171 Ethanol, Methanol, and Ethylene Glycol
 Ethanol Intoxication
Ethanol Intoxication
Ethanol is rapidly absorbed by the gastrointestinal tract and distributed throughout body water.1 The blood ethanol concentration (in mg/dL) resulting from a one-time dose can be estimated from the volume (in mL) of ingested alcoholic beverage, the fractional concentration of ethanol (by volume) in the beverage, and body weight (in kg), by the following equation:
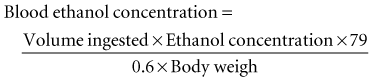
Metabolism
Between 2% and 10% of ingested ethanol is excreted intact by the kidneys and lungs, but the major fraction is metabolized by hepatic alcohol dehydrogenase (ADH) to acetaldehyde by the following reaction2:
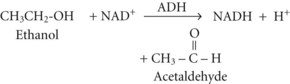
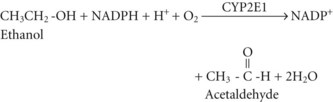

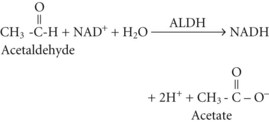
Acetate can then enter the tricarboxylic acid cycle and ultimately be metabolized to carbon dioxide (CO2) and water. Polymorphisms in the dehydrogenase enzymes can result in increased production rates or diminished metabolic clearance of acetaldehyde. As a consequence, some individuals experience marked vasodilation, facial flushing, tachycardia, and other unpleasant symptoms after ethanol consumption because of the effects of excessive acetaldehyde accumulation. Alleles leading to this reaction are particularly prevalent in persons of Chinese or Japanese descent but are uncommon in Caucasians.2
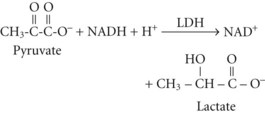
The resulting increase in blood lactate level is usually small, however, and the presence of lactic acidosis should prompt consideration of an alternative cause such as circulatory shock or seizures.3
Clinical Manifestations
Acute intoxication can induce cardiac dysrhythmias, particularly atrial fibrillation. As denoted by the descriptive sobriquet, “holiday heart syndrome,” this phenomenon frequently occurs during an alcoholic binge. A variety of neurologic abnormalities are associated with chronic alcoholism, including Wernicke-Korsakoff syndrome, chronic cerebellar ataxia, Marchiafava-Bignami syndrome, and central pontine myelinolysis.4 Wernicke encephalopathy can manifest as lethargy, confusion, truncal ataxia, nystagmus, and ophthalmoplegia, whereas Korsakoff dementia manifests as retentive memory impairment, confabulation, and learning deficits.5
Acutely, ethanol has well-known, dose-dependent inebriating and sedating effects (Table 171-1), although remarkable variability in this relationship is observed in some individuals.4 These central nervous system (CNS) effects appear to be at least partly caused by interference with N-methyl-D-aspartate receptor and perhaps γ-aminobutyric acid receptor function.4,6,7 The cognitive, behavioral, perceptual, and psychomotor effects of ethanol intoxication play a causative role in a substantial proportion of deaths and injuries involving motor vehicle–related trauma, accidental drownings, residential fires, homicides, and suicides. The legal driving threshold for blood ethanol concentration is 80 mg/dL in the United States for operators aged 21 years or older. Tachycardia, mydriasis, diaphoresis, hypotension, and hypothermia can occur in cases of marked intoxication. Blood ethanol concentrations of approximately 350 mg/dL have been associated with fatal outcomes, although many patients have survived much higher levels, including one subject who reportedly survived a level of 1500 mg/dL.8
TABLE 171-1 Relationship Between Blood Ethanol Concentration and Clinical Manifestations*
| Blood Ethanol Concentration (mg/dL) | Clinical Manifestations |
|---|---|
| <30 | Little demonstrable effect |
| 30-50 | Mild euphoria, minimal central nervous system effects, subjective sensation of cutaneous warmth |
| 50-80 | Relaxation, jocularity, gregariousness, cutaneous flushing, prolongation of reaction time |
| 80-100 | Statutory intoxication in many jurisdictions |
| 100-200 | Loquacity, animation, exuberance, exaggerated emotional responses, uninhibited behavior, impaired judgment |
| 200-300 | Sedation interrupted by periods of boisterous or antisocial behavior, nausea, emesis, dysarthria, horizontal nystagmus, impaired visual pursuit, diplopia, ataxia |
| 300-400 | Unstable station and gait, incoherent speech, somnolence, impairment of protective airway reflexes, incontinence, obtundation, stupor |
| >400 | Coma, loss of protective reflexes, respiratory depression, death |
* This information serves only as an imperfect guide, because considerable variability and overlap is possible, and individuals with chronic heavy ethanol exposure often develop learned tolerance.
Laboratory Manifestations
Blood ethanol concentration correlates at least approximately with the manifestations of intoxication (see Table 171-1). In chronic alcoholic subjects, a blood ethanol concentration below 250 mg/dL is an unlikely explanation for alterations in consciousness and should prompt a search for an alternative cause.8 Numerous other blood test abnormalities can be seen in intoxicated subjects, particularly in patients with chronic ethanol abuse: hyponatremia, hypokalemia, hypomagnesemia, hypophosphatemia, hypoglycemia, thrombocytopenia, and coagulopathy. Elevated activities of various circulating enzymes including amylase, lipase, creatine phosphokinase, transaminases, and γ-glutamyl transpeptidase, can occur as a reflection of alcohol-induced pancreatitis, rhabdomyolysis, hepatitis, or cirrhosis. The latter can also result in hyperbilirubinemia and hypoalbuminemia.
Treatment
In the absence of associated illness or injury (Table 171-2), mild to moderate intoxication requires no special treatment other than abstinence and a period of observation. Regardless of the degree of intoxication, withdrawal precautions are recommended for chronic imbibers, particularly those with a history of heavy chronic use or alcohol withdrawal manifestations. The treatment of severe ethanol intoxication is largely supportive. As with any patient who presents to the hospital in an unconscious state, initial empirical treatment should include IV thiamine, dextrose, and naloxone, once adequate airway, ventilation, and perfusion are ensured. Gastric lavage and activated charcoal administration are of dubious value for hastening removal of ethanol from the body.9–12
TABLE 171-2 Concomitant or Complicating Disorders Associated with Alcohol Intoxication or Withdrawal
| Alcoholic hepatitis | Hypoglycemia |
| Aspiration pneumonitis | Hypothermia |
| Circulatory shock (due to dehydration or hemorrhage) | Infections (e.g., pneumonia, meningitis) |
| Cirrhosis | Intracranial hemorrhage (e.g., subdural hematoma) |
| Coagulopathy | Pancreatitis |
| Dehydration | Peripheral neuropathy |
| Drug overdose or other toxic ingestion | Psychosis |
| Electrolyte derangements | Rhabdomyolysis |
| Gastrointestinal hemorrhage (due to gastritis, peptic ulcer disease, esophageal varices, hemorrhoids, or Mallory-Weiss tear) | Seizures |
| Sepsis | |
| Head injury | Thrombocytopenia |
| Heat stroke | Vitamin deficiency (folate, thiamine, other B vitamins) |
| Hepatic encephalopathy | Wernicke-Korsakoff syndrome |
A thorough evaluation for common associated illnesses and injuries should include physical and laboratory examinations for evidence of head, neck, and somatic trauma, rhabdomyolysis, pancreatitis, hepatic dysfunction, coagulopathy, blood dyscrasias, and fluid and electrolyte derangements. Accordingly, routine laboratory testing should include a complete blood count, prothrombin and partial thromboplastin times, serum assays for electrolytes (including sodium, potassium, chloride, total CO2 content, magnesium, and phosphorus), glucose, liver and kidney function tests, and amylase, lipase, transaminases, and creatine phosphokinase activities. Screening for alternative or concomitant intoxications or overdoses is occasionally fruitful.13 Identification of metabolic acidosis should prompt investigation for alcoholic ketoacidosis, lactic acidosis, renal failure, and relevant toxic ingestions, particularly methanol and ethylene glycol. Microbiological cultures are indicated if there are signs of serious infection.
Intravenous thiamine and a multivitamin preparation containing folate are routinely administered to hospitalized patients with alcohol intoxication or withdrawal. Parenteral thiamine (50 or 100 mg) is given during the initial phase of management, regardless of the level of sensorium, to prevent or treat Wernicke-Korsakoff syndrome.5
Oxygenation may be assessed either by pulse oximetry or by arterial blood gas analysis, and supplemental oxygen should be provided as necessary. Administration of vitamin K, fresh frozen plasma, or platelet transfusions may be necessary if there is gastrointestinal or other hemorrhage and coagulopathy or severe thrombocytopenia. The level of consciousness should be monitored periodically. Hemodialysis has been employed and is effective at removing ethanol from the body, but in general, this modality poses greater risks than simply providing supportive care and allowing physiologic ethanol elimination. Its use might be warranted in rare cases of profound life-threatening ethanol intoxication, or if there are other reasons for dialysis.14,15
 Alcoholic Ketoacidosis
Alcoholic Ketoacidosis
Metabolism
Although the precise metabolic mechanisms that lead to the development of AKA are incompletely understood, several mechanisms appear to be operative. Abnormal insulin and counterregulatory hormone levels occur,16 but the disorder is distinct from simple starvation and diabetes mellitus. Ethanol results in inhibition of gluconeogenesis and depletion of glycogen stores, leading to low glucose availability, particularly when coupled with fasting. Hypoglycemia causes release of epinephrine, cortisol, and growth hormone, as well as decreased insulin production; these are all factors that favor ketone synthesis. Ethanol metabolism results in a surfeit of acetate and NADH, which promotes lactate and ketone production. Marked ketonemia results in acidosis and ketonuria. The latter causes osmotic diuresis, intravascular volume depletion, and electrolyte losses. Thus, starvation, dehydration, excessive acetate production, an altered redox state, hormonal imbalances, and perhaps genetic predisposition are all potentially involved.17
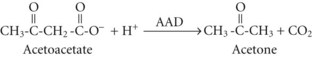
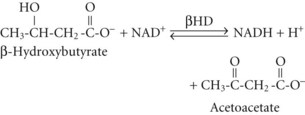
In both AKA and diabetic ketoacidosis (DKA), β-hydroxybutyrate is quantitatively the more important molecule. However, the ratio of β-hydroxybutyrate to acetoacetate tends to be higher in AKA (typically 5 : 1 but sometimes exceeding 10 : 1),18 compared with DKA (typically 3 : 1).
Clinical Manifestations
AKA characteristically develops 24 to 72 hours after an alcoholic debauch as the blood ethanol concentration is declining, during which time the subject ceases ethanol consumption and has little or no caloric intake. Gastrointestinal symptoms predominate and include anorexia, nausea, epigastric pain, and vomiting.19,20 The subject usually has a temporary aversion to food and alcoholic beverages and complains of malaise. On physical examination, there is a clear sensorium in most cases. The odor of acetone may be detectable on the subject’s breath. Tachypnea or Kussmaul respirations may be evident if there is marked acidemia. Tachycardia and other signs of volume depletion may be apparent. In some cases, manifestations of underlying cirrhosis (e.g., jaundice, ascites, ecchymoses, hemorrhoids) or other disorders commonly associated with chronic alcohol abuse (see Table 171-2) may be present.
Laboratory Manifestations
The key laboratory findings in AKA are metabolic acidosis, ketonemia, and ketonuria in the presence of a normal, low, or mildly elevated blood glucose concentration. Ethanol may be detectable in the blood, but it is not a requirement for the diagnosis and is frequently not detectable by the time the patient presents to the hospital. If the acidosis is clinically significant, elevation of the serum anion gap is expected. Other causes of metabolic acidosis must be excluded. Simple starvation can cause mild ketoacidosis, but with simple starvation the serum total CO2 content or bicarbonate concentration generally remains above 18 mmol/L. DKA and renal failure are readily excluded by routine blood glucose and creatinine measurements. Lactic acidosis may be suggested by the associated clinical setting (e.g., seizures, hypotension), but it should be excluded by direct assay. Mild degrees of hyperlactatemia can occur in AKA, but concentrations greater than 3 mmol/L should prompt consideration of occult hypoperfusion, seizures, or another cause. Occult toxic ingestions also require exclusion, particularly ingestions of methanol, ethylene glycol, and salicylate intoxication.15,21–24 Ingestion of exogenous acetone or isopropanol can cause marked ketosis due to acetonemia, but in isolation these intoxications are not associated with anion gap elevation or metabolic acidosis unless the poisoning is severe enough to cause seizures or circulatory shock, thereby resulting in lactic acidosis.
Because vomiting and dehydration are frequent manifestations in AKA, metabolic alkalosis can complicate the acid-base derangement. The combination of metabolic acidosis (from ketoacidosis) and metabolic alkalosis (from vomiting and volume contraction) can result in arterial pH and blood gas values that underestimate the severity of one or both of these metabolic disturbances. For example, mild metabolic alkalosis can be obscured by the presence of moderate or severe metabolic acidosis. Rarely, both metabolic processes are present and of approximately equal severity. In this situation, blood pH and bicarbonate concentration can be within normal limits despite the acid-base disturbances.23 Or, the metabolic alkalosis can predominate and obscure the acidosis. The serum anion gap can aid in detecting these situations. An abnormally high anion gap suggests metabolic acidosis even if no acid-base disorder is evident by arterial blood gas analysis. In the face of a wide serum anion gap, the quotient of the delta anion gap (i.e., the subject’s anion gap minus the average normal anion gap) divided by the delta bicarbonate (i.e., the average normal bicarbonate concentration minus the subject’s blood bicarbonate concentration) should equal unity in organic metabolic acidoses if there is no metabolic alkalosis.25 A quotient well above unity (e.g., >1.2) is evidence of concomitant metabolic alkalosis.
Treatment
Alternative explanations for the metabolic acidosis should be promptly excluded.24 As in acute alcohol intoxication, the initial assessment should focus on identifying relevant alternative, underlying, or complicating illnesses or injuries that may require specific urgent therapy. Although patients with AKA sometimes have severe metabolic acidemia, the acid-base disturbance usually responds rapidly to IV hydration and ample dextrose administration.17 Rapid infusion of 50 mL of 50% dextrose is indicated if hypoglycemia is identified. Five percent dextrose in normal saline is infused IV, at a high rate initially, to correct any hypovolemia or hypoglycemia and provide substrate for metabolic correction of the ketoacidosis. Thereafter, dextrose-containing normal or half-normal saline can be substituted at a high maintenance infusion rate, titrated to ongoing fluid losses. Ample dextrose administration is key to reversing the metabolic acidosis. The blood glucose concentration should be monitored frequently to allow detection of recurrent hypoglycemia or any intolerance to the provided glucose load.
 Ethanol Withdrawal
Ethanol Withdrawal
Ethanol withdrawal is common among hospitalized patients, either as a primary reason for admission or as a development during hospitalization for some other illness or injury. It is a potentially fatal syndrome that occurs after abrupt discontinuation of ethanol in individuals who regularly consume ethanol-containing beverages. Although in most cases it occurs after complete abstinence, it can also occur in the face of ongoing ethanol consumption if the level of ethanol intake is substantially decreased. The pathophysiology is incompletely understood but probably involves changes in neurotransmitter levels and alterations in neurotransmitter receptor function, as well as elevated circulating catecholamine levels.6,7,26,27 A number of disorders should be of particular consideration in the differential diagnosis of alcohol withdrawal (see Table 171-2). The mortality rate associated with advanced stages of alcohol withdrawal can exceed 15%.28,29
Clinical Manifestations
The syndrome is traditionally classified into four stages, although the stages do not always follow the indicated sequence, and not every patient develops every stage.29 The time of development of each stage is also quite variable, and overlaps can occur. A typical temporal sequence is described.
The first stage occurs 6 to 24 hours or more after the last drink or after a somewhat longer period of markedly decreased ethanol intake. Manifestations include anxiety, restlessness, decreased attention, tremulousness, insomnia, and craving for alcoholic beverages. Stage 2, which occurs about 24 hours after the onset of abstinence, is characterized by hallucinations, misperceptions, irritability, and vivid dreams.30 Hallucinations may be auditory, but more often they are visual or tactile. Formication, the delusional sensation of insects crawling on the skin, and vivid or threatening visual hallucinations are particularly common. During this stage, the patient may appear otherwise lucid or somewhat confused, hypervigilant, and easily startled or misled. In stage 3, which commonly occurs 7 to 48 hours after cessation of drinking, seizures occur, usually of the grand mal variety.4 The seizures classically manifest as a cluster of brief tonic-clonic convulsions, at one time referred to as “rum fits.” They are more likely to occur in subjects with a history of repeated withdrawal episodes.32 A relatively lucid interval ranging from hours to 2 or 3 days is sometimes seen between stages 3 and 4. Stage 4 manifests 2 to 6 days or more after initiation of abstinence and consists of a global confusional state associated with signs of neuronal excitation and severe autonomic hyperactivity. Vernacular usage notwithstanding, the term delirium tremens specifically refers to stage 4 of withdrawal. Only a small minority of individuals with alcohol withdrawal develop delirium tremens. Tremors, hallucinations, and seizures are common during this stage. As is characteristic of delirium in general, the degree of confusion and disorientation can wax and wane. Hyperadrenergic manifestations may include diaphoresis, flushing, mydriasis, tachycardia, hypertension, and low-grade fever.4
Treatment
Initial steps in management include ensuring that a patent airway is present and that ventilation, oxygenation, and perfusion are adequate; establishing IV access; and excluding serious coexisting or complicating disorders. Subsequent treatment focuses mainly on judiciously titrated sedation and vigilant monitoring for progression of the syndrome or development of complications. All patients with alcohol withdrawal are given prophylactic multivitamin supplements including parenteral thiamine and folate, and fluid deficits and electrolyte deficiencies are corrected.33 Routine administration of magnesium sulfate in the absence of hypomagnesemia has not been shown to be beneficial.34,35 Prophylaxis against deep vein thrombosis is recommended.
The agent of choice is a benzodiazepine given orally in milder cases or IV in more severe withdrawal states.30,33,36–38 Limited evidence suggests that symptom-triggered dosing is superior to fixed-schedule benzodiazepine dosing.39 Individualized dosing requires the expert judgment of an experienced clinician, but practicality often necessitates substitution of protocol-driven dosing schemes. These typically use a quantitative assessment scale such as the Revised Clinical Institute Withdrawal Assessment Scale for Alcohol to score the degree of withdrawal manifestations.40,41 Lorazepam can be administered IV in incremental doses, starting with 1 or 2 mg, followed by intermittent (e.g., every 2-6 hours) IV dosing or a continuous IV infusion (e.g., initiated at 1 mg/h and titrated to effect).29,42 Alternatively, midazolam can be employed, beginning with 2 to 4 mg by IV injection, followed by 2 mg/h by continuous IV infusion, which may be titrated to effect. Diazepam is another option, given initially in titrated doses of 5 to 10 mg at intervals as frequent as every 10 minutes if necessary until a calm but awake level of consciousness is achieved. Subsequent dosing at 5 to 20 mg every 4 to 6 hours is typically required with this agent. Prolonged administration of diazepam can lead to prolonged duration of sedation due to accumulation of the parent drug and an active metabolite, both of which have long half-lives. This effect is less likely to occur with lorazepam.
Oral benzodiazepines have been employed commonly in mild cases of withdrawal that do not require IV sedation.30,31 These agents also can be used in more serious cases after the severe manifestations have abated and parenteral benzodiazepines are no longer required. Typical oral chlordiazepoxide dosage is 25 to 100 mg every 6 to 12 hours. Intramuscular administration is sometimes employed, but it entails a less predictable dose-response due to erratic absorption, and there is the potential for a depot effect.
Other sedative-hypnotic drugs can be effective but are not considered first-line therapeutic agents.33,36 Barbiturates have a long history of successful use. The most commonly used agent is phenobarbital, which can be difficult to titrate because of its long duration of action. The shorter-acting barbiturate, pentobarbital, also has been employed. Oral ethanol and, in the past, paraldehyde have been used but have been discouraged, in part because of the risks of aspiration and gastric irritation, but also because their use can be interpreted as reinforcing the acceptability of using alcoholic beverages, either in general or for treatment of withdrawal symptoms. The latter criticism has also been directed at the use of ethanol administered IV for this purpose. A randomized trial examining IV ethanol administration for alcohol withdrawal prophylaxis in trauma ICU patients found no advantage compared to benzodiazepine management.43 Propofol is effective, but it is not a first-line agent and is not recommended unless an endotracheal tube is in place and mechanical ventilation is used.29 Regardless of the specific sedative agent employed, appropriate dose titration is crucial. The goal is to ameliorate the manifestations of withdrawal without causing excessive sedation. Sedation should be titrated with the use of an objective sedation scale such as the Ramsay Sedation Scale,44 the Riker Sedation-Agitation Scale,45 or the Richmond Agitation-Sedation Scale.46 The goal should be to achieve a calm awake state or, if that is not feasible, a state of light somnolence from which the patient can easily be aroused and is able to respond verbally.
Clonidine may be administered if hyperautonomic symptoms are prominent.47–49 Typical oral dosing is 0.1 to 0.2 mg every 6 to 12 hours. β-Adrenergic receptor blockers are not recommended for routine use, but barring contraindications, they may be considered in selected cases as adjunctive agents for controlling severe hyperadrenergic manifestations. Haloperidol and other neuroleptic agents are not routinely used, because they can lower the threshold for seizures. In selected cases, haloperidol may be used in conjunction with benzodiazepines for marked agitation or hallucinations, but this agent or similar drugs should probably not be used as monotherapy.36
Seizure precautions should be instituted for all patients in withdrawal. Withdrawal seizures are managed primarily with benzodiazepines, which usually are effective at the doses used for sedation.42 In refractory cases, higher doses may be necessary but may necessitate endotracheal intubation and mechanical ventilation. Concomitant use of other anticonvulsants also can be considered. Barbiturates may be used for this purpose, but phenytoin is usually ineffective unless the seizures are due to a specific cause other than alcohol withdrawal, such as underlying epilepsy or a complicating acute disorder of the CNS (e.g., meningitis, head trauma).33,50,51 In such cases, phenytoin is usually the anticonvulsant of choice. A variety of other anticonvulsant and sedative drugs have been studied for potential use in treating alcohol withdrawal, including valproic acid, baclofen, γ-hydroxybutyrate, gabapentin, oxcarbazepine, and carbamazepine. However, data on safety and efficacy are limited, particularly for hospitalized patients and those with comorbid illness.52–59
 Methanol Intoxication
Methanol Intoxication
Methanol, also known as wood alcohol, is a clear, colorless liquid having a faint alcoholic odor. It is widely used in laboratories and industry as a solvent and synthetic precursor. It is also a constituent or vehicle in numerous commercially available products for residential use (Box 171-1).15 Methanol is also used as a denaturant to intentionally render ethanol unfit for consumption. The minimum lethal dose of methanol is highly variable, reportedly ranging from less than 10 mL to more than 500 mL. This variability may result from multiple factors including the degree of concomitant ethanol intoxication, the presence of folate deficiency, and perhaps other factors.
More than 2000 cases of methanol exposure, most of which are accidental, are reported annually by the American Association of Poison Control Centers.60,61–63 Intentional ingestion can represent a suicidal gesture or attempt, but it more commonly occurs among desperate alcoholics who have no access to ethanol-containing beverages and are either unaware or heedless of the risks of consuming methanol. There are individual cases of surreptitious poisoning in which an individual prepares a small volume of an alcoholic drink intentionally laced with methanol with malice aforethought for the intended victim. More often, malicious intent is absent, and the goal is simply illicit production of a small or large volume of alcoholic beverage, with methanol used because of its availability or under a mistaken notion that it will serve as a more potent but still potable inebriant. Sharing or black-market distribution of these illicit concoctions has resulted in periodic epidemics of methanol intoxication, sometimes involving hundreds of unwitting subjects.64–68 There are also rare reports of dermal or inhalational exposure causing intoxication, but most cases involve oral ingestion.69
Metabolism
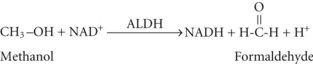
and then rapidly to formic acid, depicted here as its dissociation products, formate and a hydrogen ion70:
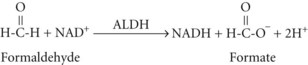
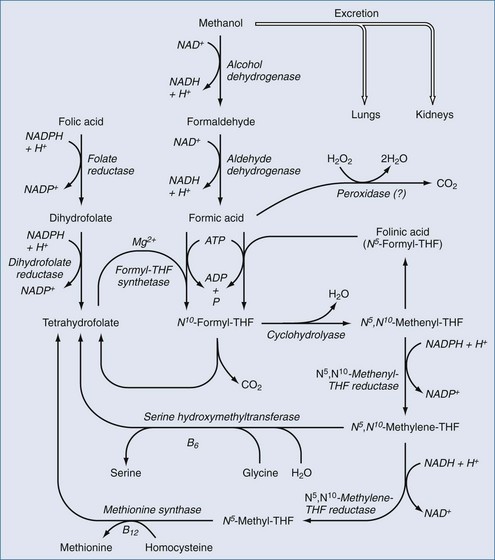
Formic acid production can result in metabolic acidosis. Independent of the acidosis, formic acid inhibits cytochrome oxidase and has direct neurotoxic effects, particularly affecting the retina and optic nerves.68,71–76 Small amounts of methanol are present as congeners in fermented alcoholic beverages.77 Small amounts are also formed during the metabolism of certain fruits and vegetables and by metabolism of the artificial sweetener, aspartame.78,79 However, the quantity of methanol available or formed from these sources is small, and there are enzyme systems present in the body that can convert these small amounts of formate to harmless CO2 (Figure 171-1). The large amounts of formate produced in serious cases of methanol intoxication overwhelm these enzymes, resulting in toxic accumulation of formate. Certain nonhuman mammalian species have enzymes with much higher activity for metabolism of formate; even large quantities of methanol are nontoxic to these species. Methanol ingested by these species is still converted to formaldehyde and formate, but these toxins are rapidly metabolized to CO2 so that significant formate accumulation does not occur. The enzymes that convert formate to CO2 require folinic acid, the activated form of folic acid, as an obligate cofactor.70
Clinical Manifestations
Like ethanol, methanol has dose-dependent sedating and inebriating effects that manifest shortly after ingestion, but methanol is less potent in this regard. Both alcohols also have similar gastrointestinal irritant effects that can provoke nausea, vomiting, abdominal pain, gastritis, hematemesis, and pancreatitis, although methanol may be more potent in this regard. Methanol ingestion can lead to additional CNS manifestations that are not observed with ethanol intoxication, which can sometimes provide helpful clinical clues in cases of occult methanol intoxication.66,68 These more specific manifestations are caused by formate, the end product of methanol metabolism. There is a characteristic delay, usually 12 to 24 hours, between ingestion and development of these manifestations, and this delay is attributable to the relatively slow conversion of methanol to formaldehyde. Delayed CNS manifestations can include cerebral edema, seizures, signs of meningeal irritation, and cerebral infarction (particularly infarction of basal ganglia).80 However, the most specific clinical findings are ocular and range from mildly blurred vision to visual field defects or tunnel vision to complete and sometimes permanent blindness.66,68 Other possible ocular manifestations include scotomata, scintillations, papilledema, and loss of pupillary light reflexes. Most survivors recover visual function, but permanent visual deficits occur in as many as a third of patients with serious intoxication. If the metabolic acidosis is severe, it can result in Kussmaul respirations and dyspnea. In the most severe cases of poisoning, profound acidosis, respiratory failure, and circulatory shock intervene. Severe global brain injury and brain death can also occur.
Laboratory Manifestations
The clinical laboratory can be helpful by providing clues to the diagnosis in cases of occult intoxication and by corroborating cases with a clear history of methanol ingestion. The serum total CO2 content may be abnormally low as a consequence of metabolic acidosis due to formic acid production. The dissociation product of formic acid, formate, is negatively charged, and can widen the serum anion gap. Arterial blood gas analysis can corroborate the presence of metabolic acidosis. Metabolic acidosis associated with a wide serum anion gap has a limited number of causes, the most common of which are lactic acidosis, ketoacidosis, and renal failure.23,24 These other causes of wide-gap metabolic acidosis are easily excluded by measuring the concentrations in blood of lactate, ketones, glucose, and creatinine. Certain toxins (e.g., propylene glycol) can result in lactic acidosis by direct metabolic conversion of the parent compound to lactate. More commonly, lactic acidosis can occur in association with any toxic exposure or drug overdose that causes seizures or circulatory shock (e.g., iron, isoniazid). The metabolic acidosis seen in methanol intoxication is mainly due to formic acid formation but can also be due in part to lactic acidosis secondary to these other mechanisms. Analogous to ethanol metabolism, conversion of methanol to formaldehyde and formic acid leads to a reducing environment in cells, which tends to increase lactate concentration. By inhibiting cytochromes, formate also may interfere with normal aerobic metabolism and lead to an increase in anaerobic glycolysis with resulting lactic acidosis. Therefore, hyperlactatemia does not exclude methanol poisoning. A few other toxic agents besides methanol, notably ethylene glycol and salicylates, can directly cause a wide anion gap metabolic acidosis. Although measurement of plasma formate concentration would seem to be a rational method to confirm the diagnosis of methanol poisoning, this assay is rarely available in hospital laboratories.81
Life-threatening methanol poisoning can result in profound metabolic acidosis which sometimes is refractory to large doses of sodium bicarbonate. However, even with severe methanol exposure, metabolic acidosis may be absent if testing is performed within a few hours after the ingestion.81 In these cases, the plasma methanol level may be very high, but the slow rate of its metabolism has not allowed for appreciable conversion to formic acid. Therefore, in the presence of a compatible history for toxic alcohol ingestion, the absence of a wide anion gap or hypobicarbonatemia should not be regarded as excluding the possibility of methanol poisoning.
A potentially useful screening test for recognition of methanol exposure early in its course is the serum osmolality gap. Serum osmolality is determined by the concentration of osmotically active solutes, or osmoles.82,83 Osmotic activity is directly proportional to the osmole concentration of a solution, which is directly proportional to the mass concentration of the solute and inversely proportional to the solute’s molecular weight. Therefore, to have an appreciable effect on osmolality, a solute must be present at relatively high mass concentration and have a relatively low molecular weight. For example, albumin is present at relatively large mass concentrations in serum, normally averaging about 4000 mg/dL, in comparison with urea, which normally averages only about 10 mg/dL. However, albumin has a far higher molecular weight (approximately 69,000 daltons, compared with 60 daltons for urea), making its osmolar concentration less than 1 mOsm/L. The elemental ions sodium and chloride are present in appreciable mass concentration, and their atomic weight is comparatively low (23 and 35 daltons, respectively), making them quantitatively important serum osmoles. Therefore, total serum osmolality normally comprises sodium, low atomic or molecular weight anions, plus urea and glucose; although many other osmoles are present in serum, their collective contribution is comparatively small. Based on these principles, serum osmolality may be estimated by the following formula83:
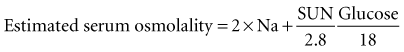
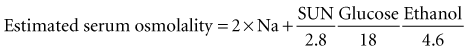
Here, the units for ethanol are mg/dL, and the divisor is based on the molecular weight of ethanol, 46 daltons. These millimolar concentration units technically provide an estimate of serum osmolarity; however, for practical purposes, they can be equated to millimolal units and designated osmolality (i.e., milliosmoles per kilogram of water). Just as ethanol can appreciably affect serum osmolality, so too can methanol.84,85 The serum osmolality may be estimated from the formula shown and compared with a more direct measurement of serum osmolality; the difference between the two results affords a method for detecting and crudely quantifying the concentration of exogenous osmoles such as methanol. This is accomplished by means of the following formula:
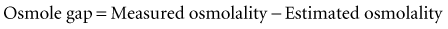
Measured serum osmolality is determined in most clinical chemistry laboratories by analysis of the freezing point of the sample. Freezing point represents a colligative property of solutions that is depressed in proportion to osmolality, regardless of the chemical nature of the osmoles. This method, therefore, allows an empirical assessment of osmolality. The normal serum osmole gap is typically less than 10 mOsm/kg H2O with this formula. Appreciable elevation of the osmole gap suggests the presence of an exogenous osmole (e.g., methanol). The only toxins that can appreciably affect the osmole gap are those that have a low molecular weight and can accumulate in relatively high concentration in the blood. A number of other exogenous compounds besides methanol meet these criteria, including ethylene glycol, acetone, isopropanol, propylene glycol, and acetonitrile, all of which have been reported to increase osmolality and the osmole gap.15,23,82,83
The constellation of laboratory findings that includes metabolic acidosis along with abnormal widening of both the serum anion gap and the serum osmole gap provides presumptive or corroborative evidence of methanol (or ethylene glycol) poisoning in compatible clinical settings. However, the serum osmole gap is not foolproof, and it has important limitations. False-positive results have been described in cases of circulatory shock, DKA or AKA, the hyperglycemic nonketotic dehydration syndrome, chronic renal failure, and multiple organ system failure.83 False-negative results can occur if the ingestion involved a small but still potentially lethal volume of methanol. When assessing the serum osmole gap, it is important to ensure that all relevant measurements are made from the same serum specimen to minimize variability due to temporal changes in individual analyte concentrations. Some clinical chemistry laboratories assay serum osmolality by the dew point or vapor pressure method. For technical reasons, this method yields spuriously low osmolality readings in the presence of ethanol, methanol, and other volatile alcohols, and therefore it should not be used to assess the osmole gap.83
Methanol assays are available in many clinical chemistry laboratories and provide a direct assessment of methanol concentration in serum samples. This test is not definitive, because patients who present late after methanol intake may have metabolized much or all of the ingested alcohol, although the toxic byproducts may be present in appreciable concentration.86 The delay between ingestion and presentation represents another factor that may explain the wide range of blood methanol concentrations reportedly associated with fatal outcome.87 Methanol assay results should be interpreted in conjunction with assessments of acid-base status, serum anion gap, and serum osmole gap, as well as the history and clinical findings.
Treatment
As with any toxic ingestion, the patient’s airway and ventilation must be immediately assessed and, if necessary, adequate support provided. Circulatory shock is treated with fluid resuscitation, inotropic support, and vasopressor agents, as appropriate. Whether the patient is initially unstable or not, close monitoring of vital signs, cardiopulmonary status, and neurologic status is indicated. Vomiting should not be induced because of the risk of aspiration and the lack of demonstrable benefit. Gastric lavage is unlikely to be of value unless the patient presents within 1 hour after ingestion. Activated charcoal is also of dubious benefit unless there is a concomitant toxic ingestant.9,10,12,88–93 However, co-intoxication with another drug or toxin should be considered routinely. Accordingly, naloxone should be administered if the subject is unconscious. Blood and urine samples should be obtained for toxicologic screening. As in acute ethanol intoxication, complicating and occult underlying comorbid disorders must be considered (see Table 171-2).
Specimens should also be obtained for diagnostic laboratory tests. However, because specific toxicologic identification is not available on site at all hospital laboratories, antidotal therapy should not be delayed if there is an obvious history of methanol ingestion.8,70,93,94 Even if “stat” testing is available, methanol intoxication may not be considered in occult cases until routine laboratory test results are obtained and reveal unexplained metabolic acidosis. In such cases, the preliminary laboratory test results in conjunction with a compatible setting and perhaps physical findings may allow a presumptive diagnosis to be made and antidotal treatment to be initiated. Treatment predicated on the presumptive diagnosis can be stopped if further studies convincingly argue against methanol intoxication. Symptomatic poisoned patients require ICU admission for frequent monitoring of vital signs and level of consciousness and to provide specific antidotal treatment, which consists of ethanol or fomepizole administration, hemodialysis, and folate administration.
Ethanol has been the conventional form of antidotal pharmacotherapy for methanol intoxication. The principle is that the enzymes, alcohol dehydrogenase and aldehyde dehydrogenase, have higher affinity for ethanol than for methanol, and ethanol thereby serves as an effective competitive inhibitor.95–98 As a result, conversion of methanol to formaldehyde and formate is significantly slowed in the presence of ethanol, allowing methanol to be excreted by the kidneys and lungs, and by hemodialysis if that modality is employed. If inhibition is incomplete, the body may be able to safely eliminate the much smaller amounts of formaldehyde and formate that are metabolically produced from the methanol. Indications for ethanol therapy include a serum methanol concentration greater than 20 mg/dL or a history or strong clinical suspicion of methanol ingestion in conjunction with either an elevated osmole gap or evidence of metabolic acidosis (e.g., arterial blood pH < 7.30 and bicarbonate < 20 mmol/L).
A loading dose is given so as to rapidly effect maximal enzyme inhibition. The goal is to achieve a serum ethanol level of 100 to 150 mg/dL. Based on the volume of distribution of ethanol (0.6-0.7 L/kg in men, slightly less in women and elderly subjects, and less in obese subjects) and a target serum ethanol concentration of 100 mg/dL, the necessary loading dose is theoretically 600 mg/kg in terms of absolute ethanol. Given the specific gravity of absolute ethanol (0.79), this is equivalent to a dose of 0.76 mL/kg in terms of absolute ethanol. Absolute (i.e., 100%) ethanol is unlikely to be available in a hospital formulary. Oral loading can be accomplished using 100 proof liquor, which is 50% ethanol by volume (equivalent to 40 g/dL), at a dose of 1.5 mL/kg. Alternatively, IV loading using a 5% (volume/volume) solution of ethanol in dextrose and water (i.e., an ethanol concentration of 4 g/dL by weight/volume) would require 15 mL/kg, typically administered over 1 hour. The dosing calculations described frequently underestimate the ethanol dose necessary to achieve the target level. Loading doses of 700 mg/kg given IV, or even higher doses if given orally, are more likely to achieve the goal initially.1 If the patient’s current ethanol concentration is already at or above the targeted level due to co-ingestion of ethanol, no ethanol loading dose is required. A proportionately lower loading dose is used in patients with a preexisting subtherapeutic blood ethanol concentration.
Fomepizole (4-methylpyrazole) is a newer therapeutic alternative to ethanol.99–102 The indications for fomepizole use are the same as for ethanol therapy. Like ethanol, fomepizole inhibits alcohol dehydrogenase, but it is considerably more costly than ethanol. Nevertheless, fomepizole has supplanted ethanol at many centers, owing to its advantage of being easier to dose and titrate and because it has no sedative effects. Frequent serial blood ethanol assays are avoided. Compared with oral dosing of ethanol, there is no risk of nausea, vomiting, gastritis, or abdominal pain with fomepizole. Compared with IV ethanol administration, there is less risk of overhydration.
Severe methanol poisoning can be associated with profound metabolic acidosis in some cases. Traditionally, sodium bicarbonate was a staple part of the treatment for most causes of metabolic acidosis, but lack of demonstrable efficacy has tempered its routine use, particularly in the treatment of lactic acidosis and DKA. There are laboratory animal data and anecdotal clinical reports ascribing benefit to bicarbonate administration in cases of alcohol or glycol poisoning. Specifically, administration of bicarbonate is claimed to be capable of reversing ocular manifestations and lowering mortality, but controlled clinical trials are lacking. There also is evidence that undissociated formic acid is more toxic than the dissociation product, formate; increasing the extracellular fluid pH favors conversion of formic acid to formate.103 Given the potential severity of the acidosis and the likely benefit of alkali therapy, sodium bicarbonate is recommended for subjects with an arterial pH less than 7.30, although intentional alkalemia is not advocated.
Ethanol and fomepizole minimize conversion of methanol to its toxic metabolites, but these forms of pharmacotherapy do not hasten elimination of methanol from the body. Methanol is excreted by the kidneys and lungs, but only slowly. Hemodialysis can effectively and more rapidly remove methanol and its toxic metabolites from the body. Charcoal or resin hemoperfusion techniques are not effective, and peritoneal dialysis is recommended only if hemodialysis is not available. Hemodialysis is recommended as a supplement to ethanol or fomepizole in patients with serious degrees of methanol intoxication. Serious intoxication is defined by the presence of metabolic acidosis, a serum methanol level above 50 mg/dL, any type of subjective or objective ocular findings, or other findings that indicate severe poisoning. Hemodialysis also is recommended if there is renal impairment. As previously noted, fomepizole and ethanol dosing must be altered during hemodialysis. Methods have been described to incorporate ethanol into the dialysate to facilitate maintaining therapeutic ethanol levels during hemodialysis.104 The endpoint for dialysis is a serum methanol level less than 20 mg/dL and normalization of the anion gap, indicating clearance of formate. Direct measurement of plasma formate would be a logical method of monitoring if rapid assays were available.
In humans and certain nonhuman primates, formate is only slowly metabolized, allowing the development of acidosis and ocular pathology if substantial amounts of methanol are ingested. Monkeys given large doses of folinic or folic acid before or after methanol administration had lower formate levels and less toxicity than control animals.105 Based on these and other experimental data, large doses of folic or folinic acid are recommended in clinical methanol poisoning. Typical recommendations are to administer 50 mg of folinic or folic acid IV every 4 to 6 hours. Folic acid must be reduced to tetrahydrofolate before it can serve as a cofactor for metabolizing formate. Folinic acid does not require reduction and therefore is the preferred form of the vitamin when available.
 Ethylene Glycol Intoxication
Ethylene Glycol Intoxication
Ethylene glycol is a clear, colorless, almost odorless, sweet-tasting, viscous liquid that is commonly used as the main constituent in most formulations of permanent automotive antifreeze. It also finds use in a variety of commercially available automotive fluids and paint products (Box 171-2), and it is used industrially as a solvent and synthetic precursor. Like methanol, it is occasionally ingested, either intentionally as an ethanol substitute or accidentally. More than 5000 cases of ethylene glycol exposure have been reported annually by the American Association of Poison Control Centers in recent years.60,61,62 Based on limited anecdotal data, the lethal dose in humans has been estimated at 1 to 2 mL/kg, but there are case reports of fatalities after lower doses and survival after higher doses.
Metabolism
The metabolism of ethylene glycol is more complicated than that of methanol.106,107 As with methanol, the parent compound is only minimally toxic, but its metabolites are very toxic. Also, in common with methanol, the initial step in metabolism is catalyzed by alcohol dehydrogenase (Figure 171-2). The action of alcohol dehydrogenases converts ethylene glycol to glycoaldehyde, which can be converted further to glyoxal. Both glycoaldehyde and glyoxal are metabolized first to glycolic acid, then more slowly to glyoxylic acid, and finally to oxalic acid. Glycoaldehyde and glyoxylate have demonstrable nephrotoxicity in isolated rodent renal tubular segments, whereas glycolate, oxalate, and ethylene glycol do not.108 Glycolate and probably some of the other metabolites are also neurotoxic. Oxalic acid can precipitate as calcium oxalate crystals within various tissues, including notably the renal parenchyma and tubules.
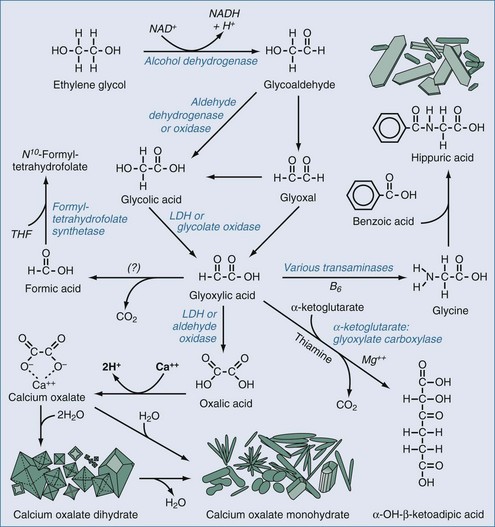
Clinical Manifestations
The initial effects involve the CNS and typically manifest within 30 minutes to 12 hours after ingestion.15 The CNS manifestations of ethylene glycol poisoning can range from effects that are similar to those seen with acute ethanol intoxication, such as excitement, confusion, disorientation, and ataxia, to signs of CNS depression, such as lethargy, stupor, or coma. Nausea, vomiting, myoclonus, and seizures also can occur. Cranial nerve deficits including nystagmus, ophthalmoplegia, facial palsy, dysarthria, and dysphagia have been reported. There are also rare case reports of pupillary abnormalities and changes in visual acuity, but these findings are not characteristic; if they do occur, they may be the result of co-ingestion of methanol. Classically, the second phase manifests 12 to 24 hours after ingestion and consists of cardiorespiratory effects which may include dyspnea and a Kussmaul respiratory pattern secondary to metabolic acidosis or pulmonary edema. The latter can result in frank respiratory failure necessitating endotracheal intubation and mechanical ventilation. Tachycardia, hypotension, frank circulatory shock, coma, and death also can occur during this phase. The third phase, which usually takes 1 to 3 days to manifest, consists of renal failure, either oliguric or nonoliguric, due to acute tubular necrosis. Flank pain also can occur. The time course of each phase of intoxication is variable, and overlap is frequent.
Laboratory Manifestations
Laboratory findings are similar to those seen in methanol poisoning. Detection of ethylene glycol in serum provides definitive evidence of the diagnosis. However, if the patient presents late and significant metabolism of the toxic agent has occurred, the measured concentration may not represent the patient’s peak ethylene glycol level. After ingestion of a substantial quantity of ethylene glycol, metabolic acidosis due to metabolic breakdown of the parent compound occurs during the first phase of intoxication.86 The acidosis may be severe and is principally caused by glycolic acid accumulation.109–112 Dissociation of this acid results in the accumulation of glycolate, which leads to an increase in the serum anion gap.
Measurement of the plasma glycolate concentration is a rational method of assessment, but clinical availability of the assay is lacking.81 The blood lactate concentration may be elevated because of the reducing intracellular milieu induced by ethylene glycol metabolism or as a manifestation of complicating seizures or circulatory shock. Lactate levels also may be artifactually elevated to a substantial degree because of the cross-reactivity of glycolate with lactate in certain automated lactate analyzers.113 The serum osmole gap may be elevated due to high blood levels of ethylene glycol and its metabolites. Because the molecular weight of ethylene glycol (62 daltons) is higher that of methanol (32 daltons), the osmole gap is less affected by a given amount (by weight) of ethylene glycol ingested or by a given blood level (by weight/volume) compared with methanol.84 Therefore, the osmole gap is more likely to yield a false-negative result after ingestion of ethylene glycol, compared with a similar mass amount of methanol.
There are two notable laboratory findings that may be seen in ethylene glycol poisoning; these are findings not observed in methanol poisoning. The first is calcium oxalate crystalluria.114 Oxalate produced by ethylene glycol metabolism chelates calcium, forming crystals and potentially producing hypocalcemia in the process (see Figure 171-2). Two crystalline forms of this organic salt can occur. One is calcium oxalate dihydrate, also known as weddellite. These crystals have a characteristic octahedral shape, making them relatively easy to distinguish from various nonoxalate forms of crystalluria. The second form is calcium oxalate monohydrate, also known as whewellite. These crystals can be polymorphic; they can appear as monoclinic prisms or assume a needle-like, dumbbell-shaped, ovoid, or hempseed-like appearance. Hippurate crystalluria also has been described, but it can be difficult to morphologically discriminate hippurate from some forms of whewellite by light microscopy.106 The finding of oxalate crystalluria corroborates the diagnosis; however, these crystals occasionally can be seen in the urine in the absence of ethylene glycol exposure, so their presence is not proof of glycol poisoning. On the other hand, because crystalluria does not uniformly occur after ethylene glycol ingestion, its absence does not exclude the diagnosis.
The other potential finding is fluorescence of the urine on exposure to ultraviolet radiation.115,116 This finding is present when the ingested formulation of ethylene glycol contains fluorescein, a fluorescent dye added to many automotive antifreeze solutions to facilitate identification of cooling system leaks and to mitigate accidental confusion with potable liquids. The fluorescein is excreted in the urine and fluoresces yellow-green on exposure to ultraviolet light, such as from a Wood’s lamp (commonly used in emergency departments and ophthalmology clinics to detect corneal lesions after topical application of fluorescein to the eye). False-positive results have been described due to other fluorescent substances in urine (e.g., carotene, carbamazepine, niacin, benzodiazepine metabolites) and from certain types of glass or plastic specimen containers that have a high degree of native fluorescence.115,117 False-negative results may occur if more than 4 hours has elapsed since the ingestion—that is, sufficient time for the fluorescein to be excreted, at least by some individuals. A false-negative result is obviously expected if the ingested ethylene glycol formulation did not contain fluorescein or involved a small volume. False-negative results can occur if the urine pH is less than 4.5, but this may be circumvented by urine pH testing followed by upward titration of the specimen’s pH if necessary. Owing to interfering factors and the limited ability of untrained examiners to detect fluorescence, clinical decision making should not hinge on this test in isolation.118
Treatment
With a few exceptions, the treatment of ethylene glycol poisoning is the same as for methanol intoxication. Gastric lavage may have some efficacy, but only if it is performed within 1 hour after the ingestion. Activated charcoal is not effective unless there is an amenable concomitant toxic ingestion.12,119 Ethanol98,120,121 or fomepizole99–102 is administered to slow the conversion of the glycol to toxic intermediates; sodium bicarbonate is given if there is significant metabolic acidosis (e.g., arterial pH < 7.30); and hemodialysis is used in cases of serious intoxication to speed elimination of the parent compound and toxic metabolites. Ethanol or fomepizole is recommended if the serum ethylene glycol concentration is above 20 mg/dL. However, ethylene glycol assays are not available at all institutions, and inhibitor treatment should be initiated while awaiting definitive identification of the glycol if there is presumptive evidence of intoxication.8,94,106 This evidence can include a clear history of recent ethylene glycol ingestion or strong clinical suspicion of ingestion in conjunction with either an elevated osmole gap, evidence of metabolic acidosis (e.g., arterial blood pH < 7.30 and bicarbonate < 20 mmol/L), or oxalate crystals in the urine. Dosing of ethanol and fomepizole is the same as for methanol intoxication. Fomepizole can be recommended over ethanol if the sensorium is depressed. Inhibitor treatment is continued until the serum ethylene glycol level falls below 20 mg/dL.
Although there is some evidence that formic acid may be produced as a minor product of ethylene glycol metabolism, it probably does not play a significant role in the pathophysiology of this form of poisoning. Therefore, folate administration has not been routinely recommended. However, there is more convincing evidence that glyoxylate may be metabolized to nontoxic products by enzyme systems that rely on other vitamin cofactors, specifically pyridoxine (vitamin B6) and thiamine (see Figure 171-2). Providing supplements of pyridoxine (e.g., 50 mg IV every 6 hours) and thiamine (e.g., 100 mg IV every 6 hours) could hasten elimination of toxic intermediates, although evidence of efficacy is quite limited.106 Given the low toxicity of these vitamins, both are recommended. Magnesium is a necessary cofactor for the enzymatic degradation of glyoxylate, and supplemental magnesium should be given if there is hypomagnesemia.
Key Points
Amato L Amato L, Minozzi S, Vecchi S, et al. Benzodiazepines for alcohol withdrawal. Cochrane Database Syst Rev 2010;3:CD005063.
Barceloux DG, Bond GR, Krenzelok EP, et al. American Academy of Clinical Toxicology practice guidelines on the treatment of methanol poisoning. J Toxicol Clin Toxicol. 2002;40:415-446.
Brent J, McMartin K, Phillips S, et al. Fomepizole for the treatment of ethylene glycol poisoning. N Engl J Med. 1999;340:832-838.
Kruse JA, Cadnapaphornchai P. The serum osmole gap. J Crit Care. 1994;9:185-197.
Winter ML, Ellis MD, Snodgrass WR. Urine fluorescence using a Wood’s lamp to detect the antifreeze additive sodium fluorescein: a qualitative adjunctive test in suspected ethylene glycol ingestions. Ann Emerg Med. 1990;19:663-667.
1 Cobaugh DJ, Gibbs M, Shapiro DE, et al. A comparison of the bioavailabilities of oral and intravenous ethanol in healthy male volunteers. Acad Emerg Med. 1999;6:984-988.
2 Lentner C, editor. Geigy Scientific Tables, 8th ed, vol 4. Basel: Ciba-Geigy, 1986;289-297.
3 MacDonald L, Kruse JA, Levy D, et al. Lactic acidosis and acute ethanol intoxication. Am J Emerg Med. 1994;12:32-35.
4 Charness ME, Simon RP, Greenberg DA. Ethanol and the nervous system. N Engl J Med. 1989;321:442-454.
5 Harper CG, Giles M, Finlay-Jones R. Clinical signs in the Wernicke-Korsakoff complex: A retrospective analysis of 131 cases diagnosed at necropsy. J Neurol Neurosurg Psychiatry. 1986;49:341-345.
6 Tsai G, Coyle JT. The role of glutamatergic neurotransmission in the pathophysiology of alcoholism. Annu Rev Med. 1998;49:173-184.
7 Hu X-J, Ticku MK. Chronic ethanol treatment upregulates the NMDA receptor function and binding in mammalian cortical neurons. Brain Res Mol Brain Res. 1995;30:347-356.
8 Church AS, Witting MD. Laboratory testing in ethanol, methanol, ethylene glycol, and isopropanol toxicities. J Emerg Med. 1997;15:687-692.
9 Pollack CVJr, Jorden RC, Carlton FB, et al. Gastric emptying in the acutely inebriated patient. J Emerg Med. 1992;10:1-5.
10 Krenzelok EP. New developments in the therapy of intoxications. Toxicol Lett. 2002;127:299-305.
11 Hultén BÅ, Heath A, Mellstrand T, et al. Does alcohol absorb to activated charcoal. Hum Toxicol. 1985;5:211-212.
12 Nejman G, Hoekstra J, Kelley M. Gastric emptying in the poisoned patient. Am J Emerg Med. 1990;8:265-269.
13 Baskin LB, Morgan DL. Drugs detected in patients suspected of acute intoxication. Texas Med. 1997;93:50-58.
14 Atassi WA, Noghnogh AA, Hariman R, et al. Hemodialysis as a treatment of severe ethanol poisoning. Int J Artif Organs. 1999;22:18-20.
15 Kruse JA. Methanol, ethylene glycol, and related intoxications. In: Carlson RW, Geheb MA, editors. Principles and Practice of Medical Intensive Care. Philadelphia: WB Saunders; 1993:1714-1723.
16 Umpierrez GE, DiGirolamo M, Tuvlin JA, et al. Differences in metabolic and hormonal milieu in diabetic- and alcohol-induced ketoacidosis. J Crit Care. 2000;15:52-59.
17 Cuevas-Korensky C, Kruse JA. Alcoholic ketoacidosis. In: Kruse JA, Parker MM, Carlson RW, et al, editors. Companion to Principles and Practice of Medical Intensive Care. Philadelphia: WB Saunders; 1996:383-386.
18 Tanaka M, Miyazaki Y, Ischikawa S, et al. Alcoholic ketoacidosis associated with multiple complications: report of 3 cases. Intern Med. 2004;43:955-959.
19 Fulop M. Alcoholic ketoacidosis. Endocrinol Metab Clin North Am. 1993;22:209-219.
20 Cuevas-Korensky CE, Kruse JA, Carlson RW. Characteristic findings in patients with alcoholic ketoacidosis. Chest. 1991;100:81S.
21 Cisek J, Kruse JA. Salicylate intoxication. In: Kruse JA, Parker MM, Carlson RW, et al, editors. Companion to Principles and Practice of Medical Intensive Care. Philadelphia: WB Saunders; 1996:549-553.
22 Kruse JA. Critical care crises. Clinical Case Studies in Respiratory Medicine. 1998;4:33-40.
23 Kruse JA. Clinical utility and limitations of the anion gap. Int J Intensive Care. 1997;4:51-66.
24 Kruse JA. Acid-base interpretations. In: Prough DS, Traystman RJ, editors. Critical Care: State of the Art, vol 14. Anaheim, CA. Society of Critical Care Medicine, and Baltimore: Williams & Wilkins; 1993:275-297.
25 Kruse JA. Use of the anion gap in intensive and care and emergency medicine. In: Vincent J-L, editor. Yearbook of Intensive Care and Emergency Medicine. New York: Springer-Verlag; 1994:685-696.
26 Dodd P. Neural mechanisms of adaptation in chronic ethanol exposure and alcoholism. Alcohol Clin Exp Res. 1996;20:151A-156A.
27 Airaksinen MM, Peura P. Mechanisms of alcohol withdrawal syndrome. Med Biol. 1987;65:105-112.
28 Spies CD, Dubisz N, Neumann T, et al. Therapy of alcohol withdrawal syndrome in intensive care unit patients following trauma: Results of a prospective, randomized trial. Crit Care Med. 1996;24:414-422.
29 Carlson RW, Raj JM. Alcohol withdrawal syndrome. In: Kruse JA, Fink MP, Carlson RW, editors. Saunders Manual of Critical Care. Philadelphia: WB Saunders; 2003:240-243.
30 Lohr RH. Treatment of alcohol withdrawal in hospitalized patients. Mayo Clin Proc. 1995;70:777-782.
31 Kumar CN, Andrade C, Murthy P. A randomized, double-blind comparison of lorazepam and chlordiazepoxide in patients with uncomplicated alcohol withdrawal. J Stud Alcohol Drugs. 2009;70:467-474.
32 Lechtenberg R, Worner TM. Seizure risk with recurrent alcohol detoxification. Arch Neurol. 1990;47:535-538.
33 Erstad BL, Cotugno CL. Management of alcohol withdrawal. Am J Health Syst Pharm. 1995;52:697-709.
34 Jermain DM, Crismon ML, Nisbet RB. Controversies over the use of magnesium sulfate in delirium tremens. Ann Pharmacother. 1992;26:650-652.
35 Wilson A, Vulcano B. A double-blind, placebo-controlled trial of magnesium sulfate in the ethanol withdrawal syndrome. Alcohol Clin Exp Res. 1984;8:542-545.
36 Mayo-Smith MF. Pharmacological management of alcohol withdrawal: A meta-analysis and evidence-based practice guideline. American Society of Addiction Medicine Working Group on Pharmacological Management of Alcohol Withdrawal. JAMA. 1997;278:144-151.
37 Holbrook AM, Crowther R, Lotter A, et al. Meta-analysis of benzodiazepine use in the treatment of acute alcohol withdrawal. CMAJ. 1999;160:649-655.
38 Amato L, Minozzi S, Vecchi S, et al. Benzodiazepines for alcohol withdrawal (review). Cochrane Database Syst Rev 2010;3:CD005063.
39 Daeppen JB, Gache P, Landry U, et al. Symptom-triggered vs fixed-schedule doses of benzodiazepine for alcohol withdrawal: a randomized treatment trial. Arch Intern Med. 2002;162:1117-1121.
40 Phillips S, Haycock C, Boyle D. Development of an alcohol withdrawal protocol. CNS collaborative exemplar. Clin Nurse Spec. 2006;20:190-198.
41 Sullivan JK, Sykora K, Schneiderman J, et al. Assessment of alcohol withdrawal: the revised clinical institute withdrawal assessment for alcohol scale (CIWA-Ar). Br J Addict. 1989;84:1353-1357.
42 D’Onofrio G, Rathlev NK, Ulrich AS, et al. Lorazepam for the prevention of recurrent seizures related to alcohol. N Engl J Med. 1999;340:915-919.
43 Weinberg JA, Magnotti LJ, Fischer PE, et al. Comparison of intravenous ethanol versus diazepam for alcohol withdrawal prophylaxis in the trauma ICU: results of a randomized trial. J Trauma Injury Infect Crit Care. 2008;64:99-104.
44 Ramsay MA, Savage TM, Simpson BRJ, et al. Controlled sedation with alphaxalone-alphadolone. Br Med J. 1974;2:656-659.
45 Riker RR, Picard JT, Fraser GL. Prospective evaluation of the sedation-agitation scale for adult critically ill patients. Crit Care Med. 1999;27:1325-1329.
46 Ely EW, Truman B, Shintani A, et al. Monitoring sedation status over time in ICU patients: Reliability and validity of the Richmond Agitation-Sedation Scale. JAMA. 2003;289:2983-2991.
47 Baumgartner GR, Rowen RC. Clonidine versus chlordiazepoxide in the management of acute alcohol withdrawal syndrome. Arch Intern Med. 1987;147:1223-1226.
48 Baumgartner GR, Rowen RC. Transdermal clonidine versus chlordiazepoxide in alcohol withdrawal: A randomized, controlled clinical trial. South Med J. 1991;84:312-321.
49 Dobrydnjov I, Axelsson K, Berggren L, et al. Intrathecal and oral clonidine as prophylaxis for postoperative alcohol withdrawal syndrome: a randomized double-blinded study. Anesth Analg. 2004;98:738-744.
50 Chance JF. Emergency department treatment of alcohol withdrawal seizures with phenytoin. Ann Emerg Med. 1991;20:520-522.
51 Rathlev NK, D’Onofrio G, Fish SS, et al. The lack of efficacy of phenytoin in the prevention of recurrent alcohol-related seizures. Ann Emerg Med. 1994;23:513-518.
52 Minozzi S, Amato L, Vecchi S, et al. Anticonvulsants for alcohol withdrawal. Cochrane Database Syst Rev 2010;3:CD005064.
53 Leone MA, Vigna-Taglianti F, Avanzi G, et al. Gamma-hydroxybutyrate (GHB) for treatment of alcohol withdrawal and prevention of relapses. Cochrane Database Syst Rev 2010;2:CD006266.
54 Croissant B, Loeber S, Diehl A, et al. Oxcarbazepine in combination with tiaprid in inpatient alcohol-withdrawal–a RCT. Pharmacopsychiatry. 2009;42:175-181.
55 Addolorato G, Leggio L, Abenavoli L, et al. Baclofen in the treatment of alcohol withdrawal syndrome: a comparative study vs diazepam. Am J Med. 2006;119:276.e13-276.e18.
56 Anton RF, Myrick H, Baros AM, et al. Efficacy of a combination of flumazenil and gabapentin in the treatment of alcohol dependence. J Clin Psychoparmacol. 2009;29:334-342.
57 Lum E, Gorman SK, Slavik RS. Valproic acid management of acute alcohol withdrawal. Ann Pharmacother. 2006;40:441-448.
58 Leggio L, Kenna GA, Swift RM. New developments for the pharmacological treatment of alcohol withdrawal syndrome. A focus on non-benzodiazepine GABAergic medications. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32:1106-1117.
59 Myrick H, Malcolm R, Randall PK, et al. A double-blind trial of gabapentin versus lorazepam in the treatment of alcohol withdrawal. Alcohol Clin Exp Res. 2009;33:1582-1588.
60 Bronstein AC, Spyker DA, Cantilena LRJr, et al. 2008 Annual Report of the American Association of Poison Control Centers’ National Poison Data System (NPDS): 26th Annual Report. Clin Toxicol. 2009;47:911-1084.
61 Litovitz TL, Klein-Schwartz W, White S, et al. 1999 Annual Report of the American Association of Poison Control Centers Toxic Exposure Surveillance System. Am J Emerg Med. 2000;18:517-574.
62 Litovitz TL, Klein-Schwartz W, White S, et al. 2000 Annual Report of the American Association of Poison Control Centers Toxic Exposure Surveillance System. Am J Emerg Med. 2001;19:337-395.
63 Davis LE, Hudson D, Benson BE, et al. Methanol poisoning exposures in the United States: 1993-1998. Clin Toxicol. 2002;40:499-505.
64 Pincus F. Die Massenerkrankungen im städtischen Asyl für Obdachlose in Berlin 24, bis 31 December 1911. Med Klin. 1912;1:41.
65 Swartz RD, Millman RP, Billi JE, et al. Epidemic methanol poisoning: Clinical biochemical analysis of a recent episode. Medicine (Baltimore). 1981;60:373-382.
66 Bennett ILJr, Cary FH, Mitchell GL, et al. Acute methyl alcohol poisoning: A review based on experiences in an outbreak of 323 cases. Medicine (Baltimore). 1953;32:4314-4363.
67 Naraqi S, Dethlefs RF, Slobodniuk RA, et al. An outbreak of acute methyl alcohol intoxication. Aust N Z J Med. 1979;9:65-68.
68 Benton CDJr, Calhoun FPJr. The ocular effects of methyl alcohol poisoning: Report of a catastrophe involving 320 persons. Am J Ophthalmol. 1953;36:1677-1685.
69 Aufderheide TP, White SM, Brady WJ, et al. Inhalational and percutaneous methanol toxicity. Ann Emerg Med. 1993;22:1916-1918.
70 Kruse JA. Methanol poisoning. Intensive Care Med. 1992;18:391-397.
71 Martin-Amat G, McMartin KE, Hayreh SS, et al. Methanol poisoning: Ocular toxicity produced by formate. Toxicol Appl Pharmacol. 1978;45:201-208.
72 Eells JT, Salzman MM, Lewandowski MF, et al. Formate-induced alterations in retinal function in methanol-intoxicated rats. Toxicol Appl Pharmacol. 1996;140:58-69.
73 Ferrari LA, Arado MG, Nardo CA, et al. Post-mortem analysis of formic acid disposition in acute methanol intoxication. Forensic Sci Int. 2003;133:152-158.
74 Hayreh MS, Hayreh SS, Baumbach GL, et al. Methyl alcohol poisoning III. Ocular toxicity. Arch Ophthalmol. 1977;95:1851-1858.
75 Seme MT, Summerfelt P, Neitz J, et al. Differential recovery of retinal function after mitochondrial inhibition by methanol intoxication. Investig Ophthalmol Vis Sci. 2001;42:834-841.
76 Tephly TR. The toxicity of methanol. Life Sci. 1991;48:1031-1041.
77 Malandain H, Cano Y. Serum methanol in the absence of methanol ingestion. Ann Emerg Med. 1996;28:102-103.
78 Stegink LD, Brummel MC, McMartin K, et al. Blood methanol concentrations in normal adult subjects administered abuse doses of aspartame. J Toxicol Environ Health. 1981;7:281-290.
79 Leon AS, Hunninghake DB, Bell C, et al. Safety of long-term large doses of aspartame. Arch Intern Med. 1989;149:2318-2324.
80 Chattopadhyay S, Chandra P. Images in clinical medicine. Putaminal necrosis. N Engl J Med. 2007;356:e23.
81 Fraser AD, Coffin L, Worth D. Drug and chemical metabolites in clinical toxicology investigations: The importance of ethylene glycol, methanol and cannabinoid metabolite analyses. Clin Biochem. 2002;25:501-511.
82 Kruse JA. Diagnosis and treatment of hyperosmolar emergencies. In: Vincent J-L, editor. 2000 Yearbook of Intensive Care and Emergency Medicine. New York: Springer-Verlag; 2000:544-559.
83 Kruse JA, Cadnapaphornchai P. The serum osmole gap. J Crit Care. 1994;9:185-197.
84 Lushine KA, Harris CR, Holger JS. Methanol ingestion: Prevention of toxic sequelae after massive ingestion. J Emerg Med. 2003;24:433-436.
85 Walker JA, Schwartzbard A, Krauss EA, et al. The missing gap: A pitfall in the diagnosis of alcohol intoxication by osmometry. Arch Intern Med. 1986;146:1843-1844.
86 Fraser AD, Coffin L. Optimal screening and monitoring of methanol and ethylene glycol poisoning by formic acid and glycolic acid. Ther Drug Monit. 2001;23:486.
87 Yayci N, Aùgritmis H, Turla A, et al. Fatalities due to methyl alcohol intoxication in Turkey: An 8-year study. Forensic Sci Int. 2003;131:36-41.
88 Kulig K, Duffy JP, Linden CH, et al. Toxic effects of methanol, ethylene glycol, and isopropyl alcohol. Top Emerg Med. 1984;6:14.
89 Decker WJ, Corby DG, Hilburn RE, et al. Adsorption of solvents by activated charcoal, polymers, and mineral sorbents. Vet Hum Toxicol. 1981;23(Suppl 1):44-45.
90 Whalen JE, Richards CJ, Ambre J. Inadequate removal of methanol and formate using the sorbent based regeneration hemodialysis delivery system. Clin Nephrol. 1979;11:318-321.
91 Szabuniewicz M, Bailey EM, Wiersig DO. A new regimen for the treatment of ethylene glycol poisoning. IRCS Med Sci. 1975;3:102.
92 Cooney DO. The treatment of ethylene glycol poisoning with activated charcoal. IRCS Med Sci. 1977;5:265.
93 Barceloux DG, Bond GR, Krenzelok EP, et al. American Academy of Clinical Toxicology practice guidelines on the treatment of methanol poisoning. J Toxicol Clin Toxicol. 2002;40:415-446.
94 Kearney J, Rees S, Chiang W. Availability of serum methanol and ethylene glycol levels: A national survey. J Toxicol Clin Toxicol. 1997;35:509.
95 McCoy HG, Cipolle RJ, Ehlers SM, et al. Severe methanol poisoning: Application of a pharmacokinetic model for ethanol therapy and hemodialysis. Am J Med. 1979;67:804-807.
96 Bartlett GR. Inhibition of methanol oxidation by ethanol in the rat. Am J Physiol. 1950;163:619-621.
97 Zatman LJ. The effect of ethanol on the metabolism of methanol in man. Biochem J. 1946;40:67-68.
98 Peterson CD, Collins AJ, Himes JM, et al. Pharmacokinetics during therapy with ethanol and hemodialysis. N Engl J Med. 1981;304:21-23.
99 Brent J, McMartin K, Phillips S, et al. Fomepizole for the treatment of ethylene glycol poisoning. N Engl J Med. 1999;340:832-838.
100 Mbia J-JE, Guérit J-M, Haufroid V, et al. Fomepizole therapy for reversal of visual impairment after methanol poisoning: A case documented by visual evoked potentials investigation. Am J Ophthalmol. 2002;134:914-916.
101 Druteika DP, Zed PJ, Ensom MHH. Role of fomepizole in the management of ethylene glycol toxicity. Pharmacotherapy. 2002;22:365-372.
102 Sivilotti MLA, Burns MJ, McMartin KE, et al. Toxicokinetics of ethylene glycol during fomepizole therapy: Implications for management. Ann Emerg Med. 2000;36:114-125.
103 Liesivuori J, Savolainen H. Methanol and formic acid toxicity: Biochemical mechanisms. Pharmacol Toxicol. 1991;69:157-163.
104 Chow MT, Di Silvestro VA, Yung CY, et al. Treatment of acute methanol intoxication with hemodialysis using an ethanol-enriched, bicarbonate-based dialysate. Am J Kidney Dis. 1997;30:568-570.
105 Noker PE, Eells JT, Tephly TR. Methanol toxicity: Treatment with folic acid and 5-formyl tetrahydrofolic acid. Alcohol Clin Exp Res. 1980;4:378-383.
106 Kruse JA. Ethylene glycol intoxication. J Intensive Care Med. 1992;7:234-243.
107 Rajagopal G, Ramakrishnan S. Hepatic metabolism of ethylene glycol and its relevance to ethanol as antidote in EG toxicity. Ind J Pharmacol. 1994;26:108-111.
108 Poldelski V, Johnson A, Wright S, et al. Ethylene glycol-mediated tubular injury: Identification of critical metabolites and injury pathways. Am J Kidney Dis. 2001;38:339-348.
109 Jacobsen D. Organic acids in ethylene glycol intoxication. Ann Intern Med. 1986;105:799-800.
110 Jacobsen D, Ostby N, Bredesen JE. Studies on ethylene glycol poisoning. Acta Med Scand. 1982;212:11-15.
111 Jacobsen D, Ovrebo S, Ostborg J, et al. Glycolate causes the acidosis in ethylene glycol poisoning and is effectively removed by hemodialysis. Acta Med Scand. 1984;216:409-416.
112 Hewlett TP, McMartin KE, Lauro AJ, et al. Ethylene glycol poisoning: The value of glycolic acid determinations for diagnosis and treatment. Clin Toxicol. 1986;24:389-402.
113 Morgan TJ, Clark C, Clague A. Artifactual elevation of measured plasma L-lactate concentration in the presence of glycolate. Crit Care Med. 1999;27:2177-2179.
114 Morfin J, Chin A. Images in clinical medicine. Urinary calcium oxalate crystals in ethylene glycol intoxication. N Engl J Med. 2005;353:e21.
115 Winter ML, Ellis MD, Snodgrass WR. Urine fluorescence using a Wood’s lamp to detect the antifreeze additive sodium fluorescein: A qualitative adjunctive test in suspected ethylene glycol ingestions. Ann Emerg Med. 1990;19:663-667.
116 Davis DP, Bramwell KJ, Hamilton RS, et al. Ethylene glycol poisoning: Case report of a record-high level and a review. J Emerg Med. 1997;15:653-657.
117 Casavant MJ, Shah MN, Battels R. Urine fluorescence: Is it a good test for ethylene glycol ingestion? Pediatrics. 2002;109:345.
118 Wallace KL, Suchard JR, Curry SC, et al. Diagnostic use of physicians’ detection of urine fluorescence in a simulated ingestion of sodium fluorescein-containing antifreeze. Ann Emerg Med. 2001;38:49-54.
119 Barceloux DG, Krenzelok EP, Olson K, et al. American Academy of Clinical Toxicology practice guidelines on the treatment of ethylene glycol poisoning. J Toxicol Clin Toxicol. 1999;37:537-560.
120 Peterson DI, Peterson JE, Hardinge MG, et al. Experimental treatment of ethylene glycol poisoning. JAMA. 1963;186:955-957.
121 Nunamaker DM, Medway W, Berg P. Treatment of ethylene glycol poisoning in the dog. J Am Vet Med Assoc. 1971;159:310-314.
