Fetal Skin Development
Steven B. Hoath, Theodora Mauro
Introduction
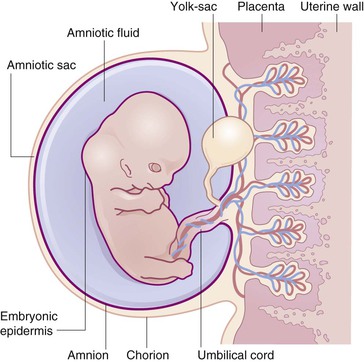
The skin simultaneously contacts a changing environment and provides closure for the body. It is a cellular and molecular interface, which plays a critical functional role in neurobehavior and perception.1 The skin is embryologically continuous with the nervous system via involution and with the amnion via simple lateral extension. Thus, with regard to the fetoplacental unit, the skin is an ‘internal’ organ marking the boundary between the developing nervous system and the fetal membranes surrounding the liquor amnii (Fig. 1.1).
Few organs carry such capacity to convey and evoke emotion. The experiments of Hooker and Humphrey, for example, provide unequivocal video evidence that localized tactile stimulation of the skin of the human embryo as early as 8–9 weeks’ gestation will evoke a reflex response.2,3 Thus, even early in development, the skin cannot be considered apart from the developing central nervous system and other fetal organs, nor, as will be demonstrated in this chapter, from the changing dynamics of its aqueous environment and maternal enclosure.
Fetal skin development occurs in a unique physiological world. The normal intrauterine environment is profoundly hypoxic, a condition aptly described by the eminent fetal physiologist, Joseph Barcroft, as ‘Mt Everest in utero’.4 Fetal wounds heal with little or no scarring.5 Touch, the first sense to develop in all vertebrates,6 occurs in a milieu where stimuli are muted and receptor pathways may be immature.7 The electrical impedance of the fetal skin is inconstant and increases markedly in mid-gestation.8 The fetus develops in an environment of relative immune privilege9 and critical protective functions of the skin, which are unnecessary prenatally, must be operative immediately at birth, e.g., protection from environmental trauma, infection, cold, and xeric stress.10
Comparison of human fetal skin development to other well-studied animal models highlights important and peculiar differences. Humans, for example, have a relatively long gestation (Table 1.1 ). Human infants have a prolonged postnatal period of vulnerability when maternal bonding and skin-to-skin contact figure prominently. According to Brazelton, the human neonate, in contrast to other species, is relatively precocious in sensory capabilities and relatively helpless in motor skills.11 This developmental disparity places a premium on understanding the intrauterine maturation of the skin as a sensory organ and the impact of cutaneous function on the developing nervous system.12 Dermatologists and skin scientists will note that humans are distinguished from other primates, not merely by opposable thumbs and a large, versatile brain but most obviously by a body surface, which is strikingly furless, vulnerable, and characterized by an expanse of well-developed interfollicular epidermis, earning humans the designation, the ‘Naked Ape’.13
). Human infants have a prolonged postnatal period of vulnerability when maternal bonding and skin-to-skin contact figure prominently. According to Brazelton, the human neonate, in contrast to other species, is relatively precocious in sensory capabilities and relatively helpless in motor skills.11 This developmental disparity places a premium on understanding the intrauterine maturation of the skin as a sensory organ and the impact of cutaneous function on the developing nervous system.12 Dermatologists and skin scientists will note that humans are distinguished from other primates, not merely by opposable thumbs and a large, versatile brain but most obviously by a body surface, which is strikingly furless, vulnerable, and characterized by an expanse of well-developed interfollicular epidermis, earning humans the designation, the ‘Naked Ape’.13
TABLE 1.1
Gestation lengths7
| Species | Gestation in days (approx.) |
| Laboratory mouse | 20.5 |
| Domestic rat | 21.5 |
| Domestic rabbit | 32 |
| Dog | 57–63 |
| Domestic cat | 63 |
| Guinea pig | 68 |
| Sheep | 147 |
| Monkey (rhesus) | 165 |
| Man | 265 |
(Adapted from Bradley RM, Mistretta CM. Fetal sensory receptors. Physiological Reviews 1975; 55(3):352–382.)
In contrast to term gestation, the border of viability is the gestational age when the prematurely delivered fetus can survive in the extrauterine environment. In humans, this border, dependent in part upon medical intervention, exhibits racial and gender differences and varies from country to country, but is typically placed at 23–25 weeks’ gestation.14 This is parenthetically the time of formation of the epidermal barrier, i.e., that skin structure critical for postnatal transition and survival.15,16
Although typically outside the purview of traditional dermatology, the infant at birth benefits from desirable surface characteristics, including skin suppleness, softness, and smoothness as well as pheromonal influences, which positively influence maternal bonding and caregiver support.1,17,18 This positive perception of infant skin emphasizes the skin–brain connection and cannot be dismissed from dermatological inquiries in the newborn period. The quintessential maternal–infant bonding experience in mammals, i.e., breast-feeding, involves intimate skin-to-skin contact between mother and infant and the production of milk by an ectodermal, skin-based glandular derivative.19
In addition to normal physiological development, pathophysiological events may arise in utero with subsequent need for medical intervention. Innate immune mechanisms to combat chorioamnionitis, for example, are important in the last trimester, to forestall systemic inflammatory responses in the fetus which carry long-term neurological sequelae.10,20,21 Birth trauma from intrauterine events such as amniotic bands or iatrogenic incidents may arise along with a panoply of gene defects, leading to congenital skin disorders. Later chapters detail specific dermatological conditions, which have their roots in utero. New tools of molecular investigation and the possibility of intrauterine therapies are exciting new fields of research. Finally, recognition of the body surface as a critical interface for receiving and delivering care transcends specific diseases and includes a plethora of important functions such as skin adhesion, monitoring, topical wound care, bathing, cleansing, emolliency, and microbiome support.1,22 All of these skin-based functions have their beginning in the transition of the fetus to the infant at birth.
Timing of embryonic and fetal development
Important morphologic events in intrauterine skin development are illustrated in Table 1.2. In Table 1.2, estimated gestational age (EGA) refers to the system used in basic embryology texts and by researchers to refer to the age of the fetus.24 In this system, fertilization occurs on day 1. However, the dating system used by obstetricians and other clinicians as a convenient method for staging pregnancy defines day 1 as the first day of the last menstrual period (LMP) and is synonymous with menstrual age.25 In this dating system, fertilization occurs on approximately day 14. Thus, a woman who is 14 weeks’ pregnant (LMP) is carrying a 12-week-old fetus (EGA).
TABLE 1.2
Landmarks in human fetal skin development relevant to prenatal diagnosis23
| Structure or event | Estimated gestational age (weeks) |
| Epidermal stratification and expression of K5, K14 and K1, K10 | 6 |
| Presence of melanocytes and Langerhans’ cells in the epidermis | 8 |
| Formation of complete hemidesmosomes, anchoring filaments, and anchoring fibrils | 8–10 |
| Formation of the nail primordium | 10 |
| Initiation of hair follicles | 12 |
| Initiation of eccrine sweat glands on the palms and soles | 10–12 |
| Delineation of papillary and reticular dermis | 11–12 |
| Formation of adipose tissue in hypodermis | 15 |
| Follicular keratinization | 15 |
| Interfollicular keratinization | 22–24 |
| Formation of eccrine sweat glands on the body | 24–26 |
(Adapted from Holbrook KA, Smith LT, Elias S. Prenatal diagnosis of genetic skin disease using fetal skin biopsy samples. Archives of Dermatology 1993; 129(11):1437–1454.)
From a functional point of view, fetal skin development can be divided into three temporally overlapping stages – organogenesis, histogenesis, and maturation26 – that correspond roughly to the embryonic period (0–60-plus days); the early fetal period (60 days to 5 months); and the late fetal period (5–9 months) of development. The first stage, organogenesis, involves the specification of ectoderm lateral to the neural plate to become epidermis and the allocation of subsets of mesenchymal and neural crest cells to become dermis. During this stage, embryonic ectoderm and mesoderm become physically apposed, and they initiate the signaling cross-talk necessary for basement membrane and subsequent skin appendage (hair, nail, and sweat gland) formation.
The second stage, histogenesis, is characterized by dramatic morphologic changes in the presumptive skin, including epidermal stratification, epidermal appendage involution and differentiation, mesenchymal subdivision of the dermis and hypodermis, and vascular neogenesis. The third stage, maturation, entails the functional evolution of these skin components, so that they provide adequate thermoregulatory capacity, surface tensile strength, and barrier function for postnatal survival in the harsh, arid, nonsterile extrauterine environment. The remainder of this chapter highlights selected events of structural development of the skin in utero along with important physiological and clinical correlates.
Epidermis
Embryonic development
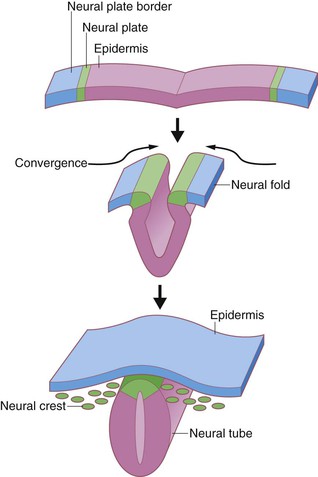
During the third week after fertilization, the human embryo undergoes gastrulation, a complex process of involution and cell redistribution that generates the three primary embryonic germ layers: endoderm, mesoderm, and ectoderm.24 Shortly after gastrulation, the ectoderm is further subdivided into neuroectoderm, a medial strip parallel to the long axis of the developing embryo, and presumptive epidermis on either side of this strip. Neurulation results in infolding of the embryonic ectoderm to become the neural tube and subsequent brain and spinal cord (Fig. 1.2). The extraembryonic ectoderm (lateral to the epidermis) becomes the amnion lining the amniotic sac. The early presumptive epidermis is a loosely associated single cell layer.29 By 6 weeks’ EGA (8 weeks’ LMP), the surface ectoderm covering most regions of the body already consists of basal cells and more superficial periderm cells (Fig. 1.3).30,31 The periderm layer is a transient embryonic layer that does not participate in the production of definitive epidermal progenitors. The presumptive epidermis at these early stages is not considered a true stratified epithelium.
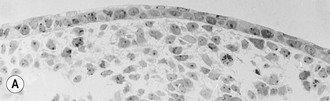
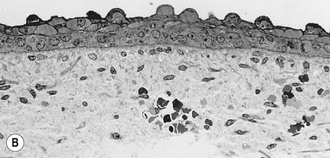
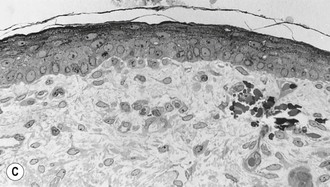
The basal cells of the embryonic epidermis display morphologic and biochemical features similar – but not identical – to basal cells of later developmental stages. Embryonic basal cells are slightly more columnar than later fetal basal cells and lack morphologically distinct hemidesmosomes.32,33 Matrix adhesion molecules critical for histogenesis and signal transduction, such as E- and P-cadherins and integrins β-1 and β-4, exhibit spatially and temporally coordinated expression in the developing epidermis.34 The keratin pair K8/K18, typically found in simple epithelial cells, is the first pair expressed in embryogenesis35 and may represent the oldest phylogenetic keratins.36 Keratins involved in higher order tonofilament formation such as K5/K14 can also be identified.
Periderm cells of the embryonic epidermis are larger and flatter than the underlying basal cells. As such, periderm cells have been termed a ‘pavement epithelium’.30,37 Apical surfaces in contact with the amniotic fluid are studded with microvilli. Their lateral surfaces in contact with adjacent peridermal cells are sealed with tight junctions, possibly precluding passive – but not active – diffusion of fluids across this outer layer of the embryo. Periderm cells, like the embryonic basal cells, express the stratified epithelial keratins K5 and K14, but also express simple epithelial keratins K8, K18, and K19.38,39 Towards the end of the second trimester these superficial cells are eventually sloughed.40
Early fetal development
By the end of 8 weeks’ gestation (10 weeks’ LMP), the basic components of most organ systems have been laid down and hematopoietic production has shifted to the bone marrow. This marks the classic division between embryonic and fetal development, and it corresponds to initial epidermal stratification and the formation of the third ‘intermediate’ layer between the two pre-existing cell layers (Fig. 1.3). Cells in the intermediate layer of the early fetal epidermis express the K1/10 skin differentiation-type keratin markers, as well as the desmosomal protein desmoglein 3, which is also known as the pemphigus vulgaris antigen.41,42 Moreover, intermediate filaments and desmosomal junctions are more abundant in this layer than in the basal or periderm layers. In contrast to the spinous cells of the mature nonwounded epidermis, cells within the intermediate layer remain highly proliferative.43,44 Over the next several weeks, more layers are gradually added to this intermediate zone of the developing epidermis, such that by 22–24 weeks’ EGA, the epidermis contains four to five layers in addition to terminal differentiation of the periderm with formation of a cross-linked cornified envelope (Fig. 1.4).45
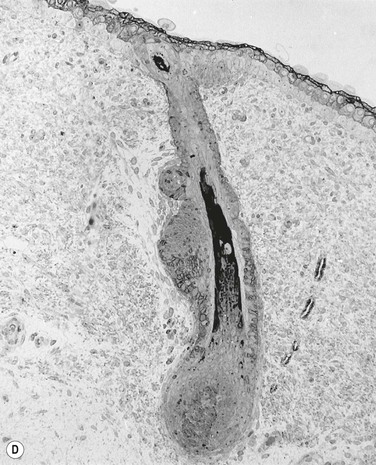
After the onset of stratification, the basal layer also displays characteristic morphologic and biochemical changes. Basal cells become more cuboidal and begin to synthesize other keratin peptides, including K6, K8, K19, and the K6/K16 hyperproliferative pair.38,39 This latter keratin pair is not normally expressed in mature interfollicular epidermis but is upregulated in response to wounding and hyperproliferative conditions.46 During early fetal development, the basal cell layer also begins to express the hemidesmosomal proteins BPA1 and BPA2, and to secrete collagen types V and VII, the latter being the major component of the anchoring fibrils of the dermis.41,47–49 DNA-labeling studies indicate that by 80–90 days’ EGA, a distinct subset of slow-cycling cells exists within the basal cell population, suggesting that an epidermal stem cell population has already been set aside at these early stages.44
Late fetal development
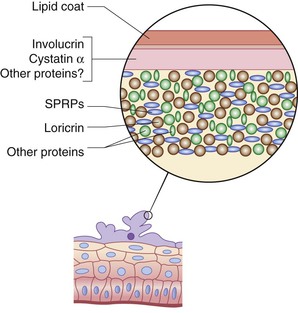
Maturation of the epidermis during late fetal development is characterized by the generation of granular and stratum corneum (SC) layers, the formation of a water-impermeable barrier, and the sloughing of the periderm. Keratinization, the terminal differentiation seen in the stratum granulosum (SG) and SC, is initiated first in the skin appendages between 11 and 15 weeks’ EGA, and extends to the interfollicular epidermis from about 22–24 weeks’ EGA.38,39 During the third trimester, the cornified cell layers increase in number, aiding in the formation of a barrier.
The prenatal epidermal water permeability barrier was previously thought to be derived almost entirely from lipid secreted from cells of the outer SG and processed to highly hydrophobic species in the SC interstices, controlled not only by the intrinsic program of epidermal barrier development but also by prenatal exposure to fetal or maternal hormones,50 nutrient gradients,51 or by air exposure at birth. In postnatal humans and rodents, this epidermal permeability barrier is formed by polar lipids secreted from the SG that are then processed into impermeant lamellar bilayers.52–56 In contrast, since prior freeze-fracture electron microscopy studies showed only discontinuous tight junction (TJ) strands in adult mouse epidermis,57 it was widely thought that in contrast to amphibian skin or other ‘tight’ mammalian epithelium (such as kidney), TJ played only a minor role in the epidermal water permeability barrier.
Although congenital human skin diseases caused by mutations in TJ are rare,58 common diseases such as atopic dermatitis have been linked with acquired TJ dysfunction.59 Further, studies show that claudin-1 deficient mice suffer barrier defects leading to death soon after birth.60 Involucrin-Cldn6 (Inv-Cldn6) transgenic mice also display skin barrier defects, the severity of which is dependent upon the level of Cldn6 over-expression.61 These results suggest that TJ also plays an important role in forming the epidermal permeability barrier during the prenatal period, or in regulating the subsequent development of the lipid barrier.
Clinical relevance
Gross defects in early epidermal specification and organogenesis are rarely observed in the neonate, probably because they are incompatible with fetal survival. Using mice as an animal model system, researchers demonstrated that obliteration of the p63 gene precludes the formation of most multilayered epithelia in the body, leading to perinatal lethality due to loss of skin barrier function. Humans who carry mutations in this gene still retain some functionality and therefore display less severe alterations in their epidermis and appendages (see below).
In contrast, congenital defects in epidermal maturation are not uncommon, as they do not usually impinge on in utero survival. Lamellar ichthyosis (see Chapter 19) is usually inherited in an autosomal recessive manner and in 30% of patients is caused by mutations in the gene encoding epidermal transglutaminase,62–64 the enzyme that cross-links submembranous proteins to form the insoluble cornified envelope of the SC. In its absence, large, dark polygonal scales form over the entire body, and at birth, the infant may be transiently wrapped in a waxy, collodion-like membrane.65 A similar clinical presentation can be seen in patients homozygous for mutations in the ABCA12 gene, which encodes an ATP-binding cassette thought to be important for lipid trafficking across keratinocyte membranes. Infants with the more severe ‘harlequin ichthyosis’ (see Chapter 19) are born encased in armor-like, thickened, adherent SC which cracks upon exposure to air.66–68 This extreme variant also appears to be due to mutations in the ABCA12 gene.
In contrast to the permanent manifestations of genetic defects, the inadequate epidermal keratinization and maturation of the premature epidermis are transient. Immaturity of the SC, especially in infants born before 28 weeks’ EGA (30 weeks’ LMP), places these neonates at increased risk for dehydration, excessive penetration of topical drugs or other chemicals, and infection from organisms newly colonizing the skin (see Chapters 4 and 5).69–74 In general, even full-term newborns display a somewhat reduced barrier function, and continued maturation occurs over the first few weeks of life, such that by 3 weeks of age, the newborn’s SC is structurally and functionally equivalent to that of the adult; maturation is accelerated in the premature infant, although the duration may be longer in extremely premature infants.69,75
Specialized cells within the epidermis
Two major immigrant cells – melanocytes and Langerhans’ cells – populate the epidermis during early embryonic development. Melanocytes are derived from a subset of neuroectoderm cells, the neural crest, which forms along the dorsal neural tube and gives rise to a variety of cell types, including many tissues of the face and peripheral autonomic neurons.27 Neural crest cells destined to become melanocytes migrate away from the neural tube within the mesenchyme subjacent to the presumptive epidermis. They migrate as semicoherent clones laterally and then ventrally around the trunk to the thoraco-abdominal midline, anteriorly over the scalp and face, and distally along the extremities. Postnatally, the embryonic paths taken by these partially coherent clones can be readily visualized in patients with banded pigmentary dyscrasias following Blaschko’s lines, such as the disorders classified as hypomelanosis of Ito, and linear and whorled hypermelanosis (see Chapters 23 and 24).76,77
Melanocytes are first detected within the epidermis of the human embryo at approximately 50 days’ EGA, recognized by their dendritic morphology and their specific immunoreactivity.78 Even at these early developmental timepoints, the density of melanocytes is quite high (1000 cells/mm2).79 The density increases further around the time of epidermal stratification (80–90 days’ EGA) and initiation of appendageal development. Between 3 and 4 months EGA, depending on body site and the race of the fetus, melanin (visible pigment) production becomes detectable, and by 5 months, melanocytes begin transferring melanosomes to the keratinocytes, a process that will continue after birth.80–82 Although all melanocytes are in place at birth and melanogenesis is well under way, the skin of the newborn infant is not fully pigmented and will continue to darken over the first several months. This is most apparent in individuals with darker skin.
Langerhans’ cells, the other major immigrant population, are detectable within the epidermis by 40 days’ EGA.83 Similar to melanocytes, the early embryonic Langerhans’ cells do not yet possess the specialized organelles characteristic of mature cells, but can be distinguished from other epidermal cells by their dendritic morphology, immunopositive reaction for the HLA-DR surface antigen, and high levels of ATPase activity. After the transition from embryo to fetus, they begin to express the CD1 antigen on their surface and to produce characteristic granules of mature Langerhans’ cells.83,84 Although the extent of dendritic processes from individual Langerhans’ cells increases during the second trimester, the total number of cells remains low and only increases to typical adult numbers in the third trimester.85
Another distinct subset of cells within the basal cell layer are Merkel cells, which are highly innervated neuroendocrine cells involved in mechanoreception. Merkel cells can be round or dendritic, and are found at particularly high densities in volar skin. They are frequently associated with epidermal appendageal structures and are occasionally detected within the dermis. Their distinguishing morphologic and immunohistochemical features are cytoplasmic dense-core granules, keratin 18, and neuropeptide expression, which can be detected as early as 8–12 weeks’ EGA in palmoplantar epidermis and at slightly later times in interfollicular skin.86 Recent keratin expression data, as well as transplant studies, suggest that Merkel cells are derived from pluripotent keratinocytes, rather than neural progenitors such as neural crest, but the results are not conclusive.86–88
Clinical relevance
Many clinical defects are known to affect normal pigmentation. Defects in melanoblast migration, proliferation, and/or survival occur in several clinical syndromes, and many of the genetic mutations responsible for these defects have been identified (see Chapter 23). Failure of an adequate number of melanoblasts to completely supply distal points on their embryonic migration path occurs in the different types of Waardenburg syndrome, as well as in piebaldism, resulting in depigmented patches on the central forehead, central abdomen, and extremities. These defects are associated with mutations in several different genes, including genes encoding transcription factors, such as Pax3 and MITF, as well as membrane receptors and their ligands, such as endothelin 3, endothelin-receptor B, and c-kit.89–91 In albinism, however, melanocyte development is normal, but production of pigment or melanin is inadequate. The most severe form of oculocutaneous albinism results from null mutations in the gene encoding tyrosinase, the rate-limiting enzyme in the production of melanin. Less severe forms of albinism are caused by mutations in tyrosinase alleles, which lead to partial loss of function, as well as by mutations in other genes encoding proteins important in melanin assembly in melanosomes or transport.92
Nerves and vasculature
Development of the cutaneous innervation closely parallels that of the vascular system in terms of its pattern, rate of maturation, and organization. Nerves of the skin consist of somatic sensory and sympathetic autonomic fibers, which are predominantly small and unmyelinated. Localized somatosensory reflexes can first be elicited using von Frey hairs in the perioral area of the 7.5-week fetus, at which time free nerve endings can be visualized beneath the epithelium.7,93,94 The growing sensory nerve endings, therefore, are presumably sensitive to mechanical stimuli transmitted through the fetal skin. Cauna and Mannan have suggested that the developing nerve plexus as a whole functions as a receptor in early development to be superseded by more definitive end organs later in gestation.95
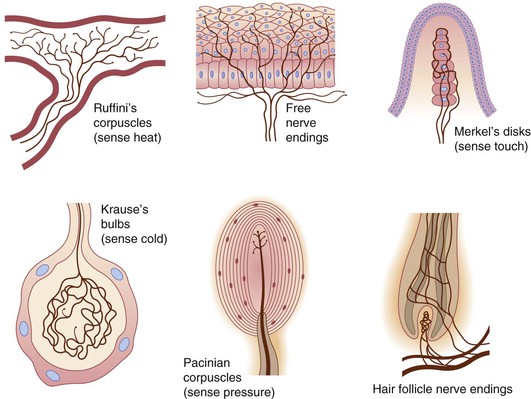
Various specialized nerve endings have been well-documented in embryonic and fetal skin (Fig. 1.5).7 Work by Paus, Slominski, and others has highlighted novel neuroimmunoendocrine functions of postnatal skin.99,100 The stress hormone cortisol, for example, can be manufactured de novo by the human hair follicle101 and is present on preterm SC.102 These exciting new developments place a premium on better understanding the skin–brain connection during normal development, as well as in conditions such as the neurocristopathies, neurofibromatosis, incontinentia pigmenti, tuberous sclerosis and other diseases highlighted in later chapters. The importance of early fetal somatic (i.e., muscle) innervation to normal skin morphogenesis is the recent demonstration that lack of the fetus-specific component of the acetylcholine receptor produces the extensive dermal webbing seen in multiple pterygium syndrome.103
The development of the cutaneous vascular system is dynamic and dependent on gestational age, body site, and function, among other factors. Vessels of the endoderm–mesoderm interface form through the in-situ differentiation of endothelial cells (vasculogenesis).104,105 Originally, they form horizontal plexuses within the subpapillary and deep reticular dermis, which are interconnected by groups of vertical vessels. This vascular framework has been elegantly reconstructed by the use of computer graphics to illustrate the complexity that already exists by 45–50 days’ EGA.106 Such structure does not remain constant even throughout fetal life, but varies depending on the body region and gestational age, as well as on the presence of hair follicles and glands that may require an increased blood supply. Furthermore, vascular emergence and development correlate directly with the particular tissue, determined specifically by the influences of pressure and function.
Regional variation also depends on gestational age. Blood vessels have been identified in fetal skin as early as 9 weeks’ EGA. At this stage, they help delineate the dermal–hypodermal junction. By 3 months, the distinct horizontal and vertical networks have formed. And by 5 months, vasculogenesis has largely ceased and the formation of the complex vascular plexus is initiated by angiogenesis, the budding and migration of endothelium from pre-existing vessels. With increasing gestational age, the superficial architecture becomes more organized, culminating at birth in an extensive capillary network responsible for the skin redness often observed in the newborn. Within the first few postnatal months the complexity decreases as skin surface area increases, lanugo hairs are lost, and sebaceous gland activity decreases. It is during this time that the rate of skin growth is greatest. By approximately 3 months of age, the vascular patterns more closely resemble those of the adult.
Not only do the number and caliber of the blood vessels change over time, so too does the direction of blood flow. Considering the dynamic nature of this circulatory system, it is not surprising that, of the malformations and tumors seen in newborns, vascular anomalies are the most common.
Errors in neurovascular morphogenesis likely lead to several relatively common syndromes such as Klippel–Trenaunay, Sturge–Weber and PHACE syndromes. See Chapters 21 and 22 for further discussion of these topics.
Dermis and subcutis
Overview
The fully developed dermis is characterized by complex interwoven collagen and elastic fibers enmeshed in a proteoglycan matrix. Fibroblasts, mast cells, and macrophages are scattered throughout the dermis, and nerve fibers and vascular networks course through it, dividing it into distinct domains. In contrast, the embryonic dermis is quite cellular and amorphous, lacking organized extracellular fibers. Embryonic mesenchymal cells capable of differentiating into a wide variety of cell types are embedded in a highly hydrated gel, rich in hyaluronic acid. Moreover, only a few nerve fibers have reached this peripheral location, and vessels have not evolved into their mature patterns. During the course of fetal development, this so-called cellular dermis, which is conducive to cell migration and tissue remodeling, is transformed into the fibrillar dermis of the adult, which provides increased strength, resilience, and structural support.107
Embryonic dermal development
The specification and allocation of dermal mesenchymal cells are rather complex and not well understood. The cell of origin for the presumptive dermis depends on its anatomic location. The dermis of the face is derived from neural crest cells; that of the dorsal trunk is derived from the dermatomyotome portion of the differentiated somite; and the dermis of the limbs is derived from the lateral plate (somatic) mesoderm.107,108 Regional patterning of the skin and differences in the type and quality of the epidermal appendages produced in the older fetus might in part reflect these early differences in dermal cell precursors. In addition, signaling from adjacent tissues plays a critical role.109,110
By 6–8 weeks’ EGA, the presumptive dermal cells already underlie the epidermis. However, there is, as yet, no sharp demarcation between cells giving rise to skin dermis and those giving rise to musculoskeletal elements. Electron microscopic (EM) studies of the presumptive dermis at these stages demonstrate fine filaments, but rarely fibers.111 Although most protein components of collagen fibers and some microfibrillar components of elastin fibers (fibrillin) are synthesized by the embryonic dermal cells, the proteins are not yet assembled into large, rigid fibers.49,112 Moreover, the ratio of collagen III to collagen I is 3 : 1, the reverse of that in the adult.49,113,114
Fetal dermal development
After embryonic–fetal transition at 60 days, the presumptive dermis is distinguishable from the underlying skeletal condensations. Moreover, within the dermis, there is a progressive change in matrix organization and cell morphology, such that by 12–15 weeks, the fine interwoven mesh of the papillary dermis adjacent to the epidermis can be distinguished from the deeper, more fibrillar reticular dermis.49,112 Large collagen fibers accumulate in the reticular dermis during the second and third trimesters. Definitive elastin fibers first become detectable by EM studies around 22–24 weeks’ EGA,115 although both the microfibrillar protein fibrillin and the microfibrillar structures, which are morphologically similar to elastin-associated microfibrils of the adult, can be detected at earlier stages.112 By the end of gestation, the dermis is thick and well organized, but is still much thinner than in the adult and has a higher water content, reminiscent of the fetal dermis. Dermal maturation is marked by increasing tensile strength and the transition from a nonscarring to a scarring response after wounding. Thus, fetal skin biopsies tend to heal with little evidence of the surgical event. This has obvious clinical implications, and the molecular controls critical for nonscarring fetal wound healing are an area of active research (Table 1.3).5,116,117
TABLE 1.3
Comparison of fetal wound healing profile with postnatal wound healing5
| Growth factors | Fetal regenerative phenotype | Postnatal scar formation |
| bFGF | Lower | Higher |
| PDGF | Lower | Higher |
| VEGF | Higher | Lower |
| TGF-β | ||
| TGF-β1 | Low levels | High levels |
| TGF-β2 | Low levels | High levels |
| TGF-β3 | High levels | Low levels |
| Inflammatory response | ||
| Inflammatory cell | Minimal | High levels leukocytes, macrophages, mast cells infiltrate |
| Cytokines | ||
| Proinflammatory: IL-6, IL-8 | Low levels | High levels |
| Anti-inflammatory: IL-10 | High levels | Low levels |
| Extracellular matrix | ||
| Collagen | ||
| Histology | Fine, reticular weave | Thick, rope-like bundles |
| Type III collagen | High levels | Low levels |
| Deposition | Immediate | Delayed |
| Cross-linking | Low levels | High levels |
| TGF-β1-stimulated deposition | Absent | Present |
| Hyaluronan | ||
| Expression | High levels – Persistent expression | Low levels – Transient expression |
| Molecular weight | High | Low |
| HA receptors (fibroblast) | High levels | Low levels |
| Mechanical force | ||
| Myofibroblast (day 14) | Absent | Present |
| Stem cells | ||
| MSC | High levels | Lower levels |
| Dot cells | Present | Absent |
bFGF, basic fibroblast growth factor; HA, hyaluronan; MSC, mesenchymal stem cell; PDGF, platelet-derived growth factor; TGF, transforming growth factor; VEGF, vascular endothelial growth factor.
(Adapted from Leung A, Crombleholme TM, Keswani SG. Fetal wound healing: implications for minimal scar formation. Current Opinion in Pediatrics 2012; 24(3):371–378.)
Clinical relevance
Congenital defects in the specification and development of the dermis are probably incompatible with survival to term, although there are a few exceptions. Infants with restrictive dermopathy disorder, which is characterized by a thin, flat dermis, lack of elastic tissue fibers, and shortened appendageal structures, do survive to birth but then die in the neonatal period, partly because of insufficient elaboration of the dermis.118,119 This disorder is caused by mutations in either the LaminA gene or the gene encoding the LaminA processing enzyme. Another syndrome characterized by inadequate dermal development is Goltz syndrome (focal dermal hypoplasia).65,120 This is an X-linked dominant condition caused by mutations in the PORCN gene, in which most males who inherit the mutation on their single X chromosome die in utero. In contrast, females are functional mosaics as a result of random X-inactivation early in embryogenesis, and those with Goltz syndrome display areas of dermal hypoplasia where the mutant X is active. These bands of dermal hypoplasia follow Blaschko’s lines and alternate with bands of normal dermal development where the normal X is active.121,122
Development of the hypodermis and adipose tissue
The hypodermis can be delineated by 50–60 days’ EGA.26 It is a distinct region that is separated from the overlying cellular dermis by a plane of thin-walled vessels. Toward the end of the first trimester, the sparse matrix of the hypodermis can be distinguished morphologically from the slightly denser, more fibrous matrix of the dermis.107,123 In the second trimester, mesenchymally derived preadipocytes begin to differentiate and accumulate lipids124 and by the third trimester, the more mature adipocytes are aggregated into large lobules of fat divided by fibrous septa. In addition to a passive fuel reserve for the body, recent evidence supports an active endocrine role for adipose tissue with effects on vascular and immune function.125 An example is the gene that encodes leptin, whose abnormal regulation has been implicated in the pathogenesis of obesity.
Combined dermoepidermal structures
Dermoepidermal junction
The dermoepidermal junction (DEJ) is the region where the epidermis and dermis abut. In the broadest definition, it includes the specialized extracellular matrix on which the basal keratinocytes sit, known as the basement membrane, as well as the basal-most portion of the basal cells and the superficial-most portion of the dermis. Importantly, both dermal and epidermal compartments contribute to the molecular synthesis, assembly, and integration of this region.
A simple basement membrane, separating the dermis and epidermis, can be discerned as early as 8 weeks’ EGA. The basic protein constituents common to all basement membranes can already be detected immunohistochemically at this stage.32,47,126 These include collagen IV, laminin, and heparin sulfate and proteoglycans.
Specialized components of the DEJ do not appear until after the embryonic–fetal transition, around the time of initial epidermal stratification.32,47,126 With a few exceptions, all basement membrane antigens are in place by the end of the first trimester.26 As discussed, the α6 and β4 integrin subunits are expressed quite early by embryonic basal cells.32 However, they do not become localized to the basal surface until after 9.5 weeks, which is coincident with the time when bullous pemphigoid antigens are first detected immunohistochemically and hemidesmosomes are recognized ultrastructurally.32,33,47,127 Similarly, anchoring filaments and anchoring fibrils, the basement membrane components that mediate basal cell attachment to extracellular matrix, are recognizable by 9 weeks’ EGA.26,33 Collagen VII, the anchoring fibril protein, is detected slightly earlier, at 8 weeks.33 Recent experimental data have delineated many of the molecular interactions crucial for connecting the cytoskeletal networks of the basal cells with the extracellular filamentous networks important in matrix adhesion (Fig. 1.6 ) (see Chapters 10 and 11).
) (see Chapters 10 and 11).
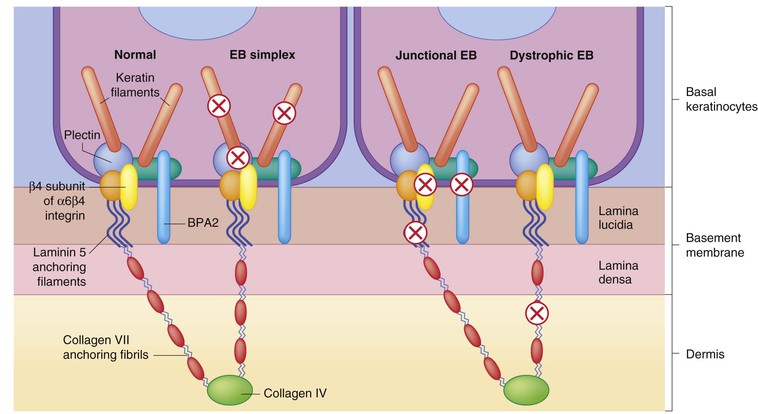
Clinical applications
Several congenital skin blistering diseases result from mutations in genes encoding DEJ components (see Chapters 10 and 11).128 The severity of the disorder, the exact plane of tissue separation, and the involvement of nonskin tissues depend in part upon which proteins are mutated. Because these blistering disorders are associated with a high postnatal morbidity and mortality they are frequent candidates for prenatal testing, identifying the responsible genetic mutation by chorionic villus sampling (CVS) or amniocentesis.
Development of appendages
Skin appendages (hair, nails, sweat and mammary glands in mammals, and feathers and scales in birds and reptiles) all consist of two distinct components: an epidermal component that elaborates the differentiated end-product, such as the hair or nail, and the dermal component that regulates specification and differentiation of the appendage. Fetal development of these structures depends on rigidly choreographed, collaborative interactions between early epidermis and dermis.107,129,130 Defects in dermal induction or specification of the overlying ectoderm, or in the ectoderm’s responses to these instructions, result in aberrant development, as has been demonstrated in genetic studies and transplant experiments in animal model systems.107,129–131 Moreover, the demonstration that defects in human homologs of mouse hairless, LMX1B, and tabby genes result in clinically significant developmental abnormalities in humans confirms the relevance of such animal studies to our understanding of human skin appendage development.132–136
Hair follicle and sebaceous gland development
Hair follicle formation begins on the head and then spreads caudally and ventrally in waves, resulting in regularly spaced rows and whorls of follicles.137,138 The first morphologic evidence of follicle formation in humans, the ectodermal placode, is the focal crowding of small groups of basal keratinocytes at regularly spaced intervals, starting between 75 and 80 days on the face and scalp.137–140 Slightly later in development, mesenchymal cell clusters are observed beneath these ectodermal placodes. Although morphologically similar to other dermal fibroblasts, these clustered mesenchymal cells are biochemically distinguishable based on their continued expression of certain molecular markers, such as nerve growth factor receptor (NGFR).26 On the trunk at approximately 80 days’ EGA, a cluster of basal epidermal cells thickens and begins to bud downward into the dermis, forming the early hair germs.140,141 Transplant studies in other species indicate that ectodermal budding requires an induction signal from the underlying mesenchymal cells. The cells of the early ectodermal bud or placode then respond with their own signal, which elicits a second mesenchymal signal. This second signal directs the species-specific type of mesenchymal appendage that will ultimately develop.107,129
The next stage of hair development involves further proliferation and resulting downward elongation of the ectodermal bud, forming the so-called hair peg.138 At 12–14 weeks’ EGA, the hair peg develops a widened bulb at its base that flattens and then invaginates, engulfing the subjacent clustered mesenchymal cells, which become the follicular dermal papilla. In addition to the widened bulb at the base, two other bulges form along the length of the developing follicle, which is now termed the bulbous hair peg (see Fig. 1.3).138,142 The uppermost bulge is the presumptive sebaceous gland, and the middle bulge, which forms at approximately one-third the distance from the follicular base, is the site of the future insertion of the arrector pili muscle and is one location of multipotent stem cells, which give rise to all the progenitors necessary for regeneration of the lower portion of the follicle during postnatal follicular cycling, as well as to cells capable of replenishing the overlying epidermal covering in the event of extensive surface wounds or burns.143
Maturation of the hair peg into a definitive follicle is a complex process involving the formation of a patent hair canal and the elaboration of at least six distinct concentric rings of cells.144 The most peripheral ring of ectodermal cells makes up the outer root sheath, whose upper portion is continuous with the interfollicular epidermis and undergoes a similar process of keratinization. The lower portion of the outer root sheath, in contrast, does not form a granular layer or classic SC. The inner root sheath forms just internal to the outer root sheath. The cells in this sheath do form a granular layer through the keratin proteins, and keratin-aggregating products produced here differ from those produced by the normal epidermis. Cells in this inner root sheath arise from self-renewing progenitor cells at the base of the follicle, which differentiate as they move upward toward the skin surface surrounding the hair shaft. Likewise, the three internal concentric layers of the hair shaft – cuticle, cortex, and medulla (from outer to inner) – arise from the matrix cells at the base of the follicle. These deep matrix cells sit on the basement membrane ‘mat,’ along the concavity of the hair follicle invagination in close proximity to the dermal papillae mesenchymal cells.
By 19–21 weeks’ EGA, the hair canal has fully formed and the scalp hairs are visible just above the surface of the fetal epidermis.137,145,146 They continue to lengthen until 24–28 weeks, when they shift from the active growing phase (anagen) to the short-lived degenerative phase (catagen) and then to the resting phase (telogen).26,147 They then re-enter the active growing stage (second anagen), and the first wave of hairs is shed into the amniotic fluid as the new hairs grow out. Cycling through active and inactive phases continues for all hairs throughout the life of an individual,148 although cycles for individual hairs become asynchronous postnatally. The maintenance of a tight anatomic relationship between dermal papilla cells and the cycling ectodermal portion of the hair follicle is critical for follicular self-renewal, and the inability to maintain this relationship results in a form of inherited alopecia, in which hair neogenesis is normal but, after the first resting phase, cycling is aberrant.132
Perinatally the second wave of fine lanugo hairs is shed. With subsequent cycles, hairs increase in diameter and coarseness, forming first vellus and then adult-type terminal hair shafts on the scalp and brow.147 During adolescence, vellus hairs of androgen-sensitive areas undergo a similar transition to terminal-type hair follicles.
Sebaceous gland maturation occurs in parallel with that of the follicle proper and begins between 13 and 16 weeks’ EGA.149 Lipogenic cells produced by the outer proliferative layer of the sebaceous gland progressively accumulate lipid/sebum until they terminally differentiate, which results in their disintegration and the release of their products into the upper portion of the newly formed hair canal.150,151 The synthesis and secretion of sebum is accelerated in the second and third trimesters under the influence of maternal steroids and/or fetal adrenal dihydroepidandrosterone.152–154 Of note, the first evidence of the human epidermal permeability barrier in utero is in the vicinity of the pilosebaceous unit.15 Sebaceous lipids are also a prominent component of vernix caseosa.155 Fetal corneocytes associated with vernix may also derive from the pilosebaceous unit. Sebaceous gland hyperplasia and activity generally becomes quiescent during the first few postnatal months of life but may persist in cases of infantile acne (see Chapter 7).
Nail development
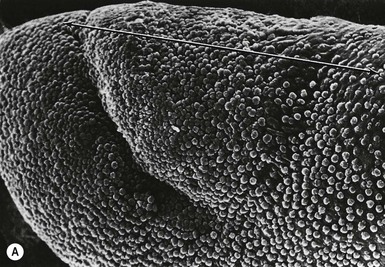
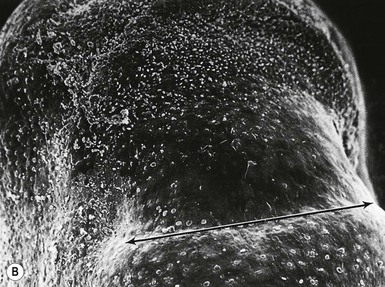
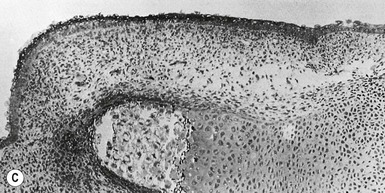
The first evidence of nail formation is delineation of the flat surface of the future nail bed on the dorsal digit tip at 8–10 weeks,112,156 slightly earlier than the initiation of hair follicle development (Fig. 1.7 ). Along the proximal boundary of the early nail field a wedge of ectoderm buds inward at an oblique angle to the surface, forming the proximal nail fold. The presumptive nail matrix cells, which will give rise to the differentiated nail plate, reside on the ventral (deeper) side of the proximal invagination. At around 11 weeks, the dorsal surface of the nail bed begins to keratinize, a process similar to subsequent keratinization of the embryonic epidermis.157–159 In the 4th month, the definitive nail plate grows out distally from the proximal fold, replacing the embryonic cornified layers, and completely covers the nail bed by the 5th month. Keratinization of the nail resembles that of the epidermis except that nail terminal differentiation, like hair shaft differentiation, involves the synthesis of distinct keratins and keratin-aggregating proteins normally not expressed in epidermis.36 The keratins found in hairs and nails provide greater structural stability and rigidity than those in epidermis.
). Along the proximal boundary of the early nail field a wedge of ectoderm buds inward at an oblique angle to the surface, forming the proximal nail fold. The presumptive nail matrix cells, which will give rise to the differentiated nail plate, reside on the ventral (deeper) side of the proximal invagination. At around 11 weeks, the dorsal surface of the nail bed begins to keratinize, a process similar to subsequent keratinization of the embryonic epidermis.157–159 In the 4th month, the definitive nail plate grows out distally from the proximal fold, replacing the embryonic cornified layers, and completely covers the nail bed by the 5th month. Keratinization of the nail resembles that of the epidermis except that nail terminal differentiation, like hair shaft differentiation, involves the synthesis of distinct keratins and keratin-aggregating proteins normally not expressed in epidermis.36 The keratins found in hairs and nails provide greater structural stability and rigidity than those in epidermis.
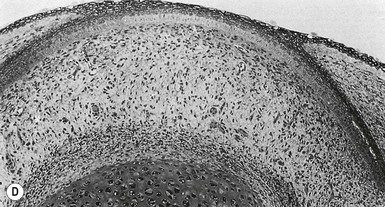
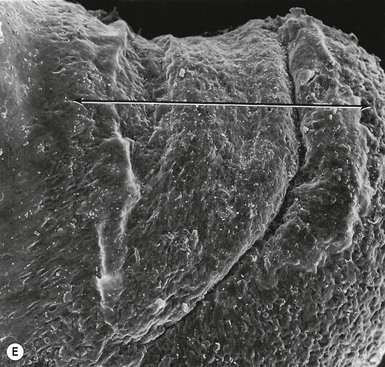
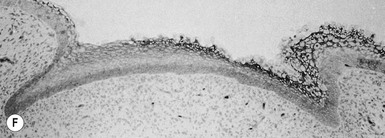
Eccrine and apocrine sweat gland development
The first morphologic indicators of palmoplantar eccrine gland development are the formation of large mesenchymal bulges or pads on the volar surfaces of the hands and feet between 55 and 65 days’ EGA, and the induction of parallel ectodermal ridges overlying these bulges between 12 and 14 weeks (Fig. 1.8).160,161 The curves and whorls that these ridges adopt are intimately related to the size and shape of the embryonic volar pads and result in the characteristic dermatoglyphic patterns, or ‘fingerprints,’ which can be visualized on the surface of the digit tips in the fifth month.160,162 In contrast to those of most other animal species, the volar mesenchymal pads in humans regress by the third trimester.
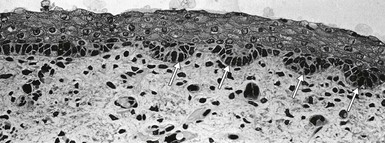
Individual eccrine gland primordia bud at regularly spaced intervals along the ectodermal ridges, elongating as cords of cells into the pad mesenchyme. By 16 weeks, the glandular regions at the terminal portion of this downgrowth have formed, and the secretory and myoepithelial components become discernible. Canalization of the dermal components of the glands occurs by loss of desmosome adhesion along the innermost ectodermal surfaces with maintenance of lateral adhesion between cells of the duct and gland walls. This process is complete by 16 weeks, whereas the opening of the epidermal portion of the duct by vesicular fusion and autolysis and keratinization of the wall are not complete until 22 weeks’ EGA.163,164 Although the primary ectodermal ridges are established quite early in embryonic development, secondary ridges form at later stages and the complexity of the undulating DEJ increases further at late fetal stages and postnatally with the formation of dermal papillae protruding into the overlying epidermis.
In contrast to volar eccrine glands, interfollicular eccrine glands and apocrine glands do not begin budding until the 5th month of gestation.145,165 Apocrine sweat glands, like sebaceous glands, usually bud from the upper portion of a hair follicle, whereas eccrine sweat glands arise independently. Over the next several weeks, the glandular cords of cells elongate. By 7 months’ EGA, the clear cells and mucin-secreting dark cells characteristic of apocrine glands are distinguishable. At birth, the secretory portions of nonvolar sweat glands remain high in the dermis but postnatally extend progressively down to the subcutis. The apocrine gland functions transiently in the third trimester and then becomes quiescent in the neonate,166,167 whereas the eccrine gland does not appear to function in utero but progressively reaches functional maturity postnatally.168
Ectodermal appendages: clinical relevance
Genetic studies in mice have suggested that mutations in the genes that direct early regional patterning of the mammalian embryo can have a profound impact on later skin appendage specification and differentiation.109,110,169,170
One such gene, LMX1B, which encodes a homeobox-containing transcription factor, acts several stages before the initiation of appendageal downgrowths and is important in distal limb patterning.169 Experimental ablation of LMX1B function in mice results in the transformation of dorsal limb musculoskeletal elements and dermal structures to a more ventral (volar side) phenotype. Viewed from the side, Lmx1b mutant mouse limbs appear perfectly symmetrical, with the paw pads present on both the ventral and dorsal aspects. Mutations in LMX1B have been observed in at least some people with the autosomal dominant disease nail–patella syndrome.133 These patients display a much less dramatic limb phenotype, characterized by aberrant or absent development of the nails and patellae (elbows). This milder effect on dorsal limb structures reflects the fact that individuals with nail–patella syndrome are heterozygotes. Thus, they carry a single defective copy of LMX1B, together with a wild-type copy, resulting in only partial loss of gene function.
Several other genes have been identified that appear to regulate skin appendage formation. Positional cloning strategies have been used to identify genes affected in several different types of ectodermal dysplasias, a heterogeneous group of disorders defined by their involvement of hair, nails, glands, and/or teeth. The most common type of ectodermal dysplasia, anhidrotic (hypohidrotic) ectodermal dysplasia, is caused by mutations in the EDA or EDAR genes, which encode a ligand/receptor pair related to TNF signaling components.136 The gene encoding MSX1, another homeobox-containing transcription factor, has been shown to be mutated in familial tooth agenesis, which affects not only tooth development but also the formation of nails and hair.171,172
Studies in mice suggest that other classic embryonic patterning genes play a direct role in epidermal appendage development, including genes encoding components of the Notched, Wnt, and Sonic hedgehog signaling pathways.130,173 The importance of such genes in human skin homeostasis has already been demonstrated by the finding that PATCHED is the tumor suppressor gene mutated in nevoid basal cell carcinoma (Gorlin) syndrome, and that it and other genes of the pathway are frequently mutated in spontaneous basal cell carcinomas.174–176 Indeed, several genes involved in tumor formation are critical regulators during embryonic development. Finally, it is important to consider potential teratogenic effects on normal skin appendage development. Dilantin, for example, can cause broadening of the nail associated with distortions in the underlying distal phalange.
Clinical paradigms
Innate immune function
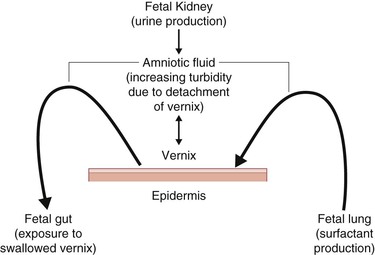
A critical intrauterine function of the skin is the provision of innate immune protection from exogenous infection. Preterm delivery is highly associated with ascending infection from the vaginal vault and subsequent chorioamnionitis. When the infection cannot be confined to the amniotic sac, the fetus responds with a well-coordinated systemic inflammatory response linked to long-term neurological sequelae.10,21,22 During the last trimester, increasing levels of pulmonary surfactant are present in the amniotic fluid as measured by lung lamellar body counts.177 Significantly, analysis of these lamellar bodies show a paucity of contamination with lamellar bodies derived from the epidermis. Vernix caseosa, produced in part by the pilosebaceous apparatus, is present on the skin surface in the last trimester, interacts with pulmonary surfactant with subsequent detachment,178 and is swallowed by the fetus with potential effects on the developing gut (Fig. 1.9).179 Pulmonary surfactant and vernix caseosa both contain significant innate immune modulators and provide a first line of defense against microbial invasion of the amniotic fluid.10,180
Prenatal diagnosis of congenital skin disorders
A number of inherited skin disorders are compatible with in utero survival but are life-threatening or result in severe morbidity after birth. Often, these disorders can be diagnosed during the first or second trimesters of pregnancy. Box 1.1 lists a number of diagnostic modalities for detecting skin diseases prenatally. Candidates for prenatal testing include those fetuses with an affected sibling or other family member. Importantly, the need for prenatal testing depends on the familial relationship, the mode of inheritance of the disorder in question, and in some cases, the sex of the fetus. DNA from parents and from both affected and unaffected siblings should be analyzed before conception to determine the exact mutational event responsible for the disorder in the relevant pedigree, and the likelihood that the fetus will in fact inherit the disease.188 With this in mind, prenatal and genetic counseling should be a critical component of early interventional care of infants affected with severe genodermatoses.183
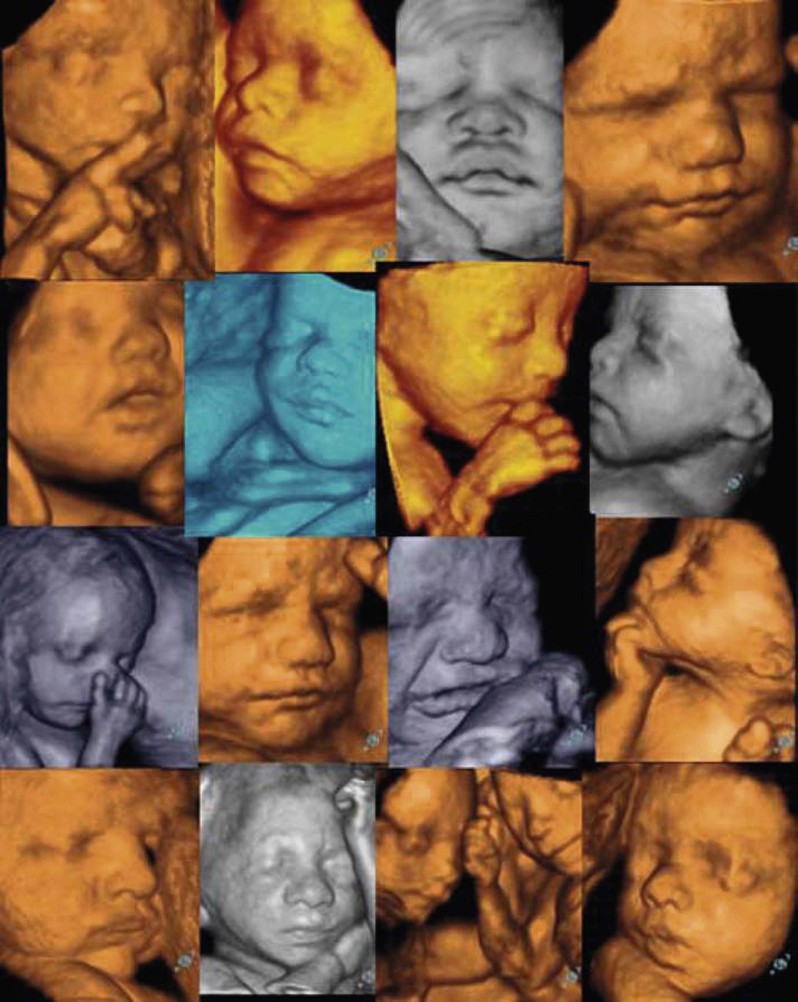
In the past, prenatal diagnosis of inherited disorders often relied on fetal skin biopsies performed between 19 and 22 weeks’ EGA.23,188 The procedure is carried out under ultrasound guidance, and multiple biopsies must be taken, although the number of biopsies and the sites from which they are taken depend on the disorder for which the fetus is at risk. In some disorders, such as those in which keratinization of the interfollicular dermis is not yet complete, analysis of the developing appendageal structures is required for accurate diagnosis.31 Fortunately, because of the identification of the genetic mutations responsible for many of these disorders, diagnosis can be made using cells obtained from CVS at 8–10 weeks’ EGA, or amniocentesis at 16–18 weeks’ EGA.184 The obvious advantage of these procedures is that they can be performed early in the pregnancy with minimal risk to the mother and fetus. A parallel development in high technology in obstetrics includes 3D/4D ultrasound imaging, wherein computer reconstruction of real-time images allows an unprecedented view of the body surface and fetal behavior (Fig. 1.10).189 The spatial resolution of this technique is remarkable,190 opening the door to a possible ‘prenatal dermatology’.
Fundamental knowledge of those skin disorders whose etiologies are genetic, is crucial for the practicing pediatric dermatologist. The number of genodermatoses for which the gene mutation is known increases yearly (Table 1.4 ). Data on genetic test availability, provided by both commercial and research laboratories, are provided by GeneTest (http://www.genetests.org), a database for healthcare providers. Information regarding the identity of genes critical for specific disorders, as well as whether laboratories exist that conduct relevant testing, can also be accessed through the website for Online Mendelian Inheritance in Man (OMIM), at: http://www.ncbi.nlm.nih.gov/omim/.
). Data on genetic test availability, provided by both commercial and research laboratories, are provided by GeneTest (http://www.genetests.org), a database for healthcare providers. Information regarding the identity of genes critical for specific disorders, as well as whether laboratories exist that conduct relevant testing, can also be accessed through the website for Online Mendelian Inheritance in Man (OMIM), at: http://www.ncbi.nlm.nih.gov/omim/.
TABLE 1.4
Genodermatoses associated with allelic variations in known genes181
| Genetic skin disorder | Illustrative disease state or condition | Chapter |
| Epidermolysis bullosa (EB) | EB simplex variants, junctional EB variants, dystrophic EB variants – recessive and dominant, transient bullous dermolysis of the newborn | 10, 11 |
| Disorders of keratinization | ||
| Ichthyoses | Congenital hemidysplasia with ichthyosiform erythroderma and limb defects (CHILD) syndrome, keratitis–ichthyosis–deafness (KID) syndrome, lamellar ichthyosis, Netherton syndrome, Refsum syndrome, X-linked chondrodysplasia punctata (Conradi–Hunerman syndrome), X-linked recessive ichthyosis | 19 |
| Palmoplantar keratodermas | Pachyonychia congenita, striate palmoplantar keratoderma variants, Vohwinkel syndrome variants, Vorner syndrome | |
| Follicular keratosis | Darier–White disease, Hailey–Hailey disease | |
| Erythrokeratodermias | Erythrokeratodermia variabilis, progressive symmetric erythrokeratodermia | |
| Disorders with abnormal pigmentation | Chediak–Higashi syndrome, Hermansky–Pudlak syndrome variants, McCune–Albright syndrome, neurofibromatosis variants, oculocutaneous albinism variants, Peutz–Jeghers syndrome, piebaldism, Waardenburg syndrome variants | 23, 24 |
| Disorders associated with malignancy | Cowden syndrome, dyskeratosis congenita, Gardner syndrome, nevoid basal cell carcinoma syndrome (Gorlin syndrome), tuberous sclerosis complex, xeroderma pigmentosum | 24, 29 |
| Disorders of ectodermal appendages including ectodermal dysplasias | Cartilage hair hypoplasia, familial incontinentia pigmenti, hidrotic and X-linked hypohidrotic ectodermal dysplasias, monilethrix, nail–patella syndrome | 29, 32 |
| Connective tissue defects | Cutis laxa, Ehlers–Danlos syndrome and variants, Fabry disease, Marfan syndrome, Menkes kinky hair syndrome, pseudoxanthoma elasticum, Williams syndrome | 29, 31 |
| Vascular and lymphatic disorders | Hereditary lymphedema type I, hyperkeratotic cutaneous capillary venous malformations, Osler–Rendu–Weber syndrome, glomerulovenous malformations, venous malformations | 20, 21 |
| Porphyrias | Congenital erythropoietic porphyria, erythropoietic protoporphyria (EPP), porphyria cutanea tarda | 20 |
| Disorders associated with immunodeficiency | Omenn syndrome, Wiskott–Aldrich syndrome | 18 |
| Miscellaneous disorders | Bloom syndrome, Cockayne syndrome, Rothmund Thompson syndrome, hereditary angioedema, Werner syndrome | 20, 29 |
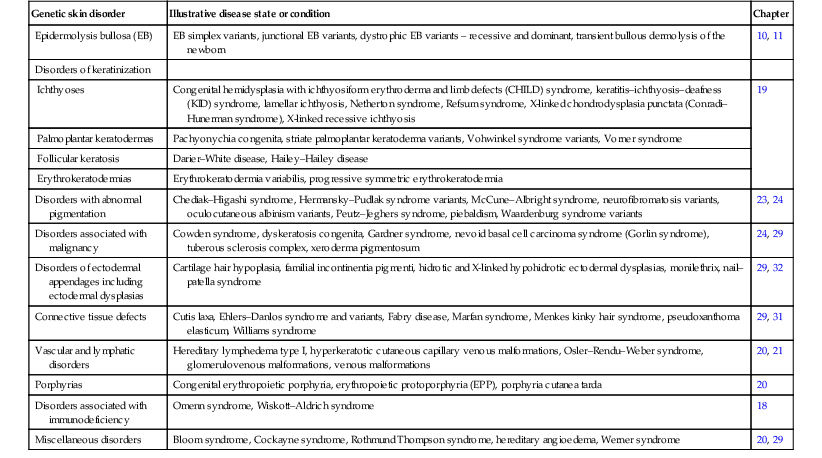
(Adapted from Irvine AD, McLean WH. The molecular genetics of the genodermatoses: progress to date and future directions. British Journal of Dermatology 2003; 148:1–13.)
Gestational age assessment
Throughout this chapter, there has been a conscious effort to highlight the close embryological, physiological, and perceptual–behavioral connections between the skin and the brain. Dermatology and neurology, more than other clinical specialties, place a high priority on the traditional physical examination.191 This focus on the body surface and associated neurobehavior has a parallel in the assessment of gestational age. The commonly used Ballard exam consists of a quantitative bedside evaluation of a set of neurological signs and physical findings, which yield a score commensurate with gestational age between 20 and 40+ weeks’ gestation.192 The physical scores are largely indicative of selected markers of cutaneous development (Table 1.5). The accurate determination of gestational age is important for organizing care expectations in the newborn nursery and intensive care unit. Subsequent chapters will address the care of the neonate and associated skin disorders of the term and preterm infant.
TABLE 1.5
Physical maturity scores used to determine gestational age in premature and term infants192
| Physical maturity sign | Score | ||||||
| –1 | 0 | 1 | 2 | 3 | 4 | 5 | |
| Skin | Sticky, friable, transparent | Gelatinous, red, translucent | Smooth, pink, visible veins | Superficial peeling and/or rash, few veins | Cracking pale areas, rare veins | Parchment, deep cracking, no vessels | Leathery, cracked, wrinkled |
| Lanugo | None | Sparse | Abundant | Thinning | Bald areas | Mostly bald | |
| Plantar surface | Heel–toe 40–50 mm: – 1; <40 mm: – 2 | >50 mm, no crease | Faint red marks | Anterior transverse crease only | Creases ant. ⅔ | Creases over entire sole | |
| Breast | Imperceptible | Barely perceptible | Flat areola, no bud | Stippled areola, 1–2 mm bud | Raised areola, 3–4 mm bud | Full areola, 5–10 mm bud | |
| Eye/Ear | Lids fused loosely: – 1; tightly: – 2 | Lids open, pinna flat, stays folded | Sl. curved pinna; soft; slow recoil | Well-curved pinna; soft but ready recoil | Formed and firm instant recoil | Thick cartilage ear stiff | |
| Genitals (male) | Scrotum flat, smooth | Scrotum empty, faint rugae | Testes in upper canal, rare rugae | Testes descending, few rugae | Testes down, good rugae |
Testes pendulous, deep rugae | |
| Genitals (female) | Clitoris prominent and labia flat | Prominent clitoris and small labia minora | Prominent clitoris and enlarging minora | Majora and minora equally prominent | Majora large, minora small | Majora cover clitoris and minora | |
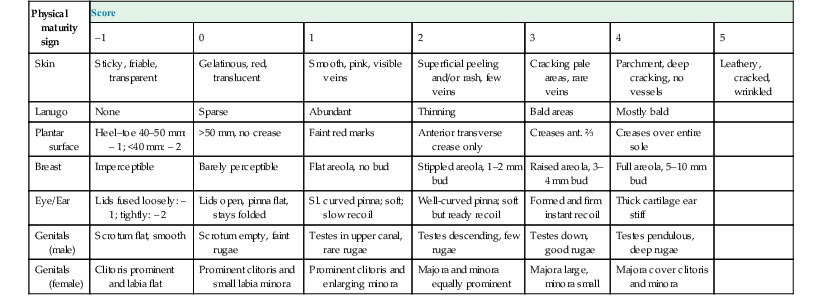
This common bedside tool assigns a score to various physical and neurological criteria, the sum of all of which is then extrapolated to estimate the gestational age of the baby with an effective range of 20–44 weeks (LMP). The scoring relies on the intrauterine changes that the fetus undergoes during its maturation. Whereas the neurological criteria (not shown) depend mainly upon muscle tone, the physical criteria reflect topological skin-based signs.192
(Adapted from the scoring system from Ballard JL, Khoury JC, Wedig K, Wang L, Eilers-Walsman BL, Lipp R. New Ballard Score, expanded to include extremely premature infants. Journal of Pediatrics 1991; 119(3):417–423.)
Acknowledgments
The authors wish to acknowledge contributors to earlier versions of this chapter: Cynthia Loomis, Tamara Koss, and David Chu. We are also grateful to Karen Holbrook for her pioneering morphological studies on the ultrastructure of human fetal skin.
Access the full reference list at ExpertConsult.com 
Figures 6 and 7 are available online at ExpertConsult.com 
Tables 1 and 4 are available online at ExpertConsult.com 
