Chapter 16
Fetal and Newborn Cardiopulmonary Physiology
Elizabeth A. Hughes and Christine K. Sperle
After reading this chapter, you will be able to:
• Describe the events that occur during each of the five stages of fetal lung development
• Explain how prematurity might lead to respiratory failure in the newborn
• Explain how gas exchange occurs between fetal and maternal blood
• Describe how alterations in maternal-fetal physiology affect fetal development
• Identify the key anatomical differences between infant and adult airways
• Describe the physiological responses to heat loss in infants
• Explain how infants generate heat
• Explain how the fetal cardiovascular system forms and becomes functional early in gestation
• Identify the factors that determine whether a fetus is viable for postnatal life
• Describe the key anatomical and physiological differences between fetal and neonatal circulation
• Describe the key events that occur during transition from fetal to extrauterine life
atrioventricular septal defect (AVSD)
congenital diaphragmatic hernia
extracorporeal life support (ECLS)
patent ductus arteriosus (PDA)
respiratory distress syndrome (RDS)
transient tachypnea of the newborn
Development of the Respiratory System
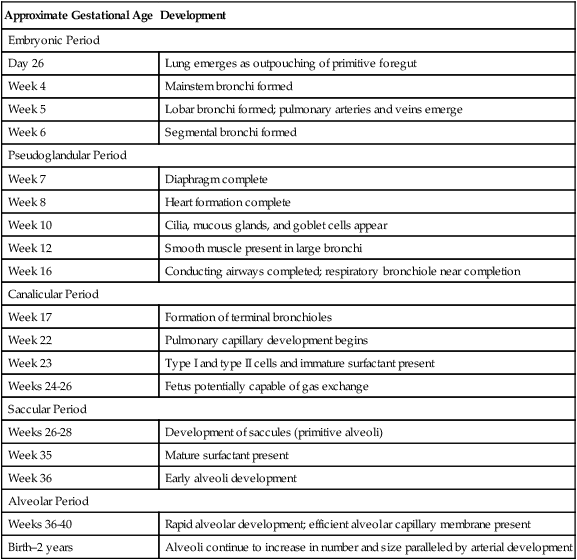
The development of the respiratory system progresses along a predetermined series of structural changes throughout gestation. Organs of the lower respiratory tract (larynx, trachea, bronchi, and lungs) begin to develop during the fourth week of gestation and continue to grow throughout early childhood. Development of the lung occurs in five overlapping stages: (1) the embryonic period, during which the trachea and major bronchi are formed; (2) the pseudoglandular period, during which the remaining conducting airways develop; (3) the canalicular period, in which the vascular bed and framework of the acinus are developed; (4) the saccular period, during which the terminal airways widen and form cylindrical structures known as saccules; and (5) the alveolar period, in which the alveoli are developed. Table 16-1 summarizes these events of development.
TABLE 16-1
Summary of Pulmonary Development
| Approximate Gestational Age | Development |
| Embryonic Period | |
| Day 26 | Lung emerges as outpouching of primitive foregut |
| Week 4 | Mainstem bronchi formed |
| Week 5 | Lobar bronchi formed; pulmonary arteries and veins emerge |
| Week 6 | Segmental bronchi formed |
| Pseudoglandular Period | |
| Week 7 | Diaphragm complete |
| Week 8 | Heart formation complete |
| Week 10 | Cilia, mucous glands, and goblet cells appear |
| Week 12 | Smooth muscle present in large bronchi |
| Week 16 | Conducting airways completed; respiratory bronchiole near completion |
| Canalicular Period | |
| Week 17 | Formation of terminal bronchioles |
| Week 22 | Pulmonary capillary development begins |
| Week 23 | Type I and type II cells and immature surfactant present |
| Weeks 24-26 | Fetus potentially capable of gas exchange |
| Saccular Period | |
| Weeks 26-28 | Development of saccules (primitive alveoli) |
| Week 35 | Mature surfactant present |
| Week 36 | Early alveoli development |
| Alveolar Period | |
| Weeks 36-40 | Rapid alveolar development; efficient alveolar capillary membrane present |
| Birth–2 years | Alveoli continue to increase in number and size paralleled by arterial development |
The embryo consists of three primary germ layers from which all tissues, organs, and organ systems arise (Figure 16-1). The endoderm is the innermost germ layer, the mesoderm is the middle layer, and the ectoderm is the outermost layer. Table 16-2 lists the structures that arise from each of the three germ layers. The epithelium of the respiratory tract originates from the endoderm, whereas supporting structures such as connective tissue and muscle arise from the mesoderm.
TABLE 16-2
Structures Arising from the Three Embryonic Layers
| Ectoderm | Mesoderm | Endoderm |
| Central nervous system Peripheral nervous system Sensory epithelia Glandular tissues Epidermal tissues Teeth |
Cardiovascular system Connective tissue Bone, cartilage, muscle Kidney and spleen tissue Reproductive tissues Serous linings |
Epithelial tissue Respiratory, digestive, and urinary systems Large glands: tonsils, thyroid, thymus Auditory structures |
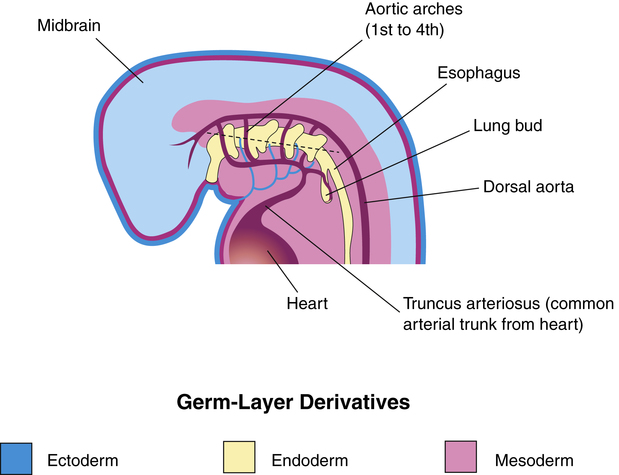
Embryonic Period
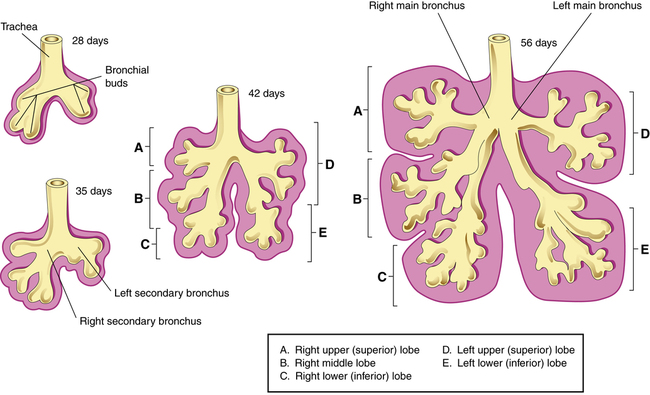
The embryonic period begins approximately 26 days after conception.1–4 During this period, the lung begins to emerge as an outpouching of the primitive foregut (see Figure 16-1). This outpouching elongates and separates into two bronchial buds and the trachea. The right lung bud parallels the esophagus and branches further into three additional buds. The left lung bud is directed more laterally and divides into two additional buds. Thus, the asymmetry of the main bronchi present in the adult is established. The right and left lung buds continue to grow and branch into segmental and subsegmental bronchi.
By the end of the embryonic period (near the sixth week of gestation), the major bronchi are formed, consisting of 10 right branches and 9 left branches (Figure 16-2).1–4 During this time, the pulmonary arteries and veins emerge from the developing heart. The diaphragm also develops during this stage and is formed by about the seventh gestational week.1–4 The diaphragm separates abdominal contents from the thorax. Failure of the diaphragm to form completely can result in congenital diaphragmatic hernia, allowing abdominal organs to enter the pleural cavity and compress the lungs and possibly lead to severe underdevelopment, or hypoplasia, of the lung.
The dorsal portion of the foregut evolves into the esophagus. The formation of the tracheoesophageal septum separates the esophagus from the trachea (Figure 16-3). Teratogens (agents that disturb fetal development, such as drugs, infection, or chemicals) can produce congenital anomalies and may cause injury to the embryo during this stage of development, such as tracheoesophageal fistula (an abnormal opening between the trachea and the esophagus), choanal atresia (occlusion of the passageway between the nose and the pharynx), and pulmonary hypoplasia (underdevelopment of the lung).
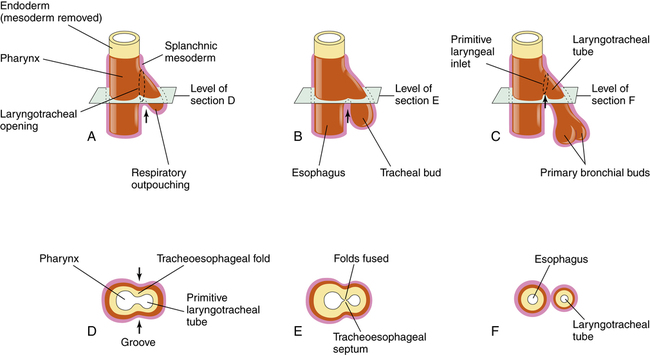
Pseudoglandular Period
The pseudoglandular period begins about the sixth week and continues into the sixteenth week of gestation.1–3 The lung resembles a gland at this stage, giving rise to the name pseudoglandular. Branching and division of the tracheobronchial tree continue to occur asymmetrically and dichotomously (branching into two parts) forming the conducting airways. By the end of this period, the formation of respiratory bronchioles is nearly complete. Cilia, mucous glands, and goblet cells begin to appear in the epithelial lining at approximately 10 weeks’ gestation.1–4 Absence or dysfunction of cilia can impair mucous transport and lead to chronic respiratory infections (see Chapter 1). Smooth muscle, which originates from the mesoderm, also appears during this stage and is present in the large bronchi by the twelfth week. Any event that alters the development of smooth muscle, cartilage, or vascular structures during this stage can lead to pulmonary disorders in infancy. A fetus born prematurely during this period is unable to survive.
Canalicular Period
The third stage of development, or the canalicular period, begins at approximately 17 weeks and continues through 26 weeks’ gestation.1–4 During this stage, the terminal bronchioles subdivide further to form the basic structure of the acinus, or gas-exchanging unit.1–4 Capillaries begin to establish a network around the alveoli allowing for limited gas exchange by 22 weeks.1–4 At approximately 23 weeks’ gestation, type I and type II alveolar cells begin to differentiate. Type I cells are important in the development of the alveolar capillary membrane, whereas type II cells are involved in surfactant production. By the end of the canalicular stage, some thin-walled primitive alveoli have developed, and the lung tissue is well vascularized. A fetus born prematurely at the end of this stage (approximately 24 to 26 weeks’ gestation) is potentially capable of air breathing and may survive with intensive medical care. The immature respiratory system often requires mechanical ventilatory assistance or surfactant replacement therapy or both. (See Chapter 3 for further discussion of surfactant replacement therapy.)
Saccular Period
The saccular period begins around 26 weeks of gestation.1–4 During this stage of development, the terminal airways continue to widen and form cylindrical structures called saccules. These saccules subdivide to form subsaccules, which eventually develop into alveoli. By the end of this stage, the blood-air barrier is established allowing for adequate gas exchange for the fetus if it is born prematurely. Alveolar cells continue to differentiate into type I and type II cells, with type II cells synthesizing pulmonary surfactant. Surfactant is a complex mixture of phospholipids and proteins that lines the inner walls of the alveoli and offsets surface tension forces at the air-liquid interface of the alveoli (see Chapter 3). Surfactant production begins at approximately 20 weeks’ gestation and is present in only small amounts in infants born prematurely. The production of surfactant increases during gestation and reaches adequate levels to support air breathing during the last two weeks of pregnancy. Mature surfactant contains the phospholipid phosphatidylglycerol (PG), which is required for normal surfactant function. PG first appears at about 35 weeks of gestation and increases to peak levels at term.1–3 Immature surfactant has insufficient PG and is easily inhibited by hypoxia, hypothermia, and acidosis. Thus, premature infants, especially infants born before 35 weeks’ gestation, are susceptible to developing respiratory distress syndrome (RDS) because of surfactant deficiency. Maternal administration of prenatal corticosteroids, which induce surfactant production, and the administration of exogenous surfactant directly into the newborn lungs after birth can decrease the severity of RDS and the need for mechanical ventilation.
Alveolar Period
The final period of lung development, the alveolar period, begins at approximately 36 weeks’ gestation and extends through infancy and childhood.1–4 During this stage, alveoli continue to develop and mature at a rapid rate, and pulmonary surfactant production increases. The full-term fetus has developed numerous alveoli with mature surfactant, creating an efficient alveolar capillary gas-exchange membrane. Although the lungs are completely developed, they do not take on a respiratory function until the moment of birth.
The lung at birth is not a miniature adult lung; it continues to develop and grow well into childhood. During the first years of life, alveoli develop rapidly paralleled by the expansion of the pulmonary capillary bed. As body weight increases, alveolar development increases proportionally, matched by an increase in the lungs’ oxygen uptake. At birth, approximately 50 million alveoli are present; by age 8 years, this number has increased to about 300 million alveoli.1–3 This explains why children who sustain lung injury during the neonatal period (birth to 28 days) can seemingly “outgrow” their lung disease. Alveolar development is usually complete by age 8; from this time on, alveoli increase in size until thoracic growth is complete.1–4 Collateral pathways such as the canals of Lambert and pores of Kohn are poorly developed in young children. Consequently, children less than 10 years of age are more likely to develop airway obstruction and atelectasis than older children.5
Factors Affecting Fetal Development
Fetal Lung Fluid
The fetal lung has no respiratory function before birth; it is a secretory organ that produces approximately 250 to 350 mL of fluid per day.4 Fetal lung fluid is present by the sixth week of gestation and is derived from alveolar epithelium secretions. Most of the fluid produced remains in the lung; however, some is swallowed, and some is emitted into the amniotic fluid during periodic opening of the larynx when the fetus makes breathing movements. Lung fluid is constantly produced and plays a significant role in determining the size and shape of the developing air space. The composition of fetal lung fluid differs from the composition of amniotic fluid. Fetal lung fluid is higher in sodium and chloride concentrations; lower in pH; and lower in bicarbonate, potassium, and protein concentrations.4 Lung fluid also contains components of pulmonary surfactant and other fluids from alveolar epithelial cells; its presence in amniotic fluid aids the clinician in determining the degree of lung maturity.
Although the presence of fetal lung fluid is essential for normal development of the lung, the switch from placental to pulmonary gas exchange at birth requires rapid removal of this fluid from the newborn lung. The production of lung fluid decreases shortly before term to approximately 65% of previous values. In addition, lung fluid is absorbed during labor so that only about 35% of the original lung fluid volume needs to be cleared during delivery.4 At the time of birth, the infant’s inspiratory effort generates an enormous amount of negative intrapulmonary pressure to fill the lungs with air. With the first few breaths, most fetal lung fluid is expelled; the remaining lung fluid is cleared by absorption through the pulmonary vasculature and lymphatic systems.
Fetal breathing movements begin at approximately 10 weeks of gestation and increase in strength and frequency as gestation progresses, increasing to a rate of 30 to 70 breathing efforts per minute during the last 10 weeks.4 This early respiratory activity contributes to the regulation of lung fluid and aids in the stretch of lung tissue, which influences lung growth. Fetal breathing movements appear to originate from the diaphragm and are thought to be necessary for training and developing the respiratory muscles so that they can generate enough force to overcome the surface tension of airless alveoli during the initial breath. The absence of lung fluid and breathing movements during fetal development results in an underdeveloped lung.2
Maternal-Fetal Gas Exchange
Shortly after the fertilized egg implants in the uterine wall, the placenta begins to develop with small finger-like projections invading the endometrial lining of the uterus. These projections, called chorionic villi, continue to branch further into the endometrium, creating irregular pockets around the villi called intervillous spaces (Figure 16-4). Maternal blood fills these spaces supplying oxygen and nutrients to the fetus. As the fetus matures, villi increase in number, expanding the surface area for gas exchange.
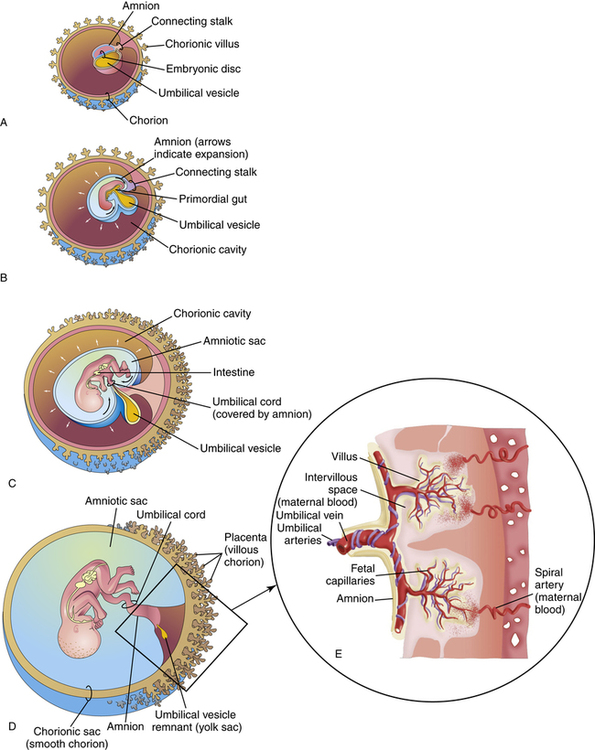
Maternal blood enters the intervillous space through the spiral arteries (see Figure 16-4). At this point, the diffusion of oxygen, carbon dioxide, and metabolic products occurs between maternal and fetal blood. After this exchange, maternal blood exits the intervillous spaces through venous channels. Oxygenated fetal blood leaves the chorionic villi via capillaries that merge into a single umbilical vein.
The placenta is not a very efficient gas-exchange organ. The PO2 of maternal blood in the intervillous spaces is about 50 mm Hg, but blood leaving the placenta through the umbilical vein to oxygenate the fetus has a PO2 value of only about 30 mm Hg. Fetal blood returning through the umbilical arteries to the placenta for reoxygenation has a PO2 of only about 19 mm Hg.2 The maximum PO2 experienced by the fetus is about 30 mm Hg. The primary factor limiting oxygen transport to the fetus is blood flow. Any factor diminishing uterine or fetal blood flow results in fetal hypoxia and intrauterine growth retardation (IUGR). Severe reduction in fetal blood flow may result in fetal asphyxia and death. Table 16-3 shows normal blood gas values of the umbilical arteries and veins in a full-term fetus.
TABLE 16-3
Normal Blood Gas Values in a Term Fetus
| PO2 (mm Hg) | PCO2 (mm Hg) | pH | |
| Umbilical artery | 19 | 47 | 7.36 |
| Umbilical vein | 30 | 43 | 7.39 |

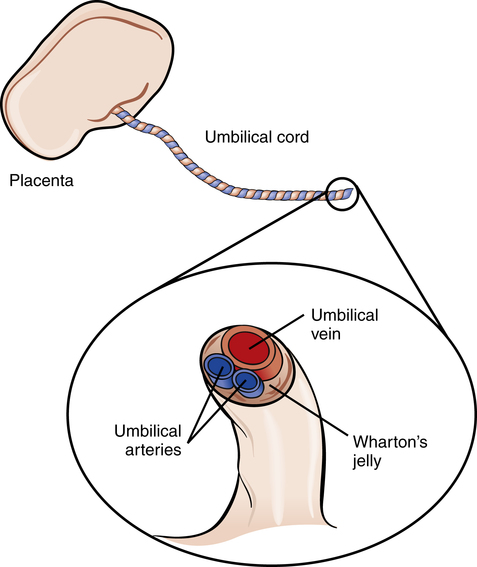
A tough gelatinous material called Wharton’s jelly surrounds the umbilical vessels, which prevents the cord from kinking and occluding blood flow to the fetus (Figure 16-5). After birth, the umbilical vessels remain open for a short time, allowing vascular access for fluid infusion and blood sampling.
Fetal Hemoglobin
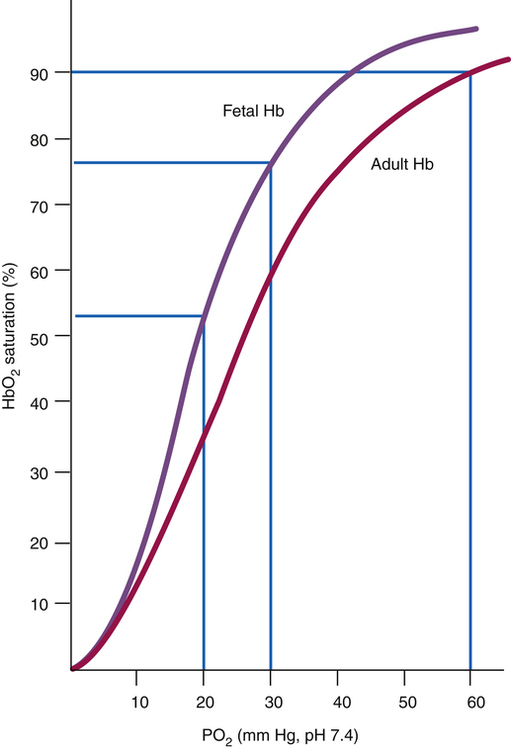
Fetal blood oxygen tension is low compared with values seen after birth. As stated previously, the most oxygenated fetal blood is found in the umbilical vein with a PO2 of approximately 30 mm Hg. Nevertheless, fetal tissues are adequately oxygenated for the following reasons: (1) the unique blood flow through fetal circulation results in increased blood flow to vital organs (liver, heart, and brain); (2) the fetus has an increased number of red blood cells and hemoglobin compared with the adult; (3) and fetal hemoglobin (HbF) has an increased affinity for oxygen compared with adult hemoglobin.6
HbF is the predominant form of hemoglobin in the fetus at approximately eight weeks of gestation; HbF continues to increase until the third trimester when its concentration begins to decline gradually. At 24 weeks of gestation, HbF constitutes about 90% of the total hemoglobin, whereas at birth, HbF represents approximately 70% of the total hemoglobin.4 The production of HbF decreases rapidly after birth, and by 6 to 12 months of age, HbF has been replaced by adult hemoglobin (HbA).4
HbF has a greater affinity for oxygen than HbA. This increase in oxygen affinity is associated with a left shift of the fetal oxyhemoglobin dissociation curve (Figure 16-6), which means that oxygen binds more readily to HbF. The placental transfer of oxygen from maternal blood to fetal blood is thus facilitated, allowing the fetus to survive in a relatively hypoxic environment. Therefore, a PO2 of 30 mm Hg in the fetus corresponds to a hemoglobin saturation of 75% to 80% (see Chapter 8 for further discussion of the oxyhemoglobin equilibrium curve). Maternal hypoxia or hyperventilation can have profound effects on fetal oxygenation; in the case of maternal hyperventilation, the resulting alkalosis shifts the maternal oxygen-hemoglobin equilibrium curve to the left (greater hemoglobin affinity for oxygen), decreasing oxygen availability to the fetus and potentially causing fetal distress.4
Amniotic Fluid
Amniotic fluid, a clear liquid produced by the fetal membranes and the fetus, appears early and surrounds the fetus throughout pregnancy. Amniotic fluid is composed mainly of water; however, its composition changes throughout gestation. During the first half of gestation, amniotic fluid is similar to maternal and fetal serum; in the second half, it is similar to dilute fetal urine with added phospholipids from the fetal lung.4 Amniotic fluid is continuously secreted and reabsorbed throughout gestation with a volume reaching approximately 1000 mL at full term. The fetus swallows this fluid, which is absorbed by the digestive tract and excreted as urine. Amniotic fluid serves many important functions. It protects the fetus from trauma by cushioning any blows or impacts to the maternal abdomen, and it allows the fetus to move freely, permitting fetal growth and development. In addition, amniotic fluid helps control the temperature of the fetus by maintaining a relatively constant thermal environment.
As stated previously, the fetus periodically exhibits shallow respiratory chest movements or “fetal breathing.” This normal occurrence moves amniotic and fetal lung fluid into and out of the oropharynx. Vagal stimulation and hypoxic stress caused by cord compression may cause rectal sphincter muscles to relax, resulting in release of meconium (fetal bowel contents) into the amniotic fluid. Deep gasping movements of the distressed fetus may cause amniotic fluid and meconium to be aspirated into the infant’s lung. Meconium aspiration usually leads to respiratory distress after birth because of airway obstruction or chemical irritation; this is known as meconium aspiration syndrome. Meconium is found in amniotic fluid in approximately 12% of all births.4
Amniocentesis involves the withdrawal of a sample of amniotic fluid for laboratory analysis. It is performed to test for lung maturity and to aid in the diagnosis of fetal abnormalities. The proportions of lecithin and sphingomyelin (L/S ratio) and the amount of phosphatidylglycerol in amniotic fluid are predictors of lung maturation (see Chapter 3 for further discussion).
Postnatal Anatomical and Physiological Considerations
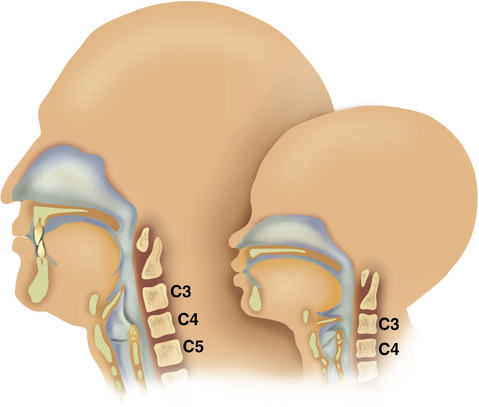
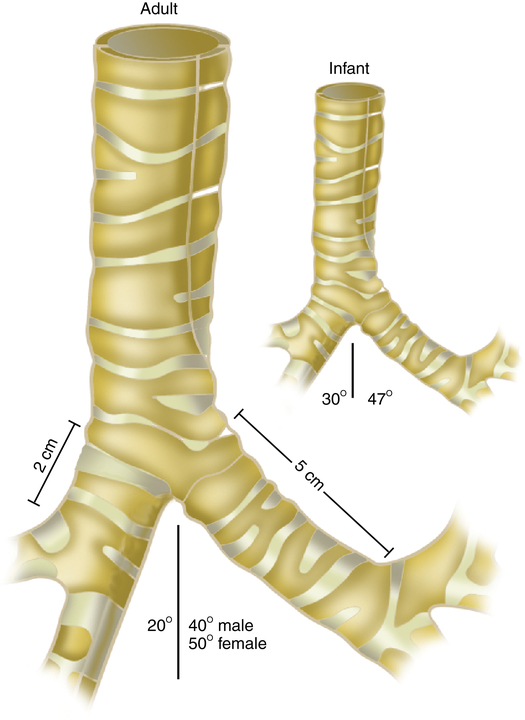
All infant airway structures are smaller and narrower than in the adult; the infant’s upper airway mucosa is thinner and more easily traumatized. The infant larynx is positioned higher in the neck and is more cone-shaped than it is in the adult; the cricoid ring is the narrowest portion of the upper airway in the infant (Figure 16-7). The newborn trachea is shorter, narrower, softer, and more collapsible than the trachea of the adult (Figure 16-8). Because of the small diameter of the infant airway, a minor degree of mucosal edema can very quickly produce significant airway narrowing and increased airflow resistance.
The chest wall of the infant is very compliant with the ribs positioned more horizontally than in the adult. The infant’s ribs and sternum are mostly cartilage offering little support to the chest wall, and intercostal muscles are immature and fatigue easily causing the infant to rely more heavily on the diaphragm for respiration; this can cause the chest and abdomen to move in opposite directions (paradoxical breathing) during respiratory distress. Signs of respiratory distress and high airway resistance in the infant include inspiratory nasal flaring, expiratory grunting, inspiratory intercostal retractions, and paradoxical breathing. Table 16-4 shows normal vital signs of full-term neonates at birth.
TABLE 16-4
Timelines in Normal and Abnormal Cardiac Development
| Normal Time | Developmental Events | Malformations Arising during Period |
| 18 days | Horseshoe-shaped cardiac primordium appears | Lethal mutations |
| 20 days | Bilateral cardia primordial fuse | Cardia bifida (experimental) |
| Cardiac jelly appears | ||
| Aortic arch is forming | ||
| 22 days | Heart is looping into S shape | Dextrocardia |
| Heart begins to beat | ||
| Dorsal mesocardium is breaking down | ||
| Aortic arches I and II are forming | ||
| 24 days | Atria are beginning to bulge | |
| Right and left ventricles act like two pumps in series | ||
| Outflow tract is distinguished from right ventricle | ||
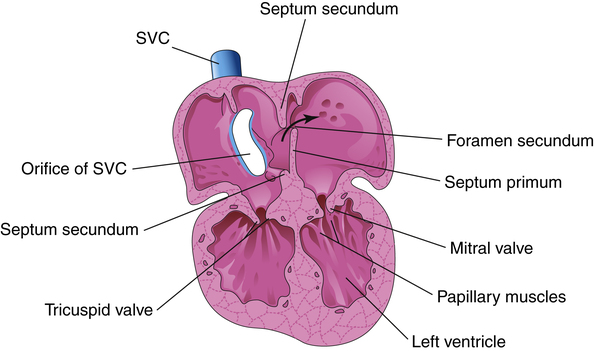
| Late in week 4 | Sinus venosus is being incorporated into right atrium Endocardial cushions appear Septum primum appears between right and left atria Muscular interventricular septum is forming Truncoconal ridges are forming Aortic arch I is regressing Aortic arch III is forming Aortic arch IV is forming |
Venous inflow malformations Persistent atrioventricular canal Common atrium Common ventricle Persistent truncus arteriosus |
| Early in week 5 | Endocardial cushions are coming together, forming right and left atrioventricular canals Further growth of interatrial septum primum and muscular interventricular septum occurs Truncus arteriosus is dividing into aorta and pulmonary artery Atrioventricular bundle is forming; there is possible neurogenic control of heartbeat Pulmonary veins are being incorporated into atrium Aortic arches I and II have regressed Aortic arches III and IV have formed Aortic arch VI is forming |
Persistent atrioventricular canal Muscular ventricular septal defects Transposition of the great vessels; aortic and pulmonary stenosis or atresia Aberrant pulmonary drainage |
| Late in week 5–early week 6 | Endocardial cushions fuse Interatrial foramen secundum is forming Interatrial septum primum is almost contacting endocardial cushions Membranous part of interventricular septum starts to form Semilunar valves begin to form |
Low atrial septal defects Membranous interventricular septal defects Aortic and pulmonary vascular stenosis |
| Late in week 6 | Interatrial foramen secundum is large | High atrial septal defects |
| Interatrial septum secundum starts to form | Tricuspid or mitral valve stenosis or atresia | |
| Atrioventricular valves and papillary muscles are forming | Membranous interventricular septal defects | |
| Interventricular septum is almost complete | Membranous interventricular septal defect | |
| Coronary circulation is becoming established | ||
| 8-9 weeks | Membranous part of interventricular septum is complete | Membranous interventricular septal defect |
From Carlson BM: Human embryology and developmental biology, ed 3, St Louis, 2004, Mosby.
Brown fat metabolism is the most important means of heat production in the newborn infant. This special fat is located between the scapulae; around the muscles and blood vessels of the neck, axillae, and mediastinum; and around the kidneys and adrenal glands. Brown fat begins to appear at approximately 26 weeks’ gestation and continues to develop until 3 to 5 weeks after birth, accounting for one tenth of the adipose tissue in term infants.4 Brown fat stores are much lower in preterm infants and almost nonexistent in infants of very low birth weight.
Several factors lead to increased heat loss in the newborn infant. Infants have a very large surface area-to-body mass ratio, they have little subcutaneous fat, and they have a relatively large head. For these reasons, heat loss is proportionately greater in infants than in adults. The infant’s thin skin with blood vessels near the surface provides poor insulation, leading to further heat loss. In addition, shivering, which is a major method of heat gain in adults, is not well developed in newborns. Hypothermia, or cold stress, is a serious condition that can be fatal if not detected and treated early. Hypothermia occurs when heat is lost at a greater rate than it can be replaced. Clinical manifestations of cold stress include tachypnea, peripheral vasoconstriction, pallor, skin mottling, and metabolic acidosis. Cold stress may occur when the infant’s core body temperature decreases to less than 36° C.7 Therefore, the clinician should ensure that the infant is placed in a neutral thermal environment and is kept warm and dry; this minimizes heat loss, improves survival, and decreases metabolic demands (see Clinical Focus 16-3).
Fetal and Neonatal Cardiovascular Development and Physiology
The cardiovascular system is the first organ system to develop and function in the fetus, appearing at about 18 or 19 days of gestation. The rapidly growing embryo requires an efficient method to transport oxygen and nutrients; cardiac contractions begin by day 22, and blood flow can be observed by Doppler ultrasonography by gestational week 4.3 The primitive heart and its relationship to the fetal cardiovascular system is shown in Figure 16-9. Understanding this development is of value to students when studying the various cardiac anomalies discussed later in this chapter.
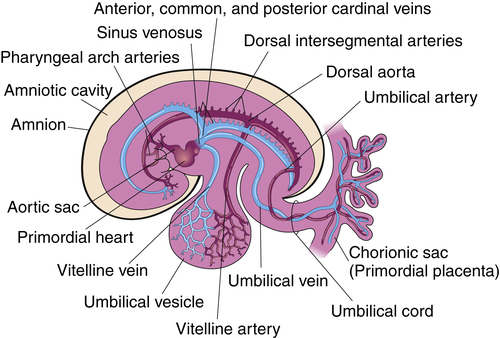
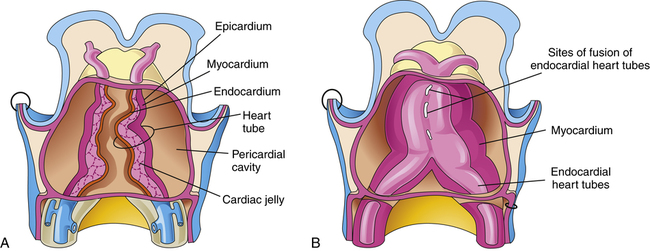
The cardiovascular system arises from the mesoderm layer of the embryo (see Table 16-2). The first identifiable features are cell clusters that can induce blood vessel formation (angiogenic cells) arranged on both sides of the embryo’s central axis.8 Late in gestational week 3, a pair of cardiac tubes forms from the combination of these cell clusters. Fusion of these heart tubes creates a single, three-layer endocardial heart tube (Figure 16-10). The inner layer of the heart tube becomes the internal endothelial lining of the heart—the endocardium—whereas the primitive myocardium becomes the muscular, myocardial layer of the heart.4 These layers are separated by a middle layer of gelatinous connective tissue referred to as cardiac jelly, which participates later in the separation of the heart chambers and formation of the atrioventricular valves.9 The epicardium, also known as the visceral pericardium, is part of the outer layer of the heart and develops from mesothelial cells that arise from the area of the sinus venosus.3
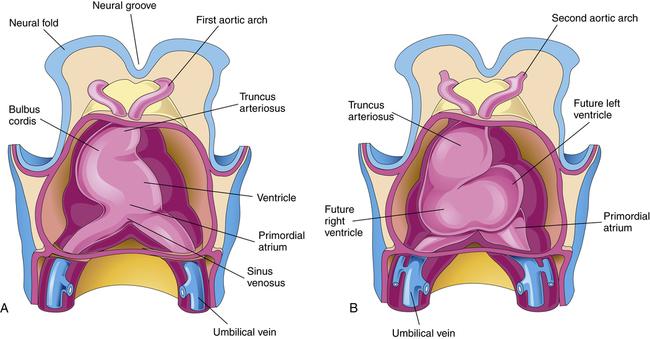
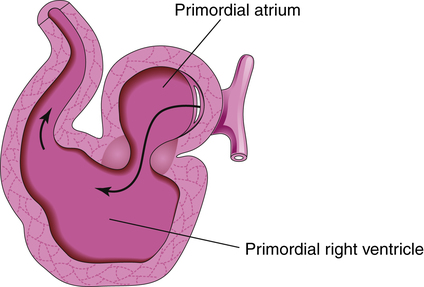
The developing heart lengthens and bends, giving rise to three recognizable structures—the bulbus cordis, the ventricle, and the primordial (primitive) atrium—from which the right ventricle, the left ventricle, and the atria arise respectively (Figure 16-11, A). The truncus arteriosus is identifiable shortly thereafter, and it divides into the pulmonary artery and aorta sometime during gestational week six (Figure 16-11, B). The fetal heart continues to bend in an S-shaped pattern, pushing the atrial bulge in a superior direction (Figure 16-12). If this process of bending and looping (sometimes referred to as cardiac looping) progresses abnormally, cardiac malformation can occur (Table 16-4 describes malformations that may arise during cardiac development). By the end of the fourth gestational week, the external appearance of the fetal heart is similar to the appearance of a mature heart; however, internal structures are incomplete. Development of the atrial and ventricular septa and the atrioventricular (tricuspid and bicuspid) and semilunar (pulmonary and aortic) valves begins between days 25 and 30 and continues through weeks 7 and 8 (Figure 16-13), during which time blood begins to flow through the fetal circulation pathway (explained later).
A heart rate of approximately 100 beats per minute may be detected by gestational week five and increases to an average rate of 140 beats per minute before term delivery.4,10 By gestational week 12, the fetal heart is positioned normally in the chest and has developed into a miniature version of the adult heart. The fetal heart doubles in size between gestational weeks 12 and 17 and triples in size by week 21.11 The cardiac conduction system (see Chapter 18, Transmission of Impulses through the Heart, for a complete description) also develops early, with the sinoatrial node and atrioventricular node evident by gestational week five.12
Fetal Circulation
The design of the fetal cardiovascular system meets the needs of the developing fetus and allows for the transition to neonatal circulation that occurs after birth. Figure 16-14 illustrates fetal blood flow, including the three fetal shunts that must close after birth. As mentioned earlier, the placenta, a highly vascular, low-resistance organ, supplies oxygen-rich blood to the fetus via the umbilical vein. Blood from the umbilical vein flows first to the liver where 18% to 30% of oxygenated blood is shunted around the liver, through the ductus venosus (the first shunt) into the inferior vena cava (IVC). Blood flow through the ductus venosus increases in response to fetal distress; flow through this shunt normally decreases in the near-term infant, which helps to supply the liver with additional nutrients.4 Because of the angle at which the IVC enters the right atrium, most of the oxygenated blood supply is directed through the opening between the right and left atrium, the foramen ovale (the second shunt) to the left atrium, where it mixes with a small amount of poorly oxygenated blood returning from the lungs through the pulmonary veins. This partially oxygenated blood mixture flows down into the left ventricle and out of the ascending aorta to perfuse myocardial and cerebral circulations. Blood with poor oxygen content coming from the superior vena cava (SVC) mixes with oxygenated blood from the IVC and because of the anatomical orientation of the SVC, blood from it tends to preferentially flow through the tricuspid valve into the right ventricle. This moderately oxygenated blood is pumped through the pulmonary valve into the pulmonary artery where only about 13% to 25% of the blood enters the pulmonary circulation to perfuse the developing lungs; the remaining 75% to 90% bypasses the lungs through the third shunt, the ductus arteriosus, which connects the pulmonary artery with the descending aorta.13
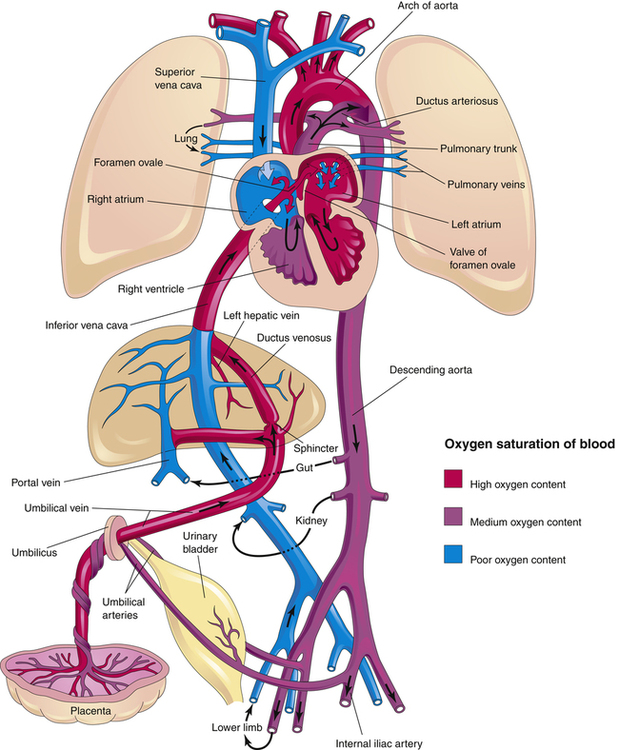
The fetal lung requires little blood flow because it does not participate in gas exchange. Fetal pulmonary vascular resistance (PVR) is increased because the fluid-filled lungs have a very low PO2, leading to hypoxic pulmonary vasoconstriction and consequently, an increase in the medial muscle layer of pulmonary arteries.14 The high PVR explains why 75% to 90% of the pulmonary artery blood flows preferentially through the ductus arterious, a path of much less resistance. The ductal blood mingles with descending aortic blood that supplies the lower extremities. Two umbilical arteries, branching from the iliac arteries, carry blood back to the placenta for reoxygenation and carbon dioxide removal (see Figure 16-14). The three fetal shunts are necessary for fetal cardiovascular function, but it is equally necessary that they close after birth.
Transition to Extrauterine Life
Respiratory Transition
As stated previously, the production of fetal lung fluid decreases shortly before term delivery, leaving approximately 35% of the original lung volume to be removed by the squeeze effect on the chest during vaginal delivery and by absorption via pulmonary capillaries and the lymphatic system after birth. Before delivery, the fetal lung contains 10 to 30 mL/kg of fluid, which is comparable to newborn functional residual capacity (FRC).4 Infants born by cesarean section and infants born after a precipitous (rapid) delivery do not benefit from vaginal chest compression and may experience retention of fetal lung fluid. Such fluid retention can result in short-term respiratory distress known as transient tachypnea of the newborn.4
After vaginal delivery, the thorax is decompressed and expands, creating a negative intrathoracic pressure that facilitates air entry into the lungs. With the first breaths, a newborn must generate pressures of −20 to −70 cm H2O to overcome the surface tension of airless alveoli and the viscosity of residual lung fluid.15 As additional alveoli inflate with subsequent breaths, surface tension is reduced and transpulmonary pressures decrease, allowing the FRC to be established within a few hours of birth. A premature infant born with underdeveloped pulmonary surfactant has great difficulty establishing the FRC because of the continued need for high transpulmonary pressures to overcome the surface tension of collapsed alveoli; this predisposes the premature infant to the development of respiratory distress syndrome (see Clinical Focus 16-1).
A transient period of asphyxia occurs after the umbilical cord is clamped and placental blood flow ceases; this lack of oxygen helps activate chemoreceptors that stimulate breathing efforts. The sensory stimulation experienced by the neonate when transitioning from the warm, moist intrauterine environment to the cooler, drier delivery room, coupled with handling and drying further stimulate breathing. Approximately 10% of newborns require some assistance to begin breathing at birth, usually in the form of oxygen administration or positive pressure ventilation, or both.7 Box 16-1 summarizes conditions that may necessitate these interventions, known as neonatal resuscitation.
Circulatory Transition
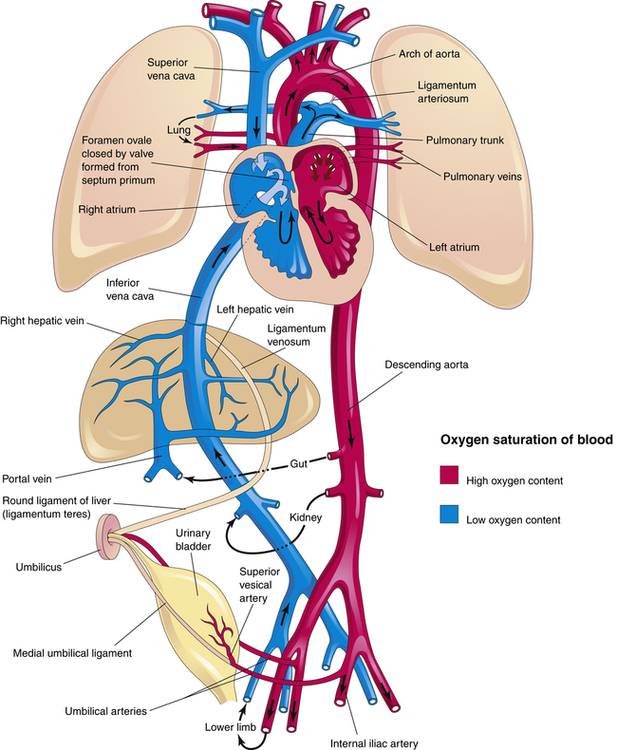
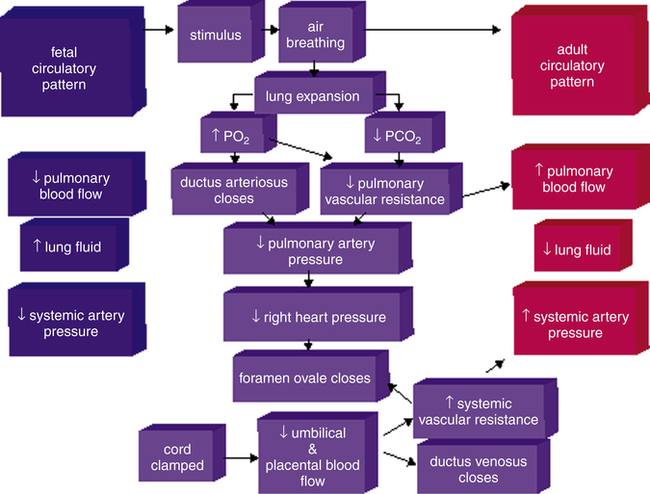
The transition from fetal to neonatal circulation is characterized by a series of dramatic cardiovascular changes. The fetus prepares for this transition by increasing the expression of pulmonary vasodilators, nitric oxide synthase, and soluble guanylate cyclase (see Chapter 6, Humoral Agents and Nitric Oxide) late in gestational life.16 Figure 16-15 illustrates neonatal circulation. Shortly after delivery, the umbilical cord is clamped, eliminating the low-resistance placenta from the circulation; this decreases blood flow to the right atrium from the umbilical vein, resulting in a decrease in pressure on the right side of the heart. Clamping of the cord also stops blood flow from the descending aorta into the placenta, increasing left-sided pressures and systemic vascular resistance (SVR). As a result,
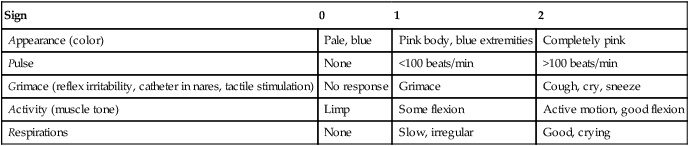
CLINICAL FOCUS 16-4 Use of Apgar Scoring in Newborn Assessment
left-sided heart pressures become greater than right-sided heart pressures. This reversal in pressure gradients physically pushes and closes the flaplike valve between the atria, functionally closing the foramen ovale. Anatomical closure of the foramen ovale occurs as tissue grows around the opening; this process may not be complete for weeks to months. Occasionally, the foramen remains patent into adulthood.17
Elimination of blood flow through the umbilical circulation also results in closure of the ductus venosus within one week of delivery. The ductus venosus becomes the ligamentum venosum. As shown in Figure 16-16, PVR decreases after birth because of pulmonary vessel dilation associated with the ever increasing PO2 and decreasing PCO2 caused by ventilation—that is, ventilation removes the stimulus for hypoxic pulmonary vasoconstriction. Consequently, more blood flows to the lungs from the right ventricle, decreasing blood flow through the ductus arteriosus. Anatomical closure of the ductus arteriosus begins before birth, when endothelial tissue (known as intimal mounds) forms in the ductal lumen.13 At birth, smooth muscle surrounding the ductus arteriosus begins to constrict because ventilation elevates blood oxygen levels, which in turn inhibits the synthesis of prostaglandins responsible for ductal patency during fetal life. Normally, the ductus arteriosus anatomically closes two to four weeks after delivery and becomes the ligamentum arteriosum by the twelfth week of postnatal life.3,13 Hypoxia and acidosis may delay closure of the ductus arteriosus, maintaining a pathway that allows blood to flow between the pulmonary artery and aorta. This condition is known as patent ductus arteriosus (PDA) and is discussed later in this chapter.
Infants born prematurely with underdeveloped surfactant (RDS) or with cardiopulmonary compromise (e.g., infection, meconium aspiration) may be unable to generate the high pressures required to expand collapsed alveoli at birth; this results in low PaO2 and persistent pulmonary vasoconstriction, sometimes referred to as persistent fetal circulation. PVR remains elevated in these infants, causing continued right-to-left shunting through the foramen ovale and ductus arteriosus, similar to conditions in fetal life. Unoxygenated venous blood bypasses the lungs through the fetal anatomical shunts and flows directly into the systemic circulation, causing significant arterial hypoxemia. This condition, commonly known as persistent pulmonary hypertension of the newborn (PPHN), manifests in the first 12 hours after birth, causing respiratory distress and arterial hypoxemia that is often unresponsive to oxygen therapy (refractory hypoxemia) because the blood supply does not come in contact with the lungs. Nevertheless, oxygen therapy is used in an attempt to reduce PVR. Some infants require more aggressive therapy, such as extracorporeal life support (ECLS), also known as extracorporeal membrane oxygenation (ECMO), which is a process whereby the blood is oxygenated outside of the body via a mechanical membrane oxygenator. Nitric oxide therapy may also be used to reduce PVR (see Clinical Focus 16-5). Table 16-5
TABLE 16-5
Circulatory Transition at Birth
| Structure | Fetal Circulation | Newborn Circulation |
| Placenta | Site of gas exchange between fetal and maternal vessels | Placental oxygenation ceases when umbilical cord is clamped |
| Foramen ovale | Allows blood to be shunted from right to left | Closes against intraatrial septum–atrium, bypassing the right ventricle and fetal lungs shortly after birth |
| Ductus arteriosus | Provides shunt for blood from pulmonary artery to descending aorta, bypassing fetal lungs | Constricts soon after birth in most infants in response to ↑ PO2 levels |
| Aorta | Mixes oxygenated blood and unoxygenated blood and directs it to the rest of the body | Delivers oxygenated blood from left ventricle to the rest of the body |
| Lungs | No gas exchange; receive minimal flow owing to high PVR | Gas exchange |
summarizes the major cardiovascular changes that occur during transition to extrauterine life. Many congenital cardiac defects require a PDA for survival, in which case prostaglandin therapy may be initiated to keep the ductus open. These conditions are described in the next section.
Congenital Cardiac Defects
Congenital cardiac defects are present at birth and are usually caused by developmental abnormalities occurring during gestational weeks three through eight.18 Approximately 1% of infants are born with some congenital heart defect, with a higher incidence in premature infants.18 Defects described in this section account for most cardiac anomalies and fall into three major categories: acyanotic (nonhypoxemic) disorders, cyanotic (hypoxemic) disorders, and obstructive disorders. Acyanotic disorders shunt already oxygenated blood from the left side of the heart to the right side of the heart, increasing pulmonary blood flow. This additional blood flow overloads the pulmonary circulation, increasing the workload of the right ventricle. Cyanotic disorders shunt deoxygenated blood from the right side of the heart to the left side of the heart, causing serious arterial hypoxemia and possible cyanosis. Acyanotic disorders may progress to cyanotic disorders if they are severe and uncorrected. Obstructive disorders create blood flow limitation within the vessels or at the level of the heart valves. A complete discussion of congenital cardiac defects is beyond the scope of this chapter, but important characteristics of the most common defects are listed subsequently.
Acyanotic Disorders
Atrial Septal Defect
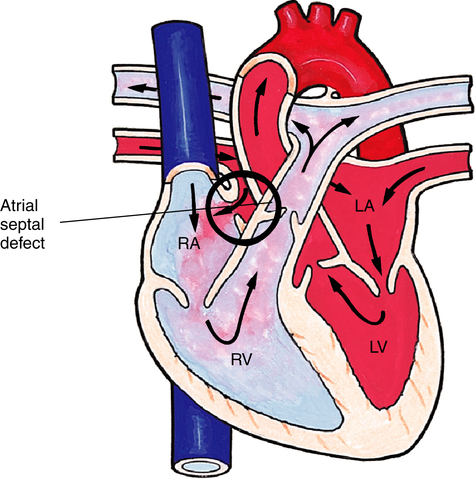
An atrial septal defect (ASD) is an abnormal opening in the atrial septum that allows blood to flow between the left atria and right atria (Figure 16-17). Atrial septal defect is one of the most common congenital cardiac anomalies but is usually asymptomatic and undetected until adulthood, when increased left-to-right blood flow can result in right atrial and ventricular hypertrophy. Surgical or catheter-based closure is an option in severe cases.
Ventricular Septal Defect
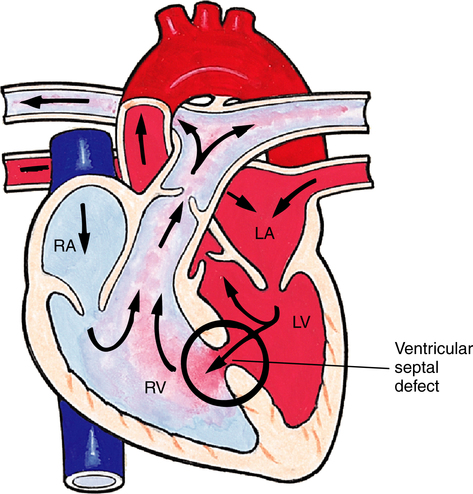
Failure of the septum between the right and left ventricles to close is known as a ventricular septal defect (VSD) and is the most common type of congenital heart defect (Figure 16-18).18 VSD may occur as an isolated defect or in association with other defects, such as tetralogy of Fallot. Symptoms depend on the size of the defect, which shunts blood from the left ventricle (higher pressure) to the right ventricle (lower pressure), causing right ventricular hypertrophy and pulmonary hypertension. Left untreated, a VSD may result in right-sided heart pressures exceeding left-sided heart pressures, reversing the direction of the shunt and causing cyanosis. Small VSDs may close on their own, whereas larger defects require surgical or catheter-based closure.
Patent Ductus Arteriosus
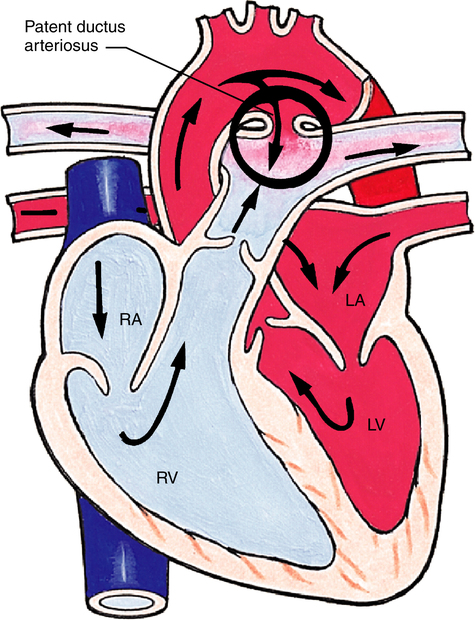
Patent ductus arteriosus (PDA) occurs when the ductus arteriosus, the fetal shunt between the pulmonary artery and the aorta, does not close after birth (Figure 16-19). The size of the PDA determines how much blood is shunted from the aorta into the pulmonary artery or, in reverse, from the pulmonary artery into the aorta. The latter occurs when PVR exceeds SVR (often seen in hypoxic premature infants), creating a cyanosis-producing defect. A large PDA shunts significant volumes of blood into the pulmonary circulation, causing pulmonary hypertension. Infants requiring treatment have two options: (1) administration of indomethacin (Indocin) or ibuprofen lysine (NeoProfen) to block the production of ductal prostaglandin (allowing ductal smooth muscle constriction) or (2) surgical intervention, either with a percutaneous transcatheter ductal closure (PTDC) device or by surgical closure.19,20
Atrioventricular Septal Defect
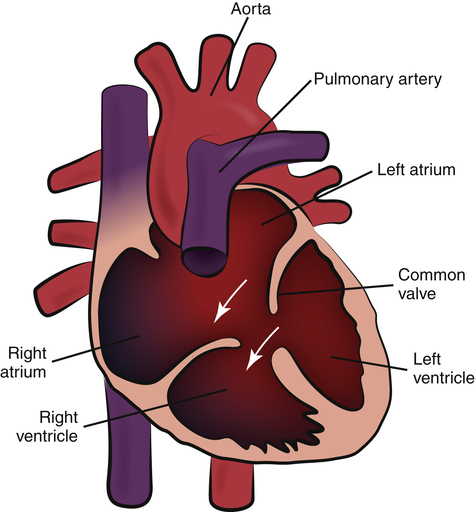
Atrioventricular septal defect (AVSD), also called atrioventricular canal (AV canal) defect, results from failure of the septa between the atria and ventricles to develop (Figure 16-20). Varying degrees of tricuspid and mitral valve malformation are also present, resulting in one large chamber or “hole” that allows blood to mix freely between all four chambers. The resultant left-to-right shunt increases pulmonary blood flow and causes volume hypertrophy of all chambers. The scooped-out appearance of the heart gives rise to the term AV canal defect, which is one of the most common types of heart defect present in patients with Down syndrome. Surgical repair is possible.21
Cyanotic Disorders
Tetralogy of Fallot
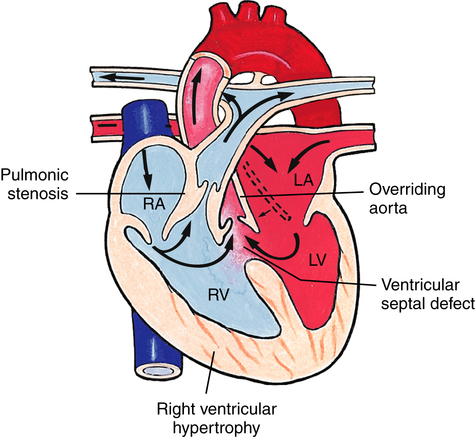
Tetralogy of Fallot is the most common cyanotic congenital heart defect.18 The four defects that characterize this anomaly are (1) pulmonary stenosis (narrowing), (2) VSD, (3) right ventricular hypertrophy, and (4) overriding or dextroposition of the aorta (Figure 16-21). The amount of blood flow to the lungs depends on the severity of obstruction to flow through the pulmonary artery. With mild narrowing, enough blood may flow through the pulmonary artery to keep right-sided pressures lower than left-sided pressures, and the shunt will be left-to-right through the VSD (no cyanosis). Severe narrowing creates high resistance to pulmonary blood flow, increasing PVR over SVR, causing right-to-left shunting across the VSD and into the overriding aorta. Infants with tetralogy of Fallot may exhibit a “boot-shaped” heart on chest x-ray secondary to right ventricular hypertrophy caused by high PVR. Periods of cyanosis may be mild or severe, depending on the degree of pulmonary stenosis. Surgical repair is recommended, with the type of repair and timing of the surgery determined by the specific details of the defect. Infants with severe pulmonary stenosis require prostaglandin therapy to maintain adequate pulmonary blood flow through a PDA.
Transposition of the Great Arteries
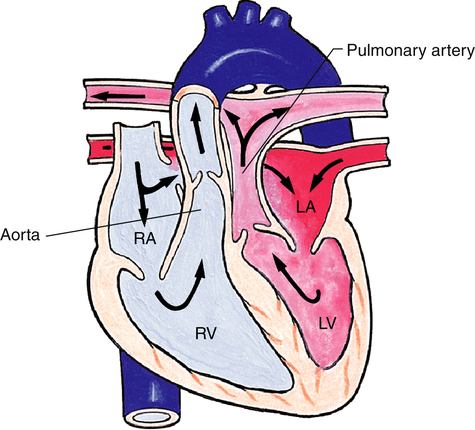
Transposition of the great arteries (TGA) causes severe cyanosis that is usually evident shortly after birth. In TGA, the aorta arises from the right ventricle, and the pulmonary artery arises from the left ventricle, creating two parallel circulations (Figure 16-22). Deoxygenated blood flows from the right atrium, through the right ventricle to the aorta, out into the systemic circulation and then back to the right atrium. Oxygenated blood returning to the left atrium through the pulmonary veins travels through the left ventricle, into the pulmonary circulation, and back to the left atrium.
Anomalous Venous Return
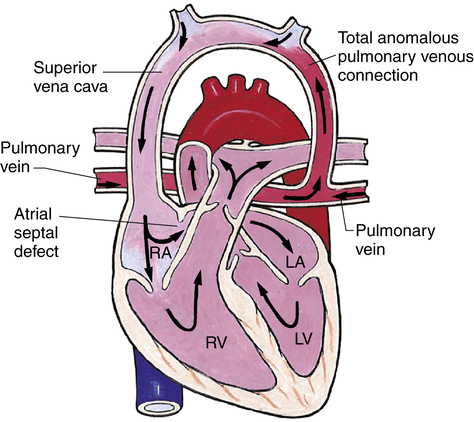
In anomalous venous return, oxygenated blood returning from the lungs is carried by the pulmonary veins abnormally to the right atrium instead of the left atrium (Figure 16-23). The result is mixing of pulmonary and systemic blood in the right atrium, increasing pressures on the right side of the heart. An ASD must be present for oxygenated blood to move across to the left side of the heart and into systemic circulation. These infants are usually cyanotic and must have early surgical intervention.
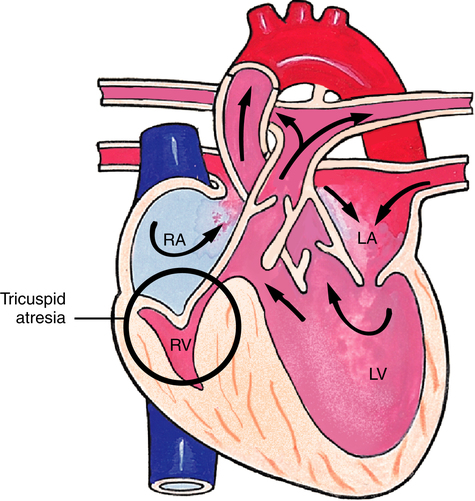
Tricuspid Atresia
Complete occlusion of the tricuspid valve and hypoplastic (underdeveloped) right ventricle characterizes tricuspid atresia (Figure 16-24). Blood cannot flow from the right atrium into the right ventricle, so circulation is maintained by a right-to-left shunt through either an ASD or patent foramen ovale. A VSD is also present, allowing small amounts of blood to be directed to the right ventricle and pulmonary circulation. With this disorder, cyanosis is usually present at birth. Numerous surgical options are available for treatment. Preoperative management includes prostaglandin administration to maintain ductal patency and enhance pulmonary blood flow.
Truncus Arteriosus
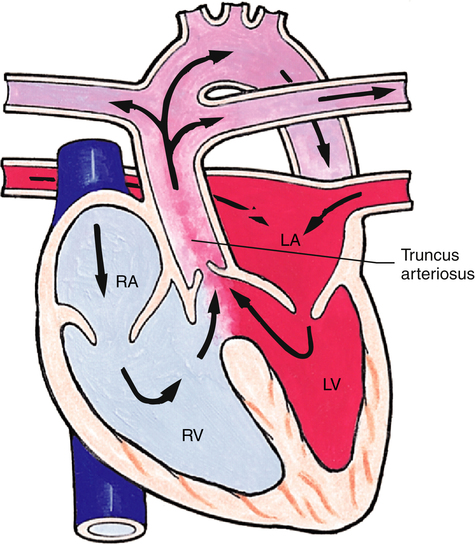
Truncus arteriosus is a defect resulting from failure of the embryonic truncus arteriosus to separate into the aorta and pulmonary artery (Figure 16-25). A single great artery overlies the left and right ventricles, receiving blood through a large VSD. Pulmonary and systemic blood mix and circulate throughout the body, resulting in cyanosis. Surgical treatment involves separating the common vessel into two separate vessels and closing the VSD.
Obstructive Congenital Anomalies
Coarctation of the Aorta
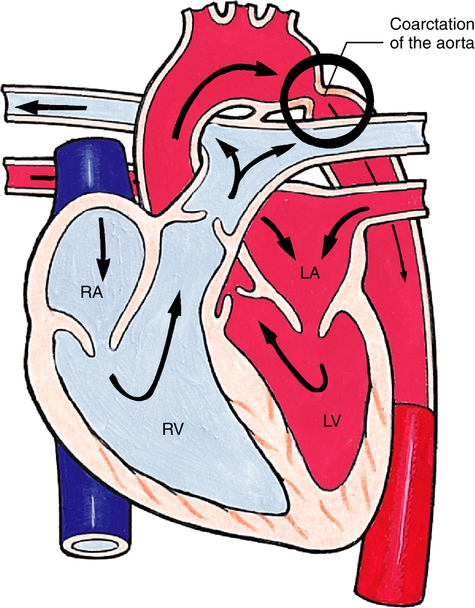
A constricted area of the aortic lumen is known as coarctation of the aorta (Figure 16-26). Narrowing may occur at any point along the aorta; however, in 90% of the cases, the constriction is found directly opposite the ductal entrance into the aorta.3 Clinical manifestations depend on the severity of obstruction. Blood pressure is higher in the upper extremities than in the lower extremities; heart murmurs (fluttering sound heard with a stethoscope, associated with blood moving through a narrowed lumen) can be heard; and cardiomegaly (enlarged heart) may be seen on chest x-ray. Cardiomegaly occurs because of an increased workload caused by high blood flow resistance through the narrowed aorta. Coarctation of the aorta may occur in association with other cardiac defects such as PDA, VSD, ASD, or mitral regurgitation. Treatment is usually surgical repair or sometimes balloon dilation of the constricted area (balloon angioplasty).
Aortic Stenosis
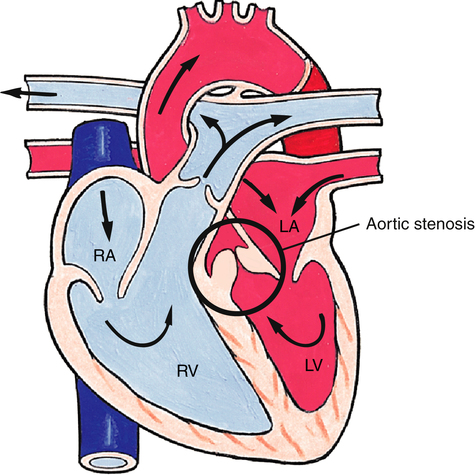
Narrowing found above, below, or at the level of the aortic valve obstructs flow out of the left ventricle and is the cause of the congenital heart defect known as aortic stenosis (Figure 16-27). Clinical manifestations depend on the degree of stenosis. Resistance to blood flow through the aorta results in increased left ventricular work and eventual hypertrophy. Left ventricular pressures may become high enough to cause elevation of pulmonary venous and pulmonary capillary pressures, causing pulmonary edema and congestive heart failure. Numerous options exist for surgical repair, including balloon dilation of the stenotic aortic valve, removal of the stenotic area of the aortic valve, and repair of the area above the valve with a patch to widen the narrowed area.
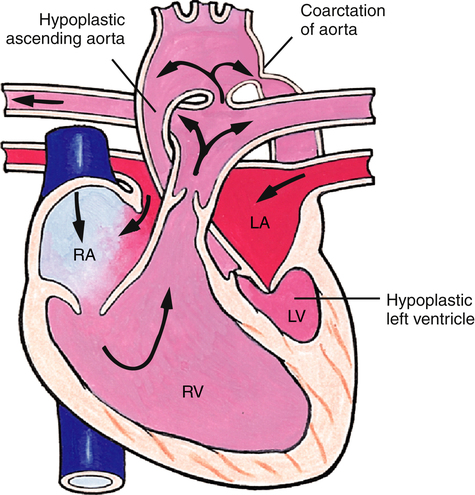
Hypoplastic Left Heart Syndrome
Hypoplastic left heart syndrome includes (1) hypoplastic left ventricle, (2) aortic and mitral valve stenosis or atresia, and often (3) hypoplasia of the ascending aorta (Figure 16-28). Blood flow through the left ventricle is severely compromised, necessitating a PDA to supply the systemic circulation and an ASD to allow pulmonary venous blood to flow into the right atrium. Infants born with hypoplastic left heart syndrome appear gray and hypotensive shortly after birth and die without surgical intervention or heart transplantation.