[level-membership-for-anesthesiology-category]
26 Essentials of Nephrology
Renal Physiology
When renal blood flow is adjusted for body surface area, it doubles during the first 2 weeks of postnatal life and continues to increase until it reaches adult values by the age of 2 years (see Figs. 6-10 and 6-11).1,2 Increased blood flow results from an increase in cardiac output and a decrease in renal vascular resistance. Paralleling these changes, the GFR, when adjusted for body surface area, also doubles over the first 2 weeks of postnatal life and continues to increase until it reaches adult values by the age of 1 to 2 years. The initial GFR and the rate of increase correlate with gestational age at birth. For example, the GFR of an infant of 28 weeks gestation is half of that of a full-term infant (see Figs. 6-10 and 6-11).3 An estimate of GFR can be made from the serum creatinine concentration and the height of the child according to the following formula4,5:

In the equation, k is 0.45 for infants, 0.55 for children, and 0.7 for adolescent boys. The serum creatinine concentration, especially in the first days of life, reflects the maternal serum creatinine concentration and therefore cannot be used to predict neonatal renal function until at least 2 days after birth.6
Fluids and Electrolytes
The kidney regulates total body sodium balance and maintains normal extracellular and circulating volumes.7 The adult kidney filters 25,000 mEq of sodium per day, but it excretes less than 1% through extremely efficient resorption mechanisms along the nephron. The proximal tubule resorbs 50% to 70%, the ascending limb of the loop of Henle resorbs about 25%, and the distal nephron accounts for 10% of the filtered load of sodium. Several hormones, including renin, angiotensin II, aldosterone, and atrial natriuretic peptide, and changes in circulating volume play roles in maintaining sodium balance.8
Serum osmolality is tightly regulated through changes in arginine vasopressin (AVP) release and the appreciation of thirst.9–11 AVP, also called antidiuretic hormone, is synthesized in the hypothalamus and stored in the posterior pituitary, where it is released in response to an increasing plasma osmolality. AVP is also released in response to a decreased circulating volume or hypotension, including those responses to nausea, vomiting, and possibly opioids. AVP binds to receptors in the collecting duct, increasing the permeability of the tubules to water and leading to increased water resorption and concentrated urine. Neonates are much less able to conserve or excrete water compared with older children, rendering the fluid management and volume issues important tasks of the pediatric anesthesiologist in this young age-group.12
The regulation of serum potassium is managed by the kidney and depends on the concentration of plasma aldosterone. Aldosterone binds to receptors on cells in the distal nephron, increasing the secretion of potassium in the urine. Neonates are much less efficient at excreting potassium loads compared with adults, and the normal range of serum potassium concentrations is therefore greater in neonates; Table 26-1 provides the normal values.13 Potassium regulation is affected by the acid-base status; excretion of potassium increases in the presence of alkalosis and decreases in the presence of acidosis. Causes of hyperkalemia and hypokalemia are presented in Tables 26-2 and 26-3, respectively.
| Age | Serum Potassium Range (mEq/L) |
|---|---|
| 0-1 month | 4.0-6.0 |
| 1 month-2 years | 4.0-5.5 |
| 2-17 years | 3.8-5.0 |
| >18 years | 3.2-4.8 |
TABLE 26-2 Causes of Hyperkalemia
TABLE 26-3 Causes of Hypokalemia
Acid-Base Balance
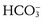 ) lost in the neutralization of acid generated by the normal combustion of food, especially protein, and the formation of bone. New bicarbonate is generated by the cells of the distal nephron by decomposing the carbonic acid (H2CO3) formed from water (H2O) and carbon dioxide (CO2) by carbonic anhydrase. The protons (H+) that are generated from this process are pumped into the lumen of the collecting duct, where they combine with hydrogen phosphate (
) lost in the neutralization of acid generated by the normal combustion of food, especially protein, and the formation of bone. New bicarbonate is generated by the cells of the distal nephron by decomposing the carbonic acid (H2CO3) formed from water (H2O) and carbon dioxide (CO2) by carbonic anhydrase. The protons (H+) that are generated from this process are pumped into the lumen of the collecting duct, where they combine with hydrogen phosphate (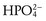 ) or ammonia (NH3) generated by the catabolism of amino acids, mainly glutamine, in the tubule cells.
) or ammonia (NH3) generated by the catabolism of amino acids, mainly glutamine, in the tubule cells.Infants, especially neonates, maintain a slightly acidotic pH (7.37) and decreased plasma bicarbonate concentration (22 mEq/L) compared with older children and adults (pH = 7.39; plasma bicarbonate = 24 to 28 mEq/L).14 Neonates can maintain acid-base homeostasis but are limited in their ability to respond to an acid load.15 This is especially true for preterm infants. This reduced plasma 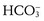 concentration in infants is the result of a reduced threshold, or the plasma concentration at which
concentration in infants is the result of a reduced threshold, or the plasma concentration at which 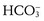 is no longer completely resorbed by the kidney.
is no longer completely resorbed by the kidney.
Disease States
Acute Renal Failure and Acute Kidney Injury
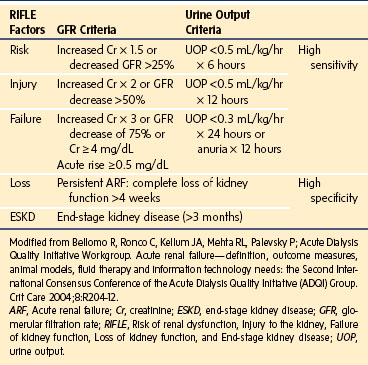
Acute kidney injury (AKI) has almost replaced the traditional term acute renal failure, which was used in reference to the subset of patients with an acute need for dialysis. With the recognition that even modest increases in serum creatinine are associated with a dramatic impact on the risk for mortality, the clinical spectrum of acute decline in GFR is broader. The minor deteriorations in GFR and kidney injury are captured in a working clinical definition of kidney damage that allows early detection and intervention and uses AKI as a replacement for the term ARF. The term ARF is preferably restricted to patients who have AKI and need renal replacement therapy.16 The prognosis of AKI is assessed in part by the use of the RIFLE criteria, which include three severity categories (i.e., Risk, Injury, and Failure) and two clinical outcome categories (Loss and End-stage renal disease) (Table 26-4).
Etiology and Pathophysiology
AKI is often multifactorial in origin or the result of several distinct insults. To treat AKI, it is important to understand its causes and pathophysiology. The causes of AKI are varied but in general can be classified as follows (Table 26-5):



| Prerenal Failure | Renal Failure | Postrenal Failure |
|---|---|---|
| Hypovolemia Volume loss Gastrointestinal, renal losses Sequestration (burns, postoperative) |
Acute glomerulonephritis Postinfectious Membranoproliferative glomerulonephritis Rapidly progressive glomerulonephritis Glomerulonephritis due to systemic disease (e.g., HUS, DIC, SLE) |
Obstruction Intrinsic (papillary necrosis due to diabetes, sickle cell disease, or analgesic nephropathy) Intrarenal abnormalities, ureteral obstruction, obstruction of the bladder or urethra Extrinsic (tumor compression, lymphadenopathy) |
| Hypotension Shock Vasodilators |
Acute interstitial nephritis Drug-induced hypersensitivity (penicillin) Infections |
|
| Decreased effective blood flow Low cardiac output Cirrhosis Nephrotic syndrome |
Tubular disease ATN (ischemic, nephrotoxic) Intratubular obstruction (uric acid, oxalate) |
|
| Renal hypoperfusion Use of ACE inhibitors NSAIDs Hepatorenal syndrome |
Cortical necrosis Gram-negative sepsis Hemorrhage Shock |
|
| Vascular occlusion Thromboembolic phenomenon Aortic dissection Renal vein thrombosis (dehydration, hypercoagulable state, neoplasm) |
Acute renal failure Toxins Organic solvents Heavy metals Insecticides Hemoglobin Myoglobin |
|
| Chronic renal failure Chronic interstitial nephritis Chronic glomerulonephritis Chronic glomerulosclerosis Nephrocalcinosis Obstructive uropathy Hypertension |
ACE, Angiotensin-converting enzyme; ATN, acute tubular necrosis; DIC, disseminated intravascular coagulation; HUS, hemolytic uremic syndrome; NSAIDs, nonsteroidal antiinflammatory drugs; SLE, systemic lupus erythematosus.
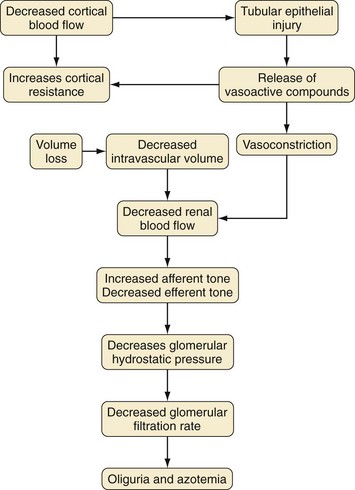
Prerenal insults are a common cause of AKI, accounting for up to 70% of all cases. Prerenal failure usually results from extracellular fluid loss, such as from gastroenteritis, burns, hemorrhage, or excessive diuresis. It also occurs in the setting of cardiac failure or sepsis. The common feature of this condition is diminished renal perfusion. In response to the reduction in flow, there is a compensatory increase in afferent tone, which decreases the GFR and increases the retention of salt and water. The net effect of these events is a drastic reduction in urine volume, often resulting in oliguria. If the underlying problem is recognized and treated aggressively, progressive renal insufficiency may be averted. Nonsteroidal antiinflammatory drugs, angiotensin-converting enzyme (ACE) inhibitors, and angiotensin receptor blockers can aggravate prerenal azotemia by further reducing glomerular capillary pressure and the GFR.17
The exact pathophysiology of AKI remains unclear, but several factors have been identified.18 There is a profound vasoconstriction in the initial phase of AKI that contributes to the reduced GFR (Fig. 26-1). Factors implicated in increased vasoconstriction include increased activity of the renin-angiotensin and the adrenergic systems and endothelial dysfunction with increased endothelin release and decreased nitric oxide synthesis. However, therapeutic interventions to increase vasodilatation, such as prostaglandin and dopamine infusions, ACE inhibitors, calcium channel blockers, and endothelin receptor antagonists, have not significantly improved established AKI.19
Another factor in the pathogenesis of AKI is renal tubule cell injury that is a direct result of a nephrotoxic agent or from an ischemic insult (Fig. 26-2). Cellular injury leads to sloughing of the brush border, swelling, mitochondrial condensation, disruption of cellular architecture, and loss of adhesion to the basement membrane with shedding of cells into the tubular lumen.20 These changes, which occur within minutes of an ischemic event, contribute to the decreased GFR by obstructing the lumen of the tubule.21 These cellular changes allow the filtrate to leak back into the peritubular blood, reducing the excretion of solutes and the effective GFR.
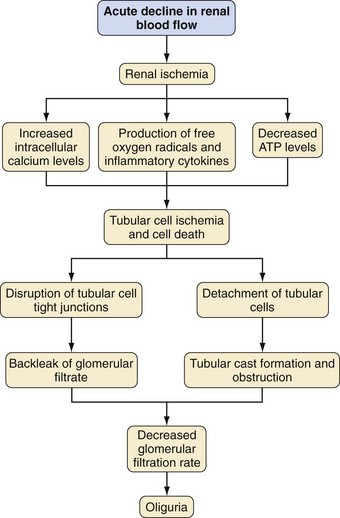
Some of these cell derangements in AKI, such as a decrease in ATP concentrations,21 cell membrane injury by reactive oxygen molecules,22 and increased intracellular calcium levels from changes in membrane phospholipid metabolism, lead to cell death. Reactive oxygen molecules also stimulate the production of cytokines and chemokines that play a role in cell injury and vasoconstriction.
Infiltrating neutrophils, recruited during reperfusion injury after renal ischemia, mediate parenchymal damage.23 Reperfusion injury increases intracellular adhesion molecule 1 (ICAM-1) on endothelial cells promoting the adhesion of circulating neutrophils and their eventual infiltration into the parenchyma. Neutrophils then release reactive oxygen molecules, elastases, proteases, and other enzymes that lead to further tissue injury.
Diagnostic Procedures
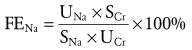
UNa and SNa are urine and serum sodium concentrations, and UCr and SCr are the urine and serum creatinine concentrations, respectively. In prerenal azotemia, the FENa is usually less than 1% for adults and children and less than 2.5% for infants. In established AKI from ischemia and nephrotoxins, but not acute glomerulonephritis, the FENa is usually increased above 1%. Diuretics confound the interpretation of this test.
Therapeutic Interventions
Therapeutic interventions in children with AKI should be aimed at the underlying cause and at improving renal function and urine flow. Children with AKI due to hypovolemia should be fluid resuscitated with at least 20 mL/kg over 30 to 60 minutes of normal saline or a balanced salt solution. For children with significant hypotension, an alternative choice is a colloid-containing solution. Children with oliguria due to hypovolemia usually respond within 4 to 6 hours with increased urine output. Although there are anecdotal reports supporting low-dose dopamine in AKI, clinical trials have not shown a benefit from dopamine in preventing or improving AKI.24
Diuretics have been commonly used to treat oliguric AKI. There are several theoretical reasons why mannitol, furosemide, or other loop diuretics may ameliorate AKI. Diuretics may convert oliguric AKI to nonoliguric AKI. Loop diuretics decrease energy-driven transport in the loop of Henle, and this may protect cells in regions of hypoperfusion. However, neither mannitol nor loop diuretics can predictably convert an oliguric patient with AKI to a polyuric patient. Diuretics have not been shown in clinical studies to influence renal recovery, need for dialysis, or survival in patients with AKI.25,26 Diuretics should be used only after the circulating volume has been adequately restored and should be stopped if there is no early response.
Dopamine has been widely used to prevent and manage AKI. In low doses (0.5 to 2.0 µg/kg/min), dopamine increases renal plasma flow, GFR, and renal sodium excretion by activating dopaminergic receptors. Infusion rates in excess of 3 µg/kg/min stimulate α-adrenergic receptors on systemic arterial resistance vasculature causing vasoconstriction; cardiac β1-adrenergic receptors increasing cardiac contractility, heart rate, and cardiac index; and β2-adrenergic receptors on systemic arterial resistance vasculature causing vasodilatation. In a meta-analysis of 24 studies and 854 patients, dopamine did not prevent renal failure, alter the need for dialysis, or change the mortality rate.27 In a randomized clinical trial of low-dose dopamine in 328 critically ill patients, dopamine did not change the duration or severity of the renal failure, need for dialysis, or mortality.28 From these data, the routine use of low-dose dopamine in patients with AKI cannot be supported.
Several other agents that were useful in experimental models of AKI have been investigated but not shown clinical success. Atrial natriuretic peptide increases GFR in animal models of AKI by increasing renal perfusion pressure and sodium excretion. Initial studies demonstrated some benefit in patients with AKI,29 especially oliguric AKI,30 but a subsequent study of 222 patients with oliguric AKI revealed no statistical difference between patients treated with atrial natriuretic peptide and placebo in terms of the need for dialysis or mortality.31 Insulin-like growth factor 1 has been beneficial in animal models of AKI, presumably by potentiating cell regeneration. However, in a multicenter, placebo-controlled trial enrolling 72 patients with AKI, insulin-like growth factor 1 did not speed recovery, decrease the need for dialysis, or alter the mortality rate.32 Thyroxine abbreviates the course of experimental acute renal failure but had no effect on the duration of renal failure in patients and increased mortality threefold (by suppression of thyroid-stimulating hormone).33
In patients with severe AKI, renal replacement therapy through dialysis is life sustaining. The indications for initiation of dialytic therapy are persistent hyperkalemia, volume overload refractory to diuretics, severe metabolic acidosis, and overt signs and symptoms of uremia such as pericarditis and encephalopathy. Many nephrologists advocate for initiation of dialysis if the BUN value approaches 100 mg/dL or even earlier, especially in the oliguric patient, although this has not proved to alter outcome. A retrospective study that compared early (BUN <60 mg/dL) versus late (BUN >60 mg/dL) initiation of dialysis in 100 adult patients suggested that early initiation improved survival.34 However, the timing of the initiation of dialysis remains an unresolved question.
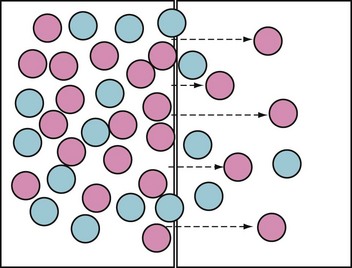
Although the modalities are technically different, they are based on the same principles (Fig. 26-3). The aim of all renal replacement therapies is to promote the removal of nitrogenous wastes (i.e., urea), excess fluid, and excess solute, especially potassium. This is achieved by exposing blood to a salt solution (i.e., dialysate), with the two separated by a semipermeable membrane. The movement of solute occurs by diffusion (i.e., solute moves across the membrane in response to a concentration gradient) and ultrafiltration (i.e., osmotic or hydrostatic pressures). The rate of removal of water and solute waste depends on membrane characteristics (i.e., pore size and selectivity), diffusion, and ultrafiltration.35
Peritoneal Dialysis
Peritoneal dialysis has a long history as a renal replacement therapy in children.36 It is relatively simple and easily performed even in small infants. Although not as efficient as hemodialysis, it is best done continuously to control solute and water balance. In contrast to hemodialysis, it is much less likely to cause hemodynamic instability. Peritoneal dialysis involves instilling dialysate fluid into the peritoneum for a set period and then draining the fluid and replacing it with fresh dialysate. This cycling removes waste products by diffusion and water by ultrafiltration as a consequence of a high glucose concentration in the dialysate. The efficacy of peritoneal dialysis depends on the volume instilled and the number of cycles per day. Most children with acute renal failure can be managed with 1- to 2-hour cycles of 5- to 30-mL/kg dwell volumes. Children with chronic renal failure are managed with greater cycle times and larger dwell volumes. The amount of fluid removed can be varied by changing the concentration of the glucose in the dialysate. Short-term peritoneal dialysis can be accomplished with a nontunneled catheter, but dialysis that should continue beyond 3 to 5 days is best achieved with a subcutaneously tunneled cuffed catheter to minimize the risk of peritonitis.
Chronic Renal Failure
The loss of functioning renal mass results in a compensatory increase in filtration by the remaining renal tissue.37 For example, after a unilateral nephrectomy, there is a demonstrable increase in the GFR and evidence of contralateral renal hypertrophy within the first 48 hours. By 2 to 4 weeks, the GFR has returned to 80% of normal, and there is no clinical evidence of renal dysfunction. With the loss of 50% to 75% of renal mass, there is an increase in the residual function to 50% to 80% of normal and often little evidence of clinical renal insufficiency. When the residual renal function decreases to 30% to 50% of normal, the term chronic renal insufficiency applies. At this point, acute illness and other stress states may result in acidosis, hyperkalemia, and dehydration. It is only when the residual function decreases to less than 30% of normal that the term chronic renal failure is used. At this point, electrolyte abnormalities begin to appear, and more importantly, there is limited ability of the kidney to adjust to variations in fluid and electrolyte homeostasis. The term uremia refers to the symptoms of anorexia, nausea, lethargy, and somnolence that develop as a result of chronic renal failure. Uremia ultimately results in death unless dialysis therapy or renal transplantation is performed. Initiating dialysis or transplanting a kidney is referred to as end-stage renal disease care.
Stage I: The GFR is normal (>90 mL/min/1.73 m2), but there may be evidence of chronic renal disease, including an abnormal urinalysis, hypertension, or abnormal renal ultrasound results.
Stage II: The GFR of 60 to 89 mL/min/1.73 m2 indicates mild kidney damage with a reduction in the GFR.
Stage III: The GFR of 30 to 59 mL/min/1.73 m2 indicates a moderate reduction in the GFR.
Stage IV: The GFR of 15 to 29 mL/min/1.73 m2 indicates a severe decline in the GFR, often accompanied by electrolyte or metabolic derangements.
Stage V: The GFR of less than 15 mL/min/1.73 m2 indicates kidney failure requiring renal replacement therapy.
Stage VI: Patients are receiving dialysis or are transplant recipients.
Hyperkalemia is a major problem in chronic renal failure.38 In contrast, significant hypokalemia is unusual in the absence of potassium restriction, alkalosis, or diuretic therapy. Hyperkalemia can result from an extrinsic potassium load, but it also can be caused by fasting or acidosis, in which case the source of the potassium is the intracellular compartment. This can be a particular problem when a child is fasted before surgery and can be ameliorated by an infusion of glucose and insulin. Drugs that can cause hyperkalemia in renal failure include spironolactone, β-adrenergic blockers, and ACE inhibitors. When clinically significant hyperkalemia develops in a child with chronic renal failure, the first-line therapy is to stabilize the myocardium with exogenous calcium and then to redistribute the potassium into the intracellular compartment with insulin and glucose. To deliver the same dose of ionized calcium, three times as much calcium gluconate (mg/kg) must be given than calcium chloride. All doses of calcium are optimally delivered through a central venous access line because calcium infusions are irritating to peripheral veins and can cause necrosis of the skin if extravasation occurs. More definitive correction of hyperkalemia is accomplished by removing potassium from the body using dialysis or Kayexalate. At eight times the usual asthma dose, nebulized albuterol has been effective in redistributing potassium intracellularly, whereas sodium bicarbonate (NaHCO3) administration has not been effective (Table 26-6).
| Treatment | Dosage |
|---|---|
| Stabilization of Myocardium | |
| Calcium and bicarbonate | Calcium gluconate: 10% 30-100 mg/kg IV or Calcium chloride: 10% 10-33 mg/kg IV Sodium bicarbonate: 1 mEq/kg IV if acidotic |
| Shifting of Potassium to Intracellular Space | |
| Insulin and glucose | Insulin: 0.1-0.3 unit/kg or 0.1 unit/kg/hr infusion Glucose: D50 1-2 mL/kg or D25 2-4 mL/kg IV or D5 1-2 mL/kg/hr |
| Albuterol | Albuterol: 2.5-5 mg/mL nebulization |
| Decreasing Total Body Potassium | |
| Sodium polystyrene sulfonate (Kayexalate) | 1 g/kg up to 40 g every 4 hours PO or PR |
| Furosemide (diuretic) | 0.5 mg/kg up to 40 mg |
D, Dextrose; IV, intravenous; PO, per os (oral); PR, per rectum (suppository).
Metabolic acidosis is common in patients with chronic renal failure.39 The metabolic acidosis is associated with a normal anion gap in moderate renal insufficiency, but with severe renal insufficiency, there is retention of phosphate, sulfate, and organic acids, resulting in an elevated anion gap. The primary cause of metabolic acidosis in chronic renal failure is the inability of the remaining proximal renal tubules to increase ammonium formation to keep pace with the loss of renal mass. The kidney becomes unable to generate the 1 to 3 mEq/kg/day of new bicarbonate that is necessary to compensate for that lost to buffer endogenous acid production. Previous studies have suggested a major role for decreased resorption of bicarbonate by the proximal renal tubule in chronic renal failure. Although this may occur in the presence of volume overload, severe secondary hyperparathyroidism, and disorders such as Fanconi syndrome, it is not a major mechanism causing acidosis in chronic renal failure. Except for severe phosphate depletion, decreased excretion of phosphate as a titratable acid normally does not contribute to metabolic acidosis.
One of the earliest manifestations of chronic renal failure is secondary hyperparathyroidism.40 Secondary hyperparathyroidism, which results from inadequate formation of 1,25-(OH)2 vitamin D (i.e., 1,25-dihydroxyvitamin D3 or calcitriol), develops in moderate renal insufficiency in the presence of normal serum concentrations of calcium and phosphorus. With more severe renal insufficiency, overt hypocalcemia and hyperphosphatemia often develop. Hypocalcemia is caused by decreased calcium absorption from the gastrointestinal tract as a result of a true deficiency of 1,25-(OH)2 vitamin D. Diminished release of calcium from bone occurs as a result of resistance to the action of parathyroid hormone, and calcium and phosphate are deposited in soft tissues as a consequence of hyperphosphatemia.
Hematologic Problems
One of the most common manifestations of chronic renal failure is anemia. The anemia of chronic renal failure results from impaired erythropoiesis, hemolysis, and bleeding. Of these, impaired erythropoiesis is most important and usually the result of a deficiency of erythropoietin production. Erythropoietin is synthesized and secreted by peritubular cells in the renal cortex in response to decreased tissue oxygenation. It acts on receptors on the erythroid burst-forming units and erythroid colony-forming units. With loss of renal mass, erythropoietin secretion does not respond adequately to hypoxia, and anemia results. Children with chronic renal failure are now routinely treated with recombinant erythropoietin.41,42 Current recommendations are to treat children with chronic renal failure in whom the hematocrit is less than 30%, starting at a dosage of 50 to 150 units/kg intravenously three times per week. When a target hematocrit of 36% is reached, a maintenance dosage of about 75 units/kg is instituted. Subcutaneous administration of erythropoietin is also effective, can be given only once each week, and obviates the need for intravenous injections. Doses greater than 150 units/kg increase the hematocrit faster than smaller doses, but both therapies take 4 to 8 weeks to reach target hematocrit values of 33% to 36%. The most common cause for failure of erythropoietin is concurrent iron deficiency. Children who are scheduled for erythropoietin therapy should begin oral iron, vitamin C, and folic acid 2 to 3 weeks in advance to ensure adequate iron and folic acid stores to facilitate erythropoiesis. Current recommendations are to maintain serum ferritin concentrations above 250 ng/mL and transferrin saturation above 25%. Other causes for failure of erythropoietin to increase or maintain the hematocrit are occult infections, hemolysis, aluminum overload, severe hyperparathyroidism, and occult bleeding. Complications of erythropoietin therapy include worsening of hypertension and a possible increased incidence of thrombosis of polytetrafluoroethylene vascular grafts.
Cardiovascular Complications
Cardiovascular disease is the most common cause of death in patients receiving long-term dialysis, including children.43 Patients with chronic renal failure can have abnormalities of the pericardium, myocardium, cardiac valves, and coronary arteries. Another cardiac manifestation, pericarditis, has long been recognized as a complication of uremia. Pericarditis was once considered a sign of the terminal phase of uremia, but it occurs in 15% of children receiving dialysis and can be symptomatic or clinically silent. In nondialyzed uremic patients with pericarditis, intensive dialysis often results in its resolution within about 2 weeks. Some children require surgical procedures such as pericardiocentesis, pericardial drainage with a catheter or through a pericardial window, or pericardiectomy.
Causes of Chronic Renal Failure
The causes of chronic renal insufficiency and failure can be correlated with age (Table 26-7). The chronic renal failure that is commonly encountered in early infancy results largely from congenital anomalies or perinatal asphyxia. Later in childhood, renal failure may result from dysplasia, or acquired lesions, whereas those affected in adolescence may have deterioration of function related to acquired disease, manifestation of inherited disease, or secondary lesions resulting from other illnesses (e.g., systemic lupus erythematosus, sickle cell disease) or their treatments.
| Infancy (Congenital Anomalies) | Childhood | Adolescence |
|---|---|---|
| Prune-belly syndrome | Dysplasia | Focal segmental glomerulosclerosis |
| Congenital obstruction | Agenesis | Membranoproliferative glomerulonephritis |
| Posterior urethral valves | Autosomal dominant PKD | Secondary glomerulonephritis |
| Multicystic dysplasia | Reflux nephropathy | Systemic lupus erythematosus |
| Agenesis | Obstruction | Sickle cell disease |
| Autosomal recessive PKD | Focal segmental glomerulosclerosis | HIV-associated nephropathy |
| Reflux nephropathy | Membranoproliferative glomerulonephritis | Diabetes mellitus Vasculitis Hemolytic uremic syndrome Henoch-Schönlein purpura Interstitial nephritis Malignancy |
HIV, Human immunodeficiency virus; PKD, polycystic kidney disease.
Preoperative Preparation of the Child with Renal Dysfunction
Perioperative renal dysfunction may occur in children with normal renal function when subjected to perioperative insults such as hypoperfusion from hypotension or hypovolemia. Preexisting renal insufficiency compounds this risk, and precautions to preserve renal perfusion must be taken.44 Associated risk factors include hypovolemia leading to vasoconstriction, nephrotoxic agents such as contrast media, embolic events in cases involving arterial vessel cross-clamping, renal ischemia, and inflammation. Perioperative renal failure is associated with mortality rates of 60% to 90%, and it is therefore important to avoid factors that may augment preexisting renal dysfunction.45,46 A large national database has shown that 1% of all adult patients who underwent general surgical procedures developed postoperative AKI47; the patients at greatest risk were older men (≥56 years old). These data may not be applicable to children, but an increased incidence of AKI among children with congestive heart failure, hypertension, preoperative renal insufficiency, or ascites may help to identify children who are also at risk. Identification of those at risk is not a trivial exercise because postoperative AKI increased postoperative morbidity threefold and postoperative mortality fivefold.
Preoperative Laboratory Evaluation
Abnormal potassium concentrations often occur in children with renal failure; acceptable limits depend on the status of the child and the trends in the potassium concentration over time. Chronic hypokalemic or hyperkalemic states are less likely to have cardiac effects than acute changes. Acute hypokalemia reduces arrhythmia threshold and increases cardiac excitability. Acute hyperkalemia may result in life-threatening arrhythmias from electrical conduction suppression. A child with chronic renal failure whose serum potassium concentrations are chronically 5.5 to 6.0 mEq/L does not need correction of the hyperkalemia, whereas a child with an acute increase to a potassium concentration greater than 5.5 mEq/L requires intervention before the surgical procedure and anesthesia. Existing acidosis must be taken into consideration in determining total body potassium concentrations, with the understanding that acute acidosis promotes extracellular hyperkalemia at a rate of 0.5 mEq/L for every decrease in pH of 0.1 unit. Treatment of hyperkalemia has several options (see Table 26-6).
Hemoglobin, hematocrit, and platelet counts should be part of the preoperative evaluation. Anemia is a common finding in children with renal disease, and morbidity and mortality are associated with hemoglobin concentrations less than 11 g/dL in adult patients with renal failure.48,49 This relationship was based on the effect of anemia on the incidence of left ventricular hypertrophy and associated morbidity and may be less of a concern in children. Recombinant erythropoietin reduces the risks of cardiac compromise from left ventricular hypertrophy by increasing the hemoglobin to normal values.50,51 Blood transfusion is generally not indicated if the hematocrit is more than 25%.
Although not proved to be reliable markers of renal injury, concentrations of serum cystatin C, which reflects the GFR better than serum creatinine, and urinary neutrophil gelatinase-associated lipocalin, which is produced in response to injury by tubular cells, may be used more in the future to provide information preoperatively.52,53
Medications
Children with renal failure may require that their medications be adjusted in the perioperative period. These medications typically include antihypertensives, and although proceeding with elective surgery with moderate hypertension may be acceptable, severe or labile hypertension should be controlled before surgery. Induction of anesthesia may cause hypotension in children with chronic hypertension, although preloading with balanced salt solution may offset this effect. There is a temporal relationship between adults who take ACE inhibitors for blood pressure control on the day of surgery and hypotension at induction of anesthesia and cardiac arrest.54 Moderate hypotension was significantly more frequent in patients who discontinued their ACE inhibitor within 10 hours of their anesthetic induction compared with those who had not taken their medication for more than 10 hours before induction.54 Additional studies and opinion leaders suggest that ACE inhibitors should be stopped the day before surgery to prevent hypotension after induction of anesthesia, although the hypotension can be easily managed, especially during total intravenous anesthesia in adults.55–57 All other antihypertensives, immunosuppressives, and steroids should be continued. Most other medications can be safely held until they can be resumed postoperatively.
Intraoperative Management
Special Considerations
Children with chronic renal failure frequently present with serious medical problems that complicate anesthesia when surgery is required.58,59 These problems stem mainly from fluid and electrolyte abnormalities, complications of chronic renal failure such as anemia and hypertension, and differences in the pharmacokinetics of anesthetic agents in children with renal failure. Although several empirical measures have been advocated for renal protection in the perioperative period, a Cochrane review concluded that no interventions, whether pharmacologic or otherwise, protected the kidney in the perioperative period.60
Fluids and Blood Products
In the child with renal insufficiency, fluid management requires a balanced approach. The child must receive adequate hydration to prevent further renal deterioration in an otherwise injured kidney. Children with renal failure and a history of hypertension are at risk for hypotension and hypertension and require some degree of fluid resuscitation for stability. However, they also may have hypoalbuminemia with low oncotic pressure that puts them at risk for pulmonary edema. Ideally, if the child is euvolemic, standard fluid therapy based on typical surgical fluid management may ensue. Fluid overload must be avoided in all anuric children and in outpatients. Although common sense and years of practice suggest that normal saline is preferable to lactated Ringer solution due to the potassium load in the latter, there is evidence to the contrary. In a series of adults who underwent kidney transplantation with normal saline or lactated Ringer solution, 19% of the patients in the saline group had a potassium concentration of 6 mEq/L and 31% had a metabolic acidosis that required treatment, compared with none for both metabolic disorders in the lactated Ringer group.61 Consideration should be given to returning to lactated Ringer solution for renal failure patients.
Anesthetic Agents
Induction of anesthesia may be carried out safely as long as the child is euvolemic and the pharmacokinetics and pharmacodynamics of the induction agent are understood and accounted for. Anesthetic agents may be affected by the presence of anemia, acidosis, and altered drug binding due to hypoproteinemia in children with renal disease. Antihypertensives such as ACE inhibitors, particularly in combination with diuretics, may lead to profound hypotension.62
The dose of propofol to induce anesthesia using the bispectral index and clinical signs to indicate the state of hypnosis in adult patients with renal failure were significantly greater than in those without renal disease.63 This was attributed to a larger volume of distribution in renal failure patients, consistent with previous studies of thiopental.64 Anemia is another contributing factor. It may indirectly cause a greater plasma volume and greater cardiac output. When propofol is delivered as an infusion, no significant differences in pharmacokinetic or pharmacodynamic parameters have been observed.65
There are insufficient data on the use of inhalational anesthetics for induction in children with renal impairment. For maintenance of anesthesia in adults, desflurane and isoflurane do not further impair renal function in those with preexisting renal disease.66 Sevoflurane at low flows is associated with increased circuit concentrations of compound A, which is nephrotoxic in rats.67,68 In adult patients with normal renal function, low-flow sevoflurane anesthesia has been associated with mild, transient proteinuria but no changes in BUN, creatinine level, or creatinine clearance.69 In adults with renal insufficiency, low-flow sevoflurane has been shown to be as safe as low-flow isoflurane in terms of kidney function.68 Overall, sevoflurane is considered safe in patients with renal disease, but low flows should be avoided. Because desflurane is minimally metabolized (rate of 0.2%) in vivo, it may be preferred even at very low flows (1 L/min).
Neuromuscular blocking drugs (NMBDs) have evolved over the years to provide a choice of relaxants for use in children with renal disease. Children with chronic renal failure may have existing autonomic neuropathy and associated delayed gastric emptying that puts them at risk for aspiration. Along with renal implications, aspiration should be anticipated when choosing a NMBD for airway management. Succinylcholine is often avoided in children with renal failure because of its well-known propensity for increasing serum potassium. However, succinylcholine does not increase the plasma potassium concentration in patients with renal failure any more than in patients with normal renal function (0.5 to 0.8 mEq/L of potassium).70,71 Plasma potassium concentration is chronically increased in renal failure, which means that the intracellular and extracellular potassium concentrations are in equilibrium. This contrasts with patients with acute hyperkalemia in whom the intracellular and extracellular potassium concentrations are not in equilibrium, which predisposes them to ventricular arrhythmias if succinylcholine is given. In the latter case, succinylcholine is relatively contraindicated, whereas in the former case, it is not contraindicated.
The pharmacodynamics of NMBDs in children with renal insufficiency merit consideration. The onset time of rocuronium in children with renal failure (139 seconds) was significantly greater than in the control children (87.3 seconds). This difference was attributed to a greater volume of distribution and decreased serum albumin concentrations and possibly to a reduced cardiac output in children with renal failure who were taking antihypertensives. The slower onset time of rocuronium in children with renal failure must be considered when a rapid-sequence intubation is required. The duration of action of rocuronium in children with end-stage renal disease and normal renal function is similar.72 The time to recover a train-of-four ratio of 70% in children with renal failure was 28.9 minutes, and in those without renal failure, it was 29.4 minutes. The clearance of rocuronium is decreased in children with renal failure.73 Vecuronium has an increased duration of action in adults with renal failure.74
NMBDs such as atracurium and cisatracurium are ideal choices for children with renal insufficiency because their elimination is independent of the kidney. Despite the fact that atracurium and cisatracurium undergo spontaneous degradation by plasma esterase and Hofmann elimination, neuromuscular blockade should be monitored.75 With appropriate monitoring and dosing, atracurium, cisatracurium, vecuronium, and rocuronium are acceptable NMBDs in children with renal disease and provide reliable durations of action after a single bolus dose. However, depending on how rocuronium and vecuronium are administered during a case, their accumulation may ultimately affect their duration of action.75
If a prolonged neuromuscular blockade occurs, hypermagnesemia should be ruled out. In this case, calcium may be administered to help antagonize the blockade. The elimination of neostigmine may be delayed beyond elimination of atropine or glycopyrrolate, and muscarinic effects such as bradycardia, increased secretions, or bronchospasm may occur postoperatively after antagonism. Sugammadex, a selective relaxant binding agent, has reduced clearance in adults with severe renal failure.76 The clearance of sugammadex was only 5.5 mL/min in renal failure patients but 95.2 mL/min in the control group. Despite this difference in pharmacokinetics, sugammadex can rapidly and effectively reverse the effects of rocuronium in patients with renal failure.77
Remifentanil may be a preferred choice for a maintenance opioid in the intraoperative period in children with renal insufficiency because of its rapid metabolism by nonspecific blood and tissue esterases. The pharmacokinetics and pharmacodynamics of remifentanil are not altered in patients with renal disease, but the principal metabolite of remifentanil has reduced elimination,78,79 which is not expected to be clinically important. Doses of other opioids should be reduced by 30% to 50% to avoid unexpected respiratory depression in children with chronic renal failure. Active metabolites of morphine and meperidine can likewise accumulate in patients with renal failure, whereas those of fentanyl and sufentanil do not. The latter opioids are preferable on that basis alone.80–84 Prolonged antagonism of opioid effects with naloxone can be expected in renal failure patients.
Postoperative Concerns
Because of ischemic tissue injury, it is possible that preexisting metabolic acidosis and hyperkalemia may worsen in the postoperative period. In children who have renal failure, careful monitoring of electrolytes during and after the procedure may prevent untoward emergencies. When clinically significant hyperkalemia develops in a child with chronic renal failure, treatment is imperative (see Table 26-6).
Acute hypertension can be treated with a variety of intravenous and oral medications (Table 26-8). Because the therapy for acute symptomatic hypertension should be directed toward rapid normalization of blood pressure, prompt and effective therapy must be initiated, often before the cause has been discerned. The rate of change of blood pressure can be just as important as its absolute level in the pathogenesis of hypertensive emergencies. Blood pressure itself may be a poor determinant of the severity of the clinical situation and the need for aggressive parenteral therapy. The decision to use aggressive parenteral therapy should be based on an absolute number and on clinical findings that define the situation as emergent. When it is determined that aggressive therapy is indicated, several antihypertensive agents can be used safely in children in the acute setting. The drugs most often chosen are potent vasodilators, such as hydralazine, diazoxide, or nitroprusside. There has been widespread use of nicardipine for acute hypertension in children, and they have the advantages of intravenous administration, safety, and rapid onset of action.
| Drug | Dose | Side Effects |
|---|---|---|
| Sodium nitroprusside | 1-10 µg/kg/min | Possible cyanide and thiocyanate toxicity, acute hypotension |
| Enalaprilat* | 0.01-0.06 mg/kg/day every 6 hours | Hypotension, angioedema, anaphylactoid reaction |
| Labetalol | 0.4-3 mg/kg/hr or 0.2-1 mg/kg every 10 minutes |
Bradycardia |
| Nicardipine | 0.5-5 µg/kg/min | Acute hypotension |
Driessen JJ, Robertson EN, Van Egmond J, Booij LH. Time-course of action of rocuronium 0.3 mg/kg in children with and without end-stage renal failure. Paediatr Anaesth. 2002;12:507–510.
Kheterpal S, Trember KK, Heung M, et al. Development and validation of an acute kidney injury risk index for patients undergoing general surgery: results from a national data set. Anesthesiology. 2009;110:505–515.
This study is significant because it looks at patients with acute kidney injury undergoing surgery.
Petroni KC, Cohen NH. Continuous renal replacement therapy: anesthetic implications. Anesth Analg. 2002;94:1288–1297.
Sear JW. Kidney dysfunction in the postoperative period. Br J Anaesth. 2005;95:20–32.
Zaccharias M, Gilmore ICS, Herbison GP, Sivalingam P, Walker RJ. Interventions for protecting renal function in the perioperative period. Cochrane Database Syst Rev. 4, 2008. CD003590
1 Fawer C-L, Torrado A, Guignard J-P. Maturation of renal function in full-term and premature neonates. Helv Paediat Acta. 1979;34:11–21.
2 Aperia A, Borberger O, Elinder G, Herin P, Zetterström R. Postnatal development of renal function in pre-term and full-term infants. Acta Paediatr Scand. 1981;70:183–187.
3 Rhodin MM, Anderson BJ, Peters AM, et al. Human renal function maturation: a quantitative description using weight and postmenstrual age. Pediatr Nephrol. 2009;24:67–76.
4 Schwartz GJ, Haycock GB, Edelmann CM, Jr., Spitzer A. A simple estimate of glomerular filtration rate in children derived from body length and plasma creatinine. Pediatrics. 1976;58:259–263.
5 Schwartz GJ, Feld LG, Langford DJ. A simple estimate of glomerular filtration rate in full-term infants during the first year of life. J Pediatr. 1984;104:849–854.
6 Guignard JP, Drukker A. Why do newborn infants have a high plasma creatinine? Pediatrics. 1999;103:e49.
7 Sonnberg H. Renal regulation of salt balance: a primer for nonpurists. Pediatr Nephrol. 1990;4:354–357.
8 Tulassay T, Seri I, Rascher W. Atrial natriuretic peptide and extracellular volume contraction after birth. Acta Paediatr Scand. 1987;76:444–446.
9 Robertson GL. Physiology of ADH secretion. Kidney Int. 1988;32(Suppl 21):20–26.
10 Baylis PH. Osmoregulation and control of vasopressin secretion in healthy humans. Am J Physiol. 1987;253:R671–R678.
11 Rowe JW, Shelton RL, Helderman JH. Influence of the emetic reflex on vasopressin release in man. Kidney Int. 1979;16:729–735.
12 Svenningsen NW, Aronson AS. Postnatal development of renal concentrating capacity as estimated by the DDAVP-test in normal and asphyxiated neonates. Biol Neonate. 1974;25:230–241.
13 Tudvad F, McNamara H, Barnett HL. Renal response of premature infants to administration of bicarbonate and potassium. Pediatrics. 1954;13:4–16.
14 Edelmann CM, Jr., Rodriguez-Soriano J, Boichis H, et al. Renal bicarbonate reabsorption and hydrogen ion excretion in normal infants. J Clin Invest. 1967;46:1309–1317.
15 Svenningsen NW, Lindquist B. Postnatal development of renal hydrogen ion excretion capacity in relationship to age and protein intake. Acta Paediatr Scand. 1974;63:721–731.
16 Van Biesen W, Vanholder R, Lameire N. Defining acute renal failure: RIFLE and beyond. Clin J Am Soc Nephrol. 2006;1:1314–1319.
17 Hricik DE, Dunn MJ. Angiotensin-converting enzyme inhibitor induced renal failure: causes, consequences, and diagnostic uses. J Am Soc Nephrol. 1990;1:845–858.
18 Seigel NJ, Van Why SK, Devarajan P, Gaudio KM. Pathogenesis of acute renal failure. In: Barratt TM, Avner ED, Harmon WE, eds. Pediatric nephrology. 4th ed. Baltimore: Lippincott Williams & Wilkins; 2004:1223–1232.
19 Schreier RW, Wang W, Poole B, Mitra A. Acute renal failure: definitions, diagnosis, pathogenesis, and therapy. J Clin Invest. 2004;114:5–14.
20 Myers BD, Chui F, Hilberman M, Michaels AS. Transtubular leakage of glomerular filtrate in human acute renal failure. Am J Physiol. 1979;237:F319–F325.
21 Avison MJ, van Waard A, Stromski ME, et al. Metabolic alterations in the kidney during ischemic acute renal failure. Semin Nephrol. 1989;9:98–101.
22 Andreoli SP. Reactive oxygen molecules, oxidant injury and renal disease. Pediatr Nephrol. 1991;5:733–742.
23 Lauriat S, Linas SL. The role of neutrophils in acute renal failure. Semin Nephrol. 1998;18:498–504.
24 Denton MD, Chertow GM, Brady HR. “Renal-dose” dopamine for the treatment of acute renal failure: scientific, rationale, experimental studies and clinical trails. Kidney Int. 1996;50:4–14.
25 Brown CB, Ogg CS, Cameron JS. High dose furosemide in acute renal failure: a controlled trial. Clin Nephrol. 1981;15:90–96.
26 Shilliday IR, Quinn KJ, Allison ME. Loop diuretics in the management of acute renal failure: a prospective, double blind, placebo controlled, randomized study. Nephrol Dial Transplant. 1997;12:2592–2596.
27 Kellum JA, Decker JM. Use of dopamine in acute renal failure: a meta-analysis. Crit Care Med. 2001;29:1526–1531.
28 Bellamo R, Chapman M, Finfer S, et al. Low-dose dopamine in patients with early renal dysfunction: a placebo-controlled randomized trial. Australian and New Zealand Intensive Care Society Clinical Trials Group. Lancet. 2000;356:2139–2143.
29 Rahman SN, Kim GE, Mathew AS, et al. Effects of atrial natriuretic peptide in clinical acute renal failure. Kidney Int. 1994;45:1731–1738.
30 Allgren RL, Marbury TC, Rahman SN, et al. Anaritide in acute tubular necrosis. Auriculin Anaritide Acute Renal Failure Study. N Engl J Med. 1997;336:828–834.
31 Lewis J, Salem MM, Chertow GM, et al. Atrial natriuretic factor in oliguric acute renal failure. Am J Kidney Dis. 2000;36:767–774.
32 Hirschberg R, Kopple J, Lipsett P, et al. Multicenter clinical trial of recombinant human insulin-like growth factor I in patients with acute renal failure. Kidney Int. 1999;55:2423–2432.
33 Acker CG, Singh AR, Flick RP, et al. A trial of thyroxine in acute renal failure. Kidney Int. 2000;57:293–298.
34 Gettings LG, Reynolds HN. Outcome in post-traumatic acute renal failure when continuous renal replacement therapy is applied early vs. late. Intensive Care Med. 1999;25:805–813.
35 Goldstein S, Jabs K. Hemodialysis. In: Barratt TM, Avner ED, Harmon WE, eds. Pediatric nephrology. 4th ed. Baltimore: Lippincott Williams & Wilkins; 2004:1395–1410.
36 Warady BA, Fivush BA, Alexander SR. Peritoneal dialysis. In: Barratt TM, Avner ED, Harmon WE, eds. Pediatric nephrology. 4th ed. Baltimore: Lippincott Williams & Wilkins; 2004:1375–1394.
37 Fine LG, Kurtz I, Woolf AS, et al. Pathophysiology and nephron adaptation in chronic renal failure. In: Schrier RW, Gotschalk CW, eds. Diseases of the kidney. 5th ed. Boston: Little, Brown; 1993:2703.
38 Allon M. Treatment and prevention of hyperkalemia in end-stage renal disease. Kidney Int. 1993;43:1197–1209.
39 Warnock DG. Uremic acidosis. Kidney Int. 1988;34:278–287.
40 Sanchez CP, Goodman WG, Salusky IB. Osteodystrophy. In: Barratt TM, Avner ED, Harmon WE, eds. Pediatric nephrology. 4th ed. Baltimore: Lippincott Williams & Wilkins; 1999:1231.
41 Eschbach JW. Erythropoietin: the promise and the facts. Kidney Int. 1994;44:S70–S76.
42 Warady BA, Jabs K. New hormones in the therapeutic arsenal of chronic renal failure: growth hormone and erythropoietin. Pediatr Clin North Am. 1995;42:1551–1577.
43 Harnett JD, Parfrey PS. Cardiac disease in uremia. Semin Nephrol. 1994;14:245–252.
44 Sear JW. Kidney dysfunction in the postoperative period. Br J Anaesth. 2005;95:20–32.
45 Levy EM, Viscoli CM, Horwitz RI. The effect of acute renal failure on mortality: a cohort analysis. JAMA. 275, 1996. 1489–1484
46 Mangano CM, Diamonstone LS, Ramsey JG, et al. Renal dysfunction after myocardial revascularization: risk factors, adverse outcomes and hospital utilization. Ann Intern Med. 1998;128:194–203.
47 Kheterpal S, Trember KK, Heung M, et al. Development and validation of an acute kidney injury risk index for patients undergoing general surgery: results from a national data set. Anesthesiology. 2009;110:505–515.
48 Mann JF. What are the short-term and long-term consequences of anaemia in CRF patients? Nephrol Dial Transplant. 1999;14:29–36.
49 Berweck S, Hennig L, Sternberg C, et al. Cardiac mortality prevention in uremic patients: therapeutic strategies with particular attention to complete correction of renal anemia. Clin Nephrol. 2000;53:80–85.
50 Winearls CG. Recombinant human erythropoietin: 10 years of clinical experience. Nephrol Dial Transplant. 1998;13:3–8.
51 Donato H. Erythropoietin: an update on the therapeutic use in newborn infants and children. Expert Opin Pharmacother. 2005;6:723–734.
52 Hojs R, Bevc S, Ekart R, Gorenjak M, Puklavek L. Serum cystatin C as an endogenous marker of renal function in patients with mild to moderate impairment of kidney function. Nephrol Dial Transplant. 2006;21:1855–1862.
53 Wheeler DS, Devarajan P, Ma Q, et al. Serum neutrophil gelatinase-associated lipocalin (NGAL) as a maker of acute kidney injury in critically ill children with septic shock. Crit Care Med. 2008;36:1297–1303.
54 Comfere T, Sprung J, Kumar MM, et al. Angiotensin system inhibitors in a general surgical population. Anesth Analg. 2005;100:636–644.
55 Smith I, Jackson I. Beta-blockers, calcium channel blockers, angiotensin converting enzyme inhibitors and angiotensin receptor blockers: should they be stopped or not before ambulatory anaesthesia? Curr Opin Anesthesiol. 2010;23:687–690.
56 Wolf A, McGoldrick KE. Carrdiovascular pharmacotherapeutic considerations in patients undergoing anesthesia. Cardiol Rev. 2011;19:12–16.
57 Schulte E, Ziegler D, Philippi-Hohne C, et al. Angiotensin-converting enzyme inhibition and blood pressure response during total intravenous anaesthesia for minor surgery. Acta Anaesthiol Scand. 2011;55:435–443.
58 Sladen RN. Anesthetic considerations for the patient with renal failure. Anesthesiol Clin North Am. 2000;18:863–882.
59 Petroni KC, Cohen NH. Continuous renal replacement therapy: anesthetic implications. Anesth Analg. 2002;94:1288–1297.
60 Zacharias M, Conlon NP, Herbison GP, et al. Interventions for protecting renal function in the perioperative period. Cochrane Database Syst Rev. 4, 2008. CD003590
61 O’Malley CM, Frumento RJ, Hardy MA, et al. A randomized, double-blind comparison of lactated Ringer’s solution and 0.9% NaCl during renal transplantation. Anesth Analg. 2005;100:1518–1524.
62 Behnia R, Molteni A, Igic R. Angiotensin-converting enzyme inhibitors: mechanisms of action and implications in anesthesia practice. Curr Pharm Design. 2003;9:763–776.
63 Goyal P, Puri GD, Pandey CK, Srivastva S. Evaluation of induction doses of propofol: comparison between endstage renal disease and normal renal function patients. Anaesth Intensive Care. 2002;30:584–587.
64 Christensen JH, Andersen F, Jansen J. Pharmacokinetics and pharmacodynamics of thiopental in patients undergoing renal transplantation. Acta Anaesthesiol Scand. 1983;27:513–518.
65 Ickx B, Cockshott ID, Barvais L, et al. Propofol infusion for induction and maintenance of anaesthesia in patients with end-stage renal disease. Br J Anaesth. 1998;81:854–860.
66 Litz RJ, Hubler M, Lorenz W, Meier VK, Albrecht DM. Renal responses to desflurane and isoflurane in patients with renal insufficiency. Anesthesiology. 2002;97:1133–1136.
67 Tsukamoto N, Hirabayashi Y, Shimizu R, Mitsuhata H. The effects of sevoflurane and isoflurane anesthesia on renal tubular function in patients with moderately impaired renal function. Anesth Analg. 1996;82:909–913.
68 Conzen PF, Kharasch ED, Czerner SFA, et al. Low-flow sevoflurane compared with low-flow isoflurane anesthesia in patients with renal insufficiency. Anesthesiology. 2002;97:578–584.
69 Higuchi H, Sumita S, Wada S, et al. Effects of sevoflurane and isoflurane on renal function and on possible markers of nephrotoxicity. Anesthesiology. 1998;89:307–322.
70 Thapa S, Brull SJ. Succinylcholine-induced hyperkalemia in patients with renal failure: an old question revisited. Anesth Analg. 2000;92:237–241.
71 Yentis SM. Suxamethonium and hyperkalemia. Anaesth Intensive Care. 1990;18:92–101.
72 Driessen JJ, Robertson EN, Van Egmond J, Booij LH. Time-course of action of rocuronium 0.3 mg/kg in children with and without endstage renal failure. Paediatr Anaesth. 2002;12:507–510.
73 Cooper RA, Mirakhur RK, Wierda JM, Maddineni VR. Pharmacokinetics of rocuronium bromide in patients with and without renal failure. Eur J Anesth. 1995;11:43–44.
74 Lynam DP, Cronnelly R, Castagnoli KP, et al. The pharmacodynamics and pharmacokinetics of vecuronium in patients anesthetized with isoflurane with normal renal function or with renal failure. Anesthesiology. 1988;62:227–231.
75 Della Rocca G, Pompei L, Coccia C, et al. Atracurium, cisatracurium, vecuronium and rocuronium in patients with renal failure. Minerva Anestesiol. 2003;69:605–612.
76 Staals LM, Snoeck MMJ, Driessen JJ, et al. Reduced clearance of rocuronium and sugammadex in patients with severe to end-stage renal failure: a pharmacokinetic study. Br J Anaesth. 2010;104:31–39.
77 Staals LM, Snoeck MM, Driessen JJ, et al. Multicentre, parallel-group, comparative trial evaluating the efficacy and safety of sugammadex in patients with end-stage renal failure or normal renal function. Br J Anaesth. 2008;101:492–497.
78 Hoke JF, Schlugman D, Dershwitz M, et al. Pharmacokinetics and pharmacodynamics of remifentanil in persons with renal failure compared with healthy volunteers. Anesthesiology. 1997;87:533–541.
79 Pitsiu M, Wilmer A, Bodenham A, et al. Pharmacokinetics of remifentanil and its major metabolite, remifentanil acid, in ICU patients with renal impairment. Br J Anaesth. 2004;92:493–503.
80 Davis PJ, Stiller RL, Cook DR, Brandom BW, Davin-Robinson KA. Pharmacokinetics of sufentanil in adolescent patients with chronic renal failure. Anesth Analg. 1988;67:268–271.
81 Chauvin M, Lebrault C, Levron JC, Duvaldestin P. Pharmacokinetics of alfentanil in chronic renal failure. Anesth Analg. 1987;66:53–56.
82 Chan GL, Matzke GR. Effects of renal insufficiency on the pharmacokinetics and pharmacodynamics of opioid analgesics. Drug Intell Clin Pharm. 1987;21:773–783.
83 Davis PJ, Stiller RL, Cook DR, et al. Effects of cholestatic hepatic disease and chronic renal failure on alfentanil pharmacokinetics in children. Anesth Analg. 1989;68:579–583.
84 Osborne R, Jel S, Grebenik K, Trew D, Slevin M. The pharmacokinetics of morphine and morphine glucuronides in kidney failure. Clin Pharmacol Ther. 1993;54:158–167.
[/level-membership-for-anesthesiology-category][not-level-membership-for-anesthesiology-category]
26 Essentials of Nephrology
Renal Physiology
When renal blood flow is adjusted for body surface area, it doubles during the first 2 weeks of postnatal life and continues to increase until it reaches adult values by the age of 2 years (see Figs. 6-10 and 6-11).1,2 Increased blood flow results from an increase in cardiac output and a decrease in renal vascular resistance. Paralleling these changes, the GFR, when adjusted for body surface area, also doubles over the first 2 weeks of postnatal life and continues to increase until it reaches adult values by the age of 1 to 2 years. The initial GFR and the rate of increase correlate with gestational age at birth. For example, the GFR of an infant of 28 weeks gestation is half of that of a full-term infant (see Figs. 6-10 and 6-11).3 An estimate of GFR can be made from the serum creatinine concentration and the height of the child according to the following formula4,5:
In the equation, k is 0.45 for infants, 0.55 for children, and 0.7 for adolescent boys. The serum creatinine concentration, especially in the first days of life, reflects the maternal serum creatinine concentration and therefore cannot be used to predict neonatal renal function until at least 2 days after birth.6
Fluids and Electrolytes
The kidney regulates total body sodium balance and maintains normal extracellular and circulating volumes.7 The adult kidney filters 25,000 mEq of sodium per day, but it excretes less than 1% through extremely efficient resorption mechanisms along the nephron. The proximal tubule resorbs 50% to 70%, the ascending limb of the loop of Henle resorbs about 25%, and the distal nephron accounts for 10% of the filtered load of sodium. Several hormones, including renin, angiotensin II, aldosterone, and atrial natriuretic peptide, and changes in circulating volume play roles in maintaining sodium balance.8
Serum osmolality is tightly regulated through changes in arginine vasopressin (AVP) release and the appreciation of thirst.9–11 AVP, also called antidiuretic hormone, is synthesized in the hypothalamus and stored in the posterior pituitary, where it is released in response to an increasing plasma osmolality. AVP is also released in response to a decreased circulating volume or hypotension, including those responses to nausea, vomiting, and possibly opioids. AVP binds to receptors in the collecting duct, increasing the permeability of the tubules to water and leading to increased water resorption and concentrated urine. Neonates are much less able to conserve or excrete water compared with older children, rendering the fluid management and volume issues important tasks of the pediatric anesthesiologist in this young age-group.12
The regulation of serum potassium is managed by the kidney and depends on the concentration of plasma aldosterone. Aldosterone binds to receptors on cells in the distal nephron, increasing the secretion of potassium in the urine. Neonates are much less efficient at excreting potassium loads compared with adults, and the normal range of serum potassium concentrations is therefore greater in neonates; Table 26-1 provides the normal values.13 Potassium regulation is affected by the acid-base status; excretion of potassium increases in the presence of alkalosis and decreases in the presence of acidosis. Causes of hyperkalemia and hypokalemia are presented in Tables 26-2 and 26-3, respectively.
| Age | Serum Potassium Range (mEq/L) |
|---|---|
| 0-1 month | 4.0-6.0 |
| 1 month-2 years | 4.0-5.5 |
| 2-17 years | 3.8-5.0 |
| >18 years | 3.2-4.8 |
TABLE 26-2 Causes of Hyperkalemia
TABLE 26-3 Causes of Hypokalemia
Acid-Base Balance
Infants, especially neonates, maintain a slightly acidotic pH (7.37) and decreased plasma bicarbonate concentration (22 mEq/L) compared with older children and adults (pH = 7.39; plasma bicarbonate = 24 to 28 mEq/L).14 Neonates can maintain acid-base homeostasis but are limited in their ability to respond to an acid load.15 This is especially true for preterm infants. This reduced plasma concentration in infants is the result of a reduced threshold, or the plasma concentration at which is no longer completely resorbed by the kidney.
Disease States
Acute Renal Failure and Acute Kidney Injury
Acute kidney injury (AKI) has almost replaced the traditional term acute renal failure, which was used in reference to the subset of patients with an acute need for dialysis. With the recognition that even modest increases in serum creatinine are associated with a dramatic impact on the risk for mortality, the clinical spectrum of acute decline in GFR is broader. The minor deteriorations in GFR and kidney injury are captured in a working clinical definition of kidney damage that allows early detection and intervention and uses AKI as a replacement for the term ARF. The term ARF is preferably restricted to patients who have AKI and need renal replacement therapy.16 The prognosis of AKI is assessed in part by the use of the RIFLE criteria, which include three severity categories (i.e., Risk, Injury, and Failure) and two clinical outcome categories (Loss and End-stage renal disease) (Table 26-4).
Etiology and Pathophysiology
AKI is often multifactorial in origin or the result of several distinct insults. To treat AKI, it is important to understand its causes and pathophysiology. The causes of AKI are varied but in general can be classified as follows (Table 26-5):
| Prerenal Failure | Renal Failure | Postrenal Failure |
|---|---|---|
| Hypovolemia Volume loss Gastrointestinal, renal losses Sequestration (burns, postoperative) |
Acute glomerulonephritis Postinfectious Membranoproliferative glomerulonephritis Rapidly progressive glomerulonephritis Glomerulonephritis due to systemic disease (e.g., HUS, DIC, SLE) |
Obstruction Intrinsic (papillary necrosis due to diabetes, sickle cell disease, or analgesic nephropathy) Intrarenal abnormalities, ureteral obstruction, obstruction of the bladder or urethra Extrinsic (tumor compression, lymphadenopathy) |
| Hypotension Shock Vasodilators |
Acute interstitial nephritis Drug-induced hypersensitivity (penicillin) Infections |
|
| Decreased effective blood flow Low cardiac output Cirrhosis Nephrotic syndrome |
Tubular disease ATN (ischemic, nephrotoxic) Intratubular obstruction (uric acid, oxalate) |
|
| Renal hypoperfusion Use of ACE inhibitors NSAIDs Hepatorenal syndrome |
Cortical necrosis Gram-negative sepsis Hemorrhage Shock |
|
| Vascular occlusion Thromboembolic phenomenon Aortic dissection Renal vein thrombosis (dehydration, hypercoagulable state, neoplasm) |
Acute renal failure Toxins Organic solvents Heavy metals Insecticides Hemoglobin Myoglobin |
|
| Chronic renal failure Chronic interstitial nephritis Chronic glomerulonephritis Chronic glomerulosclerosis Nephrocalcinosis Obstructive uropathy Hypertension |
ACE, Angiotensin-converting enzyme; ATN, acute tubular necrosis; DIC, disseminated intravascular coagulation; HUS, hemolytic uremic syndrome; NSAIDs, nonsteroidal antiinflammatory drugs; SLE, systemic lupus erythematosus.
Prerenal insults are a common cause of AKI, accounting for up to 70% of all cases. Prerenal failure usually results from extracellular fluid loss, such as from gastroenteritis, burns, hemorrhage, or excessive diuresis. It also occurs in the setting of cardiac failure or sepsis. The common feature of this condition is diminished renal perfusion. In response to the reduction in flow, there is a compensatory increase in afferent tone, which decreases the GFR and increases the retention of salt and water. The net effect of these events is a drastic reduction in urine volume, often resulting in oliguria. If the underlying problem is recognized and treated aggressively, progressive renal insufficiency may be averted. Nonsteroidal antiinflammatory drugs, angiotensin-converting enzyme (ACE) inhibitors, and angiotensin receptor blockers can aggravate prerenal azotemia by further reducing glomerular capillary pressure and the GFR.17
The exact pathophysiology of AKI remains unclear, but several factors have been identified.18 There is a profound vasoconstriction in the initial phase of AKI that contributes to the reduced GFR (Fig. 26-1). Factors implicated in increased vasoconstriction include increased activity of the renin-angiotensin and the adrenergic systems and endothelial dysfunction with increased endothelin release and decreased nitric oxide synthesis. However, therapeutic interventions to increase vasodilatation, such as prostaglandin and dopamine infusions, ACE inhibitors, calcium channel blockers, and endothelin receptor antagonists, have not significantly improved established AKI.19
Another factor in the pathogenesis of AKI is renal tubule cell injury that is a direct result of a nephrotoxic agent or from an ischemic insult (Fig. 26-2). Cellular injury leads to sloughing of the brush border, swelling, mitochondrial condensation, disruption of cellular architecture, and loss of adhesion to the basement membrane with shedding of cells into the tubular lumen.20 These changes, which occur within minutes of an ischemic event, contribute to the decreased GFR by obstructing the lumen of the tubule.21 These cellular changes allow the filtrate to leak back into the peritubular blood, reducing the excretion of solutes and the effective GFR.
Some of these cell derangements in AKI, such as a decrease in ATP concentrations,21 cell membrane injury by reactive oxygen molecules,22 and increased intracellular calcium levels from changes in membrane phospholipid metabolism, lead to cell death. Reactive oxygen molecules also stimulate the production of cytokines and chemokines that play a role in cell injury and vasoconstriction.
Infiltrating neutrophils, recruited during reperfusion injury after renal ischemia, mediate parenchymal damage.23 Reperfusion injury increases intracellular adhesion molecule 1 (ICAM-1) on endothelial cells promoting the adhesion of circulating neutrophils and their eventual infiltration into the parenchyma. Neutrophils then release reactive oxygen molecules, elastases, proteases, and other enzymes that lead to further tissue injury.
Diagnostic Procedures
UNa and SNa are urine and serum sodium concentrations, and UCr and SCr are the urine and serum creatinine concentrations, respectively. In prerenal azotemia, the FENa is usually less than 1% for adults and children and less than 2.5% for infants. In established AKI from ischemia and nephrotoxins, but not acute glomerulonephritis, the FENa is usually increased above 1%. Diuretics confound the interpretation of this test.
Therapeutic Interventions
Therapeutic interventions in children with AKI should be aimed at the underlying cause and at improving renal function and urine flow. Children with AKI due to hypovolemia should be fluid resuscitated with at least 20 mL/kg over 30 to 60 minutes of normal saline or a balanced salt solution. For children with significant hypotension, an alternative choice is a colloid-containing solution. Children with oliguria due to hypovolemia usually respond within 4 to 6 hours with increased urine output. Although there are anecdotal reports supporting low-dose dopamine in AKI, clinical trials have not shown a benefit from dopamine in preventing or improving AKI.24
Diuretics have been commonly used to treat oliguric AKI. There are several theoretical reasons why mannitol, furosemide, or other loop diuretics may ameliorate AKI. Diuretics may convert oliguric AKI to nonoliguric AKI. Loop diuretics decrease energy-driven transport in the loop of Henle, and this may protect cells in regions of hypoperfusion. However, neither mannitol nor loop diuretics can predictably convert an oliguric patient with AKI to a polyuric patient. Diuretics have not been shown in clinical studies to influence renal recovery, need for dialysis, or survival in patients with AKI.25,26 Diuretics should be used only after the circulating volume has been adequately restored and should be stopped if there is no early response.
Dopamine has been widely used to prevent and manage AKI. In low doses (0.5 to 2.0 µg/kg/min), dopamine increases renal plasma flow, GFR, and renal sodium excretion by activating dopaminergic receptors. Infusion rates in excess of 3 µg/kg/min stimulate α-adrenergic receptors on systemic arterial resistance vasculature causing vasoconstriction; cardiac β1-adrenergic receptors increasing cardiac contractility, heart rate, and cardiac index; and β2-adrenergic receptors on systemic arterial resistance vasculature causing vasodilatation. In a meta-analysis of 24 studies and 854 patients, dopamine did not prevent renal failure, alter the need for dialysis, or change the mortality rate.27 In a randomized clinical trial of low-dose dopamine in 328 critically ill patients, dopamine did not change the duration or severity of the renal failure, need for dialysis, or mortality.28 From these data, the routine use of low-dose dopamine in patients with AKI cannot be supported.
Several other agents that were useful in experimental models of AKI have been investigated but not shown clinical success. Atrial natriuretic peptide increases GFR in animal models of AKI by increasing renal perfusion pressure and sodium excretion. Initial studies demonstrated some benefit in patients with AKI,29 especially oliguric AKI,30 but a subsequent study of 222 patients with oliguric AKI revealed no statistical difference between patients treated with atrial natriuretic peptide and placebo in terms of the need for dialysis or mortality.31 Insulin-like growth factor 1 has been beneficial in animal models of AKI, presumably by potentiating cell regeneration. However, in a multicenter, placebo-controlled trial enrolling 72 patients with AKI, insulin-like growth factor 1 did not speed recovery, decrease the need for dialysis, or alter the mortality rate.32 Thyroxine abbreviates the course of experimental acute renal failure but had no effect on the duration of renal failure in patients and increased mortality threefold (by suppression of thyroid-stimulating hormone).33
In patients with severe AKI, renal replacement therapy through dialysis is life sustaining. The indications for initiation of dialytic therapy are persistent hyperkalemia, volume overload refractory to diuretics, severe metabolic acidosis, and overt signs and symptoms of uremia such as pericarditis and encephalopathy. Many nephrologists advocate for initiation of dialysis if the BUN value approaches 100 mg/dL or even earlier, especially in the oliguric patient, although this has not proved to alter outcome. A retrospective study that compared early (BUN <60 mg/dL) versus late (BUN >60 mg/dL) initiation of dialysis in 100 adult patients suggested that early initiation improved survival.34 However, the timing of the initiation of dialysis remains an unresolved question.
Although the modalities are technically different, they are based on the same principles (Fig. 26-3). The aim of all renal replacement therapies is to promote the removal of nitrogenous wastes (i.e., urea), excess fluid, and excess solute, especially potassium. This is achieved by exposing blood to a salt solution (i.e., dialysate), with the two separated by a semipermeable membrane. The movement of solute occurs by diffusion (i.e., solute moves across the membrane in response to a concentration gradient) and ultrafiltration (i.e., osmotic or hydrostatic pressures). The rate of removal of water and solute waste depends on membrane characteristics (i.e., pore size and selectivity), diffusion, and ultrafiltration.35
