venous-to-arterial, and arterial–arterial) between the fetuses. TTTS occurs when these connections lead to unbalanced blood flow from one twin to the other. As a result of the transfusion of blood from the donor twin to the recipient twin through this unbalanced flow, hemodynamic compromise can occur in either or both twins. The donor twin suffers from a low flow state manifesting initially as oligohydramnios and possibly resulting in high-output cardiac failure or ischemia to the brain and kidneys. Conversely, the recipient twin has fluid overload (polyhydramnios), and may develop congestive heart failure and hydrops. The hallmark of TTTS is oligohydramnios in the donor twin and polyhydramnios in the recipient twin, both of which must be present to make the diagnosis. Often there is size discordance between the twins with the donor being smaller than the recipient.
Quintero described four stages of TTTS (Table 10-1).62 Advanced stages of the disease are evidenced by progressive discordance in fluid volumes with the donor becoming ‘stuck’ in its amniotic sac due to a lack of amniotic fluid. This is followed by worsening cardiac changes in the recipient twin and hydrops. If left untreated, TTTS carries an 80–90% mortality rate for both twins. In addition, in monochorionic twins, if one twin dies, the other is at risk for neurologic injury due to a sump phenomenon in the placenta which leads to temporary hypotension and ischemia in the surviving twin.63–65
TABLE 10-1
Four Stages of Twin–Twin Transfusion Syndrome
| Stage | Description |
| Stage I | Polyhydramnios (DVP > 8 cm) with oligohydramnios (DVP < 2 cm) with bladders present in both twins |
| Stage II | Bladder not visible in the donor twin |
| Stage III | Changes in umbilical cord or ductus venosus end-diastolic flow; tricuspid regurgitation in the recipient twin |
| Stage IV | Evidence of hydrops in either twin |
| Stage V | Fetal death |
Clinicians have attempted a variety of treatments aimed at achieving improved outcome in one or both twins. Historically, high-volume amnioreduction in the polydramniotic sac has been the primary therapy. Because polyhydramnios can incite labor, the initial aim of amnioreduction is to reduce uterine volume to decrease the risk of preterm labor. In the International Amnioreduction Registry, high-volume amnioreduction resulted in a survival rate in at least one twin of almost 60%.66
Several groups have introduced fetoscopic guidance to laser ablate the intertwin vascular connections. This approach has largely replaced amnioreduction. This can be done either nonselectively by ablating all intertwin connections, or selectively by ablating only the arteriovenous connections with flow in the causative direction. Fetoscopic laser ablation is performed percutaneously using a 3 mm fetoscope with a side channel for irrigation and insertion of a laser. Two large prospective trials have compared amnioreduction to laser ablation of intertwin vessels. A European trial enrolled 70 women in the amnioreduction arm and 72 women in the laser ablation arm. The trial was stopped early after interim analysis showed a clear survival advantage for laser therapy: 76% vs 51% single survivor and 36% vs 26% for dual survivors.67 A North American trial was also stopped early after randomizing 42 mothers (20 in the amnioreduction arm and 22 in the laser ablation cohort) because of reluctance among referring physicians to send patients to participating centers for randomization due to a strong bias for laser ablation.68 There was no survival benefit to either intervention in this study which was under-powered due to the early termination of the trial. A Cochrane review and meta-analysis also favored laser ablation for TTTS with an overall survival of 66% for laser ablation compared to 48% for amnioreduction.69 When laser ablation is not available or not possible for technical reasons, amnioreduction is an appropriate alternative.
Whether or not every case of TTTS requires intervention is controversial. At UCSF, favorable outcomes with expectant management for stage I TTTS have been found, and laser ablation is only offered in those cases with stage II or more advanced TTTS. In fact, we have identified that the presence of an arterial-arterial anastomosis is protective (by serving as a pop-off valve). Of 639 placentas evaluated at our center, only 5% of those with an arterial-arterial anastomosis had true TTTS.70
Twin Reversed Arterial Perfusion
The normal twin is put at risk for high-output heart failure and hydrops as it has to maintain blood flow throughout the entire placenta as well as to the acardiac twin. The vascular flow in the acardiac twin is characteristically reversed. The natural history of TRAP is greater than a 50% mortality in the pump twin due to hydrops.71,72 The risk of hydrops increases as the mass of the acardiac twin increases relative to the normal twin. Generally, intervention is needed when there is evidence of hydrops in the pump twin, or when the estimated fetal weight of the acardiac twin is 50% or more relative to the twin functioning as the pump.
Multiple approaches have been used to separate the vascular connections in TRAP pregnancies: open hysterotomy and delivery, fetoscopic ligation, bipolar cautery, harmonic scalpel division, thermal coagulation, and laser coagulation. At UCSF, RFA is used to coagulate the umbilical cord insertion site on the acardiac twin’s abdomen.73–75 RFA was originally designed for ablation of solid tumors, but its small size and effective coagulation has been ideal for this application.76 The most recent UCSF review identified 29 patients who underwent RFA between 18 and 24 weeks gestation.5 Survival was 92% percent overall.
Other Complications of Monochorionic Twins
Unequal placental sharing occurs because there is no predetermined organization for each umbilical cord insertion to ensure that each twin has an equal share of the placenta. When one of the twins has an eccentric cord insertion, their growth can be adversely affected when their demand exceeds what their share of the placenta can provide. This results in intrauterine growth restriction (IUGR) and eventual growth discordance between the twins (defined as discordant weights >20%). In fact, the growth-restricted twin can develop oligohydramnios raising the suspicion for TTTS, but the distinction is that the normal twin will have normal amniotic fluid volume. The growth-restricted twin can become distressed leading to preterm labor and extreme prematurity which can also adversely affect the normal twin. The other scenario is IUFD. When this occurs, there is transient shunting with hypotension in the normal twin that can lead to permanent neurologic injury in 20–40% of cases. These pregnancies require close monitoring to ensure there is no evidence of TTTS and that growth is appropriate. Selective RFA of the growth-restricted twin has been offered to protect the normal twin. At UCSF, survival of the normal twin has been 87% without any reported adverse neurologic outcomes.70
The corollary to unequal sharing is polyhydramnios affecting a recipient-like twin (PART). In this condition, one of the twins has polyhydramnios which can raise a concern for TTTS. However, the other twin does not have oligohydramnios and therefore does not meet criteria for TTTS.70 Similarly, pregnancies affected by PART require close surveillance. Currently, there is no fetal intervention recommended for PART, but underlying causes for the polyhydramnios should be sought and treated as needed.
Myelomeningocele
MMC, or spina bifida, is characterized by an open neural tube and exposed spinal canal elements. MMC can occur anywhere along the spine, but most commonly occurs in the lumbar or cervical vertebral levels. Complications include neurologic deficits with motor and somatosensory abnormalities which correspond to the level of the spinal defect. In addition, autonomic function is commonly affected with an inability to control bladder or bowel function. Also, nearly all patients with MMC develop the Arnold–Chiari II malformation of the hindbrain and most will require ventriculoperitoneal (VP) shunting for hydrocephalus. Unlike patients that have historically been considered for fetal intervention, fetuses with MMC are generally born alive and healthy. However, the attendant morbidity from the neurologic abnormalities is severe. Up to 30% of patients die before reaching adulthood due to respiratory, urinary, or central nervous system complications. Standard current therapy for MMC is postnatal repair of the spinal defect followed by extensive rehabilitation.77
The rationale for fetal intervention in MMC is the ‘two-hit’ hypothesis, where the first hit is the original neural tube defect that results in an open spinal canal. The second hit is postulated to be trauma to the exposed neural elements while the fetus is in utero.78,79 It is this second hit that may be ameliorated by fetal intervention and early closure.80 The results of animal and preliminary human studies showed improved neurologic outcomes and a decreased need for VP shunting with prenatal closure.79,81–84 These promising findings prompted a multi-institutional prospective randomized trial known as the management of myelomeningocele study (MOMS) that compared open fetal repair with postnatal repair.85
Fetal repair in the MOMS trial was performed using an open hysterotomy (Fig. 10-4) with primary repair or the use of skin allografts for large defects as had been previously described.86–89 The study’s power analysis indicated 200 patients were required. However the study was terminated early, after 183 patients, because of the clear advantage to prenatal repair compared to postnatal repair for the primary outcome variable which was need for VP shunting. At 12 months, only 68% of the prenatally repaired group met study criteria for VP shunt placement compared to 98% in the postnatal group. Furthermore, neurologic function favored the prenatal repair group, with 42% walking without assistance at 30 months compared to 21% in the postnatal repair cohort.85 Not only were these results a milestone in the evolving treatment of MMC, but this was the first nonlethal anomaly for which fetal surgery has been shown to be beneficial.
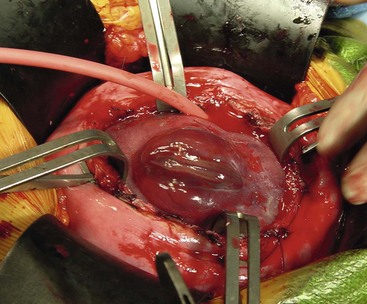
FIGURE 10-4 The myelomeningocele is exposed after maternal hysterectomy. The defect is closed by pediatric neurosurgeons using an operating microscope.
The benefit of prenatal MMC repair is associated with risks. In the MOMS trial, there was a 38% incidence of preterm labor. Also, the mean gestational age in the prenatal repair group was 34 weeks compared to 37 weeks in the postnatal group. Additionally, 46% had premature rupture of membranes contributing to earlier delivery. There was a 10% rate of uterine dehiscence, and fetal death occurred in 3% of the prenatal repair group.85 For these reasons, families require extensive counseling to ensure that they fully understand the risks and benefits. Currently, less invasive methods for the treatment of MMC are being pursued to minimize morbidity. One promising area may be fetoscopic coverage of the defect for temporary protection followed by definitive closure postnatally.90
Hydronephrosis and Low Urinary Tract Obstruction
Hydronephrosis is a common prenatal diagnosis. In most cases of minimal hydronephrosis, there will be complete resolution. However, 10% will have progression and require postnatal evaluation.91 Therefore, in the setting of minimal hydronephrosis, an ultrasound should be obtained in the third trimester to determine if there has been resolution or progression which will help guide the need for postnatal evaluation.
More severe cases of hydronephrosis suggest a ureteropelvic junction (UPJ) obstruction, a ureterovesical junction obstruction, or an obstructing ureterocele. In these scenarios, when unilateral, there is no indication for fetal intervention. Bilateral hydronephrosis is a more significant problem which can be complicated by oligohydramnios which leads to fatal pulmonary hypoplasia. Fortunately, bilateral UPJ obstruction rarely leads to oligohydramnios and the need for fetal intervention with shunting.92
Lower urinary tract obstruction (LUTO) can be due to urethral atresia or, most commonly, posterior urethral valves (PUV). The fetus presents with a classic constellation of three ultrasound findings: a dilated, keyhole shaped bladder; bilateral megaureters; and bilateral hydronephrosis.93 The fetus with LUTO is at high risk for the development of oligohydramnios and subsequent pulmonary hypoplasia that is fatal in the immediate postnatal period. Additionally, the pressure that results from the obstruction leads to dysplastic changes in the kidney that predisposes the surviving neonate to at least a 20–30% risk of developing end-stage renal disease in their lifetime.94–96 The chronic distention of the bladder also results in a 45% incidence of neurogenic bladder, although most of these babies have some degree of permanent bladder dysfunction.96
The timing of intervention in the absence of oligohydramnios is controversial. Imaging and fetal urine electrolyte studies are not definitive in identifying those fetuses at risk for renal dysplasia. Fetal urine electrolytes can be useful when obtained beyond 20 weeks gestation and are easily assessed by a percutaneous aspirate from the dilated fetal bladder.97 However, the initial tap may be a misrepresentation because the urine has been in the bladder for some time. Thus, serial taps can be more helpful in determining the presence of renal dysplasia, especially if the β2-microblogulin is elevated.97 Established normal values for fetal urine electrolytes are outlined in Table 10-2.
TABLE 10-2
Normal values for fetal urine electrolytes
| Measurement | Normal Values |
| Osm | <210 mEq/L |
| Na | <100 mEq/L |
| Cl | <90 mEq/L |
| Ca | <2 mmol/L |
| PO4 | <2 mmol/L |
| β2-microglobulin | <2 mg/L |
Abdominal Wall Defects
Gastroschisis is often diagnosed prenatally by ultrasound and usually has an elevated maternal serum α-fetoprotein (which can also be elevated in omphalocele).98 Attention has been given to the presence of bowel dilation or bowel wall thickening on prenatal ultrasound as an early series of ten patients indicated this represented intestinal injury.99 However, several subsequent series since have failed to find any correlation between bowel dilation or bowel wall thickening and outcomes.100–102 Current recommendations are not to deliver early based on the presence of bowel dilation or bowel wall thickening.
Pregnancies complicated by gastroschisis require close monitoring. Nearly three-quarters of fetuses with gastroschisis are affected by IUGR. Preterm labor may be precipitated by polyhydramnios. The risk for IUFD is as high as 10%, particularly in the third trimester.103 Contrary to popular belief, gastroschisis is not an indication for cesarean section, and vaginal delivery should be offered in the absence of another indication for cesarean delivery.104
Similar to MMC, clinicians have theorized that intestinal injury and the intestinal dysmotility associated with gastroschisis may be related to injury while the bowel is exposed to the amniotic fluid. Initial investigations have centered on amnio exchange where the amniotic fluid is replaced with sterile saline.105 While these investigators reported subjectively favorable findings of less serosal inflammation, the postnatal outcomes were not different. Currently, some investigators have applied the same principle of early coverage to gastroschisis and have proposed fetoscopic closure. This has been attempted in a lamb model, but this approach has not been well studied.106 Given long-term follow-up data and the quality of life studies that have found that most gastroschisis patients are essentially normal by two years of age, aggressive pursuit of fetal intervention for gastroschisis will probably only benefit those patients who develop short-gut syndrome, either anatomically or functionally.107 Unfortunately, it has not been possible to identify these patients prenatally.
Stem Cells and Gene Therapy
Gene therapy for prenatally identifiable diseases is being actively pursued for specific disorders. The rationale behind in utero therapy with stem cells and/or virally directed genes includes halting the progression of disease in the fetus during gestation as well as taking advantage of the developing immune system of the fetus, thus potentially negating postnatal problems of tolerance, rejection, and graft versus host disease.108
Specific issues for in utero treatment of genetic diseases include the timing of diagnosis and therapy, how to deliver the stem cells or genes, the sources of the stem cells, and the longevity of treatment. With the advent of chorionic villus sampling, genetic diseases can now be identified in the first trimester. Timing of potential treatments is crucial to take advantage of the possible ‘pre-immune’ status of the fetus, making fetuses potentially more receptive to exogenous genes or cells. Several investigators have utilized hematopoietic stem cells (HSC) as a vector in an attempt to induce chimerism to treat the diseases.109–111 Others have investigated the use of retroviral vectors to insert genetic material into the fetus.112,113 This approach reduces the problem of obtaining the large numbers of stem cells needed to create even a modest amount of chimerism. Other approaches include using maternal stem cells or genetic material as studies have demonstrated early cross-trafficking of maternal cells in the fetus.
Diseases that are a candidate for this approach include hematologic, immunologic, metabolic, and neurologic abnormalities (Box 10-1). To date, there have been over 30 reports of in utero therapy utilizing HSCs with limited success. Until recently, the only durable treatment has been in patients with preexisting immunologic defects.114,115 However a recent study from UCSF applied HSCs in the treatment of four patients with Pelizaeus–Merzbacher disease, a demyelinating leukodystrophy, with promising results in three of the patients who have demonstrated modest improvements in neurologic function as well as durable engraftment.116
References
1. Harrison, MR, Golbus, MS, Filly, RA, et al. Fetal surgery for congenital hydronephrosis. New Engl J Med. 1982; 306:591–593.
2. Farrell, JA, Albanese, CT, Jennings, RW, et al. Maternal fertility is not affected by fetal surgery. Fetal Diagn Ther. 1999; 14:190–192.
3. VanderWall, KJ, Meuli, M, Szabo, Z, et al. Percutaneous access to the uterus for fetal surgery. J Laparoendosc Surg. 1996; 6:S65–S67.
4. Sydorak, RM, Feldstein, V, Machin, G, et al. Fetoscopic treatment for discordant twins. J Pediatr Surg. 2002; 37:1736–1739.
5. Lee, H, Wagner, AJ, Sy, E, et al. Efficacy of radiofrequency ablation for twin-reversed arterial perfusion sequence. Am J Obstet Gynecol. 2007; 196:459e1–4.
6. De Buck, F, Deprest, J, Van de Velde, M. Anesthesia for fetal surgery. Curr Opin Anaesthesiol. 2008; 21:293–297.
7. Harrison, MR, Anderson, J, Rosen, MA, et al. Fetal surgery in the primate I. Anesthetic, surgical, and tocolytic management to maximize fetal-neonatal survival. J Pediatr Surg. 1982; 17:115–122.
8. Rosen, MA. Anesthesia for fetal procedures and surgery. Yonsei Med J. 2001; 42:669–680.
9. Logan, JW, Rice, HE, Goldberg, RN, et al. Congenital diaphragmatic hernia: A systematic review and summary of best-evidence practice strategies. J Perinatol. 2007; 27:535–549.
10. Moya, FR, Lally, KP. Evidence-based management of infants with congenital diaphragmatic hernia. Semin Perinatol. 2005; 29:112–117.
11. Doyle, NM, Lally, KP. The CDH Study Group and advances in the clinical care of the patient with congenital diaphragmatic hernia. Semin Perinatol. 2004; 28:174–184.
12. Metkus, AP, Filly, RA, Stringer, MD, et al. Sonographic predictors of survival in fetal diaphragmatic hernia. J Pediatr Surg. 1996; 31:148–152.
13. Jani, J, Nicolaides, KH, Keller, RL, et al. Observed to expected lung area to head circumference ratio in the prediction of survival in fetuses with isolated diaphragmatic hernia. Ultrasound Obstet Gynecol. 2007; 30:67–71.
14. Cruz-Martinez, R, Castanon, M, Moreno-Alvarez, O, et al. Usefulness of lung-to-head ratio and intrapulmonary Doppler in predicting neonatal morbidity in fetuses with congenital diaphragmatic hernia treated with fetoscopic tracheal occlusion. Ultrasound Obstet Gynecol. 2012; 41:59–65.
15. Jani, JC, Benachi, A, Nicolaides, KH, et al. Prenatal prediction of neonatal morbidity in survivors with congenital diaphragmatic hernia: A multicenter study. Ultrasound Obstet Gynecol. 2009; 33:64–69.
16. Coakley, FV, Lopoo, JB, Lu, Y, et al. Normal and hypoplastic fetal lungs: Volumetric assessment with prenatal single-shot rapid acquisition with relaxation enhancement MR imaging. Radiology. 2000; 216:107–111.
17. Barnewolt, CE, Kunisaki, SM, Fauza, DO, et al. Percent predicted lung volumes as measured on fetal magnetic resonance imaging: A useful biometric parameter for risk stratification. J Pediatr Surg. 2007; 42:193–197.
18. Victoria, T, Bebbington, MW, Danzer, E, et al. Use of magnetic resonance imaging in prenatal prognosis of the fetus with isolated left congenital diaphragmatic hernia. Prenatal Diagnosis. 2012; 32:715–723.
19. Adzick, NS, Harrison, MR, Flake, AW. Experimental studies on prenatal treatment of congenital anomalies. Br J Hosp Med. 1985; 34:154–159.
20. Adzick, NS, Outwater, KM, Harrison, MR, et al. Correction of congenital diaphragmatic hernia in-utero. IV. An early gestational fetal lamb model for pulmonary vascular morphometric analysis. J Pediatr Surg. 1985; 20:673–680.
21. Adzick, NS, Harrison, MR, Glick, PL, et al. Diaphragmatic hernia in the fetus: Prenatal diagnosis and outcome in 94 cases. J Pediatr Surg. 1985; 20:357–361.
22. Harrison, MR, Adzick, NS, Flake, AW, et al. Correction of congenital diaphragmatic hernia in-utero. VI. Hard-earned lessons. J Pediatr Surg. 1993; 28:1411–1418.
23. Harrison, MR, Adzick, NS, Longaker, MT, et al. Successful repair in utero of a fetal diaphragmatic hernia after removal of herniated viscera from the left thorax. New Eng J Med. 1990; 322:1582–1584.
24. Harrison, MR, Adzick, NS, Bullard, KM, et al. Correction of congenital diaphragmatic hernia in utero VII: A prospective trial. J Pediatr Surg. 1997; 32:1637–1642.
25. Hedrick, MH, Ferro, MM, Filly, RA, et al. Congenital high airway obstruction syndrome (CHAOS): A potential for perinatal intervention. J Pediatr Surg. 1994; 29:271–274.
26. Carmel, JA, Friedman, F, Adams, FH, et al. Fetal tracheal ligation and lung development. Am J Dis Child. 1965; 109:452–457.
27. DiFiore, JW, Fauza, DO, Slavin, R, et al. Experimental fetal tracheal ligation and congenital diaphragmatic hernia: A pulmonary vascular morphometric analysis. J Pediatr Surg. 1995; 30:917–924.
28. DiFiore, JW, Fauza, DO, Slavin, R, et al. Experimental fetal tracheal ligation reverses the structural and physiological effects of pulmonary hypoplasia in congenital diaphragmatic hernia. J Pediatr Surg. 1994; 29:248–257.
29. Hedrick, MH, Estes, JM, Sullivan, KM, et al. Plug the lung until it grows (PLUG): A new method to treat congenital diaphragmatic hernia in-utero. J Pediatr Surg. 1994; 29:612–617.
30. Harrison, MR, Adzick, NS, Flake, AW, et al. Correction of congenital diaphragmatic hernia in utero VIII: Response of the hypoplastic lung to tracheal occlusion. J Pediatr Surg. 1996; 31:1339–1346.
31. Hirose, S, Harrison, MR. The ex-utero intrapartum treatment (EXIT) procedure. Semin Neonatol. 2003; 8:207–214.
32. Harrison, MR, Mychaliska, GB, Albanese, CT, et al. Correction of congenital diaphragmatic hernia in utero IX: Fetuses with poor prognosis (liver herniation and lung-to-head ratio) can be saved by fetoscopic temporary tracheal occlusion. J Pediatr Surg. 1998; 33:1017–1023.
33. Harrison, MR, Keller, RL, Hawgood, SB, et al. A randomized trial of fetal endoscopic tracheal occlusion for severe fetal congenital diaphragmatic hernia. New Engl J Med. 2003; 349:1916–1924.
34. Saddiq, WB, Piedboeuf, B, Laberge, JM, et al. The effects of tracheal occlusion and release on type II pneumocytes in fetal lambs. J Pediatr Surg. 1997; 32:834–838.
35. Cannie, MM, Jani, JC, De Keyzer, F, et al. Evidence and patterns in lung response after fetal tracheal occlusion: Clinical controlled study. Radiology. 2009; 252:526–533.
36. Hirose, S, Farmer, DL, Lee, H, et al. The ex-utero intrapartum treatment procedure: Looking back at the EXIT. J Pediatr Surg. 2004; 39:375–380.
37. Jani, JC, Nicolaides, KH, Gratacos, E, et al. Severe diaphragmatic hernia treated by fetal endoscopic tracheal occlusion. Ultrasound Obstet Gynecol. 2009; 34:304–310.
38. Jelin, E, Lee, H. Tracheal occlusion for fetal congenital diaphragmatic hernia: The ultrasound experience. Clin Perinatol. 2009; 36:349–361.
39. Adzick, NS, Harrison, MR, Glick, PL, et al. Fetal cystic adenomatoid malformation: Prenatal diagnosis and natural history. J Pediatr Surg. 1985; 20:483–488.
40. Adzick, NS. Open fetal surgery for life-threatening fetal anomalies. Semin Fetal Neonatal Med. 2010; 15:1–8.
41. Schott, S, Mackensen-Haen, S, Wallwiener, M, et al. Cystic adenomatoid malformation of the lung causing hydrops fetalis: Case report and review of the literature. Arch Gynecol Obstet. 2009; 280:293–296.
42. Kunisaki, SM, Barnewolt, CE, Estroff, JA, et al. Large fetal congenital cystic adenomatoid malformations: Growth trends and patient survival. J Pediatr Surg. 2007; 42:404–410.
43. Ierullo, AM, Ganapathy, R, Crowley, S, et al. Neonatal outcome of antenatally diagnosed congenital cystic adenomatoid malformations. Ultrasound Obstet Gynecol. 2005; 26:150–153.
44. Hsieh, CC, Chao, AS, Chang, YL, et al. Outcome of congenital cystic adenomatoid malformation of the lung after antenatal diagnosis. Int J Gynaecol Obstet. 2005; 89:99–102.
45. Crombleholme, TM, Coleman, B, Hedrick, HL, et al. Cystic adenomatoid malformation volume ratio predicts outcome in prenatally diagnosed cystic adenomatoid malformation of the lung. J Pediatr Surg. 2002; 37:331–338.
46. Miller, JA, Corteville, JE, Langer, JC. Congenital cystic adenomatoid malformation in the fetus: Natural history and predictors of outcome. J Pediatr Surg. 1996; 31:805–808.
47. Wilson, RD, Baxter, JK, Johnson, MP, et al. Thoracoamniotic shunts: Fetal treatment of pleural effusions and congenital cystic adenomatoid malformations. Fetal Diagn Ther. 2004; 19:413–420.
48. Wilson, RD, Baxter, JK, Johnson, MP, et al. Thoracoamniotic shunts: Fetal treatment of pleural effusions and congenital cystic adenomatoid malformations. Fetal Diagn Ther. 2004; 19:413–420.
49. Hedrick, HL, Flake, AW, Crombleholme, TM, et al. History of fetal diagnosis and therapy: Children’s Hospital of Philadelphia experience. Fetal Diagn Ther. 2003; 18:65–82.
50. Adzick, NS, Harrison, MR. Management of the fetus with a cystic adenomatoid malformation. World J Surg. 1993; 17:342–349.
51. Adzick, NS, Harrison, MR, Flake, AW, et al. Fetal surgery for cystic adenomatoid malformation of the lung. J Pediatr Surg. 1993; 28:806–812.
52. Tsao, K, Hawgood, S, Vu, L, et al. Resolution of hydrops fetalis in congenital cystic adenomatoid malformation after prenatal steroid therapy. J Pediatr Surg. 2003; 38:508–510.
53. Curran, PF, Jelin, EB, Rand, L, et al. Prenatal steroids for microcystic congenital cystic adenomatoid malformations. J Pediatr Surg. 2010; 45:145–150.
54. Peranteau, WH, Wilson, RD, Liechty, KW, et al. Effect of maternal betamethasone administration on prenatal congenital cystic adenomatoid malformation growth and fetal survival. Fetal Diagn Ther. 2007; 22:365–371.
55. French, NP, Hagan, R, Evans, SF, et al. Repeated antenatal corticosteroids: Size at birth and subsequent development. Am J Obstet Gynecol. 1999; 180:114–121.
56. Rodriguez, MA, Cass, DL, Lazar, DA, et al. Tumor volume to fetal weight ratio as an early prognostic classification in fetal sacrococcygeal teratoma. J Pediatr Surg. 2011; 46:1182–1185.
57. Shue EH, Bolouri MS, Jelin EB, et al. Tumor metrics and morphology predict poor outcome in prenatally diagnosed sacrococcygeal teratoma. J Pediatr Surg. In Press.
58. Westerburg, B, Feldstein, VA, Sandberg, PL, et al. Sonographic prognostic factors in fetuses with sacrococcygeal teratoma. J Pediatr Surg. 2000; 35:322–326.
59. Hedrick, HL, Flake, AW, Crombleholme, TM, et al. Sacrococcygeal teratoma: Prenatal assessment, fetal intervention, and outcome. J Pediatr Surg. 2003; 39:430–438.
60. Ruano, R, Duarte, S, Zugaib, M. Percutaneous laser ablation of sacrococcygeal teratoma in a hydropic fetus with severe heart failure—too late for a surgical procedure? Fetal Diagn Ther. 2009; 25:26–30.
61. Sebire, NJ, Snijders, RJ, Hughes, K, et al. The hidden mortality of monochorionic twin pregnancies. Br J Obstet Gynaecol. 1997; 104:1203–1207.
62. Quintero, R, Morales, W, Allen, M, et al. Staging of twin-twin transfusion syndrome. J Perinatol. 1999; 19:550–555.
63. Fusi, L, Gordon, H. Twin pregnancy complicated by single intrauterine death. Problems and outcome with conservative management. Br J Obstet Gynaecol. 1990; 97:511–516.
64. Fusi, L, McParland, P, Fisk, N, et al. Acute twin-twin transfusion: A possible mechanism for brain-damaged survivors after intrauterine death of a monochorionic twin. Obstet Gynecol. 1991; 78:517–520.
65. Berghella, V, Kaufmann, M. Natural history of twin-twin transfusion syndrome. J Reprod Med. 2001; 46:480–484.
66. Roberts, D, Gates, S, Kilby, M, et al. Interventions for twin-twin transfusion syndrome: A Cochrane review. Ultrasound Obstet Gynecol. 2008; 31:701–711.
67. Senat, MV, Deprest, J, Boulvain, M, et al. Endoscopic laser surgery versus serial amnioreduction for severe twin-to-twin transfusion syndrome. New Engl J Med. 2004; 351:136–144.
68. Crombleholme, TM, Shera, D, Lee, H, et al. A prospective, randomized, multicenter trial of amnioreduction vs. selective fetoscopic laser photocoagulation for the treatment of severe twin-twin transfusion syndrome. Am J Obstet Gynecol. 2007; 197:396 e1–9.
69. Rossi, AC, D’Addario, V. Laser therapy and serial amnioreduction as therapy for twin-twin transfusion syndrome: A metaanalysis and review of literature. Am J Obstet Gynecol. 2008; 198:147–152.
70. Rand, L, Lee, H. Complicated monochorionic twin pregnancies: Updates in fetal diagnosis and treatment. Clin Perinatol. 2009; 36:417–430.
71. Van Allen, MI, Smith, DW, Shepard, TH. Twin reversed arterial perfusion (TRAP) sequence: A study of 14 twin pregnancies with acardius. Semin Perinatol. 1983; 7:285–293.
72. Goh, A, Loke, HL, Tan, KW. The ‘TRAP’ sequence–life threatening consequences to the pump twin. Singapore Med J. 1994; 35:329–331.
73. Quintero, R, Munoz, H, Hasbun, J, et al. [Fetal endoscopic surgery in a case of twin pregnancy complicated by reversed arterial perfusion sequence (TRAP sequence)]. Rev Chil Obstet Ginecol. 1995; 60:112–117.
74. Hecher, K, Hackeloer, BJ, Ville, Y. Umbilical cord coagulation by operative microendoscopy at 16 weeks’ gestation in an acardiac twin. Ultrasound Obstet Gynecol. 1997; 10:130–132.
75. Tan, TY, Sepulveda, W. Acardiac twin: A systematic review of minimally invasive treatment modalities. Ultrasound Obstet Gynecol. 2003; 22:409–419.
76. Tsao, K, Feldstein, VA, Albanese, CT, et al. Selective reduction of acardiac twin by radiofrequency ablation. Am J Obstet Gynecol. 2002; 187:635–640.
77. Hirose, S, Farmer, DL. Fetal surgery for myelomeningocele. Clin Perinatol. 2009; 36:431–438.
78. Heffez, DS, Aryanpur, J, Hutchins, GM, et al. The paralysis associated with myelomeningocele: Clinical and experimental data implicating a preventable spinal cord injury. Neurosurgery. 1990; 26:987–992.
79. Meuli, M, Meuli-Simmen, C, Yingling, CD, et al. Creation of myelomeningocele in-utero: A model of functional damage from spinal cord exposure in fetal sheep. J Pediatr Surg. 1995; 30:1028–1033.
80. Walsh, DS, Adzick, NS, Sutton, LN, et al. The rationale for in-utero repair of myelomeningocele. Fetal Diagn Ther. 2001; 16:312–322.
81. Meuli, M, Meuli-Simmen, C, Yingling, CD, et al. In-utero repair of experimental myelomeningocele saves neurological function at birth. J Pediatr Surg. 1996; 31:397–402.
82. Bruner, JP, Tulipan, N, Paschall, RL, et al. Fetal surgery for myelomeningocele and the incidence of shunt-dependent hydrocephalus. JAMA. 1999; 282:1819–1825.
83. Sutton, LN, Adzick, NS, Bilaniuk, LT, et al. Improvement in hindbrain herniation demonstrated by serial fetal magnetic resonance imaging following fetal surgery for myelomeningocele. JAMA. 1999; 282:1826–1831.
84. Farmer, DL, von Koch, CS, Peacock, WJ, et al. In-utero repair of myelomeningocele: Experimental pathophysiology, initial clinical experience, and outcomes. Arch Surg. 2003; 138:872–878.
85. Adzick, NS, Thom, EA, Spong, CY, et al. A randomized trial of prenatal versus postnatal repair of myelomeningocele. N Engl J Med. 2011; 364:993–1004.
86. Hirose, S, Meuli-Simmen, C, Meuli, M. Fetal surgery for myelomeningocele: Panacea or peril? World J Surg. 2003; 27:87–94.
87. Johnson, MP, Sutton, LN, Rintoul, N, et al. Fetal myelomeningocele repair: Short-term clinical outcomes. Am J Obstet Gynecol. 2003; 189:482–487.
88. Bruner, JP, Tulipan, NE, Richards, WO. Endoscopic coverage of fetal open myelomeningocele in-utero. Am J Obstet Gynecol. 1997; 176:256–257.
89. Bruner, JP, Tulipan, NB, Richards, WO, et al. In-utero repair of myelomeningocele: A comparison of endoscopy and hysterotomy. Fetal Diagn Ther. 2000; 15:83–88.
90. Fontecha, CG, Peiro, SL, Sevilla, JJ, et al. Fetoscopic coverage of experimental myelomeningocele in sheep using a patch with surgical sealant. Eur J Obstet Gynecol Reprod Biol. 2011; 156:171–176.
91. Morin, L, Cendron, M, Crombleholme, TM, et al. Minimal hydronephrosis in the fetus: Clinical significance and implications for management. J Urol. 1996; 155:2047–2049.
92. Flake, AW, Adzick, NS, Harrison, MR, et al. Ureteropelvic junction obstruction. J Pediatr Surg. 1986; 21:1058–1064.
93. Mahoney, BS, Callen, PW, Filly, RA. Sonographic evaluation of renal dysplasia. Radiology. 1984; 152:143–149.
94. Heikkla, J, Holmberg, C, Kyllonen, L, et al. Long-term risk of end-stage renal disease in patients with posterior urethral valves. J Urol. 2011; 186:2392–2396.
95. Caione, P, Nappo, SG. Posterior urethral valves: Long-term outcome. Pediatr Surg Int. 2011; 27:1027–1035.
96. Biard, JM, Johnson, MP, Carr, MC, et al. Long-term outcomes in children treated by prenatal vesicoamniotic shunting for lower urinary tract obstruction. Obstet Gynecol. 2005; 106:503–508.
97. Nicolini, U, Fisk, NM, Rodeck, CH, et al. Fetal urine biochemistry: An index of renal maturation and dysfunction. Br J Obstet Gynaecol. 1992; 99:46–50.
98. Carroll, SG, Kuo, PY, Kyle, PM, et al. Fetal protein loss in gastroschisis as an explanation of associated morbidity. Am J Obstet Gynecol. 2001; 184:1297–1301.
99. Bond, SJ, Harrison, MR, Filly, RA, et al. Severity of fetal midgut herniation: Normal size criteria and correlation with crown-rump length. J Ultrasound Med. 1993; 12:251–254.
100. Lenke, RR, Persutte, WH, Nemes, J. Ultrasonographic assessment of intestinal damage in fetuses with gastroschisis: Is it of clinical value? Am J Obstet Gynecol. 1990; 163:995–998.
101. Sipes, SL, Weiner, CP, Williamson, RA, et al. Fetal gastroschisis complicated by bowel dilatation: An indication for imminent delivery? Fetal Diagn Ther. 1990; 5:100–103.
102. Alsulyman, OM, Monteiro, H, Ouzounian, JG, et al. Clinical significance of prenatal ultrasonographic intestinal dilatation in fetuses with gastroschisis. Am J Obstet Gynecol. 1996; 175:982–984.
103. Crawford, RAF, Ryan, G, Wright, VM, et al. The importance of serial biophysical assessment of fetal wellbeing in gastroschisis. Br J Obstet Gynaecol. 1992; 99:899–902.
104. Langer, JC. Abdominal wall defects. World J Surg. 2003; 27:117–124.
105. Aktug, T, Erdag, G, Kargi, A, et al. Amnio-allantoic fluid exchange for the prevention of intestinal damage in gastroschisis: An experimental study on chick embryos. J Pediatr Surg. 1995; 30:384–387.
106. Kohl, T, Tchatcheva, K, Stressiq, R, et al. Is there a therapeutic role for fetoscopic surgery in the prenatal treatment of gastroschisis? A feasibility study in sheep. Surg Endosc. 2009; 23:1499–1505.
107. Koivusalo, A, Lindahl, H, Rintala, RJ. Morbidity and quality of life in adult patients with a congenital abdominal wall defect: A questionnaire survey. J Pediatr Surg. 2002; 37:1594–1601.
108. Wagner, AM, Schoeberlein, A, Surbek, D. Fetal gene therapy: Opportunities and risks. Adv Drug Deliv. 2009; 61:813–821.
109. Burt, R, Testor, A, Craig, R, et al. Hematopoietic stem cell transplantation for autoimmune disease: What have we learned? J Autoimmun. 2008; 30:116–120.
110. Verda, L, Kim, D, Ikehara, S, et al. Hematopoietic mixed chimerism derived from allogeneic embryonic stems cells prevents autoimmune diabetes mellitus in NOD mice. Stem Cells. 2008; 26:381–386.
111. Shizuru, J, Weissman, I, Kernoff, R, et al. Purified hematopoietic stem cell grafts induce tolerance to alloantigens and can mediate positive and negative T cell selection. Proc Natl Acad Sci. 2000; 97:9555–9560.
112. Moreno, R, Rosal, M, Cabero, L, et al. Feasibility of retroviral vector-mediated in-utero gene transfer to the fetal rabbit. Fetal Diagn Ther. 2005; 20:485–493.
113. Ekhterae, D, Crumbleholme, T, Karson, E, et al. Retroviral vector-mediated transfer of the bacterial neomycin resistance gene into fetal and adult sheep and human hematopoietic progenitors in vitro. Blood. 1990; 75:365–369.
114. Hayashi, S, Flake, AW. In-utero hematopoietic stem cell therapy. Yonsei Med J. 2001; 42:615–629.
115. Shaaban, AF, Flake, AW. Fetal hematopoietic stem cell transplantation. Semin Perinatol. 1999; 23:515–523.
116. Gupta, N, Henry, RG, Strober, J, et al. Neural stem cell engraftment and myelination in the human brain. Sci Transl Med. 4, 2012.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
Fetal Therapy
The first open fetal surgical procedure was performed at the University of California, San Francisco (UCSF) in 1982.1 Since then, more than 515 fetal interventions have been performed at UCSF over the past 30 years without maternal mortality. Over the past 30 years, a number of advances have been developed to allow a broader application of fetal intervention. These techniques, including maternal hysterotomy, minimal-access fetoscopy, and percutaneous fetal access, were initially tested and validated in animal models. More recently, in utero therapy has shown to be beneficial for non-life-threatening conditions such as myelomeningocele (MMC). In this chapter, we will present an overview of the current state of fetal surgery and will review specific fetal problems outlining current management strategies.
General Principles
The primary morbidity following fetal surgery has been, and remains, preterm labor resulting in premature delivery, usually between 25 and 35 gestational weeks. Preventing preterm labor after fetal intervention remains problematic. Complications can also arise from endotracheal intubation, general anesthesia, epidural and spinal anesthesia, blood transfusion, premature rupture of membranes, chorioamniotic separation, chorioamnionitis, and placental abruption. Long-term morbidity from the hysterotomy includes infertility, uterine rupture with the current and future pregnancies, and mandatory cesarean section with future pregnancies. Notably, in reviewing our experience, subsequent fertility following fetal intervention has been good.2
Fetal Access
Ultrasound-guided percutaneous procedures are performed through small skin incisions on the mother’s abdominal wall. During these operations, real-time ultrasound is needed to visualize the fetal and maternal anatomy.3 Catheters and shunts can be inserted into the fetus to drain cystic masses, ascites, or pleural fluid into the amniotic space. In addition, radio frequency ablation (RFA) probes can be deployed into the amniotic space to treat various twin gestational anomalies. The needles used to place these catheters, as well as the RFA device, are approximately 1.5–2 mm in diameter, minimizing morbidity to the mother and irritation of the uterus.4,5
Open fetal procedures require general anesthesia with a combination of preoperative indomethacin and high mean alveolar concentration of inhalational agents to maintain uterine relaxation.6–8 An epidural is also inserted for postoperative analgesia.
Anomalies Amenable to Fetal Surgery
Congenital Diaphragmatic Hernia
Despite significant advances in neonatal respiratory support, survival for children born with congenital diaphragmatic hernia (CDH) remains only 60–70% throughout the USA. Additionally, survival for prenatally diagnosed CDH may be as low as 25% due to IUFD and stillborns that are not included in conventional postnatal survival data.9–11 This high mortality rate has made CDH a primary area of interest for the development of effective prenatal intervention. In fact, improving outcomes specifically for CDH was a significant driving force in the genesis of fetal surgery at UCSF.
Prognostic Criteria
One of the key elements in developing fetal intervention for CDH has been identifying what factors will identify those fetuses at the greatest risk for a poor outcome. The factors most consistently associated with a poor outcome on prenatal ultrasound are (1) the presence of liver herniation into the chest; and (2) a low lung-to-head ratio (LHR). In our experience, survival has been 100% in fetuses with CDH that do not have liver herniation on prenatal ultrasound and 56% in fetuses with CDH and liver herniation into the chest.12 The LHR is calculated as the area of the contralateral lung at the level of the cardiac atria divided by the head circumference. This LHR value has been shown to statistically correlate with survival: 100% survival with an LHR greater than 1.35, 61% survival with an LHR between 0.6 and 1.35, and 0% survival with an LHR less than 0.6.12
While the LHR has been a reliable predictor of outcomes at our center, other institutions have suggested the LHR does not account for discrepant growth rates between the head and lung during gestation and therefore may not be reliable at certain gestational ages.13,14 To account for this, the observed to expected LHR (OE LHR) has been proposed. The OE LHR is represented as a percentage of what the expected LHR would be in a normal fetus of the same gestational age. For left-sided defects, an OE LHR <25% is associated with an 18% survival whereas an OE LHR >45% correlates with 89% survival.13,15
Magnetic resonance imaging (MRI) for volumetric measurement of the lungs is a promising modality for prognosis with CDH.16 MRI can be used to calculate the percent-predicted lung volume (PPLV). Results for PPLV have varied. In one study, a PPLV >20% was associated with 100% survival whereas survival was only 40% when PPLV was <15%.17 In another study, a PPLV <25% was associated with a 13% survival and a PPLV >35% correlated with 83% survival.18 MRI can also be used to determine the percentage of liver herniation, although the prognostic value of this finding is still being investigated.18
Fetal Interventions
CDH and its effect on fetal lung development has been studied in animal models.19,20 In the fetal lamb model, compression of the lungs, either with an intrathoracic balloon or by creation of a diaphragmatic hernia, results in uniformly fatal pulmonary hypoplasia. However, in utero correction of the compressing lesion leads to sufficient lung growth and development, which improves postnatal survival.20
This concept of early, in utero correction of CDH has been studied and applied in humans.21,22 Fetal surgery for CDH initially involved open repair of the diaphragmatic defect. The first successful case was reported in 1990 which demonstrated the feasibility of open fetal repair using a two-step approach which involved creation of an abdominal silo to accommodate the reduced viscera and prevent compression of the umbilical vessels.23 This initial success was followed by a prospective trial at UCSF comparing open fetal surgery to postnatal repair in severe cases of prenatally diagnosed CDH. However, in this study, there was no difference in survival or in the need for extracorporeal membranous oxygenation (ECMO) between fetal repair and postnatal repair.22,24 Concordant with this effort, investigators at UCSF observed that fetuses with congenital high airway obstruction syndrome (CHAOS) had pulmonary hyperplasia.25 Also, fetal tracheal occlusion had been shown to cause pulmonary hyperplasia.26 In this condition, the lung parenchyma creates fluid that is ‘exhaled’ by the fetus. Occluding the trachea causes a build-up of this fluid and subsequent pulmonary hyperplasia.27,28 The inability to improve outcomes with open fetal repair for severe cases of CDH led to an interest in this physiologic process.29
The first eight patients were treated with open hysterotomy and tracheal occlusion with a metallic clip.30 This approach proved to be problematic for several reasons. First, the open hysterotomy led to significant prematurity due to premature labor. Second, the use of clips was associated with tracheal stenosis and also required a stringent delivery plan—which was later described as the ex utero intrapartum treatment (EXIT) procedure—whereby the fetus was exposed through a hysterotomy and maintained on utero–placental circulation while the clip was removed and a patent airway established prior to delivering the baby.31 However, outcomes with this approach were poor with only a 15% survival rate.30
Ongoing advancements in fetal surgery led to fetoscopic balloon placement for tracheal occlusion (Fig. 10-1). This technique has the advantages of being less invasive, a lower risk of tracheal stenosis, and the balloon being much easier to remove, although still necessitating an EXIT procedure. Results in the first eight cases were favorable with a 75% survival rate compared to a 38% survival rate in historical, case-matched controls managed with postnatal repair.32
FIGURE 10-1 This schematic diagram shows the method of fetoscopic tracheal occlusion. A fetoscope is placed into the fetal mouth, the airway is identified, and a balloon is inserted into the trachea by using both fetoscopic and ultrasonographic visualization.
These early results led to an National Institutes of Health (NIH) funded, prospective randomized trial comparing in utero fetoscopic tracheal occlusion to standard postnatal care for fetuses diagnosed with severe left-sided CDH (liver up and LHR <1.4) and no other detectable anomalies. However, results of the trial showed no difference in survival between the tracheal occlusion group and the standard postnatal care group (73% vs 77%, respectively).33 Unexpectedly, the survival in the postnatal repair group was considerably greater when compared to historical controls. Although this study did not demonstrate a difference in survival between the prenatal intervention group and the postnatal group, the results of this trial demonstrate the tremendous importance of proper randomized controlled trials for novel fetal surgical procedures.
Further data regarding fetal tracheal occlusion have suggested that temporary, short-term reversible tracheal occlusion may be preferable to a longer duration of occlusion. Animal models of fetal tracheal occlusion have demonstrated that long-term tracheal occlusion can be deleterious to type II pneumocytes (the cells that secrete surfactant) and that this adverse effect is not seen with a shorter duration of tracheal occlusion.34 To test the hypothesis that temporary fetal tracheal occlusion is better, Deprest et al. studied patients undergoing fetal tracheal balloon occlusion who also had the balloon removed prenatally to limit the duration of occlusion.35 In this group of patients, improved lung growth was evident on fetal MRI and was also associated with improved postnatal survival. While reversal of the tracheal occlusion requires a second maternal and fetal intervention for balloon removal, it obviates the need for an EXIT procedure at birth. Early results have been favorable.31,36
These promising findings with temporary tracheal occlusion have led to its current application in Europe. The European FETO consortium has reported a 48% survival rate among 210 cases of severe CDH treated with temporary fetal tracheal occlusion, and the Eurofetus group is currently sponsoring a prospective fetal tracheal occlusion trial that seeks to determine the ideal time and duration for tracheal occlusion.37 Our group at UCSF is currently offering reversible, fetal tracheal balloon occlusion for fetuses with liver herniation in the chest and an LHR of <1.0, as these babies continue to have a very high mortality.38 This study has Food and Drug Administration oversight, and involves percutaneous placement of a fetoscopic tracheal balloon between 26 and 28 weeks gestation, with removal of the balloon via a second percutaneous fetoscopic procedure between 32 and 34 weeks.
Neoplasms
Fortunately, fetal neoplasms are rare. When they do occur, most are benign. However, if they become large enough, they can impede venous return to the heart or cause high-output heart failure via arteriovenous shunting. Such shunting can lead to non-immune fetal hydropic changes such as polyhydramnios, placentomegaly, skin and scalp edema, and pleural, pericardial, and peritoneal fluid accumulation. When only one compartment is involved, this is considered early fetal hydrops; when two or more compartments are affected, then true hydrops is present. If left untreated, hydrops is nearly always fatal.39–40 The two most common prenatally diagnosed neoplasms that cause nonimmune fetal hydrops are congenital pulmonary airway malformations (CPAM) and sacrococcygeal teratomas (SCT).
Congenital Pulmonary Airway Malformations
CPAMs are pulmonary lesions with a broad range of clinical presentations. This new terminology includes congenital cystic adenomatoid malformations (CCAM) and bronchopulmonary sequestrations. CCAMs are much more likely than sequestrations to cause nonimmune fetal hydrops. CCAMs are characterized by an overgrowth of respiratory bronchioles with the formation of cysts of various sizes.41–44 Most fetuses diagnosed with a CCAM develop normally, and can be followed with serial ultrasound studies. These asymptomatic patients then undergo standard, postnatal resection. A small percentage of patients with the prenatal diagnosis of CCAM will develop non-immune hydrops.43,44
Various measurements have been developed to predict which fetuses are at risk for developing hydrops. The most accepted measurement is the CCAM volume ratio (CVR), defined as the product of the three longest measurements of the lesion on ultrasound multiplied by the constant 0.52, and then divided by the head circumference. Crombleholme and colleagues identified a CVR of 1.6 as a cut-off for an increased likelihood of developing hydrops.45 When the CVR is <1.6, there is only a 2% risk of developing hydrops. When the CVR is >1.6, there is an 80% chance of developing hydrops.
CCAMs that are predominantly microcystic have a more predictable course than the macrocystic ones. Microcystic or solid CCAMs undergo steady growth that tends to plateau at 26 to 28 weeks gestation. At this point, fetal growth exceeds that of the CCAM. For this reason, patients with microcystic or solid CCAMs should be followed closely up to 26 to 28 weeks gestation at which point the interval between ultrasound examinations can be lengthened if the pregnancy has been otherwise uncomplicated. In contrast, macrocystic CCAMs undergo abrupt enlargement due to rapid fluid accumulation in a dominant cyst. Therefore, macrocystic CCAMs require close follow-up with serial ultrasound throughout the duration of the pregnancy.42,46
If a fetus develops hydrops at a viable gestational age, early delivery should be considered. Hydropic fetuses who are not yet viable outside the uterus, and have a dominant macrocystic lesion, are appropriate candidates for a thoracoamniotic shunt.47 Needle drainage alone has not been found to be an effective therapy as rapid re-accumulation of fluid in the cyst necessitates repeat intervention. In the largest single-center experience with thoracoamniotic shunts, shunting led to a mean 51% volume reduction in the size of the lesion and a 70% survival rate.48 Other institutions have reported similar survival rates.49 Despite shunting, these babies can still have significant respiratory distress at birth and should be delivered at a tertiary referral center.
Open fetal thoracotomy and CCAM resection is an option in the pre-viable fetus with a microcystic or solid lesion. This is performed through an open hysterotomy. A thoracotomy is made through the fifth intercostal space, and the lobe containing the CCAM is identified and exteriorized through the incision (Fig. 10-2). The pulmonary hilar structures are then mass ligated using an endoloop or endoscopic stapler. The thoracotomy is then closed in layers.50–51
FIGURE 10-2 These photographs depict an infant with a large left upper lobe CCAM undergoing in utero left lobectomy. (A) The infant’s left arm is visualized. Note the maternal hysterotomy and the left fetal thoracotomy (with retractors inserted) through the fifth intercostal space. (B) The left upper lobe containing the CCAM has been identified and exteriorized through the thoracotomy incision. The pulmonary hilar structures were mass ligated using an endoloop. (C) The fetal thoracotomy incision (arrow) has been closed. (D) The left upper lobe specimen containing the CCAM is seen.
In a group of 120 patients with the prenatal diagnosis of CPAM from UCSF and Children’s Hospital of Philadelphia (CHOP), 79 had no evidence of hydrops.51 Of these, 76 were followed expectantly and all survived. Three fetuses without evidence of hydrops and with large dominant cysts underwent thoracoamniotic shunting. All three fetuses survived. Twenty-five hydropic fetuses were followed with no intervention. All mothers delivered prematurely and all fetuses died perinatally. Sixteen fetuses with hydrops underwent intervention: 13 underwent open fetal surgery while three underwent thoracoamniotic shunting. Two of the three survived in the group that underwent shunt insertion and eight of 13 survived in the open fetal surgery group.
Despite positive results with open fetal resection in the hydropic fetus, there has been a shift away from this therapy in the last five years due to the efficacy of maternal steroids. This finding was discovered serendipitously at UCSF during the preparation of several hydropic fetuses for open fetal surgery.52 In these cases, maternal steroids were administered to enhance fetal lung maturity. Preoperative ultrasound studies showed resolution of the hydrops and those fetuses survived to delivery and beyond. Thirteen patients with microcystic CCAMs, nine of which were complicated by hydrops, had an overall survival rate of 85% with resolution of hydrops in seven of nine fetuses.53 CHOP has reported a series of 11 patients, five of which had hydrops, and all survived after receiving steroids.54
Currently, we recommend maternal betamethasone for fetuses with nonimmune hydrops or a CVR >1.6. Steroids can be re-dosed, but repeated administration of maternal steroids beyond three to five courses can result in untoward effects such as reduced birth weight.55 It is widely accepted that steroids are most effective for predominantly microcystic or solid lesions as this is the component of the malformation that responds to steroids. Macrocystic lesions are less likely to respond.
