Familial endocrine disease
genetics, clinical presentation and management
Introduction
A brief overview of clinical endocrine genetics
The growth, replication and differentiation of cells are regulated by many different genes. When these genes become damaged – or ‘mutated’ – cell proliferation may become disordered and give rise to a tumour, whether benign or malignant. The majority of tumours result from acquired genetic damage which accumulates in a complex stepwise, age-related fashion. Some tumours, however, result from a germ-line – usually inherited – gene mutation. This can give rise to a familial tumour predisposition syndrome (Fig. 4.1), and the familial endocrine diseases discussed on the following pages are examples of such syndromes. They are typically characterised by predisposition to one or more tumours arising in endocrine and some neural crest-derived tissues, both benign (functional and non-functional endocrine tumours) and malignant (e.g. medullary thyroid cancer), and often separated by many years. Some individuals and families, however, only ever manifest with one tumour type: familial medullary thyroid cancer and familial hyperparathyroidism, for example.
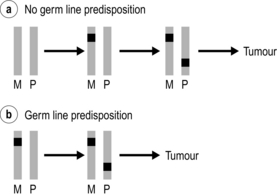
Figure 4.1 Single gene predisposition to endocrine tumours: ‘two-hit’ hypothesis. A stereotypical ‘tumour gene’ is represented by a grey bar. A mutation is denoted by a black square. Most individuals (a) will inherit two normal copies (‘alleles’) of the gene, one from their mother (M) and one from their father (P). Over time, one allele may become damaged (the first hit), but the remaining allele needs to be damaged (the second hit) in order to trigger a tumour. The probability of this process happening more than once in an individual is low, so the development of second primary endocrine tumours is rare. In individuals with a germ-line predisposition (b) the first hit is either inherited from a parent or occurs as a sporadic event during parental spermatogenesis or oogenesis. Again, a second hit affecting the second allele is required to trigger tumour development. Statistically, this process is more likely to happen more than once, giving rise to metachronous endocrine tumours.
• Identification of a disease-causing mutation: a change in the gene sequence that has a predictable deleterious effect on gene function or protein chemistry and is therefore believed to be the cause of the proband’s disease.
• Identification of a variant of uncertain significance (VUS): a change in the gene sequence that usually results in an amino acid change in the corresponding protein but which has an unpredictable effect on that protein. In this situation, further investigations may clarify whether the variant is causally related to the phenotype. A VUS should never form the basis of a predictive genetic test (see below).
• No mutation or VUS identified: the mutation detection rate for a given gene in a particular clinical context is rarely 100%; in other words, the disease-causing mutation is undetectable in a proportion (usually small) of individuals with classical disease. Failure to identify a mutation has three possible implications:
• The diagnosis is correct but the gene mutation has not been identified. For example, a small proportion of probands with classical MEN1 have a mutation that cannot be detected using current technology (see under MEN1). This may also be a particular problem in diseases that can be caused by mutations in a number of different genes (e.g. familial paraganglioma/phaeochromocytoma), and should prompt the question ‘have we tested the correct gene?’
• The diagnosis is correct but the phenotype is not caused by a germ-line gene mutation, for example extra-adrenal paraganglioma/phaeochromocytoma.
• The diagnosis is not correct. For example, an 80-year-old woman with primary hyperparathyroidism, acromegaly and no family history of endocrine disease is likely to have a normal MEN1 genetic test result. The term phenocopy is used in this case to describe someone with coincidental ‘common’ endocrine problems that mimic MEN1.
Multiple endocrine neoplasia type 1 (MEN1)
MEN1 is an autosomal dominant familial syndrome characterised by the development of multiple and metachronous endocrine and non-endocrine tumours (Table 4.1). Approximately 10% of cases arise de novo, without a prior family history of the syndrome.1 The precise prevalence of MEN1 is unclear. This in part refects variability in disease expression, even though penetrance may be high. The hallmark features of MEN1 are endocrine tumours of the pituitary, pancreas and parathyroid.
Table 4.1
Clinical features of multiple endocrine neoplasia type 1 (MEN1)
| Tumour/site* | Hormonal/other characteristics* |
| Parathyroid adenoma (90%) | |
| Enteropancreatic islet tumour (30–80%) | NF (80%)Gastrinoma (40%) Pancreatic polypeptidoma (20%) Insulinoma (10%) Glucagonoma VIPoma Somatostatinoma ACTHoma (rare) GRFoma (rare) |
| Anterior pituitary tumour (10–60%) | Prolactinoma (20%) NF (6%) GHoma (5%) ACTHoma (2%) |
| Foregut carcinoid | Gastric ECL tumour (10%) Thymic carcinoid (2–8%) Bronchial carcinoid (2%) |
| Adrenocortical tumour | Non-functioning adenoma (25%) Adrenocortical carcinoma (rare) Hyperaldosteronism (rare) |
| Cutaneous manifestations | Lipoma (30%) Angiofibroma (85%) Collagenoma (70%) |
*Values in brackets are estimates of penetrance of given characteristic at age 40.
Genetics
MEN1 is associated with heterozygous germ-line loss of function mutations in the MEN1 gene located on chromsome 11q13.2 Endocrine tumours from patients with MEN1 demonstrate loss of heterozygosity for the MEN1 locus, indicating that tumour formation is dependent on the development of a second somatic mutation in the wild-type allele (Fig. 4.1). MEN1 therefore acts as a tumour suppressor gene. Heterozygous MEN1 mutant mice develop tumours mimicking the human phenotype.3 The MEN1 gene encodes a 67-kDa protein – menin – which has multiple functional domains (Fig. 4.2). Menin can influence a number of key cellular processes including transcription, DNA repair and cytoskeletal function. Menin is known to bind several signalling proteins including JunD and Smad3. Recent data have highlighted menin’s role in the regulation of key developmental genes through influences on histone methylation.4,5
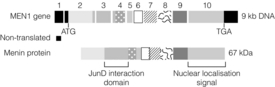
Figure 4.2 Schematic representation of MEN1 gene and menin protein, indicating functional domains of menin protein.
• Mutation in a non-coding, regulatory region of the MEN1 (e.g. promoter).
• Presence of a whole exon deletion or duplication. Most mutation searching strategies now include an exon dosage assay.
• Disease mediated though an alternative MEN1 locus.
• Phenocopy – this refers to the chance ocurrence of two or more endocrine pathologies (both of which can be seen as part of MEN1) in the same person. One of these is usually primary hyperparathyroidism, which is a common sporadic condition, and the patient is usually over the age of 50.
MEN1 exhibits variable penetrance and variable expressivity (see above). Not all features of MEN1 will occur in a single patient or indeed a single family. Some families exhibit only hyperparathyroidism.1 There is considerable variation in age-related tumour penetrance and no clear genotype–phenotype correlation. It is therefore difficult to predict with any degree of accuracy the natural history of MEN1 in an individual or within a family.6
Presentation
Enteropancreatic islet tumours
The prevalence of enteropancreatic islet tumours in patients with MEN1 may be as high as 80%, although the majority of such tumours are clinically silent and non-functional. Functional tumours can present in the second decade of life. Many a symptomatic patients have radiologically detectable tumours by the third decade. Tumours can arise throughout the pancreas and the duodenal submucosa. They are commonly multicentric, metachronous, and range in size and characteristics from micro- and macroadenomas to invasive and metastatic carcinoma. The prognosis of these tumours may relate to specific somatic molecular changes.7
Pituitary tumours
The prevalence of pituitary tumours in MEN1 is uncertain, due to the range of patients and methods employed in the majority of studies to date. A large European multicentre study of 324 patients with MEN1 found pituitary tumours in 42% of cases.8 The most common pituitary lesion is prolactinoma. There are few prospective data on age-related penetrance of pituitary disease. However, MEN1-associated pituitary macroadenoma has occurred as early as 5 years of age.9
Foregut carcinoids
MEN1-associated foregut carcinoid tumours are found in the thymus, stomach and bronchi. They are not generally hormonally active, and do not present with carcinoid syndrome. Their true prevalence is unclear. Gastric enterochromaffin-like (ECL) tumours are generally discovered at endoscopy. They exhibit loss of heterozygosity at the MEN1 gene locus and are promoted by hypergastrinaemia. Thus, they generally arise in MEN1 patients with gastrinoma. They can regress with normalisation of gastrin levels after surgical excision of gastrinoma.10 Thymic carcinoid disease has been highlighted as a major cause of mortality in MEN1. However, relatively little is known about its natural history. A prospective study of 85 patients with MEN1 found an incidence of 8% over a mean follow-up period of 8 years.11 Patients were all male, and most had no symptoms of the tumour at the time of detection. Interestingly, 4 of 7 of the tumours did not show somatic loss of heterozygosity at the MEN1 locus, raising questions as to the mechanism of tumour development. Serum chromogranin A was elevated in 6 of 7 tumours. Mean time interval between diagnosis of MEN1 and development of thymic carcinoid was 19 years. It may be that as early mortality reduces in MEN1 due to improved surgical and medical treatment, this relatively late expression of the disease increases in prevalence and impact.
Adrenocortical tumours
Adrenocortical disease occurs in 20–40% of patients with MEN1. It is unusual in patients who do not have pancreatic disease. Pathology may include diffuse hyperplasia, solitary adenoma and carcinoma.12 Disease can be bilateral. Excess hormone secretion is rare, and the majority of lesions are detected on routine radiological monitoring.13
Cutaneous manifestations
A variety of cutaneous pathologies are now firmly established as components of MEN1. Cutaneous lipomas are often nodular and multicentric. Visceral lipomas have also been described. Cutaneous manifestations of MEN1 are useful clinically in the presymptomatic diagnosis of MEN1 in affected families.14
Diagnosis
The application of diagnostic DNA analysis has altered the phenotypic spectrum of MEN1, revealing both asymptomatic individuals and those with atypical phenotypes. DNA analysis does not always provide answers, however, as illustrated by phenocopies: the association of an endocrine tumour that has a low population prevalence – such as growth hormone (GH)-secreting pituitary tumour – with PHP could represent MEN1 or MEN1 phenocopy. Recent data suggest that mutations in the MEN1 coding region are infrequent in those patients without a family history of MEN1 who develop this combination of endocrinopathies.15 Absence of an MEN1 mutation may therefore be difficult to interpret, particularly if the patient is young and there is no supportive family history.
Management
MEN1 is associated with premature death, most commonly (30%) through metastatic islet cell tumours.16 Advances in the medical management of gastrinoma and hyperparathyroidism may result in a paradoxical increase in cumulative morbidity from other facets of the condition in the coming decade. The principal organs involved in MEN1 are difficult to screen for early tumours, and prophylactic surgery is either not appropriate or has not been shown to prevent the development of tumour (cervical thymectomy).11 The challenge is therefore to improve morbidity and mortality through targeted surgical and medical interventions as directed by surveillance and molecular screening programmes that aim to detect disease expression at an early stage in an inclusive manner.17
Primary hyperparathyroidism
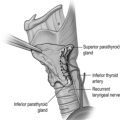
PHP in MEN1 is characterised by asynchronous involvement of all parathyroid glands. However, there remains debate as to the optimum type and timing of parathyroid surgery. Subtotal parathyroidectomy for PHP in MEN1 is associated with a surgical cure rate (as defined by the number of patients not hypercalcaemic) of 60% at 10 years and 51% at 15 years.18 The alternative, total parathyroidectomy with or without autograft, is associated with postoperative hypoparathyroidism and lifelong treatment with vitamin D analogues. Preoperative imaging and minimally invasive approaches may be difficult because of the need to examine all four glands. Transcervical thymectomy is recommended at the time of parathyroidectomy.
Enteropancreatic islet tumours
Surgery is the main treatment for patients with insulinoma in MEN1 (Figs 4.3 and 4.4). All other syndromes of hormone excess due to enteropancreatic tumours respond well to medical therapy with proton-pump inhibitors (gastrinoma) or somatostatin analogues (VIPoma). The timing of surgery in the management of these conditions is debated.
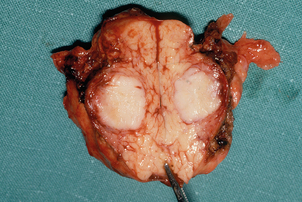
Gastrinomas in MEN1 are often multifocal and small, and can be situated in the duodenum. Extensive pancreatic–duodenal surgery can be associated with significant morbidity. Surgery for gastrinoma in MEN1 is frequently not curative, in part due to the multifocal nature of the problem.19 Furthermore, metastatic disease is found at surgery in a substantial number of patients in whom it is not apparent preoperatively.20 Nevertheless, the outcome of patients treated surgically for locally advanced disease can be the same as those with limited disease. Indeed, there are data that demonstrate that surgery is beneficial in increasing disease-related survival and decreasing advanced disease in Zollinger–Ellison syndrome.21

A subset of MEN1 patients with gastrinoma has aggressive disease and decreased survival.22 Features associated with aggressive tumour behaviour include:
• diagnosis of MEN1 before 35 years of age;
• onset of gastrinoma at 27 years or younger;
Aggressive antitumour treatment in this group needs to be considered.
The standard surgical approach other than for gastrinoma is spleen-preserving distal pancreatectomy (Fig. 4.5) and intraoperative bidigital palpation, coupled with intraoperative ultrasound and enucleation of any tumour found in the pancreatic head and duodenal submucosa. Surgery for gastrinoma should include duodenotomy.23 A Whipple procedure may be considered for tumours at the pancreatic head. Preoperative localisation of the target lesion with corroborative intraoperative ultrasound is useful in planning the appropriate approach. This can be important in the management of functional tumours as the pancreas and duodenum may contain multiple abnormalities, leading to uncertainty as to which of several lesions is the source of excess hormone production. Surgery prompted by abnormal biochemistry but in the absence of any scan-detected lesion should be considered to prevent malignant transformation of microadenomas. Distal 80% subtotal pancreatectomy should be considerd for risk modification in any paitent undergoing surgery for localised islet-cell tumour in MEN1.24
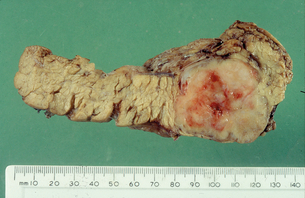
Pituitary tumours
Pituitary tumours should be managed in the same manner as in isolated pituitary disease. Prolactinomas should be treated with dopamine agonists, with biochemical and radiological confirmation of response. Normalisation of prolactin levels without tumour shrinkage suggests misdiagnosis of a non-functioning pituitary adenoma with secondary hyperprolactinaemia. Non-functioning tumours should be treated with surgery. GH-secreting adenomas are best treated with primary surgery followed by consideration of external beam radiotherapy and somatostatin analogue therapy for persistent disease.14
Foregut carcinoids
The optimum management of this generally late expression of MEN1 is unclear. Resection of bronchial carcinoid is usually required to make the diagnosis. Long-term follow-up is then required to check for recurrence. The natural history and malignant potential of ECL gastric carcinoids is unclear. Thymic carcinoid tumours are generally asymptomatic when detected through radiological screening, and can behave aggressively. Relapse is common after surgery, and the optimum adjuvant medical and radiotherapeutic approaches are not yet established.11
Surveillance and screening

Biochemical and radiological surveillance
Biochemical and radiological screening should be offered to all patients with a diagnosis of MEN1, to asymptomatic relatives found to harbour an MEN1-defining MEN1 mutation on genetic testing, and to those found to be at risk through linkage studies (Table 4.2). First-degree relatives of those patients with MEN1 in whom an MEN1 mutation has not been found should also be offered screening pending the outcome of promoter and exon dosage analyses. Biochemical and radiological screening should commence in early childhood, balancing age-dependent penetration, sensitivity of specific studies in specific age groups, and the inconvenience caused by the process. Screening should be lifelong for those patients with MEN1, those known to harbour MEN1-defining MEN1 mutations, and those defined as ‘at risk’ by haplotype and linkage studies. It should continue to the age of 50 in those kindreds in whom no genetic risk stratification is possible.
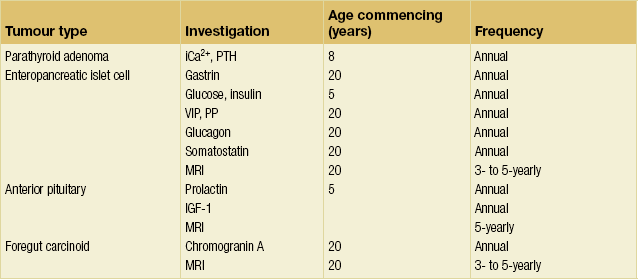
MEN1: differential diagnosis
Familial isolated pituitary adenoma (FIPA)
Pituitary adenomas can occur in MEN1 and Carney syndrome (see below), as well as familial isolated pituitary adenoma (FIPA) syndrome. Indeed, familial acromegaly has been recognised for many years. FIPA is an autosomal dominant condition with variable penetrance: 15–25% of FIPA families harbour heterozygous mutations in the aryl hydrocarbon receptor-interacting (AIP) gene; in the remaining 80% of families the causative gene – or genes – remain(s) unknown.25,26
Presentation: FIPA is characterised by early-onset pituitary adenoma, particularly somatotrophs, lactotrophs and somatolactotrophs. Corticotrophs, gonadotrophs and non-functioning pituitary adenomas (NFPAs) have also been described. Age of onset is younger than in sporadic pituitary adenomas, particularly so in families with AIP mutations.25 FIPA-related somatotroph adenomas appear to behave aggressively and response to somatostatin analogue therapy is poor.
Management: Identification of a mutation in the AIP gene not only serves to confirm the diagnosis but allows accurate cascade screening of the family. Mutation analysis of AIP is available in several laboratories worldwide. Individual features of the syndrome should be managed as in sporadic disease. Presentation of cortisol excess may be atypical and indolent. Diagnosis of FIPA syndrome should trigger periodic clinical, biochemical and radiological screening for additional features with the aim of reducing associated morbidity. However, there is currently no clear consensus as to the age this should commence, the method or the frequency of review/surveillance.
Familial intestinal carcinoid
This appears to be very rare. Germ-line sequence variants in the SDHD gene have been reported in a small series of apparently sporadic intestinal carcinoids, although it is not clear whether these represent disease-causing mutations.27 This may be akin to the identification of SDHD mutations in individuals presenting with apparently sporadic paraganglioma/ phaeochromocytoma (see below).
Multiple endocrine neoplasia type 2
• MEN2A – MTC (90%), phaeochromocytoma (50%) and PHP (20–30%).
• MEN2A with cutaneous lichen amyloidosis.
• MEN2A with Hirschsprung’s disease (HD).
• Familial medullary thyroid cancer (FMTC) – at least 10 or more carriers or affected cases of MTC in a kindred over the age of 50 with no clinical or detectable evidence of other features of MEN2.
• MEN2B – MTC, phaeochromocytoma, decreased upper/lower body ratio, marfanoid habibitus, gastrointestinal and mucosal ganglioneuromatosis.
Genetics
MEN2 is associated with heterozygous gain of function mutations in the RET gene found on Ch10q11.2. The RET gene codes for a membrane-associated tyrosine kinase with an extracellular cadherin-like domain and two independent intracellular tyrosine kinase (TK) domains (Fig. 4.6). RET protein is expressed by a range of neuroendocrine cell types including the adrenal medulla, thyroid C-cells and parathyroid. In normal physiology, extracellular signals lead to RET dimerisation, triggering TK domain phosphorylation and a downstream signal transduction cascade leading to cell growth and differentiation. Gain of function mutations found in MEN2 produce constitutive activation of the RET signal transduction cascade outwith normal control processes.28,29
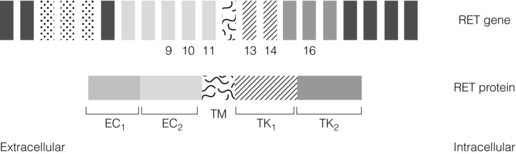
Figure 4.6 Schematic representation of MEN2 gene and RET protein, highlighting domain structure of RET protein.
In contrast to MEN1, there is a partial genotype– phenotype correlation in MEN2. For the majority of families with MEN2A and FMTC the mutations in RET affect cysteine residues in the extracellular domain of the RET protein. The exact position of the cysteine residue involved by any particular mutation affects the likelihood of the phenotype being either MEN2A or FMTC. Virtually all mutations in MEN2A are found in exons 10 and 11 of the RET gene. For FMTC, mutations may be found in exons 13–15 as well as some in exons 10 and 11. For MEN2B, 95% have a mutation in exon 16 (codon 918), at a site that is prone to somatic mutation in sporadic MTC (Fig. 4.7).30 There are data to suggest that there may be additional modifying factors, such as key RET single nucleotide polymorphisms (SNPs), that impact on disease expression within a given genotype. These may be particularly relevant in the situation of RET mutations that result in relatively weak constitutive activation.31,32
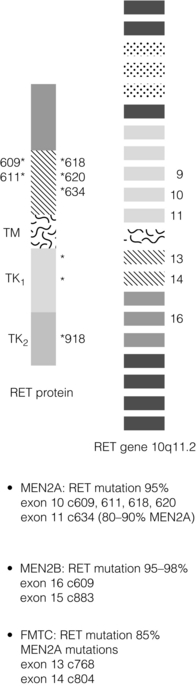
Presentation
Medullary thyroid cancer
MTC has been the first manifestation of MEN2 in most kindreds. It can present in the first decade of life with intrathyroidal, locally advanced or disseminated disease. Historically, MTC has been the major cause of morbidity and mortality in MEN2. Current management approaches will alter this natural history (see later). MTC in MEN2 is preceded by C-cell hyperplasia. Recent data have highlighted a 6.6-year window between development of MTC and progression to nodal metastases in MEN2A patients harbouring the most common (codon 634) RET mutation.33
Management
New cases of MEN2 presenting with MTC should be treated by thyroidectomy with central or more widespread node dissection, depending on pre- and perioperative staging. Thyroidectomy for MEN2B should include central node dissection. However, the aim of surgical management encompasses and is focused increasingly on prevention of MTC. Surgery for MTC in MEN2 should be performed before the age at which malignant progression occurs.34 Historically, this decision was based on basal and stimulated levels of the hormone calcitonin, produced by C-cells of the thyroid and a valuable tumour marker for MTC. However, this approach has an unacceptable sensitivity and specificity. Decisions on the timing of thyroidectomy in new cases of MEN2 without apparent MTC at presentation (such as those cases detected through genetic screening) should follow a stratified approach based on the genotype–phenotype relationships linking specific RET mutations with a specific natural history of MTC. Such an approach balances the earliest age at which MTC can present in association with a given RET genotype against the potential surgical morbidity of thyroidectomy at a young age (Fig. 4.8).
Figure 4.8 Operative view at the conclusion of a prophylactic thyroidectomy for MEN2B in a child aged 18 months.

Patients are assigned to one of three risk bands:
• Risk level 1 – all patients with MEN2B; patients with RET mutations involving codons 883, 918 and 922.
• Risk level 2 – patients with RET mutations involving codons 611, 618, 620 and 634.
• Risk level 3 – patients with RET mutations involving codons 609, 768, 790, 791, 804 and 891.
Patients in risk level 1 should undergo thyroidectomy in the first year of life, and preferably in the first 6 months of life. Those in risk level 2 should undergo thyroidectomy before the age of 5 years. There is no consensus on the optimum approach to patients in risk level 3. MTC presents at an older age in this group, and is commonly less aggressive. Recent data suggest that thyroidectomy need not take place before the age of 10 years, and that central node dissection is unnecessary before 20 years. The cumulative experience on which such recommendations are based remains limited.33,35
Accumulating experience suggests that a relatively conservative approach, involving serial monitoring, may be appropriate for some families harbouring ‘milder’ RET mutations.36 Some have proposed that this approach can be supported by using serial pentagastrin stimulation tests to assist decision-making on the timing of surgery.37
Persistence of elevated calcitonin levels following primary surgery should trigger radiological staging with computed tomography (CT) or magnetic resonance imaging (MRI). 111In pentetreotide scanning can detect somatostatin receptor-positive disease. Flurodopamine positron emission tomography (PET) is an additional sensitive modality for detection of occult recurrent MTC.38 Local recurrent or residual disease is the most common cause of persistently elevated tumour markers following primary treatment. In the absence of widespread distant disease, re-operation should be considered. If more distant metastatic disease is found, repeat surgery for tumour debulking should be considered for control of local pressure symptoms or those due to humoral factors secreted by the tumour.
Standard chemotherapy regimens are not particularly effective in the management of systemic metastatic disease. Novel agents targeting angiogenesis and components of the RET signalling pathway may prove to be beneficial in patients with disseminated disease.39 External beam radiotherapy can be used for the palliative treatment of bone metastases. However, metastatic MTC can remain asymptomatic, and conservative approaches to management coupled with regular biochemical surveillance of tumour load can result in good quality of life for many years.
Phaeochromocytoma
The principles of diagnosis and intervention should be similar to those applied to sporadic disease (see Chapter 3). However, it is important to exclude active phaeochromocytoma in any patient with suspected or established disease prior to surgical intervention for a separate or linked condition, in early pregnancy and prior to labour.

Analysis of timed overnight urine metanephrines, together with plasma metanephrines, provides the highest degree of sensitivity and specificity in biochemical diagnostic and surveillance programmes.40 Plasma metanephrines alone may generate significant false-positive screening data. Positive biochemistry should trigger appropriate imaging studies with MRI, supported by radionuclear imaging if necessary.
Surveillance and screening
RET analysis should also be considered in all patients presenting with apparently sporadic single MEN2-related tumour features. For example, a significant proportion of individuals presenting with medullary carcinoma of the thyroid are found to have an RET mutation, whereas a much smaller proportion of individuals with adrenal phaeochromocytoma have an RET mutation.41,42 Failure to detect an RET mutation in a sporadic MTC patient leaves a residual probability of MEN2/FMTC, although in practice this is small. If DNA testing is to be offered in this context, it is important that issues of consent and data disclosure are addressed carefully in view of the potential consequences of identifying germ-line RET mutations for both the inidividual and other family members.
Biochemical and radiological surveillance

• New patients presenting with two synchronous or metachronous features of MEN2.
• Patients presenting with a single manifestation of MEN2 who have a first-degree relative with an endocrine feature of MEN2.
• Infants presenting with gastrointestinal or mucosal features of MEN2B.
• Patients presenting with MTC.
• Infants presenting with Hirschsprung’s disease and a family history suggestive of MTC.
MEN2: differential diagnosis
Familial phaeochromocytoma/paraganglioma
Genetics: Familial paraganglioma may result from predisposing mutations in an increasing number of genes. These genes fall into two main groups:
1. Those that prevent degradation of the transcription factor HIF (hypoxia-inducible factor), a key regulator of the cellular response to hypoxia. Mutations in VHL, SDHAF2, SDHB, SDHC and SDHD result in overactivity of HIF-mediated processes, resulting in overexpression of angiogenesis factors and tumour formation.43
2. Those that result in abnormal expression of genes involved in RNA synthesis, protein production, and signalling in molecular pathways involved in apoptosis. The two genes in which this has been particularly studied are RET and NF1.
The molecular mechanisms for mutations in the KIF1B and TMEM127 genes are not well understood.44,45 Heterozygous loss of function germ-line mutations in SDHB, C and D have also ben identified in patients with the diad of paraganglioma and gastrointestinal stromal tumour (the Carney–Stratakis syndrome). This is an autosomal dominant condition with incomplete penetrance. Why some patients express this diad while others only express paraganglioma remains to be determined.46
Presentation: Familial paraganglioma syndromes can present with tumour in the head and neck, chest or abdomen. Not all paragangliomas are secretory. Only 5% of those occurring in the head and neck (such as those arising from the carotid body) are thought to secrete catecholamines and thus present with local symptoms. Functional paragangliomas and phaeochromocytomas present in the same manner as in sporadic disease, though the development of effective screening programmes in affected families is likely to lead to increasing detection in the asymptomatic phase.
Patients with familial paraganglioma due to mutations in the genes encoding succinate dehydrogenase subunits B, C and D (SDHB, SDHC, SDHD) can develop both phaeochromocytoma and paraganglioma.47 Paraganglioma in SDHB-related disease is usually intrathoracic or intra-abdominal. Malignant behaviour is relatively common. In contrast, paraganglioma in SDHD-related disease is generally confined to the head and neck and is usually biochemically silent. Familial paraganglioma due to mutations in the gene encoding SDHC presents with non-functioning head and neck tumours and/or phaeochromocytoma.48
Age-related, and site-specific, penetrance for SDHB and SDHD is shown in Tables 4.3 and 4.4; these figures may be an overestimate since they are derived from cross-sectional data in referral populations. Penetrance of SDHD and SDHAF2 mutations appears to be dependent upon parent of origin, a phenomenon known as ‘genomic imprinting’ (Fig. 4.9). The disease is not expressed if the mutation is inherited from a female, although exceptions have been reported.51–53
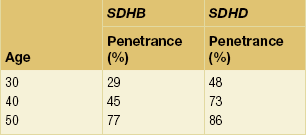
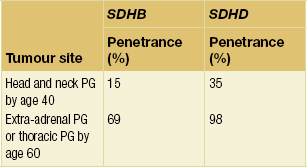
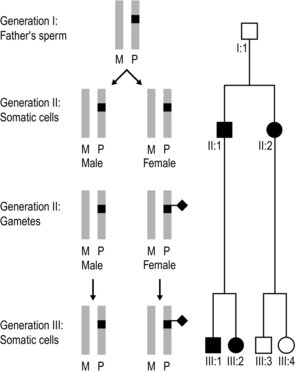
Figure 4.9 Imprinting effect in SDHD and SDHAF2-associated familial paragangliomatosis. A pair of grey bars represents the two alleles of the SDHD (or SDHAF2) gene; M denotes the allele inherited from a person’s mother and P the mutant copy inherited from a person’s father. The mutation is ‘silenced’ following transmission through a female (indicated by a black diamond), so that the disease will only express itself if the mutant allele has been inherited from a male. In this example, all individuals shown in the pedigree above can be assumed to have inherited the mutant allele. Individual I:1 has a maternally-derived mutant SDHD/SDHAF2 allele and is disease free. In his sperm he removes the maternal imprint. II:1 and II:2 therefore inherited their mutant allele from their father, and they manifest disease. II:1’s sperm are not imprinted, so III:1 and III:2 manifest disease. II:2 reapplies a maternal imprint in her oocytes so that individuals III:3 and III:4 do not manifest disease, even though they have inherited the gene mutation. III:3’s children have a 50% risk of inheriting the mutant allele and, if they do, they are highly likely to develop paragangliomas. In contrast, III:4’s children have a 50% risk of inheriting the mutant allele but will not manifest disease.
Management: Functional tumours should be removed if possible. Partially excised locally aggressive and metastatic disease may benefit from treatment with [131I]metaiodobenzylguanidine (MIBG).54 Excision of non-functional tumours should be considered if there are significant local symptoms, or radiological evidence of growth on serial monitoring. The metachronous nature of the condition means that recurrences and the development of additional tumours are common.
Genetic testing: Index cases with phaeochromocytoma or paraganglioma – whether familial or apparently sporadic – should be offered diagnostic mutation analysis of predisposing genes. As a minimum, this should include SDHB and SDHD in addition to RET and VHL. Mulitigene assays including SDHAF2, TMEM127, KIF1B and MAX are available in many countries. The results help to define the risk of functional tumour to the patient and in developing genetic screening programmes for other members of the kindred. SDH gene mutations may be found in some 30% of patients presenting with apparent sporadic head and neck paraganglioma.55,56,57 Absence of a mutation may not exclude familial disease; all genetic test results should be interpreted in the context of full clinical and family history data. SDHB immunohistochemistry has recently been shown to be an effective way of identifying tumours that result from germ-line SDH gene mutation; this is helpful in individuals found to have novel sequence variants whose significance cannot be inferred from sequence data alone.58
Biochemical and radiological surveillance: Patients with SDHB, C and D mutations should undergo annual biochemical screening for functional tumours with urine or plasma metanephrines and plasma chromogranin A. At-risk relatives should also be offered biochemical surveillance unless the disease-causing mutation is known in the family and they have had a negative predictive test result. Non-functional tumours are only detectable through clinical and radiological assessment. Optimum strategies for imaging these patients have not yet been established.
SDHB mutations have been identified in a small proportion of families in which susceptibility to renal cell carcinoma segregates as an autosomal dominant trait.59 The opposite also appears to be true: individuals with an SDHB mutation ascertained through a family history of paraganglioma are also at risk of renal cell carcinoma; papillary thyroid cancer has also been described but does not appear to be a common feature.60 Renal surveillance has been suggested for patients with SDHB mutations, although the best screening modality has yet to be confirmed.
Carney–Stratakis syndrome
Heterozygous loss of function germ-line mutations in SDHB, C and D has also been identified in patients with the diad of paraganglioma and gastrointestinal stromal tumour (GIST), known as the Carney–Stratakis syndrome.61 This is a rare autosomal dominant condition with incomplete penetrance. GISTs in this syndrome demonstrate loss of SDHB immunostaining,62 which is a helpful diagnostic test in cases who have not yet developed a paraganglioma. Why some patients express this diad while others only express paraganglioma remains to be determined.
Familial hyperparathyroidism (FHP) syndromes
Familial isolated hyperparathyroidism (FIHP)
FIHP is a rare autosomal dominant disorder characterised by uniglandular or multiglandular hyperparathyroidism in the absence of other endocrine disease and without evidence of jaw tumours.64 Recent data suggest that at least 20% of kindreds thought to have FIHP have inactivating mutations in MEN1, suggesting that a significant proportion of FIHP may represent a distinct variant of MEN1.65 Whether more intensive surveillance will detect other features of MEN1 in these kindreds over time remains unclear. FIHP in some kindreds may thus be a prelude to MEN1 or a skewed variant of MEN1.
Mutations in the CDC73 gene have also been described in some families with FIHP; the same gene is also implicated in the hyperparathyroidism–jaw tumour syndrome (see below).66 A further FIHP gene is thought to lie on chromosome 2, although the gene itself has yet to be identified.
Familial hypocalciuric hypercalcaemia (FHH) and neonatal severe hyperparathyroidism (NSHP)
FHH is inherited as an autosomal dominant condition, and is characterised by lifelong mild to moderate hypercalcaemia that is generally asymptomatic, and normal-range values of parathyroid hormone (PTH). It is caused in many families by heterozygous loss-of-function mutation in the CASR gene, which encodes the calcium-sensing receptor. Gain-of-function mutations cause familial hypoparathyroidism, which is not considered further here. Identification of a mutation in this gene in an FHH family not only serves to confirm the diagnosis but allows accurate cascade screening of the family.67
NSHP is usually caused by homozygous (the same mutation in both alleles) or compound heterozygous (different mutations in each of both alleles) mutations in the CASR gene (and is therefore an autosomal recessive disease), although de novo heterozygous mutations (i.e. new dominant mutations in the affected child) have been reported.68 The disease presents in the first week of life with anorexia, constipation, hypotonia and respiratory distress. There is severe hypercalcaemia (total calcium concentration 3.5–7.7 mmol/L), often with hypermagnesaemia. PTH can be significantly elevated. Skeletal radiology shows demineralisation and typical features of severe hyperparathyroidism. As NSHP can result from recessively inherited CASR mutations, there may be a history or biochemical evidence of FHH in one or both parents.
Familial hyperparathyoidism–jaw tumour syndrome (FHP-JT)
FHP-JT is an autosomal dominant condition caused in many families by mutations in the CDC73 gene (also known as HPRT2). Individuals with FHP-JT manifest variably with hyperparathyroidism, caused in most cases by parathyroid hyperplasia (cystic adenomas have been reported), and ossifying tumours (fibromas) of the mandible and maxilla.69 Polycystic kidney disease has also been described in families with this condition. Hyperparathyroidism presents as in sporadic cases. Jaw and maxillary tumours can be occult, and may only be apparent on screening by orthopantogram. Increased awareness of this condition has led to its recognition as the underlying problem in kindreds previously thought to have familial isolated hyperparathyroidism.70
Both somatic and germ-line mutations in CDC73 have been identified in patients with apparently sporadic parathyroid carcinoma, suggesting that some patients with this unusual tumour may represent a phenotypic variant of FHP-JT that behaves as a rare but typical ‘two-hit’ tumour predisposition syndrome.71
Von Hippel–Lindau disease
Von Hippel–Lindau (VHL) disease is an autosomal dominant familial syndrome characterised by the metachronous development of multiple benign and malignant tumours. It may occur in an individual as the result of a new mutation. Incidence is of the order of 1 in 40 000 and there is variable penetrance and expression.72 Key features are central nervous system haemangioblastoma, renal cell carcinoma and phaeochromocytoma. A number of additional lesions are recognised (Table 4.5).
Table 4.5
Clinical characteristics of von Hippel–Lindau (VHL) disease: age at presentation and frequency of expression
| Tumour | Age at presentation (years) | Frequency of expression (%) |
| Retinal haemangioblastoma | 1–67 | 25–60 |
| Cerebellar haemangioblastoma | 9–78 | 44–72 |
| Brainstem haemangioblastoma | 12–46 | 10–25 |
| Spinal cord haemangioblastoma | 12–66 | 13–50 |
| CNS haemangioblastoma (miscellaneous) | < 1 | |
| Renal cell carcinoma or cysts | 16–67 | 25–60 |
| Phaeochromocytoma | 5–58 | 10–20 |
| Pancreatic tumour or cysts | 5–70 | 35–70 |
| Endolymphatic sac tumours | 12–50 | 10 |
| Epididymal cystadenoma | Unknown | 25–60 |
| Broad ligament cystadenoma | Unknown | Unknown |
Genetics
VHL disease results from a germ-line mutation in the VHL tumour suppressor gene situated at the chromosomal locus 3p25-26 (Fig. 4.10). The products of the VHL gene (a 213-amino-acid, 18-kDa protein and a truncated 160-amino-acid, 18-kDa protein arising from an alternative translational start site) are important components in the pathway targeting intracellular proteins for degradation via proteasomes as part of the integrated cellular response to hypoxia. The tumours seen in VHL are vascular with pronounced angiogenesis. Their cells exhibit over-expression of vascular endothelial growth factor (VEGF). Production of VEGF is mediated by a pathway of hypoxia detection involving the VHL protein and the elongin complex. Many hypoxia-inducible genes are controlled by hypoxia-inducible factor (HIF). HIF is composed of an α subunit and a β subunit. The HIF α subunit is degraded if oxygen is present; this requires functioning VHL protein.
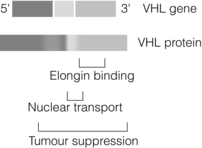
Figure 4.10 Schematic representation of VHL gene and protein, highlighting functional domains of the protein.
VHL disease has been divided into four subtypes on the basis of clinical presentation, as depicted in Box 4.1. To date, endolymphatic sac tumours and cystadenomas of the epididymis and broad ligament have not been assigned to a specific disease subtype. Within this classification there is evidence of genotype–phenotype correlation. Patients with type 1 VHL are most likely to have deletions or premature termination mutations. Those with type 2 VHL are more likely to have missense mutations.73 Expression of subtype phenotype tends to be consistent within a given family. Mutations in VHL are found in the majority of families with VHL disease.
Presentation
VHL disease has two major endocrine manifestations: phaeochromocytoma and pancreatic islet-cell tumours.74 Phaeochromocytoma associated with VHL disease is pathologically distinct from that occurring as part of MEN2. Tumours have a thick vascular capsule, and contain small to medium-sized tumour cells interspersed with multiple small blood vessels. There is no evidence of adrenomedullary hyperplasia outwith the tumour, as can be found in MEN2.75 Clinical presentation of phaeochromocytoma is similar to that in sporadic and other familial forms. However, compared with tumours associated with MEN2, patients presenting with phaeochromocytoma as part of VHL disease have fewer symptoms. This clinical observation correlates with lower tumour catecholamine content and reduced expression of tyrosine hydroxylase.76 Increasingly, presentation is with asymptomatic disease detected through routine biochemical and radiological screening. Tumours can be multiple and extra-adrenal.
Diagnosis
Diagnosis of phaeochromocytoma in VHL disease follows the principles established in sporadic and other forms of familial disease: clinical suspicion, genetic and biochemical testing, and radiological localisation. Phaeochromocytoma associated with VHL disease has a predominantly noradrenergic phenotype. Urine catecholamine excretion can be normal, as can plasma metanephrines. A combination of elevated plasma normetanephrines together with normal plasma metanephrines is highly suggestive of VHL-associated phaeochromocytoma.76 Localisation of biochemical disease can employ MRI, CT and radioisotope scanning.77 Adrenal and extra-adrenal masses detected on routine radiological surveillance for renal cell carcinoma should trigger appropriate testing to exclude phaeochromocytoma, with initial biochemical testing followed by further complementary radiological or radioisotope studies.
Treatment
Surgical and non-surgical treatments for phaeochromocytoma in VHL disease follow the same principles as outlined in sporadic and other forms of the tumour (see Chapter 3).
Surveillance and screening
Index cases should be offered genetic testing. These data may help to guide subtype classification and will enable cascade predictive genetic testing within the wider family. VHL gene analysis should form part of the assessment of patients with apparent sporadic phaeochromocytoma,42 although the genotype–phenotype correlation is not robust enough to enable the broader phenotype to be predicted with accuracy in those patients presenting found to have VHL mutations. Comparisons of the relative effectiveness of molecular and clinical approaches in this situation are required.
Neurofibromatosis type 1 (NF1)
NF178 is an autosomal dominant multisystem disorder with predominant neurological, cutaneous, ophthalmic and skeletal manifestations. Prevalence is estimated at 1 in 3500. Fifty per cent of cases are sporadic, and the disease is usually 100% penetrant by 5 years of age. NF1 can be segmental, due to a postzygotic somatic mutation. Expression is variable.
Two or more of the following criteria are required for a diagnosis of NF1:
• six or more café-au-lait macules > 5 mm prepubertal or > 15 mm postpubertal;
• axillary or inguinal freckling;
• two or more neurofibromas or a single plexiform neurofibroma;
• sphenoid wing dysplasia, thinning of long cortical bones or pseudoarthrosis;
Familial non-medullary thyroid cancer syndromes
PTEN hamartoma tumour syndrome
Mutations in the PTEN gene cause a number of phenotypes that are collectively known as the PTEN hamartoma tumour syndrome.79 Cowden’s syndrome (CS) – the commonest presentation – is an autosomal dominant inherited cancer syndrome, originally described in adults and characterised by three main groups of abnormalities:
1. Multiple benign tumours, including hamartomatous gastrointestinal polyps, benign thyroid disease (adenomatous nodules and follicular adenomas in addition to hypothyroidism or hyperthyroidism), benign breast disease and orofacial skin lesions.
2. Predisposition to breast adenocarcinoma, thyroid cancer (non-medullary, particularly follicular) and endometrial adenocarcinoma.
3. Other features such as macrocephaly, mild to moderate learning difficulties and occasionally Lhermitte–Duclos disease (LDD), an unusual condition of cerebellar ganglion cell hypertrophy that causes ataxia and seizures.
The International Cowden Syndrome Consortium has defined operational criteria for the diagnosis of CS (Box 4.2). In the absence of a family history of CS, a diagnosis can be made on mucocutaneous findings alone if any of the following criteria are met:
• six or more facial papules of which at least three are trichilemmoma;
• facial papules and oral mucosal papillomatosis;
Expression of CS is varied. Penetrance is age dependent, increasing from less than 10% under the age of 20 years to nearly 100% for cutaneous stigmata by the third decade. Thyroid abnormalities occur in 50–67% of CS patients, with a lifetime thyroid cancer risk of around 10%. Benign breast disease affects up to 67% of women; the lifetime breast cancer risk is 85%, with 50% penetrance by age 50. The lifetime endometrial cancer risk is approximately 28%.80
Bannayan–Zonana syndrome (also called Ruvalcaba–Mhyre–Smith or Bannayan–Riley–Ruvalcaba syndrome) is a rarer manifestation of PTEN mutation, described in children, that presents as an autosomal dominant condition characterised by intestinal polyps, haemangiomas and lipomas, café-au-lait patches on the penis and macrocephaly.81 Other features are breast cancer, lipid storage disorder, protein-losing enteropathy and thyroid disease including thyroid cancer. CS and Bannayan–Zonana syndrome have both been shown to be caused by mutations in PTEN. There are some reports of both occurring in the same family as differing manifestations of the same PTEN mutation.82 It is not understood why penetrance and expression of mutations in PTEN can be so variable.
Surveillance and screening
Genetic testing: Diagnostic mutation amalysis of the PTEN gene is now routine. Most diagnostic laboratories offer sequence analysis alone, which will identify a pathogenic mutation in 80% of people who fulfil the diagnostic criteria for CS; research-based promoter sequencing may identify a further 10%. The mutation detection rate in non-CS PTEN hamartoma syndromes is considerably less, presumably as a result of aetiological heterogeneity.79 Identification of a pathogenic mutation allows cascade genetic testing in relatives.
Surveillance: Once a diagnosis of CS has been made, individuals and affected relatives should have endocrine screening with thyroid palpation as part of an annual medical review. The value of routine ultrasound surveillance of the thyroid has not been established. Recommendations for breast and endometrial surveillance are variable.79
Familial papillary thyroid cancer
Between 3% and 13% of patients with papillary thyroid cancer (PTC) have a relative affected by papillary, follicular or mixed papillary/follicular thyroid cancer. The fact that telomeres appear to be shorter in some families with a cluster of non- medullary thyroid cancers (PTC), compared to sporadic cases, suggests that some family clusters are the result of a discrete inherited cancer predisposition syndrome.83 Familial PTC is often more aggressive than its sporadic counterpart, and in keeping with other tumour predisposition syndromes, the disease can be multifocal.
While somatic rearrangements of RET and NTRK1 are common findings in PTC, the genetic basis for familial disease remains unclear. Putative susceptibility loci have been mapped to chromosomes 2q21 and 19p13.2, although the genes themselves remain elusive.84,85
Familial adenomatous poylposis (FAP)
FAP (also sometimes referred to as Gardner syndrome) is an autosomal dominant disorder caused by mutations in the APC gene and characterised by the occurrence of multiple gastrointestinal adenomatous polyps in association with osteomas, epidermoid cysts, desmoid tumours and retinal pigmentation. Hepatoblastomas and adenomas of the upper gastrointestinal tract and pancreas are more unusual components of the syndrome. Expression is variable, though the disease is usually penetrant in the third decade. In families with classical disease, colorectal adenocarcinoma is usual by early adulthood unless prophylactic colectomy can be undertaken. FAP is associated with an increased risk of thyroid neoplasia, particularly for women. However, the risk is sufficiently low (affecting approximately 1% of those with FAP) that – apart from an awareness of the risk – it is unnecessary to organise a screening programme of the thyroid gland.86 There is some evidence of familial aggregation of thyroid cancer in FAP: for such rare families it is important to raise awareness and consider screening.
Familial adrenocortical disease
Familial predisposition to adrenocortical carcinoma
Adrenocortical carcinoma (ACC) is very unusual in children and young adults. When it does occur in childhood, a tumour predisposition syndrome is likely. ACC in this context is often a manifestation of the Li–Fraumeni syndrome: an autosomal dominant familial cancer syndrome caused by heterozygosity for germ-line loss of function mutations in the TP53 tumour supressor gene.87 In a series of 14 such cases, nine were shown to be due to TP53 and two were likely to have TP53 mutations that could not be identified. The one case not due to a TP53 mutation occurred in a child with Beckwith–Wiedemann syndrome, a familial cancer predisposition syndrome resulting in over-expression of the paracrine growth factor insulin-like growth factor 2 (IGF-2). ACC has also been described as a rare feature of FAP (see above), although there remains some doubt as to whether this represents a true or apparent phenomenon.
Carney syndrome
Carney syndrome is a multiple neoplasia syndrome with cardiac, cutaneous, endocrine and nervous system manifestations.88 It is inherited in an autosomal dominant manner. Sixty per cent of affected kindreds harbour an inactivating mutation in the tumour suppressor gene PRKAR1A, which codes for the type 1α regulatory subunit of protein kinase A.89 A second Carney syndrome gene has been localised to chromosome 2, but the gene itself has not been identified.
Presentation
• Spotty pigmentation – hypermelanosis; lentigines; blue naevi; combined naevi.
• Myxomas – cardiac (any chamber and possibly multiple); cutaneous; breast; oral cavity.
• Endocrinopathy – Cushing’s syndrome due to primary pigmented nodular adrenal disease; GH-secreting pituitary tumour; large-cell Sertoli cell tumour of testis; Leydig cell tumour; thyroid tumours; ovarian cysts.
• Psammomatous melanotic schwannoma – sympathetic chain; gastrointestinal tract.
Familial isolated PPNAD and familial isolated atrial myxoma represent rare familial disorders in which a single manifestation of Carney syndrome segregates as a familial trait. PPNAD families have been described with mutations in the PRKAR1A, PDE11A or PDE8B genes.90 Familial isolated atrial myxoma families have been described with mutations in the PRKAR1A gene.91
Management
Approximately 60% of patients with Carney syndrome have a mutation in the PRKAR1A gene.92 Identification of a mutation in this gene not only serves to confirm the diagnosis but allows accurate cascade screening of the family. Mutation analysis of PRKAR1A is available in several laboratories worldwide. Individual features of the syndrome should be managed as in sporadic disease. Presentation of cortisol excess may be atypical and indolent. Diagnosis of Carney syndrome should trigger periodic clinical, biochemical and radiological screening for additional features, with the aim of reducing associated morbidity.
Familial ACTH-independent adrenal hyperplasia
The hypothalamo-pituitary regulation of glucocorticoid production is mediated through ACTH binding to its cognate G-protein-coupled receptor on the plasma membrane of steroidogenic cells of the zona fasciculata and reticularis of the adrenal cortex. Introduction of other, non-ACTH G-protein-coupled receptors to the regulatory pathway controlling steroidogenesis within the adrenal cortex would uncouple the process from the negative feedback loops that maintain normal glucocorticoid production. ACTH-independent macronodular adrenal hyperplasia (AIMAH) is an endogenous form of adrenal Cushing syndrome characterised by multiple bilateral adrenocortical nodules resulting from the ectopic expression of G-protein-coupled receptors on adrenocortical cells that activate steroidogenesis but are not under the influence of negative feedback.93 Although some familial cases have been reported, nearly all AIMAH cases appear to be sporadic, arising from somatic mutation of the GNAS1 gene and constitutive activation of the G-protein.94 Bilateral adrenocortical nodular hyperplasia can also be found in McCune–Albright syndrome, which is also caused by mutation in the GNAS1 gene. The cause of familial AIMAH has not been elucidated.
Familial hyperaldosteronism
Presentation
FHA1 constitutes 1–3% of all cases of primary hyperaldosteronism.95 Unlike other forms of hyperaldosteronism, it is present from birth and has no gender bias. It is characterised by moderate to severe hypertension and elevated aldosterone/renin ratios (though this is not specific). Many patients are normokalaemic. The diagnosis should be considered in any patient presenting with hypertension under the age of 25 years. A strong family history of hypertension is not always apparent. There may be a prominent family history of haemorrhagic stroke. Many patients do not respond to conventional antihypertensive agents, or develop hypokalaemia on potassium-wasting diuretics. The cause is a well-documented fusion of the regulatory component of the CYP11B1 (steroid hydroxylase) gene to the coding region of the adjacent CYP11B2 (aldosterone synthase) gene, which effectively couples aldosterone synthesis to a steroid-responsive control element.96 Diagnostic genetic testing for this fusion gene is widely available. Diagnosis is supported by suppression of aldosterone to undetectable levels during a low-dose dexamethasone suppression test (0.5 mg dexamethasone 6-hourly for 48 hours), giving rise to the alternative terms for this condition of glucocorticoid-remediable aldosteronism and steroid-suppressible hyperaldosteronism.
FHA2 is not suppressible by dexamethasone and is mechanistically distinct from FHA1. It is caused by germ-line mutations in the potassium ion channel gene KCNJ2.97 It is clinically, biochemically and pathologically indistinguishable from non-familial primary hyperaldosteronism, in which somatic KCNJ2 mutations have also been described. Mutations appear to cause increased sodium conductance and cell depolarisation in adrenal glomerulosa cells, resulting in calcium influx, which signals aldosterone production and cellular proliferation.
Management
Management of FHA2 should follow the same principles as that of primary hyperaldosteronism, balancing surgical and medical approaches dependent upon localisation studies against the response to antihypertensive therapy with amiloride, mineralocorticoid antagonists and/or dihydropyridine calcium-channel blockers (see Chapter 3).
References
1. Hannan, F.M., Nesbit, M.A., Christie, P.T., et al, Familial isolated primary hyperparathyroidism caused by mutaions of the MEN1 gene. Nat Clin Pract Endocrinol Metab 2008; 4:53–58. 18084346
2. Chandrasekharappa, S.C., Guru, S.C., Mannickam, P., et al, Positional cloning of the gene for multiple endocrine neoplasia type 1. Science 1997; 276:404–406. 9103196
3. Bertolino, P., Tong, W-M., Galendo, D., et al, Heterozygous Men1 mutant mice develop a range of endocrine tumours mimicking multiple endocrine neoplasia type 1. Mol Endocrinol 2003; 17:1880–1892. 12819299
4. Kaji, H., Canaff, L., Lebrun, J.J., et al, Inactivation of menin, a Smad3-interacting protein, blocks transforming growth factor type beta signalling. Proc Natl Acad Sci U S A 2001; 98:3837–3842. 11274402
5. Scacheri, P.C., Davis, S., Odom, D.T., et al, Genome-wide analysis of menin binding provides insights into MEN1 tumorigenesis. PLoS Genet. 2006;2(4):e51. 16604156
6. Macens, A., Schaaf, L., Karges, W., et al, Age-related penetrance of endocrine tumours in multiple endocrine neoplasia type 1 (MEN1): a multicentre study of 258 gene carriers. Clin Endocrinol (Oxf) 2007; 67:613–622. 17590169
7. Guo, S.S., Wu, A.Y., Sawicki, M.P., Deletion of chromosome 1, but not mutation of MEN-1, predicts prognosis in sporadic pancreatic endocrine tumours. World J Surg 2002; 26:843–847. 11960210
8. Verges, B., Boureille, F., Goudet, P., et al, Pituitary disease in MEN type 1 (MEN1): data from the France–Belgium MEN1 multicenter study. J Clin Endocrinol Metab 2002; 87:457–465. 11836268
9. Stratakis, C.A., Schussheim, D.H., Freedman, S.M., et al, Pituitary macroadenoma in a 5-year old: an early expression of multiple endocrine neoplasia type 1. J Clin Endocrinol Metab 2000; 85:4776–4780. 11134142
10. Richards, M.L., Gauger, P., Thompson, N.W., et al, Regression of type II gastric carcinoids in multiple endocrine neoplasia type 1 patients with Zollinger–Ellison syndrome after surgical excision of all gastrinomas. World J Surg 2004; 28:652–658. 15383867
11. Gibril, F., Chen, Y-J., Schrump, D., et al, Prospective study of thymic carcinoids in patients with multiple endocrine neoplasia type 1. J Clin Endocrinol Metab 2003; 88:1066–1081. 12629087
12. Langer, P., Cupisti, K., Bartsch, D.K., et al, Adrenal involvement in MEN type 1. World J Surg 2002; 26:891–896. 12016472
13. Burgess, J.R., Harle, R.A., Tucker, P., et al, Adrenal lesions in a large kindred with multiple endocrine neoplasia type 1. Arch Surg 1996; 131:699–702. 8678766
14. Brandi, L.B., Gagel, R.F., Angeli, A., et al, Guidelines for the diagnosis and therapy of MEN type 1 and type 2. J Clin Endocrinol Metab 2001; 86:5658–5671. 11739416
15. Hai, N., Aoki, N., Shimatsu, A., et al, Clinical features of multiple endocrine neoplasia type 1 (MEN1) phenocopy without germline MEN1 gene mutations: analysis of 20 Japanese sporadic cases without MEN1. Clin Endocrinol (Oxf) 2000; 52:509–518. 10762295
16. Dean, P.G., van Heerden, J.A., Farley, D.R., et al, Are patients with multiple endocrine neoplasia type 1 prone to premature death? World J Surg 2000; 24:1437–1441. 11038219
17. Skogseid, B., Multiple endocrine neoplasia type 1. Br J Surg 2003; 90:383–385. 12673737
18. Arnalsteen, L.C., Alesina, P.F., Quiereux, J.L., et al, Long term results of less than total parathyroidectomy for hyperparathyroidism in multiple endocrine neoplasia type 1. Surgery 2002; 132:1119–1124. 12490864
19. Norton, J.A., Fraker, D.L., Alexander, H.R., et al, Surgery to cure the Zollinger–Ellison syndrome. N Engl J Med 1999; 341:644–653. 10564689
20. Norton, J.A., Alexander, H.R., Fraker, D.L., et al, Comparison of surgical results in patients with advanced and limited disease with multiple endocrine neoplasia type 1 and Zollinger–Ellison syndrome. Ann Surg 2001; 234:495–505. 11573043
21. Norton, J.A., Fraker, D.L., Alexander, H.R., et al, Surgery increases survival in patients with gastrinoma. Ann Surg 2006; 244:410–419. 16926567
22. Gibril, F., Venzon, D.J., Ojeaburu, J.V., et al, Prospective study of the natural history of gastrinoma in patients with MEN1: definition of an aggressive and a nonaggressive form. J Clin Endocrinol Metab 2001; 86:5282–5293. 11701693
23. Gauger, P.G., Scheiman, J.M., Wamsteker, E-J., et al, Endoscopic ultrasound helps to identify and resect MEN-1 endocrine pancreatic tumours at an early stage. Br J Surg 2003; 90:748–754. 12808627
24. Akerström, G., Hessman, O., Hellman, P., et al, Pancreatic tumours as part of the MEN-1 syndrome. Best Pract Res Clin Gastroenterol 2005; 19:819–830. 16253903
25. Chahal, H.S., Chapple, J.P., Frohman, L.A., et al, Clinical, genetic and molecular characterization of patients with familial isolated pituitary adenomas (FIPA). Trends Endocrinol Metab 2010; 21:419–427. 20570174
26. Toledo, R.A., Lourenco, D.M., Jr., Toledo, S.P., Familial isolated pituitary adenoma: evidence for genetic heterogeneity. Front Horm Res 2010; 38:77–86. 20616498
27. Kytola, S., Nord, B., Elder, E.E., et al, Alterations of the SDHD gene locus in midgut carcinoids, Merkel cell carcinomas, pheochromocytomas, and abdominal paragangliomas. Genes Chromosomes Cancer 2002; 34:325–332. 12007193
28. Ullrich, A., Schlessinger, J., Signal transduction by receptors with tyrosine kinase activity. Cell 1990; 61:203–212. 2158859
29. Santoro, M., Carlomango, F., Romano, A., et al, Activation of RET as a dominant transforming gene by germ-line mutations of MEN2A and MEN2B. Science 1995; 267:381–383. 7824936
30. Eng, C., Clayton, D., Schuffenecker, I., et al, The relationship between specific RET proto-oncogene mutations and disease phenotype in multiple endocrine neoplasia type 2. JAMA. 1996;276(19):1575–1579. 8918855
31. Weber, F., Eng, C., Editorial: germline variants within RET – clinical utility or scientific playtoy? J Clin Endocrinol Metab 2005; 90:6334–6336. 16275981
32. Tamanaha, R., Cleber, P., Camacho, C.P., et al, Y791F RET mutation and early onset medullary thyroid carcinoma in a Brazilian kindred: evaluation of phenotype-modifying effect of germline variants. Clin Endocrinol (Oxf) 2007; 67:806–808. 17610518
33. Machens, A., Nicolli-Sire, P., Hoegel, J., et al, Early malignant progression of hereditary medullary thyroid cancer. N Engl J Med 2003; 349:1517–1525. 14561794 This paper outlines the evidence base for the timing of thyroidectomy in patients with MEN2 based on the age of expression of extrathyroidal MTC. Such data are key to the basis of clinical approaches to management of the condition based on early molecular diagnostics and tailored intervention.
34. Kahraman, T., de Groot, J.W.B., Rou, W.E.C., et al, Acceptable age for prophylactic surgery in children with multiple endocrine neoplasia type 2a. Eur J Surg Oncol 2003; 29:331–335. 12711285
35. Sherman, S.I., Thyroid carcinoma. Lancet 2003; 361:501–511. 12583960
36. Vestergard, P., Vestergard, E.M., Brockstedt, H., et al, Codon Y791F mutation in a large kindred: is prophylactic thyroidectomy always indicated? World J Surg 2007; 31:996–1001. 17483988
37. Costante, G., Meringolo, D., Durante, C., et al, Predictive value of serum calcitonin levels for preoperative diagnosis of medullary thyroid carcinoma in a cohort of 5817 consecutive patients with thyroid nodules. J Clin Endocrinol Metab 2007; 92:450–455. 17119000
38. Gourgiotis, L., Sarlis, N.J., Reynolds, J.C., et al, Localization of medullary thyroid carcinoma metastasis in a multiple endocrine neoplasia type 2A patient by 6-[18F]-fluorodopamine positron emission tomography. J Clin Endocrinol Metab 2003; 88:637–641. 12574193
39. Sclumberger, M., Carlomagno, F., Baudin, E., et al, Novel therapeutic approaches to treat medullary thyroid carcinoma. Nat Clin Pract Endocrinol Metab 2008; 4:22–32. 18084343
40. Grossman, A., Pacak, K., Sawka, A., et al, Biochemical diagnosis and localization of phaeochromocytoma. Can we reach a consensus? Ann N Y Acad Sci 2006; 1073:332–347. 17102103 This paper outlines a consensus approach established as an outcome of the first International Phaeochromocytoma Workshop, presenting the relative merits of a number of alternative testing strategies.
41. Ciampi, R., Romei, C., Cosci, B., et al, Chromosome 10 and RET gene copy number alterations in hereditary and sporadic Medullary Thyroid Carcinoma. Mol Cell Endocrinol 2012; 348:176–182. 21867742
42. Neumann, H.P.H., Bausch, B., McWhinney, S.R., et al, Germ-line mutations in non-syndromic phaeochromocytoma. N Engl J Med 2002; 346:1459–1466. 12000816
43. Dahia, P.L., Ross, K.N., Wright, M.E., et al, A HIF1alpha regulatory loop links hypoxia and mitochondrial signals in pheochromocytomas. PLoS Genet 2005; 1:72–80. 16103922
44. Yeh, I.T., Lenci, R.E., Qin, Y., et al, A germline mutation of the KIF1B beta gene on 1p36 in a family with neural and nonneural tumors. Hum Genet 2008; 124:279–285. 18726616
45. Qin, Y., Yao, L., King, E.E., et al, Germline mutations in TMEM127 confer susceptibility to pheochromocytoma. Nat Genet 2010; 42:229–233. 20154675
46. McWhinney, S.R., Pasini, B., Stratakis, C.A., Familial gastrointestinal stromal tumours and germ-line mutations. N Engl J Med 2007; 357:1054–1056. 17804857
47. Astuti, D., Latif, F., Dallol, A., et al, Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial phaeochromocytoma and to familial paraganglioma. Am J Hum Genet 2002; 69:49–54. 12046006
48. Peczkowska, M., Cascon, A., Prejbbisz, A., et al, Extra-adrenal and adrenal pheochromocytomas associated with a germline SDHC mutation. Nat Clin Pract Endocrinol Metab 2008; 4:1111–1115. 18091718
49. Benn, D.E., Gimenez-Roqueplo, A.P., Reilly, J.R., et al, Clinical presentation and penetrance of pheochromocytoma/paraganglioma syndromes. J Clin Endocrinol Metab 2006; 91:827–836. 16317055
50. Neumann, H.P., Pawlu, C., Peczkowska, M., European–American Paraganglioma Study Group, Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations. JAMA 2004; 292:943–951. 15328326
51. Astuti, D., Douglas, F., Lennard, T.W.J., et al, Germline SDHD mutation in familial phaeochromocytoma. Lancet 2001; 357:1181–1182. 11323050
52. Pigny, P., Vincent, A., Cardot, Bauters C., et al, Paraganglioma after maternal transmission of a succinate dehydrogenase gene mutation. J Clin Endocrinol Metab 2008; 93:1609–1615. 18211978
53. Yeap, P.M., Tobias, E.S., Mavraki, E., Molecular analysis of pheochromocytoma after maternal transmission of SDHD mutation elucidates mechanism of parent-of-origin effect. J Clin Endocrinol Metab 2011; 96:E2009–E2013. 21937622
54. Kaltsas, G.A., Mukherjee, J.J., Foley, R., et al, Treatment of metastatic phaeochromocytoma with 131I-metaiodobenzylguanidine (MIBG). Endocrinologist 2003; 13:321–333. 11453952
55. Baysal, B.E., Willet-Brozick, J.E., Lawrence, E.C., et al, Prevalence of SDHB, SDHC and SDHD germline mutations in clinic patients with head and neck paragangliomas. J Med Genet 2002; 39:178–183. 11897817
56. Taschner, P.E., Jansen, J.C., Baysal, B.E., et al, Nearly all hereditary paragangliomas in the Netherlands are caused by two founder mutations in the SDHD gene. Genes Chromosomes Cancer 2001; 31:274–281. 11391798
57. Bayley, J.P., van Minderhout, I., Weiss, M.M., et al, Mutation analysis of SDHB and SDHC: novel germline mutations in sporadic head and neck paraganglioma and familial paraganglioma and/or pheochromocytoma. BMC Med Genet 2006; 7:1. 16405730
58. van Nederveen, F.H., Gaal, J., Favier, J., et al, An immunohistochemical procedure to detect patients with paraganglioma and phaeochromocytoma with germline SDHB, SDHC, or SDHD gene mutations: a retrospective and prospective analysis. Lancet Oncol 2009; 10:764–771. 19576851
59. Ricketts, C., Woodward, E.R., Killick, P., et al, Germline SDHB mutations and familial renal cell carcinoma. J Natl Cancer Inst 2008; 100:1260–1262. 18728283
60. Vanharanta, S., Buchta, M., McWhinney, S.R., et al, Early-onset renal cell carcinoma as a novel extraparaganglial component of SDHB-associated heritable paraganglioma. Am J Hum Genet 2004; 74:153–159. 14685938
61. Carney, J.A., Stratakis, C.A., Familial paraganglioma and gastric stromal sarcoma: a new syndrome distinct from the Carney triad. Am J Med Genet 2002; 108:132–139. 11857563
62. Gaal, J., Stratakis, C.A., Carney, J.A., et al, SDHB immunohistochemistry: a useful tool in the diagnosis of Carney–Stratakis and Carney triad gastrointestinal stromal tumors. Mod Pathol 2011; 24:147–151. 20890271
63. Stratakis, C.A., Carney, J.A., The triad of paragangliomas, gastric stromal tumours and pulmonary chondromas (Carney triad), and the dyad of paragangliomas and gastric stromal sarcomas (Carney–Stratakis syndrome): molecular genetics and clinical implications. J Intern Med 2009; 266:43–52. 19522824
64. Online Mendelian Inheritance in Man, OMIM®. Baltimore, MD: Johns Hopkins University. MIM Number: 145000. World Wide Web URL: http://omim.org/entry/145000; [accessed 22.05.12].
65. Hannan, F.M., Nesbit, M.A., Christie, P.T., et al, Familial isolated primary hyperparathyroidism caused by mutations of the MEN1 gene. Nat Clin Pract Endocrinol Metab 2008; 4:53–58. 18084346
66. Perrier, N.D., Villablanca, A., Larsson, C., et al, Genetic screening for MEN-1 mutations in families presenting with familial primary hyperparathyroidism. World J Surg 2002; 26:907–913. 12016470
67. Online Mendelian Inheritance in Man, OMIM®. Baltimore, MD: Johns Hopkins University. MIM Number: 145980. World Wide Web URL: http://omim.org/entry/145980; [accessed 22.05.12].
68. Online Mendelian Inheritance in Man, OMIM®. Baltimore, MD: Johns Hopkins University. MIM Number: 239200. World Wide Web URL: http://omim.org/entry/239200; [accessed 22.05.12].
69. Chen, J.D., Morrison, C., Zhang, C., et al, Hyperparathyroidism–jaw tumour syndrome. J Intern Med 2003; 253:634–642. 12755959
70. Cetani, F., Pardi, E., Giovannetti, A., et al, Genetic analysis of the MEN1 and HPRT2 locus in two Italian kindreds with familial isolated hyperparathyroidism. Clin Endocrinol (Oxf) 2002; 56:457–464. 11966738
71. Shattuck, T.M., Valimaki, S., Obara, T., et al, Somatic and germ-line mutations of the HRPT2 gene in sporadic parathyroid carcinoma. N Engl J Med 2003; 349:1722–1729. 14585940
72. Lonser, R.R., Glen, G.M., Walther, M., et al, Von Hippel–Lindau disease. Lancet 2003; 361:2059–2067. 12814730
73. Friedrich, C.A., Genotype–phenotype correlation in von Hippel Lindau syndrome. Hum Mol Genet. 2001;10(7):763–767. 11257110
74. Hes, F.J., Hoppener, J.W.M., Lips, C.J.M., Phaeochromocytoma in von Hippel–Lindau disease. J Clin Endocrinol Metab 2003; 88:969–974. 12629069
75. Koch, C.A., Mauro, D., Walhter, M.M., et al, Phaeochromocytoma in von Hippel–Lindau disease: distinct histopathologic phenotype compared to phaeochromocytoma in multiple endocrine neoplasia type 2. Endocr Pathol 2002; 13:17–27. 12114747
76. Eisenhofer, G., Walther, M.M., Huynh, T-T., et al, Phaeochromocytomas in von Hippel–Lindau syndrome and multiple endocrine neoplasia type 2 display distinct biochemical and clinical phenotypes. J Clin Endocrinol Metab 2001; 86:1999–2008. 11344198
77. Grossman, A., Pacak, K., Sawka, A., et al, Biochemical diagnosis and localization of phaeochromocytoma. Can we reach a consensus? Ann N Y Acad Sci 2006; 1073:332–347. 17102103
78. Friedman, J.M., Neurofoibromatosis 1. GeneReviews at GeneTests Medical Genetics Information Resource (database online). University of Washington, Seattle©, 1997–2012. Available at. http://www.ncbi.nlm.nih.gov/books/NBK1109/ [[accessed 22.05.12]].
79. Eng, C., PTEN hamartoma tumor syndrome. GeneReviews at GeneTests Medical Genetics Information Resource (database online). University of Washington, Seattle©, 1997–2012. Available at. http://www.ncbi.nlm.nih.gov/books/NBK1488/
80. Tan, M.H., Mester, J., Ngeow, J., et al, Lifetime cancer risks in individuals with germline PTEN mutations. Clin Cancer Res 2012; 18:400–407. 22252256
81. Gujrati, M., Thomas, C., Zelby, A., et al, Bannayan Zonana syndrome: a rare autosomal dominant syndrome with multiple lipomas and haemangiomas: a case report and review of the literature. Surg Neurol 1998; 50:164–168. 9701122
82. Celebi, J.T., Tsou, H.C., Chen, F.F., et al, Phenotypic findings of Cowden syndrome and Bannayan–Zonana syndrome in a family associated with a single germline mutation in PTEN. J Med Genet 1999; 36:360–364. 10353779
83. Capezzone, M., Cantara, S., Marchisotta, S., et al, Telomere length in neoplastic and non-neoplastic tissues of patients with familial and sporadic papillary thyroid cancer. J Clin Endocrinol Metab 2011; 96:E1852–E1856. 21865371
84. Online Mendelian Inheritance in Man, OMIM®. Baltimore, MD: Johns Hopkins University. MIM Number: 606240. World Wide Web URL: http://omim.org/entry/606240; [accessed 22.05.12].
85. Online Mendelian Inheritance in Man, OMIM®. Baltimore, MD: Johns Hopkins University. MIM Number: 603386. World Wide Web URL: http://omim.org/entry/603386; [accessed 22.05.12].
86. Bulow, C., Bulow, S., Group LCP, Is screening for thyroid carcinoma indicated in familial adenomatous polyposis. Int J Colorectal Dis 1997; 12:240–242. 9272455
87. Schneider, K., Garber, J., Li Fraumeni syndrome. GeneReviews at GeneTests Medical Genetics Information Resource (database online). ©University of Washington, Seattle, 1997–2012. Available at. http://www.ncbi.nlm.nih.gov/books/NBK1311/ [[accessed 22.05.12]].
88. Carney, J.A., Discovery of the Carney complex, a familial lentiginosis–multiple endocrine neoplasia syndrome: a medical odyssey. Endocrinologist 2003; 13:23–30. 16682939
89. Stratakis, C.A., Kirschner, L.S., Carney, J.A., Clinical and molecular features of the Carney complex: diagnostic criteria and recommendations for patient evaluation. J Clin Endocrinol Metab 2001; 86:4041–4046. 11549623
90. Online Mendelian Inheritance in Man, OMIM®. Baltimore, MD: Johns Hopkins University. MIM Number: 610489. World Wide Web URL: http://omim.org/entry/610489; [accessed 22.05.12].
91. Online Mendelian Inheritance in Man, OMIM®. Baltimore, MD: Johns Hopkins University. MIM Number: 255960. World Wide Web URL: http://omim.org/entry/255960; [accessed 18.02.13].
92. Groussin, L., Jullian, E., Perlemoine, K., et al, Mutations of the PRKAR1A gene in Cushing’s syndrome due to sporadic primary pigmented nodular adrenocortical disease. J Clin Endocrinol Metab 2002; 87:4324–4329. 12213893
93. Vezzosi, D., Cartier, D., Régnier, C., et al, Familial adrenocorticotropin-independent macronodular adrenal hyperplasia with aberrant serotonin and vasopressin adrenal receptors. Eur J Endocrinol 2007; 156:21–31. 17218722
94. Fragoso, M.C.B.V., Domenice, S., Latronico, A.C., et al, Cushing’s syndrome secondary to adrenocorticotropin-independent macronodular adrenocortical hyperplasia due to activating mutations of GNAS1 gene. J Clin Endocrinol Metab 2003; 88:2147–2151. 12727968
95. Jackson, R.V., Lafferty, A., Torpy, D.J., et al, New genetic insights in familial hyperaldosteronism. Ann N Y Acad Sci 2002; 970:77–88. 12381543
96. Lifton, R.P., Dluhy, R.G., Powers, M., et al, A chimaeric 11-beta-hydroxylase/aldosterone synthase gene causes glucocorticoid-remediable aldosteronism and human hypertension. Nature 1992; 355:262–265. 1731223
97. Choi, M., Scholl, U.I., Yue, P., et al, K+ channel mutations in adrenal aldosterone-producing adenomas and hereditary hypertension. Science 2011; 331:768–772. 21311022