[level-membership-for-anesthesiology-category]
28 Essentials of Hepatology
Anatomy
Hepatic developmental changes occur throughout gestation. Hepatic hematopoiesis develops in utero at 5 to 6 weeks gestation, followed closely by protein synthesis.1 The ability to metabolize carbohydrates and lipids begins by 10 weeks gestation, followed closely by the development of drug metabolizing systems.
During the neonatal period, liver function is immature in its ability to metabolize and clear xenobiotics. Factors believed to affect the clearance of medications include hepatic blood flow and the developmental status of hepatic transport and enzyme systems. Size alone does not account for this observed degree of immaturity, because the fetal and neonatal liver account for a greater percentage of body weight than the adult counterpart (3.6% of body weight versus 2.4% in adults).2 The neonatal liver contains approximately 20% fewer hepatocytes than the adult liver, and the cells are almost one half of the size of adult hepatocytes. These structural features may play some role in the functional deficiencies exhibited by infant livers. Cellular growth and hypertrophy of the liver continue at a rapid pace into young adulthood.
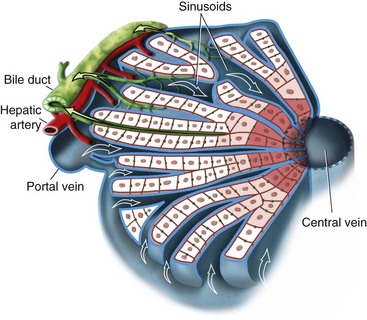
The structural unit of the liver parenchyma is the lobule, a hub-and-spoke structure with the central vein serving as the hub that is bordered by portal tracts, which contain a bile duct and tributaries of the portal vein and hepatic artery. While the mixed venous and arterial blood flows from the portal tract to the central vein, bile flows in the opposite direction through a canalicular matrix that then enters the bile ductule in the portal tract. The functional unit of the liver is the hepatic acinus, which is centered on the portal track and extends in three concentric zones (i.e., zones of Rappaport) outward to the central vein (Fig. 28-1). The more central zones (zones 1 and 2) are most active in oxidative processes, whereas the distal zone 3, which is closer to the central vein, is more dependent on glycolysis and more susceptible to ischemic and toxic injury.
Principles of Hepatic Drug Metabolism
Lipid solubility, an important and desired feature of most administered drugs, allows passive diffusion across cellular membranes. Lipophilic drugs are also difficult to excrete. They have a propensity to accumulate in the body’s fat stores and are bound to proteins in plasma, thereby limiting renal excretion. Renal and biliary excretion of lipid-soluble compounds can result in their resorption across their respective membranes. A major role of the liver is to transform lipid-soluble drugs into water-soluble compounds that become easily excreted metabolites. An interesting example of the need for this biotransformation is the anesthetic compound thiopental, which if not transformed into its less lipophilic counterpart would have a plasma half-life of approximately 25 years.1
The primary family of liver enzymes assigned the task of metabolizing these exogenous substances is the cytochrome P-450 (CYP) family. P designates a red pigment, which is related to a heme molecule and absorbs light at a wavelength of 450 nm.3 The primary reactions involved in the drug biotransformation and metabolism are hydroxylation and conjugation. Hydroxylation prepares the metabolite for conjugation (i.e., phase II reaction). The CYP family of enzymes is responsible for most phase I reactions, and the members were first thought to be chemically similar to mitochondrial cytochromes.
Phase I Reactions
The CYP enzymes likely evolved as a mechanism by which the host was able to protect itself from toxins ingested from the environment. Most enzymes involved in hepatic drug metabolism are categorized in three distinct families: CYP1, CYP2, and CYP3. Each family is further divided into subfamilies that are designated with capital letters and numbered in the order in which they were discovered. The CYP enzymes are generally conserved across species, but their regulation and catalytic activity vary among species, which highlights the challenges associated with laboratory analysis of drug metabolism.3
Genetic and nongenetic factors contribute to the variability in the enzymatic activity seen across all CYP enzymes.4 Approximately 5% of Caucasian populations lack CYP2D6 activity, which is associated with altered metabolism of some drugs.5 For example, a lack of CYP2D6 activity enhances the effect of drugs such as haloperidol and metoprolol that require the enzyme for efficient metabolism, whereas codeine, which is metabolized to morphine by CYP2D6, provides little analgesia in a child with CYP2D6 deficiency.6
Nongenetic factors that influence CYP activity include concomitant disease states, malnutrition,7 and exposure to a host of pharmacologic and naturally occurring compounds. Many drugs can inhibit or stimulate the enzyme system (Table 28-1). Inhibition of the CYPs occurs when drugs compete for the same enzyme. The degree to which this competition becomes clinically significant depends on five factors: the relative amount of the specific CYP, the concentrations of each drug, the degree of pharmacologically active metabolite generated through this system, the importance of the enzyme in elimination of the drugs, and the therapeutic index of the drug.3
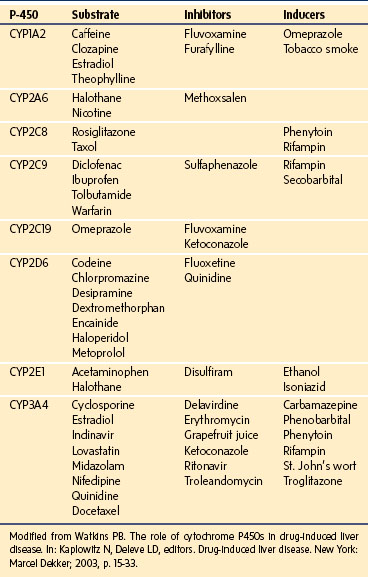
Enhanced CYP expression occurs after amplified transcription of the specific gene that is induced by a variety of compounds. For example, rifampin and phenytoin induce CYP3A4 by binding the cytosolic human pregnenolone-X receptor (hPXR) or the steroid xenobiotic receptor (SXR).8 The activated receptor translocates into the nucleus, where it binds the regulator elements of the CYP3A4 gene and promotes increased transcription of CYP3A4, which can lead to toxic levels of intermediate compounds, as is the case with erythromycin, or to subtherapeutic levels of cyclosporine in transplant recipients.9
Cytochrome P-450 Activity
The superfamily of CYP enzymes is divided into subfamilies based on sequence homology and on demonstration of broad substrate specificities. An important example is the CYP3A subfamily, which is the most abundant group of cytochromes involved in the metabolism of xenobiotics. The three identified isoforms are CYP3A4, CYP3A5, and CYP3A7. CYP3A4 is the most abundant single enzyme in the human liver, accounting for the metabolism of approximately 50% of clinically used pharmaceuticals.10 CYP3A5 is more commonly found in the kidneys and lungs and to a lesser degree in the liver. CYP3A7 is the predominant isoform in the neonatal liver but is replaced after birth by CYP3A4. Given its critical involvement in hepatic biotransformation of xenobiotics, the CYP3A family of enzymes is used to study and estimate hepatic drug clearance in various age and gender groups.
Changes in the distribution and activity of the CYP enzyme systems occur with hepatic growth and maturation (Fig. 28-2). The CYP3A family is homogeneously distributed across the liver parenchyma in the fetal liver and shortly after birth. However, during postnatal growth, expression of the CYP3A protein shifts toward the centrilobular region of the acinus (i.e., Rappaport zone 1). By adulthood, expression of CYP2A becomes increasingly limited to the zone 1 and zone 2 hepatocytes, with sparse expression occurring in zone 3.11 Other examples of developmental changes in the CYP system include the CYP2C and CYP3A3/4 subfamilies, which have negligible expression in the fetus but have increased expression in the first few weeks of life.12,13 CYP2D6 reaches adult activity levels within 1 month chronological age; variability thereafter is determined primarily by genetic polymorphisms.13a
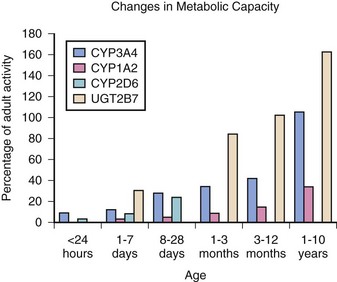
FIGURE 28-2 Changes in metabolic capacity versus percentage of adult activity.
(Modified from Kearns G, Abdel-Rahman SM, Alander SW, et al. Developmental pharmacology—drug disposition, action, and therapy in infants and children. N Engl J Med 2003;349:1157-67.)
Changes in activity of CYP enzyme families and subfamilies have correlated with drug clearance. For example, midazolam clearance correlates with changes in CYP3A4 activity, with decreased clearance in fetal and neonatal livers and with adult clearance rates achieved by 3 months of age.14,15 In contrast, CYP3A7 activity peaks at about 1 week postpartum and steadily diminishes during the first year of life, reaching approximately 10% of fetal liver activity by adulthood.
Phase II Reactions
Conjugation of lipophilic compounds decreases their lipid solubility and facilitates renal excretion.16,17 Conjugation reactions (i.e., glucuronidation, sulfation, glutathione conjugation, acetylation, and methylation) are decreased in infants compared with adults.18
Glucuronidation is catalyzed by uridine 5′-diphosphate (UDP)–glucuronosyltransferase (UGT) family of enzymes, which are derived from assimilation of proteins from separate genes or alternate splicing from single-gene transcripts.19 UGT enzymes are responsible for the metabolism of several drugs, including phenols, estrogens, and opioids (see Chapter 6). As with the CYP enzymes, individual UGT enzymes demonstrate substrate specificity and can act in concert to metabolize single compounds. Glucuronidation is not fully active in infants and can place this population at risk for toxic drug accumulation.20 Hepatic UGT enzyme concentrations are reduced during fetal and early postnatal development. At 3 months of age, the levels of many UGTs are 25% of those in adults.21
UGT1A enzyme activity, which is involved in the conjugation of bilirubin and ethinylestradiol, is decreased in the fetus, but it increases to adult levels within 3 to 6 months after a term delivery.22 UGT1A6, which conjugates acetaminophen and naproxen, has 10% of adult activity in the fetus and neonate, and it achieves only 50% of adult activity by 6 months of age.18 UGT2B7 is active in the metabolism of the nonsteroidal antiinflammatory drugs naloxone, codeine, and lorazepam. Fetal activity of this enzyme approaches 10% to 20% of adult levels, with a rapid increase to adult levels by 2 months of age.23
Sulfation is accomplished by sulfotransferases, a family of cytosolic enzymes that are divided into two categories: catechol and phenol sulfotransferase. These enzymes conjugate inorganic sulfate from 3′-phosphoadenosine-5′-sulfophosphate (PAPS) with compounds containing functional hydroxyl groups.16 The catechol transferases develop earlier in fetal life than the phenol counterparts and appear to exhibit decreased activity in the developing neonate. Although specific sulfotransferase substrates require identification, the activity of these enzymes is increased in fetuses and neonates and theorized to be an efficient conjugation pathway in this age group.
Glutathione S-transferases (GSTs) conjugate glutathione with a broad spectrum of lipophilic and electrophilic compounds. The family of GSTs is composed of up to five different groups in various classes designated µ, α, θ, and π, which are derived from at least three genetic loci.24 Tissue-specific expression of these enzymes has been demonstrated, with the liver expressing the greatest amount of protein. Variable time-dependent expression has also been shown, with α- and π-class GSTs having enhanced expression between 16 and 24 weeks gestation, whereas only the α-class enzymes predominate in the neonate and adult liver.25 The hepatic π-class enzymes disappear from their hepatocellular location by 6 months of age and can be found only in the epithelial cells of the biliary canaliculi. Variations in the developmental expression of this class of enzymes have made it challenging to fully appreciate what is likely a multitude of clinical interactions.25
Acetylation reactions are catalyzed by N-acetyltransferases, which transfer an acetyl group from acetyl coenzyme A to a variety of substrates (e.g., p-aminobenzoic acid, p-aminosalicylic acid, procainamide). Two genes, NAT1 and NAT2, are responsible for yielding two specific enzymes with different allelic forms. Despite having 87% sequence homology, these enzymes exhibit different substrate specificities.26 Both are cytosolic enzymes involved in the biotransformation of several drugs and the bioactivation of several human carcinogens. NAT1 is present in multiple fetal and postnatal tissues and accounts for the most N-acetyltransferase substrate metabolism in children younger than 1 year of age. NAT2 is located primarily in the liver and becomes the dominant acetylator after 1 year of age. NAT2 has polymorphisms with enzyme kinetics that differentiate patients with slow or rapid acetylation capabilities. Infants younger than 1 year of age usually are slow acetylators; subsequent age-dependent alterations lead an individual’s targeted acetylator status.27 Individuals who are genetically destined for rapid acetylation manifest this feature by 2 to 4 years of age.
Anesthetic Agents
Inhalational Anesthetic Metabolism
Inhalational anesthetics are poorly metabolized, with 15% to 20% of halothane undergoing biotransformation. Although both oxidative and reductive pathways are involved, the primary pathway of metabolism of halothane is through oxidation to a reactive intermediate, trifluoroacetyl chloride,28 which then undergoes glutathione conjugation.29
Isoflurane is metabolized by CYP2E1 to a limited extent (0.2%).30 Isoflurane is excreted as inorganic fluoride and trifluoroacetic acid after oxidative metabolism in the liver.31 Desflurane is the least metabolized (0.02%) of the volatile anesthetics, which is approximately 10% of the rate of isoflurane.32 Both are metabolized in the liver along similar paths because the urinary metabolite for desflurane, trifluoroacetic acid, is the same as for isoflurane.
Only 2% to 5% of inhaled sevoflurane is metabolized in humans by means of the hepatic CYP2E1 enzyme, as occurs for the other ether anesthetics.33 Oxidation of sevoflurane generates the intermediate formyl fluoride, a highly reactive species thought to generate liver protein adducts. Carbon dioxide and inorganic fluoride are released through this oxidative mechanism, and the final product is hexafluoroisopropanol, which undergoes glucuronide conjugation and is further excreted in the urine (see Chapter 6).34
Neuromuscular Blocking Drugs
Neuromuscular blockade is achieved by depolarizing or nondepolarizing neuromuscular blocking agents (see Chapter 6). The only depolarizing agent still in use is short-acting suxamethonium (i.e., succinylcholine).35 Succinylcholine is hydrolyzed completely by plasma cholinesterases, which are synthesized by the liver.36 Enzyme activity varies with age, but it is decreased in liver disease and has been used as a metric of liver prognosis.37
Nondepolarizing muscle relaxants are divided into aminosteroids (i.e., pipecuronium, pancuronium, vecuronium, and rocuronium) and benzylisoquinolinium diesters (i.e., doxacurium, atracurium, and cisatracurium besylate). Renal and hepatic diseases affect their safety and efficacy.38 Hepatic elimination depends on protein binding, hepatic blood flow, and drug extraction. The volume of drug distribution is increased in hepatic disease, leading to a slower onset and prolonged effect. In children with cholestatic liver disease, such as biliary atresia, uptake of these compounds by the liver is decreased, which decreases plasma clearance and prolongs their effects.39,40 Approximately 75% of the administered dose is bound to plasma proteins, with most bound to albumin. Despite these problems, children with liver disease and a correspondingly low albumin concentration are at minimal risk for the adverse effects from this group of drugs.41
Rocuronium is an analogue of vecuronium with a more rapid onset of action. Unlike other aminosteroids, which largely undergo renal excretion, only 12% to 22% of rocuronium is cleared through the kidney.42 In patients with hepatic disease, the volume of distribution of rocuronium increased by 33% compared with healthy controls.43 In patients undergoing liver transplantation, the clearance of rocuronium was only slightly reduced by the diseased native liver compared with the functioning allograft and healthy individuals.44 Reduced infusion requirements of rocuronium during liver transplantation may indicate graft dysfunction.45
Hepatic elimination of the benzyl isoquinolinium agents is poorly understood, but hepatic dysfunction has been observed to affect their efficacy. Despite increased concentrations in biliary secretions, the pharmacokinetics of doxacurium are unchanged in the presence of hepatocellular injury, but recovery indices are prolonged.46 Although no longer available in the United States, mivacurium is a mixture of three stereoisomers. Clearance of the trans-trans and cis-trans isomers is reduced in patients with liver disease,47 whereas clearance of the cis-cis isomer is unchanged. Prolonged recovery occurs as a result of a decrease in plasma cholinesterases in significant liver disease.47 Clearance of cisatracurium is decreased, although patients with liver disease experience no delay in recovery and minimal delay in onset of action. Children with liver disease, regardless of cause, usually tolerate this class of compounds, and there is no observed toxicity.48
Sedatives, Opioids, and Liver Disease
The commonly used sedatives midazolam, propofol, and ketamine undergo hepatic metabolism through oxidation and conjugation. These compounds are all lipid soluble, and their effects are altered by liver disease.49 Midazolam has an increased clearance rate that depends in part on hepatic blood flow. In children with cirrhosis, the clearance of midazolam is halved with a corresponding doubling of the half-life compared with healthy controls.50 With 95% to 97% bound to albumin, diseases that decrease the concentration of albumin dramatically increase the free fraction of drug, which leads to a greater effect.51,52 Propofol is eliminated primarily (>88%) in the urine, and hepatocellular injury does not alter its pharmacokinetics.53 Ketamine is also highly lipophilic and undergoes metabolism in the liver. However, unlike the other drugs described, it does so through methylation, and its clearance is minimally affected by liver dysfunction.54
Clinical effects from opioids result from their binding to opioid receptors. A greater serum concentration of opioid binds a greater number of receptors, resulting in a correspondingly greater opioid effect. Hepatic clearance and protein binding govern the serum concentration of an opioid.55 Most opioids are oxidized in the liver. However, morphine and buprenorphine undergo glucuronidation, and remifentanil is metabolized by plasma and tissue esterases, enzymes that are mature in term neonates. The ability of the diseased liver to oxidize opioids is diminished, leading to increased oral bioavailability due to decreased first-pass metabolism and decreased drug clearance. The clearance of morphine, despite being metabolized by glucuronidation, is also negatively affected by the presence of cirrhosis.56
Clearance of drugs that are highly extracted by the liver, such as meperidine and morphine, depend on hepatic blood flow.57,58 Clearance (CL) is a function of hepatic blood flow (QH) and the extraction ratio (EH), describing the ability of the liver to efficiently remove the drug from the circulation:
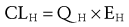
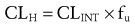
In the formula, fu is the fraction of unbound drug and CLINT describes the metabolic activity. When fu is small as with methadone (<0.1), clearance is mainly affected by reduced enzyme capacity.55
The pharmacology of the opioids is variably affected by liver disease. The analgesic effect of codeine, obtained after its demethylation to morphine, is reduced in the presence of liver disease. Liver disease can affect glucuronidation and thereby decrease clearance and prolong the half-life of morphine.59 The protein binding and clearance of alfentanil are reduced in the presence of hepatic dysfunction,60,61 whereas the half-life and volume of distribution of methadone are increased.62 The pharmacokinetics of fentanyl, remifentanil, and sufentanil are unchanged in the presence of significant hepatic dysfunction.63–65 Because the degree of hepatic dysfunction exhibits interindividual variability, the level of adverse events of these medications also varies. Careful monitoring of children with liver disease is required when administering opioids.
Anesthetic Effects on Hepatic Cellular Functions
Carbohydrates
Glucose metabolism is influenced by the availability of the substrate, the rate of entry into cells, and the ability of the target organ to convert glucose into energy or to use it to synthesize fats. In healthy individuals, hepatic glucose production accounts for most whole-body glucose production and is narrowly regulated directly and indirectly by insulin.66 Insulin directly inhibits gluconeogenesis and glycogenolysis by binding to insulin receptors in the liver, thereby diminishing hepatic glucose production.67 Of the glucose available to the liver, about 50% undergoes glycolysis and is converted to energy. Between 30% and 40% is converted to fat for storage, and 10% to 20% is shunted to glycogen.68 Anesthetics inhibit glucose uptake by hepatocytes, an action referred to as the anti-insulin effect of anesthesia.69 Although all inhalational anesthetics exhibit this tendency, halothane has the greatest impact on serum glucose, with isoflurane and sevoflurane having lesser effects.70 Inhalational anesthetics at 1 to 2 minimal alveolar concentrations (MACs) inhibit glucose uptake by up to 50%. The combined effects of anesthetics and stress from surgery or trauma increase the serum glucose concentration, which is one reason why glucose-containing intravenous fluids are no longer recommended for most healthy children undergoing elective surgery.
Protein Synthesis
The impact of anesthesia and surgery on protein synthesis and metabolism is poorly understood. Albumin, a large, soluble, single polypeptide protein with a molecular weight of 66 kD, is an important protein synthesized by the liver. Between 6 and 12 g of albumin are produced each day, and production can increase twofold to threefold according to the individual’s needs. Albumin functions as a binding and transport protein and maintains colloid oncotic pressure. Hepatic dysfunction may result in decreased synthesis of albumin and other proteins. Anesthetics may also inhibit protein synthesis. Diethyl ether causes reversible inhibition of protein synthesis in rat hepatocytes,71 whereas halothane and enflurane block protein synthesis in a dose-dependent manner.72 Halothane, sevoflurane, and enflurane inhibit protein synthesis and secretion, which may be an early indicator of hepatic cytotoxic injury.
Bilirubin Metabolism
Bilirubin, the end product of heme metabolism, is converted to unconjugated bilirubin by macrophages in the spleen and bone marrow and transported to the liver bound to plasma albumin. Unconjugated bilirubin is transported into the hepatocyte by bile acids and bacterial endotoxins. In the hepatocyte, bilirubin is conjugated with glucuronate, taurine, and to a lesser extent, glucose by glucuronyl transferase. Deficiencies of glucuronyl transferase manifest clinically in Gilbert syndrome, with more severe forms seen in Crigler-Najjar syndrome types I and II. Gilbert syndrome affects 2% to 13% of individuals, who have increases in bilirubin associated with stress or fasting. Although postoperative jaundice has been described, Gilbert syndrome has not been associated with serious adverse effects in the perioperative anesthesia period.73 Patients with Crigler-Najjar syndrome can be managed safely by minimizing their exposure to drugs that may displace bilirubin from albumin and by using anesthetics that undergo minimal hepatic metabolism.74
Hepatotoxicity
Increases in serum aminotransferase concentrations and bilirubin up to two times the upper limit of normal occur commonly in the postoperative period.75,76 These increases are typically self-limited and inconsequential. However, serious liver injury can be caused by anesthetics.77 The Drug-Induced Liver Injury Network, established by the National Institutes of Health in 2003, has and continues to standardize the nomenclature and causality assessment of drug-induced liver injury (DILI).78 Diagnosis of DILI is entertained when increases in alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase, γ-glutamyl transferase (GGT), and bilirubin occur coincident with xenobiotic exposure. Patterns of injury are classified as hepatocellular, with predominantly increased ALT and AST levels; cholestatic, with increases in alkaline phosphatase, GGT, and bilirubin; and mixed pattern, with features of both hepatocellular and cholestatic injury. Differences between predictable and unpredictable DILI and the associated clinical and histologic variability led to the clinical pearl known as the Hy rule, named after Hyman Zimmerman: If increased serum aminotransferase levels (greater than three times the upper limit of normal) and cholestasis (bilirubin greater than two times the upper limit of normal) without evidence of biliary obstruction occur at the same time in the setting of DILI, a mortality rate of at least 10% can be expected.79
Halothane hepatitis is well described and used as a model for hepatotoxicity of all inhalational anesthetics.80–85 Patients who develop halothane hepatitis typically have no history of preexisting liver disease, alcohol use, or concurrent exposure to other hepatotoxic drugs. The incidence is small, with 1 case reported for every 10,000 to 30,000 patients, making it difficult to clinically establish a causal link between exposure to halothane and the development of liver injury. Although halothane hepatitis can occur in all age-groups, the most common demographic is the obese, middle-aged woman. Although liver injury can occur after a single exposure, recurrent exposure to halothane, particularly when the interval between exposures is brief, increases the risk of liver injury and, in some cases, of liver failure. Health care workers exposed to halothane are also at increased risk through occupational exposure.86
The clinical symptoms observed in patients with halothane-induced liver injury are nonspecific and include malaise, fatigue, and anorexia. Patients may have subtle gastrointestinal symptoms such as nausea and abdominal pain. Some patients develop fever (albeit delayed), nonspecific rash, and arthralgias. Increases in serum transaminases are observed after exposure, with some patients becoming jaundiced. Jaundice can occur as late as 28 days after exposure. Many who develop halothane hepatitis do not progress to liver failure, and the inflammation usually resolves.87 If continued exposure to halothane does not occur, chronic liver disease usually does not develop. However, in a few selected patients, fulminant hepatic failure does occur, and the mortality rate associated with developing halothane-induced hepatic necrosis is 20% to 50%.88 A mild form of DILI occurs in up to 30% of patients after exposure. They tend to be asymptomatic and have increased transaminase levels, but they lack the serologic markers for immune activation, a feature common among those who develop significant liver disease.84–86,89–90
Histologic findings associated with halothane hepatitis include primarily centrilobular necrosis, although a range of findings from multifocal necrosis to massive fulminant necrosis is described. Ballooning degeneration, steatosis, and fibrosis have also been documented; in a small subset of patients, there are electron microscopic findings of mitochondrial injury. The histology of halothane hepatitis has a significant overlap with features of acute viral hepatitis and toxic hepatitis from other pharmaceutical exposures.91
Patient-specific metabolic and immunologic factors combine to account for this unpredictable hepatotoxicity. Variable expression and activation of CYP2E1 and the GSTs likely account for the different degrees of liver injury. Immunologic factors probably play a role in this process and may account for the individual variability in effects. The halothane-induced liver injury is associated with circulating antibodies to tissue antigens, peripheral eosinophilia, and the finding of halothane-induced liver neoantigens.92–94
Evidence suggests that molecular mimicry exists between the trifluoroacetylated proteins and the lipoyl-lysine regions of the E2 subunits (i.e., dihydrolipoyl transacetylase) of the mitochondrial pyruvate dehydrogenase complex because they are epitopes recognized by the antibodies in patients with halothane hepatitis.95,96 Molecular mimicry may account for immunologic tolerance to the trifluoroacetylated proteins resulting from anesthetic metabolism, providing an explanation for the lack of tissue-specific injury observed in most patients exposed to these compounds.97 Various levels of expression of the E2 component of the pyruvate dehydrogenase complex have been observed, with abnormally low levels seen in tissue specimens from patients with halothane hepatitis. It is postulated that individuals with low levels of the E2 subunit of the pyruvate dehydrogenase complex are more likely to develop injury because of a decrease in immune tolerance of triacetylated proteins.97 However, many thousands of children have been safely anesthetized with halothane, and its use has proved to be very safe for most children. Concern about the rare occurrence of halothane hepatitis should not discourage the use of this anesthetic agent in children.
The obesity epidemic and associated nonalcoholic fatty liver disease and nonalcoholic steatohepatitis have raised concerns that these patients may be at increased risk for DILI.98 Obesity and hypercholesterolemia can induce CYP2E1, which may facilitate development of liver injury.99 Few data are available to inform the best practice of anesthesia management for the obese child, and adequately powered prospective studies are needed.100
Enflurane, isoflurane, desflurane, and sevoflurane have also been associated with liver injury. Agents such as enflurane, isoflurane, and desflurane are associated with a lesser degree of toxic hepatitis, possibly because they are metabolized to a lesser extent than halothane.101 Case reports have documented that each is capable of producing liver injury. Enflurane, for example, was associated with the development of severe liver disease, with an estimated incidence of 1 case per 800,000 exposed patients. The clinicopathologic features are similar when compared with halothane-induced disease, including histologic findings, delayed onset of jaundice, and a history of previous exposure to the anesthetic. There is also a correspondingly high mortality rate.102 It is thought that previous exposure to halothane leads to an increased risk of disease, occurring through previous cross-sensitization among chemicals.
Isoflurane has been associated with development of liver disease, although significantly less frequently than other agents. Features of isoflurane hepatic injury mimic those found with halothane. Detection of hepatic metabolite–modified neoantigens that occur in a small percentage of patients demonstrates an immune-mediated pathogenesis similar to that of halothane.103 Desflurane-associated liver injury has been described, and the finding of modified liver neoantigens supports an immune-mediated mechanism of injury similar to that of the other inhalation anesthetics.104,105 Hepatic disease after exposure to sevoflurane has been reported, but an immunologic correlation, as seen with the other inhalational drugs, has not been established.106
Perioperative Considerations in Liver Disease
Patients with known or suspected liver disease should be assessed for hepatocellular and bile duct injury, coagulopathy, ascites, and encephalopathy. Hepatopulmonary syndrome and portopulmonary hypertension are rare but important complications in children with known chronic liver disease, and they may serve as relative or absolute contraindications to elective surgery.107 Perioperative mortality is greatest among those who have acute hepatitis compared with those with chronic liver disease.108–110 The physiologic stress associated with surgical procedures decreases portal blood flow. Liver disease may decrease the degree to which hepatic artery blood flow can compensate for reduced portal blood flow,111 which can increase the risk of developing ischemic injury.
Parenteral nutrition–associated liver disease (PNALD) occurs in children requiring parenteral nutrition for longer than 60 days. Children with short bowel syndrome are at greatest risk for PNALD. Aside from the disturbances to hepatic and portal blood flows seen in children with liver disease, those exposed to total parenteral nutrition (TPN) are at increased risk for perioperative glucose derangements.112–114 Frequent perioperative glucose monitoring with adjustment of dextrose infusion appears to be the standard of care, which is associated with various rates of TPN discontinuation. A survey administered to members of the Study Group of Pediatric Anesthesia found that approximately 50% of respondents checked glucose levels as often as every 1 to 2 hours.115 They also found that 19% discontinued TPN and administered a glucose-containing solution, whereas 35% decreased the fluids to one half of maintenance rates and 33% continued maintenance rates unchanged.115
Bjorkman S. Prediction of drug disposition in infants and children by means of physiologically based pharmacokinetic (PBPK) modeling: theophylline and midazolam as model drugs. Br J Clin Pharmacol. 2004;59:691–704.
Kharasch ED, Hankins D, Mautz D, Thummel KE. Identification of the enzyme responsible for oxidative halothane metabolism: implication for prevention of halothane hepatitis. Lancet. 1996;347:1367–1371.
Watkins PB. The role of cytochrome P450s in drug induced liver disease. In: Kaplowitz N, Deleve LD, eds. Drug-induced liver disease. New York: Marcel Dekker; 2003:15–33.
1 Zhao R, Duncan SA. Embryonic development of the liver. Hepatology. 2005;41:956–967.
2 Haddad S, Restieri C, Krishnan K. Characterization of age-related changes in body weight and organ weights from birth to adolescence in humans. J Toxicol Environ Health A. 2001;64:453–464.
3 Watkins PB. The role of cytochrome P450’s in drug induced liver disease. In: Kaplowitz N, Deleve LD, eds. Drug-induced liver disease. New York: Marcel Dekker; 2003:15–33.
4 Neville KA, Becker ML, Goldman JL, Kearns GL. Developmental pharmacogenomics. Pediatr Anesth. 2011;21:255–265.
5 Morike K, Platten HP, Mikus G, Klotz U. Variability in the frequency of cytochrome P450-2D6 (CYP2D6) deficiency. Br J Clin Pharmacol. 1998;46:87.
6 Lurcott G. The effects of the genetic absence and inhibition of CYP2D6 on the metabolism of codeine and its derivatives, hydrocodone and oxycodone. Anesth Prog. 1998;45:154–156.
7 Rendic S, Guengerich FP. Update information on drug metabolism systems—2009, part II: summary of information on the effects of diseases and environmental factors on human cytochrome P450 (CYP) enzymes and transporters. Curr Drug Metab. 2010;11:4–84.
8 Lehmann JM, Mckee DD, Watson MA, et al. The human orphan nuclear receptor PXR is activated by compounds that regulate CYP3A4 gene expression and cause drug interactions. J Clin Invest. 1998;102:1016–1023.
9 Guengerich FP. Role of cytochrome P450 enzymes in drug-drug interactions. Adv Pharmacol. 1997;43:7–35.
10 Benet LZ, Kroetz DL, Sheiner LB. Pharmacokinetics: the dynamics of drug absorption, distribution, and elimination. In: Hardman JG, Limbard LE, Molinoff PB, Ruddon RW, Gilman AG, eds. Goodman and Gilman’s the pharmacological basis of therapeutics. New York: McGraw-Hill; 1996:3–29.
11 Ratanasavanh D, Beaune P, Morel F, et al. Intralobular distribution and quantitation of cytochrome P-450 enzymes in human liver as a function of age. Hepatology. 1991;13:1142–1151.
12 Lacroix D, Sonnier M, Moncion A, Cheron G, Cresteil T. Expression of CYP3A in the human liver—evidence that the shift between CYP3A7 and CYP3A4 occurs immediately after birth. Eur J Biochem. 1997;247:625–634.
13 Treluyer JM, Bueret G, Cheron G, Sonnier M, Cresteil T. Developmental expression of CYP2C and CYP2C-dependent activities in the human liver: in-vivo/in-vitro correlation and inducibility. Pharmacogenetics. 1997;7:441–452.
13a Stevens JC, Marsh SA, Zaya MJ, et al. Developmental changes in human liver CYP2D6 expression. Drug Metab Dispos. 2008;36:1587–1593.
14 Bjorkman S. Prediction of drug disposition in infants and children by means of physiologically based pharmacokinetic (PBPK) modelling: theophylline and midazolam as model drugs. Br J Clin Pharmacol. 2005;59:691–704.
15 Tateishi T, Watanabe M, Nakure H, et al. CYP3A activity in European American and Japanese men using midazolam as an in vivo probe. Clin Pharmacol Ther. 2001;69:333–339.
16 Alcorn J, McNamara PJ. Ontogeny of hepatic and renal systemic clearance pathways in infants: part II. Clin Pharmacokinet. 2002;41:1077–1094.
17 Alcorn J, McNamara PJ. Ontogeny of hepatic and renal systemic clearance pathways in infants: part I. Clin Pharmacokinet. 2002;41:959–998.
18 Krauer B, Dayer P. Fetal drug metabolism and its possible clinical implications. Clin Pharmacokinet. 1991;21:70–80.
19 Mackenzie PI, Owens IS, Burchell B, et al. The UDP glycosyltransferase gene superfamily: recommended nomenclature update based on evolutionary divergence. Pharmacogenetics. 1997;7:255–269.
20 de Wildt SN, Kearns GL, Leeder JS, van den Anker JN. Glucuronidation in humans. Pharmacogenetic and developmental aspects. Clin Pharmacokinet. 1999;36:439–452.
21 Coughtrie MW, Burchell B, Leakey JE, Hume R. The inadequacy of perinatal glucuronidation: immunoblot analysis of the developmental expression of individual UDP-glucuronosyltransferase isoenzymes in rat and human liver microsomes. Mol Pharmacol. 1988;34:729–735.
22 Onishi S, Kawade N, Itoh S, Isobe K, Sugiyma S. Postnatal development of uridine diphosphate glucuronyltransferase activity towards bilirubin and 2-aminophenol in human liver. Biochem J. 1979;184:705–707.
23 Leakey JE, Hume R, Burchell B. Development of multiple activities of UDP-glucuronyltransferase in human liver. Biochem J. 1987;243:859–861.
24 Mannervik B, Awasthi YC, Board PG, et al. Nomenclature for human glutathione transferases. Biochem J. 1992;282(Pt 1):305–306.
25 Matthew J, Cattan AR, Hall AG, et al. Glutathione S-transferases in neonatal liver disease. J Clin Pathol. 1992;45:679–683.
26 Ohsako S, Deguchi T. Cloning and expression of cDNAs for polymorphic and monomorphic arylamine N-acetyltransferases from human liver. J Biol Chem. 1990;265:4630–4634.
27 Szorady I, Santa A, Veress I. Drug acetylator phenotypes in newborn infants. Biol Res Pregnancy Perinatol. 1987;8(Pt 1):23–25.
28 Cohen EN. Metabolism of the volatile anesthetics. Anesthesiology. 1971;35:193–202.
29 Kharasch ED, Hankins D, Mautz D, Thummel KE. Identification of the enzyme responsible for oxidative halothane metabolism: implications for prevention of halothane hepatitis. Lancet. 1996;347:1367–1371.
30 Holaday DA, Fiserova-Bergerova V, Latto IP, Zumbiel MA. Resistance of isoflurane to biotransformation in man. Anesthesiology. 1975;43:325–332.
31 Hitt BA, Mazze RI, Cousins MJ, et al. Metabolism of isoflurane in Fischer 344 rats and man. Anesthesiology. 1974;40:62–67.
32 Sutton TS, Koblin DD, Gruenke LD, et al. Fluoride metabolites after prolonged exposure of volunteers and patients to desflurane. Anesth Analg. 1991;73:180–185.
33 Holaday DA, Smith FR. Clinical characteristics and biotransformation of sevoflurane in healthy human volunteers. Anesthesiology. 1981;54:100–106.
34 Kharasch ED. Biotransformation of sevoflurane. Anesth Analg. 1995;81(Suppl):S27–S38.
35 Ritter DM, Rettke SR, Ilstrup DM, Burritt MF. Effect of plasma cholinesterase activity on the duration of action of succinylcholine in patients with genotypically normal enzyme. Anesth Analg. 1988;67:1123–1126.
36 Davis L, Britten JJ, Morgan M. Cholinesterase. its significance in anaesthetic practice. Anaesthesia. 1997;52:244–260.
37 Garcia-Ayllon MS, Silveyra MX, Candela A, et al. Changes in liver and plasma acetylcholinesterase in rats with cirrhosis induced by bile duct ligation. Hepatology. 2006;43:444–453.
38 Atherton DP, Hunter JM. Clinical pharmacokinetics of the newer neuromuscular blocking drugs. Clin Pharmacokinet. 1999;36:169–189.
39 Lebrault C, Duvaldestin P, Hezel D, Chauvin M, Guesnon P. Pharmacokinetics and pharmacodynamics of vecuronium in patients with cholestasis. Br J Anaesth. 1986;58:983–987.
40 Parker CJ, Hunter JM. Pharmacokinetics of atracurium and laudanosine in patients with hepatic cirrhosis. Br J Anaesth. 1989;62:177–183.
41 Wierda JM, Beaufort AM, Kleef UW, Smeulers NJ, Agosto S. Preliminary investigations of the clinical pharmacology of three short-acting non-depolarizing neuromuscular blocking agents, Org 9453, Org 9489 and Org 9487. Can J Anaesth. 1994;41:213–220.
42 van den Broek L, Wierda JM, Smeulers NJ, et al. Clinical pharmacology of rocuronium (Org 9426): study of the time course of action, dose requirement, reversibility, and pharmacokinetics. J Clin Anesth. 1994;6:288–296.
43 Magorian T, Wood P, Caldwell J, et al. The pharmacokinetics and neuromuscular effects of rocuronium bromide in patients with liver disease. Anesth Analg. 1995;80:754–759.
44 Fisher DM, Ramsay MA, Hein HA, et al. Pharmacokinetics of rocuronium during the three stages of liver transplantation. Anesthesiology. 1997;86:1306–1316.
45 Gao L, Ramzan I, Baker B. Rocuronium plasma concentrations during three phases of liver transplantation: relationship with early postoperative graft liver function. Br J Anaesth. 2002;88:764–770.
46 Cook DR, Freeman JA, Lai AA, et al. Pharmacokinetics and pharmacodynamics of doxacurium in normal patients and in those with hepatic or renal failure. Anesth Analg. 1991;72:145–150.
47 Head-Rapson AG, Devlin JC, Parker CJ, Hunter JM. Pharmacokinetics of the three isomers of mivacurium and pharmacodynamics of the chiral mixture in hepatic cirrhosis. Br J Anaesth. 1994;73:613–618.
48 De Wolf AM, Freeman JA, Scott VL, et al. Pharmacokinetics and pharmacodynamics of cisatracurium in patients with end-stage liver disease undergoing liver transplantation. Br J Anaesth. 1996;76:624–628.
49 Gan TJ. Pharmacokinetic and pharmacodynamic characteristics of medications used for moderate sedation. Clin Pharmacokinet. 2006;45:855–869.
50 MacGilchrist AJ, Birnie GG, Cook A, et al. Pharmacokinetics and pharmacodynamics of intravenous midazolam in patients with severe alcoholic cirrhosis. Gut. 1986;27:190–195.
51 Fragen RJ. Pharmacokinetics and pharmacodynamics of midazolam given via continuous intravenous infusion in intensive care units. Clin Ther. 1997;19:405–419. discussion 367-8
52 Horn E, Nesbit SA. Pharmacology and pharmacokinetics of sedatives and analgesics. Gastrointest Endosc Clin N Am. 2004;14:247–268.
53 Fulton B, Sorkin EM. Propofol: an overview of its pharmacology and a review of its clinical efficacy in intensive care sedation. Drugs. 1995;50:636–657.
54 Geisslinger G, Hering W, Kamp HD, Vollmers KO. Pharmacokinetics of ketamine enantiomers. Br J Anaesth. 1995;75:506–507.
55 Tegeder I, Lötsch J, Geisslinger G. Pharmacokinetics of opioids in liver disease. Clin Pharmacokinet. 1999;37:17–40.
56 Stanski DR, Greenblatt DJ, Lowenstein E. Kinetics of intravenous and intramuscular morphine. Clin Pharmacol Ther. 1978;24:52–59.
57 Klotz U, McHorse TS, Wilkinson GR, Schenker S. The effect of cirrhosis on the disposition and elimination of meperidine in man. Clin Pharmacol Ther. 1974;16:667–675.
58 McHorse TS, Klotz U, Wilkinson G, Schenker S. Impaired elimination of meperidine in patients with liver disease. Trans Assoc Am Physicians. 1974;87:281–287.
59 Mazoit JX, Sandouk P, Zetlaoui P, Scherrmann JM. Pharmacokinetics of unchanged morphine in normal and cirrhotic subjects. Anesth Analg. 1987;66:293–298.
60 Bower S, Sear JW, Roy RC, Carter RF. Effects of different hepatic pathologies on disposition of alfentanil in anaesthetized patients. Br J Anaesth. 1992;68:462–465.
61 Ferrier C, Marty J, Bouffard Y, et al. Alfentanil pharmacokinetics in patients with cirrhosis. Anesthesiology. 1985;62:480–484.
62 Novick DM, Kreek MJ, Arns PA, et al. Effect of severe alcoholic liver disease on the disposition of methadone in maintenance patients. Alcohol Clin Exp Res. 1985;9:349–354.
63 Chauvin M, Ferrier C, Haberer JP, et al. Sufentanil pharmacokinetics in patients with cirrhosis. Anesth Analg. 1989;68:1–4.
64 Dershwitz M, Hoke JF, Rosow CE, et al. Pharmacokinetics and pharmacodynamics of remifentanil in volunteer subjects with severe liver disease. Anesthesiology. 1996;84:812–820.
65 Haberer JP, Schoeffler P, Couderc F, Duvaldstin P. Fentanyl pharmacokinetics in anaesthetized patients with cirrhosis. Br J Anaesth. 1982;54:1267–1270.
66 DeFronzo RA, Bonadonna RC, Ferrannini E. Pathogenesis of NIDDM: a balanced overview. Diabetes Care. 1992;15:318–368.
67 Fisher SJ, Kahn CR. Insulin signaling is required for insulin’s direct and indirect action on hepatic glucose production. J Clin Invest. 2003;111:463–468.
68 Peterson KF, Cline GW, Gerard DP, et al. Contribution of net hepatic glycogen synthesis to disposal of an oral glucose load in humans. Metabolism. 2001;50:598–601.
69 Tanaka T, Nabatame H, Tanifuji Y. Insulin secretion and glucose utilization are impaired under general anesthesia with sevoflurane as well as isoflurane in a concentration independent manner. J Anesth. 2005;19:277–281.
70 Saho S, Kadota Y, Sameshima T, et al. The effects of sevoflurane anesthesia on insulin secretion and glucose metabolism in pigs. Anesth Analg. 1997;84:1359–1365.
71 Bessesen A, Morland J. Effects of anaesthetics on protein synthesis in isolated rat hepatocytes: inhibition by diethyl ether in contrast to no influence by pentobarbital and fentanyl. Acta Pharmacol Toxicol (Copenh). 1985;57:23–29.
72 Aune H, Bessesen A, Olsen H, Moreland J. Acute effects of halothane and enflurane on drug metabolism and protein synthesis in isolated rat hepatocytes. Acta Pharmacol Toxicol (Copenh). 1983;53:363–368.
73 Nag DS, Sinha N, Samaddar DP, Mahanty PR. General anesthesia in a patient with Gilbert’s syndrome. J Anaesthesiol Clin Pharmacol. 2011;27:253–255.
74 Robards C, Brull S. The anesthetic implications of Crigler-Najjar syndrome. Anesth Analg. 2007;104:435–436.
75 Halevy A, Gold-Deutch R, Negri M, et al. Are elevated liver enzymes and bilirubin levels significant after laparoscopic cholecystectomy in the absence of bile duct injury? Ann Surg. 1994;219:362–364.
76 Yachimski PS, Friedman LS. Risk of surgery in patients with liver disease. In: Hancygier H, ed. Clinical hepatology. Heidelberg: Springer-Verlag; 2010:1383–1399.
77 Kharasch ED. Adverse drug reactions with halogenated anesthetics. Clin Pharmacol Ther. 2008;84:158–162.
78 Fontana RJ, Seeff LB, Andrade J, et al. Standardization of nomenclature and causality assessment in drug-induced liver injury: summary of a clinical research workshop. Hepatology. 2010;52:730–742.
79 Zimmerman HJ. Hepatotoxicity: the adverse effects of drugs and other chemicals on the liver. New York: Appleton-Century-Crofts; 1978.
80 Kenna JG. Mechanism, pathology, and clinical presentation of hepatotoxicity of anesthetic agents. In: Kaplowitz N, Delve LD, eds. Drug-induced liver disease. New York: Marcel Dekker; 2003:405–423.
81 Biebuyck JF, Lund P. Effects of halothane and other anesthetic agents on the concentrations of rat liver metabolites in vivo. Mol Pharmacol. 1974;10:474–483.
82 Davis JE. Fatal hepatic necrosis associated with halothane anesthesia. Am J Obstet Gynecol. 1972;112:967–971.
83 de Groot H, Noll T. Halothane hepatotoxicity: relation between metabolic activation, hypoxia, covalent binding, lipid peroxidation and liver cell damage. Hepatology. 1983;3:601–606.
84 Klion FM, Schaffner F, Popper H. Hepatitis after exposure to halothane. Ann Intern Med. 1969;71:467–477.
85 Ray DC, Drummond GB. Halothane hepatitis. Br J Anaesth. 1991;67:84–99.
86 Neuberger J, Vergani D, Mieli-Vergani G, Davis M, Williams R. Hepatic damage after exposure to halothane in medical personnel. Br J Anaesth. 1981;53:1173–1177.
87 Moult PJ, Sherlock S. Halothane-related hepatitis: a clinical study of twenty-six cases. Q J Med. 1975;44:99–114.
88 Sherlock S. Halothane hepatitis. Lancet. 1978;2:364–365.
89 Kenna JG, Neuberger J, Mieli-Vergani G, Mowat AP, Williams R. Halothane hepatitis in children. Br Med J (Clin Res Ed). 1987;294:1209–1211.
90 Wright R, Eade OE, Chisholm M, et al. Controlled prospective study of the effect on liver function of multiple exposures to halothane. Lancet. 1975;1:817–820.
91 Benjamin SB, Goodman ZD, Ishak KG, Zimmerman J, Irey NS. The morphologic spectrum of halothane-induced hepatic injury: analysis of 77 cases. Hepatology. 1985;5:1163–1171.
92 Eliasson E, Kenna JG. Cytochrome P450 2E1 is a cell surface autoantigen in halothane hepatitis. Mol Pharmacol. 1996;50:573–582.
93 Kenna JG, Martin JL, Satoh H, Pohl LR. Factors affecting the expression of trifluoroacetylated liver microsomal protein neoantigens in rats treated with halothane. Drug Metab Dispos. 1990;18:788–793.
94 Nguyen C, Rose NR, Njoku DB. Trifluoroacetylated IgG4 antibodies in a child with idiosyncratic acute liver failure after first exposure to halothane. J Pediatr Gastroenterol Nutr. 2008;47:199–202.
95 Farrell G, Prendergast D, Murray M. Halothane hepatitis. Detection of a constitutional susceptibility factor. N Engl J Med. 1985;313:1310–1314.
96 Gut J, Christen U, Frey N, Koch V, Stoffler D. Molecular mimicry in halothane hepatitis: biochemical and structural characterization of lipoylated autoantigens. Toxicology. 1995;97:199–224.
97 Gut J, Christen U, Huwyler J, Bürgin M, Kenna JG. Molecular mimicry of trifluoroacetylated human liver protein adducts by constitutive proteins and immunochemical evidence for its impairment in halothane hepatitis. Eur J Biochem. 1992;210:569–576.
98 Begriche K, Massart J, Robin MA, Borgne-Sanchez A, Fromenty B. Drug-induced toxicity on mitochondria and lipid metabolism: mechanistic diversity and deleterious consequences for the liver. J Hepatol. 2011;54:773–794.
99 Khemawoot P, Yokogawa K, Shimada T, Miyamoto K. Obesity-induced increase of CYP2E1 activity and its effect on disposition kinetics of chlorzoxazone in Zucker rats. Biochem Pharmacol. 2007;73:155–162.
100 Baines D. Anaesthetic considerations for the obese child. Paediatr Respir Rev. 2011;12:144–147.
101 Christ DD, Satoh H, Kenna JG, Pohl LR. Potential metabolic basis for enflurane hepatitis and the apparent cross-sensitization between enflurane and halothane. Drug Metab Dispos. 1988;16:135–140.
102 Lewis JH, Zimmerman HJ, Ishak KG, Mullick FG. Enflurane hepatotoxicity: a clinicopathologic study of 24 cases. Ann Intern Med. 1983;98:984–992.
103 Sinha A, Clatch RJ, Stuck G, Blumenthl SA, Patel SA. Isoflurane hepatotoxicity: a case report and review of the literature. Am J Gastroenterol. 1996;91:2406–2409.
104 Anderson JS, Rose NR, Martin JL, Eger EI, Njoku DB. Desflurane hepatitis associated with hapten and autoantigen-specific IgG4 antibodies. Anesth Analg. 2007;104:1452–1453. table of contents
105 Cote G, Bouchard S. Hepatotoxicity after desflurane anesthesia in a 15-month-old child with Mobius syndrome after previous exposure to isoflurane. Anesthesiology. 2007;107:843–845.
106 Ogawa M, Doi K, Mitsufuji T, Satoh K, Takatori T. [Drug-induced hepatitis following sevoflurane anesthesia in a child]. Masui. 1991;40:1542–1545.
107 O’Leary JG, Yachimski PS, Friedman LS. Surgery in the patient with liver disease. Clin Liver Dis. 2009;13:211–231.
108 Hanson KM, Johnson PC. Local control of hepatic arterial and portal venous flow in the dog. Am J Physiol. 1966;211:712–720.
109 Harville DD, Summerskill WH. Surgery in acute hepatitis. Causes and effects. JAMA. 1963;184:257–261.
110 Powell-Jackson P, Greenway B, Williams R. Adverse effects of exploratory laparotomy in patients with unsuspected liver disease. Br J Surg. 1982;69:449–451.
111 Runyon BA. Surgical procedures are well tolerated by patients with asymptomatic chronic hepatitis. J Clin Gastroenterol. 1986;8:542–544.
112 Heine G, Bines JE. New approaches to parenteral nutrition in infants and children. J Paediatr Child Health. 2002;38:433–437.
113 Klein S, Kinney J, Jeejeebhoy KN, et al. Nutrition support in clinical practice: review of published data and recommendations for future research directions. Summary of a conference sponsored by the National Institutes of Health, American Society for Parenteral and Enteral Nutrition, and American Society for Clinical Nutrition. Am J Clin Nutr. 1997;66:683–706.
114 Meadows N. Monitoring and complications of parenteral nutrition. Nutrition. 1998;14:806–808.
115 Ayers J, Graves SA. Perioperative management of total parenteral nutrition, glucose containing solutions, and intraoperative glucose monitoring in paediatric patients: a survey of clinical practice. Pediatr Anesth. 2001;11:41–44.
[/level-membership-for-anesthesiology-category][not-level-membership-for-anesthesiology-category]
28 Essentials of Hepatology
Anatomy
Hepatic developmental changes occur throughout gestation. Hepatic hematopoiesis develops in utero at 5 to 6 weeks gestation, followed closely by protein synthesis.1 The ability to metabolize carbohydrates and lipids begins by 10 weeks gestation, followed closely by the development of drug metabolizing systems.
During the neonatal period, liver function is immature in its ability to metabolize and clear xenobiotics. Factors believed to affect the clearance of medications include hepatic blood flow and the developmental status of hepatic transport and enzyme systems. Size alone does not account for this observed degree of immaturity, because the fetal and neonatal liver account for a greater percentage of body weight than the adult counterpart (3.6% of body weight versus 2.4% in adults).2 The neonatal liver contains approximately 20% fewer hepatocytes than the adult liver, and the cells are almost one half of the size of adult hepatocytes. These structural features may play some role in the functional deficiencies exhibited by infant livers. Cellular growth and hypertrophy of the liver continue at a rapid pace into young adulthood.
The structural unit of the liver parenchyma is the lobule, a hub-and-spoke structure with the central vein serving as the hub that is bordered by portal tracts, which contain a bile duct and tributaries of the portal vein and hepatic artery. While the mixed venous and arterial blood flows from the portal tract to the central vein, bile flows in the opposite direction through a canalicular matrix that then enters the bile ductule in the portal tract. The functional unit of the liver is the hepatic acinus, which is centered on the portal track and extends in three concentric zones (i.e., zones of Rappaport) outward to the central vein (Fig. 28-1). The more central zones (zones 1 and 2) are most active in oxidative processes, whereas the distal zone 3, which is closer to the central vein, is more dependent on glycolysis and more susceptible to ischemic and toxic injury.
Principles of Hepatic Drug Metabolism
Lipid solubility, an important and desired feature of most administered drugs, allows passive diffusion across cellular membranes. Lipophilic drugs are also difficult to excrete. They have a propensity to accumulate in the body’s fat stores and are bound to proteins in plasma, thereby limiting renal excretion. Renal and biliary excretion of lipid-soluble compounds can result in their resorption across their respective membranes. A major role of the liver is to transform lipid-soluble drugs into water-soluble compounds that become easily excreted metabolites. An interesting example of the need for this biotransformation is the anesthetic compound thiopental, which if not transformed into its less lipophilic counterpart would have a plasma half-life of approximately 25 years.1
The primary family of liver enzymes assigned the task of metabolizing these exogenous substances is the cytochrome P-450 (CYP) family. P designates a red pigment, which is related to a heme molecule and absorbs light at a wavelength of 450 nm.3 The primary reactions involved in the drug biotransformation and metabolism are hydroxylation and conjugation. Hydroxylation prepares the metabolite for conjugation (i.e., phase II reaction). The CYP family of enzymes is responsible for most phase I reactions, and the members were first thought to be chemically similar to mitochondrial cytochromes.
Phase I Reactions
The CYP enzymes likely evolved as a mechanism by which the host was able to protect itself from toxins ingested from the environment. Most enzymes involved in hepatic drug metabolism are categorized in three distinct families: CYP1, CYP2, and CYP3. Each family is further divided into subfamilies that are designated with capital letters and numbered in the order in which they were discovered. The CYP enzymes are generally conserved across species, but their regulation and catalytic activity vary among species, which highlights the challenges associated with laboratory analysis of drug metabolism.3
Genetic and nongenetic factors contribute to the variability in the enzymatic activity seen across all CYP enzymes.4 Approximately 5% of Caucasian populations lack CYP2D6 activity, which is associated with altered metabolism of some drugs.5 For example, a lack of CYP2D6 activity enhances the effect of drugs such as haloperidol and metoprolol that require the enzyme for efficient metabolism, whereas codeine, which is metabolized to morphine by CYP2D6, provides little analgesia in a child with CYP2D6 deficiency.6
Nongenetic factors that influence CYP activity include concomitant disease states, malnutrition,7 and exposure to a host of pharmacologic and naturally occurring compounds. Many drugs can inhibit or stimulate the enzyme system (Table 28-1). Inhibition of the CYPs occurs when drugs compete for the same enzyme. The degree to which this competition becomes clinically significant depends on five factors: the relative amount of the specific CYP, the concentrations of each drug, the degree of pharmacologically active metabolite generated through this system, the importance of the enzyme in elimination of the drugs, and the therapeutic index of the drug.3
Enhanced CYP expression occurs after amplified transcription of the specific gene that is induced by a variety of compounds. For example, rifampin and phenytoin induce CYP3A4 by binding the cytosolic human pregnenolone-X receptor (hPXR) or the steroid xenobiotic receptor (SXR).8 The activated receptor translocates into the nucleus, where it binds the regulator elements of the CYP3A4 gene and promotes increased transcription of CYP3A4, which can lead to toxic levels of intermediate compounds, as is the case with erythromycin, or to subtherapeutic levels of cyclosporine in transplant recipients.9
Cytochrome P-450 Activity
The superfamily of CYP enzymes is divided into subfamilies based on sequence homology and on demonstration of broad substrate specificities. An important example is the CYP3A subfamily, which is the most abundant group of cytochromes involved in the metabolism of xenobiotics. The three identified isoforms are CYP3A4, CYP3A5, and CYP3A7. CYP3A4 is the most abundant single enzyme in the human liver, accounting for the metabolism of approximately 50% of clinically used pharmaceuticals.10 CYP3A5 is more commonly found in the kidneys and lungs and to a lesser degree in the liver. CYP3A7 is the predominant isoform in the neonatal liver but is replaced after birth by CYP3A4. Given its critical involvement in hepatic biotransformation of xenobiotics, the CYP3A family of enzymes is used to study and estimate hepatic drug clearance in various age and gender groups.
Changes in the distribution and activity of the CYP enzyme systems occur with hepatic growth and maturation (Fig. 28-2). The CYP3A family is homogeneously distributed across the liver parenchyma in the fetal liver and shortly after birth. However, during postnatal growth, expression of the CYP3A protein shifts toward the centrilobular region of the acinus (i.e., Rappaport zone 1). By adulthood, expression of CYP2A becomes increasingly limited to the zone 1 and zone 2 hepatocytes, with sparse expression occurring in zone 3.11 Other examples of developmental changes in the CYP system include the CYP2C and CYP3A3/4 subfamilies, which have negligible expression in the fetus but have increased expression in the first few weeks of life.12,13 CYP2D6 reaches adult activity levels within 1 month chronological age; variability thereafter is determined primarily by genetic polymorphisms.13a
FIGURE 28-2 Changes in metabolic capacity versus percentage of adult activity.
(Modified from Kearns G, Abdel-Rahman SM, Alander SW, et al. Developmental pharmacology—drug disposition, action, and therapy in infants and children. N Engl J Med 2003;349:1157-67.)
Changes in activity of CYP enzyme families and subfamilies have correlated with drug clearance. For example, midazolam clearance correlates with changes in CYP3A4 activity, with decreased clearance in fetal and neonatal livers and with adult clearance rates achieved by 3 months of age.14,15 In contrast, CYP3A7 activity peaks at about 1 week postpartum and steadily diminishes during the first year of life, reaching approximately 10% of fetal liver activity by adulthood.
Phase II Reactions
Conjugation of lipophilic compounds decreases their lipid solubility and facilitates renal excretion.16,17 Conjugation reactions (i.e., glucuronidation, sulfation, glutathione conjugation, acetylation, and methylation) are decreased in infants compared with adults.18
Glucuronidation is catalyzed by uridine 5′-diphosphate (UDP)–glucuronosyltransferase (UGT) family of enzymes, which are derived from assimilation of proteins from separate genes or alternate splicing from single-gene transcripts.19 UGT enzymes are responsible for the metabolism of several drugs, including phenols, estrogens, and opioids (see Chapter 6). As with the CYP enzymes, individual UGT enzymes demonstrate substrate specificity and can act in concert to metabolize single compounds. Glucuronidation is not fully active in infants and can place this population at risk for toxic drug accumulation.20 Hepatic UGT enzyme concentrations are reduced during fetal and early postnatal development. At 3 months of age, the levels of many UGTs are 25% of those in adults.21
UGT1A enzyme activity, which is involved in the conjugation of bilirubin and ethinylestradiol, is decreased in the fetus, but it increases to adult levels within 3 to 6 months after a term delivery.22 UGT1A6, which conjugates acetaminophen and naproxen, has 10% of adult activity in the fetus and neonate, and it achieves only 50% of adult activity by 6 months of age.18 UGT2B7 is active in the metabolism of the nonsteroidal antiinflammatory drugs naloxone, codeine, and lorazepam. Fetal activity of this enzyme approaches 10% to 20% of adult levels, with a rapid increase to adult levels by 2 months of age.23
Sulfation is accomplished by sulfotransferases, a family of cytosolic enzymes that are divided into two categories: catechol and phenol sulfotransferase. These enzymes conjugate inorganic sulfate from 3′-phosphoadenosine-5′-sulfophosphate (PAPS) with compounds containing functional hydroxyl groups.16 The catechol transferases develop earlier in fetal life than the phenol counterparts and appear to exhibit decreased activity in the developing neonate. Although specific sulfotransferase substrates require identification, the activity of these enzymes is increased in fetuses and neonates and theorized to be an efficient conjugation pathway in this age group.
Glutathione S-transferases (GSTs) conjugate glutathione with a broad spectrum of lipophilic and electrophilic compounds. The family of GSTs is composed of up to five different groups in various classes designated µ, α, θ, and π, which are derived from at least three genetic loci.24 Tissue-specific expression of these enzymes has been demonstrated, with the liver expressing the greatest amount of protein. Variable time-dependent expression has also been shown, with α- and π-class GSTs having enhanced expression between 16 and 24 weeks gestation, whereas only the α-class enzymes predominate in the neonate and adult liver.25 The hepatic π-class enzymes disappear from their hepatocellular location by 6 months of age and can be found only in the epithelial cells of the biliary canaliculi. Variations in the developmental expression of this class of enzymes have made it challenging to fully appreciate what is likely a multitude of clinical interactions.25
Acetylation reactions are catalyzed by N-acetyltransferases, which transfer an acetyl group from acetyl coenzyme A to a variety of substrates (e.g., p-aminobenzoic acid, p-aminosalicylic acid, procainamide). Two genes, NAT1 and NAT2, are responsible for yielding two specific enzymes with different allelic forms. Despite having 87% sequence homology, these enzymes exhibit different substrate specificities.26 Both are cytosolic enzymes involved in the biotransformation of several drugs and the bioactivation of several human carcinogens. NAT1 is present in multiple fetal and postnatal tissues and accounts for the most N-acetyltransferase substrate metabolism in children younger than 1 year of age. NAT2 is located primarily in the liver and becomes the dominant acetylator after 1 year of age. NAT2 has polymorphisms with enzyme kinetics that differentiate patients with slow or rapid acetylation capabilities. Infants younger than 1 year of age usually are slow acetylators; subsequent age-dependent alterations lead an individual’s targeted acetylator status.27 Individuals who are genetically destined for rapid acetylation manifest this feature by 2 to 4 years of age.
Anesthetic Agents
Inhalational Anesthetic Metabolism
Inhalational anesthetics are poorly metabolized, with 15% to 20% of halothane undergoing biotransformation. Although both oxidative and reductive pathways are involved, the primary pathway of metabolism of halothane is through oxidation to a reactive intermediate, trifluoroacetyl chloride,28 which then undergoes glutathione conjugation.29
Isoflurane is metabolized by CYP2E1 to a limited extent (0.2%).30 Isoflurane is excreted as inorganic fluoride and trifluoroacetic acid after oxidative metabolism in the liver.31 Desflurane is the least metabolized (0.02%) of the volatile anesthetics, which is approximately 10% of the rate of isoflurane.32 Both are metabolized in the liver along similar paths because the urinary metabolite for desflurane, trifluoroacetic acid, is the same as for isoflurane.
Only 2% to 5% of inhaled sevoflurane is metabolized in humans by means of the hepatic CYP2E1 enzyme, as occurs for the other ether anesthetics.33 Oxidation of sevoflurane generates the intermediate formyl fluoride, a highly reactive species thought to generate liver protein adducts. Carbon dioxide and inorganic fluoride are released through this oxidative mechanism, and the final product is hexafluoroisopropanol, which undergoes glucuronide conjugation and is further excreted in the urine (see Chapter 6).34
Neuromuscular Blocking Drugs
Neuromuscular blockade is achieved by depolarizing or nondepolarizing neuromuscular blocking agents (see Chapter 6). The only depolarizing agent still in use is short-acting suxamethonium (i.e., succinylcholine).35 Succinylcholine is hydrolyzed completely by plasma cholinesterases, which are synthesized by the liver.36 Enzyme activity varies with age, but it is decreased in liver disease and has been used as a metric of liver prognosis.37
Nondepolarizing muscle relaxants are divided into aminosteroids (i.e., pipecuronium, pancuronium, vecuronium, and rocuronium) and benzylisoquinolinium diesters (i.e., doxacurium, atracurium, and cisatracurium besylate). Renal and hepatic diseases affect their safety and efficacy.38 Hepatic elimination depends on protein binding, hepatic blood flow, and drug extraction. The volume of drug distribution is increased in hepatic disease, leading to a slower onset and prolonged effect. In children with cholestatic liver disease, such as biliary atresia, uptake of these compounds by the liver is decreased, which decreases plasma clearance and prolongs their effects.39,40 Approximately 75% of the administered dose is bound to plasma proteins, with most bound to albumin. Despite these problems, children with liver disease and a correspondingly low albumin concentration are at minimal risk for the adverse effects from this group of drugs.41
Rocuronium is an analogue of vecuronium with a more rapid onset of action. Unlike other aminosteroids, which largely undergo renal excretion, only 12% to 22% of rocuronium is cleared through the kidney.42 In patients with hepatic disease, the volume of distribution of rocuronium increased by 33% compared with healthy controls.43 In patients undergoing liver transplantation, the clearance of rocuronium was only slightly reduced by the diseased native liver compared with the functioning allograft and healthy individuals.44 Reduced infusion requirements of rocuronium during liver transplantation may indicate graft dysfunction.45
[/not-level-membership-for-anesthesiology-category]
