[level-membership-for-cardiovascular-category]63
Exercise-Induced Arrhythmias
Exercise-Induced Atrial Arrhythmias
Exercise-Induced Ventricular Arrhythmias
Outflow Tract Ventricular Tachycardia
Idiopathic Left Ventricular Tachycardia
Arrhythmogenic Right Ventricular Dysplasia
Catecholaminergic Polymorphic Ventricular Tachycardia
Antiarrhythmics and Exercise-Induced Ventricular Arrhythmias
Several physiological changes that occur during exercise may precipitate cardiac arrhythmias. Activation of the sympathetic nervous system results in an increase in circulating catecholamines.1 Increased automaticity and enhanced triggered activity may increase the likelihood of arrhythmias. The presence of premature beats during exercise can lead to initiation of reentrant supraventricular and ventricular arrhythmias. Important factors in arrhythmogenicity include electrolyte shifts, baroreceptor activation, myocardial stretch, ischemia, and genetic predisposition.
Exercise can increase potassium levels, decrease pH, and raise catecholamines.2 These catecholamines may counterbalance the harmful cardiac effects of hyperkalemia and acidosis and improve action potential characteristics in potassium-depolarized ventricular myocytes.3 In normal myocardial tissue, hyperkalemia decreases the incidence of norepinephrine-induced arrhythmias. However in ischemic or infarcted tissue, hyperkalemia and catecholamines may jointly potentiate arrhythmias. The heart is also at increased risk in the post-exercise period. During this time, plasma potassium is low and adrenergic tone is high. An abnormal regulation of sympathovagal balance and electrolytes in recovery, compounded with ischemia, may increase the susceptibility to arrhythmias.1 Despite the myriad of physiological changes that occur in exercise, in the absence of structural or electrical heart disease sudden death due to arrhythmias is extremely rate.
Exercise-Induced Atrial Arrhythmias
Exercise-induced atrial arrhythmias are less common than ventricular arrhythmias. In the Baltimore Longitudinal Study of Aging, 1383 asymptomatic volunteers aged 20 to 94 years underwent exercise testing.4 Exercise treadmill–induced supraventricular arrhythmia (ETISVA) was noted in 85 subjects (6%). An eightfold increase in the relative risk of developing lone atrial fibrillation (AF) was noted in subjects with exercise-induced ETISVA. In a 5.7-year follow-up, 10% of 85 subjects with ETISVA developed AF or paroxysmal supraventricular tachycardia (SVT). Therefore, ETISVA may be a marker for AF or paroxysmal SVT during follow-up.
The relationship between exercise and atrial fibrillation is particularly relevant to athletes. The overall risk for AF is significantly higher in athletes than in controls (odds ratio 5.29, 95% confidence interval [CI] 3.57 to 7.85, P = .0001).5 Endurance sports increase preload, which increases atrial pressure and therefore shortens atrial refractory periods and increases the dispersion of atrial refractoriness. Increased vagal tone in athletes, sympathetic surges during exercise, and fluid and electrolyte changes during exercise may also contribute to the development of atrial arrhythmias.6 Anatomically, athletes may have larger left atrial dimensions and fibrosis secondary to chronic systemic inflammation from excessive endurance exercise.7
In patients with no structural heart disease, treatment is generally targeted at addressing the trigger for the arrhythmia. A reduction in exercise intensity or duration is often highly effective in reducing arrhythmia burden.8 However, many patients, especially competitive athletes, may be unwilling or unable to reduce or refrain from exercise. In these cases, β-blockers, antiarrhythmics, and catheter ablation can be considered.
In a study of 5375 patients with known or suspected coronary artery disease (CAD), 24% of patients developed atrial ectopy, 3.4% developed SVT, and 0.8% developed AF upon treadmill testing.9 Exercise treadmill–induced supraventricular arrhythmias were not predictive of any end point.
Exercise-Induced Ventricular Arrhythmias
Apparently Healthy Subjects
Asymptomatic patients without prior evidence of CAD have been noted to have variable rates of ventricular ectopy. In the Advisory Group for Aerospace Research and Development study of 1640 healthy aviators, the prevalence of PVCs (other than single or occasional) increased with age: 6.6% for ages 20 to 29, 7.6% for ages 30 to 39, and 13.1% for ages 40 to 53.10 The percentages of patients with three or more consecutive PVCs were 0.8% for ages 20 to 29, 1.0% for ages 30 to 39, and 3.5% for ages 40 to 53. In another study of 597 male and 325 female healthy adult volunteers, only 1.1% of the patients had exercise treadmill–induced ventricular arrhythmias (ETIVAs).11 These episodes were typically asymptomatic, short, and limited to 3 to 6 beats, usually near peak exercise. The prognostic significance of ventricular ectopy in asymptomatic healthy individuals remains controversial.
Coronary Artery Disease
In a population of veterans referred for exercise stress testing, the risk of mortality in patients with resting (pre-exercise) PVCs and ETIVA was increased.12 The combination of rest PVCs and ETIVA carries the highest risk.1 These variables were independent predictors of cardiovascular mortality after adjustment for other clinical and exercise test variables, which included exercise-induced ischemia. However it is possible that additional adjustment for left ventricular functional abnormalities or coronary disease burden might have mitigated the association. Regardless, patients found to have arrhythmias during exercise testing should undergo an evaluation of their left ventricular function.
Coronary Anomalies
Congenital coronary anomalies are implicated in 10% to 20% of all deaths in young athletes.13 The right coronary artery arising from the left coronary sinus is more common than the left coronary artery arising from the anterior sinus, although the latter is a more common cause of sudden death. Of the four types of anomalous left coronary arteries, the interarterial type is the only type that places the patient at increased risk for sudden death. In this variant, the left coronary artery arises from the right cusp and passes anteriorly between the aorta and the right ventricular outflow tract (RVOT). Similarly, an interarterial course of an anomalous right coronary artery that arises from the left cusp would also put patients at risk for sudden cardiac death (SCD). However, the larger territory supplied by the left coronary artery results in increased risk over right coronary artery anomalies. SCD associated with or shortly after vigorous exercise is very unusual after the patient is >35 years of age.
Angelini et al report that the incidence of anomalous coronary arteries was 1.07% (right anomalous coronary from the left coronary sinus in 0.92%; left anomalous coronary from the right coronary sinus in 0.15%).14 Davis et al. report a prevalence of 0.17% for anomalous origins of coronary arteries among 2388 children and adolescents.15 Among 1686 coronary anomalies found in 126,595 adult coronary angiograms, Yamanaka and Hobbs reported an incidence of 0.17% for anomalous left coronary arteries and 0.107% for anomalous right coronary arteries.16
The mechanism by which the coronary anomaly causes SCD is hypothesized to be a sudden occlusion of the vessel that may involve damage, thrombosis, or spasm, resulting in severe myocardial ischemia and ventricular tachycardia/fibrillation.17 During exercise, increased dP/dt and stroke volume may result in increased systolic expansion of the proximal aorta and pulmonary artery, which could collapse the proximal anomalous coronary artery. Because exercise results in a greater percentage of time spent in systole, which is when compression occurs, SCD most frequently occurs with or shortly after exercise.
Other coronary anomalies may also be implicated in arrhythmias and sudden death. Patients with a single coronary artery that divides into all three major branches are at risk for SCD during athletic activity.18,19 Patients with hypoplasia of portions of the coronary tree or with coronary fistulas, or the small minority of patients with an anomalous left coronary artery arising from the pulmonary artery (ALCAPA) who reached adulthood, would also be at risk for exercise-induced arrhythmias.
Outflow Tract Ventricular Tachycardia
Outflow tract VT should be considered in patients with a structurally normal heart and a QRS in VT that features a left bundle branch block morphology and an inferior axis. Most outflow tract VTs originate from the RVOT (80%), and the remainder originate from the left ventricular outflow tract (LVOT).20 RVOT VT has a left bundle branch QRS morphology and an inferior axis, with an R/S transition typically in V3 or V4. LVOT VT may also have left bundle branch QRS morphology but with small R waves in V1 and an earlier R/S transition. Lerman and collegues have determined that outflow tract VTs are the product of triggered activity secondary to cyclic adenosine monophosphate–mediated delayed afterdepolarizations. 21 The VT is adrenergically mediated and is sensitive to perturbations that lower intracellular calcium such as adenosine and verapamil. The diagnosis of idiopathic VT is one of exclusion; therefore other causes of left bundle branch block (LBBB) pattern VT should be considered.
Clinically, outflow tract VT accounts for the vast majority of idiopathic VTs. The age of presentation is usually 30 to 50 years, and patients usually have a benign clinical course.20 The most common complaint among patients is palpitations (48% to 80%), followed by presyncope or light-headedness (28% to 50%). Syncope is rare (<10%) and SCD is extremely rare. However, some patients may develop cardiomyopathy from incessant repetitive monomorphic VT or from a high burden of ventricular premature complexes (VPCs). Ablation of the focus usually normalizes left ventricular function in a few months.
The spectrum of outflow tract VT includes three clinical subtypes.22 Patients may have repetitive monomorphic VPCs, repetitive nonsustained monomorphic VT, or exercise-induced sustained VT. Patients may present with a predominance of one type; however, significant overlap has been noted, and it is believed that the subtypes share the same cellular mechanism. Generally, outflow tract tachycardias are provoked by exercise, and treadmill testing is useful in reproducing the clinical VT. Approximately 70% of patients who present with sustained VT will have VT induced by exercise testing.22 However, in patients who present with monomorphic nonsustained ventricular tachycardia (NSVT) or VPCs on monitoring, exercise testing may induce sustained VT in only 10%. Overall, exercise testing reproduces VT in less than 50% of patients with clinical VT. As a result, exercise testing may not be a reliable indicator of β-blocker or antiarrhythmic efficacy, and ambulatory monitoring would be an appropriate adjunct.
Two responses of outflow tract VT to exercise testing have been reported. In the first case, VT occurs during acceleration of the heart rate with exercise. A progression from VPCs to salvos of NSVT to sustained VT may be observed. In contrast, patients with repetitive monomorphic VT may have suppression of their VT during exercise and development of VT during the recovery phase of exercise.23 These responses indicate that a critical window of heart rates is required for VT initiation. This cycle length dependence of ventricular ectopy may also be observed on ambulatory monitoring. 23,24
In general, patients with exercise-induced outflow tract VTs have no structural heart disease. In contrast, patients with post-infarction septal VTs will have a history of coronary disease and myocardial infarction. Bundle branch reentry VT is usually seen in the setting of structural heart disease, most commonly a dilated cardiomyopathy.25 Patients with antidromic atrioventricular reciprocating tachycardia (AVRT) using an atriofascicular bypass tract might also demonstrate an LBBB morphology VT, although the axis is usually leftward.26 The triggered activity of outflow tract VT also differs from the reentry mechanisms of post-infarction VT, bundle branch reentry VT, and antidromic AVRT using an atriofascicular bypass tract. Although they characteristically have exercise-induced VT, patients with catecholaminergic polymorphic ventricular tachycardia (CPVT) will have polymorphic ventricular ectopy and will manifest bidirectional VT. Last, outflow tract morphology VTs may be a manifestation of arrhythmogenic right ventricular dysplasia (ARVD), although the morphology of VT is often varied in ARVD.
In addition to exercise, ventricular ectopy, including sustained VT, can be provoked by emotional stress. High levels of sympathetic tone contribute to arrhythmogenicity. This is also illustrated in the circadian variations in episodes of VT, ventricular runs (2 to 4 beats), and VPCs. Hayashi et al. demonstrated that peaks for these ventricular arrhythmias occurred around 7 AM and 6 PM.27 β-Blockers completely eliminated VT episodes and blunted ventricular runs; however rates of single VPCs were similar to those before β-blockade therapy.
Idiopathic Left Ventricular Tachycardia
Approximately 10% of idiopathic VTs originate from the fascicles of the left ventricle. These arrhythmias are referred to as fascicular VT or verapamil-sensitive VT.28 This condition should be considered in the differential diagnosis of right bundle branch block (RBBB) with left anterior fascicular block pattern VT. Less commonly, a left posterior block pattern may be seen (5% to 10%).28 The usual age of presentation is between 15 and 40 years, and patients usually have normal resting electrocardiogram (ECG) and left ventricular function.29 Although verapamil-sensitive VT was originally described at rest, VT is sensitive to exercise or emotional stress, is frequently precipitated by exercise, and may be seen during or after exertion.
This reentrant arrhythmia is believed to involve the posterior Purkinje system with the left posterior fascicle as one limb and abnormal Purkinje tissue with slow, decremental conduction as the other limb.30 In the small percentage of patients with RBBB and a right inferior axis VT (left posterior block pattern), the left anterior fascicle is thought to be the involved Purkinje system and the site of ventricular exit.31 The circuit is sensitive to catecholamines as evidenced by induction by exercise, and induction during electrophysiological testing is facilitated by isoproterenol infusion. Whereas adenosine has no effect on the arrhythmia, administration of verapamil slows the VT rate and then terminates it.32
Patients suspected to have verapamil-sensitive VT should undergo an evaluation to exclude structural heart disease. An echocardiogram, a stress test, and/or cardiac catheterization may be indicated depending on the clinical suspicion for coronary disease. Medical treatment for verapamil-sensitive VT involves verapamil; however patients with severe or recurrent symptoms can undergo catheter-based ablation with high rates of success.33
Arrhythmogenic Right Ventricular Dysplasia
Ventricular arrhythmias can occur during exercise, and SCD is thought to be due to acceleration of VT with degeneration into ventricular fibrillation. The most common arrhythmia is sustained or nonsustained monomorphic VT from the right ventricle, therefore manifesting a left bundle branch pattern. Usually the VT seen in ARVD will be of left bundle branch morphology with a superior rather than inferior axis. However, preferential or isolated outflow tract involvement has been described. Exercise testing may demonstrate monomorphic VT in up to 50% to 60% of patients. Exercise is thought to impart increased stress on the right ventricle and may increase right ventricular dilatation and worsen the manifestations of ARVD. Accordingly, patients with ARVD should not engage in competitive sports or endurance training and should not participate in any activities that cause palpitations, presyncope, or syncope.34
The diagnosis of ARVD consists of the fulfillment of multiple criteria.35 ECG findings suggestive of ARVD include the presence of right ventricular (RV) conduction delay, precordial T wave inversions, and epsilon waves.35,36 The presence of notching in the QRS complex of the LBBB VT may also suggest ARVD.37 Treadmill exercise testing may be used to elicit the characteristic LBBB morphology of ventricular tachycardia of ARVD. Nonsustained or sustained VT of LBBB morphology with a superior axis fulfills a major criterion for the diagnosis of ARVD, whereas an inferior axis fulfills a minor criterion.35 Other corroborative data are needed for the diagnosis of ARVD, and the usual diagnostic work-up for ARVD consists of a signal-averaged ECG, echocardiogram, cardiac magnetic resonance imaging (MRI), a detailed family history, and, if needed, invasive electroanatomic mapping of the right ventricle.
In addition to exercise restriction, treatment for ARVD involves implantation of an implantable cardioverter-defibrillator (ICD) in patients deemed at high risk for arrhythmic events and for secondary prevention of SCD.38 Medical management with antiarrhythmics or sotalol may be used in those who are not candidates for ICD therapy and in those with frequent shocks. Radiofrequency ablation also plays a role in patients with recurrent ventricular arrhythmias.
Hypertrophic Cardiomyopathy
Described in further detail in Chapter 86, hypertrophic cardiomyopathy (HCM) is a disease of left ventricular hypertrophy with a spectrum of clinical manifestations and hemodynamic abnormalities that are most often caused by mutations in one of several sarcomere genes. The mechanism of SCD in HCM patients is likely multifactorial but ultimately results from ventricular tachyarrhythmias. The arrhythmia is thought to result from electrical instability and distorted electrophysiological propagation from the disorganized arrangement of cardiac muscle cells of HCM. Findings of increased SCD during the afternoon hours in athletes support an exercise-induced mechanism.34 This is also supported by findings of sinus tachycardia preceding VT/VF events in stored ICD events, which suggest that high sympathetic drive is proarrhythmic when a susceptible substrate is present.39
Although NSVT during Holter monitoring has been demonstrated to be a useful prognostic factor, ventricular arrhythmias induced during exercise testing have not been well established as a prognostic indicator of SCD. In a study of 263 patients with HCM who underwent exercise testing, 3.0% of patients had new nonsustained atrial arrhythmias and 4.2% had nonsustained ventricular arrhythmias.40 In a larger series by Gimeno et al. of 1380 patients referred to a cardiomyopathy clinic, only 27 (2.0%) had NSVT (defined as 3 or more consecutive beats at a rate of ≥120 beats/min) or VF during exercise testing.41 Although NSVT was not frequent, researchers found that the hazard ratio for exercise-induced NSVT/VF (HR = 3.14) in predicting SCD or appropriate ICD discharge was higher than that of all other predictors (HR = 2.57 for NSVT on Holter; HR = 1.79 for family history of SCD). High rates of SCD or ICD discharge in patients with exercise-induced ventricular arrhythmia suggest that treadmill exercise may be a useful tool for assessing SCD risk in HCM.
Prevention of ventricular tachyarrhythmias and prevention of SCD involve the use of ICDs.42 The risk of SCD may also be reduced with septal myectomy in patients with symptomatic LVOT obstruction, although septal ablation has not been found to yield similar results.43 Pharmacologic agents such as antiarrhythmic drugs or negative inotropes have little impact on the incidence of SCD. Last, because of the potential risk of SCD associated with exercise in HCM patients, activity restriction is advised.34
Long QT Syndrome
Detailed discussion of long QT syndrome (LQTS) is found in other chapters; however, we will discuss here some of the distinguishing features of LQT-1 that pertain to exercise. LQT-4 and LQT-7 will be discussed in the following section, given some overlapping features with CPVT. The most common LQTS is LQT-1, which is the type whose events are most often triggered by adrenergic stimuli and exercise. Events in LQT-2 patients are less often triggered by adrenergic stimulation, and events in LQT-3 patients usually occur during rest or sleep.44 LQT-1 should be considered in patients who have a prolonged QTc and a history of syncope with stress. The T wave morphology in LQT-1 is typically broad-based and high in amplitude. This contrasts with the precordial biphasic T wave pattern of LQT-2 and the long isoelectrical ST-T segment of LQT-3.
Arrhythmic events in LQT-1 generally occur at relatively elevated heart rates. Tan et al. observed an average sinus heart rate of 98 beats/min before torsades de pointes for LQT-1 in contrast to an average of 73 beats/min for LQT-2.45 The preceding TdP in LQT-1 is not pause dependent, unlike that in LQT-2 patients.46
The defect in LQT-1 is a mutation in KCNQ1, resulting in loss-of-function in IKs.47 At fast heart rates, IKs is the most important current that shortens the action potential.48 As a result, the dysfunctional IKs in LQT-1 patients results in an inability to appropriately shorten QTc during exercise.47 This particular response to exercise and adrenergic stimulation is of particular interest in exercise stress testing and epinephrine QT stress testing.
Observation of a paradoxical prolongation of the uncorrected QT interval by >30 ms during infusion of low-dose epinephrine suggests a diagnosis of LQT-1.49 Epinephrine QT stress testing has been demonstrated to have a positive predictive value of 76% and a negative predictive value of 96% for LQT-1, even when the baseline ECG has a normal QTc.49
Ackerman and associates analyzed 243 treadmill tests of patients studied for LQTS and reported good diagnostic accuracy of treadmill testing for the diagnosis of LQT-1.50 During exercise, QTc decreases in LQT-2, LQT-3, and control patients. In contrast, LQT-1 patients essentially have unchanged QTc durations. Additionally, the QTc of concealed LQT-1 patients actually increased at peak exercise (Figure 63-1). Investigators found that an absolute QTc ≥460 ms during the recovery phase or a paradoxical increase in QTc, which was defined as (QTc at 3 minutes of recovery) − (QTc baseline) ≥30 ms, reliably distinguished patients with manifest or concealed LQT-1 from those with LQT-2 and LQT-3.
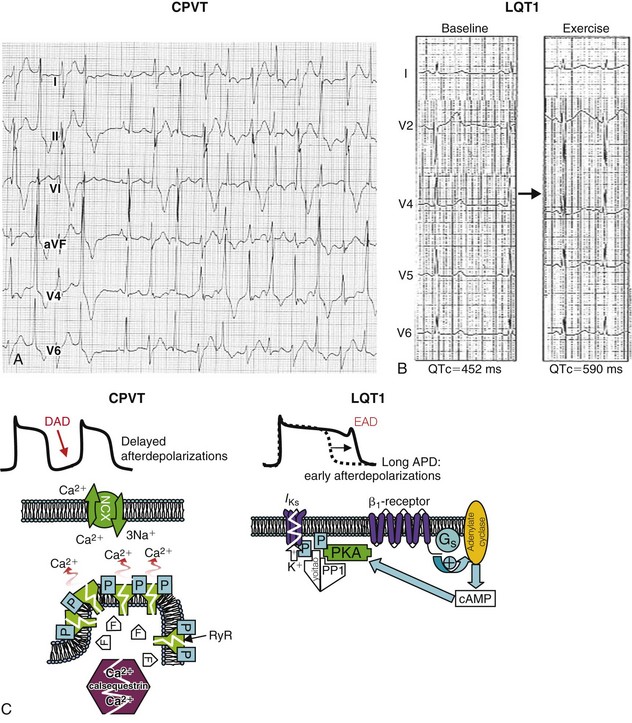
Figure 63-1 Exercise response in catecholaminergic polymorphic ventricular tachycardia (CPVT) and long QT syndrome type 1 (LQT-1).
A, Bidirectional ventricular tachycardia (alternating QRS axis) during exercise in a patient with CPVT. B, Paradoxical QT prolongation during exercise in a patient with LQT-1. C, Mechanisms of arrhythmogenesis. In CPVT (left), mutations (white zigzag line) in either the ryanodine receptor (RyR) or the calsequestrin gene make calcium leak. Excess calcium is transported by the Na+/Ca2+ exchanger (NCX), which brings in three sodium ions for each calcium, thereby generating a slow depolarization (transient inward current) that can reach threshold and generate a delayed afterdepolarization (DAD). In long QT syndrome (right), a mutated potassium channel (zigzag line) does not enhance IKs on phosphorylation, and the action potential prolongs, which can lead to early afterdepolarizations (EADs). APD, Action potential duration; cAMP, cyclic adenosine monophosphate; PKA, protein kinase A; PP1, protein phosphatase 1. (A, From Francis J, Sankar V, Nair VK, Priori SG: Catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm 2:550-554, 2005. B, From Takenaka K, Ai T, Shimizu W, et al: Exercise stress test amplifies genotype-phenotype correlation in the LQT1 and LQT2 forms of the long-QT syndrome. Circulation 107:838-844, 2003.)
Catecholaminergic Polymorphic Ventricular Tachycardia
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is a rare inherited syndrome that is characterized by adrenergically induced supraventricular and ventricular arrhythmias in structurally normal hearts and is associated with syncope and sudden death. It typically manifests in children and adolescents and provokes symptoms in up to 80% of patients younger than 40 years of age.51 SCD is often the first manifestation of the disease, which carries a 30% to 40% overall mortality by age 40.51
The basis for the overwhelming majority of cases of CPVT is a mutation in the cardiac ryanodine receptor RyR2, which is inherited in an autosomal dominant fashion.52 Mutations in the ryanodine receptor are implicated in the uncontrolled release of calcium from the sarcoplasmic reticulum in cardiomyocytes during electrical diastole, which produces delayed afterdepolarizations and cardiac arrhythmias.53 Genetic screening allows successful genotyping of CPVT in approximately 50% to 60% of patients with a definitive clinical diagnosis of CPVT.
The hallmark of CPVT is bidirectional ventricular tachycardia, where the beat-to-beat axis rotates 180 degrees each beat (see Figure 63-1). This contrasts with torsades de pointes, where the QRS axis gradually and chaotically rotates around the baseline. However, a small minority of CPVT patients have been noted to show irregular polymorphic VT. The vast majority of the initiating beat of VT has been localized to the RVOT.54 The second most common source was the LVOT. The morphology of the initiating beat of bidirectional VT is generally reproducible in 80% of patients with multiple episodes of VT.
In general, CPVT patients have a normal ECG at baseline but may demonstrate prominent U waves and slower resting heart rates. A normal QT interval is useful in distinguishing CPVT from long QT patients. However, some long QT patients exhibit borderline QT prolongation, and a couple of long QT syndromes share CPVT-type phenotypes. Adrenergically triggered bidirectional VT is also seen in long QT-4 (a mutation in the ANK2 gene, which codes for cardiac ankyrin-B) and long QT-7, otherwise known as Andersen-Tawil syndrome (a mutation in KCNJ2, which codes for a potassium channel). LQT-4 is associated with mild QTc prolongation, AF, sinus bradycardia, sinus node dysfunction, and polyphasic U waves. Andersen-Tawil syndrome is characterized by QT prolongation, prominent U waves, facial dysmorphisms, and periodic paralysis. The bidirectional VT rate in Anderson-Tawil syndrome has been reported to be much slower than in CPVT.55 Additional features of these syndromes, along with genetic testing, help distinguish them from CPVT.
A definitive diagnosis of CPVT depends on the finding of bidirectional VT in exercise ECG, Holter monitoring, or isoproterenol infusion, with a family or personal history of syncope during exercise or emotional stress.55 Infusion of isoproterenol can also be used to elicit the characteristic arrhythmias, although exercise testing is preferred. Monomorphic or polymorphic PVCs are insufficient for a diagnosis of CPVT, and further testing or monitoring is required for a diagnosis.
The main and most effective therapy for CPVT is β-blockade.38 Follow-up exercise testing is advised as a method of assessing the adequacy of β-blocker dosage. Compelling evidence indicates that the addition of flecainide at doses of 150 to 200 mg/day is also effective in suppressing ventricular arrhythmias in CPVT.56 Implantation of an ICD is indicated in patients who have survived cardiac arrest and who have ventricular arrhythmias despite maximally tolerated β-blockade. Finally, left cervical sympathetic denervation should be considered for patients in whom β-blockers and ICDs are insufficient.57 Patients with CPVT are advised against competitive exercise, high-stress occupations, and substances that would increase sympathetic tone.
Antiarrhythmics and Exercise-Induced Ventricular Arrhythmias
Class I sodium channel blocking antiarrhythmic drugs, such as flecainide and propafenone, can cause ventricular reentry through a use-dependent mechanism. QRS duration increases progressively with increasing levels of exercise while taking flecainide.58 A QRS increase of approximately 15% to 20% is consistent with pharmacologic effect; however further increases in QRS duration would warrant dose reduction or discontinuation of the drug. Provocation of ventricular tachycardia, preceded by excessive QRS widening, has been documented on treadmill exercise testing.59 Guidelines recommend that the QRS duration should not prolong by more than 50% from baseline and that a QRS duration of <120 ms should be maintained.60 To ensure that the QRS duration does not exceed these parameters at higher heart rates, treadmill testing may be employed to determine the degree of maximal QRS prolongation during exercise after the patient is fully loaded with flecainide or propafenone.
References
1. Beckerman, J, Mathur, A, Stahr, S, et al. Exercise-induced ventricular arrhythmias and cardiovascular death. Ann Noninvasive Electrocardiol. 2005; 10:47–52.
2. Beckerman, J, Wu, T, Jones, S, et al. Exercise test-induced arrhythmias. Prog Cardiovasc Dis. 2005; 47:285–305.
3. Paterson, DJ. Antiarrhythmic mechanisms during exercise. J Appl Physiol. 1996; 80:1853–1862.
4. Maurer, MS, Shefrin, EA, Fleg, JL. Prevalence and prognostic significance of exercise-induced supraventricular tachycardia in apparently healthy volunteers. Am J Cardiol. 1995; 75:788–792.
5. Abdulla, J, Nielsen, JR. Is the risk of atrial fibrillation higher in athletes than in the general population? A systematic review and meta-analysis. Europace. 2009; 11:1156–1159.
6. Turagam, MK, Velagapudi, P, Kocheril, AG. Atrial fibrillation in athletes. Am J Cardiol. 2012; 109:296–302.
7. Pelliccia, A, Maron, BJ, Di Paolo, FM, et al. Prevalence and clinical significance of left atrial remodeling in competitive athletes. J Am Coll Cardiol. 2005; 46:690–696.
8. Furlanello, F, Bertoldi, A, Dallago, M, et al. Atrial fibrillation in elite athletes. J Cardiovasc Electrophysiol. 1998; 9(8 Suppl):S63–S68.
9. Bunch, TJ, Chandrasekaran, K, Gersh, BJ, et al. The prognostic significance of exercise-induced atrial arrhythmias. J Am Coll Cardiol. 2004; 43:1236–1240.
10. Froelicher, VF, Allen, M, Lancaster, MC. Maximal treadmill testing of normal USAF aircrewmen. Aerosp Med. 1974; 45:310–315.
11. Fleg, JL, Lakatta, EG. Prevalence and prognosis of exercise-induced nonsustained ventricular tachycardia in apparently healthy volunteers. Am J Cardiol. 1984; 54:762–764.
12. Partington, S, Myers, J, Cho, S, et al. Prevalence and prognostic value of exercise-induced ventricular arrhythmias. Am Heart J. 2003; 145:139–146.
13. Corrado, D, Basso, C, Rizzoli, G, et al. Does sports activity enhance the risk of sudden death in adolescents and young adults? J Am Coll Cardiol. 2003; 42:1959–1963.
14. Angelini P, ed. Coronary Artery Anomalies: A Comprehensive Approach. Lippincott Williams & Wilkins: Baltimore, MD, 1999:200.
15. Davis, JA, Cecchin, F, Jones, TK, et al. Major coronary artery anomalies in a pediatric population: Incidence and clinical importance. J Am Coll Cardiol. 2001; 37:593–597.
16. Yamanaka, O, Hobbs, RE. Coronary artery anomalies in 126,595 patients undergoing coronary arteriography. Cathet Cardiovasc Diagn. 1990; 21:28–40.
17. Cheitlin, MD, MacGregor, J. Congenital anomalies of coronary arteries: Role in the pathogenesis of sudden cardiac death. Herz. 2009; 34:268–279.
18. Mofrad, PS, Weigold, G, Clavijo, LC. Sudden cardiac death in athlete with anomalous single coronary artery. Cardiovasc Revasc Med. 2005; 6:89–90.
19. Choi, JH, Kornblum, RN. Pete Maravich’s incredible heart. J Forensic Sci. 1990; 35:981–986.
20. Iwai, S, Cantillon, DJ, Kim, RJ, et al. Right and left ventricular outflow tract tachycardias: Evidence for a common electrophysiologic mechanism. J Cardiovasc Electrophysiol. 2006; 17:1052–1058.
21. Lerman, BB. Response of nonreentrant catecholamine-mediated ventricular tachycardia to endogenous adenosine and acetylcholine: Evidence for myocardial receptor-mediated effects. Circulation. 1993; 87:382–390.
22. Kim, RJ, Iwai, S, Markowitz, SM, et al. Clinical and electrophysiological spectrum of idiopathic ventricular outflow tract arrhythmias. J Am Coll Cardiol. 2007; 49:2035–2043.
23. Lerman, BB, Stein, KM, Markowitz, SM, et al. Ventricular arrhythmias in normal hearts. Cardiol Clin. 2000; 18:265–291.
24. Josephson, ME. Clinical Cardiac Electrophysiology: Techniques and Interpretations, ed 4. Baltimore, MD: Lippincott Williams & Wilkins; 2008.
25. Blanck, Z, Dhala, A, Deshpande, S, et al. Bundle branch reentrant ventricular tachycardia: Cumulative experience in 48 patients. J Cardiovasc Electrophysiol. 1993; 4:253–262.
26. Bardy, GH, Fedor, JM, German, LD, et al. Surface electrocardiographic clues suggesting presence of a nodofascicular Mahaim fiber. J Am Coll Cardiol. 1984; 3:1161–1168.
27. Hayashi, H, Fujiki, A, Tani, M, et al. Circadian variation of idiopathic ventricular tachycardia originating from right ventricular outflow tract. Am J Cardiol. 1999; 84:99–101.
28. Issa, ZF, Miller, JM, Zipes, DP. Clinical arrhythmology and electrophysiology, ed 1. Philadelphia: Saunders; 2009.
29. Lerman, BB, Stein, KM, Markowitz, SM. Mechanisms of idiopathic left ventricular tachycardia. J Cardiovasc Electrophysiol. 1997; 8:571–583.
30. Aiba, T, Suyama, K, Aihara, N, et al. The role of Purkinje and pre-Purkinje potentials in the reentrant circuit of verapamil-sensitive idiopathic LV tachycardia. Pacing Clin Electrophysiol. 2001; 24:333–344.
31. Zipes, D, Jalife, J. Cardiac Electrophysiology: From Cell to Bedside: Expert Consult—Online and Print, ed 5. Philadelphia: Saunders; 2009.
32. Iwai, S, Lerman, BB. Management of ventricular tachycardia in patients with clinically normal hearts. Curr Cardiol Rep. 2000; 2:515–521.
33. Nakagawa, H, Beckman, KJ, McClelland, JH, et al. Radiofrequency catheter ablation of idiopathic left ventricular tachycardia guided by a Purkinje potential. Circulation. 1993; 88:2607–2617.
34. Maron, BJ, Ackerman, MJ, Nishimura, RA, et al. Task Force 4: HCM and other cardiomyopathies, mitral valve prolapse, myocarditis, and Marfan syndrome. J Am Coll Cardiol. 2005; 45:1340–1345.
35. Marcus, FI, McKenna, WJ, Sherrill, D, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia (ARVC/D). Circulation. 2010; 121:1533–1541.
36. Gaita, F, Giustetto, C, Di Donna, P, et al. Long-term follow-up of right ventricular monomorphic extrasystoles. J Am Coll Cardiol. 2001; 38:364–370.
37. Hoffmayer, KS, Machado, ON, Marcus, GM, et al. Electrocardiographic comparison of ventricular arrhythmias in patients with arrhythmogenic right ventricular cardiomyopathy and right ventricular outflow tract tachycardia. J Am Coll Cardiol. 2011; 58:831–838.
38. Zipes, DP, Camm, AJ, Borggrefe, M, et al. ACC/AHA/ESC 2006 guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. J Am Coll Cardiol. 2006; 48:e247–e346.
39. Cha, Y-M, Gersh, BJ, Maron, BJ, et al. Electrophysiologic manifestations of ventricular tachyarrhythmias provoking appropriate defibrillator interventions in high-risk patients with hypertrophic cardiomyopathy. J Cardiovasc Electrophysiol. 2007; 18:483–487.
40. Drinko, JK, Nash, PJ, Lever, HM, et al. Safety of stress testing in patients with hypertrophic cardiomyopathy. Am J Cardiol. 2004; 93:1443–1444.
41. Gimeno, JR, Tomé-Esteban, M, Lofiego, C, et al. Exercise-induced ventricular arrhythmias and risk of sudden cardiac death in patients with hypertrophic cardiomyopathy. Eur Heart J. 2009; 30:2599–2605.
42. Gersh, BJ, Maron, BJ, Bonow, RO, et al. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Developed in collaboration with American Association for Thoracic Surgery, American Society of Echocardiography, American Society of Nuclear Cardiology, Heart Failure Society of America, Heart Rhythm Society, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons. J Am Coll Cardiol. 2011; 58:e212–e260.
43. McLeod, CJ, Ommen, SR, Ackerman, MJ, et al. Surgical septal myectomy decreases the risk for appropriate implantable cardioverter defibrillator discharge in obstructive hypertrophic cardiomyopathy. Eur Heart J. 2007; 28:2583–2588.
44. Schwartz, PJ, Priori, SG, Spazzolini, C, et al. Genotype-phenotype correlation in the long-QT syndrome: Gene-specific triggers for life-threatening arrhythmias. Circulation. 2001; 103:89–95.
45. Tan, HL, Bardai, A, Shimizu, W, et al. Genotype-specific onset of arrhythmias in congenital long-QT syndrome: Possible therapy implications. Circulation. 2006; 114:2096–2103.
46. Marban, E, Robinson, SW, Wier, WG. Mechanisms of arrhythmogenic delayed and early afterdepolarizations in ferret ventricular muscle. J Clin Invest. 1986; 78:1185–1192.
47. Swan, H, Viitasalo, M, Piippo, K, et al. Sinus node function and ventricular repolarization during exercise stress test in long QT syndrome patients with KvLQT1 and HERG potassium channel defects. J Am Coll Cardiol. 1999; 34:823–829.
48. Faber, GM, Rudy, Y. Action potential and contractility changes in [Na(+)](i) overloaded cardiac myocytes: A simulation study. Biophys J. 2000; 78:2392–2404.
49. Vyas, H, Hejlik, J, Ackerman, MJ. Epinephrine QT stress testing in the evaluation of congenital long-QT syndrome: Diagnostic accuracy of the paradoxical QT response. Circulation. 2006; 113:1385–1392.
50. Horner, JM, Horner, MM, Ackerman, MJ. The diagnostic utility of recovery phase QTc during treadmill exercise stress testing in the evaluation of long QT syndrome. Heart Rhythm. 2011; 8:1698–1704.
51. Mohamed, U, Napolitano, C, Priori, SG. Molecular and electrophysiological bases of catecholaminergic polymorphic ventricular tachycardia. J Cardiovasc Electrophysiol. 2007; 18:791–797.
52. Laitinen, PJ, Brown, KM, Piippo, K, et al. Mutations of the cardiac ryanodine receptor (RyR2) gene in familial polymorphic ventricular tachycardia. Circulation. 2001; 103:485–490.
53. Liu, N, Colombi, B, Memmi, M, et al. Arrhythmogenesis in catecholaminergic polymorphic ventricular tachycardia: insights from a RyR2 R4496C knock-in mouse model. Circ Res. 2006; 99:292–298.
54. Sumitomo, N, Harada, K, Nagashima, M, et al. Catecholaminergic polymorphic ventricular tachycardia: Electrocardiographic characteristics and optimal therapeutic strategies to prevent sudden death. Heart. 2003; 89:66–70.
55. Brugada, J, Brugada, P. Clinical Approach to Sudden Cardiac Death Syndromes, ed 1. New York: Springer; 2010.
56. van der Werf, C, Kannankeril, PJ, Sacher, F, et al. Flecainide therapy reduces exercise-induced ventricular arrhythmias in patients with catecholaminergic polymorphic ventricular tachycardia. J Am Coll Cardiol. 2011; 57:2244–2254.
57. Wilde, AAM, Bhuiyan, ZA, Crotti, L, et al. Left cardiac sympathetic denervation for catecholaminergic polymorphic ventricular tachycardia. N Engl J Med. 2008; 358:2024–2029.
58. Ranger, S, Talajic, M, Lemery, R, et al. Amplification of flecainide-induced ventricular conduction slowing by exercise: A potentially significant clinical consequence of use-dependent sodium channel blockade. Circulation. 1989; 79:1000–1006.
59. Falk, RH. Flecainide-induced ventricular tachycardia and fibrillation in patients treated for atrial fibrillation. Ann Intern Med. 1989; 111:107–111.
60. ACC/AHA/ESC. 2006 guidelines for the management of patients with atrial fibrillation: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the European Society of Cardiology Committee for Practice Guidelines (writing committee to revise the 2001 guidelines for the management of patients with atrial fibrillation). Developed in collaboration with the European Heart Rhythm Association and the Heart Rhythm Society. Circulation. 2006; 114:e257–e354.
[/level-membership-for-cardiovascular-category][not-level-membership-for-cardiovascular-category]63
Exercise-Induced Arrhythmias
Exercise-Induced Atrial Arrhythmias
Exercise-Induced Ventricular Arrhythmias
Outflow Tract Ventricular Tachycardia
Idiopathic Left Ventricular Tachycardia
Arrhythmogenic Right Ventricular Dysplasia
Catecholaminergic Polymorphic Ventricular Tachycardia
Antiarrhythmics and Exercise-Induced Ventricular Arrhythmias
Several physiological changes that occur during exercise may precipitate cardiac arrhythmias. Activation of the sympathetic nervous system results in an increase in circulating catecholamines.1 Increased automaticity and enhanced triggered activity may increase the likelihood of arrhythmias. The presence of premature beats during exercise can lead to initiation of reentrant supraventricular and ventricular arrhythmias. Important factors in arrhythmogenicity include electrolyte shifts, baroreceptor activation, myocardial stretch, ischemia, and genetic predisposition.
Exercise can increase potassium levels, decrease pH, and raise catecholamines.2 These catecholamines may counterbalance the harmful cardiac effects of hyperkalemia and acidosis and improve action potential characteristics in potassium-depolarized ventricular myocytes.3 In normal myocardial tissue, hyperkalemia decreases the incidence of norepinephrine-induced arrhythmias. However in ischemic or infarcted tissue, hyperkalemia and catecholamines may jointly potentiate arrhythmias. The heart is also at increased risk in the post-exercise period. During this time, plasma potassium is low and adrenergic tone is high. An abnormal regulation of sympathovagal balance and electrolytes in recovery, compounded with ischemia, may increase the susceptibility to arrhythmias.1 Despite the myriad of physiological changes that occur in exercise, in the absence of structural or electrical heart disease sudden death due to arrhythmias is extremely rate.
Exercise-Induced Atrial Arrhythmias
Exercise-induced atrial arrhythmias are less common than ventricular arrhythmias. In the Baltimore Longitudinal Study of Aging, 1383 asymptomatic volunteers aged 20 to 94 years underwent exercise testing.4 Exercise treadmill–induced supraventricular arrhythmia (ETISVA) was noted in 85 subjects (6%). An eightfold increase in the relative risk of developing lone atrial fibrillation (AF) was noted in subjects with exercise-induced ETISVA. In a 5.7-year follow-up, 10% of 85 subjects with ETISVA developed AF or paroxysmal supraventricular tachycardia (SVT). Therefore, ETISVA may be a marker for AF or paroxysmal SVT during follow-up.
The relationship between exercise and atrial fibrillation is particularly relevant to athletes. The overall risk for AF is significantly higher in athletes than in controls (odds ratio 5.29, 95% confidence interval [CI] 3.57 to 7.85, P = .0001).5 Endurance sports increase preload, which increases atrial pressure and therefore shortens atrial refractory periods and increases the dispersion of atrial refractoriness. Increased vagal tone in athletes, sympathetic surges during exercise, and fluid and electrolyte changes during exercise may also contribute to the development of atrial arrhythmias.6 Anatomically, athletes may have larger left atrial dimensions and fibrosis secondary to chronic systemic inflammation from excessive endurance exercise.7
In patients with no structural heart disease, treatment is generally targeted at addressing the trigger for the arrhythmia. A reduction in exercise intensity or duration is often highly effective in reducing arrhythmia burden.8 However, many patients, especially competitive athletes, may be unwilling or unable to reduce or refrain from exercise. In these cases, β-blockers, antiarrhythmics, and catheter ablation can be considered.
In a study of 5375 patients with known or suspected coronary artery disease (CAD), 24% of patients developed atrial ectopy, 3.4% developed SVT, and 0.8% developed AF upon treadmill testing.9 Exercise treadmill–induced supraventricular arrhythmias were not predictive of any end point.
Exercise-Induced Ventricular Arrhythmias
Apparently Healthy Subjects
Asymptomatic patients without prior evidence of CAD have been noted to have variable rates of ventricular ectopy. In the Advisory Group for Aerospace Research and Development study of 1640 healthy aviators, the prevalence of PVCs (other than single or occasional) increased with age: 6.6% for ages 20 to 29, 7.6% for ages 30 to 39, and 13.1% for ages 40 to 53.10 The percentages of patients with three or more consecutive PVCs were 0.8% for ages 20 to 29, 1.0% for ages 30 to 39, and 3.5% for ages 40 to 53. In another study of 597 male and 325 female healthy adult volunteers, only 1.1% of the patients had exercise treadmill–induced ventricular arrhythmias (ETIVAs).11 These episodes were typically asymptomatic, short, and limited to 3 to 6 beats, usually near peak exercise. The prognostic significance of ventricular ectopy in asymptomatic healthy individuals remains controversial.
Coronary Artery Disease
In a population of veterans referred for exercise stress testing, the risk of mortality in patients with resting (pre-exercise) PVCs and ETIVA was increased.12 The combination of rest PVCs and ETIVA carries the highest risk.1 These variables were independent predictors of cardiovascular mortality after adjustment for other clinical and exercise test variables, which included exercise-induced ischemia. However it is possible that additional adjustment for left ventricular functional abnormalities or coronary disease burden might have mitigated the association. Regardless, patients found to have arrhythmias during exercise testing should undergo an evaluation of their left ventricular function.
Coronary Anomalies
Congenital coronary anomalies are implicated in 10% to 20% of all deaths in young athletes.13 The right coronary artery arising from the left coronary sinus is more common than the left coronary artery arising from the anterior sinus, although the latter is a more common cause of sudden death. Of the four types of anomalous left coronary arteries, the interarterial type is the only type that places the patient at increased risk for sudden death. In this variant, the left coronary artery arises from the right cusp and passes anteriorly between the aorta and the right ventricular outflow tract (RVOT). Similarly, an interarterial course of an anomalous right coronary artery that arises from the left cusp would also put patients at risk for sudden cardiac death (SCD). However, the larger territory supplied by the left coronary artery results in increased risk over right coronary artery anomalies. SCD associated with or shortly after vigorous exercise is very unusual after the patient is >35 years of age.
Angelini et al report that the incidence of anomalous coronary arteries was 1.07% (right anomalous coronary from the left coronary sinus in 0.92%; left anomalous coronary from the right coronary sinus in 0.15%).14 Davis et al. report a prevalence of 0.17% for anomalous origins of coronary arteries among 2388 children and adolescents.15 Among 1686 coronary anomalies found in 126,595 adult coronary angiograms, Yamanaka and Hobbs reported an incidence of 0.17% for anomalous left coronary arteries and 0.107% for anomalous right coronary arteries.16
The mechanism by which the coronary anomaly causes SCD is hypothesized to be a sudden occlusion of the vessel that may involve damage, thrombosis, or spasm, resulting in severe myocardial ischemia and ventricular tachycardia/fibrillation.17 During exercise, increased dP/dt and stroke volume may result in increased systolic expansion of the proximal aorta and pulmonary artery, which could collapse the proximal anomalous coronary artery. Because exercise results in a greater percentage of time spent in systole, which is when compression occurs, SCD most frequently occurs with or shortly after exercise.
Other coronary anomalies may also be implicated in arrhythmias and sudden death. Patients with a single coronary artery that divides into all three major branches are at risk for SCD during athletic activity.18,19 Patients with hypoplasia of portions of the coronary tree or with coronary fistulas, or the small minority of patients with an anomalous left coronary artery arising from the pulmonary artery (ALCAPA) who reached adulthood, would also be at risk for exercise-induced arrhythmias.
Outflow Tract Ventricular Tachycardia
Outflow tract VT should be considered in patients with a structurally normal heart and a QRS in VT that features a left bundle branch block morphology and an inferior axis. Most outflow tract VTs originate from the RVOT (80%), and the remainder originate from the left ventricular outflow tract (LVOT).20 RVOT VT has a left bundle branch QRS morphology and an inferior axis, with an R/S transition typically in V3 or V4. LVOT VT may also have left bundle branch QRS morphology but with small R waves in V1 and an earlier R/S transition. Lerman and collegues have determined that outflow tract VTs are the product of triggered activity secondary to cyclic adenosine monophosphate–mediated delayed afterdepolarizations. 21 The VT is adrenergically mediated and is sensitive to perturbations that lower intracellular calcium such as adenosine and verapamil. The diagnosis of idiopathic VT is one of exclusion; therefore other causes of left bundle branch block (LBBB) pattern VT should be considered.
Clinically, outflow tract VT accounts for the vast majority of idiopathic VTs. The age of presentation is usually 30 to 50 years, and patients usually have a benign clinical course.20 The most common complaint among patients is palpitations (48% to 80%), followed by presyncope or light-headedness (28% to 50%). Syncope is rare (<10%) and SCD is extremely rare. However, some patients may develop cardiomyopathy from incessant repetitive monomorphic VT or from a high burden of ventricular premature complexes (VPCs). Ablation of the focus usually normalizes left ventricular function in a few months.
The spectrum of outflow tract VT includes three clinical subtypes.22 Patients may have repetitive monomorphic VPCs, repetitive nonsustained monomorphic VT, or exercise-induced sustained VT. Patients may present with a predominance of one type; however, significant overlap has been noted, and it is believed that the subtypes share the same cellular mechanism. Generally, outflow tract tachycardias are provoked by exercise, and treadmill testing is useful in reproducing the clinical VT. Approximately 70% of patients who present with sustained VT will have VT induced by exercise testing.22 However, in patients who present with monomorphic nonsustained ventricular tachycardia (NSVT) or VPCs on monitoring, exercise testing may induce sustained VT in only 10%. Overall, exercise testing reproduces VT in less than 50% of patients with clinical VT. As a result, exercise testing may not be a reliable indicator of β-blocker or antiarrhythmic efficacy, and ambulatory monitoring would be an appropriate adjunct.
Two responses of outflow tract VT to exercise testing have been reported. In the first case, VT occurs during acceleration of the heart rate with exercise. A progression from VPCs to salvos of NSVT to sustained VT may be observed. In contrast, patients with repetitive monomorphic VT may have suppression of their VT during exercise and development of VT during the recovery phase of exercise.23 These responses indicate that a critical window of heart rates is required for VT initiation. This cycle length dependence of ventricular ectopy may also be observed on ambulatory monitoring. 23,24
In general, patients with exercise-induced outflow tract VTs have no structural heart disease. In contrast, patients with post-infarction septal VTs will have a history of coronary disease and myocardial infarction. Bundle branch reentry VT is usually seen in the setting of structural heart disease, most commonly a dilated cardiomyopathy.25 Patients with antidromic atrioventricular reciprocating tachycardia (AVRT) using an atriofascicular bypass tract might also demonstrate an LBBB morphology VT, although the axis is usually leftward.26 The triggered activity of outflow tract VT also differs from the reentry mechanisms of post-infarction VT, bundle branch reentry VT, and antidromic AVRT using an atriofascicular bypass tract. Although they characteristically have exercise-induced VT, patients with catecholaminergic polymorphic ventricular tachycardia (CPVT) will have polymorphic ventricular ectopy and will manifest bidirectional VT. Last, outflow tract morphology VTs may be a manifestation of arrhythmogenic right ventricular dysplasia (ARVD), although the morphology of VT is often varied in ARVD.
In addition to exercise, ventricular ectopy, including sustained VT, can be provoked by emotional stress. High levels of sympathetic tone contribute to arrhythmogenicity. This is also illustrated in the circadian variations in episodes of VT, ventricular runs (2 to 4 beats), and VPCs. Hayashi et al. demonstrated that peaks for these ventricular arrhythmias occurred around 7 AM and 6 PM.27 β-Blockers completely eliminated VT episodes and blunted ventricular runs; however rates of single VPCs were similar to those before β-blockade therapy.
Idiopathic Left Ventricular Tachycardia
Approximately 10% of idiopathic VTs originate from the fascicles of the left ventricle. These arrhythmias are referred to as fascicular VT or verapamil-sensitive VT.28 This condition should be considered in the differential diagnosis of right bundle branch block (RBBB) with left anterior fascicular block pattern VT. Less commonly, a left posterior block pattern may be seen (5% to 10%).28 The usual age of presentation is between 15 and 40 years, and patients usually have normal resting electrocardiogram (ECG) and left ventricular function.29
[/not-level-membership-for-cardiovascular-category]
