22 Essentials of Neurology and Neuromuscular Disorders
General Considerations
The term neurologic disorder encompasses a wide variety of conditions that may have extremely mild or very serious effects (Table 22-1).1–5 These disorders are likely to be associated with a degree of physical, cognitive, or a combined disability. Many children with severe physical disabilities have normal intelligence and are competent to make decisions about treatment options. Those who are mildly cognitively impaired may wish to be consulted about treatment choices; adolescents in particular must be involved in decision making.6 This approach is especially important in managing individuals with chronic disorders who are accustomed to thinking about health issues and who often have strong and well-defined opinions about how they wish to be treated.7
| Condition | Prevalence | Reference |
|---|---|---|
| Cerebral palsy: all types | 2.2 per 1000 live births | 1 |
| Epilepsy | 5-10 per 1000 all ages | 2 |
| Central nervous system tumors | 1-5 per 1000 all ages | 3 |
| Neuromuscular disorders (all ages) | 1 per 2900 total population | 4 |
| Congenital myopathy | 1 per 28,600 | |
| Duchenne muscular dystrophy | 1 per 3500 | |
| Myotonic dystrophy | 1 per 12,000 | |
| Limb girdle dystrophy | 1 per 90,000 | |
| Spinal muscular atrophy | 1 per 74,000 | |
| Mitochondrial disorders | 11.5 per 100,000 all ages | 5 |
When planning anesthesia for these individuals, physicians must become knowledgeable about them and their conditions. Assumptions should not be made about their level of comprehension or about how they and their parents view the choices available.8 It was formerly acceptable practice for children with chronic neurologic disorders to be excluded from the full range of therapeutic options, but it is now essential that all options are included.9
Parents of children with chronic disorders of all types are usually accustomed to dealing with health care situations and often have thought carefully about the implications of various treatments. They know the child best and are usually most qualified to make decisions by proxy. Widespread use of the Internet has assisted many parents in becoming knowledgeable about their child’s condition and about potential interventions.10 Their aims are usually entirely appropriate, and this knowledge may assist physicians in their collaboration with children and parents in determining optimal management. Occasionally, the information has been obtained from an unreliable source and may be inaccurate or inappropriate for a particular child.11 Misinformation can precipitate difficult situations for professionals, especially when there is a perceived disparity between the desires of the parents and those of the child or what the clinician considers to be in the child’s best interest. The surgical team members usually are most involved in the process of obtaining informed consent, but the anesthesiologist must participate in the dialogue because anesthesia may be the part of treatment that carries the greatest risk.
Children with neurologic disorders usually have a regular physician overseeing their care, and this person should participate in the decision-making process. For elective procedures, the surgeon should establish contact with the child’s regular pediatric specialist and anesthesiologist as soon as surgery is contemplated, inform them about the proposed operation, and seek their opinions to optimize perioperative management. Cognitive, communication, and behavioral problems; coexisting diseases; and drug therapy that may influence management of the anesthesia for these patients should be evaluated at an early stage.12 Some children may be receiving long-term respiratory support, including cough assist devices and home ventilation, and all current therapies should be considered.
Oral communication may be difficult for some children with neurologic disorders. Use of age-appropriate assisted communication devices may help to ensure open communication between physician and patient.12 Parents’ opinions and attempts to protect their children should be respected and understood as a consequence of previous experiences, stress, frustration, anger, and probably guilt.13 Each procedure has to be assessed in terms of morbidity, mortality, and probability of improving quality of life. All aspects must be clearly and objectively discussed with parents or guardians and informed consent forms signed.
Static Neurologic Disorders
Cerebral Palsy
Cerebral palsy is a disorder of movement, muscle tone, or posture that is caused by injury or abnormal development of the immature brain. It has a prevalence of 1 case per 500 live births.14 Eighty percent of cases are acquired prenatally and have no obvious cause, although they are associated with prematurity low birth weight, intrauterine growth retardation, intracranial hemorrhage, and trauma. Birth complications, including asphyxia, are estimated to account for another 6% of cases. Postnatal causes of cerebral palsy (10%) usually arise from infectious causes (e.g., bacterial meningitis, viral encephalitis), trauma (e.g., motor vehicle collisions, falls, child abuse), or metabolic disturbance (e.g., hyperbilirubinemia).15
Classification depends on the severity, distribution, and thenature of the motor deficit. With the advent of modern imaging techniques, including magnetic resonance imaging (MRI), understanding the pathogenesis of cerebral palsy has advanced dramatically.16–18 Clinical features are classified most commonly according to the type of motor deficit, its distribution, and the severity of the deficit (Table 22-2). Involvement of a single limb is referred to as monoparesis, of both limbs on one side of the body as hemiparesis, of both lower limbs as diparesis, of three limbs as triparesis, and of all four limbs as tetraparesis (i.e., quadriparesis). The motor deficit may manifest as hypotonia, spasticity, or extrapyramidal features such as choreoathetoid/dystonic movements or ataxia. A descriptive classification includes neurologic deficit and distribution (e.g., spastic diparesis, dystonic hemiparesis).
Children with cerebral palsy require neuroimaging to confirm the diagnosis and underlying cause of the condition. Although some centers use oral sedation (i.e., midazolam or chloral hydrate) for neuroimaging, many children require general anesthesia. They may require multiple anesthesias throughout their lifetimes because of the associated comorbidities (i.e. respiratory, gastrointestinal, neuromuscular, and orthopedic), common surgical conditions, and problems unique to cerebral palsy that require treatment.19
Multisystem Comorbidities
Most children with cerebral palsy have clinically significant oromotor dysfunction, and when associated with gastroesophageal reflux, it may lead to recurrent aspiration, decreased respiratory reserve, esophageal stenosis, and malnutrition.20 Frequently employed procedures include fundoplication, gastrostomy, and esophageal dilation treatment.12 Immobility, underhydration, and poor diet predispose patients to bowel stasis and constipation, which may become severe, with fecal impaction occurring occasionally. Malnutrition may depress immune responses, and electrolyte imbalance and anemia are common. Preoperative assessment of these parameters is essential.
Pulmonary complications are common causes of death in cerebral palsy. Aspiration associated with gastroesophageal reflux is the leading cause, and it may be exacerbated by excessive oral secretions, bulbar dysfunction, recurrent respiratory infection, and chronic lung disease.21,22 Scoliosis may also restrict pulmonary function, with cardiopulmonary involvement depending on the curve pattern and the severity of the curve (see Chapter 30).12
Orthopedic operations are the most frequently performed procedures in children with cerebral palsy.12 Procedures include tendon releases to ease contractures, femoral osteotomy, and hip adductor and iliopsoas releases.23 The trend in orthopedic surgery is to perform multiple procedures involving tenotomies or osteotomies at different levels of all extremities during a single general anesthesia, rather than staging them during multiple operations.12,19 Scoliosis often requires surgery to prevent further deterioration in lung function and to stabilize the spine to facilitate ambulation and sitting. Spinal fusion is considered in all children with progressive curves greater than 40 to 50 degrees.24
Approximately 30% of children with cerebral palsy have epilepsy. It is more common in spastic hemiplegia and less common in the ataxic and choreoathetotic forms. Generalized and focal seizures frequently occur. Anticonvulsants should be maintained until the surgery date (given the morning of surgery) and restarted as soon as possible in the postoperative period.12,25
Anesthesia Considerations for Cerebral Palsy
The many multisystem comorbidities and therapies specific to children with cerebral palsy must be understood to minimize perioperative complications. Risk factors include an inability to walk, severe neurologic deficit, major cognitive dysfunction, severe scoliosis, malnutrition, and the presence of a gastrostomy or tracheostomy.19 Severely compromised children can be optimally managed postoperatively with admission to the pediatric intensive care unit to provide analgesia with comprehensive monitoring, maximum support, and aggressive respiratory care, and after they are stabilized, they can be transferred to a setting with less intense monitoring.
Most of these children have above-average intelligence. They have the same emotional and cognitive concerns as others about undergoing anesthesia, including preoperative anxiety that may require premedication. Children with contractures, especially in the upper extremities, may present a challenge for establishing intravenous access. If gastroesophageal reflux is not controlled, consideration should be given to rapid-sequence induction. Although these children have a neuromuscular disorder, intravenous succinylcholine yields only a normal release of potassium,26 despite evidence of proliferation of extrajunctional acetylcholine receptors.27 Maintenance of and emergence from anesthesia requires special considerations, including the possibility of a reduced minimal alveolar concentration (MAC),28 resistance to neuromuscular blocking agents,29 and reduced bispectral index (BIS) measurements.30 If vomiting is likely to occur, the airway must be protected.
These children have normal responses to pain, which should be managed as if they were unaffected by cerebral palsy. Caudal or epidural analgesia may be a reasonable approach for perioperative pain management if the child does not have a ventricular-peritoneal shunt. Management and assessment of perioperative pain in children with neurocognitive impairment is addressed in Chapter 43.
Malformations of the Nervous System
Malformations are common in pediatric neurologic practice and a frequent cause of early mortality. The appearance of the neural plate shows that the central nervous system (CNS) develops very rapidly in the 2-week embryo and continues until several years after birth. The cause of CNS malformations is largely uncertain, but timing appears to be more important than the nature of the insult in producing the specific type of malformation. Causative agents include maternal drugs such as sodium valproate, which is associated with neural tube defects (NTDs); infections such as cytomegalovirus, which can cause various cerebral lesions, depending on the time in gestation of the infection; toxins such as alcohol; vitamin deficiency (e.g., folic acid), and genetic disorders. Historically, because diagnostic investigation was limited, postmortem examination was required to demonstrate the neuropathologic changes causing the clinical disorder. MRI now can provide adequate images to enable a diagnosis in many instances (e.g., cortical dysplasia).31
Neural Tube Defects: Cranial and Spinal Dysraphism
The cause of NTDs is multifactorial, with genetic and environmental factors being the most important. Approximately 10% of NTDs are caused by chromosomal abnormalities such as trisomies (i.e., 18, 13, and 21), triploidy, and Turner syndrome. Preconceptual folic acid supplementation has reduced the prevalence of NTDs by 30% to 50%.32 Along with antenatal ultrasound examination, screening is done for increased maternal serum levels of α-fetoprotein, reduced human chorionic gonadotropin levels, and reduced unconjugated estriol levels; termination of pregnancy in cases that test positive has further reduced the prevalence of NTDs.33
Anencephaly is a lethal disorder that results from failure of the neural tube to form. This leads to disorganization of neural elements and the absence of skull formation.31 Some deep cerebral structures may remain intact, and the brainstem may develop normally. With the latter, normal respiration and cardiovascular functions may develop, enabling the infant to survive for hours or days after birth. Other structures in the head and brain, including the eyes, face, and pituitary gland, may not develop normally.
Encephalocele is a herniation of neural tissue and meninges out of the skull through deficient skin and bone (see Fig. 24-11, B). They are frequently associated with other cerebral malformations, such as agenesis of the corpus callosum. Encephaloceles found anteriorly are associated with underlying brain, orbital structures, or pituitary gland anomaly. Posteriorly, encephaloceles are associated with cerebral or cerebellar tissue that herniates through a bony defect in the posterior cranium. Intranasal encephaloceles may be difficult to detect. These defects carry a poor prognosis for long-term survival. Most infants die, and in survivors, severe neurodevelopmental disability is common. Most of these children have hydrocephalus.34
Spina bifida refers to a group of conditions in which there is abnormal or incomplete formation of the midline structures over the back (see Fig. 24-11, A, in Chapter 24).31 Skin, bony, and neural elements may be involved singly or in combination. Congenital malformations of the spinal cord may exist in isolation or in association with brain anomalies. These defects may present at birth, as in the case of the more severe and open lesions (i.e., spina bifida) or be identified later in childhood if the skin overlying the spinal defect is intact (e.g., spina bifida occulta). Those who develop a Chiari malformation may present with cervical cord or bulbar deficits, placing them at risk for respiratory embarrassment (see Figs. 24-12 and 24-13). Children with spinal cord lesions are at increased risk for sensory deficits, making meticulous skin care and positioning essential to prevent pressure sores and damage to neuropathic joints.
When the defect is identified at birth, it is optimally managed in a specialist center by a multidisciplinary team (i.e., pediatrician, neurologist, neurosurgeon, orthopedic surgeon, and others) who can anticipate, prevent, and treat complications and assist in the child’s long-term care. Children with dysraphism often develop postoperative hydrocephalus because of disrupted CSF flow and require a ventricular-peritoneal shunt. Long-term complications, including paraparesis, neurogenic bladder and bowel, renal insufficiency, trophic limb changes, pressure sores, joint contractures, and scoliosis, may require surgical repair and future intervention. These children are also at risk for latex allergy. From the outset, all children with NTDs should be considered latex sensitive and undergo management in a latex-safe environment. The anesthesia considerations for NTDs are presented in Chapter 24.
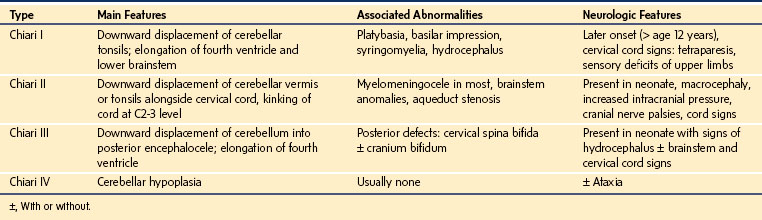
Chiari Malformation
Chiari malformations of the nervous system may coexist with other anomalies and manifest in the neonatal period or later in the early decades of life (Table 22-3).
Syringomyelia
Syringomyelia results from a glial cell–lined cavitation within the spinal cord. Diagnosis has been greatly simplified by the use of MRI, which provides images of the spinal cord and the tubular fluid-filled space within.31 The pathogenesis of syringomyelia remains unclear. It complicates several conditions, such as rare familial cases, congenital malformations, trauma, and meningeal infection. It has been reported as a coincidental finding in normal individuals.
Hydrocephalus
Hydrocephalus results from overproduction or impaired drainage of CSF from the brain.34 In practice, overproduction is an uncommon source of hydrocephalus; these cases most often result from tumors of the choroid plexus. Obstructed CSF drainage is the far more common basis. Causes of hydrocephalus include intraventricular hemorrhage, Arnold-Chiari malformation, brain tumor, congenital obstruction, and myelomeningocele.
Surgical treatment for hydrocephalus involves insertion of a drainage system to shunt CSF from the brain to another site in the body (see Fig. 24-10). The anesthesia considerations for treating hydrocephalus are discussed in Chapter 24.
Disorders of Ventral Induction
Holoprosencephaly is a cephalic disorder in which the forebrain of the embryo fails to develop into discrete hemispheres with normal connections.31 There are three types:
1. Lobar: There is almost complete separation of the hemispheres, and the corpus callosum is almost absent.
2. Semilobar: The two hemispheres are divided posteriorly, with interhemispheric connections present anteriorly. The corpus callosum is absent anteriorly, and the thalami are fused in the midline.
3. Alobar: An undivided and small forebrain with a dorsal sac may contain some cortex. Severe facial defects may include cyclopia (i.e., single orbit with fused globes), cebocephaly (i.e., single nostril), and a midline cleft lip.
Disorders of Cortical Development
Malformations of the cerebral cortex are many and varied. Although uncommon, development of lissencephaly or agyria (i.e., smooth cortex), pachygyria (i.e., thickened cortex), and polymicrogyria (i.e., multiple, small gyri) depends on the stage of embryogenesis affected.31 MRI has advanced the identification of these features and their classification.
Progressive Neurologic Disorders
Primary Brain Tumors
The incidence of primary brain tumors is 2.6 per 100,000 children, and they account for more than 20% of all childhood malignancies. One third of these tumors occur before 5 years of age, and 75% occur before 10 years of age.35 Two thirds are located infratentorially and one third supratentorially (Table 22-4).36
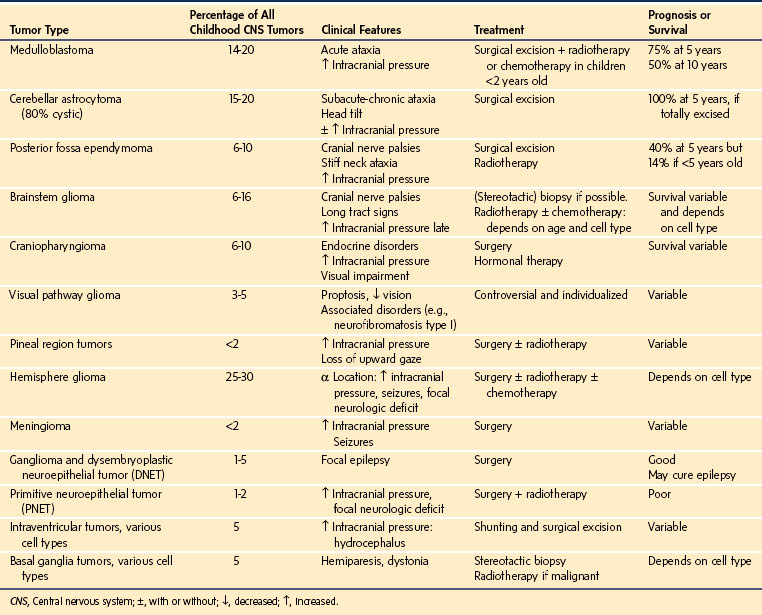
Pathologic classification, which is based on the cell of origin and degree of malignancy, extends from grade I (benign) to grade 4 (malignant).37 The patient’s presentation depends on the site of the tumor. Infants typically display irritability, failure to thrive, and macrocephaly, whereas older children develop headache, nausea, vomiting, seizures, gait disturbances, and visual deficits. If the lesion is rapidly expanding and is accompanied by significant cerebral edema or obstructs CSF drainage, the intracranial pressure (ICP) will increase. Occasionally, hemorrhage may occur into the tumor, causing a dramatic progression of signs and symptoms, mandating emergency treatment. Ultimately, brainstem decompensation and death ensue if the lesion is not treated.
The 5-year survival rate of greater than 60% for primary brain tumors largely reflects improved imaging, aggressive surgery, and evidence-based therapy.38 Unfortunately, children who survive CNS tumors frequently have permanent neurologic deficits, including epilepsy, learning disabilities, visual or hearing impairment, and growth and endocrine disorders. Short-term and long-term follow-up evaluations by specialist teams are required, along with careful emotional and social support for children and their families. Some children have genetic predispositions for CNS tumors, such as neurofibromatosis (i.e., schwannomas of the spinal cord, peripheral nerve tumors, skeletal deformities, carcinoid syndrome, and multiple endocrine neoplasia, including pheochromocytoma) and tuberous sclerosis (i.e., brain tumors, cardiac rhabdomyomas, renal abnormalities, and hepatoma), and they require genetic analysis and long-term follow-up.
Tumors of the spinal cord are rare in childhood. They may be benign or malignant and sited within the cord (intramedullary) or outside (extramedullary). Symptoms and signs may initially be nonspecific and vague, especially in young children. This may delay the diagnosis and increase the risk of spinal cord compression. Delayed relief of compression may cause vascular compromise, which may lead to total and irreversible paralysis of the limbs, bladder, and bowel and permanent, severe disability. Diagnosis is best made by MRI of the cord, which provides details of the lesion and adjacent structures without the risk of further decompensation, a problem raised by the use of myelography in the past.36
Metabolic Disease
These diseases may cause a static encephalopathy but more often produce a progressive course with loss of physical and intellectual skills. Epilepsy, especially myoclonus, is common, as is loss of vision, neuropathy, deafness, or involvement of other organ systems (particularly cardiac). Some neurometabolic diseases are associated with intellectual deficits and some with physical deficits; systemic features may be prominent, and neurologic signs are common (Table 22-5).
TABLE 22-5 Neurometabolic Disorders
There are three main groups of neurometabolic diseases39:
1. Those with a known enzymatic defect, including disorders of amino acid metabolism (e.g., phenylketonuria), peroxisomal disorders (e.g., adrenoleukodystrophy), and lysosomal storage disorders (e.g., Tay-Sachs disease)
2. Those with abnormal storage accumulation in CNS cells, including lysosomal storage disorders and mucopolysaccharidoses
3. Those with no identified biochemical defect (e.g., Cockayne syndrome), a heterogeneous group that is shrinking as research identifies the biochemical defects
Disorders that cause lactic acidosis are the most common of these diseases, although many individuals who harbor the genetic substrate for one of these disorders may not express it or may come to diagnosis very late in life.40 The features of these diseases are extremely varied, and although the neuropathologic, biochemical, and imaging abnormalities are well recognized, a precise diagnosis may remain elusive.
Neuromuscular Disorders
Neuromuscular disorders are caused by an abnormality of any component of the lower motor neuron system: anterior horn cell in the spinal cord, axon, neuromuscular junction, or muscle fiber (Fig. 22-1).41 The cardinal features are weakness of skeletal muscles that is proximal, distal, or generalized in distribution, hypotonia, and reduced deep tendon reflexes. True fatigability suggests a defect of the neuromuscular junction. Neuropathy is characterized by distal weakness and sensory deficit. Joint contractures, scoliosis, and respiratory and cardiac involvement are common complications, and some conditions are associated with cognitive deficits.
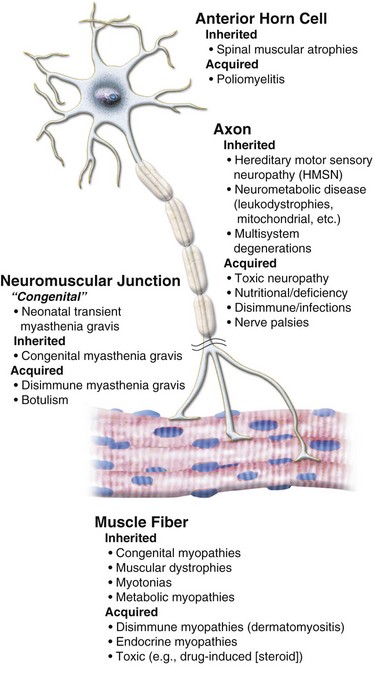
FIGURE 22-1 Diagram of a lower motor neuron.
(Modified from Dubowitz V. Muscle disorders in childhood. Philadelphia: WB Saunders; 1978. Courtesy A. Moosa, MD.)
Disorders of the Anterior Horn Cell
Spinal Muscular Atrophies
The spinal muscular atrophies (SMAs) are a group of disorders in which there is progressive degeneration of the anterior horns of the spinal cord and death of motor neurons. They are inherited as an autosomal recessive trait. The type of SMA (I, II, or III) is determined by the age of onset and the severity of symptoms.41 The diagnosis is a clinical one, but confirmation by molecular testing for the survival of motor neuron gene (SMN) in a blood sample is now possible, and electrophysiology and muscle biopsy are no longer necessary.42 Curative treatment is not available, but much can be done to improve duration and quality of life, especially for mildly affected children.
Type I SMA (i.e., Werdnig-Hoffmann disease) manifests at or soon after birth in most cases. The infant may appear neurologically normal at first, but the typical picture soon emerges. Parents may first notice an abnormal breathing pattern as the intercostal muscles are affected, and the respiratory pattern becomes diaphragmatic, with a bell-shaped chest observed clinically and radiographically. The infant is usually very alert and interactive but severely weak and floppy. There is good facial expression and normal eye movements, but the tongue fasciculates, and the tendon reflexes are absent. There is no cardiac involvement. Weakness, hypotonia, and bulbar involvement lead to progressive respiratory insufficiency and swallowing dysfunction, which are frequently complicated by episodes of aspiration. Most children die within the first 2 years of life, mainly due to respiratory complications. Management is essentially palliative, with gentle physiotherapy to keep the limbs flexible and special care with feeding. Noninvasive nocturnal ventilation is increasingly used, but invasive ventilation through a tracheostomy is not considered appropriate in most medical centers. This issue has provoked considerable debate in the literature.43
Type II SMA is an intermediate form, and it is the most prevalent. Because the weakness is milder, many children survive for years with meticulous multidisciplinary therapy, orthopedic and respiratory management, and care with nutrition. Onset occurs at 6 to 18 months of age. The clinical signs are similar to those of type I SMA, and the children are bright, intelligent, and particularly verbal. Type II patients usually achieve independent sitting at some stage, but these children never bear weight or walk. Respiratory infections are a particular problem, and noninvasive nocturnal respiratory support (i.e., biphasic or continuous positive airway pressure) with a facemask and portable ventilator is well tolerated by children and parents. This improves well-being and enables remarkably full activity despite weakness.44,45
Some children with type II SMA develop feeding difficulties because of weakness of bulbar musculature or as a complication of chronic nocturnal hypoventilation. Good nutrition is essential for health, and supplementation and gastrostomy feeding may be required.46
Type III SMA (i.e., Kugelberg-Welander disease) is a mild variant with similar signs of flaccid and areflexic weakness of lower more so than upper limbs and with proximal predominance. These children attain independent walking, although it may be later than normal and tenuous. There is often deterioration around the time of puberty, when the growth spurt causes the previously precariously balanced muscle groups to become dysfunctional. Obesity and joint contracture also may affect the situation. Aggressive measures to keep the child walking are often effective and delay or prevent scoliosis and lower limb contracture.47 Respiratory support may be necessary for nocturnal hypoventilation. Prognosis for long-term survival is good.
Other SMA variants exist in childhood. A rare type (SMA with respiratory disease) has severe diaphragmatic involvement and respiratory failure in infancy.48 Some variants have associated features such as cerebellar atrophy, distal involvement, or additional myopathic features.
Anesthesia Considerations for Spinal Muscular Atrophies
Children with SMA present for a variety of surgical procedures, including gastrostomy placement, tracheostomy, and spinal surgery, and they may also require anesthesia for diagnostic evaluation.49 Rigorous preanesthesia evaluation is essential; perioperative care needs to be tailored to each child’s needs, and postoperative respiratory support may be required. A variety of anesthesia techniques have been used successfully with and without muscle relaxants. Various degrees of sensitivity to nondepolarizing relaxants have been described, and it seems prudent to avoid or reduce the dose of these agents. If they are used, neuromuscular function should be assessed continuously and the effect of the relaxant antagonized at the conclusion of surgery. Spinal and epidural anesthesia and postoperative epidural analgesia have been used without adverse effects; however, the potential for respiratory depression may be increased with the addition of neuraxial opioids.
Poliomyelitis
Poliomyelitis is a highly contagious, infectious disease caused by poliovirus, a human enterovirus. Most poliovirus infections are asymptomatic. If symptomatic, it occurs in two phases: an acute, nonspecific, febrile illness followed by aseptic meningitis and acute, flaccid, lower motor neuron paralysis. It typically manifests asymmetrically and may affect any muscle group. In children with respiratory involvement, lifelong ventilation may be needed. Fortunately, this situation is almost unheard of in the Western world because polio has been almost eradicated by immunization.50
Axonal Disorders
Hereditary Neuropathies
Hereditary neuropathies are a group of inherited disorders that affect the peripheral nervous system. There are four main subcategories: hereditary motor and sensory neuropathy (most common is Charcot-Marie-Tooth disease),51 hereditary sensory neuropathy, hereditary motor neuropathy, and hereditary and autonomic sensory neuropathy.
Clinical presentation depends on the subtype and can occur at any age. Children usually present with disorders of gait or foot deformity, or they may come to the attention of a neurologist or geneticist through an affected parent. Clinical signs are usually confined to the lower limbs in the early years and may lead to orthopedic intervention before diagnosis. Some neuropathies are associated with multisystem involvement, including cardiac, autonomic, and respiratory systems, and a complete preoperative assessment of these children is essential.52
Peripheral neuropathy is a component of various neurometabolic disorders in which there is involvement of other parts of the nervous system or of other organs. For example, in the leukodystrophies, demyelination affects central and peripheral axons, giving a clinical picture of combined upper and lower neuron features. There are many neurometabolic disorders (e.g., mucopolysaccharidoses, sphingolipidoses, urea cycle disorders, organic disorders, amino acid disorders) with many variants, and presentation can occur at all ages from birth to adulthood. In children with mitochondrial diseases, peripheral neuropathy is often found, along with myriad other features.53
Multisystem degenerations are often genetically determined disorders of the nervous system that rarely begin in childhood.52 Features may include dementia, epilepsy, extrapyramidal signs, brainstem dysfunction, vision and hearing impairment, anterior horn cell involvement, and peripheral neuropathy. They tend to have a progressive course.
Acquired Disorders of the Peripheral Nerves
Guillain-Barré Syndrome
Guillain-Barré syndrome is an acute demyelinating disorder that causes progressive weakness usually 2 to 4 weeks after an illness with features of a viral infection or after immunization.33 The inflammatory lesions in the peripheral nerves typically are associated with loss of myelin. In severe cases, the axons are damaged, a feature that is also seen in some acute cases without significant demyelination. The precise pathologic process has not been delineated, but various antibodies, immune complexes, and complement components have been found, suggesting causal heterogeneity.
Guillain-Barré syndome is diagnosed clinically, and the diagnosis is reinforced by finding an increased protein concentration in the CSF (despite a normal cell count) and abnormalities on nerve conduction studies. The studies may confirm demyelination by showing lowered nerve conduction velocity with distal delay, axonal involvement by low-amplitude action potentials, and in early cases, abnormality or absence of the H reflex, indicating absence of the spinal reflex arc. MRI of the spine may show thickening and contrast enhancement of the nerve roots and cauda equina.54
Patients with very mild symptoms that do not interfere with activities of daily living can be observed for deterioration without treatment. Corticosteroids are ineffective and may delay recovery. Intravenous immunoglobulin and plasmapheresis are the current treatment options.53
Supportive care in the intensive care unit may be needed for children with severe bulbar or respiratory weakness.55 Children with axonal neuropathies are usually slower to recover motor function than those with demyelination, and our impression is that early treatment with intravenous immunoglobulin may alter the course.
Chronic Inflammatory Demyelinating Polyneuropathy
Chronic inflammatory demyelinating polyneuropathy is extremely rare in childhood, and it is usually confined to older age groups. It manifests in a subacute or relapsing and remitting pattern with prominent sensory involvement. diagnostic investigations are similar to those for acute Guillain-Barré syndrome. Corticosteroids, intravenous immunoglobulin, and plasma exchange have been effective.55 Although these treatments may be effective in the short term, recurrences may take place, and the usual pattern is that of a chronic, disabling, and relapsing and remitting condition that does not threaten longevity but interferes significantly with the quality of life.56
Nerve Palsies
Like neuropathies, nerve palsies are uncommon in childhood.51 The most common palsy encountered in children is neonatal or congenital facial nerve palsy. Cranial nerve palsies, especially those that involve the eye muscles, are usually related to intracranial disease such as increased ICP in children, but they may also be isolated findings that result from viral infections. In the latter instances, recovery is the norm.
Peripheral nerve palsies such as carpal tunnel syndrome have been reported in childhood.57 They may complicate severe juvenile arthritis58 or storage disorders such as mucopolysaccharidoses. Because symptoms may be difficult to elicit from children at the best of times, identifying palsies may be problematic in severely learning disabled children. Specialist care may require routine assessment of nerve conduction and possible surgical decompression. Concern has been expressed regarding the emergence of carpal tunnel syndrome as a type of repetitive strain injury in children due to excessive use of video games.59 Case reviews refer to other types of repetitive strain injury, such as basketball training and skiing, as etiologic factors.60
Disorders of the Neuromuscular Junction
Myasthenia Gravis
Myasthenia gravis is a disorder of the neuromuscular junction. The autoimmune disorder is characterized by one of several lesions of acetylcholine-mediated transmission.61 Weakness results from antibodies that block acetylcholine receptors, inhibiting the excitatory effects of acetylcholine at the neuromuscular junction. This reduction in the number of acetylcholine receptors results in a characteristic pattern of progressively reduced muscle strength with repeated use and recovery of muscle strength after a period of rest.
Neonatal Transient Myasthenia Gravis
Neonatal myasthenia gravis is caused by placental transfer of antibodies to acetylcholine receptors from an affected or previously affected mother. The infant may present with feeding difficulties or respiratory dysfunction.62 Treatment, which involves anticholinesterase medications, must be tailored to the nature and severity of the weakness, and intensive support occasionally is required. This form of myasthenia gravis is a transient disorder for which treatment is temporary and the risk of recurrence small. It should be anticipated in the offspring of any woman who has active myasthenia gravis or a history of the disorder, because cases have been described in infants of mothers in remission from myasthenia gravis.
Congenital Myasthenic Syndromes
Congenital myasthenia gravis represents several genetically determined defects of the neuromuscular junction that have in common an exercise-induced weakness of skeletal muscle.63 Fourteen genetic mutations have been identified, but many cases of congenital myasthenia gravis do not have an identified mutation. Inheritance of mutations usually follows an autosomal recessive pattern.
Although rare, congenital myasthenia gravis should be considered in the differential diagnosis of any neonate or infant who presents with motor problems (e.g., weakness, hypotonia, fatigability), eye signs (e.g., ptosis, ophthalmoplegia, pupillary abnormalities), and respiratory insufficiency (e.g., recurrent apneas, ventilator dependence). Late-onset muscle weakness has been reported in adolescence or early adulthood. Diagnosis may be difficult because the classic features of myasthenia gravis, including responses to anticholinesterase medications, may be absent.64 A Tensilon test, electromyography with repetitive nerve stimulation, a muscle biopsy, and molecular analysis in a specialist center should be sought to confirm the diagnosis.
Juvenile Myasthenia Gravis
Treatment of myasthenia gravis varies with the type and biochemical characteristics. Careful titration of pyridostigmine is needed. Corticosteroids and surgical thymectomy are indicated for patients with immunologic abnormalities.65
Anesthesia Considerations for Myasthenia Gravis
The anesthesiologist may become involved in the management of children with myasthenia gravis for several reasons66:
 Children may suffer a crisis requiring mechanical ventilation or when large-bore central access is attempted to facilitate plasma exchange transfusion.
Children may suffer a crisis requiring mechanical ventilation or when large-bore central access is attempted to facilitate plasma exchange transfusion.

 They may undergo elective or emergency surgery unrelated to their myasthenia.
They may undergo elective or emergency surgery unrelated to their myasthenia.
The activity of nondepolarizing muscle relaxants is increased as their duration of action is increased. Unfortunately, the degree to which the sensitivity is increased is somewhat unpredictable and depends on an interaction between the severity of the disease (e.g., level of acetylcholine receptor antibodies) and the efficacy of treatment. Inhalational anesthetic agents inhibit neuromuscular transmission, and these effects may be exaggerated in myasthenia gravis patients. However, no clinically significant postoperative neuromuscular depression has been demonstrated with isoflurane, sevoflurane, or desflurane. These potential neuromuscular effects are not shared by propofol, making total intravenous anesthesia (TIVA), in theory at least, the technique of choice for these children. For all but the most minor surgery in the patient with stable myasthenia gravis without significant respiratory or bulbar compromise, a tracheal tube and intermittent positive-pressure ventilation are likely to be required. If possible, intubation of the trachea should be performed without the use of muscle relaxants. Tracheal intubation with inhalational anesthetics alone or propofol with a short-acting opioid has been described in these children.67
Disorders of Muscle Fibers
Myopathies
Congenital Myopathies
Congenital myopathies are rare disorders that have extreme variations in the type and severity of features (Table 22-6).70 The myopathies may manifest at any age, but in many cases, they become apparent only in adulthood, challenging the congenital designation.71 In infancy, the usual features are weakness and hypotonia (i.e., the floppy baby).72 Although these are inherited disorders, there is often no family history, and because many cases are autosomal dominant or X-linked recessive, the carrier parent may have subclinical signs of a myopathy.
| Disorder | Genetics |
|---|---|
| Central core disease | 19q13, RYR1, ryanodine receptor |
| Multicore or minicore disease | 1p36, SEPN1 |
| Nemaline myopathy | 2q21.2-q22, NEB, nebulin |
| Desmin myopathy | 2q35, DES, desmin |
| Myotubular or centronuclear myopathy | Xq27.3-q28, MTM1, myotubularin 1 |
Data from http://neuromuscular.wustl.edu/syncm.html (accessed June 2012); http://www.genenames.org (accessed June 2012).
Children usually present with ptosis, facial weakness, limb weakness (proximal more than distal), and there may be evidence of respiratory muscle weakness and cardiomyopathy. In some cases, the extraocular muscles may be affected with external ophthalmoplegia. Joint contractures and scoliosis can also develop. Infants often have characteristic facies, with a long and narrow face, prominent ears, and a narrow, high-arched palate. Deep tendon reflexes are reduced or absent. The diagnosis usually is confirmed by characteristic findings on muscle biopsy. Molecular analysis is available for many of these myopathies in specialist laboratories.73
Paralleling the clinical features, the course of congenital myopathies varies greatly. Many individuals are mildly affected and may come to diagnosis only if a more severely affected child is diagnosed. All parents, even those without symptoms, must be carefully examined by a neurologist with expertise in neuromuscular disease to detect very mild weakness of particular muscles. For a parent who demonstrates weakness, it may help to perform a muscle biopsy and molecular testing to confirm the diagnosis. For many children with congenital myopathy, there is little or no deterioration in muscle strength as the child ages, and many children experience gradual improvement in muscle strength and a reduction in symptoms. However, weakness is severe in some cases, and clinical complications, including deglutition difficulties and respiratory failure, limit survival, although these features are not specific to congenital myopathies.74,75
Support of respiration and nutrition may be necessary. Minimally invasive methods such as noninvasive nocturnal ventilation by facemask are indicated to assist with management at home. Regular passive movements and careful management of posture and positioning to prevent contractures, especially scoliosis, is essential. Meticulous care of skin, joints, bowels, and teeth help to avoid the need for more invasive management. A complete evaluation of cardiovascular and respiratory status is required before anesthesia and surgery.76
Malignant hyperthermia (MH) is a disorder of skeletal muscle that manifests in response to anesthetic triggering agents. Central core disease is a myopathy closely associated with MH (see Chapter 40), with extreme variation in the severity of its features. MH and central core disease are primarily disorders of calcium regulation in skeletal muscle. Genetic testing identifies mutations in the ryanodine receptor gene (RYR1) in most affected individuals, accounting for their susceptibility to MH.77 The RYR1 gene encodes the channel that controls calcium release from the sarcoplasmic reticulum in skeletal muscle and to a lesser extent in other organ systems. RYR1 abnormalities alter the channel kinetics for calcium inactivation, and calcium buildup causes excessive skeletal muscle contraction due to disinhibition of the normal actin-myosin interaction. The level of adenosine triphosphate (ATP) decreases, leading to anaerobic and aerobic metabolism and to acidosis. Other effects of MH include heat production, sympathetic nervous system activation, hyperkalemia, tachycardia, muscle rigidity, tachypnea, disseminated intravascular coagulation, fever, myoglobinuria, and multiorgan dysfunction and failure.
The in vitro contracture test (IVCT) was developed to confirm susceptibility to MH by studying the contracture of muscle fibers in response to triggers such as caffeine and halothane.78 This is an invasive and expensive investigation because an open muscle biopsy is required, and very few specialized centers perform this procedure. Most individuals with central core myopathy may be susceptible to MH, as demonstrated by IVCT, which has been the only means to confirm MH susceptibility until the emergence of molecular testing.79 DNA analysis can provide a fairly reliable test for susceptibility to MH for 60% of affected individuals who have had an IVCT but for only 20% of those who have not had an IVCT (see Chapter 40).80 However, the complexity of the situation is such that assessment and advice by a specialist is recommended rather than immediately embarking on DNA analysis so that the child and family may benefit from appropriate investigation and interpretation. Any individual with central core disease and his or her family should be informed about the clinical situation, diagnosis, and possible risks and complications, including MH, and should be offered a specialized investigation. An individual who may be susceptible to MH should carry information to inform medical staff in an emergency.81 Any elective operative procedure must be meticulously planned in advance with the full involvement of the anesthesiologist.
Metabolic Myopathies
Metabolic myopathies (Table 22-7) are uncommon and complex. Most affected children have multisystem involvement of sufficient severity to mask the myopathic features. The most important concerns for the anesthesiologist are the risk of metabolic instability during surgery and the need for close liaison with the child’s pediatrician to plan fluid and electrolyte balance and nutritional management.81,82
Mitochondrial Disorders Underlying Myopathies
Diagnosis relies on characteristic clinical features, an understanding of mitochondrial genetics, and a logical, informed approach to investigations. Abnormalities of the mitochondrial genome are extremely common, and they can cause devastating phenotypes or be completely subclinical. Assessment and diagnosis are challenging.5
Neurologic symptoms and signs are common. However, they tend to be varied and include myopathy, neuropathy, strokelike episodes, ataxia, dementia, epilepsy, migraine, sensorineural deafness, and pigmentary retinopathy. The more common clinical scenarios in childhood are outlined in Table 22-8. Involvement of other organ systems, such as diabetes mellitus, gastrointestinal disease, and cardiomyopathy, may coexist or dominate the clinical picture. Evidence of mitochondrial dysfunction, such as lactic acidosis, increased CSF protein, and ragged red fibers on muscle biopsy, may or may not exist. Assessment of the child with atypical and otherwise unexplained features should include a search for evidence of mitochondrial abnormalities. The clinical details determine the investigative plan, which should include cerebral imaging with MRI and MRS (to detect lactate peaks), blood biochemistry (i.e., creatine kinase, lactate, and glucose levels), tests of urinary amino and organic acids, cardiologic assessment (i.e., chest radiography, electrocardiogram, and echocardiogram), electroencephalography, and exercise testing and neurophysiology, leading to mitochondrial studies of blood and muscle tissue. Muscle biopsy investigations include histopathology, electron microscopy, respiratory chain enzymology, and molecular analysis of mitochondrial DNA. Further investigation with respiratory chain enzymes and molecular genetic analysis is indicated in some cases.83
| Syndrome | Features |
|---|---|
| Alper-Huttenlocher, neuronal degeneration | Catastrophic onset of epilepsy, paralysis, ataxia, dementia, visual impairment, liver disease; death usually in months |
| Leigh encephalomyeloneuropathy | Brainstem signs predominant; relapsing/remitting or steadily progressive to death |
| Infantile myopathy and lactic acidosis | Hypotonia in infancy, feeding difficulties, respiratory problems, cardiomyopathy; fatal and nonfatal forms |
| Leber hereditary optic neuropathy (LHON) | Progressive visual loss in childhood, cardiac arrhythmias, dystonia |
| Kearns-Sayre (KSS) | Progressive external ophthalmoplegia, pigmentary retinopathy, deafness, heart block, choreoathetosis and ataxia, myopathy, endocrine disorders |
| NARP | Neuropathy, ataxia, retinitis pigmentosa |
| MELAS | Mitochondrial encephalomyopathy, lactic acidosis, stroke-like episodes, dementia |
| MERFF | Myoclonic epilepsy, myopathy with ragged red fibers (muscle Gomori trichrome stain), ataxia |
| MNGIE | Myoneurogenic gastrointestinal encephalopathy |
Treatment of mitochondrial disorders includes management of specific features such as antiepileptic therapy and efforts to maintain stability of metabolic pathways. Vitamins and other supplements such as coenzyme Q10 (i.e., ubiquinone), riboflavin, thiamine, and carnitine are referred to as the mitochondrial cocktail and are used in most of the mitochondrial disorders. Use of the cocktail is largely based on the assumption that higher doses of these agents may improve mitochondrial energy generation.84 Arginine has been used in the treatment of patients with mitochondrial encephalopathy with lactic acidosis and stroke-like episodes (MELAS).85
Anesthesia Considerations for Myopathies
The challenge for the anesthesiologist is to maintain metabolic stability and prevent complications. In children prone to lactic acidosis, intravenous fluids that contain lactate should be avoided, whereas administration of glucose-containing solutions is essential in any but the shortest of procedures to avoid hypoglycemia.82 In all children with mitochondrial disorders, the preoperative fasting period should be kept to a minimum to avoid hypovolemia and depletion of glucose stores. Stresses that may provoke increased energy requirements, such as perioperative pain, hypothermia, or hyperthermia, must be minimized. Children with Kearns-Sayre syndrome must have adequate assessment and perioperative monitoring because they may have cardiac conduction abnormalities as well as myopathy, diabetes, proximal renal tubular acidosis, and other multisystemic abnormalities.84
Inhalational and intravenous forms of anesthesia have been used successfully in children with mitochondrial disorders. Few serious complications have occurred, and none related to anesthesia were reported in two retrospective reviews of perioperative complications after general anesthesia in children with mitochondrial myopathies.82,86 Inhalational anesthetics have been used without consequences in these children, and there is no evidence that this population has a greater susceptibility to MH than normal children.87 However, mitochondrial disorders result from a variety of molecular changes and have varied responses to anesthetics.88
All inhalational anesthetics and propofol depress mitochondrial function at several levels. It has been suggested that children who develop metabolic acidosis and myocardial failure after propofol infusions for extended periods (more than 5 mg/kg/hr for more than 48 hours) have a subclinical form of a mitochondrial disorder. Although propofol infusion syndrome (PRIS) impairs mitochondrial oxidation and transport of free fatty acids in a manner not unlike that in mitochondrial myopathies, there is no evidence that the two disorders are linked. In a review of 61 patients with PRIS, seven (four children and three adults) developed PRIS during anesthesia.89 Impaired tissue perfusion (as seen in sepsis) may be a common underlying mechanism. Propofol interferes with fatty acid metabolism and the respiratory chain, in much the same manner as other anesthetics and medications, suggesting that its use during general anesthesia for children with mitochondrial myopathies is reasonable, and it has been used widely.90,91 Evidence suggests that any anesthesia technique may be used in children with mitochondrial myopathies.
Muscular Dystrophies
Muscular dystrophies represent a group of more than 30 inherited disorders of muscle that may occur in infancy, childhood, or adulthood (Table 22-9). The word dystrophy implies a destructive progressive process, and although this is the characteristic clinical course for many of those afflicted with muscular dystrophies, the course is extremely slow or has no muscle strength deterioration for others.92
| Disorder | Genetics |
|---|---|
| Myotonic dystrophy | |
| DM1 (98%), autosomal dominant | 9q13.3, DMPK or DM1, dystrophia myotonica |
| DM2 (2%), autosomal dominant | 3q21, CNBP (formerly ZNF9) |
| Congenital Muscular Dystrophies | |
| Autosomal recessive | |
| Fukuyama type | 9q31-q33, FKTN, fukutin |
| Walker-Warburg | 9q34.1, POMT1 |
| Merosin negative | 6q22-q23, LAMA2 |
| Merosin positive | 21q22.3, COL6A1, COL6A2 |
| Ulrich | 2q37, COL6A3 |
| X-linked recessive dystrophinopathies | |
| Duchenne and Becker types | Xp21.2, DMD, dystrophin |
| Emery-Dreifuss muscular dystrophy | |
| X-linked recessive | Xq27.3-q28, EMD, emerin |
| Autosomal dominant | 1q22, LMNA, lamin A/C |
| Limb girdle muscular dystrophies | |
| Autosomal recessive | 12 currently characterized |
| Autosomal dominant | 7 currently characterized |
| Facioscapulohumeral muscular dystrophy | 4q35, FSHMD1A |
| Autosomal dominant | |
| Distal myopathies | 5q31.2, MATR3, matrin 3 |
| Oculopharyngeal | 14q11.2, PABPN1 (formerly PABP2) |
Data from http://neuromuscular.wustl.edu/syncm.html (accessed June 2012); http://www.genenames.org (accessed June 2012).
This area of neurology has advanced greatly due to developments in molecular genetics and to a lesser extent in muscle pathology. Duchenne muscular dystrophy was the first inherited disorder for which the causative gene and defective protein were identified.92 Characterization of defective proteins leads to advances in immunohistochemistry that can assist diagnosis using a muscle biopsy. Detection of genetic mutations enables the diagnosis to be determined using a blood specimen in most instances, removing the need for muscle biopsy in many children. The mutated gene and molecular site of the defect have been identified for most of the common disorders, enabling much more focused management and informed counseling of families.93
The characteristic clinical features of muscular dystrophy include muscle weakness, the distribution of which varies from type to type; contractures, sluggish deep tendon reflexes, and involvement of respiratory and heart musculature. Other features, including learning disabilities, deafness, and ophthalmologic disorders, often exist. The creatine kinase level may be normal or increased to a minor or major degree; it provides an important marker for some disorders, such as the dystrophinopathies, in which it is raised on the order of 100 times normal. Diagnosis rests on careful clinical assessment, creatine kinase determination, molecular analysis, and muscle biopsy. Although there is no specific curative treatment for any of these disorders, advances in understanding the molecular genetic defect and proteins responsible for a disease have focused the search for an effective pharmacologic treatment to reverse the clinical signs and symptoms.94,95
For an infant who presents with severe weakness and hypotonia, signs of facial weakness, reduced alertness, and reduced respiratory effort raise the possibility of a congenital myotonic dystrophy (DM1), which is usually inherited from the mother in an autosomal dominant fashion. Clinical suspicion of DM1 may be confirmed by molecular testing for the affected gene (DMPK), which codes for myotonic dystrophy protein kinase. This protein is expressed predominantly in skeletal muscle, and the gene is located on the long arm of chromosome 19. This information negates the need for a muscle biopsy. A similar infant with good facial muscle movement, alertness, areflexia, and a diaphragmatic respiratory pattern indicating intercostal weakness is most likely to have SMA. The molecular test for the survival motor neuron is indicated to avoid the need for a muscle biopsy. Similar principles may be applied to children who present later in childhood. For example, a boy with gait abnormalities and a creatine kinase level 10 to 100 times normal is most likely to have a dystrophinopathy, and targeted molecular genetic testing can avoid the need for a biopsy.96
Myotonic Dystrophy
Myotonic dystrophy is the most common inherited neuromuscular disorder in the general population, and many cases remain subclinical or undiagnosed.4 All degrees of severity are possible, from the almost immobile infant with the congenital variant to the elderly adult with minimal motor weakness. Severity of the muscle weakness correlates with the molecular defect.
These children are susceptible to prolonged recovery from anesthesia, and care should be taken with the use of sedative medications and neuromuscular blocking agents.97 Sensitivity to succinylcholine is increased, and maximal use of regional nerve blocks or local infiltration with local anesthetic agents should be employed whenever possible to minimize the need for opioids.
Most children with congenital or later-onset myotonic dystrophy have some degree of learning difficulties, which may be severe. One or the other parent is affected to a greater or lesser extent. In congenital cases (DM1), the mother is always the affected parent and, in most instances, is not aware of the disorder, the diagnosis is made by the pediatrician or pediatric neurologist who sees the child.98 The infant does not demonstrate signs of myotonia, and a muscle biopsy is not helpful for the diagnosis at this age. The finding of facial weakness and clinical myotonia in the mother strongly suggests this disorder in a weak and floppy neonate.99 The neonatal literature suggests that shaking the maternal hand (i.e., mother is unable to easily release her grip) may be sufficient to allow the pediatrician to make this diagnosis.100 However, because it is possible to confirm the diagnosis by molecular testing of blood from the infant and mother, it is no longer necessary to rely on clinical expertise in muscle strength testing. Genetic counseling is essential and should be offered to the wider family circle. It is not unusual to uncover a large kindred with previously unsuspected disease in this situation.
These infants may require intensive care in the neonatal period, including respiratory support, but they tend to improve with age. However, significant learning disability is the rule, and the input of the multidisciplinary child development team followed by special schooling is the norm. Specific conditions requiring surgery include scoliosis and joint contractures, and perioperative care must be carefully planned.98
Dystrophinopathies
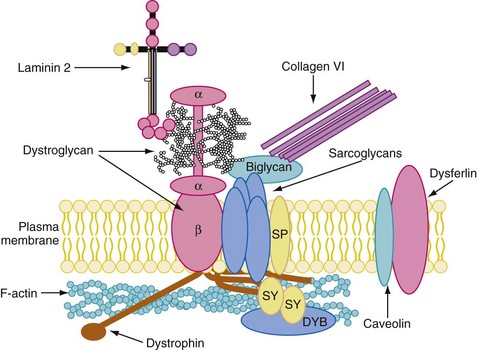
The next most commonly encountered dystrophies are the dystrophinopathies. Duchenne muscular dystrophy (DMD) is the more severe phenotype, and Becker muscular dystrophy (BMD) is the milder phenotype. Both dystrophies are inherited in an X-linked recessive pattern and affect boys almost exclusively.101 These disorders are caused by a deficiency of dystrophin (i.e., less than 3% of the normal content in DMD), a muscle membrane protein essential to the skeletal and cardiac muscle cytoskeleton and to neural tissue. Dystrophin reinforces the inner strength of the myocyte during lateral stretching and is involved in signal transduction (Fig. 22-2). DMD occurs as a result of mutations, mainly deletions in the dystrophin gene (DMD, locus Xp21.2).96
For children with DMD, glucocorticoids are the only available medication that slows the decline in muscle strength and function, which reduces the risk of scoliosis and stabilizes pulmonary function.102 Cardiac function may also improve; the limited data indicate a slower decline in echocardiographic measures of cardiac dysfunction.103,104 Prednisone is commonly used, although deflazacort is an alternative used in some countries and is probably just as effective. The most effective prednisolone regimen appears to be 0.75 mg/kg/day. International guidelines for initiation of steroids and other pharmacologic agents and the implementation of multidisciplinary care are clearly outlined in a Consensus Statement on Standard of Care for Congenital Muscular Dystrophies.105
If lower limb contractures exist despite range-of-motion exercises and splinting, surgery can be considered in some scenarios.106 The approach must be strictly individualized.105 Surgical intervention is frequently used to treat lower limb contractures and enable rehabilitation in long leg orthoses so that the ambulatory phase may be prolonged.107 This is usually indicated for children between 8 and 12 years of age, and it is well tolerated and successful if supported by a specialist team, but it may increase the burden of care for parents and families.108
Patients not treated with glucocorticoids have a 90% chance of developing significant progressive scoliosis109 and a small chance of developing vertebral compression fractures due to osteoporosis. Although glucocorticoids can reduce the risk of scoliosis,110,111 the risk of vertebral fracture is increased.112,113 Spinal care should involve an experienced spinal surgeon and comprises scoliosis monitoring, support of spinal and pelvic symmetry, and spinal extension by the wheelchair seating system. Children receiving glucocorticoids should be monitored for painful vertebral body fractures. Corrective spinal surgery improves posture and seating options, eliminates pain due to vertebral fracture from osteoporosis, and slows the rate of respiratory decline.109,114 The procedure is fraught with difficulty due to postoperative respiratory weakness and possible cardiac dysfunction.114 Careful preparation and liaison among the pediatrician or neurologist, surgeon, and anesthesiologist is essential; a plan for postoperative management, including the provision of intensive care, should be made.81
Facioscapular muscular dystrophy is less often encountered in childhood, and specific surgical and anesthesia issues are limited. There are several phenotypes in childhood, including a severe neonatal form and a variably progressive childhood type that may be associated with sensorineural deafness. Irregular alignment between phenotype and genotype implies inconsistent expression of the molecular defect.115,116 These children may benefit from surgery to fix the scapulae, which improves functionality of the upper limbs.117,118
Limb girdle syndromes (no longer a single entity; see Table 22-9) occur infrequently. Some have cardiac and respiratory implications and require preoperative assessment. In Emery-Dreifuss muscular dystrophy (EDMD), a syndrome in which cardiac conduction defects and dysrhythmias are common, syncope is usually the presenting complaint. These children are followed by a specialist team that includes a cardiologist.81
Anesthesia Considerations for Muscular Dystrophies
Despite extensive documentation that inhalational anesthetics or succinylcholine, or both, can trigger life-threatening rhabdomyolysis in children with muscular dystrophy, this problem persists.119–121 Case reports have described rhabdomyolysis, hyperkalemia, and cardiac arrest with or without the use of succinylcholine in children with DMD. No single inhalational anesthetic appears to be without blame.
The defect in dystrophinopathies results from the lack of the membrane-stabilizing protein dystrophin. It has been suggested that the addition of another destabilizing agent, such as an inhalational anesthetic, predisposes these children to mild or severe rhabdomyolysis, hyperkalemia, and death. Those most at risk for these complications are younger children, some of whom are undiagnosed until the perioperative rhabdomyolysis develops and establishes the diagnosis. Adolescents in whom muscle breakdown has waned and muscle mass has been lost and replaced by fatty infiltration have had uneventful perioperative courses when exposed to inhalational anesthetic agents and succinylcholine. Although succinylcholine is infrequently used today, the mortality rate associated with a succinylcholine-induced cardiac arrest is 30%.122
Although there is insufficient evidence to contraindicate inhalational anesthetics in children with muscular dystrophy, total intravenous anesthesia (TIVA) has emerged as an alternative to inhalational anesthetics in them.108 In the absence of a compelling reason, it seems prudent to avoid inhalational anesthetics in children with DMD.121,123 At the same time, without knowledge of the minimum concentration of inhalational anesthetic that triggers muscle breakdown, there is little justification for cleaning anesthetic workstations as in cases of MH. However, in certain clinical situations, such as a child with DMD and a difficult airway for whom an intravenous technique may be contraindicated or who refuses establishing IV access awake, brief exposure to an inhalational anesthetic to secure the airway seems reasonable. The anesthesia technique could then be converted to TIVA. The child should be carefully monitored for the signs of rhabdomyolysis (i.e., serum K+ level) and the urine for myoglobin to detect a subclinical reaction, even if the risk is low, to avoid sequelae.123
Rhabdomyolysis caused by anesthetic agents may mimic MH.124 The risk of MH in DMD children is the same as in the general population.125 Hyperkalemic arrhythmias are most effectively reversed by rapid intravenous administration of calcium (10 mg/kg of calcium chloride), which may be repeated until the arrhythmias abate. Other therapies, including hyperventilation, administration of bicarbonate, albuterol, and insulin and glucose, are also recommended. The hyperkalemia associated with acute rhabdomyolysis may be refractory to the usual treatments and may require a prolonged resuscitation (see Chapter 8).
Undiagnosed Myopathy
The child with a possible myopathy (e.g., floppy, hypotonic, motor developmental delay) but without a definitive diagnosis can pose a problem to the anesthesiologist.126 A careful history and physical examination should be carried out. Blood creatine kinase and lactate concentrations should be evaluated, and the child’s pediatrician or pediatric neurologist contacted for advice. If a progressive muscular dystrophy cannot be excluded (in the presence of an increased creatine kinase level), it may be prudent to avoid inhalational anesthetics, although their careful use for induction may be considered. An increased creatine kinase level in an asymptomatic infant or child may suggest a progressive muscular dystrophy, and inhalational anesthetics should be used with caution unless DMD or BMD are excluded.
Epilepsy
Epilepsy is a common disorder in childhood, with a prevalence of 0.5% to 1% in the school-aged population. It is defined as the tendency to have recurrent spontaneous seizures. A seizure is a sudden, excessive, uncontrolled electrical discharge of cortical neurons.127
Any part or all of the cerebral cortex may be involved, and the manifestations of seizures are numerous. The first and most important step in managing epilepsy is to establish a correct diagnosis by determining whether the episodes are epileptic, classifying the episodes, and identifying the epilepsy syndrome that matches the clinical features (Table 22-10).
Data from the International League against Epilepsy. Available at http://www.ilae.org (accessed June 2012).



 Electroencephalographic features
Electroencephalographic features
 Interval since the last seizure
Interval since the last seizure
 Current medication, timing of the last dose, and blood levels
Current medication, timing of the last dose, and blood levels
The details of the epilepsy syndrome can guide further investigations, help to plan treatment, and educate the family regarding the prognosis.128
Management is directed at treating the cause if possible and in preventing seizures using antiepileptic drugs. Medication is chosen according to the seizure type and epilepsy syndrome, taking into account the age of the child, associated disorders, and other maintenance medications129 Most of the current antiepileptic drugs are licensed for use in childhood, although there are limitations for some drugs, especially in younger children. The aim of the treatment is to eliminate all seizures with the least number of antiepileptic drugs at the minimum doses. The American Epilepsy Society guidelines and practice parameters are available at www.aesnet.org, and the U.K. Epilepsy guidelines are available at www.nice.org.uk/CG020.
 Anti-seizure medication missed due to perioperative fasting
Anti-seizure medication missed due to perioperative fasting
 Epileptogenic anesthetics (e.g., enflurane)
Epileptogenic anesthetics (e.g., enflurane)

 Electrolyte disturbance (e.g., hyponatremia)
Electrolyte disturbance (e.g., hyponatremia)
 A direct effect of neurosurgery on the brain
A direct effect of neurosurgery on the brain



Certain disorders that cause epilepsy may be associated with other medical conditions, such as cardiorespiratory deficits, nutritional difficulties, and learning disabilities.130
Preparation for surgery should include a thorough review of the child’s clinical status, including consultation with the physician who manages the child’s epilepsy. For most minor elective procedures, there is no need to miss or omit any medication in the perioperative period, and parents should be advised to give regular medications as usual on the morning of surgery and anesthesia. Careful scheduling of the time of surgery may facilitate this because many children receive antiepileptic drugs on twice-daily regimens, with doses given at 8 am and 8 pm. If surgery is scheduled for late morning or early afternoon, no medication need be missed. For children with complex epilepsy, such as those undergoing neurosurgery for the epilepsy itself, careful preoperative assessment is imperative.131 In managing surgery and anesthesia in children with epilepsy, the aim is to prevent seizures and enable smooth and effective treatment (Table 22-11).
TABLE 22-11 Perioperative Medication Management for Children with Epilepsy
Ensure regular medications up to fasting.
Use intravenous preparations of regular drugs (phenytoin, phenobarbitone, valproate, benzodiazepine), if possible; use same doses two or three times daily.
If regular drugs not possible, administer the following:
If enteral administration is possible, reestablish regular maintenance and wean intravenous phenytoin dose.
Aicardi J. Diseases of the nervous system in childhood, 3rd ed. London: Mac Keith Press; 2009.
This is a superb, comprehensive textbook that is well illustrated and extensively referenced.
Dubowitz V, Sewry CA. Muscle biopsy: a practical approach, 3rd ed. Philadelphia: WB Saunders; 2007.
Emery AELH. The muscular dystrophies. Oxford: Oxford University Press; 2002.
Hoffmann GF, Johannes Zschocke J, Nyhan WL. Inherited metabolic disease: a clinical approach. Berlin: Springer-Verlag; 2010.
This textbook provides an excellent review of an extremely complex subject.
Klinger W, Lehmann-Horn F, Jurgat-Rott K. Complications of anaesthesia in neuromuscular disorders. Neuromuscul Disord. 2005;15:195–206.
The article reviews the topic for anesthesiologists.
Neuromuscular Disease Center, Washington University at St. Louis, St. Louis. Available at http://neuromuscular.wustl.edu (accessed June 2012)
This is a web-based review of all childhood neuromuscular disorders.
1 Dolk H, Parkes J, Hill N. Trends in the prevalence of cerebral palsy in Northern Ireland, 1981-1997. Dev Med Child Neurol. 2006;48:406–412.
2 Sander J, Shorvon S. Epidemiology of the epilepsies. J Neurol Neurosurg Psychiatry. 1996;61:433–443.
3 Aicardi J. Diseases of the nervous system in childhood, 2nd ed. London: Mac Keith Press; 1998. p. 822-3
4 Hughes MI, Hicks EM, Nevin NC, et al. The prevalence of inherited neuromuscular disease in Northern Ireland. Neuromuscul Disord. 1996;6:69–73.
5 Chinnery PF, Howell N, Andrews RM, et al. Clinical mitochondrial genetics. J Med Genet. 1999;36:425–436.
6 De Lourdes Levy M, Larcher V, Kurz R. Ethics Working Group of the Confederation of European Specialists in Paediatrics (CESP). Informed consent/assent in children. Statement of the Ethics Working Group of the Confederation of European Specialists in Paediatrics (CESP). Eur J Pediatr. 2003;162:629–633.
7 Van Staden CW, Kruger C. Incapacity to give informed consent owing to mental disorder. J Med Ethics. 2003;29:41–43.
8 Alderson P, Sutcliffe K, Curtis K. Children as partners with adults in their medical care. Arch Dis Child. 2006;91:300–303.
9 Rowse VL. Consent in severely disabled children: informed or an infringement of their human rights? J Child Health Care. 2007;11:70–78.
10 Boston M, Ruwe E, Duggins A, et al. Internet use by parents of children undergoing outpatient otolaryngology procedures. Arch Otolaryngol Head Neck Surg. 2005;131:719–722.
11 Mathur S, Shanti N, Brkaric M, et al. Surfing for scoliosis: the quality of information available on the Internet. Spine. 2005;30:2695–2700.
12 Nolan J, Chalkiadis GA, Low J, et al. Anesthesia and pain management in cerebral palsy. Anesthesia. 2000;55:32–41.
13 Ong LC, Afifah I, Sofiah A, et al. Parenting stress among mothers of Malaysian children with cerebral palsy: predictors of child- and parent-related stress. Ann Trop Paediatr. 1998;18:301–307.
14 Paneth N. Establishing the diagnosis of cerebral palsy. Clin Obstet Gynecol. 2008;51:742–748.
15 Taylor F. National Institute of Neurological Disorders and Stroke (U.S.), Office of Science and Health Reports. Cerebral palsy: hope through research. Bethesda, Md: The Institute; 2001. Available at http://www.ninds.nih.gov/disorders/cerebral_palsy/detail_cerebral_palsy.htm (accessed June 2012)
16 Yin R, Reddihough D, Ditchfield M, et al. Magnetic resonance imaging findings in cerebral palsy. J Paediatr Child Health. 2000;36:139–144.
17 Bax M, Tydeman C, Flodmark O. Clinical and MRI correlates of cerebral palsy: the European Cerebral Palsy Study. JAMA. 2006;296:1602–1608.
18 Shimony JS, Lawrence R, Neil JJ, Inder TE. Imaging for diagnosis and treatment of cerebral palsy. Clin Obstet Gyn. 2008;51:787–799.
19 Wongprasartsuk P, Stevens J. Cerebral palsy and anesthesia. Paediatr Anaesth. 2002;12:296–303.
20 Reilly S. Prevalence of feeding problems and oral motor dysfunction in children with cerebral palsy: a community survey. J Pediatr. 1996;129:877–882.
21 Brett EM, Scruttom D. Cerebral palsy, perinatal injury to the spinal cord and brachial plexus birth injury. In: Brett EM, ed. Paediatric neurology. New York: Churchill Livingstone; 1997:291–331.
22 Sharrock NE, Beckman JD, Inda EC, et al. Anesthesia for orthopedic surgery. In: Miller RD, ed. Miller’s anesthesia. 6th ed. Philadelphia: Churchill Livingstone; 2005:2427.
23 Stoelting R, Dierdorf SF. Diseases common to the pediatric patient. In: Stoelting R, Dierdorf SF, eds. Anesthesia and co-existing disease. 3rd ed. New York: Churchill Livingstone; 1993:599.
24 Thomson JD, Banta JV. Scoliosis in cerebral palsy: an overview and recent results. J Pediatr Orthop B. 2001;10:6–9.
25 Ershov VL, Ostreikov F. Complications of anesthesia and their prevention in children with spastic cerebral palsy during ambulatory surgery. Anesteziol Reanimatol. 1999;4:33–35.
26 Dierdorf SF, McNiece Wl, Rao CC, et al. Effect of succinylcholine on plasma potassium in children with cerebral palsy. Anesthesiology. 1985;62:88–90.
27 Theroux MC, Akins RE, Barone C, et al. Neuromuscular junctions in cerebral palsy. Presence of extrajunctional acetylcholine receptors. Anesthesiology. 2002;96:330–335.
28 Frei FJ, Haemmerle MH, Brunner R, Kern C. Minimum alveolar concentration for halothane in children with cerebral palsy and severe mental retardation. Anaesthesia. 1997;52:1056–1060.
29 Moorthy SS, Krishna G, Dierdorf SF. Resistance to vecuronium in patients with cerebral palsy et al. Anesth Analg. 1991;73:275–277.
30 Choudhry DK, Brenn R. Bispectral index monitoring: a comparison between normal children with quadriplegic cerebral palsy. Anesth Analg. 2002;95:1582–1585.
31 Aicardi J. Malformations of the central nervous system. In: Aicardi J, ed. Diseases of the nervous system in childhood. 2nd ed. London: Mac Keith Press; 1998:69–130.
32 Rader JI, Schneeman BO. Prevalence of neural tube defects, folate status, and folate fortification of enriched cereal-grain products in the United States. Pediatrics. 2006;117:1394–1399.
33 Pitkin RM. Folate and neural tube defects. Am J Clin Nutr. 2007;85:285S–288S.
34 Aicardi J. Hydrocephalus and nontraumatic pericerebral collections. In: Aicardi J, ed. Diseases of the nervous system in childhood. 2nd ed. London: Mac Keith Press; 1998:187–209.
35 Surveillance, Epidemiology and End Results (SEER), Cancer statistics review 1973-1998. Available at http://seer.cancer.gov/csr/1973_1998/ (accessed June 2012)
36 Aicardi J. Tumors of the central nervous system and other space-occupying lesions. In: Aicardi J, ed. Diseases of the nervous system in childhood. 2nd ed. London: Mac Keith Press; 1998:491–533.
37 Packer RJ. Childhood tumors. Curr Opin Neurol. 1997;10:464–470.
38 Pollack IF. Pediatric brain tumors. Semin Surg Oncol. 1999;16:73–90.
39 Aicardi J. Metabolic diseases. In: Aicardi J, ed. Diseases of the nervous system in childhood. 2nd ed. London: Mac Keith Press; 1998:245–322.
40 Lyon G, Adams RD, Kolodny EH. Neurology of hereditary metabolic diseases of children, 2nd ed. New York: McGraw-Hill; 1996.
41 Dubowitz V. Disorders of the lower motor neurone. In: Dubowitz V, ed. Muscle disorders in childhood. Philadelphia: WB Saunders; 1978:146–190.
42 Aicardi J. Diseases of the motor neurone. In: Aicardi J, ed. Diseases of the nervous system in childhood. 2nd ed. London: Mac Keith Press; 1998:699–711.
43 Bush A, Fraser J, Jardine E, et al. Respiratory management of the infant with type 1 spinal muscular atrophy. Arch Dis Child. 2005;90:709–711.
44 Wallgren-Pettersson C, Bushby K, Mellies U, et al. 117th ENMC workshop: ventilatory support in congenital neuromuscular disorders—congenital myopathies, congenital muscular dystrophies, congenital myotonic dystrophy and SMA (II). Naarden, The Netherlands, 4-6 April 2003. Neuromuscul Disord. 2004;14:56–69.
45 Petrone A, Cutrera R. Noninvasive ventilation in children with spinal muscular atrophy types 1 and 2. Am J Phys Med Rehabil. 2007;86:216–221.
46 Birnkrant DJ, Pope JF, Martin JE, Repucci AH, Eiben RM. Treatment of type I spinal muscular atrophy with noninvasive ventilation and gastrostomy feeding. Pediatr Neurol. 1998;18:407–410.
47 Rodillo E, Marini ML, Heckmatt JZ, et al. Scoliosis in spinal muscular atrophy: review of 63 cases. J Child Neurol. 1989;4:118–123.
48 Grohmann K, Varon R, Stolz P, et al. Infantile spinal muscular atrophy with respiratory distress type 1 (SMARD1). Ann Neurol. 2003;54:719–724.
49 Graham RJ, Athiraman U, Laubach AE, Sethna NF. Anesthesia and perioperative medical management of children with spinal muscular atrophy. Pediatric Anesthesia. 2009;19:1054–1063.
50 Tangermann RH, Aylward B, Birmingham M, et al. Current status of the global eradication of poliomyelitis. World Health Stat Q. 1997;50:188–194.
51 Aicardi J. Disorders of the peripheral nerves. In: Aicardi J, ed. Diseases of the nervous system in childhood. 2nd ed. London: Mac Keith Press; 1998:712–749.
52 Grunnet ML, Donaldson JO. Juvenile multisystem degeneration with motor neuron involvement and eosinophilic intracytoplasmic inclusions. Arch Neurol. 1985;142:1114–1116.
53 Hughes RA, Swan AV, Raphael JC, et al. Immunotherapy for Guillain-Barré syndrome: a systematic review. Brain. 2007;130(Pt 9):2245–2257.
54 Yikilmaz A, Coskun A. Magnetic resonance imaging of childhood Guillain-Barre syndrome. Childs Nerv Syst. 2010;26:1103–1108.
55 Lopate G, Pestronk A. Inflammatory demyelinating neuropathies. Curr Treat Options Neurol. 2011;13:131–142.
56 Rossignol E, D’Anjou G, Lapointe N, Haddad E, Vanasse M. Evolution and treatment of childhood chronic inflammatory polyneuropathy. Pediatr Neurol. 2007;36:88–94.
57 Van Meir N, De Smet L. Carpal tunnel syndrome in children. Acta Orthop Belg. 2003;69:387–395.
58 Ünal O, Özçakar L, Çetin A, Kaymak B. Severe bilateral carpal tunnel syndrome in juvenile chronic arthritis. Pediatr Neurol. 2003;29:345–348.
59 Griffiths M. Video games and health. BMJ. 2005;331:122–123.
60 Cruz Martinez A, Arpa J. Carpal tunnel syndrome in childhood: study of 6 cases. Electroencephalogr Clin Neurophysiol. 1998;109:304–308.
61 Mullaney P, Vajsar J, Smith R, Buncic JR. The natural history and ophthalmic involvement in childhood myasthenia gravis at the hospital for sick children. Ophthalmology. 2000;107:504–510.
62 Dubowitz V. Myasthenia gravis. In: Dubowitz V, ed. Muscle disorders in childhood. Philadelphia: WB Saunders; 1978:191–201.
63 Hantai D, Richard P, Koenig J, Eymard B. Congenital myasthenic syndromes. Curr Opin Neurol. 2004;17:539–551.
64 Harper CM. Congenital myasthenic syndromes. Semin Neurol. 2004;24:111–123.
65 Batocchi AP, Evoli A, Palmisani MT, et al. Early-onset myasthenia gravis: clinical characteristics and response to therapy. Eur J Pediatr. 1990;150:66–68.
66 Masters OW, Bagshaw ON. Anaesthetic considerations in paediatric myasthenia gravis. Autoimmune Dis. 2011:250561. Sep 25 [Epub ahead of print]
67 Della Rocca G, Coccia C, Diana L, et al. Propofol or sevoflurane anesthesia without muscle relaxants allow the early extubation of myasthenic patients. Can J Anesth. 2003;50:547–552.
68 Aicardi J. Infectious diseases. In: Aicardi J, ed. Diseases of the nervous system in childhood. 2nd ed. London: Mac Keith Press; 1998:373–437.
69 Martyn JAJ, Richtfeld M. Succinylcholine-induced hyperkalemia in acquired pathologic states: Etiologic factors and molecular mechanisms. Anesthesiology. 2006;104:158–169.
70 Dubowitz V. The congenital myopathies. In: Dubowitz V, ed. Muscle disorders in childhood. Philadelphia: WB Saunders; 1978:70–107.
71 Talwalkar SS, Parker JR, Heffner RR, et al. Adult central core disease: clinical, histologic and genetic aspects: case report and review of the literature. Clin Neuropathol. 2006;25:180–184.
72 Dubowitz V. The congenital myopathies. Clin Dev Med. 1980;76:33–71.
73 Jungbluth H, Sewry CA, Muntoni F. What’s new in neuromuscular disorders? The congenital myopathies. Eur J Paediatr Neurol. 2003;7:23–30.
74 Jungbluth H, Sewry C, Brown SC, et al. Minicore myopathy in children: a clinical and histopathological study of 19 cases. Neuromuscul Disord. 2000;10:264–273.
75 Ryan MM, Schnell C, Strickland CD, et al. Nemaline myopathy: a clinical study of 143 cases. Ann Neurol. 2001;50:312–320.
76 English KM, Gibbs JL. Cardiac monitoring and treatment for children and adolescents with neuromuscular disorders. Dev Med Child Neurol. 2006;48:231–235.
77 Wu S, Ibarra MC, Malicdan MC, et al. Central core disease is due to RYR1 mutations in more than 90% of patients. Brain. 2006;129(Pt 6):1470–1480.
78 Ording H, Brancadoro V, Cozzolino S, et al. In vitro contracture test for diagnosis of malignant hyperthermia following the protocol of the European MH Group: results of testing patients surviving fulminant MH and unrelated low-risk subjects. The European Malignant Hyperthermia Group. Acta Anaesthesiol Scand. 1997;41:955–966.
79 Girard T, Treves S, Voronkov E, Siegemund M, Urwyler A. Molecular genetic testing for malignant hyperthermia susceptibility. Anesthesiology. 2004;100:1076–1080.
80 Brandom BW. Genetics of malignant hyperthermia. Scientific World J. 2006;6:1722–1730.
81 Klinger W, Lehmann-Horn F, Jurgat-Rott K. Complications of anaesthesia in neuromuscular disorders. Neuromuscul Disord. 2005;15:195–206.
82 Driessen J, Willems S, Dercksen S, et al. Anesthesia-related morbidity and mortality after surgery for muscle biopsy in children with mitochondrial defects. Paediatr Anaesth. 2007;17:16–21.
83 Wong LJ. Diagnostic challenges of mitochondrial DNA disorders. Mitochondrion. 2007;7:45–52.
84 Wallace JJ, Perndt H, Skinner M. Anaesthesia and mitochondrial disease. Paediatr Anaesth. 1998;8:249–254.
85 Koga Y, Matsuishi T. MELAS and l-arginine therapy: pathophysiology of stroke-like episodes. Ann N Y Acad Sci. 2010;1201:104–110.
86 Footitt EJ, Sinha MD, Raiman JA, et al. Mitochondrial disorders and general anaesthesia: a case series and review. Br J Anaesth. 2008;100:436–441.
87 Ross AK. Muscular dystrophy versus mitochondrial myopathy: the dilemma of the undiagnosed hypotonic child [editorial]. Paediatr Anaesth. 2007;17:1–6.
88 Morgan PG, Hoppel CL, Sedensky MM. Mitochondrial defects and anesthetic sensitivity. Anesthesiology. 2002;96:1268–1270.
89 Kam PC, Cardone D. Propofol infusion syndrome. Anaesthesia. 2007;62:690–701.
90 Driessen J. Neuromuscular and mitochondrial disorders: what is relevant to the anesthesiologist. Current Opinions in Anaesthesiology. 2008;21:350–355.
91 Lerman J. Perioperative management of the paediatric patient with coexisting neuromuscular disease. Br J Anaesth. 2011;107(Suppl 1):i79–i89.
92 Emery AEH. Muscular dystrophy—an evolving concept. In: Emery AEH, ed. The muscular dystrophies. Oxford: Oxford University Press; 2001:1–9.
93 Straub V, Bushby K. The childhood limb-girdle muscular dystrophies. Semin Pediatr Neurol. 2006;13:104–114.
94 Daniele N, Richard I, Bartoli M. Ins and outs of therapy in limb girdle muscular dystrophies. Int J Biochem Cell Biol. 2007;39:1608–1624.
95 Kunkel LM, Bachrach E, Bennett RR, Guyon J, Steffen L. Diagnosis and cell-based therapy for Duchenne muscular dystrophy in humans, mice, and zebrafish. J Hum Genet. 2006;51:397–406.
96 Aicardi J. Primary muscle disease. In: Aicardi J, ed. Diseases of the nervous system in childhood. 2nd ed. London: Mac Keith Press; 1998:750–790.
97 Kroksmark AK, Ekström AB, Björck E, Tulinius M. Myotonic dystrophy: muscle involvement in relation to disease type and size of expanded CTG-repeat sequence. Dev Med Child Neurol. 2005;47:478–485.
98 White RJ, Bass SP. Myotonic dystrophy and paediatric anaesthesia. Paediatr Anaesth. 2003;13:94–102.
99 Dubowitz V. Myotonic syndromes. In: Dubowitz V, ed. Muscle disorders in childhood. Philadelphia: WB Saunders; 1978:146–190.
100 Verrijn Stuart AA, Huisman M, van Straaten HL, Bakker JC, Arabin B. “Shake hands”; diagnosing a floppy infant—myotonic dystrophy and the congenital subtype: a difficult perinatal diagnosis. J Perinat Med. 2000;28:497–501.
101 Emery AEH, de Visser M, Hoogerwaard EM. Duchenne muscular dystrophy and Becker muscular dystrophy. In: Emery A, ed. The muscular dystrophies. Oxford: Oxford University Press; 2001:55–94.
102 Manzur AY, Kuntzer T, Pike M, Swan A. Glucocorticoid corticosteroids for Duchenne muscular dystrophy. Cochrane Database Syst Rev. 1, 2008. CD003725
103 Bushby K, Finkel R, Birnkrant D, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010;9:77–93.
104 Rideau Y, Duport G, Delaubier A, et al. Early treatment to preserve quality of locomotion forchildren with Duchenne muscular dystrophy. Semin Neurol. 1995;15:9–17.
105 Wang CH, Bonnemann CG, Rutkowski A, et al. Consensus statement on standard of care for congenital muscular dystrophies. J Child Neurol. 2010;25:1559–1581.
106 Vignos PJ, Wagner MB, Karlinchak B, Katirji B. Evaluation of a program for long-term treatment of Duchenne muscular dystrophy. Experience at the University Hospitals of Cleveland. J Bone Joint Surg Am. 1996;78:1844–1852.
107 Heckmatt JZ, Dubowitz V, Hyde SA, et al. Prolongation of walking in Duchenne muscular dystrophy with lightweight orthoses: review of 57 cases. Dev Med Child Neurol. 1985;27:149–154.
108 Garralda ME, Muntoni F, Cunniff A, Caneja AD. Knee-ankle-foot orthosis in children with Duchenne muscular dystrophy: user views and adjustment. Eur J Paediatr Neurol. 2006;10:186–191.
109 Sussman M. Duchenne muscular dystrophy. J Am Acad Orthop Surg. 2002;10:138–151.
110 Alman BA, Raza SN, Biggar WD. Steroid treatment and the development of scoliosis in males with Duchenne musculardystrophy. J Bone Joint Surg Am. 2004;86:519–524.
111 Yilmaz O, Karaduman A, Topaloglu H. Prednisolone therapy in Duchenne muscular dystrophy prolongs ambulation and prevents scoliosis. Eur J Neurol. 2004;11:541–544.
112 Talim B, Malaguti C, Gnudi S, Politano L, Merlini L. Vertebralcompression in Duchenne muscular dystrophy following deflazacort. Neuromuscul Disord. 2002;12:294–295.
113 Bothwell JE, Gordon KE, Dooley JM, et al. Vertebral fractures in boys with Duchenne musculardystrophy. Clin Pediatr (Phila). 2003;42:353–356.
114 Velasco MV, Colin AA, Zurakowski D, Darras BT, Shapiro F. Posterior spinal fusion for scoliosis in duchenne muscular dystrophy diminishes the rate of respiratory decline. Spine. 2007;32:459–465.
115 Fisher J, Upadhyaya M. Molecular genetics of facioscapulohumeral muscular dystrophy (FSHD). Neuromuscul Disord. 1997;7:55–62.
116 Lunt PW, Jardine PE, Koch MC, et al. Correlation between fragment size at D4F104S1 and age at onset or at wheelchair use, with a possible generational effect, accounts for much phenotypic variation in 4q35-facioscapulohumeral muscular dystrophy (FSHD). Hum Mol Genet. 1995;4:951–958.
117 Diab M, Darras BT, Shapiro F. Scapulothoracic fusion for facioscapulohumeral muscular dystrophy. J Bone Joint Surg Am. 2005;87:2267–2275.
118 Mummery CJ, Copeland SA, Rose MR. Scapular fixation in muscular dystrophy. Cochrane Database Syst Rev. 1, 2003. CD003278
119 Rubiano R, Chang JL, Carroll J, Solbonian N, Larson CE. Acute rhabdomyolysis following halothane anesthesia without succinylcholine. Anesthesiology. 1987;67:856–857.
120 Girshin M, Mukherjee J, Clowney R, Singer LP, Wasnick J. The postoperative cardiovascular arrest of a 5-year-old male: an initial presentation of muscular dystrophy. Paediatr Anaesth. 2006;16:170–173.
121 Yemen T, McClain C. Muscular dystrophy, anesthesia and the safety of inhalational agents revisited;again. Paediatr Anaesth. 2006;16:105–108.
122 Gurnaney H, Brown A, Litman RS. Malignant hyperthermia and muscular dystrophies. Anesth Analg. 2009;109:1043–1048.
123 Hayes J, Veckemans F, Bissonnette B. Duchenne muscular dystrophy: an old anesthesia problem revisited. Paediatr Anaesth. 2008;18:100–106.
124 Goresky GV, Cox RG. Inhalational anaesthetics and Duchenne’s muscular dystrophy. Can J Anaesth. 1999;46:525–528.
125 Brownell AK. Malignant hyperthermia: relationship to other diseases. Br J Anaesth. 1988;60:303–308.
126 Veychemans F. Can inhalation agents be used in the presence of a child with myopathy? Curr Opin Anaesthesiol. 2010;23:348–355.
127 Fisher RS, van Emde Boas W, Blume W, et al. Epileptic seizures and epilepsy: definitions proposed by the International League Against Epilepsy (ILAE) and the International Bureau for Epilepsy (IBE). Epilepsia. 2005;46:470–472.
128 International League against Epilepsy, Care, resources, and education. Available at http://www.ilae.org/ (accessed June 2012)
129 Hadjiloizou SM, Bourgeois BF. Antiepileptic drug treatment in children. Expert Rev Neurother. 2007;7:179–193.
130 Septer S, Thompson ES, Willemsen-Dunlap A. Anesthesia concerns for children with tuberous sclerosis. AANA J. 2006;74:219–225.
131 Soriano SG, Bozza P. Anesthesia for epilepsy surgery in children. Childs Nerv Syst. 2006;22:834–843.