[level-membership-for-anesthesiology-category]
9 Essentials of Hematology
The Basics
Laboratory Values and Diagnostic Tests
What is a normal hematocrit or platelet count for an infant or child who comes to the operating room? Red blood cell (RBC), white blood cell, platelet, and coagulation indices evolve in various ways through late gestation, the neonatal period, infancy, and childhood (Table 9-1).
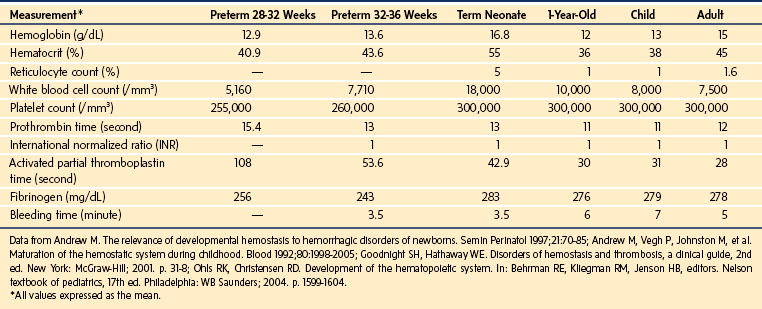
The term neonate has a relative polycythemia, reticulocytosis, and leukocytosis compared with the child. Neonatal platelet counts are similar to those of adults. Although in vitro function may be impaired for the first postnatal month, most in vivo assays of platelet function indicate normal or accelerated function. Preterm and term neonates have prolongation of the prothrombin time (PT) and activated partial thromboplastin time (aPTT) because of a relative deficiency in vitamin K–dependent factors and contact activation factors, respectively; however, concentrations of factor VIII and von Willebrand factor (vWF) are elevated.1 The international normalized ratio (INR), a normalized PT, has an average value of 1.0 for all age groups. Fibrinogen concentrations are comparable between the term neonate and adult, although neonatal fibrinogen is qualitatively dysfunctional. The plasma concentrations of many anticoagulant factors (i.e., tissue factor pathway inhibitor, antithrombin, vitamin K–dependent glycoproteins, and proteins C and S) are decreased in preterm and term neonates. The quantity and quality of plasminogen are decreased in neonates, a condition that increases the risk for thrombosis, especially in a compromised infant.1,2 Most of these differences between the neonate and older child or adult persist for 3 to 6 months postnatally.
There is no ideal single screening test to assess the bleeding risk of a child in the perioperative period. Bleeding time appears to be greater in the infant and child (and less in the neonate) than it is in the adult, but the range of values is wide and overlapping (see Table 9-1). Although this test is potentially helpful in predicting posttonsillectomy and adenoidectomy hemorrhage,3 as well as hemorrhage after percutaneous renal4 and liver5 biopsy, there is little evidence to support its use as a screening test to predict bleeding in the presence of a careful, inclusive clinical history.6,7
The thromboelastogram has been used to investigate the coagulation status of children undergoing spinal fusion,8 neurosurgical procedures,9 and cardiopulmonary bypass for cardiothoracic procedures.10 Although the thromboelastogram may provide useful information in the surgical setting to evaluate fibrinolysis, hypercoagulability, and other coagulation perturbations, its use is usually limited to clinical scenarios with dynamic coagulation changes, such as open heart surgery with cardiopulmonary bypass and liver transplantation. Platelet function analyzer (PFA-100) analysis is an additional test that is increasingly used for the assessment of platelet abnormalities, and it has the benefit of avoiding some of the difficulties of obtaining a bleeding time in children. Although several studies suggest PFA-100 analysis is equivalent or superior to the bleeding time for detecting bleeding abnormalities, there is no consensus about its role in preoperative screening.11 With the increasing use of newer agents that modify platelet function (e.g., platelet G protein–coupled receptor P2Y12 antagonists, glycoprotein GPIIb-IIIa complex antagonists), clinicians must understand that the thromboelastogram, PFA-100, and other methods for assessing platelet function may vary in their ability to monitor the effects of these agents and those of cyclooxygenase inhibitors.12
Guidelines for Transfusion
Critical analyses of the risks and benefits of transfusions in infants and children in the perioperative period have resulted in fewer transfusions. Even for infants and children in intensive care, a restrictive transfusion threshold (i.e., 7 g/dL) reduces transfusions without increasing morbidity compared with a liberal threshold (i.e., 9.5 to 10 g/dL).13 Data from the United Kingdom’s national audit of clinical transfusion, the Serious Hazards of Transfusion (SHOT), indicate that infants and children younger than 18 years of age are at greater risk for adverse transfusion-related reactions (37 and 18 in 100,000, respectively) than are adults (13 in 100,000). Most events were error related, such as administrative, laboratory, clinical judgment, and handling errors.14
Guidelines for RBC transfusion for infants and children in the perioperative setting should be consistent with those established by the American Society of Anesthesiologists Task Force on Blood Component Therapy, which propose that transfusion is not indicated for hemoglobin concentrations higher than 10 g/dL but is indicated for concentrations lower than 6 g/dL.15 When the concentration is between 6 and 10 g/dL, packed red blood cells (PRBCs) should be transfused based on the child’s vital signs, adequacy of oxygenation and perfusion, acuity and degree of blood loss, and other physiologic and surgical factors. When the concentration exceeds 10 g/dL, the decision to transfuse PRBCs to a neonate or infant should be based on the increased baseline concentrations of hemoglobin, increased oxygen consumption, increased affinity of residual fetal hemoglobin for oxygen, absolute blood volume (i.e., 85 mL/kg for a term neonate and 100 mL/kg for a preterm neonate), and other physiologic and surgical factors applicable to all children. The threshold for transfusing a healthy neonate may be 7 g/dL in some clinical settings, but it may be 12 g/dL or higher for a neonate in other settings, such as significant lung disease requiring mechanical ventilation, chronic lung disease, cyanotic congenital heart disease, or heart failure.16–18 For a preterm infant, the risks of hypovolemia, hypotension, acidosis, and postoperative apnea are magnified in the setting of operative blood loss and anemia. It is impossible to address all of the guidelines in this chapter, but many pediatric hematology and oncology consultants have clearly defined transfusion thresholds for their patient populations that should be reviewed preoperatively.
Guidelines for platelet transfusion have been published by consensus committees from France, the United Kingdom, and the United States; these reports are based on available evidence that has been gathered and critically reviewed (Table 9-2).15,19–22 Without evidence that platelet function is significantly different in the healthy infant and child, these guidelines should be applicable to these patients. The decision to transfuse platelets must take into account underlying medical conditions, platelet transfusion history, current medications, surgical bleeding, surgical interventions (e.g., cardiopulmonary bypass), and all other factors that may affect platelet function and turnover.23–27 Sevoflurane and propofol suppress28 and enhance platelet aggregation in vitro.29 Despite these effects on platelet aggregation, no change in the bleeding time has been reported, suggesting that the inhibitory effect does not impair hemostasis in vivo.30
| Medical Condition or Procedure | Platelet Count (/mm3) |
|---|---|
| Stable hematology-oncology or chronically thrombocytopenic patient | 10,000-20,000 |
| Lumbar puncture in stable leukemic child | 10,000 |
| Bone marrow aspiration or biopsy | 20,000 |
| Gastrointestinal endoscopy in cancer patient | 20,000-40,000 |
| Disseminated intravascular coagulation | 20,000-50,000 |
| Fiberoptic bronchoscopy in hematopoietic stem cell transplantation patient | 20,000-50,000 |
| Neonatal alloimmune thrombocytopenia | 30,000 |
| Major surgery | 50,000 |
| Dilutional thrombocytopenia with massive transfusion | 50,000 |
| Spinal anesthesia | 50,000 |
| Cardiopulmonary bypass | 50,000-60,000 |
| Liver biopsy | 50,000-100,000 |
| Nonbleeding preterm infant | 60,000 |
| Obstetric epidural anesthesia | 70,000-100,000 |
| Neurosurgery | 100,000 |
Data from references 19, 20, 23, 26, 27, 265.
Guidelines for the transfusion of other blood products, particularly fresh frozen plasma (FFP) and cryoprecipitate, have been established,15,26,31 and they are discussed later in the context of coagulation disorders. Indications32 for transfusing FFP are usually limited to the following:
1. Replacement of documented congenital or acquired coagulation factor deficiency when a specific sterilized or combined factor concentrate is unavailable, especially in the setting of anticipated or active bleeding
2. Acquired coagulopathy resulting from massive transfusion
3. Immediate reversal of warfarin’s effect
4. Coagulation support in disease processes such as disseminated intravascular coagulation and thrombotic thrombocytopenic purpura
5. A source of antithrombin III for children deficient of this inhibitor who require heparin
Guidelines have been established by the College of American Pathologists and other transfusion study groups for leukocyte reduction of RBC units,16 irradiation (x-ray or γ-ray) of cellular blood components,33 and administration of cytomegalovirus-seronegative RBCs (Tables 9-3 to 9-5).16 These guidelines are valuable when determining the specific choice of blood components that should be ordered and administered in the perioperative setting. For hematologic patients receiving chronic RBC transfusions, an extended phenotypic crossmatch and leukocyte reduction can decrease the risk of developing alloantibodies and transfusion reactions, especially in children of African descent if the local donor pool is primarily derived from Caucasian populations of Northern European descent.34 For oncology patients, updated specific requirements for blood products including leukocyte reduction and irradiation are often indicated and should always be reviewed with oncology specialists.
TABLE 9-3 Indications for Leukocyte-Reduced Red Blood Cell Units
Modified from Simon TL, Alverson DC, AuBuchon J, et al. Practice parameter for the use of red blood cell transfusions: developed by the Red Blood Cell Administration Practice Guideline Development Task Force of the College of American Pathologists. Arch Pathol Lab Med 1998;122:130-8.
TABLE 9-4 Indications for Irradiation of Cellular Blood Components
Modified from Simon TL, Alverson DC, AuBuchon J, et al. Practice parameter for the use of red blood cell transfusions: developed by the Red Blood Cell Administration Practice Guideline Development Task Force of the College of American Pathologists. Arch Pathol Lab Med 1998;122:130-8; Treleaven J, Gennery A, Marsh J, et al. Guidelines on the use of irradiated blood components prepared by the British Committee for Standards in Haematology blood transfusion task force. Br J Haematol 2010;152:35-51.
TABLE 9-5 Indications for Cytomegalovirus-Seronegative or Leukocyte-Reduced Red Blood Cells for Prevention of Virus Transmission
Modified from Simon TL, Alverson DC, AuBuchon J, et al. Practice parameter for the use of red blood cell transfusions: developed by the Red Blood Cell Administration Practice Guideline Development Task Force of the College of American Pathologists. Arch Pathol Lab Med 1998;122:130-8.
Hemolytic Anemias
Hereditary Spherocytosis
HS, the most common cause of inherited chronic hemolysis in North America and Northern Europe, has a prevalence of approximately 1 to 2 cases per 5000 people, if mild forms of the disease are included.35–37 First described in 1871, HS is present in many ethnic populations, but rare in African American populations. Because 75% of children inherit the disease in an autosomal dominant pattern, there is often a family history of the disorder, although autosomal recessive mutations, de novo mutations, and incomplete penetrance have been reported.37
Pathophysiology
Abnormalities in any of several erythrocyte membrane proteins, including the β subunit of spectrin, ankyrin, and band 3, can lead to HS. The variety of proteins affected and mutations observed in each gene account for the clinical heterogeneity of the disorder.36 When the erythrocyte loses surface area, it changes from a biconcave disk to a sphere, which alters its stability and flow pattern through the capillaries. The deformity leads to a loss of flexibility in the membrane, which makes it vulnerable to rupture, a condition that is worsened if the membrane surface area decreases by more than 3%.37 Damaged erythrocytes are sequestered in the splenic capillaries, which can lead to splenomegaly. The combination of intravascular and extravascular hemolysis can result in anemia, which induces extramedullary erythropoiesis. The life span of the erythrocyte is reduced from 120 days to just a few days when the RBC membrane has been deformed. If large numbers of damaged erythrocytes are lysed, unconjugated bilirubin is released into the bloodstream, which causes jaundice and possibly gallstones in as many as 60% of children.36 Membrane fragments from hemolytic reactions can lead to disseminated intravascular coagulation. Pulmonary hypertension may occur in the HS population, presumably as a result of hemolysis-induced alterations in NO metabolism.
Clinical and Laboratory Features
Children may present at any age with the triad of anemia, splenomegaly, and jaundice, which often is aggravated by concomitant viral infection. Mild, moderate, and severe forms of HS occur and are characterized by variations in laboratory results and clinical correlates. HS can manifest soon after birth and should be considered in infants who are jaundiced after the first postnatal week; resulting hyperbilirubinemia can sometimes necessitate an exchange transfusion. Mild disease occurs in 20% of children with HS; these children only occasionally present with symptomatic bilirubinate gallstones before adolescence. Approximately 5% of children have severe HS characterized by chronic anemic (hemoglobin concentration less than 8 g/dL) and a need for chronic transfusions. The course of this disease may be complicated by viral infections such as parvovirus B19 infection, which can suppress reticulocyte production37 and precipitate aplastic crises.
HS is most commonly suspected when numerous spherocytes with loss of central pallor appear on a peripheral smear. A complete blood cell count usually reveals a low hemoglobin and elevated reticulocyte count. Osmotic fragility remains the gold standard for the diagnosis of HS, but this test produces age-related results and must be performed by experienced laboratory technicians in a timely fashion. Increasingly, flow cytometry using eosin-5′-maleimide is being employed for diagnosis because it requires little blood and can be performed after overnight storage.38 As a direct result of chronic hemolysis, unconjugated bilirubin and serum lactate dehydrogenase concentrations increase, and serum haptoglobin concentrations decrease. Thrombocytopenia may develop as a result of hypersplenism.
Perioperative Considerations
Splenectomy significantly increases red cell survival in most cases and reduces the severity of the anemia and jaundice. It is usually reserved for more severe cases of HS, characterized by severe anemia that require frequent RBC transfusions, poor growth, chronic fatigue, or evidence of extramedullary hematopoiesis (e.g., frontal bossing). Splenic enlargement in a child interested in participating in contact sports is another indication.37 Splenectomy is ideally performed after the age of 6 years because of the increased risk of overwhelming infection by encapsulated organisms such as Streptococcus pneumoniae, Neisseria meningitidis, and Haemophilus influenzae type B in splenectomized younger children.39 Preoperative vaccination against these organisms is essential unless surgery is required emergently. Guidelines for the indications and duration of postoperative penicillin prophylaxis vary among institutions.36
Splenectomies in children are more frequently performed laparoscopically than by open laparotomy because the former is associated with decreased pain, quicker return of bowel function, shorter hospital stay, and improved cosmetic result. Conversion from laparoscopic to open splenectomy is necessary in fewer than 10% of cases.40 Partial splenectomies are increasingly performed because they allow retention of some immune function against bacterial infections in younger children while reducing the sequestration of spherocytes. However, residual splenic tissue can increase in size and necessitate total splenectomy at a later time.41,42 If anemia recurs after splenectomy, it may indicate the presence of accessory splenic tissue that was unrecognized initially. Transient postsplenectomy thrombocytosis marked by dramatic increases in platelet counts may also occur in children,35 in addition to a general increase in the risk of thromboembolic disease.
Gallstones occur in 21% to 63% of children with HS, but cholecystectomy is usually performed only when children are symptomatic with cholelithiasis. Children who undergo splenectomy and who also have radiographically identified gallstones may undergo concurrent cholecystectomy, whether the stones are symptomatic or not.43–45
Table 9-6 summarizes the clinical features and important perioperative considerations for the child with HS undergoing incidental or disease-related surgical procedures.
TABLE 9-6 Perioperative Concerns for Patients with Hereditary Spherocytosis
Hemoglobin, reticulocyte count, platelet count
History of transfusions and special blood requirements (e.g., extended phenotypic matching, leukocyte reduction)
History of infections, aplastic crises, and presplenectomy vaccinations
Presplenectomy antibiotic prophylaxis and immunization when indicated
Appropriate antibiotic coverage
Attention to physiologic effects of laparoscopy on circulatory and respiratory function
Potential for significant blood loss (unusual in splenectomy and cholecystectomy)
Judicious use of regional anesthesia, intramuscular medications, nasogastric tubes, nasal intubation, and other methods when platelet count is low
Limited use of medications with potential bleeding risk (e.g., ketorolac)
Glucose-6-Phosphate Dehydrogenase Deficiency
G6PD deficiency causes hemolysis in the presence of various oxidative stressors. It is the most common enzyme deficiency in humans, affecting approximately 400 million people worldwide. This enzyme deficiency is inherited in an X-linked, recessive fashion. Although males are most commonly affected, females (heterozygous or homozygous for the gene) may have clinical manifestations of the disease. More than 100 variants have been described, including a relatively mild form that affects about 10% of African American males (i.e., G6PD A−) and a more severe form that affects Italians, Greeks, and other populations in the Mediterranean, African, and Asian regions (i.e., G6PD Mediterranean).46–48 This deficiency is prevalent in geographic areas where the incidence of malaria is high, presumably because G6PD deficiency may attenuate the severity of malarial infections.
Pathophysiology
G6PD plays an important role in the hexose monophosphate/pentose phosphate shunt, which is essential for normal energy metabolism in erythrocytes. G6PD generates the reduced form of nicotinamide adenine dinucleotide phosphate (NADPH). NADPH maintains glutathione in the reduced form (GSH), which reduces peroxides and protects cells from oxidative damage in the course of normal biochemical events or in the event of excess free oxygen radical generation. Superoxide ion or hydrogen peroxide, or both, can oxidize hemoglobin, which then precipitates as insoluble membrane inclusions. These inclusions, together with the oxidative damage to cell membranes, lead to cell damage. Erythrocytes are particularly sensitive to oxidative damage because of their lack of synthetic activity. In the presence of oxidants and free radicals (e.g., produced by infection or by ingestion of certain medications and foods), this cascade of events may precipitate hemolysis in the G6PD-deficient child.46,48
Clinical and Laboratory Features
Clinical symptoms of G6PD deficiency may be deceptively variable, and they may occur in the neonatal period or in older age groups as episodic or chronic hemolytic anemia. Presenting signs include anemia and jaundice; in severe cases, these signs can be followed by lumbar and abdominal pain and by renal failure. Acute illness such as diabetic acidosis or ingestion of a variety of substances may precipitate a hemolytic event (Table 9-7). Hemolysis may range from benign and transitory to severe and life-threatening; the latter situation is more likely if the triggering agent is not eliminated or controlled. Laboratory findings include normocytic anemia, increased reticulocyte count and serum bilirubin concentration, and presence of Heinz bodies in the peripheral blood smear.
TABLE 9-7 Agents That May Precipitate Hemolysis in Patients with Glucose-6-Phosphate Dehydrogenase Deficiency
Perioperative Considerations
In the perioperative setting, G6PD deficiency does not usually cause problems if the triggering agents are avoided by susceptible children and the precipitating causes are treated or eliminated (Table 9-8). Monitoring for and treatment of possible complications are appropriate; transfusion is rarely required.
TABLE 9-8 Perioperative Concerns for Patients with Glucose-6-Phosphate Dehydrogenase Deficiency
Administration of large or excessive doses of medications such as prilocaine, benzocaine, and sodium nitroprusside may trigger hemolysis in G6PD-deficient children in the perioperative setting.46,48–50 Although these children can reduce methemoglobin that is normally produced by these agents, G6PD-deficient children may not tolerate large amounts of potent oxidizing agents (i.e., superoxide ion and hydrogen peroxide) produced by methemoglobin. Infants may be particularly susceptible to symptomatic methemoglobinemia (because of their low NADPH dehydrogenase activity) and to methemoglobin-induced hemolysis if they are G6PD deficient. Treatment of methemoglobinemia with methylene blue is contraindicated in these infants because the agent itself may precipitate hemolysis.42 Hemolysis has occurred during cardiopulmonary bypass in G6PD-deficient children,50,51 and methemoglobinemia has occurred in a child with partial G6PD deficiency after application of EMLA cream, a eutectic mixture of local anesthetics.52
Hemoglobinopathies
Sickle Cell Disease
First identified by Herrick about 100 years ago, sickle cell disease is a group of inherited hemoglobinopathies with a diverse worldwide prevalence. The disease affects about 1 in 375 African American and 1 in 20,000 Hispanic births.53 The spectrum of the disease includes sickle cell anemia (HbSS), which accounts for about 70% of the American sickle cell disease population; sickle cell/hemoglobin C disease (HbSC), accounting for about 20%; sickle cell/β-thalassemia (HbSβ), accounting for about 10%; and a host of other, uncommon sickle variants whose prevalence is increasing over time.54 HbSβ includes HbSβ+ and HbSβ0 thalassemias; the distinction depends on whether normal hemoglobin A (HbA) is expressed at all or in only a small concentration. HbSβ0, HbSC, and sickle cell/hemoglobin D disease (HbSD) and additional rare forms have the potential to sickle as severely as HbSS. Sickle cell trait (HbAS), in which approximately 40% of hemoglobin is hemoglobin S, occurs in about 8% of African Americans and in a much smaller percentage of Hispanic and other American subpopulations. The sickle gene is found commonly in Africa, Mediterranean areas, southwestern Asia, and other areas where malaria has been historically endemic and for which the gene is protective. Sickle hemoglobinopathies have many implications for perioperative care because they increase perioperative morbidity and mortality.
Pathophysiology
Hemoglobin A is composed of two α- and two β-globin chains. Hemoglobin S is caused by a mutant β-globin gene on chromosome 11, which leads to a single amino acid substitution (valine for glutamate at position 6). Replacement of negatively charged and hydrophilic glutamate by noncharged and hydrophobic valine leads to instability of the hemoglobin molecule and decreased solubility of the molecule when deoxygenated. Hemoglobin polymers form, generating long helical strands and inducing a process that leads to hemoglobin precipitation and hemolysis.55
The pathophysiologic process in sickle cell disease is much more complex than originally thought.55–57 Inflammation, vascular endothelial adhesion abnormalities, platelets, and coagulation cascade activation all contribute to vaso-occlusive episodes. The sickle red cell membrane, exposed to the destructive oxidant effects of intracellular iron, develops altered transmembrane ion transport pathways, which lead to altered permeability to sodium, potassium, and calcium, causing dehydration of the cell and irreversible sickling.58 Membrane abnormalities of phospholipid content also contribute to its deformability, and exposure of phosphatidyl serine facilitates activation of the clotting cascade. These and other factors lead to entrapment of irreversibly sickled red cells in the microcirculation, activation of coagulation and inflammatory pathways, ischemia, and infarction of tissue. At the same time, chronic intravascular hemolysis decreases production of NO, while increased scavenging decreases the bioavailability of NO. The resulting NO deficiency causes endothelial dysfunction and disease complications such as pulmonary hypertension, priapism, and skin ulceration.58,59
Clinical and Laboratory Features and Treatment
Sickle cell disease is a multisystem process involving potentially most organs of the body and often necessitating surgical intervention. Based on data collected in the early 1990s, approximately one third of patients with HbSS disease have progressive disease leading to organ dysfunction and death; about half have significant but less devastating disease; and the remainder have a reasonably stable, slowly progressive clinical course.60 Therapeutic interventions and genetic factors account in large part for the differences in outcome. Children with persistence of hemoglobin F (which itself protects against the effects of deoxygenation on red cells) and those with HbSC or HbSβ+ have fewer complications than those with HbSS or HbSβ0.
Early diagnosis and treatment of sickle cell disease have been facilitated by the widespread use of universal neonatal screening, which was first used in the state of New York in 1975. Most screening programs for sickle cell disease use isoelectric focusing of an eluate from dried blood spot samples, a technique that is also used to screen for other disorders. A few programs use high-performance liquid chromatography. Because a small percentage of children with sickle cell disease are not African American (i.e., Native American, Hispanic, and Caucasian),53 selective screening may not detect all affected infants. As of 2006, all 50 states and the District of Columbia screen all neonates for sickle hemoglobinopathies. Families of infants diagnosed with sickle trait (HbAS) on neonatal screening may not be made aware of the diagnosis, but these infants rarely develop significant clinical problems in the perioperative period.
Affected children born within the United States before universal neonatal screening and those born outside the United States and not receiving regular health care may not have received a diagnosis and appropriate care before surgery. Notwithstanding the controversy over the utility of nonselective preoperative screening,61 children at risk whose hemoglobin status is unknown preoperatively should be tested with a sickle-screening test, followed by a hemoglobin electrophoretic evaluation if screening is positive. However, infants younger than 6 months of age may have a false-negative screening test result because of presence of fetal hemoglobin, although electrophoresis is diagnostic at all ages. Children older than 10 years of age with a normal hemoglobin value, standard peripheral blood smear, and unremarkable clinical history are probably at a reduced risk for clinically significant hemoglobinopathy.62
Common clinical symptoms of sickle cell disease in children include chronic hemolytic anemia, recurrent vaso-occlusive episodes leading to pain, acute chest syndrome (ACS), infection, renal insufficiency, osteonecrosis, and cholelithiasis. Pulmonary hypertension, priapism, and skin ulcerations are related to the degree of red cell hemolysis.63 Chronic pulmonary and neurologic disease (e.g., stroke) are additional causes of significant morbidity and mortality.54 In the perioperative period, the most common complications in sickle cell children include ACS (about 10%), fever or infection (about 7%), vaso-occlusive episodes (about 5%), and transfusion-related events (about 10%).64
Chronic hemolytic anemia is a hallmark of HbSS disease. It is characterized by a baseline hemoglobin value of 5 to 9 g/dL (often more than 9 g/dL in HbSC disease), reticulocytosis (5% to 10%), and a distinctive red cell morphology observed on a peripheral smear.57 Chronic hemolysis is associated with increased red cell turnover and a propensity to form biliary stones. It may be complicated by other anemic events, such as acute splenic sequestration, typically occurring in infants and young children after a viral illness; and acute aplastic anemia, typically associated with parvovirus B19 infection. For some children, chronic and acute severe anemia are managed with RBC transfusions, although these children are prone to develop alloantibodies to RBC antigens, and untreated iron overload can lead to life-threatening cirrhosis and cardiac failure. Hydroxyurea is used to prevent vaso-occlusive episodes and end-organ damage. Most children are maintained on chronic folic acid therapy to prevent megaloblastic erythropoiesis that can result from the increased red cell production.
Vaso-occlusive episodes in sickle cell disease occur as a result of episodic microvasculature occlusions at one or more sites. The occlusive process occurs most commonly in the phalanges (i.e., dactylitis or hand-foot syndrome), long bones, ribs, sternum, spine, and the pelvis; it also can occur in the mesenteric microvasculature, producing abdominal pain that may mimic a surgical acute abdomen. Vaso-occlusive episodes are managed with hydration, warming, acute and chronic pain management (including opioids, antiinflammatory agents, and complementary modalities), and in-hospital care. It is essential to foster an ideal environment for pain control (e.g., calm, pleasant distractions, supportive personnel and objects). For children who have frequent or severe crises, oral hydroxyurea therapy has been effective in decreasing the frequency of events through several mechanisms, including inhibiting hemoglobin precipitation by increasing fetal hemoglobin concentrations, reducing the white blood cell count, modifying the inflammatory response, and facilitating NO metabolism.59,65,66 Inhaled NO may prove to be effective therapy for vaso-occlusive episodes.67
ACS is characterized by acute respiratory symptoms concurrent with a new infiltrate observed on the chest radiograph.68 ACS frequently occurs 2 to 3 days after a vaso-occlusive episode, and although its clinical presentation varies, it often includes fever, tachypnea, cough, and hypoxemia. The process may be self-limited over a period of a few days, or it may progress to respiratory failure (15%) and even death. The inconsistent presentation in part reflects the complex and variable pathogenesis of ACS. An episode may have a single or multiple causes, including infection (i.e., bacteria [often Chlamydia or Mycoplasma], viruses, and mixed flora), pulmonary fat embolism, pulmonary infarction, and pulmonary hemorrhage.69 Acute management includes supportive care and oxygen, antibiotics to cover encapsulated and atypical organisms, bronchodilators, pain control, ventilatory support as needed, and transfusion. Incentive spirometry or continuous positive airway pressure can be helpful, especially in the perioperative setting. Hydroxyurea therapy and chronic transfusion therapy decrease the frequency of ACS, whereas inhaled NO attenuates the process acutely.59,70–72 Airway reactivity is also common in children with sickle cell disease, in part due to NO deficiency, and it is responsive to bronchodilator therapy.73,74 In later life, children with sickle cell disease may develop restrictive lung disease and pulmonary hypertension as a result of repeated ACS-induced lung injury and chronic inflammation. NO deficiency, the result of decreased production, increased consumption, or altered metabolism, may also play an important role in these processes.63,68,75
Infection is a common problem in this disease because of deficits in the immunologic system and the specific effects of splenic atrophy and dysfunction that occur over the first few years of life.56 As a result of susceptibility to overwhelming infection by S. pneumoniae and H. influenzae type B, young children receive penicillin prophylaxis until 6 years of age and bacteria-specific immunizations. A host of infectious organisms have been implicated in ACS, and infection with gram-negative organisms (e.g., osteomyelitis caused by Salmonella) is common in older children and adults.69
Stroke is a devastating complication that occurs in children with sickle cell disease. A history of overt strokes is elicited in 10% or more of these children, and silent strokes occur in another approximately 15%; one fourth of children are at risk for motor or cognitive deficits at the time of presentation for surgery.76,77 A child’s first stroke often appears as early as 2 to 5 years of age.78,79 Risk factors include a reduced hemoglobin concentration, increased concentration of HbS, increased leukocyte count, and a history of dactylitis. Strokes may be precipitated by pain episodes, ACS, and infection.80 Children suffering an acute stroke are managed supportively with exchange transfusion to reduce the concentration of HbS to less than 30% and then chronic intermittent transfusions to minimize the risk of recurrence.81
The thrust of the current management of stroke is prevention. Based on the findings of the Stroke Prevention Trials (STOP 1 and 2), children are assessed annually by transcranial Doppler, and children with evidence of cerebrovascular compromise and some of those with magnetic resonance imaging abnormalities are managed with chronic transfusion therapy to minimize the risk of a stroke.82–84
Renal abnormalities in sickle cell disease develop from repeated sludging episodes, which may cause thrombosis, progressive infarction, and necrosis of the renal medulla. Proteinuria, hematuria, hyposthenuria, renal tubular acidosis, and other clinical abnormalities occur. Acute and chronic renal failure may develop.85,86 Renal dialysis and transplantation have been successful therapeutic interventions for this population.87
The clinical picture of sickle cell disease is altered to various degrees by concomitant qualitative and quantitative changes of hemoglobin. Sickle cell trait (HbAS) usually is benign, although it may be characterized by hematuria and hyposthenuria,87,88 and sickling may occur with HbAS under extremely altered physiologic circumstances (e.g., cardiopulmonary bypass).89,90 There is a small but significant risk of pulmonary emboli and sudden death with extreme exertion by individuals with HbAS, which has led to the mandatory offer of testing to all National Collegiate Athletic Association (NCAA) athletes.91
Children with HbSC disease usually have a greater baseline hemoglobin concentration and fewer complications than those with HbSS disease; delayed splenic autoinfarction reduces their risk for infection in early childhood.62 However, children with HbSC disease are more likely to have proliferative retinopathy and avascular necrosis of bone.56
Children with HbSβ0 (i.e., one sickle globin allele and one thalassemic allele expressing no β-globin) have a course identical to that of HbSS, whereas those with HbSβ+ (i.e., one sickle globin allele and one thalassemic allele expressing β-globin at a reduced level) tend to have a more benign course that is proportional to the amount of normal β-globin expression. The coexistence of hemoglobin S with α-thalassemia produces a variable clinical picture, but it may predispose children to a greater incidence of pain episodes.62
In addition to the treatment modalities mentioned earlier, many new and experimental modalities show promise in the treatment of sickle cell disease and its complications. They include induction of hemoglobin F by short-chain fatty acids such as butyrate or demethylating agents such as decitabine; membrane-active medications such as the Gardos channel inhibitors, magnesium; and antiadhesion therapies. New therapeutic targets are being identified, such a BCL11A, a zinc finger protein that plays a key role in the silencing of fetal globin genes. Reduction of BCL11A results in an induction of fetal globin.92 Hematopoietic stem cell transplantation (HSCT) is increasingly used as a curative intervention for sickle cell disease when instituted before development of organ dysfunction, but its application is limited in part by the lack of suitable donors.56,93 To circumvent this limitation, gene therapy protocols using a child’s own stem cells have been approved for use.
Perioperative Considerations
Perioperative morbidity and mortality are greater in children with sickle cell disease than in the general population. These children often require surgical procedures; the most common are cholecystectomy94; ear, nose, and throat procedures95; and orthopedic procedures (especially hip procedures for osteonecrosis).96 Placement of long-term vascular access for transfusions, antibiotics, analgesia, and other therapies is frequently performed. The Cooperative Study of Sickle Cell Disease reported that 7% of all deaths among children with this disease were related to surgery.54 Early reviews reported perioperative mortality rates as great as 10% and morbidity rates as great as 50% for children with sickle cell disease.97–100 Studies published in the 1990s indicated that the 30-day mortality rate was about 1%.101 In a group of more than 600 patients managed according to standard guidelines of care and prospectively studied, the incidence of any complication was about 30%, and the incidences of ACS and pain crisis were 10% and 5%, respectively.64 Patient factors (e.g., age, history of pulmonary disease, number of prior hospitalizations) and surgical factors (i.e., invasive or superficial) appear to affect the incidence of complications. The impact of newer interventions and technologies (e.g., laparoscopic and robotically assisted cholecystectomy and splenectomy)102–104 on perioperative morbidity and mortality rates is unclear.
The principles of optimal perioperative care are based on maintaining optimal physiologic parameters throughout the perioperative period, avoiding factors that may precipitate a sickle crisis, optimizing pain management, and close consultation among hematologists, surgeons, and anesthesiologists (Table 9-9).105 The child with sickle cell disease who is undergoing surgery should be viewed and managed primarily as a hematology patient whose care is being shared with, rather than assumed by, the surgeon and anesthesiologist during the perioperative period. Avoiding unnecessary and potentially dangerous surgical procedures (e.g., exploratory laparotomy to rule out appendicitis in a child who is experiencing an abdominal pain crisis) and minimizing perioperative complications should be the focus of the multidisciplinary care team. Based on a survey of perioperative management of sickle cell disease among anesthesiologists in North America, most anesthesiologists do consult with hematologists in all cases or on a case-by-case basis.106
TABLE 9-9 Perioperative Concerns for Patients with Sickle Cell Disease
Screening if unknown status in at-risk children
Primary management by hematology service (in most circumstances)
History of acute chest syndrome, vaso-occlusive pain crises, hospitalizations, transfusions, transfusion reactions
Neurologic assessment (e.g., strokes, cognitive limitations)
History of analgesic and other medication use
Oxygen saturation (on room air), chest radiograph
Pulmonary function tests (when appropriate)
Echocardiography (when appropriate)
Neurologic imaging (for recent changes)
Transfusion crossmatch (e.g., antibody-matched, leukocyte reduced, sickle negative)
Transfusion to correct anemia (in most circumstances)
Parenteral hydration for nil per os (NPO) status
Aggressive bronchodilator therapy
Appropriate antibiotic therapy, including presplenectomy antibiotics and immunizations (as indicated)
Maintenance of oxygenation, perfusion, normal acid–basis status, temperature, hydration
Availability of appropriately prepared blood (as indicated)
Anesthetic technique appropriate for procedure and postoperative analgesic requirements
Attention to physiologic effects of laparoscopy on circulatory and respiratory function
Appropriate antibiotic therapy
Judicious use of tourniquets, cell saver, and cardiopulmonary bypass
Management by hematology service
Monitoring for complications, especially acute chest syndrome and vaso-occlusive pain crises
Maintenance of oxygen saturation monitoring and supplementation as needed, including supplemental oxygen the first 24 hours regardless of oxygen saturation
Appropriate hydration (oral plus parenteral)
Appropriate antibiotic therapy
Incentive spirometry (possibly with continuous or bilateral positive airway pressure) and bronchodilator therapy
Although there is no evidence to support or refute many of the long-standing guidelines for perioperative care and individual practices vary widely,106 it seems appropriate to avoid the specific factors that may promote intravascular sickling: hypoxia, acidosis, hyperthermia, hypothermia, and dehydration.55 Meticulous attention to pain management is also essential because perioperative vaso-occlusive pain is common and is associated with ACS. Monitoring of vital signs throughout the perioperative period is mandatory, especially monitoring of oxygenation with pulse oximetry. Oxygen saturation as determined by pulse oximetry may underestimate measured oxygen saturation in patients with sickle cell disease, although usually not to a clinically significant degree.107,108 Because ACS, a common (10%) and potentially life-threatening complication of surgery, occurs 1 to 3 days postoperatively, it is important to extend adherence to guidelines of care into the postoperative period, regardless of the apparent well-being of the child.64 In light of the renal concentrating defect found in these patients, perioperative hydration is important to maintain and may require in-hospital preoperative care, although overhydration may compromise vulnerable cardiovascular and respiratory physiology.
Transfusion in the perioperative period remains a controversial subject.109 Transfusion of non-HbS RBCs to a child with sickle cell disease has several beneficial effects: correction of anemia, dilution of HbS red cells, compensation for blood loss, and prevention of some complications (e.g., stroke). However, transfusion is not without risks, including alloimmunization,34,110 transfusion reactions (about 7% in the perioperative period),64 infection, iron overload, time, and expense. Although there have been many reports of surgery performed safely in children with sickle cell disease without preoperative transfusion,111 uncontrolled studies indicate that preoperative transfusion does decrease the rate of perioperative complications.94,101 The Preoperative Transfusion in Sickle Cell Disease Study Group demonstrated prospectively in 604 operations (70% were cholecystectomies and otolaryngologic and orthopedic operations) that simple transfusion (i.e., correction of preoperative anemia to 10 g/dL with straight transfusion) was as effective as aggressive transfusion (i.e., lowering the preoperative HbS level to less than 30%, often with exchange transfusion) in preventing perioperative complications and was associated with fewer transfusion-related complications in children.64
To directly determine if transfusion prevents perioperative complications in the current era of surgical and anesthesia practices, an international randomized trial was initiated, the Transfusion Alternatives Preoperatively in Sickle Cell Disease (TAPS) trial. However, this trial was halted in March 2011 due to an excessive number of complications in the nontransfusion group. Although the final results of TAPS have not been published, it remains prudent to follow prior recommendations for transfusion for moderate and complicated operations in sickle cell patients. It is currently recommended that most children with HbSS undergoing most surgical procedures receive preoperative correction of anemia with simple transfusion to a hemoglobin concentration of about 10 g/dL. Children maintained on chronic transfusion programs (e.g., stroke prevention) should continue such management preoperatively. Recommendations for children with HbSC disease are less clear because these children typically maintain a baseline hemoglobin concentration at about 10 g/dL. For HbSC children who have a history of ACS, frequent pain crises, underlying pulmonary disease, or other complications, it is recommended that they receive selective preoperative exchange transfusion to reduce the HbS concentration without increasing total hemoglobin.112 Because of the high risk of alloimmunization in the sickle cell population, blood to be administered to these patients should undergo extended phenotype matching, including Rh, Cc, D, Ee, and Kell in addition to ABO84,113; leukocyte reduction; and sickle cell screening. Directed donation of blood from family members should be avoided if the child is a hematopoietic stem cell transplant candidate because it can lead to alloimmunization and later graft rejection.
Anesthetic technique does not have a clear effect on perioperative outcomes for children with sickle cell disease.114 Inhalational anesthetics do not affect the sickling process, although there is some experimental evidence suggesting that halothane may increase the viscosity of sickled blood.115 Pharmacokinetics of some agents commonly used with general anesthesia such as atracurium may be altered in this population.116 Regional anesthesia has been associated with an increased risk of postoperative complications in one retrospective study,101 but it has not been shown to affect perioperative outcome in others.94,96 Vasodilatory and analgesic properties of regional anesthesia can be effective in the management of vaso-occlusive episodes and priapism and in providing perioperative anesthetic care.117,118
Hyperventilation should be avoided because of its potential to reduce cerebral perfusion in children at an increased risk for stroke.119 The use of a tourniquet in HbSS and HbAS diseases has been questioned.120–122 However, tourniquets have been applied intraoperatively for up to 2 hours without complication, and the predominance of evidence supports their safe use as long as they are used carefully and selectively in combination with general guidelines of perioperative care (see Chapter 30).123–126 Intraoperative blood salvage using cell saver devices has been used safely in sickle cell patients,127 although there is some evidence that the salvage device itself may produce sickling in the processed blood, even sickle trait blood.128 Cardiopulmonary bypass seems to be an optimal setting in which to induce sickling, given the cold, hypoxic, acidotic, and stagnant environment created. Although there are reports of bypass surgery conducted in children with HbSS or HbAS with standard bypass procedures without transfusion,129–132 these children usually are managed with aggressive exchange transfusion before or during bypass.
Thalassemias
Pathophysiology
Anemia in thalassemia is the result of hemolysis and ineffective erythropoiesis; the latter is the result of accelerated cell apoptosis triggered in part by excess deposition of unpaired globin chains in erythroid precursors.133 Unpaired globin subunits are oxidized and form hemichromes, whose rate of formation determines rate of hemolysis. Precipitation of hemichromes leads to a complex process that includes release of toxic agents and formation of reactive oxygen species; alteration of red cell membranes, causing cells to become rigid, aggregate, and disintegrate; and activation of the coagulation process. As a result of chronic anemia and ineffective erythropoiesis, bone expansion and extramedullary hematopoiesis may develop in the liver and spleen, and marrow space expansion may occur at sites such as the cranium and paravertebral areas, thereby causing disfiguring bony changes.
Clinical and Laboratory Features and Treatment
The disease picture in α-thalassemia reflects complete loss of expression of one to four α-globin genes. As a four-gene globin deletion, it is characterized by hydrops and in utero or perinatal death unless diagnosed early and supported with in utero transfusions. As a three-gene deletion, or hemoglobin H (HbH) disease, it is relatively benign, characterized by chronic hemolytic anemia, which may be exacerbated by exposure to stress and oxidants.134,135 The few patients requiring intermittent transfusion therapy usually have a two-gene deletion along with a hemoglobin Constant Spring (HbCS) mutation. A two-gene deletion alone is characterized by mild, clinically insignificant microcytic anemia. A one-gene deletion is characterized by a silent carrier state with no anemia or microcytosis.
The clinical problems in thalassemia are those associated with chronic anemia: the physiologic response to the need for increased erythropoiesis, transfusions to maintain a hemoglobin concentration greater than 9 g/dL, iron overload from excess transfusions and pardoxical increased iron absorption, and chelation therapy.134 Clinical problems include transfusion-associated alloimmunization and infection, splenomegaly, bone abnormalities (due to extramedullary hematopoiesis, chelation therapy, and other factors), endocrine dysfunction (including hypogonadism, hypopituitarism, and diabetes mellitus), short stature, pulmonary hypertension, venous thrombosis and thromboembolism, and cardiomyopathy (primarily due to iron overload). Thalassemia patients may be hypercoagulable,136 a condition that may be exaggerated after splenectomy.136,137
Routine therapies used to treat severe diseases and to prevent complications include phenotypic matching and leukocyte reduction of transfused blood, chelation therapy, and hormone and vitamin D therapy. When an appropriate donor is available, HSCT is recommended before severe liver damage occurs because it provides a potential cure for thalassemia. To ameliorate the course of the disease, other therapies are being investigated, including administration of erythropoietin, fetal hemoglobin modifiers (e.g., hydroxyurea, butyrate), and antioxidants. Gene therapy trials are ongoing, and at least one patient has become transfusion independent.138
Perioperative Considerations
Children with moderate or severe thalassemia may require cholecystectomy, splenectomy, and vascular access placement for frequent transfusions.139 Demineralized long bones may be prone to fracture, and older children may require osteotomies for bony deformities. Bony abnormalities of the maxillofacial area may render securing the airway challenging.140 Laparoscopic and robotic techniques for cholecystectomy141 and splenectomy have been used successively in children with thalassemia, although perioperative hypertension may be a common problem in laparoscopic splenectomy.142,143 Open heart surgery requiring cardiopulmonary bypass with judicious use of sodium nitroprusside therapy has been successfully performed in a patient with HbH disease.144 Perioperative considerations and concerns for children with thalassemia, especially for those with thalassemia major, are listed in Table 9-10.
TABLE 9-10 Perioperative Concerns for Patients with Thalassemia
Transfusion crossmatch if appropriate (antibody-matched, leukocyte-reduced source for frequently transfused children)
Evaluation for endocrine dysfunction (e.g., diabetes mellitus, hypopituitarism)
Cardiac function, including echocardiogram (when appropriate)
Hepatic function, awareness of risk of cirrhosis and iron or virus-induced damage
Presplenectomy antibiotics and immunizations (when appropriate)
Thrombocytopenia
Idiopathic Thrombocytopenic Purpura
ITP is the most common cause of acute-onset thrombocytopenia in the otherwise healthy child, and it commonly manifests in the operative setting. ITP has an estimated incidence of about 4 per 100,000 children, and it is usually a benign, self-limited disorder affecting children between the ages of 2 and 10 years.35 Primary ITP has no clear predisposing cause, but secondary ITP is triggered by a drug or medical disorder. Diagnosis is by exclusion, the differential list is extensive, and response to ITP-specific treatment usually solidifies the diagnosis.
Pathophysiology
ITP is characterized by antibody-mediated clearance by tissue macrophages, resulting in thrombocytopenia (platelet count less tan 100,000/mm3) and shortened platelet survival. Antibodies may also suppress megakaryocytes and platelet development. Platelet autoantibodies may exist alone or as part of immune complexes, and they usually are immunoglobulin G (IgG) in type. They often show specificity for platelet membrane glycoproteins IIb-IIIa and Ib-IX.145 Thrombocytopenia develops when the reticuloendothelial system, typically the spleen, destroys the antibody-covered platelets.
Clinical and Laboratory Features and Treatment
Typically, ITP in children is a benign process occurring after a viral illness or immunization that manifests as petechiae of mucosal surfaces or purpura over bony prominences, thrombocytopenia, and a normal to increased mean platelet volume with increased megakaryocytes in the marrow. This process resolves within weeks or months regardless of therapy. ITP is classified as newly diagnosed (less than 3 months), persistent (3 to 12 months), and chronic (more than 12 months).146
Although platelet function in children with ITP is usually increased, treatment is often initiated only when the counts are less than 10,000 to 20,000/mm3.145 Observation with avoidance of activity that may lead to head trauma is an increasingly accepted treatment plan. Medical treatment most commonly consists of agents that decrease monocyte/macrophage-mediated destruction of antibody-coated platelets (e.g., steroids, intravenous immunoglobulin, anti-D immunoglobulins, vinca alkaloids). Agents that decrease antibody production (e.g., cyclophosphamide, anti-CD20 antibody) and investigational agents that stimulate the thrombopoietin receptor are reserved for those who demonstrate an inadequate response to initial therapy.147 Platelet transfusions are recommended only for life-threatening emergencies. Splenectomy removes a major site of platelet destruction and is recommended as an option only in chronic, symptomatic ITP or acute, life-threatening ITP unresponsive to medical treatment.148,149 This procedure, which is commonly performed noninvasively, has a success rate of about 75%.150–152
Perioperative Considerations
In view of the clinical and laboratory features of ITP, the anesthesiologist providing care for the child with ITP who is undergoing splenectomy or incidental surgery should consider the concerns listed in Table 9-11. A hematologist should be consulted to assess the need for medical therapy, including platelet transfusion, before surgery.
TABLE 9-11 Perioperative Concerns for Patients with Idiopathic Thrombocytopenia Purpura
Appropriate antibiotic coverage
Stress corticosteroid coverage
Medical therapy and platelet transfusion as above (platelets ideally administered after clamping of the splenic artery during splenectomy)
Judicious use of regional anesthesia, intramuscular medications, nasogastric tubes, nasal intubation, and other methods
Limited use of medications with potential bleeding risk (e.g., ketorolac)
Attention to physiologic effects of laparoscopy on circulatory and respiratory function
Coagulation Disorders
Screening
The clinical history of the child and family is the most essential screening tool. The family history should identify family members who have been labeled as bleeders, who have required blood transfusion unexpectedly during surgery, or who returned to surgery for unexpected postoperative bleeding. A history of maternal menorrhagia may also be significant. Suggestive signs and symptoms in a child’s medical history are easy bruising, mucosal bleeding, and in older girls, menorrhagia. Although diagnosing easy bruising is subjective, the clinician should suspect bleeding tendencies if skin bruising occurs in nontraumatized sites (e.g., trunk) or is unusually large without evidence of previous trauma. Mucosal bleeding includes epistaxis and gingival bleeding. Occasional nosebleeds can be common in children, but their clinical significance is enhanced by increased frequency, duration, bilaterality, and coexistence with abnormal bleeding from other sites. Gingival bleeding is common after tooth brushing or flossing, but its clinical significance is enhanced by spontaneous occurrence or chronicity, especially in the presence of good dental hygiene.153 A history of prolonged or excessive bleeding is important when associated with umbilical dehiscence, dental work (especially extractions), and circumcision. Although mouth injuries can produce impressive blood loss acutely in any individual, recurrent or persistent bleeding from such an injury may indicate an underlying disorder.
Von Willebrand Disease
von Willebrand disease (vWD) is considered to be one of the most common bleeding disorders, although studies suggest that the prevalence may be as low as 1 case per 10,000 people.154–157 Initially named pseudohemophilia because of an inheritance pattern that is different from that of hemophilia, vWD is the result of an abnormal amount, structure, or function of the vWF.154
Pathophysiology and Classification
Classification of vWD is essential for understanding and management of this disorder. The current classification was developed by a subcommittee on vWD through the International Society on Thrombosis and Haemostasis.155,156 The two general types are categorized as quantitative abnormalities (types 1 and 3) or qualitative abnormalities (type 2, including subtypes A, B, M, and N). All types are inherited in an autosomal dominant pattern, except types 2N and 3, which are autosomal recessive.
Because vWD is heterogeneous, clinical definitions have been proposed using categories such as mild, moderate, and severe, categories that are based on bleeding history (i.e., number of bleeding episodes) and laboratory measurement of factor concentration and activity.157 As the molecular basis of vWD becomes better understood, classification of this disease likely will change to reflect the new data. For example, Rodeghiero and colleagues proposed a practical approach to diagnosing and categorizing patients with vWD to provide optimal management.157 Their approach includes a standardized bleeding history score, focused laboratory analysis, and a trial infusion of DDAVP in certain subtypes. Categorization of patients with vWD is important for determining how they are managed in the perioperative setting.158
Clinical and Laboratory Features and Treatment
Children with vWD have traditionally been described as having prolonged bleeding times and aPTTs, but those with mild disease often have normal values. The aPTT is prolonged only if factor VIII activity is at or below a concentration that is determined by the sensitivity of the particular assay at an institution (often below 30% to 35%). The platelet functional assay (PFA-100) has better sensitivity and specificity (both near 90%) for diagnosis of the disease.159,160 The PFA-100 test, which measures closure time of an aperture on a membrane coated with collagen and adenosine diphosphate (ADP) or epinephrine, depends on vWF activity and platelet function. Because this test has some variability, its interpretation should be used in conjunction with results of other tests.161 Platelet count is typically normal in all types of vWD except type 2B.
Other laboratory tests used to delineate vWD include vWF antigen (vWF : A), which is a measure of the total level of vWF; vWF activity, often measured as a ristocetin cofactor activity (vWF : R), which is a measure of vWF binding to platelets through GPIb receptors; factor VIII coagulant activity; and vWF multimer analysis. Certain disease states have been associated with “acquired vWD” and include lymphoproliferative disorders or gammopathies (marked by antibodies to vWF), chronic renal failure, hypothyroidism, Wilms tumor, and certain congenital heart diseases, such as aortic stenosis (characterized by proteolysis of vWF multimers).159
In consultation with a hematologist, determination of appropriate treatment based on specific diagnosis and response to therapy is necessary before surgery. Treatment focuses on increasing concentrations of endogenous vWF with administration of DDAVP when possible or on replacement of factors with factor concentrates.162 DDAVP is usually effective in type 1 but less so in types 2A and 2M. DDAVP may have little or even undesired effects in some children: it may increase abnormal vWF in types 2A, 2M, and 2N; it can exacerbate thrombocytopenia in type 2B; and its repeated administration may lead to tachyphylaxis. DDAVP usually is not administered to very young children because of the risk of free water retention, hyponatremia, and central nervous system pathology, including seizures. Similarly, intravenous fluids may need to be limited after its administration to any child. Factor concentrates (including factor VIII and vWF [Humate-P or Alphanate]) typically are required for types 2B, 2N, and 3. Cryoprecipitate may be used when vWF-containing concentrates are unavailable, but it is not recommended as first-line therapy because it is not virus free, and vWF in solvent or heat-treated cryoprecipitate may be abnormal.153 Because of the complexity of response to therapies in this disease and the ever-changing availability of replacement products, determination of appropriate treatment in a particular child before surgery is crucial.158,163
Perioperative Concerns
The major preoperative concerns in children with confirmed vWD are directed toward appropriate preoperative treatment, avoidance of medications that may interfere with coagulation, and anticipation of intraoperative and postoperative bleeding (Table 9-12).162,164 All concerns should be addressed in consultation with a hematologist. Although regional anesthesia for these children usually is contraindicated, there are reports of its use without complications.165
TABLE 9-12 Perioperative Considerations for Patients with von Willebrand Disease
Consultation with hematologist: establish correct diagnosis and response to desmopressin (DDAVP); administer DDAVP or viral attenuated factor concentrates containing factor VIII and von Willebrand factor (vWF) such as Humate-P for severe vWD or for those types not responsive to DDAVP161
Determination of actual and desired factor concentrations and expected duration of postoperative therapy162
Discontinuation of any platelet-inhibiting medication (e.g., aspirin)
Judicious use of regional anesthesia, intramuscular medications, nasogastric tubes, nasal intubation, and other procedures that may cause bleeding
Limited use of medications with potential bleeding risk (e.g., ketorolac)
Coagulation profiles, including platelet counts for more invasive surgeries
Treatment of bleeding with appropriate blood products
Consider use of antifibrinolytic agents (i.e., ε-aminocaproic acid, tranexamic acid)163
Possible use of recombinant factor VIIa for severe bleeding episodes in severe vWD type 3 or patients with inhibitors
Hemophilia
Hemophilia is a group of congenital bleeding disorders caused by deficiency in factor VIII (i.e., hemophilia A, or classic hemophilia), factor IX (i.e., hemophilia B, or Christmas disease), or factor XI (i.e., hemophilia C). During the 10-year period from 1982 through 1991, the incidence of the more common varieties, hemophilia A and B, was 1 case per 5032 live male births in the United States within a six-state surveillance area; the prevalence of hemophilia A was 10.5 cases per 100,000 male births, and that of hemophilia B was 2.9 cases per 100,000 male births.166
Because of X-linked recessive inheritance of hemophilia A and B, family history is very important in establishing the diagnosis. Although boys are usually affected, girls may rarely inherit the disorder if their fathers are affected and their mothers are carriers or in instances of extreme lyonization (inactivation of an X chromosome). The daughter of an affected father is an obligate carrier with a 50% chance of passing it on to any of her sons. De novo mutations are relatively common and suspected in male patients lacking a family history.159 Hemophilia C is a mild form of hemophilia, affecting primarily Ashkenazi Jews. It is distinguished from the other two forms of hemophilia by an autosomal recessive inheritance pattern (linked through chromosome 4), lack of joint bleeding, and infrequent need for treatment. Affected female patients may notice heavy menses, and affected male patients may have frequent nosebleeds and occasionally have excessive bleeding during surgery.
Clinical and Laboratory Features and Treatment
The wide range of clinical features is similar for hemophilia A and B (Table 9-13). The severity of bleeding in these children directly relates to the degree of their deficiency.159 Children with mild or moderate hemophilia may bleed excessively only after a hemostatic challenge such as trauma or surgery, whereas children with severe hemophilia may bleed spontaneously (e.g., hemarthroses).167 Female carriers on average have 50% of normal factor concentrations and usually are asymptomatic, although they occasionally present with a clinical picture similar to that of mild cases of hemophilia.159
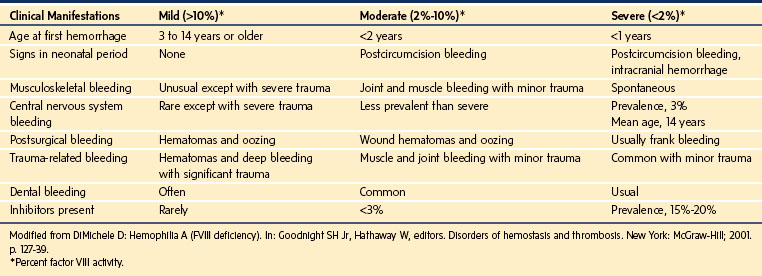
Results of a general coagulation screen are typically normal except for the aPTT, which is prolonged in proportion to the concentration of factors in the blood. The diagnosis is confirmed by measuring the specific factor concentrations.168 If hemophilia is suspected but there is no family history, testing for vWD is prudent, especially for the types that may mimic hemophilia (i.e., types 2N and 3). Because of the variable sensitivity of the aPTT to specific factor deficiencies, the ability of this test to detect carriers varies between laboratories; diagnosis of a carrier state usually requires specific factor assays.
Hemophilias A and B are treated by replacing the deficient factor concentrations. These factor concentrations should be maintained at specified levels to prevent sequelae (Table 9-14). Exposure to plasma products should be minimized. The duration of treatment should be tailored to the severity of disease. DDAVP may be effective in selected mild cases by increasing factor VIII concentrations through the release of endogenous stores. Because tachyphylaxis limits the prolonged use of DDAVP, it is typically recommended only for minor operations.167 For most cases, especially for those who require increased factor concentrations to be maintained for effective hemostasis, factor concentrates should be used. Although recombinant forms are preferable because they do not carry infectious risk, substitution with plasma-derived forms may be needed when supplies are limited.159,167 For the rare patient with hemophilia C who has excessive surgical bleeding, treatment with recombinant factor XI or FFP may be required. It is essential to consult with a hematologist to determine a customized factor treatment plan for every child with hemophilia.
| Bleeding Site | Target Concentrations for Factor VIII or IX (%) | Duration of Treatment (days)* |
|---|---|---|
| Muscle | 30-50 | 1-2 |
| Joint | 50-80 | 1-2 |
| Gastrointestinal tract | 40-60 | 10-14 |
| Oral mucosa | 30-50 | 2-3 |
| Epistaxis | 30-50 | 2-3 |
| Hematuria | 30-100 | 1-2 |
| Retroperitoneal | 80-100 | 7-10 |
| Central nervous system | 80-100 | 14 |
| Trauma or surgery | 80-100 | 14 |
*May be reduced depending on clinical circumstances and severity of disease.
Modified from Brown DL. Congenital bleeding disorders. Curr Probl Pediatr Adolesc Health Care 2005;35:38-62.
Children who have developed inhibitors to factor concentrates pose a challenge in the perioperative period. Until recently, they were denied surgery unless it was absolutely necessary, at which point they were often managed with increased concentrations of factors or a desensitization regimen. However, a review that included two randomized, controlled trials and data from the Hemophilia Research Society and the Hemophilia and Thrombosis Research Society reported effectiveness of recombinant factor VIIa (rFVIIa) for most of these patients.169 Among the randomized trials, one study compared two bolus-dosing regimens (35 versus 90 µg/kg), and the other study compared bolus dosing with continuous infusion; the greater dosing regimen was more effective for major operations. Combining data from these studies and the two Research Society databases, the overall effectiveness rate of rFVIIa in controlling bleeding approached 84%, with a low thrombotic rate of less than 1%. Such evidence is providing more options for these difficult-to-manage patients with inhibitors. Alternatively, partially activated prothrombin complex concentrates, such as factor VIII inhibitor bypass activity (FEIBA, Baxter Healthcare Corp., Westlake Village, Calif.), have been effective for patients with inhibitors undergoing surgery.170
Perioperative Concerns
The perioperative concerns in hemophilia focus on prevention and treatment of bleeding, similar to the management of patients with vWD (Table 9-15). Many consider regional anesthesia to be contraindicated for patients with hemophilia, but there are reports of its use without complications as long as factor concentrations are maintained.171
TABLE 9-15 Perioperative Concerns for Patients with Hemophilia
Consultation with hematologist, establishment of correct diagnosis
Determination and testing of treatment plan, including use of desmopressin or factors (concentrates or recombinant)
Consideration of multiple procedures performed together to reduce factor exposure
Discontinuation of any platelet-inhibiting medication (e.g., aspirin)
Judicious use of regional anesthesia, intramuscular medications, nasogastric tubes, nasal intubation, and other procedures that may cause bleeding
Limited use of medications with potential bleeding risk (e.g., ketorolac)
Follow coagulation profiles, especially factor levels (factors VIII and IX)
Anticipate and treat bleeding with appropriate blood products
Consider recombinant activated factor VII (rFVIIa) for severe bleeding
Hypercoagulability
A hypercoagulable state is a condition in which the development of thrombus is favored (i.e., thrombophilia). The condition results in an increased risk for abnormal clot formation and venous thromboembolic events (VTEs), which often are the presenting symptoms at the time of diagnosis. Thrombophilia can be acquired or congenital. The incidence of VTE among children is less than it is among adults, even in those with known congenital thrombophilic conditions,172 although neonates and adolescents are at relatively high risk in the pediatric population.173 Congenital thrombophilic conditions include factor V Leiden disorder, prothrombin gene mutation, protein C and S deficiencies, and antithrombin III deficiency.174 Risk factors for acquired thrombophilia include the presence of a central venous catheter, infection, malignancy, surgery, or trauma.173
Screening of nonoperative children with suspected hypercoagulability is controversial and not recommended for those who are asymptomatic, even those with a positive family history.172 Evidenced-based guidelines for children are lacking with regard to screening and prophylactic treatment of those with suspected hypercoagulability. Current evidence suggests pharmacologic prophylaxis in the nonoperative setting is recommended only in children on long-term home total parenteral nutrition (TPN) and those with specific complex cardiac lesions (e.g., Fontan patients).175 However, children who present for surgery with a strong family history of thromboses may benefit from screening and referral to a hematologist for management and consideration of pharmacologic prophylaxis, such as enoxaparin administered postoperatively. The use of nonpharmacologic prophylaxis, including early mobilization after surgery, adequate hydration, and compression stockings, is left to the discretion of individual providers and to institutional practice based on the child’s medical history, family history, and risk factors for VTE.
Cancer and Hematopoietic Stem Cell Transplantation
Cancer
Cancer is the second and fourth most common cause of death in children younger than 15 and 20 years of age, respectively.176,177 The most common malignancies affecting children are different from those affecting adults, and they include leukemia, brain tumors, lymphomas, and solid tumors such as sarcomas of soft tissue and bone. Embryonal tumors (e.g., neuroblastoma, Wilms tumor, retinoblastoma, medulloblastoma) are unique to early childhood. Survival rates for most pediatric cancers have improved significantly in the past several decades; more than 80% of children diagnosed with a childhood malignancy will become 5-year survivors of their cancers.178,179 The great improvements in survival for many malignancies of childhood are directly related to advances in diagnostic modalities and the large percentage of children treated on cooperative clinical trial protocols. Treatment follows these protocols and may include chemotherapy, radiation therapy, biologic modifiers, and HSCT.
Clinical and Laboratory Features and Treatment
The direct effects of tumor, even at the time of diagnosis, may contribute to morbidity and mortality. For example, a childhood tumor may manifest with increased intracranial pressure, pleural or pericardial effusion, or compression of abdominal organs. Lymphoma often manifests with symptoms associated with an anterior mediastinal mass, including respiratory distress and cardiorespiratory collapse under anesthesia.180,181 Most children with Hodgkin disease or non-Hodgkin lymphoma have mediastinal involvement at diagnosis, and almost one half have respiratory symptoms at presentation (see Chapter 13).182,183
Myelosuppression, which manifests with various degrees of anemia, thrombocytopenia, and neutropenia, is a common direct effect of cancer in children. Anemia is common at the time of diagnosis of many pediatric cancers, including 50% to 75% of children with newly diagnosed neuroblastoma, rhabdomyosarcoma, Hodgkin disease, Ewing sarcoma, and osteosarcoma184 and 80% of children with acute lymphoblastic leukemia (ALL).185 Thrombocytopenia is typically identified during the diagnosis of children with acute leukemia and is common with tumors causing bone marrow infiltration.186,187 Neutropenia is usual in children with ALL. Hyperleukocytosis is predominant in children with acute myelogenous leukemia (AML), 20% of whom present with a white blood cell count greater than 100,000/mm3.185,188 This high concentration of leukemic blasts, especially when the count is greater than 200,000/mm3, may lead to intravascular clumping and the potentially fatal condition of leukostasis.189 Myelosuppression may be a direct effect of tumor cells and marrow infiltration by tumor cells and may result from radiation therapy and chemotherapy. As a consequence of myelosuppression, children with cancer have a frequent need for transfusion of blood products and are at risk for procedure-acquired and line-related infections and for delayed wound healing after surgical procedures. Frequent hospitalizations and immune compromise predispose them to colonization and infection by nosocomial, community-acquired, antibiotic-resistant organisms and opportunistic infections. Neutropenia predisposes them to specific infections such as perirectal abscesses and typhlitis.
An uncommon but potentially fatal effect of the tumor itself is tumor lysis syndrome (TLS). TLS is most common with certain hematologic malignancies, especially ALL and Burkitt lymphoma, but it also occurs with other malignancies characterized by a high proliferative rate, large tumor burden, or high sensitivity to cytotoxic therapy. TLS is characterized by rapid and massive destruction of tumor cells and resultant massive release of phosphorus, potassium, nucleic acids, and proteins that is sufficient to cause metabolic derangements and possible renal failure and death.190,191 TLS can occur spontaneously, especially in children with AML, but it more often manifests in the setting of cytotoxic therapy, radiation therapy, fever, surgery, and anesthesia.192–197
Chemotherapy used in pediatric oncology leads to myelosuppression and to a host of important side effects that may be clinically important in the perioperative setting (Table 9-16). These effects of chemotherapy can be synergistic with toxicities of radiation therapy.198 New chemotherapeutic agents are continuously being introduced, and at academic centers, the use of investigational agents is common. Although it is impossible to provide a comprehensive review of chemotherapeutics, the most common agents that cause toxicities of particular importance to the delivery of anesthesia are briefly mentioned here, and a complete list of toxicities can be found in E-Table 9-1.
| Drug | Side Effects |
|---|---|
| l-Asparaginase | Hyperglycemia, hypersensitivity, hepatic dysfunction (secondary hypoalbuminemia and coagulopathies), pancreatitis, thrombosis, stroke |
| Bischloroethyl nitrosourea (BCNU) | Encephalopathy, hepatotoxicity, pulmonary toxicity |
| Bleomycin | Anaphylactoid reactions, fever, hyperpigmentation, nausea, vomiting, pulmonary fibrosis |
| Busulfan | Encephalopathy, hepatotoxicity, pulmonary toxicity |
| Carboplatin | Myelosuppression, nausea, vomiting, nephrotoxicity, neurotoxicity, ototoxicity |
| Cisplatin | Nausea/vomiting, nephrotoxicity, ototoxicity, peripheral neuropathy |
| Corticosteroids | Adrenal suppression, avascular necrosis, cataracts, edema, gastritis, hyperglycemia, hypertension, myopathy, osteoporosis, obesity, osteopenia, psychosis |
| Cyclophosphamide (Cytoxan) | Cardiotoxicity, hemorrhagic cystitis, myelosuppression, nausea, vomiting, syndrome of inappropriate secretion of antidiuretic hormone (SIADH) |
| Cyclosporine | Cortical blindness, electrolyte disturbances, encephalopathy, gingival hyperplasia, hemolytic uremia, hepatotoxicity, hyperlipidemia, hypertension, hirsutism, myositis, paresthesias, tremor |
| Cytarabine | Myelosuppression, mucositis, hepatitis, nausea/vomiting, neurotoxicity |
| Dactinomycin (Actinomycin D) | Nausea/vomiting, mucositis, myelosuppression, radiation recall |
| Daunorubicin (Daunomycin) Doxorubicin (Adriamycin) Idarubicin (Idamycin) |
Cardiomyopathy, mucositis, myelosuppression, red-orange urine |
| Etoposide | Hypotension, mucositis, myelosuppression, nausea, vomiting |
| Ifosfamide | Hemorrhagic cystitis, myelosuppression, nephrotoxicity, neurotoxicity |
| Melphalan | Mucositis |
| Methotrexate | Hepatotoxicity, mucositis, myelosuppression, renal failure, neurotoxicity |
| Mercaptopurine (6-MP) | Hepatotoxicity, myelosuppression |
| Mycophenolate mofetil (Cellcept) | Electrolyte disturbance, gastrointestinal toxicity, hypercholesterolemia, myelosuppression, rash |
| Procarbazine | Myelosuppression |
| Sirolimus | Hyperlipidemia, myelosuppression |
| Tacrolimus (Prograf) | Anemia, anorexia, back pain, encephalopathy, diarrhea, hyperglycemia, nephrotoxicity, pleural effusion, rash |
| Thiotepa | Neurotoxicity, mucositis |
| Thioguanine (6-TG) | Hepatotoxicity, myelosuppression |
| Total body irradiation | Dental/bony maldevelopment, gastrointestinal toxicity, hepatotoxicity, pulmonary toxicity |
| Vinblastine (Velban) | Myelosuppression, neurotoxicity, SIADH |
| Vincristine (Oncovin) | Neurotoxicity, SIADH |
Modified from Carpenter PA, Mielcarek M, Woolfrey AE. Hematopoietic cell transplantation. In: Irwin S, Rippe JM, editors. Intensive care medicine. 6th ed. Philadelphia: Lippincott Williams & Wilkins; 2008. p. 2150-68.

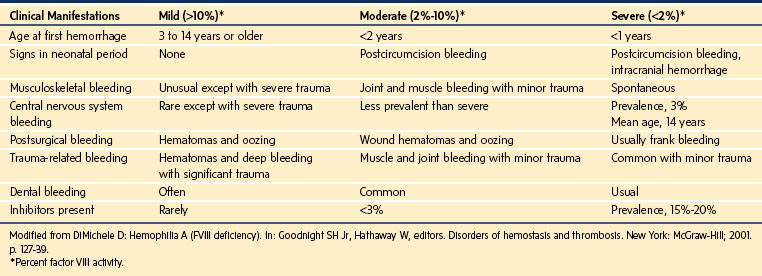
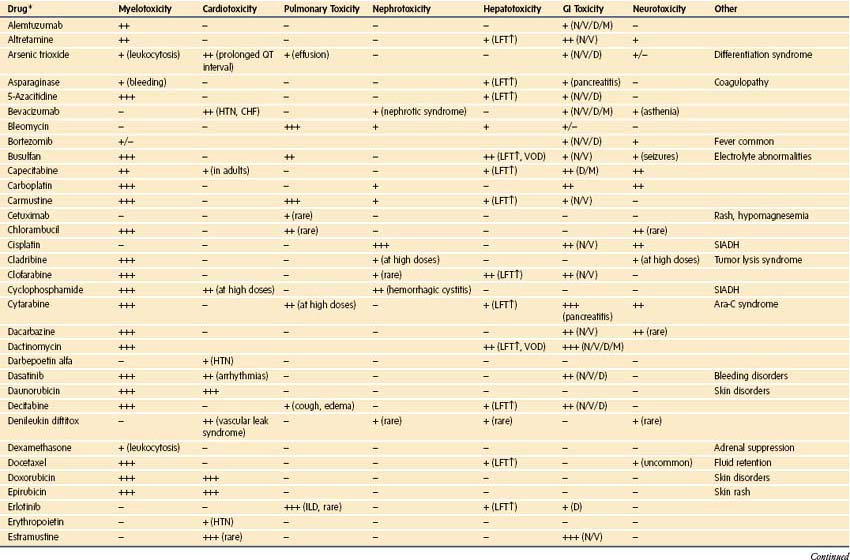
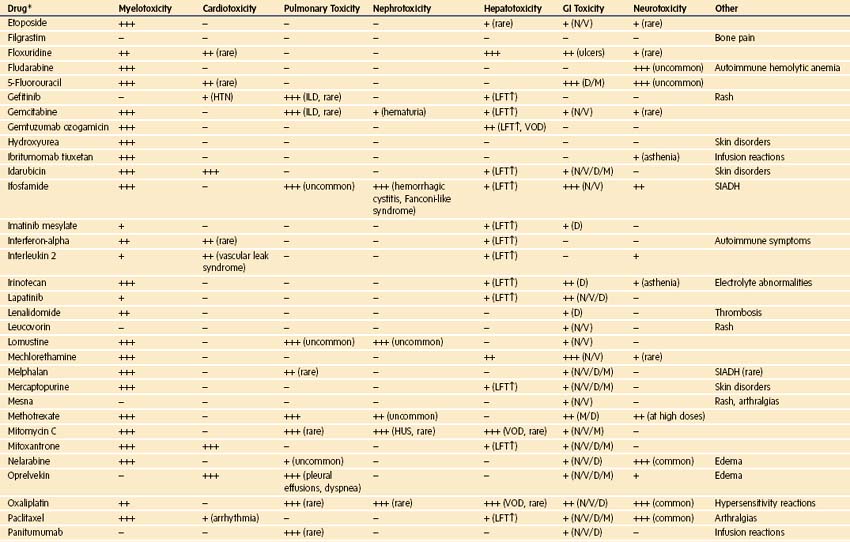
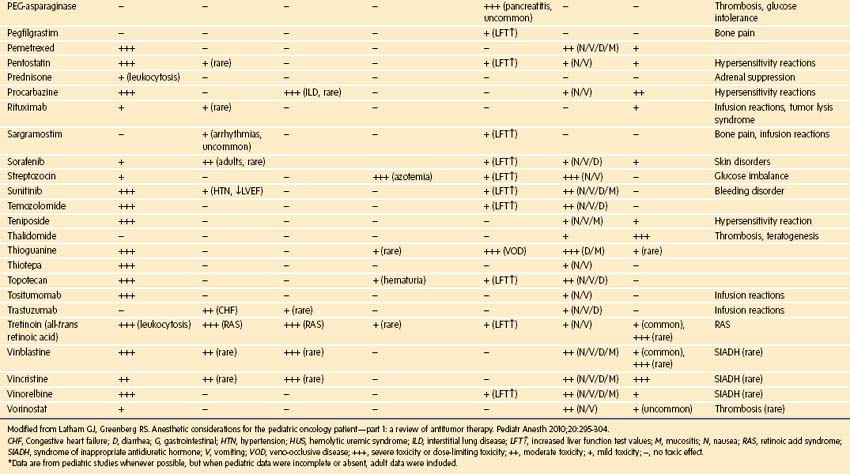
Corticosteroids are frequently used and may cause adrenal suppression, hypertension, thromboembolism, and obesity; the latter potentially renders venous access and tracheal intubation difficult. Children who have received corticosteroid therapy have a period of measurable glucocorticoid-induced adrenal suppression of 2 weeks to 8 months after such therapy.199–205 Because of this variability, administering stress-dose steroids in the first 1 to 2 months after cessation of glucocorticoids is reasonable.199 However, any child at risk of TLS should not receive corticosteroids without direct discussion with an oncologist.
The anthracycline drugs (e.g., doxorubicin, daunorubicin, idarubicin, and epirubicin) and mitoxantrone cause cardiomyopathy in a dose-dependent fashion, which is aggravated by radiation delivered to the precordial area. About 10% of children who received a cumulative dose of 300 mg/m2 or more of anthracycline develop clinically significant cardiomyopathy.206 Later studies demonstrated clinically significant cardiomyopathy after much smaller doses207,208 and an increased sensitivity in children with trisomy 21 and leukemia.209 Children receiving anthracyclines should be evaluated with echocardiography at baseline and intermittently during the course of therapy and for a prolonged period after therapy because anthracycline-induced cardiac failure can take years to fully declare itself.210,211 Physical examination may miss more than 50% of children with heart failure.212 A recent echocardiogram should be available preoperatively if a child has received anthracycline therapy and any of the following conditions are met: cumulative dose greater than 240 mg/m2, any dose received during infancy, or chest irradiation dose greater than 30 Gy with concomitant anthracycline treatment.213,214 Although uncommon, radionuclide angiocardiography may be chosen for those with poor echocardiographic windows.210,215
Several other chemotherapeutic agents are of interest to the anesthesiologist caring for a child with cancer. l-Asparaginase is associated with a 1% to 2% risk of hemorrhage or thrombosis due to deficiencies in fibrinogen, plasminogen, antithrombin III, and vWF,216–218 as well as hepatic dysfunction and acute hemorrhagic pancreatitis. Bleomycin can produce acute pneumonitis and progression to pulmonary fibrosis. Although the incidence and risk in children are not known, reports suggest that 46% of adults treated with cumulative doses greater than 400 units/m2 develop bleomycin-induced pneumonitis and 3% of these die.219–221 Studies suggest that high concentrations of inspired oxygen during or soon after bleomycin treatment may promote pulmonary toxicity and lead to postoperative respiratory distress.222–224 A medical history and physical examination are sufficient to determine whether pulmonary function testing is required preoperatively.225–227 Cisplatin and ifosfamide may cause renal tubular damage that can lead to Fanconi syndrome with electrolyte wasting. Methotrexate may cause renal failure at large doses (more than 1 mg/m2). Vinblastine and vincristine may cause peripheral neuropathies and seizures. Increasingly, biologic mediators such as retinoids are being used to modify tumor biology, and recombinant antibodies are used to directly attack tumors and modulate the immune response to tumors. The use of investigational agents and novel chemotherapeutics amplifies the importance of reviewing cases with the oncologist.
Toxicity from the effects of radiation therapy to healthy tissues is unavoidable, and the developing tissues of children are particularly susceptible to the acute and late effects of irradiation. The susceptibility of normal tissues depends on the total and fractional dose received, the sensitivity of the tissue to the dose of radiation, the volume of tissue irradiated, and time course of treatment (Table 9-17). Concurrent chemotherapy potentiates toxicity.
TABLE 9-17 Late Effects of Radiation Therapy
| Radiation Field | Late Effects | Risk Factors |
|---|---|---|
| Cranial | Neurocognitive deficits | >18 Gy, IV/IT methotrexate |
| Leukoencephalopathy | >18 Gy with IT methotrexate | |
| Growth hormone deficiency | >18 Gy | |
| Panhypopituitarism | >40 Gy | |
| Large vessel stroke | >60 Gy | |
| Second cancers | Variable | |
| Dental problems | >10 Gy | |
| Cataracts | >2-8 Gy single dose, 10-15 Gy fractionated dose | |
| Ototoxicity | >35-50 Gy | |
| Chest | Cardiac disease | |
| Coronary artery disease | >30 Gy | |
| Cardiomyopathy | >35 Gy, >25 Gy with anthracyclines | |
| Valvular disease | >40 Gy | |
| Pericardial disease | >35 Gy | |
| Arrhythmias | Unknown | |
| Thyroid disease | ||
| Hypothyroidism | >20 Gy local, >7.5 Gy TBI | |
| Hyperthyroidism | >20 Gy local, >7.5 Gy TBI | |
| Thyroid nodules, cancer | Any dose | |
| Pulmonary disease | ||
| Pulmonary fibrosis | >15-20 Gy | |
| Restrictive lung disease | Unknown | |
| Obstructive lung disease | Unknown | |
| Abdomen/pelvis | Chronic enteritis | >40 Gy |
| Gastrointestinal malignancy | Unknown | |
| Hepatic fibrosis/cirrhosis | >30 Gy | |
| Renal insufficiency | >20 Gy | |
| Bladder disease | ||
| Fibrosis | >30 Gy prepubertal, >50 postpubertal | |
| Hemorrhagic cystitis | Enhances cyclophosphamide and ifosfamide effect | |
| Bladder cancer | Unknown | |
| Gonadal dysfunction | ||
| Ovarian failure | 4-12 Gy | |
| Testicular failure | >1-6 Gy | |
| Any radiation | Skin cancer | |
| Musculoskeletal changes | ||
| Bone length discrepancy | >20 Gy | |
| Pathologic fractures | >40 Gy | |
| TBI | All the above |
Gy, Gray; IT, intrathecal; IV, intravenous; TBI, total body irradiation.
From Latham GJ, Greenberg RS. Anesthetic considerations for the pediatric oncology patient—part 1: a review of antitumor therapy. Pediatr Anesth 2010;20:295-304.
Almost every organ system is affected by cancer and its treatment. Nausea and vomiting are common. Nutrition is often compromised, along with skin integrity and bone and tooth mineralization. Mucositis can lead to difficult intubation because of pseudomembrane formation, supraglottic edema, and bleeding from the friable oral mucosa.228,229 Acute and chronic pain often require complex treatment regimens that may include opioids, nonsteroidal antiinflammatory agents, antidepressants, anxiolytics, and a wide range of complementary and alternative modalities.
Perioperative Considerations
A retrospective review of 177 children undergoing 3833 radiotherapy sessions with anesthesia reported a complication rate of 1.3%, which is comparable to that for children without cancer anesthetized with propofol.230 However, it is clear that many children with cancer who present to the operating room are gravely ill and susceptible to relatively small changes to their physiology.
All of the described considerations regarding the possible effects of pediatric cancer and its treatment may influence our strategies in the perioperative period. Children with cancer undergo a host of procedures that require anesthesia during acute and chronic phases of disease. Included among them are diagnostic tumor or lymph node biopsy; tumor resection; placement of peripheral and central venous access for treatment and nutrition; diagnostic and monitoring procedures (e.g., lumbar puncture, bone marrow aspirate and biopsy, lung and liver biopsy, skin biopsy, bronchoalveolar lavage, esophagogastroduodenoscopy); radiologic procedures (e.g., magnetic resonance imaging, computed tomography, nuclear scans, positron emission tomography); radiation therapy; and placement of pain management devices (e.g., indwelling epidural catheters). Splenectomy is performed as part of staging or management of some pediatric malignancies, and the procedure may be associated with increased risk of postoperative infection, postoperative thrombocytosis, and thrombocytopenia.231
Surgical intervention may be required for acute and potentially life-threatening emergencies in children with cancer, and the effects of specific tumors can complicate anesthesia care. Wilms tumor may be accompanied by an acquired vWD condition, and anterior mediastinal masses may produce superior vena cava obstruction, pulmonary artery compression, and tracheal obstruction.181,182 Neuroblastoma may be accompanied by pheochromocytoma-like signs and symptoms (3% of cases),232 and in advanced stages, it may cause massive hepatic enlargement. Spinal tumors may cause acute spinal cord compression, and tumor or hemorrhage in the brain may cause acute intracranial hypertension. In view of the complexity of pediatric oncologic disease, there are many considerations for the perioperative period,233 as detailed in Table 9-18.
TABLE 9-18 Perioperative Concerns in Pediatric Cancer and Hematopoietic Stem Cell Transplantation
Consultation with service primarily responsible for child’s care
Echocardiogram or chest radiograph, or both (when appropriate)
Transfusion crossmatch with specifications (e.g., cytomegalovirus seronegativity, leukocyte reduction, irradiation) determined in consultation with oncology service
Determination of blood typing requirements for all stem cell transplant recipients
History of acute and chronic pain medication use
Anxiolytic and analgesic therapy (when appropriate)
Infection prophylaxis with antibiotics
Avoidance of any marrow-suppressive medications in stem cell transplant patients
Observation of indicated isolation precautions
Attention to skin, teeth, eyes, and joints; careful positioning and padding
Sterile technique with central line access
Full-stomach precautions (when appropriate, as in graft-versus-host disease)
Appropriate hydration and maintenance of urine output
Continuation of total parenteral nutrition (and other parenteral fluids with high glucose concentration)
Avoidance of high fraction of inspired oxygen (Fio2) and restriction of hydration if prior treatment with bleomycin
Judicious use of cardiac depressants in patients with compromised cardiac function
Nausea and vomiting prophylaxis
Hematopoietic Stem Cell Transplantation
The first attempts to use bone marrow clinically to treat malignancy took place about 50 years ago.234 HSCT is a potentially curative treatment for a wide range of malignant and nonmalignant pediatric disorders. Throughout the process of HSCT, children are likely to undergo procedures that require anesthesia.
HSCT is used in the treatment of leukemia, lymphoma, solid tumors (e.g., neuroblastoma), myelodysplasia, aplastic anemia, hemoglobinopathies (including sickle cell disease and thalassemia), and congenital immune and metabolism deficiencies.235,236 Hematopoietic stem cells used in the transplantation can be obtained from bone marrow, “mobilized” peripheral blood, or umbilical cord blood. The source of the cells may be the child (autologous), an identical twin (syngeneic), or another individual (allogeneic). Allogeneic donor cells are commonly derived from HLA-identical siblings (available in about 30% of patients), but advances in matching and supportive care have improved outcomes with HLA-matched unrelated and mismatched donors.237,238 Accumulative toxicity in HSCT derives from the underlying illness and complications of past therapy, the transplant-conditioning regimen, complications resulting from long-term myelosuppression, and graft-versus-host disease and its treatment.
Clinical and Laboratory Features and Treatment
The process of HSCT involves several steps:
1. The preparative or conditioning regimen, during which high-dose chemotherapy with or without irradiation or immunomodulating agents eradicates malignancy (where present), clears marrow space for incoming stem cells, and suppresses the recipient’s immune system
2. Transplantation through infusion of hematopoietic stem cells
3. Transplant engraftment (more than 30 days after transplantation)
4. Early engraftment (30 to 100 days after transplantation)
5. Late engraftment (more than 100 days after transplantation)
Side effects and toxicities of therapeutic modalities can affect the transplant recipient throughout the process (see Tables 9-16 and 9-17). The incidence of transplantation-related morbidity and mortality depends on the patient’s age, primary disease, comorbidities, and histocompatibility between donor and recipient. Complications contributing to morbidity and mortality are related to infection, regimen-related toxicity, and alloreactivity.239
Regimen-related toxicity can involve every organ of the body through the direct and indirect effects of irradiation and chemotherapy (see Tables 9-16 and 9-17 and E-Table 9-1). Mucositis is common and can increase the risk of aspiration and airway compromise.229,240 Sinusoidal obstruction syndrome (SOS), previously called veno-occlusive disease, occurs in 10% to 40% of children after HSCT. Mortality rates range from 19% to almost 50% due to hepatorenal failure and subsequent multiple-organ failure. SOS is characterized by hepatomegaly, jaundice, and fluid retention; its presence requires careful attention to fluid balance because sodium administration must be minimized, and coagulation abnormalities and refractoriness to platelet transfusions may occur.241–243 Other gastrointestinal complications include hemorrhage,244 infection, and opioid-induced abdominal pain and distention (i.e., narcotic bowel syndrome). Acute pulmonary complications occur in 30% to 60% of HSCT patients and include infection, hemorrhage, edema, bronchiolitis obliterans, acute respiratory distress syndrome (ARDS), and idiopathic pneumonia syndrome (IPS), a noninfectious inflammatory lung process.245–247
In the early phase after HSCT, significant cardiomyopathy and arrhythmias are uncommon (5%), but sepsis, cardiovascular collapse, and heart failure are common admitting diagnoses of the 10% to 40% of children who require intensive care after HSCT.247–250 Late cardiac complications depend on the dosage of radiation and cardiotoxic chemotherapeutics used during the conditioning regimen. Acute renal failure occurs in 30% to 50% of children251,252 and warrants judicious use of fluids in these patients253; hemorrhagic cystitis is also common.254 A process of thrombotic microangiopathy similar to hemolytic uremic syndrome may occur in one fourth of children receiving cyclosporine or the calcineurin inhibitor tacrolimus.255 Central nervous system complications include infection, hemorrhage, and encephalopathy and peripheral neuropathy due to metabolic and chemotherapeutic effects.256–260
Graft-versus-host disease (GVHD) is the clinical manifestation of the recognition of recipient alloantigens by donor T cells. Acute GVHD is a common event (incidence depends on the histocompatibility of the donor and recipient) before day 100 after transplantation, and it is characterized by inflammatory dermatitis, enteritis, and hepatitis. Chronic GVHD occurs in 6% to 50% of pediatric patients and is typically recognized 100 days or more after transplantation. This broad range of incidence depends on the age of donor and recipient, gender matching, and degree of matching between donor and recipient.261 Chronic GVHD has many features of autoimmune diseases (e.g., sclerodermatous changes, dry mouth and conjunctivae, esophagitis, pulmonary dysfunction including bronchiolitis obliterans, contractures of extremities and soft tissue, alopecia, thrombocytopenia). Opportunistic infections are common during chronic GVHD. Hemolysis is an additional manifestation of alloantigenicity, the result of major and minor blood group incompatibilities between donor and recipient.
In contrast to the myeloablative procedures described previously in the context of malignant disorders, nonablative transplants are increasingly being used, particularly in nonmalignant disorders. Use of these transplants is based on the observation that in some instances, minimal myelotoxicity in conjunction with profound and prolonged immunosuppression leads to successful donor engraftment.262 This modality is mostly used in children with nonmalignant hematologic conditions, marrow failure, and immunodeficiency syndromes.
Perioperative Considerations
Surgical and procedural interventions are common throughout the transplantation process and are similar to those described earlier for children with cancer. A surgical procedure that is an integral component of HSCT is harvesting of hematopoietic stem cells from the recipient or another individual. Harvesting of bone marrow in children usually requires general anesthesia, although the procedure can be performed under spinal anesthesia with equivalent safety.263 The donor is usually placed in a prone position to extract approximately 10 mL/kg (recipient weight) of marrow from the posterior iliac crests. When stem cells are obtained from peripheral blood, placement of vascular access in children usually requires sedation or general anesthesia. There is little evidence to support the avoidance of nitrous oxide for harvesting procedures, a concern raised in the past because nitrous oxide affects methionine synthase activity and DNA synthesis.264
In view of the wide range of possible complications, the many issues enumerated earlier for children with cancer must be considered for children undergoing HSCT (see Table 9-18). Radiation, glucocorticoids, mucositis, and chronic GVHD may contribute to an airway that is friable and a neck that is scarred, thereby increasing the risks of airway trauma, dental injury, and difficult tracheal intubation. Chemotherapy and GVHD may affect the skin and make venous access difficult. Chronic GVHD can lead to sclerodermatous changes, which can profoundly restrict range of motion, and sicca syndrome, which may necessitate use of artificial tears. GVHD may alter gut motility and delay gastric emptying. Immune compromise increases the risk of infection from vascular access lines and therefore mandates meticulous technique at all times. Chemotherapy may compromise cardiac and pulmonary function, alter hepatic metabolism of medications, and limit renal excretion of medications and fluids. Immune compromise and modifications resulting from transplantation require special processing of blood components, as recommended by hematology and blood bank consultants (see Tables 9-3, 9-4, and 9-5). Only irradiated, leukocyte-reduced, cytomegalovirus-negative (when the recipient is cytomegalovirus negative) blood components should be administered, and blood typing depends on engraftment of donor antigens. It is critical to coordinate the choice of blood products with the transplant service because often the blood type changes from that of recipient to that of donor.
Guzzetta NA, Miller BE. Principles of hemostasis in children: models and maturation. Paediatr Anaesth. 2011;21:3–9.
Key NS, Derebail VK. Sickle-cell trait: novel clinical significance. Hematology. 2010;2010:418–422.
Latham GJ, Greenberg RS. Anesthetic considerations for the pediatric oncology patient—part 1: a review of antitumor therapy. Paediatr Anaesth. 2010;20:295–304.
Latham GJ, Greenberg RS. Anesthetic considerations for the pediatric oncology patient—part 2: systems-based approach to anesthesia. Paediatr Anaesth. 2010;20:396–420.
Latham GJ, Greenberg RS. Anesthetic considerations for the pediatric oncology patient—part 3: pain, cognitive dysfunction, and preoperative evaluation. Paediatr Anaesth. 2010;20:479–489.
Morley SL. Red blood cell transfusions in acute paediatrics. Arch Dis Child Educ Pract Ed. 2009;94:65–73.
Vichinsky EP, Haberkern CM, Neumayr L, et al. A comparison of conservative and aggressive transfusion regimens in the perioperative management of sickle cell disease. The Preoperative Transfusion in Sickle Cell Disease Study Group. N Engl J Med. 1995;333:206–213.
1 Guzzetta NA, Miller BE. Principles of hemostasis in children: models and maturation. Pediatr Anesth. 2011;21:3–9.
2 Ankola P, Nardi M, Karpatkin M. Functional activity of protein C in newborn infants. A report of a study and a review of the literature. Am J Pediatr Hematol Oncol. 1992;14:140–143.
3 Kang J, Brodsky L, Danziger I, Volk M, Stanievich J. Coagulation profile as a predictor for post-tonsillectomy and adenoidectomy (T + A) hemorrhage. Int J Pediatr Otorhinolaryngol. 1994;28:157–165.
4 Mattix H, Singh AK. Is the bleeding time predictive of bleeding prior to a percutaneous renal biopsy? Curr Opin Nephrol Hypertens. 1999;8:715–718.
5 Boberg KM, Brosstad F, Egeland T, Egge T, Schrumpf E. Is a prolonged bleeding time associated with an increased risk of hemorrhage after liver biopsy? Thromb Haemost. 1999;81:378–381.
6 Rodgers RP, Levin J. A critical reappraisal of the bleeding time. Semin Thromb Hemost. 1990;16:1–20.
7 Peterson P, Hayes TE, Arkin CF, et al. The preoperative bleeding time test lacks clinical benefit: College of American Pathologists’ and American Society of Clinical Pathologists’ position article. Arch Surg. 1998;133:134–139.
8 Brenn BR, Theroux MC, Dabney KW, Miller F. Clotting parameters and thromboelastography in children with neuromuscular and idiopathic scoliosis undergoing posterior spinal fusion. Spine. 2004;29:E310–E314.
9 Goobie SM, Soriano SG, Zurakowski D, McGowan FX, Rockoff MA. Hemostatic changes in pediatric neurosurgical patients as evaluated by thrombelastograph. Anesth Analg. 2001;93:887–892.
10 Miller BE, Guzzetta NA, Tosone SR, Levy JH. Rapid evaluation of coagulopathies after cardiopulmonary bypass in children using modified thromboelastography. Anesth Analg. 2000;90:1324–1330.
11 Cariappa R, Wilhite TR, Parvin CA, Luchtman-Jones L. Comparison of PFA-100 and bleeding time testing in pediatric patients with suspected hemorrhagic problems. J Pediatr Hematol Oncol. 2003;25:474–479.
12 Gibbs NM. Point-of-care assessment of antiplatelet agents in the perioperative period: a review. Anaesth Intensive Care. 2009;37:354–369.
13 Morley SL. Red blood cell transfusions in acute paediatrics. Arch Dis Child Educ Pract Ed. 2009;94:65–73.
14 Harrison E, Bolton P. Serious hazards of transfusion in children (SHOT). Pediatr Anesth. 2011;21:10–13.
15 Practice Guidelines for blood component therapy: A report by the American Society of Anesthesiologists Task Force on Blood Component Therapy. Anesthesiology. 1996;84:732–747.
16 Simon TL, Alverson DC, AuBuchon J, et al. Practice parameter for the use of red blood cell transfusions: developed by the Red Blood Cell Administration Practice Guideline Development Task Force of the College of American Pathologists. Arch Pathol Lab Med. 1998;122:130–138.
17 Gibson BE, Todd A, Roberts I, et al. Transfusion guidelines for neonates and older children. BrJ Haematol. 2004;124:433–453.
18 Voak D, Cann R, Finney RD, et al. Guidelines for administration of blood products: transfusion of infants and neonates. British Committee for Standards in Haematology Blood Transfusion Task Force. Transfus Med. 1994;4:63–69.
19 Samama CM, Djoudi R, Lecompte T, Nathan-Denizot N, Schved JF. Perioperative platelet transfusion: recommendations of the Agence Francaise de Securite Sanitaire des Produits de Sante (AFSSaPS) 2003. CanJ Anaesth. 2005;52:30–37.
20 Rebulla P. Platelet transfusion trigger in difficult patients. Transfus Clin Biol. 2001;8:249–254.
21 British Committee for Standards in Haematology, Blood Transfusion Task Force. Guidelines for the use of platelet transfusions. Br J Haematol. 2003;122:10–23.
22 Murphy MF, Brozovic B, Murphy W, Ouwehand W, Waters AH. Guidelines for platelet transfusions. British Committee for Standards in Haematology, Working Party of the Blood Transfusion Task Force. Transfus Med. 1992;2:311–318.
23 Vavricka SR, Walter RB, Irani S, Halter J, Schanz U. Safety of lumbar puncture for adults with acute leukemia and restrictive prophylactic platelet transfusion. Ann Hematol. 2003;82:570–573.
24 Lawrence JB, Yomtovian RA, Hammons T, et al. Lowering the prophylactic platelet transfusion threshold: a prospective analysis. Leuk Lymphoma. 2001;41:67–76.
25 Howard SC, Gajjar A, Ribeiro RC, et al. Safety of lumbar puncture for children with acute lymphoblastic leukemia and thrombocytopenia. JAMA. 2000;284:2222–2224.
26 Practice parameter for the use of fresh-frozen plasma, cryoprecipitate, and platelets. Fresh-Frozen Plasma, Cryoprecipitate, and Platelets Administration Practice Guidelines Development Task Force of the College of American Pathologists. JAMA. 1994;271:777–781.
27 Beilin Y, Zahn J, Comerford M. Safe epidural analgesia in thirty parturients with platelet counts between 69,000 and 98,000 mm(−3). Anesth Analg. 1997;85:385–388.
28 Dogan IV, Ovali E, Eti Z, Yayci A, Gogus FY. The in vitro effects of isoflurane, sevoflurane, and propofol on platelet aggregation. Anesth Analg. 1999;88:432–436.
29 Hirakata H, Nakamura K, Yokubol B, et al. Propofol has both enhancing and suppressing effects on human platelet aggregation in vitro. Anesthesiology. 1999;91:1361–1369.
30 Aoki H, Mizobe T, Nozuchi S, Hiramatsu N. In vivo and in vitro studies of the inhibitory effect of propofol on human platelet aggregation. Anesthesiology. 1998;88:362–370.
31 Contreras M, Ala FA, Greaves M, et al. Guidelines for the use of fresh frozen plasma. British Committee for Standards in Haematology, Working Party of the Blood Transfusion Task Force. Transfus Med. 1992;2:57–63.
32 Heller EL, Paul L. Anticoagulation management in a patient with an acquired antithrombin III deficiency. J Extra Corpor Technol. 2001;33:245–248.
33 Guidelines on gamma irradiation of blood components for the prevention of transfusion-associated graft-versus-host disease. BCSH Blood Transfusion Task Force. Transfus Med. 1996;6:261–271.
34 Vichinsky EP, Earles A, Johnson RA, et al. Alloimmunization in sickle cell anemia and transfusion of racially unmatched blood. N Engl J Med. 1990;322:1617–1621.
35 Bolton-Maggs PH. Idiopathic thrombocytopenic purpura. Arch Dis Child. 2000;83:220–222.
36 Bolton-Maggs PH, Stevens RF, Dodd NJ, et al. Guidelines for the diagnosis and management of hereditary spherocytosis. Br J Haematol. 2004;126:455–474.
37 Shah S, Vega R. Hereditary spherocytosis. Pediatr Rev. 2004;25:168–172.
38 King MJ, Behrens J, Rogers C, et al. Rapid flow cytometric test for the diagnosis of membrane cytoskeleton-associated haemolytic anaemia. Br J Haematol. 2000;111:924–933.
39 Gallagher PG. Update on the clinical spectrum and genetics of red blood cell membrane disorders. Curr Hematol Rep. 2004;3:85–91.
40 Minkes RK, Lagzdins M, Langer JC. Laparoscopic versus open splenectomy in children. J Pediatr Surg. 2000;35:699–701.
41 de Buys Roessingh AS, de Lagausie P, Rohrlich P, Berrebi D, Aigrain Y. Follow-up of partial splenectomy in children with hereditary spherocytosis. J Pediatr Surg. 2002;37:1459–1463.
42 Rice HE, Oldham KT, Hillery CA, et al. Clinical and hematologic benefits of partial splenectomy for congenital hemolytic anemias in children. Ann Surg. 2003;237:281–288.
43 Patton ML, Moss BE, Haith LR, Jr., et al. Concomitant laparoscopic cholecystectomy and splenectomy for surgical management of hereditary spherocytosis. Am Surg. 1997;63:536–539.
44 Yamagishi S, Watanabe T. Concomitant laparoscopic splenectomy and cholecystectomy for management of hereditary spherocytosis associated with gallstones. J Clin Gastroenterol. 2000;30:447.
45 Sandler A, Winkel G, Kimura K, Soper R. The role of prophylactic cholecystectomy during splenectomy in children with hereditary spherocytosis. J Pediatr Surg. 1999;34:1077–1078.
46 Martin LD, Casella ES. Anesthesia and glucose-6-phosphate dehydrogenase deficiency in a child with congenital heart disease. J Cardiothorac Vasc Anesth. 1991;5:596–599.
47 Mason PJ. New insights into G6PD deficiency. Br J Haematol. 1996;94:585–591.
48 Smith CL, Snowdon SL. Anaesthesia and glucose-6-phosphate dehydrogenase deficiency. a case report and review of the literature. Anaesthesia. 1987;42:281–288.
49 Srikanth MS, Kahlstrom R, Oh KH, et al. Topical benzocaine (Hurricaine) induced methemoglobinemia during endoscopic procedures in gastric bypass patients. Obes Surg. 2005;15:584–590.
50 Maddali MM, Fahr J. Postoperative methemoglobinemia with associated G-6-P-D deficiency in infant cardiac surgery—enigmas in diagnosis and management. Paediatr Anaesth. 2005;15:334–337.
51 Gerrah R, Shargal Y, Elami A. Impaired oxygenation and increased hemolysis after cardiopulmonary bypass in patients with glucose-6-phosphate dehydrogenase deficiency. Ann Thorac Surg. 2003;76:523–527.
52 Moretti S, Jouvet P, Schleiermacher G, et al. Pulse oximetry and methemoglobinemia. Arch.Pediatr. 1996;3:258–260.
53 Botkin JR. Research for newborn screening: developing a national framework. Pediatrics. 2005;116:862–871.
54 Platt OS, Brambilla DJ, Rosse WF, et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med. 1994;330:1639–1644. 9
55 Firth PG, Head CA. Sickle cell disease and anesthesia. Anesthesiology. 2004;101:766–785.
56 Steinberg MH. Management of sickle cell disease. N Engl J Med. 1999;340:1021–1030.
57 Stuart MJ, Nagel RL. Sickle-cell disease. Lancet. 2004;364:1343–1360.
58 Hebbel RP, Eaton JW. Pathobiology of heme interaction with the erythrocyte membrane. Semin Hematol. 1989;26:136–149.
59 Gladwin MT, Schechter AN. Nitric oxide therapy in sickle cell disease. Semin Hematol. 2001;38:333–342.
60 Powars D, Chan LS, Schroeder WA. The variable expression of sickle cell disease is genetically determined. Semin Hematol. 1990;27:360–376.
61 Crawford MW, Galton S, Abdelhaleem M. Preoperative screening for sickle cell disease in children: clinical implications. Can J Anaesth. 2005;52:1058–1063.
62 Gill FM, Sleeper LA, Weiner SJ, et al. Clinical events in the first decade in a cohort of infants with sickle cell disease. Cooperative Study of Sickle Cell Disease. Blood. 1995;86:776–783.
63 Kato GJ, Gladwin MT, Steinberg MH. Deconstructing sickle cell disease: reappraisal of the role of hemolysis in the development of clinical subphenotypes. Blood Rev. 2007;21:37–47.
64 Vichinsky EP, Haberkern CM, Neumayr L, et al. A comparison of conservative and aggressive transfusion regimens in the perioperative management of sickle cell disease. The Preoperative Transfusion in Sickle Cell Disease Study Group. N Engl J Med. 1995;333:206–213.
65 Charache S, Terrin ML, Moore RD, et al. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia. N Engl J Med. 1995;332:1317–1322.
66 Buchanan GR, DeBaun MR, Quinn CT, Steinberg MH. Sickle cell disease. Hematology Am Soc Hematol Educ Program. 2004;2004:35–47.
67 Weiner DL, Hibberd PL, Betit P, et al. Preliminary assessment of inhaled nitric oxide for acute vaso-occlusive crisis in pediatric patients with sickle cell disease. JAMA. 2003;289:1136–1142.
68 Walters MC, Nienhuis AW, Vichinsky E. Novel therapeutic approaches in sickle cell disease. Hematology Am Soc Hematol Educ Program. 2002;2002:10–34.
69 Vichinsky EP, Neumayr LD, Earles AN, et al. Causes and outcomes of the acute chest syndrome in sickle cell disease. National Acute Chest Syndrome Study Group. N Engl J Med. 2000;342:1855–1865.
70 Atz AM, Wessel DL. Inhaled nitric oxide in sickle cell disease with acute chest syndrome. Anesthesiology. 1997;87:988–990.
71 Sullivan KJ, Goodwin SR, Evangelist J, Moore RD, Mehta P. Nitric oxide successfully used to treat acute chest syndrome of sickle cell disease in a young adolescent. Crit Care Med. 1999;27:2563–2568.
72 Gladwin MT, Schechter AN, Shelhamer JH, Ognibene FP. The acute chest syndrome in sickle cell disease. Possible role of nitric oxide in its pathophysiology and treatment. Am J Respir Crit Care Med. 1999;159:1368–1376.
73 Leong MA, Dampier C, Varlotta L, Allen JL. Airway hyperreactivity in children with sickle cell disease. J Pediatr. 1997;131:278–283.
74 Koumbourlis AC, Zar HJ, Hurlet-Jensen A, Goldberg MR. Prevalence and reversibility of lower airway obstruction in children with sickle cell disease. J Pediatr. 2001;138:188–192.
75 Sullivan KJ, Kissoon N, Gauger C. Nitric oxide metabolism and the acute chest syndrome of sickle cell anemia. Pediatr Crit Care Med. 2008;9:159–168.
76 Powars D, Wilson B, Imbus C, Pegelow C, Allen J. The natural history of stroke in sickle cell disease. Am J Med. 1978;65:461–471.
77 Ohene-Frempong K, Weiner SJ, Sleeper LA, et al. Cerebrovascular accidents in sickle cell disease: rates and risk factors. Blood. 1998;91:288–294.
78 Miller ST, Macklin EA, Pegelow CH, et al. Silent infarction as a risk factor for overt stroke in children with sickle cell anemia: a report from the Cooperative Study of Sickle Cell Disease. J Pediatr. 2001;139:385–390.
79 Kinney TR, Sleeper LA, Wang WC, et al. Silent cerebral infarcts in sickle cell anemia: a risk factor analysis. The Cooperative Study of Sickle Cell Disease. Pediatrics. 1999;103:640–645.
80 Prengler M, Pavlakis SG, Prohovnik I, Adams RJ. Sickle cell disease: the neurological complications. Ann Neurol. 2002;51:543–552.
81 Pegelow CH, Adams RJ, McKie V, et al. Risk of recurrent stroke in patients with sickle cell disease treated with erythrocyte transfusions. J Pediatr. 1995;126:896–899.
82 Adams RJ, McKie VC, Hsu L, et al. Prevention of a first stroke by transfusions in children with sickle cell anemia and abnormal results on transcranial Doppler ultrasonography. N Engl J Med. 1998;339:5–11.
83 Vichinsky EP, Luban NL, Wright E, et al. Prospective RBC phenotype matching in a stroke-prevention trial in sickle cell anemia: a multicenter transfusion trial. Transfusion. 2001;41:1086–1092.
84 de Montalembert M. Management of sickle cell disease. BMJ. 2008;337:a1397.
85 Wesson DE. The initiation and progression of sickle cell nephropathy. Kidney Int. 2002;61:2277–2286.
86 Wong WY, Elliott-Mills D, Powars D. Renal failure in sickle cell anemia. Hematol Oncol Clin North Am. 1996;10:1321–1331.
87 Pham PT, Pham PC, Wilkinson AH, Lew SQ. Renal abnormalities in sickle cell disease. Kidney Int. 2000;57:1–8.
88 Atlas SA. The sickle cell trait and surgical complications. A matched-pair patient analysis. JAMA. 1974;229:1078–1080. 19
89 Kark JA, Posey DM, Schumacher HR, Ruehle CJ. Sickle-cell trait as a risk factor for sudden death in physical training. N Engl J Med. 1987;317:781–787.
90 Kark JA, Ward FT. Exercise and hemoglobin S. Semin Hematol. 1994;31:181–225.
91 Key NS, Derebail VK. Sickle-cell trait: novel clinical significance. Hematology Am Soc Hematol Educ Program. 2010;2010:418–422.
92 Sankaran VG, Menne TF, Xu J, et al. Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A. Science. 2008;322:1839–1842.
93 Mankad VN. Exciting new treatment approaches for pathyphysiologic mechanisms of sickle cell disease. Pediatr Pathol Mol Med. 2001;20:1–13.
94 Haberkern CM, Neumayr LD, Orringer EP, et al. Cholecystectomy in sickle cell anemia patients: perioperative outcome of 364 cases from the National Preoperative Transfusion Study. Preoperative Transfusion in Sickle Cell Disease Study Group. Blood. 1997;89:1533–1542.
95 Waldron P, Pegelow C, Neumayr L, et al. Tonsillectomy, adenoidectomy, and myringotomy in sickle cell disease: perioperative morbidity. Preoperative Transfusion in Sickle Cell Disease Study Group. J Pediatr Hematol Oncol. 1999;21:129–135.
96 Vichinsky EP, Neumayr LD, Haberkern C, et al. The perioperative complication rate of orthopedic surgery in sickle cell disease: report of the National Sickle Cell Surgery Study Group. Am J Hematol. 1999;62:129–138.
97 Searle JF. Anaesthesia in sickle cell states. A review. Anaesthesia. 1973;28:48–58.
98 Holzmann L, Finn H, Lichtman HC, Harmel MH. Anesthesia in patients with sickle cell disease: a review of 112 cases. Anesth Analg. 1969;48:566–572.
99 Gilbertson AA. Anaesthesia in West African patients with sickle-cell anaemia, haemoglobin SC disease, and sickle-cell trait. Br J Anaesth. 1965;37:614–622.
100 Browne RA. Anaesthesia in patients with sickle-cell anaemia. Br J Anaesth. 1965;37:181–188.
101 Koshy M, Weiner SJ, Miller ST, et al. Surgery and anesthesia in sickle cell disease. Cooperative Study of Sickle Cell Diseases. Blood. 1995;86:3676–3684.
102 Holcomb GW, 3rd., Olsen DO, Sharp KW. Laparoscopic cholecystectomy in the pediatric patient. J Pediatr Surg. 1991;26:1186–1190.
103 Seleem MI, Al-Hashemy AM, Meshref SS. Mini-laparoscopic cholecystectomy in children under 10 years of age with sickle cell disease. ANZ J Surg. 2005;75:562–565.
104 Al-Mulhim AS, Al-Mulhim FM, Al-Suwaiygh AA. The role of laparoscopic cholecystectomy in the management of acute cholecystitis in patients with sickle cell disease. Am J Surg. 2002;183:668–672.
105 Esseltine DW, Baxter MR, Bevan JC. Sickle cell states and the anaesthetist. Can J Anaesth. 1988;35:385–403.
106 Firth PG, McMillan KN, Haberkern CM, et al. A survey of perioperative management of sickle cell disease in North America. Pediatr Anesth. 2011;21:43–49.
107 Fitzgerald RK, Johnson A. Pulse oximetry in sickle cell anemia. Crit Care Med. 2001;29:1803–1806.
108 Blaisdell CJ, Goodman S, Clark K, Casella JF, Loughlin GM. Pulse oximetry is a poor predictor of hypoxemia in stable children with sickle cell disease. Arch Pediatr Adolesc Med. 2000;154:900–903.
109 Riddington C, Williamson L. Preoperative blood transfusions for sickle cell disease. Cochrane Database Syst Rev. 3, 2001. CD003149
110 Rosse WF, Gallagher D, Kinney TR, et al. Transfusion and alloimmunization in sickle cell disease. The Cooperative Study of Sickle Cell Disease. Blood. 1990;76:1431–1437.
111 Griffin TC, Buchanan GR. Elective surgery in children with sickle cell disease without preoperative blood transfusion. J Pediatr Surg. 1993;28:681–685.
112 Neumayr L, Koshy M, Haberkern C, et al. Surgery in patients with hemoglobin SC disease. Preoperative Transfusion in Sickle Cell Disease Study Group. Am J Hematol. 1998;57:101–108.
113 Castro O, Sandler SG, Houston-Yu P, Rana S. Predicting the effect of transfusing only phenotype-matched RBCs to patients with sickle cell disease: theoretical and practical implications. Transfusion. 2002;42:684–690.
114 Gross ML, Schwedler M, Bischoff RJ, Kerstein MD. Impact of anesthetic agents on patients with sickle cell disease. Am Surg. 1993;59:261–264.
115 Laasberg LH, Hedley-Whyte J. Viscosity of sickle disease and trait blood: changes with anesthesia. J Appl Physiol. 1973;35:837–843.
116 Dulvadestin P, Gilton A, Hernigou P, Marty J. The onset time of atracurium is prolonged in patients with sickle cell disease. Anesth Analg. 2008;107:113–116.
117 Yaster M, Tobin JR, Billett C, Casella JF, Dover G. Epidural analgesia in the management of severe vaso-occlusive sickle cell crisis. Pediatrics. 1994;93:310–315.
118 McHardy P, McDonnell C, Lorenzo AJ, Salle JL, Campbell FA. Management of priapism in a child with sickle cell anemia; successful outcome using epidural analgesia. Can J Anaesth. 2007;54:642–645.
119 Prengler M, Pavlakis SG, Boyd S, et al. Sickle cell disease and electroencephalogram hyperventilation. Ann Neurol. 2006;59:214–215.
120 Sarjeant JM, Callum JL. The use of tourniquets in patients with sickle cell disease. Anesth Analg. 2004;99:630.
121 Oginni LM, Rufai MB. How safe is tourniquet use in sickle-cell disease? Afr J Med Med Sci. 1996;25:3–6.
122 Willinsky JS, Lepow R. Sickle cell trait and use of the pneumatic tourniquet. A case report. J Am Podiatry Assoc. 1984;74:38–41.
123 Tobin JR, Butterworth J. Sickle cell disease: dogma, science, and clinical care. Anesth Analg. 2004;98:283–284.
124 Martin WJ, Green DR, Dougherty N, et al. Tourniquet use in sickle cell disease patients. J Am Podiatry Assoc. 1984;74:291–294.
125 Adu-Gyamfi Y, Sankarankutty M, Marwa S. Use of a tourniquet in patients with sickle-cell disease. Can J Anaesth. 1993;40:24–27.
126 Stein RE, Urbaniak J. Use of the tourniquet during surgery in patients with sickle cell hemoglobinopathies. Clinl Orthop Relat Res. 1980:231–233.
127 Fox JS, Amaranath L, Hoeltge GA, Andrish JT. Autologous blood transfusion and intraoperative cell salvage in a patient with homozygous sickle cell disease. Cleve Clin J Med. 1994;61:137–140.
128 Brajtbord D, Johnson D, Ramsay M, et al. Use of the cell saver in patients with sickle cell trait. Anesthesiology. 1989;70:878–879.
129 Metras D, Coulibaly AO, Ouattara K, et al. Open-heart surgery in sickle-cell haemoglobinopathies: report of 15 cases. Thorax. 1982;37:486–491.
130 Djaiani GN, Cheng DC, Carroll JA, Yudin M, Karski JM. Fast-track cardiac anesthesia in patients with sickle cell abnormalities. Anesth Analg. 1999;89:598–603.
131 Frimpong-Boateng K, Amoah AG, Barwasser HM, Kallen C. Cardiopulmonary bypass in sickle cell anaemia without exchange transfusion. Eur J Cardiothorac Surg. 1998;14:527–529.
132 Harban FM, Connor P, Crook R, Bingham R. Cardiopulmonary bypass for surgical correction of congenital heart disease in children with sickle cell disease: a case series. Anaesthesia. 2008;63:648–651.
133 Rund D, Rachmilewitz E. Beta-thalassemia. N Eng J Med. 2005;353:1135–1146.
134 Chui DH, Fucharoen S, Chan V. Hemoglobin H disease: not necessarily a benign disorder. Blood. 2003;101:791–800.
135 Lal A, Goldrich ML, Haines DA, et al. Heterogeneity of hemoglobin H disease in childhood. N Engl J Med. 2011;364:710–718.
136 Eldor A, Rachmilewitz EA. The hypercoagulable state in thalassemia. Blood. 2002;99:36–43.
137 Cappellini MD, Robbiolo L, Bottasso BM, et al. Venous thromboembolism and hypercoagulability in splenectomized patients with thalassaemia intermedia. Br J Haematol. 2000;111:467–473.
138 Cavazzana-Calvo M, Payen E, Negre O, et al. Transfusion independence and HMGA2 activation after gene therapy of human beta-thalassaemia. Nature. 2010;467:318–322.
139 Kitoh T, Tanaka S, Ono K, Hasegawa J, Otagiri T. Anesthetic management of a patient with beta-thalassemia intermedia undergoing splenectomy: a case report. J Anesth. 2005;19:252–256.
140 Mak PH, Ooi RG. Submental intubation in a patient with beta-thalassaemia major undergoing elective maxillary and mandibular osteotomies. Br J Anaesth. 2002;88:288–291.
141 Katz R, Goldfarb A, Muggia M, Gimmon Z. Unique features of laparoscopic cholecystectomy in beta thalassemia patients. Surg Laparosc Endosc Percutan Tech. 2003;13:318–321.
142 Suwanchinda V, Pirayavaraporn S, Yokubol B, et al. Does furosemide prevent hypertension during perioperative splenectomy in thalassemic children? J Med Assoc Thai. 1995;78:542–546.
143 Suwanchinda V, Tengapiruk Y, Udomphunthurak S. Hypertension perioperative splenectomy in thalassemic children. J Med Assoc Thail. 1994;77:66–70.
144 Rowbottom SJ, Sudhaman DA. Haemoglobin H disease and cardiac surgery. Anaesthesia. 1988;43:1033–1034.
145 Blanchette V, Carcao M. Approach to the investigation and management of immune thrombocytopenic purpura in children. Semin Hematol. 2000;37:299–314.
146 Rodeghiero F, Stasi R, Gernsheimer T, et al. Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group. Blood. 2009;113:2386–2393.
147 Cuker A, Cines DB. Immune thrombocytopenia. Hematology Am Soc Hematol Educ Program. 2010;2010:377–384.
148 George JN, Woolf SH, Raskob GE, et al. Idiopathic thrombocytopenic purpura: a practice guideline developed by explicit methods for the American Society of Hematology. Blood. 1996;88:3–40.
149 Guidelines for the investigation and management of idiopathic thrombocytopenic purpura in adults, children and in pregnancy. Br J Haematol. 2003;120:574–596.
150 Cordera F, Long KH, Nagorney DM, et al. Open versus laparoscopic splenectomy for idiopathic thrombocytopenic purpura: clinical and economic analysis. Surgery. 2003;134:45–52.
151 Wang T, Xu M, Ji L, Yang R. Splenectomy for chronic idiopathic thrombocytopenic purpura in children: a single center study in China. Acta Haematol. 2006;115:39–45.
152 Schlinkert RT, Braich TA. Laparoscopic assisted splenectomy for treatment of presumed immune thrombocytopenic purpura: initial results. Mayo Clin Proc. 1994;69:422–424.
153 Montgomery RR. Von Willebrand disease. In: Goodnight SH, Jr., Hathaway WE, eds. Disorders of hemostasis and thrombosis. 2nd ed. New York: McGraw Hill; 2001:115–126.
154 Schneppenheim R. The evolving classification of von Willebrand disease. Blood Coagul Fibrinolysis. 2005;16(Suppl 1):S3–10.
155 Sadler JE. A revised classification of von Willebrand disease. For the Subcommittee on von Willebrand Factor of the Scientific and Standardization Committee of the International Society on Thrombosis and Haemostasis. Thromb Haemost. 1994;71:520–525.
156 Sadler JE, Mannucci PM, Berntorp E, et al. Impact, diagnosis and treatment of von Willebrand disease. Thromb Haemost. 2000;84:160–174.
157 Federici AB. Clinical diagnosis of von Willebrand disease. Haemophilia. 2004;10(Suppl 4):169–176.
158 Rodeghiero F, Castaman G, Tosetto A. How I treat von Willebrand disease. Blood. 2009;114:1158–1165.
159 Brown DL. Congenital bleeding disorders. Curr Probl Pediatr Adolesc Health Care. 2005;35:38–62.
160 Dean JA, Blanchette VS, Carcao MD, et al. von Willebrand disease in a pediatric-based population–comparison of type 1 diagnostic criteria and use of the PFA-100 and a von Willebrand factor/collagen-binding assay. Thromb Haemost. 2000;84:401–409.
161 Favaloro EJ. The utility of the PFA-100 in the Identification of von Willebrand disease: a concise review. Semin Thromb Hemost. 2006;32:537–545.
162 Federici AB. Management of von Willebrand disease with factor VIII/von Willebrand factor concentrates: results from current studies and surveys. Blood Coagul Fibrinolysis. 2005;16(Suppl 1):S17–S21.
163 Nichols WL, Hultin MB, James AH, et al. von Willebrand disease (VWD): evidence-based diagnosis and management guidelines, the National Heart, Lung, and Blood Institute (NHLBI) Expert Panel report (USA). Haemophilia. 2008;14:171–232.
164 Kitchens CS. Surgery and hemostasis. In: Kitchens CS, Alving BM, Kessler CM, eds. Consultative hemostasis and thrombosis. Philadelphia: WB Saunders; 2002:463–480.
165 Stedeford JC, Pittman JA. Von Willebrand’s disease and neuroaxial anaesthesia. Anaesthesia. 2000;55:1228–1229.
166 Soucie JM, Evatt B, Jackson D. Occurrence of hemophilia in the United States. The Hemophilia Surveillance System Project Investigators. Am J Hematol. 1998;59:288–294.
167 Martlew VJ. Peri-operative management of patients with coagulation disorders. Br J Anaesth. 2000;85:446–455.
168 DiMichele D. Hemophilia A (FVIII deficiency). In: Goodnight SH, Jr., Hathaway W, eds. Disorders of hemostasis and thrombosis. New York: McGraw-Hill; 2001:127–139.
169 Valentino LA, Cooper DL, Goldstein B. Surgical experience with rFVIIa (NovoSeven) in congenital haemophilia A and B patients with inhibitors to factors VIII or IX. Haemophilia. 2011;17:579–589.
170 Lauroua P, Ferrer AM, Guerin V. Successful major and minor surgery using factor VIII inhibitor bypassing activity in patients with haemophilia A and inhibitors. Haemophilia. 2009;15:1300–1307.
171 Kang SB, Rumball KM, Ettinger RS. Continuous axillary brachial plexus analgesia in a patient with severe hemophilia. J Clin Anesth. 2003;15:38–40.
172 Tormene D, Simioni P, Prandoni P, et al. The incidence of venous thromboembolism in thrombophilic children: a prospective cohort study. Blood. 2002;100:2403–2405.
173 Jackson PC, Morgan JM. Perioperative thromboprophylaxis in children: development of a guideline for management. Paediatr Anaesth. 2008;18:478–487.
174 Cacciapuoti F. Some considerations about the hypercoagulable states and their treatments. Blood Coagul Fibrinolysis. 2011;22:155–159.
175 Cole CH. Primary prophylaxis of venous thromboembolism in children. J Paediatr Child Health. 2010;46:288–290.
176 Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–249.
177 Ries LAG, Percy CL, Bunin GR, Introduction. NIH Pub. No. 99-4649 ed. Ries LAG, Smith MA, Gurney JG, et al, eds. Cancer incidence and survival among children and adolescents: United States SEER Program 1975-1995, Bethesda, MD, National Cancer Institute SEER Program, 1999:1.
178 Horner MJ, Ries LAG, Krapcho M, SEER cancer statistics review, 1975-2006. Available at http://seer.cancer.gov/csr/1975_2006 (accessed May 2012)
179 Oeffinger KC, Mertens AC, Sklar CA, et al. Chronic health conditions in adult survivors of childhood cancer. N Engl J Med. 2006;355:1572.
180 Hammer GB. Anaesthetic management for the child with a mediastinal mass. Paediatr Anaesth. 2004;14:95–97.
181 Narang S, Harte BH, Body SC. Anesthesia for patients with a mediastinal mass. Anesthesiol Clini North Am. 2001;19:559–579.
182 King DR, Patrick LE, Ginn-Pease ME, McCoy KS, Klopfenstein K. Pulmonary function is compromised in children with mediastinal lymphoma. J Pediatr Surg. 1997;32:294.
183 Ferrari LR, Bedford RF. General anesthesia prior to treatment of anterior mediastinal masses in pediatric cancer patients. Anesthesiology. 1990;72:991–995.
184 Hockenberry MJ, Hinds PS, Barrera P, et al. Incidence of anemia in children with solid tumors or Hodgkin disease. J Pediatr Hematol Oncol. 2002;24:35–37.
185 Margolin JF, Steuber CP, Poplack DG. Acute lymphoblastic leukemia. In: Pizzo PA, Poplack DG, eds. Principles and practice of pediatric oncology. 5th ed. Philadelphia: Lippincott Williams & Wilkins; 2006:538.
186 Hastings CA, Lubin BH, Feusner J. Hematologic supportive care for children with cancer. In: Pizzo PA, Poplack DG, eds. Principles and practice of pediatric oncology. 5th ed. Philadelphia: Lippincott Williams & Wilkins; 2006:1231.
187 Malogolowkin MH, Quinn JJ, Steuber CP, Stuart SE. Clinical assessment and differential diagnosis of the child with suspected cancer. In: Pizzo PA, Poplack DG, eds. Principles and practice of pediatric oncology. 5th ed. Philadelphia: Lippincott Williams & Wilkins; 2006:145.
188 Golub TR, Arceci RJ. Acute Myelogenous Leukemia. In: Pizzo PA, Poplack DG, eds. Principles and practice of pediatric oncology. 5th ed. Philadelphia: Lippincott Williams & Wilkins; 2006:591.
189 Creutzig U, Ritter J, Budde M, Sutor A, Schellong G. Early deaths due to hemorrhage and leukostasis in childhood acute myelogenous leukemia. Associations with hyperleukocytosis and acute monocytic leukemia. Cancer. 1987;60:3071–3079.
190 Coiffier B, Altman A, Pui CH, Younes A, Cairo MS. Guidelines for the management of pediatric and adult tumor lysis syndrome: an evidence-based review. J Clin Oncol. 2008;26:2767–2778.
191 Del Toro G, Morris E, Cairo MS. Tumor lysis syndrome: pathophysiology, definition, and alternative treatment approaches. Clin Adv Hematol Oncol. 2005;3:54–61.
192 Farley-Hills E, Byrne AJ, Brennan L, Sartori P. Tumour lysis syndrome during anaesthesia. Paediatr Anaesth. 2001;11:233–236.
193 Duzova A, Cetin M, Gumruk F, Yetgin S. Acute tumour lysis syndrome following a single-dose corticosteroid in children with acute lymphoblastic leukaemia. Eur J Haematol. 2001;66:404–407.
194 Sinha R, Bose S, Subramaniam R. Tumor lysis under anesthesia in a child. Acta Anaesthesiol Scand. 2009;53:131–133.
195 Lee MH, Cheng KI, Jang RC, et al. Tumour lysis syndrome developing during an operation. Anaesthesia. 2007;62:85–87.
196 Osthaus WA, Linderkamp C, Bunte C, Juttner B, Sumpelmann R. Tumor lysis associated with dexamethasone use in a child with leukemia. Pediatr Anesth. 2008;18:268–270.
197 McDonnell C, Barlow R, Campisi P, Grant R, Malkin D. Fatal peri-operative acute tumour lysis syndrome precipitated by dexamethasone. Anaesthesia. 2008;63:652–655.
198 Carpenter PA, Mielcarek M, Woolfrey AE. Hematopoietic cell transplantation. In: Irwin S, Rippe JM, eds. Intensive care medicine. 6th ed. Philadelphia: Lippincott Williams & Wilkins; 2008:2150–2168.
199 Einaudi S, Bertorello N, Masera N, et al. Adrenal axis function after high-dose steroid therapy for childhood acute lymphoblastic leukemia. Pediatr Blood Cancer. 2008;50:537–541.
200 Kuperman H, Damiani D, Chrousos GP, et al. Evaluation of the hypothalamic-pituitary-adrenal axis in children with leukemia before and after 6 weeks of high-dose glucocorticoid therapy. J Clin Endocrinol Metab. 2001;86:2993–2996.
201 Felner EI, Thompson MT, Ratliff AF, White PC, Dickson BA. Time course of recovery of adrenal function in children treated for leukemia. J Pediatr. 2000;137:21–24.
202 Rix M, Birkebaek NH, Rosthoj S, Clausen N. Clinical impact of corticosteroid-induced adrenal suppression during treatment for acute lymphoblastic leukemia in children: a prospective observational study using the low-dose adrenocorticotropin test. J Pediatr. 2005;147:645–650.
203 Mahachoklertwattana P, Vilaiyuk S, Hongeng S, Okascharoen C. Suppression of adrenal function in children with acute lymphoblastic leukemia following induction therapy with corticosteroid and other cytotoxic agents. J Pediatr. 2004;144:736–740.
204 Petersen KB, Muller J, Rasmussen M, Schmiegelow K. Impaired adrenal function after glucocorticoid therapy in children with acute lymphoblastic leukemia. Medical and Pediatric Oncology. 2003;41:110–114.
205 Cunha Cde F, Silva IN, Finch FL. Early adrenocortical recovery after glucocorticoid therapy in children with leukemia. J Clin Endocrinol Metab. 2004;89:2797–2802.
206 van Dalen EC, van der Pal HJ, Kok WE, Caron HN, Kremer LC. Clinical heart failure in a cohort of children treated with anthracyclines: a long-term follow-up study. Eur J Cancer. 2006;42:3191–3198.
207 Hudson MM, Rai SN, Nunez C, et al. Noninvasive evaluation of late anthracycline cardiac toxicity in childhood cancer survivors. J Clin Oncol. 2007;25:3635–3643.
208 Agarwala S, Kumar R, Bhatnagar V, et al. High incidence of adriamycin cardiotoxicity in children even at low cumulative doses: role of radionuclide cardiac angiography. J Pediatr Surg. 2000;35:1786–1789.
209 O’Brien MM, Taub JW, Chang MN, et al. Cardiomyopathy in children with Down syndrome treated for acute myeloid leukemia: a report from the Children’s Oncology Group Study POG 9421. J Clin Oncol. 2008;26:414–420.
210 Shankar SM, Marina N, Hudson MM, et al. Monitoring for cardiovascular disease in survivors of childhood cancer: report from the Cardiovascular Disease Task Force of the Children’s Oncology Group. Pediatrics. 2008;121:e387–e396.
211 Jannazzo A, Hoffman J, Lutz M. Monitoring of anthracycline-induced cardiotoxicity. Ann Pharmacother. 2008;42:99–104.
212 Simbre VC, Duffy SA, Dadlani GH, Miller TL, Lipshultz SE. Cardiotoxicity of cancer chemotherapy: implications for children. Paediatr Drugs. 2005;7:187–202.
213 Sorensen K, Levitt GA, Bull C, Dorup I, Sullivan ID. Late anthracycline cardiotoxicity after childhood cancer: a prospective longitudinal study. Cancer. 2003;97:1991–1998.
214 Latham GJ, Greenberg RS. Anesthetic considerations for the pediatric oncology patient—part 3: pain, cognitive dysfunction, and preoperative evaluation. Paediatr Anaesth. 2010;20:479–489.
215 van Dalen EC, van den Brug M, Caron HN, Kremer LC. Anthracycline-induced cardiotoxicity: comparison of recommendations for monitoring cardiac function during therapy in paediatric oncology trials. Eur J Cancer. 2006;42:3199–3205.
216 Ott N, Ramsay NK, Priest JR, et al. Sequelae of thrombotic or hemorrhagic complications following l-asparaginase therapy for childhood lymphoblastic leukemia. Am J Pediatr Hematol Oncol. 1988;10:191–195.
217 Kieslich M, Porto L, Lanfermann H, et al. Cerebrovascular complications of l-asparaginase in the therapy of acute lymphoblastic leukemia. Am J Pediatr Hematol Oncol. 2003;25:484–487.
218 Homans AC, Rybak ME, Baglini RL, et al. Effect of l-asparaginase administration on coagulation and platelet function in children with leukemia. J Clin Oncol. 1987;5:811–817.
219 Sleijfer S. Bleomycin-induced pneumonitis. Chest. 2001;120:617–624.
220 Eigen H, Wyszomierski D. Bleomycin lung injury in children. Pathophysiology and guidelines for management. Am J Pediatr Hematol Oncol. 1985;7:71–78.
221 Carver JR, Shapiro CL, Ng A, et al. American Society of Clinical Oncology clinical evidence review on the ongoing care of adult cancer survivors: cardiac and pulmonary late effects. J Clin Oncol. 2007;25:3991–4008.
222 Klein DS, Wilds PR. Pulmonary toxicity of antineoplastic agents: anaesthetic and postoperative implications. Can Anaesth Soc J. 1983;30:399–405.
223 Goldiner PL, Carlon GC, Cvitkovic E, Schweizer O, Howland WS. Factors influencing postoperative morbidity and mortality in patients treated with bleomycin. BMJ. 1978;1:1664–1667.
224 Goldiner PL, Schweizer O. The hazards of anesthesia and surgery in bleomycin-treated patients. Semin Oncol. 1979;6:121–124.
225 Uderzo C, Pillon M, Corti P, et al. Impact of cumulative anthracycline dose, preparative regimen and chronic graft-versus-host disease on pulmonary and cardiac function in children 5 years after allogeneic hematopoietic stem cell transplantation: a prospective evaluation on behalf of the EBMT Pediatric Diseases and Late Effects Working Parties. Bone Marrow Transplant. 2007;39:667–675.
226 Ferry C, Gemayel G, Rocha V, et al. Long-term outcomes after allogeneic stem cell transplantation for children with hematological malignancies. Bone Marrow Transplant. 2007;40:219–224.
227 Kaya Z, Weiner DJ, Yilmaz D, Rowan J, Goyal RK. Lung function, pulmonary complications, and mortality after allogeneic blood and marrow transplantation in children. Biol Blood Marrow Transplant. 2009;15:817–826.
228 Chaimberg KH, Cravero JP. Mucositis and airway obstruction in a pediatric patient. Anesth Analg. 2004;99:59–61.
229 Drew B, Peters C, Rimell F. Upper airway complications in children after bone marrow transplantation. Laryngoscope. 2000;110:1446–1451.
230 Anghelescu DL, Burgoyne LL, Liu W, et al. Safe anesthesia for radiotherapy in pediatric oncology: St. Jude Children’s Research Hospital Experience, 2004-2006. Int J Radiat Oncol Biol Phys. 2008;71:491–497.
231 McLure HA, Trenfield S, Quereshi A, Williams J. Post-splenectomy thrombocytopenia: implications for regional analgesia. Anaesthesia. 2003;58:1106–1110.
232 Haberkern CM, Coles PG, Morray JP, Kennard SC, Sawin RS. Intraoperative hypertension during surgical excision of neuroblastoma. Case report and review of 20 years’ experience. Anesth Analg. 1992;75:854–858.
233 Latham GJ, Greenberg RS. Anesthetic considerations for the pediatric oncology patient—part 2: systems-based approach to anesthesia. Pediatr Anesth. 2010;20:396–420.
234 Thomas ED, Lochte HL, Jr., Lu WC, Ferrebee JW. Intravenous infusion of bone marrow in patients receiving radiation and chemotherapy. N Engl J Med. 1957;257:491–496.
235 Savasan S, Abella EM. Current issues in pediatric stem cell transplantation. Clin Lab Med. 2005;25:519–540.
236 Cutler C, Antin JH. An overview of hematopoietic stem cell transplantation. Clin Chest Med. 2005;26:517–527.
237 Woolfrey AE, Anasetti C, Storer B, et al. Factors associated with outcome after unrelated marrow transplantation for treatment of acute lymphoblastic leukemia in children. Blood. 2002;99:2002–2008.
238 Shaw PJ, Kan F, Woo Ahn K, et al. Outcomes of pediatric bone marrow transplantation for leukemia and myelodysplasia using matched sibling, mismatched related, or matched unrelated donors. Blood. 2010;116:4007–4015.
239 Tabbara IA, Zimmerman K, Morgan C, Nahleh Z. Allogeneic hematopoietic stem cell transplantation: complications and results. Arch Intern Med. 2002;162:1558–1566.
240 Majorana A, Schubert MM, Porta F, Ugazio AG, Sapelli PL. Oral complications of pediatric hematopoietic cell transplantation: diagnosis and management. Support Care Cancer. 2000;8:353–365.
241 Barker CC, Butzner JD, Anderson RA, Brant R, Sauve RS. Incidence, survival and risk factors for the development of veno-occlusive disease in pediatric hematopoietic stem cell transplant recipients. Bone Marrow Transplant. 2003;32:79–87.
242 Cesaro S, Pillon M, Talenti E, et al. A prospective survey on incidence, risk factors and therapy of hepatic veno-occlusive disease in children after hematopoietic stem cell transplantation. Haematologica. 2005;90:1396–1404.
243 Lee SH, Yoo KH, Sung KW, et al. Hepatic veno-occlusive disease in children after hematopoietic stem cell transplantation: incidence, risk factors, and outcome. Bone Marrow Transplant. 2010;45:1287–1293.
244 Schwartz JM, Wolford JL, Thornquist MD, et al. Severe gastrointestinal bleeding after hematopoietic cell transplantation, 1987-1997: incidence, causes, and outcome. Am J Gastroenterol. 2001;96:385–393.
245 Afessa B, Peters SG. Major complications following hematopoietic stem cell transplantation. Semin Respir Crit Care Med. 2006;27:297–309.
246 Cerveri I, Fulgoni P, Giorgiani G, et al. Lung function abnormalities after bone marrow transplantation in children: has the trend recently changed? Chest. 2001;120:1900–1906.
247 Lamas A, Otheo E, Ros P, et al. Prognosis of child recipients of hematopoietic stem cell transplantation requiring intensive care. Intensive Care Med. 2003;29:91–96.
248 Creutzig U, Diekamp S, Zimmermann M, Reinhardt D. Longitudinal evaluation of early and late anthracycline cardiotoxicity in children with AML. Pediatr Blood Cancer. 2007;48:651–662.
249 Diaz de Heredia C, Moreno A, Olive T, Iglesias J, Ortega JJ. Role of the intensive care unit in children undergoing bone marrow transplantation with life-threatening complications. Bone Marrow Transplant. 1999;24:163–168.
250 Dursun O, Hazar V, Karasu GT, et al. Prognostic factors in pediatric cancer patients admitted to the pediatric intensive care unit. J Pediatr Hematol Oncol. 2009;31:481–484.
251 Esiashvili N, Chiang KY, Hasselle MD, et al. Renal toxicity in children undergoing total body irradiation for bone marrow transplant. Radiother Oncol. 2009;90:242–246.
252 Hazar V, Gungor O, Guven AG, et al. Renal function after hematopoietic stem cell transplantation in children. Pediatr Blood Cancer. 2009;53:197–202.
253 Michael M, Kuehnle I, Goldstein SL. Fluid overload and acute renal failure in pediatric stem cell transplant patients. Pediatr Nephrol. 2004;19:91–95.
254 Seber A, Shu XO, Defor T, Sencer S, Ramsay N. Risk factors for severe hemorrhagic cystitis following BMT. Bone Marrow Transplant. 1999;23:35–40.
255 Rosenthal J, Pawlowska A, Bolotin E, et al. Transplant-associated thrombotic microangiopathy in pediatric patients treated with sirolimus and tacrolimus. Pediatr Blood Cancer. 2011;57:142–146.
256 Graus F, Saiz A, Sierra J, et al. Neurologic complications of autologous and allogeneic bone marrow transplantation in patients with leukemia: a comparative study. Neurology. 1996;46:1004–1009.
257 De La Camara R, Tomas JF, Figuera A, Berberana M, Fernandez-Ranada JM. High dose busulfan and seizures. Bone Marrow Transplant. 1991;7:363–364.
258 Vogler WR, Winton EF, Heffner LT, et al. Ophthalmological and other toxicities related to cytosine arabinoside and total body irradiation as preparative regimen for bone marrow transplantation. Bone Marrow Transplant. 1990;6:405–409.
259 Anderson NR, Tandon DS. Ifosfamide extrapyramidal neurotoxicity. Cancer. 1991;68:72–75.
260 Bechstein WO. Neurotoxicity of calcineurin inhibitors: impact and clinical management. Transpl Int. 2000;13:313–326.
261 Baird K, Cooke K, Schultz KR. Chronic graft-versus-host disease (GVHD) in children. Pediatr Clin North Am. 2010;57:297–322.
262 Baron F, Storb R. Allogeneic hematopoietic cell transplantation following nonmyeloablative conditioning as treatment for hematologic malignancies and inherited blood disorders. Mol Ther. 2006;13:26–41.
263 Burmeister MA, Standl T, Brauer P, et al. Safety and efficacy of spinal vs general anaesthesia in bone marrow harvesting. Bone Marrow Transplant. 1998;21:1145–1148.
264 Csete M. Cellular transplantation. Anesthesiol Clin North Am. 2004;22:887–901.
265 Slichter SJ. Evidence-based platelet transfusion guidelines. Hematolology Am Soc Hematol Educ Program. 2007;1:172–178.
[/level-membership-for-anesthesiology-category][not-level-membership-for-anesthesiology-category]
9 Essentials of Hematology
The Basics
Laboratory Values and Diagnostic Tests
What is a normal hematocrit or platelet count for an infant or child who comes to the operating room? Red blood cell (RBC), white blood cell, platelet, and coagulation indices evolve in various ways through late gestation, the neonatal period, infancy, and childhood (Table 9-1).
The term neonate has a relative polycythemia, reticulocytosis, and leukocytosis compared with the child. Neonatal platelet counts are similar to those of adults. Although in vitro function may be impaired for the first postnatal month, most in vivo assays of platelet function indicate normal or accelerated function. Preterm and term neonates have prolongation of the prothrombin time (PT) and activated partial thromboplastin time (aPTT) because of a relative deficiency in vitamin K–dependent factors and contact activation factors, respectively; however, concentrations of factor VIII and von Willebrand factor (vWF) are elevated.1 The international normalized ratio (INR), a normalized PT, has an average value of 1.0 for all age groups. Fibrinogen concentrations are comparable between the term neonate and adult, although neonatal fibrinogen is qualitatively dysfunctional. The plasma concentrations of many anticoagulant factors (i.e., tissue factor pathway inhibitor, antithrombin, vitamin K–dependent glycoproteins, and proteins C and S) are decreased in preterm and term neonates. The quantity and quality of plasminogen are decreased in neonates, a condition that increases the risk for thrombosis, especially in a compromised infant.1,2 Most of these differences between the neonate and older child or adult persist for 3 to 6 months postnatally.
There is no ideal single screening test to assess the bleeding risk of a child in the perioperative period. Bleeding time appears to be greater in the infant and child (and less in the neonate) than it is in the adult, but the range of values is wide and overlapping (see Table 9-1). Although this test is potentially helpful in predicting posttonsillectomy and adenoidectomy hemorrhage,3 as well as hemorrhage after percutaneous renal4 and liver5 biopsy, there is little evidence to support its use as a screening test to predict bleeding in the presence of a careful, inclusive clinical history.6,7
The thromboelastogram has been used to investigate the coagulation status of children undergoing spinal fusion,8 neurosurgical procedures,9 and cardiopulmonary bypass for cardiothoracic procedures.10 Although the thromboelastogram may provide useful information in the surgical setting to evaluate fibrinolysis, hypercoagulability, and other coagulation perturbations, its use is usually limited to clinical scenarios with dynamic coagulation changes, such as open heart surgery with cardiopulmonary bypass and liver transplantation. Platelet function analyzer (PFA-100) analysis is an additional test that is increasingly used for the assessment of platelet abnormalities, and it has the benefit of avoiding some of the difficulties of obtaining a bleeding time in children. Although several studies suggest PFA-100 analysis is equivalent or superior to the bleeding time for detecting bleeding abnormalities, there is no consensus about its role in preoperative screening.11 With the increasing use of newer agents that modify platelet function (e.g., platelet G protein–coupled receptor P2Y12 antagonists, glycoprotein GPIIb-IIIa complex antagonists), clinicians must understand that the thromboelastogram, PFA-100, and other methods for assessing platelet function may vary in their ability to monitor the effects of these agents and those of cyclooxygenase inhibitors.12
Guidelines for Transfusion
Critical analyses of the risks and benefits of transfusions in infants and children in the perioperative period have resulted in fewer transfusions. Even for infants and children in intensive care, a restrictive transfusion threshold (i.e., 7 g/dL) reduces transfusions without increasing morbidity compared with a liberal threshold (i.e., 9.5 to 10 g/dL).13 Data from the United Kingdom’s national audit of clinical transfusion, the Serious Hazards of Transfusion (SHOT), indicate that infants and children younger than 18 years of age are at greater risk for adverse transfusion-related reactions (37 and 18 in 100,000, respectively) than are adults (13 in 100,000). Most events were error related, such as administrative, laboratory, clinical judgment, and handling errors.14
Guidelines for RBC transfusion for infants and children in the perioperative setting should be consistent with those established by the American Society of Anesthesiologists Task Force on Blood Component Therapy, which propose that transfusion is not indicated for hemoglobin concentrations higher than 10 g/dL but is indicated for concentrations lower than 6 g/dL.15 When the concentration is between 6 and 10 g/dL, packed red blood cells (PRBCs) should be transfused based on the child’s vital signs, adequacy of oxygenation and perfusion, acuity and degree of blood loss, and other physiologic and surgical factors. When the concentration exceeds 10 g/dL, the decision to transfuse PRBCs to a neonate or infant should be based on the increased baseline concentrations of hemoglobin, increased oxygen consumption, increased affinity of residual fetal hemoglobin for oxygen, absolute blood volume (i.e., 85 mL/kg for a term neonate and 100 mL/kg for a preterm neonate), and other physiologic and surgical factors applicable to all children. The threshold for transfusing a healthy neonate may be 7 g/dL in some clinical settings, but it may be 12 g/dL or higher for a neonate in other settings, such as significant lung disease requiring mechanical ventilation, chronic lung disease, cyanotic congenital heart disease, or heart failure.16–18 For a preterm infant, the risks of hypovolemia, hypotension, acidosis, and postoperative apnea are magnified in the setting of operative blood loss and anemia. It is impossible to address all of the guidelines in this chapter, but many pediatric hematology and oncology consultants have clearly defined transfusion thresholds for their patient populations that should be reviewed preoperatively.
Guidelines for platelet transfusion have been published by consensus committees from France, the United Kingdom, and the United States; these reports are based on available evidence that has been gathered and critically reviewed (Table 9-2).15,19–22 Without evidence that platelet function is significantly different in the healthy infant and child, these guidelines should be applicable to these patients. The decision to transfuse platelets must take into account underlying medical conditions, platelet transfusion history, current medications, surgical bleeding, surgical interventions (e.g., cardiopulmonary bypass), and all other factors that may affect platelet function and turnover.23–27 Sevoflurane and propofol suppress28 and enhance platelet aggregation in vitro.29 Despite these effects on platelet aggregation, no change in the bleeding time has been reported, suggesting that the inhibitory effect does not impair hemostasis in vivo.30
| Medical Condition or Procedure | Platelet Count (/mm3) |
|---|---|
| Stable hematology-oncology or chronically thrombocytopenic patient | 10,000-20,000 |
| Lumbar puncture in stable leukemic child | 10,000 |
| Bone marrow aspiration or biopsy | 20,000 |
| Gastrointestinal endoscopy in cancer patient | 20,000-40,000 |
| Disseminated intravascular coagulation | 20,000-50,000 |
| Fiberoptic bronchoscopy in hematopoietic stem cell transplantation patient | 20,000-50,000 |
| Neonatal alloimmune thrombocytopenia | 30,000 |
| Major surgery | 50,000 |
| Dilutional thrombocytopenia with massive transfusion | 50,000 |
| Spinal anesthesia | 50,000 |
| Cardiopulmonary bypass | 50,000-60,000 |
| Liver biopsy | 50,000-100,000 |
| Nonbleeding preterm infant | 60,000 |
| Obstetric epidural anesthesia | 70,000-100,000 |
| Neurosurgery | 100,000 |
Data from references 19, 20, 23, 26, 27, 265.
Guidelines for the transfusion of other blood products, particularly fresh frozen plasma (FFP) and cryoprecipitate, have been established,15,26,31 and they are discussed later in the context of coagulation disorders. Indications32 for transfusing FFP are usually limited to the following:
1. Replacement of documented congenital or acquired coagulation factor deficiency when a specific sterilized or combined factor concentrate is unavailable, especially in the setting of anticipated or active bleeding
2. Acquired coagulopathy resulting from massive transfusion
3. Immediate reversal of warfarin’s effect
4. Coagulation support in disease processes such as disseminated intravascular coagulation and thrombotic thrombocytopenic purpura
5. A source of antithrombin III for children deficient of this inhibitor who require heparin
Guidelines have been established by the College of American Pathologists and other transfusion study groups for leukocyte reduction of RBC units,16 irradiation (x-ray or γ-ray) of cellular blood components,33 and administration of cytomegalovirus-seronegative RBCs (Tables 9-3 to 9-5).16 These guidelines are valuable when determining the specific choice of blood components that should be ordered and administered in the perioperative setting. For hematologic patients receiving chronic RBC transfusions, an extended phenotypic crossmatch and leukocyte reduction can decrease the risk of developing alloantibodies and transfusion reactions, especially in children of African descent if the local donor pool is primarily derived from Caucasian populations of Northern European descent.34 For oncology patients, updated specific requirements for blood products including leukocyte reduction and irradiation are often indicated and should always be reviewed with oncology specialists.
TABLE 9-3 Indications for Leukocyte-Reduced Red Blood Cell Units
Modified from Simon TL, Alverson DC, AuBuchon J, et al. Practice parameter for the use of red blood cell transfusions: developed by the Red Blood Cell Administration Practice Guideline Development Task Force of the College of American Pathologists. Arch Pathol Lab Med 1998;122:130-8.
TABLE 9-4 Indications for Irradiation of Cellular Blood Components
Modified from Simon TL, Alverson DC, AuBuchon J, et al. Practice parameter for the use of red blood cell transfusions: developed by the Red Blood Cell Administration Practice Guideline Development Task Force of the College of American Pathologists. Arch Pathol Lab Med 1998;122:130-8; Treleaven J, Gennery A, Marsh J, et al. Guidelines on the use of irradiated blood components prepared by the British Committee for Standards in Haematology blood transfusion task force. Br J Haematol 2010;152:35-51.
TABLE 9-5 Indications for Cytomegalovirus-Seronegative or Leukocyte-Reduced Red Blood Cells for Prevention of Virus Transmission
Modified from Simon TL, Alverson DC, AuBuchon J, et al. Practice parameter for the use of red blood cell transfusions: developed by the Red Blood Cell Administration Practice Guideline Development Task Force of the College of American Pathologists. Arch Pathol Lab Med 1998;122:130-8.
Hemolytic Anemias
Hereditary Spherocytosis
HS, the most common cause of inherited chronic hemolysis in North America and Northern Europe, has a prevalence of approximately 1 to 2 cases per 5000 people, if mild forms of the disease are included.35–37 First described in 1871, HS is present in many ethnic populations, but rare in African American populations. Because 75% of children inherit the disease in an autosomal dominant pattern, there is often a family history of the disorder, although autosomal recessive mutations, de novo mutations, and incomplete penetrance have been reported.37
Pathophysiology
Abnormalities in any of several erythrocyte membrane proteins, including the β subunit of spectrin, ankyrin, and band 3, can lead to HS. The variety of proteins affected and mutations observed in each gene account for the clinical heterogeneity of the disorder.36 When the erythrocyte loses surface area, it changes from a biconcave disk to a sphere, which alters its stability and flow pattern through the capillaries. The deformity leads to a loss of flexibility in the membrane, which makes it vulnerable to rupture, a condition that is worsened if the membrane surface area decreases by more than 3%.37 Damaged erythrocytes are sequestered in the splenic capillaries, which can lead to splenomegaly. The combination of intravascular and extravascular hemolysis can result in anemia, which induces extramedullary erythropoiesis. The life span of the erythrocyte is reduced from 120 days to just a few days when the RBC membrane has been deformed. If large numbers of damaged erythrocytes are lysed, unconjugated bilirubin is released into the bloodstream, which causes jaundice and possibly gallstones in as many as 60% of children.36 Membrane fragments from hemolytic reactions can lead to disseminated intravascular coagulation. Pulmonary hypertension may occur in the HS population, presumably as a result of hemolysis-induced alterations in NO metabolism.
Clinical and Laboratory Features
Children may present at any age with the triad of anemia, splenomegaly, and jaundice, which often is aggravated by concomitant viral infection. Mild, moderate, and severe forms of HS occur and are characterized by variations in laboratory results and clinical correlates. HS can manifest soon after birth and should be considered in infants who are jaundiced after the first postnatal week; resulting hyperbilirubinemia can sometimes necessitate an exchange transfusion. Mild disease occurs in 20% of children with HS; these children only occasionally present with symptomatic bilirubinate gallstones before adolescence. Approximately 5% of children have severe HS characterized by chronic anemic (hemoglobin concentration less than 8 g/dL) and a need for chronic transfusions. The course of this disease may be complicated by viral infections such as parvovirus B19 infection, which can suppress reticulocyte production37 and precipitate aplastic crises.
HS is most commonly suspected when numerous spherocytes with loss of central pallor appear on a peripheral smear. A complete blood cell count usually reveals a low hemoglobin and elevated reticulocyte count. Osmotic fragility remains the gold standard for the diagnosis of HS, but this test produces age-related results and must be performed by experienced laboratory technicians in a timely fashion. Increasingly, flow cytometry using eosin-5′-maleimide is being employed for diagnosis because it requires little blood and can be performed after overnight storage.38 As a direct result of chronic hemolysis, unconjugated bilirubin and serum lactate dehydrogenase concentrations increase, and serum haptoglobin concentrations decrease. Thrombocytopenia may develop as a result of hypersplenism.
Perioperative Considerations
Splenectomy significantly increases red cell survival in most cases and reduces the severity of the anemia and jaundice. It is usually reserved for more severe cases of HS, characterized by severe anemia that require frequent RBC transfusions, poor growth, chronic fatigue, or evidence of extramedullary hematopoiesis (e.g., frontal bossing). Splenic enlargement in a child interested in participating in contact sports is another indication.37 Splenectomy is ideally performed after the age of 6 years because of the increased risk of overwhelming infection by encapsulated organisms such as Streptococcus pneumoniae, Neisseria meningitidis, and Haemophilus influenzae type B in splenectomized younger children.39 Preoperative vaccination against these organisms is essential unless surgery is required emergently. Guidelines for the indications and duration of postoperative penicillin prophylaxis vary among institutions.36
Splenectomies in children are more frequently performed laparoscopically than by open laparotomy because the former is associated with decreased pain, quicker return of bowel function, shorter hospital stay, and improved cosmetic result. Conversion from laparoscopic to open splenectomy is necessary in fewer than 10% of cases.40 Partial splenectomies are increasingly performed because they allow retention of some immune function against bacterial infections in younger children while reducing the sequestration of spherocytes. However, residual splenic tissue can increase in size and necessitate total splenectomy at a later time.41,42 If anemia recurs after splenectomy, it may indicate the presence of accessory splenic tissue that was unrecognized initially. Transient postsplenectomy thrombocytosis marked by dramatic increases in platelet counts may also occur in children,35 in addition to a general increase in the risk of thromboembolic disease.
Gallstones occur in 21% to 63% of children with HS, but cholecystectomy is usually performed only when children are symptomatic with cholelithiasis. Children who undergo splenectomy and who also have radiographically identified gallstones may undergo concurrent cholecystectomy, whether the stones are symptomatic or not.43–45
Table 9-6 summarizes the clinical features and important perioperative considerations for the child with HS undergoing incidental or disease-related surgical procedures.
TABLE 9-6 Perioperative Concerns for Patients with Hereditary Spherocytosis
Hemoglobin, reticulocyte count, platelet count
History of transfusions and special blood requirements (e.g., extended phenotypic matching, leukocyte reduction)
History of infections, aplastic crises, and presplenectomy vaccinations
Presplenectomy antibiotic prophylaxis and immunization when indicated
Appropriate antibiotic coverage
Attention to physiologic effects of laparoscopy on circulatory and respiratory function
Potential for significant blood loss (unusual in splenectomy and cholecystectomy)
Judicious use of regional anesthesia, intramuscular medications, nasogastric tubes, nasal intubation, and other methods when platelet count is low
Limited use of medications with potential bleeding risk (e.g., ketorolac)
Glucose-6-Phosphate Dehydrogenase Deficiency
G6PD deficiency causes hemolysis in the presence of various oxidative stressors. It is the most common enzyme deficiency in humans, affecting approximately 400 million people worldwide. This enzyme deficiency is inherited in an X-linked, recessive fashion. Although males are most commonly affected, females (heterozygous or homozygous for the gene) may have clinical manifestations of the disease. More than 100 variants have been described, including a relatively mild form that affects about 10% of African American males (i.e., G6PD A−) and a more severe form that affects Italians, Greeks, and other populations in the Mediterranean, African, and Asian regions (i.e., G6PD Mediterranean).46–48 This deficiency is prevalent in geographic areas where the incidence of malaria is high, presumably because G6PD deficiency may attenuate the severity of malarial infections.
Pathophysiology
G6PD plays an important role in the hexose monophosphate/pentose phosphate shunt, which is essential for normal energy metabolism in erythrocytes. G6PD generates the reduced form of nicotinamide adenine dinucleotide phosphate (NADPH). NADPH maintains glutathione in the reduced form (GSH), which reduces peroxides and protects cells from oxidative damage in the course of normal biochemical events or in the event of excess free oxygen radical generation. Superoxide ion or hydrogen peroxide, or both, can oxidize hemoglobin, which then precipitates as insoluble membrane inclusions. These inclusions, together with the oxidative damage to cell membranes, lead to cell damage. Erythrocytes are particularly sensitive to oxidative damage because of their lack of synthetic activity. In the presence of oxidants and free radicals (e.g., produced by infection or by ingestion of certain medications and foods), this cascade of events may precipitate hemolysis in the G6PD-deficient child.46,48
Clinical and Laboratory Features
Clinical symptoms of G6PD deficiency may be deceptively variable, and they may occur in the neonatal period or in older age groups as episodic or chronic hemolytic anemia. Presenting signs include anemia and jaundice; in severe cases, these signs can be followed by lumbar and abdominal pain and by renal failure. Acute illness such as diabetic acidosis or ingestion of a variety of substances may precipitate a hemolytic event (Table 9-7). Hemolysis may range from benign and transitory to severe and life-threatening; the latter situation is more likely if the triggering agent is not eliminated or controlled. Laboratory findings include normocytic anemia, increased reticulocyte count and serum bilirubin concentration, and presence of Heinz bodies in the peripheral blood smear.
TABLE 9-7 Agents That May Precipitate Hemolysis in Patients with Glucose-6-Phosphate Dehydrogenase Deficiency
Perioperative Considerations
In the perioperative setting, G6PD deficiency does not usually cause problems if the triggering agents are avoided by susceptible children and the precipitating causes are treated or eliminated (Table 9-8). Monitoring for and treatment of possible complications are appropriate; transfusion is rarely required.
TABLE 9-8 Perioperative Concerns for Patients with Glucose-6-Phosphate Dehydrogenase Deficiency
Administration of large or excessive doses of medications such as prilocaine, benzocaine, and sodium nitroprusside may trigger hemolysis in G6PD-deficient children in the perioperative setting.46,48–50 Although these children can reduce methemoglobin that is normally produced by these agents, G6PD-deficient children may not tolerate large amounts of potent oxidizing agents (i.e., superoxide ion and hydrogen peroxide) produced by methemoglobin. Infants may be particularly susceptible to symptomatic methemoglobinemia (because of their low NADPH dehydrogenase activity) and to methemoglobin-induced hemolysis if they are G6PD deficient. Treatment of methemoglobinemia with methylene blue is contraindicated in these infants because the agent itself may precipitate hemolysis.42 Hemolysis has occurred during cardiopulmonary bypass in G6PD-deficient children,50,51 and methemoglobinemia has occurred in a child with partial G6PD deficiency after application of EMLA cream, a eutectic mixture of local anesthetics.52
Hemoglobinopathies
Sickle Cell Disease
First identified by Herrick about 100 years ago, sickle cell disease is a group of inherited hemoglobinopathies with a diverse worldwide prevalence. The disease affects about 1 in 375 African American and 1 in 20,000 Hispanic births.53 The spectrum of the disease includes sickle cell anemia (HbSS), which accounts for about 70% of the American sickle cell disease population; sickle cell/hemoglobin C disease (HbSC), accounting for about 20%; sickle cell/β-thalassemia (HbSβ), accounting for about 10%; and a host of other, uncommon sickle variants whose prevalence is increasing over time.54 HbSβ includes HbSβ+ and HbSβ0 thalassemias; the distinction depends on whether normal hemoglobin A (HbA) is expressed at all or in only a small concentration. HbSβ0, HbSC, and sickle cell/hemoglobin D disease (HbSD) and additional rare forms have the potential to sickle as severely as HbSS. Sickle cell trait (HbAS), in which approximately 40% of hemoglobin is hemoglobin S, occurs in about 8% of African Americans and in a much smaller percentage of Hispanic and other American subpopulations. The sickle gene is found commonly in Africa, Mediterranean areas, southwestern Asia, and other areas where malaria has been historically endemic and for which the gene is protective. Sickle hemoglobinopathies have many implications for perioperative care because they increase perioperative morbidity and mortality.
Pathophysiology
Hemoglobin A is composed of two α- and two β-globin chains. Hemoglobin S is caused by a mutant β-globin gene on chromosome 11, which leads to a single amino acid substitution (valine for glutamate at position 6). Replacement of negatively charged and hydrophilic glutamate by noncharged and hydrophobic valine leads to instability of the hemoglobin molecule and decreased solubility of the molecule when deoxygenated. Hemoglobin polymers form, generating long helical strands and inducing a process that leads to hemoglobin precipitation and hemolysis.55
The pathophysiologic process in sickle cell disease is much more complex than originally thought.55–57 Inflammation, vascular endothelial adhesion abnormalities, platelets, and coagulation cascade activation all contribute to vaso-occlusive episodes. The sickle red cell membrane, exposed to the destructive oxidant effects of intracellular iron, develops altered transmembrane ion transport pathways, which lead to altered permeability to sodium, potassium, and calcium, causing dehydration of the cell and irreversible sickling.58 Membrane abnormalities of phospholipid content also contribute to its deformability, and exposure of phosphatidyl serine facilitates activation of the clotting cascade. These and other factors lead to entrapment of irreversibly sickled red cells in the microcirculation, activation of coagulation and inflammatory pathways, ischemia, and infarction of tissue. At the same time, chronic intravascular hemolysis decreases production of NO, while increased scavenging decreases the bioavailability of NO. The resulting NO deficiency causes endothelial dysfunction and disease complications such as pulmonary hypertension, priapism, and skin ulceration.58,59
Clinical and Laboratory Features and Treatment
Sickle cell disease is a multisystem process involving potentially most organs of the body and often necessitating surgical intervention. Based on data collected in the early 1990s, approximately one third of patients with HbSS disease have progressive disease leading to organ dysfunction and death; about half have significant but less devastating disease; and the remainder have a reasonably stable, slowly progressive clinical course.60 Therapeutic interventions and genetic factors account in large part for the differences in outcome. Children with persistence of hemoglobin F (which itself protects against the effects of deoxygenation on red cells) and those with HbSC or HbSβ+ have fewer complications than those with HbSS or HbSβ0.
Early diagnosis and treatment of sickle cell disease have been facilitated by the widespread use of universal neonatal screening, which was first used in the state of New York in 1975. Most screening programs for sickle cell disease use isoelectric focusing of an eluate from dried blood spot samples, a technique that is also used to screen for other disorders. A few programs use high-performance liquid chromatography. Because a small percentage of children with sickle cell disease are not African American (i.e., Native American, Hispanic, and Caucasian),53 selective screening may not detect all affected infants. As of 2006, all 50 states and the District of Columbia screen all neonates for sickle hemoglobinopathies. Families of infants diagnosed with sickle trait (HbAS) on neonatal screening may not be made aware of the diagnosis, but these infants rarely develop significant clinical problems in the perioperative period.
Affected children born within the United States before universal neonatal screening and those born outside the United States and not receiving regular health care may not have received a diagnosis and appropriate care before surgery. Notwithstanding the controversy over the utility of nonselective preoperative screening,61 children at risk whose hemoglobin status is unknown preoperatively should be tested with a sickle-screening test, followed by a hemoglobin electrophoretic evaluation if screening is positive. However, infants younger than 6 months of age may have a false-negative screening test result because of presence of fetal hemoglobin, although electrophoresis is diagnostic at all ages. Children older than 10 years of age with a normal hemoglobin value, standard peripheral blood smear, and unremarkable clinical history are probably at a reduced risk for clinically significant hemoglobinopathy.62
Common clinical symptoms of sickle cell disease in children include chronic hemolytic anemia, recurrent vaso-occlusive episodes leading to pain, acute chest syndrome (ACS), infection, renal insufficiency, osteonecrosis, and cholelithiasis. Pulmonary hypertension, priapism, and skin ulcerations are related to the degree of red cell hemolysis.63 Chronic pulmonary and neurologic disease (e.g., stroke) are additional causes of significant morbidity and mortality.54 In the perioperative period, the most common complications in sickle cell children include ACS (about 10%), fever or infection (about 7%), vaso-occlusive episodes (about 5%), and transfusion-related events (about 10%).64
Chronic hemolytic anemia is a hallmark of HbSS disease. It is characterized by a baseline hemoglobin value of 5 to 9 g/dL (often more than 9 g/dL in HbSC disease), reticulocytosis (5% to 10%), and a distinctive red cell morphology observed on a peripheral smear.57 Chronic hemolysis is associated with increased red cell turnover and a propensity to form biliary stones. It may be complicated by other anemic events, such as acute splenic sequestration, typically occurring in infants and young children after a viral illness; and acute aplastic anemia, typically associated with parvovirus B19 infection. For some children, chronic and acute severe anemia are managed with RBC transfusions, although these children are prone to develop alloantibodies to RBC antigens, and untreated iron overload can lead to life-threatening cirrhosis and cardiac failure. Hydroxyurea is used to prevent vaso-occlusive episodes and end-organ damage. Most children are maintained on chronic folic acid therapy to prevent megaloblastic erythropoiesis that can result from the increased red cell production.
Vaso-occlusive episodes in sickle cell disease occur as a result of episodic microvasculature occlusions at one or more sites. The occlusive process occurs most commonly in the phalanges (i.e., dactylitis or hand-foot syndrome), long bones, ribs, sternum, spine, and the pelvis; it also can occur in the mesenteric microvasculature, producing abdominal pain that may mimic a surgical acute abdomen. Vaso-occlusive episodes are managed with hydration, warming, acute and chronic pain management (including opioids, antiinflammatory agents, and complementary modalities), and in-hospital care. It is essential to foster an ideal environment for pain control (e.g., calm, pleasant distractions, supportive personnel and objects). For children who have frequent or severe crises, oral hydroxyurea therapy has been effective in decreasing the frequency of events through several mechanisms, including inhibiting hemoglobin precipitation by increasing fetal hemoglobin concentrations, reducing the white blood cell count, modifying the inflammatory response, and facilitating NO metabolism.59,65,66 Inhaled NO may prove to be effective therapy for vaso-occlusive episodes.67
ACS is characterized by acute respiratory symptoms concurrent with a new infiltrate observed on the chest radiograph.68 ACS frequently occurs 2 to 3 days after a vaso-occlusive episode, and although its clinical presentation varies, it often includes fever, tachypnea, cough, and hypoxemia. The process may be self-limited over a period of a few days, or it may progress to respiratory failure (15%) and even death. The inconsistent presentation in part reflects the complex and variable pathogenesis of ACS. An episode may have a single or multiple causes, including infection (i.e., bacteria [often Chlamydia or Mycoplasma], viruses, and mixed flora), pulmonary fat embolism, pulmonary infarction, and pulmonary hemorrhage.69 Acute management includes supportive care and oxygen, antibiotics to cover encapsulated and atypical organisms, bronchodilators, pain control, ventilatory support as needed, and transfusion. Incentive spirometry or continuous positive airway pressure can be helpful, especially in the perioperative setting. Hydroxyurea therapy and chronic transfusion therapy decrease the frequency of ACS, whereas inhaled NO attenuates the process acutely.59,70–72 Airway reactivity is also common in children with sickle cell disease, in part due to NO deficiency, and it is responsive to bronchodilator therapy.73,74 In later life, children with sickle cell disease may develop restrictive lung disease and pulmonary hypertension as a result of repeated ACS-induced lung injury and chronic inflammation. NO deficiency, the result of decreased production, increased consumption, or altered metabolism, may also play an important role in these processes.63,68,75
Infection is a common problem in this disease because of deficits in the immunologic system and the specific effects of splenic atrophy and dysfunction that occur over the first few years of life.56 As a result of susceptibility to overwhelming infection by S. pneumoniae and H. influenzae type B, young children receive penicillin prophylaxis until 6 years of age and bacteria-specific immunizations. A host of infectious organisms have been implicated in ACS, and infection with gram-negative organisms (e.g., osteomyelitis caused by Salmonella) is common in older children and adults.69
Stroke is a devastating complication that occurs in children with sickle cell disease. A history of overt strokes is elicited in 10% or more of these children, and silent strokes occur in another approximately 15%; one fourth of children are at risk for motor or cognitive deficits at the time of presentation for surgery.76,77 A child’s first stroke often appears as early as 2 to 5 years of age.78,79 Risk factors include a reduced hemoglobin concentration, increased concentration of HbS, increased leukocyte count, and a history of dactylitis. Strokes may be precipitated by pain episodes, ACS, and infection.80 Children suffering an acute stroke are managed supportively with exchange transfusion to reduce the concentration of HbS to less than 30% and then chronic intermittent transfusions to minimize the risk of recurrence.81
The thrust of the current management of stroke is prevention. Based on the findings of the Stroke Prevention Trials (STOP 1 and 2), children are assessed annually by transcranial Doppler, and children with evidence of cerebrovascular compromise and some of those with magnetic resonance imaging abnormalities are managed with chronic transfusion therapy to minimize the risk of a stroke.82–84
Renal abnormalities in sickle cell disease develop from repeated sludging episodes, which may cause thrombosis, progressive infarction, and necrosis of the renal medulla. Proteinuria, hematuria, hyposthenuria, renal tubular acidosis, and other clinical abnormalities occur. Acute and chronic renal failure may develop.85,86 Renal dialysis and transplantation have been successful therapeutic interventions for this population.87
The clinical picture of sickle cell disease is altered to various degrees by concomitant qualitative and quantitative changes of hemoglobin. Sickle cell trait (HbAS) usually is benign, although it may be characterized by hematuria and hyposthenuria,87,88 and sickling may occur with HbAS under extremely altered physiologic circumstances (e.g., cardiopulmonary bypass).89,90 There is a small but significant risk of pulmonary emboli and sudden death with extreme exertion by individuals with HbAS, which has led to the mandatory offer of testing to all National Collegiate Athletic Association (NCAA) athletes.91
Children with HbSC disease usually have a greater baseline hemoglobin concentration and fewer complications than those with HbSS disease; delayed splenic autoinfarction reduces their risk for infection in early childhood.62 However, children with HbSC disease are more likely to have proliferative retinopathy and avascular necrosis of bone.56
Children with HbSβ0 (i.e., one sickle globin allele and one thalassemic allele expressing no β-globin) have a course identical to that of HbSS, whereas those with HbSβ+ (i.e., one sickle globin allele and one thalassemic allele expressing β-globin at a reduced level) tend to have a more benign course that is proportional to the amount of normal β-globin expression. The coexistence of hemoglobin S with α-thalassemia produces a variable clinical picture, but it may predispose children to a greater incidence of pain episodes.62
In addition to the treatment modalities mentioned earlier, many new and experimental modalities show promise in the treatment of sickle cell disease and its complications. They include induction of hemoglobin F by short-chain fatty acids such as butyrate or demethylating agents such as decitabine; membrane-active medications such as the Gardos channel inhibitors, magnesium; and antiadhesion therapies. New therapeutic targets are being identified, such a BCL11A, a zinc finger protein that plays a key role in the silencing of fetal globin genes. Reduction of BCL11A results in an induction of fetal globin.92 Hematopoietic stem cell transplantation (HSCT) is increasingly used as a curative intervention for sickle cell disease when instituted before development of organ dysfunction, but its application is limited in part by the lack of suitable donors.56,93 To circumvent this limitation, gene therapy protocols using a child’s own stem cells have been approved for use.
Perioperative Considerations
Perioperative morbidity and mortality are greater in children with sickle cell disease than in the general population. These children often require surgical procedures; the most common are cholecystectomy94; ear, nose, and throat procedures95; and orthopedic procedures (especially hip procedures for osteonecrosis).96 Placement of long-term vascular access for transfusions, antibiotics, analgesia, and other therapies is frequently performed. The Cooperative Study of Sickle Cell Disease reported that 7% of all deaths among children with this disease were related to surgery.54 Early reviews reported perioperative mortality rates as great as 10% and morbidity rates as great as 50% for children with sickle cell disease.97–100 Studies published in the 1990s indicated that the 30-day mortality rate was about 1%.101 In a group of more than 600 patients managed according to standard guidelines of care and prospectively studied, the incidence of any complication was about 30%, and the incidences of ACS and pain crisis were 10% and 5%, respectively.64 Patient factors (e.g., age, history of pulmonary disease, number of prior hospitalizations) and surgical factors (i.e., invasive or superficial) appear to affect the incidence of complications. The impact of newer interventions and technologies (e.g., laparoscopic and robotically assisted cholecystectomy and splenectomy)102–104 on perioperative morbidity and mortality rates is unclear.
The principles of optimal perioperative care are based on maintaining optimal physiologic parameters throughout the perioperative period, avoiding factors that may precipitate a sickle crisis, optimizing pain management, and close consultation among hematologists, surgeons, and anesthesiologists (Table 9-9).105 The child with sickle cell disease who is undergoing surgery should be viewed and managed primarily as a hematology patient whose care is being shared with, rather than assumed by, the surgeon and anesthesiologist during the perioperative period. Avoiding unnecessary and potentially dangerous surgical procedures (e.g., exploratory laparotomy to rule out appendicitis in a child who is experiencing an abdominal pain crisis) and minimizing perioperative complications should be the focus of the multidisciplinary care team. Based on a survey of perioperative management of sickle cell disease among anesthesiologists in North America, most anesthesiologists do consult with hematologists in all cases or on a case-by-case basis.106
TABLE 9-9 Perioperative Concerns for Patients with Sickle Cell Disease
Screening if unknown status in at-risk children
Primary management by hematology service (in most circumstances)
History of acute chest syndrome, vaso-occlusive pain crises, hospitalizations, transfusions, transfusion reactions
Neurologic assessment (e.g., strokes, cognitive limitations)
History of analgesic and other medication use
Oxygen saturation (on room air), chest radiograph
Pulmonary function tests (when appropriate)
Echocardiography (when appropriate)
Neurologic imaging (for recent changes)
Transfusion crossmatch (e.g., antibody-matched, leukocyte reduced, sickle negative)
Transfusion to correct anemia (in most circumstances)
Parenteral hydration for nil per os (NPO) status
Aggressive bronchodilator therapy
Appropriate antibiotic therapy, including presplenectomy antibiotics and immunizations (as indicated)
Maintenance of oxygenation, perfusion, normal acid–basis status, temperature, hydration
Availability of appropriately prepared blood (as indicated)
Anesthetic technique appropriate for procedure and postoperative analgesic requirements
Attention to physiologic effects of laparoscopy on circulatory and respiratory function
Appropriate antibiotic therapy
Judicious use of tourniquets, cell saver, and cardiopulmonary bypass
Management by hematology service
Monitoring for complications, especially acute chest syndrome and vaso-occlusive pain crises
Maintenance of oxygen saturation monitoring and supplementation as needed, including supplemental oxygen the first 24 hours regardless of oxygen saturation
Appropriate hydration (oral plus parenteral)
Appropriate antibiotic therapy
Incentive spirometry (possibly with continuous or bilateral positive airway pressure) and bronchodilator therapy
Although there is no evidence to support or refute many of the long-standing guidelines for perioperative care and individual practices vary widely,106 it seems appropriate to avoid the specific factors that may promote intravascular sickling: hypoxia, acidosis, hyperthermia, hypothermia, and dehydration.55 Meticulous attention to pain management is also essential because perioperative vaso-occlusive pain is common and is associated with ACS. Monitoring of vital signs throughout the perioperative period is mandatory, especially monitoring of oxygenation with pulse oximetry. Oxygen saturation as determined by pulse oximetry may underestimate measured oxygen saturation in patients with sickle cell disease, although usually not to a clinically significant degree.107,108 Because ACS, a common (10%) and potentially life-threatening complication of surgery, occurs 1 to 3 days postoperatively, it is important to extend adherence to guidelines of care into the postoperative period, regardless of the apparent well-being of the child.64 In light of the renal concentrating defect found in these patients, perioperative hydration is important to maintain and may require in-hospital preoperative care, although overhydration may compromise vulnerable cardiovascular and respiratory physiology.
Transfusion in the perioperative period remains a controversial subject.109 Transfusion of non-HbS RBCs to a child with sickle cell disease has several beneficial effects: correction of anemia, dilution of HbS red cells, compensation for blood loss, and prevention of some complications (e.g., stroke). However, transfusion is not without risks, including alloimmunization,34,110 transfusion reactions (about 7% in the perioperative period),64 infection, iron overload, time, and expense. Although there have been many reports of surgery performed safely in children with sickle cell disease without preoperative transfusion,111 uncontrolled studies indicate that preoperative transfusion does decrease the rate of perioperative complications.94,101 The Preoperative Transfusion in Sickle Cell Disease Study Group demonstrated prospectively in 604 operations (70% were cholecystectomies and otolaryngologic and orthopedic operations) that simple transfusion (i.e., correction of preoperative anemia to 10 g/dL with straight transfusion) was as effective as aggressive transfusion (i.e., lowering the preoperative HbS level to less than 30%, often with exchange transfusion) in preventing perioperative complications and was associated with fewer transfusion-related complications in children.64
To directly determine if transfusion prevents perioperative complications in the current era of surgical and anesthesia practices, an international randomized trial was initiated, the Transfusion Alternatives Preoperatively in Sickle Cell Disease (TAPS) trial. However, this trial was halted in March 2011 due to an excessive number of complications in the nontransfusion group. Although the final results of TAPS have not been published, it remains prudent to follow prior recommendations for transfusion for moderate and complicated operations in sickle cell patients. It is currently recommended that most children with HbSS undergoing most surgical procedures receive preoperative correction of anemia with simple transfusion to a hemoglobin concentration of about 10 g/dL. Children maintained on chronic transfusion programs (e.g., stroke prevention) should continue such management preoperatively. Recommendations for children with HbSC disease are less clear because these children typically maintain a baseline hemoglobin concentration at about 10 g/dL. For HbSC children who have a history of ACS, frequent pain crises, underlying pulmonary disease, or other complications, it is recommended that they receive selective preoperative exchange transfusion to reduce the HbS concentration without increasing total hemoglobin.112 Because of the high risk of alloimmunization in the sickle cell population, blood to be administered to these patients should undergo extended phenotype matching, including Rh, Cc, D, Ee, and Kell in addition to ABO84,113; leukocyte reduction; and sickle cell screening. Directed donation of blood from family members should be avoided if the child is a hematopoietic stem cell transplant candidate because it can lead to alloimmunization and later graft rejection.
Anesthetic technique does not have a clear effect on perioperative outcomes for children with sickle cell disease.114 Inhalational anesthetics do not affect the sickling process, although there is some experimental evidence suggesting that halothane may increase the viscosity of sickled blood.115 Pharmacokinetics of some agents commonly used with general anesthesia such as atracurium may be altered in this population.116 Regional anesthesia has been associated with an increased risk of postoperative complications in one retrospective study,101 but it has not been shown to affect perioperative outcome in others.94,96 Vasodilatory and analgesic properties of regional anesthesia can be effective in the management of vaso-occlusive episodes and priapism and in providing perioperative anesthetic care.117,118
Hyperventilation should be avoided because of its potential to reduce cerebral perfusion in children at an increased risk for stroke.119 The use of a tourniquet in HbSS and HbAS diseases has been questioned.120–122
[/not-level-membership-for-anesthesiology-category]
