14 Essentials of Cardiology
Syndromes, Associations, and Systemic Disorders: Cardiovascular Disease and Anesthetic Implications
Selected Vascular Anomalies and Their Implications for Anesthesia
Evaluation of the Child with a Cardiac Murmur
Basic Interpretation of the Electrocardiogram in Children
Essentials of Cardiac Rhythm Interpretation and Acute Arrhythmia Management in Children
Pacemaker Therapy in the Pediatric Age Group
Diagnostic Modalities in Pediatric Cardiology
Perioperative Considerations for Children with Cardiovascular Disease
Congenital Heart Disease
Incidence
Congenital heart disease (CHD) represents the most common form of congenital pathology, with an estimated incidence of 0.3% to 1.2% of live births.1 CHD is a major cause of morbidity and mortality during the neonatal period. However, with advances in medical and surgical management, including significant contributions related to anesthesia care, survival to adulthood is the expectation for most infants and children with CHD.2–4
A bicuspid aortic valve (Video 14-1) is the most common cardiac defect, occurring in up to 1% of the population.5,6 Ventricular septal defects (VSD) (Video 14-2)
is the most common cardiac defect, occurring in up to 1% of the population.5,6 Ventricular septal defects (VSD) (Video 14-2) represent the next most common congenital pathology,5,7–11 followed by secundum atrial septal defects (ASD) (Video 14-3)
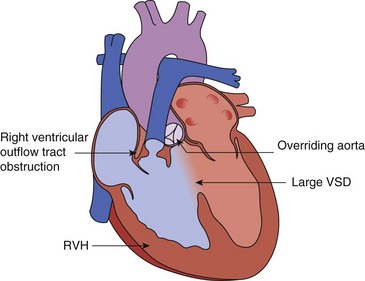
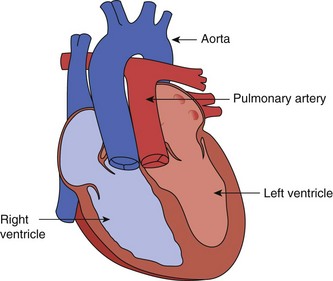
represent the next most common congenital pathology,5,7–11 followed by secundum atrial septal defects (ASD) (Video 14-3) .5,12 Among cyanotic lesions, tetralogy of Fallot (TOF) predominates, affecting almost 6% of children with CHD (Fig. 14-1).13 In the first week of life, d-transposition of the great arteries (Fig. 14-2) is the most frequently encountered cause of cardiac cyanosis; TOF is sometimes not detected until later in life because cyanosis is absent or only mild desaturation is present.
.5,12 Among cyanotic lesions, tetralogy of Fallot (TOF) predominates, affecting almost 6% of children with CHD (Fig. 14-1).13 In the first week of life, d-transposition of the great arteries (Fig. 14-2) is the most frequently encountered cause of cardiac cyanosis; TOF is sometimes not detected until later in life because cyanosis is absent or only mild desaturation is present.
Segmental Approach to Diagnosis
Several classification schemes have been proposed to characterize and categorize various congenital cardiac defects.14–20 The segmental approach to the diagnosis of CHD assumes a sequential, systematic analysis of the three major cardiac segments (i.e., atria, ventricles, and great arteries) to describe the anatomic abnormalities. The principle of this scheme is that specific cardiac chambers and vascular structures have characteristic morphologic properties that determine their identities, rather than their positions within the body.21 An organized, systematic identification of all cardiac structures or segments and their relationships (i.e., connections or alignments between segments) is carried out to define a child’s anatomy.22
The initial approach is to determine the cardiac position within the thorax and the arrangement or situs of the thoracic and abdominal organs. The cardiac position can be described in terms of its location within the thoracic cavity (Fig. 14-3) and the direction of the cardiac apex. Although the cardiac position within the thorax may be considered independent of the cardiac base-apex axis, for simplicity the following terms are used: levocardia if the heart is in the left hemithorax (as is normally the case); dextrocardia if the heart is located in the right hemithorax; and mesocardia if the heart is displaced rightward but not completely in the right thoracic cavity. An abnormal location of the heart within the thorax (i.e., cardiac malposition) may result from displacement by adjacent structures or underlying noncardiac malformations (e.g., diaphragmatic hernia, lung hypoplasia, scoliosis).
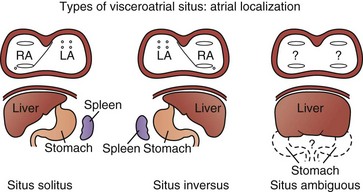
The visceral situs, or sidedness, of the abdominal organs (i.e., liver and stomach) and atrial situs are considered independently (Fig. 14-4). Visceral situs is classified as solitus (i.e., normal arrangement of viscera, with the liver on the right, stomach on the left, and a single spleen on the left), inversus (i.e., inversion of viscera, with the liver on the left and stomach on the right), or ambiguous (i.e., indeterminate visceral position). Abnormal arrangements or sidedness of the abdominal viscera, heart, and lungs suggests a high likelihood of complex cardiovascular disease. The atrial situs, atrioventricular (AV) connections, ventricular looping (i.e., position of the ventricles as a result of the direction of bending of the straight heart tube in early development), ventriculoarterial connections, and the relationship between the great vessels are then delineated.
Physiologic Classification of Defects
A physiologic classification system can facilitate understanding of the basic hemodynamic abnormalities common to a group of congenital or acquired lesions and assist in patient management (Table 14-1).23,24 Several classification schemes have been proposed, including some that categorize structural defects as simple or complex lesions, consider the presence or absence of cyanosis, or consider whether pulmonary blood flow is increased or decreased.25–27 The following approach groups pediatric heart disease into six broad categories according to the underlying physiology or common features of the pathologies.
TABLE 14-1 Physiologic Classification of Congenital Heart Disease (Representative Lesions)
Volume Overload Lesions
Volume overload lesions typically are caused by left-to-right shunting at the atrial, ventricular, or great artery levels. If the location of the left-to-right shunt is proximal to the mitral valve (e.g., ASDs, partial anomalous pulmonary venous return, unobstructed total anomalous pulmonary venous return), right heart dilation will occur. Lesions distal to the mitral valve (e.g., VSD, patent ductus arteriosus [PDA], truncus arteriosus) lead to left heart dilation. Children with AV septal defects (i.e., AV canal defects) also fit into this category. The magnitude of the shunt and resultant pulmonary-to-systemic blood flow ratio (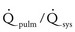 ) dictate the presence and severity of the symptoms and guide medical and surgical therapies. Diuretic therapy and afterload reduction are beneficial in controlling pulmonary overcirculation and ensuring adequate systemic cardiac output. Transcatheter approaches or surgical interventions may be required to address the primary pathology associated with ventricular volume overload (see Chapter 20).
) dictate the presence and severity of the symptoms and guide medical and surgical therapies. Diuretic therapy and afterload reduction are beneficial in controlling pulmonary overcirculation and ensuring adequate systemic cardiac output. Transcatheter approaches or surgical interventions may be required to address the primary pathology associated with ventricular volume overload (see Chapter 20).
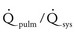 ratio, requiring diuretic therapy and manipulation of the systemic and pulmonary vascular resistances to control blood flow.
ratio, requiring diuretic therapy and manipulation of the systemic and pulmonary vascular resistances to control blood flow.Parallel Circulation
In the neonate with d-transposition of the great arteries, the pulmonary and systemic circulations operate in parallel rather than in the normal configuration in series. In this condition, the right ventricle ejects deoxygenated blood into the aorta, and the left ventricle ejects oxygenated blood into the pulmonary circulation. Mixing of blood in this setting may occur at the atrial, ventricular, or ductal levels (see Fig. 14-2). Although prostaglandin E1 therapy maintains ductal patency, balloon atrial septostomy to create or enlarge an existing restrictive interatrial communication and optimize mixing may benefit some infants. Mixing at the atrial level is considered much more effective than at the ventricular or ductal levels.
Single-Ventricle Lesions
This category is the most heterogeneous group, consisting of defects associated with AV valve atresia (i.e., tricuspid atresia), heterotaxy syndromes, and many others.28 In some cases, both atria empty into a dominant ventricular chamber (i.e., double-inlet left ventricle), and although a second rudimentary ventricle may be present, the physiology is that of a single-ventricle or univentricular heart. Other cardiac malformations with two distinct ventricles (i.e., unbalanced AV septal defect) may also be considered in the functional single-ventricle category because of associated defects that may preclude a biventricular repair. A common feature of these lesions is complete mixing of the systemic and pulmonary venous blood at the atrial or ventricular level. Another common finding is aortic or pulmonary outflow tract obstruction.
Affected children are a challenge to the practitioner and require careful delineation of their anatomy. An important goal in single-ventricle management involves optimization of the balance between the pulmonary and systemic circulations early in life. This is a critical issue because low pulmonary vascular resistance and limitation of the ventricular volume load represent a prerequisite for later palliative strategies and favorable outcomes in these children. These considerations are also relevant during anesthesia management for noncardiac surgery (see Chapter 21).26,29,30
Acquired Heart Disease
Cardiomyopathies
The term cardiomyopathy usually refers to diseases of the myocardium associated with cardiac dysfunction.31,32 They have been classified as primary and secondary forms. The most common types in children are hypertrophic, dilated or congestive, and restrictive cardiomyopathies. Other forms include left ventricular noncompaction33–35 and arrhythmogenic right ventricular dysplasia.36 Secondary forms of cardiomyopathies are those associated with neuromuscular disorders such as Duchenne muscular dystrophy, glycogen storage diseases (i.e., Pompe disease), hemochromatosis or iron overload, and mitochondrial disorders. Chemotherapeutic agents such as anthracyclines may result in dilated cardiomyopathy.37 It is important to understand the hemodynamic processes behind the myocardial disease and implications for acute and chronic management.
Hypertrophic cardiomyopathy (HCM) is characterized by ventricular hypertrophy without an identifiable hemodynamic cause that results in increased thickness of the myocardium. This accounts for almost 40% of cardiomyopathies in children.38–41 The condition represents a heterogenous group of disorders, and most of the identified genetic defects exhibit autosomal dominant inheritance patterns.42,43 This is the most common cause of sudden cardiac death (SCD) in athletes.44,45 Most children with HCM do not have systemic outflow tract obstruction (i.e., nonobstructive cardiomyopathy). It is unclear whether the few with hypertrophic obstructive cardiomyopathy, previously known as idiopathic hypertrophic subaortic stenosis, are at increased risk for SCD compared with children without obstruction.44
Most children with HCM present for evaluation of a heart murmur, syncope, palpitations, or chest pain. Occasionally, an abnormal electrocardiogram (ECG) leads to referral. An accurate family history is essential. An apical impulse is often prominent. Auscultation may reveal a systolic ejection outflow murmur that becomes louder with maneuvers that decrease preload or afterload (e.g., standing, Valsalva maneuver) or increased contractility. The murmur decreases in intensity with squatting and isometric hand grip. A mitral regurgitant murmur may also be present. The ECG meets criteria for left ventricular hypertrophy in most children (Fig. 14-5). In some, the electrocardiographic findings may be striking (Fig. 14-6). A hypertrophied, nondilated left ventricle is a diagnostic feature determined by echocardiography (Video 14-4, A and B) .45 In many children, the hypertrophy may be asymmetric (Video 14-5, A and B)
.45 In many children, the hypertrophy may be asymmetric (Video 14-5, A and B) . Echocardiography is the primary imaging modality for long-term assessment of wall thickness, ventricular dimensions, presence and severity of obstruction, systolic and diastolic function, valve competence, and response to therapy. Other diagnostic approaches such as cardiac catheterization and magnetic resonance imaging (MRI) may add helpful information in some cases.
. Echocardiography is the primary imaging modality for long-term assessment of wall thickness, ventricular dimensions, presence and severity of obstruction, systolic and diastolic function, valve competence, and response to therapy. Other diagnostic approaches such as cardiac catheterization and magnetic resonance imaging (MRI) may add helpful information in some cases.
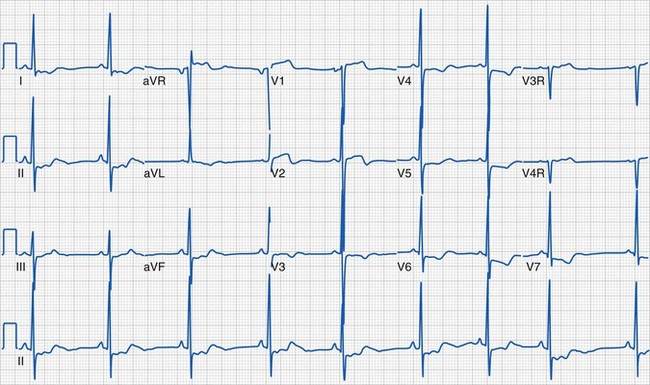
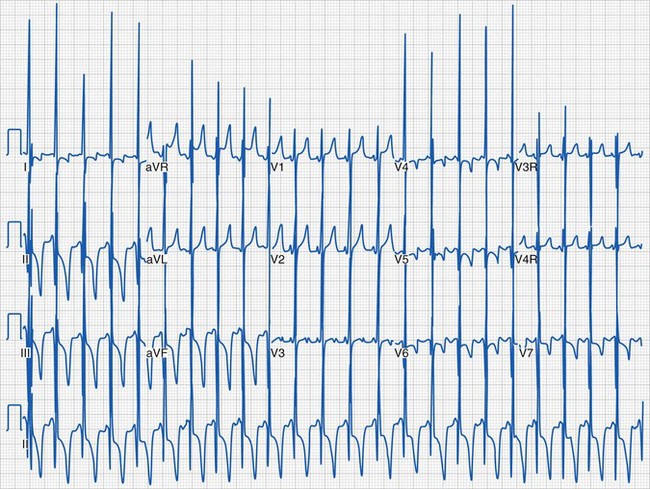
The care of children with HCM includes maintenance of adequate preload, particularly in those with dynamic obstruction. Diuretics are not indicated and often worsen the hemodynamic state by reducing left ventricular volume and increasing the outflow tract obstruction. Drugs that augment myocardial contractility (e.g., inotropic agents, calcium infusions) are not well tolerated. Patients usually undergo continuous electrocardiographic monitoring (i.e., Holter recording) and exercise testing for risk stratification.46 β-Blockers and calcium channel blockers are the primary agents for outpatient drug therapy.47 Therapies range widely and include longitudinal observation with medical management of heart failure and arrhythmias, implantation of cardioverter-defibrillators, surgical myotomy or myectomy, transcatheter alcohol septal ablation, and cardiac transplantation.
Dilated cardiomyopathy (DCM), also known as congestive cardiomyopathy, is characterized by thinning of the left ventricular myocardium, dilation of the ventricular cavity, and impaired systolic function.48–50 The broad number of etiologies range from genetic or familial forms to those caused by infections,51 metabolic derangements, toxic exposures, and degenerative disorders.52,53 Chronic tachyarrhythmias can also lead to DCM that may or may not improve after the rhythm disturbance is controlled.54–56
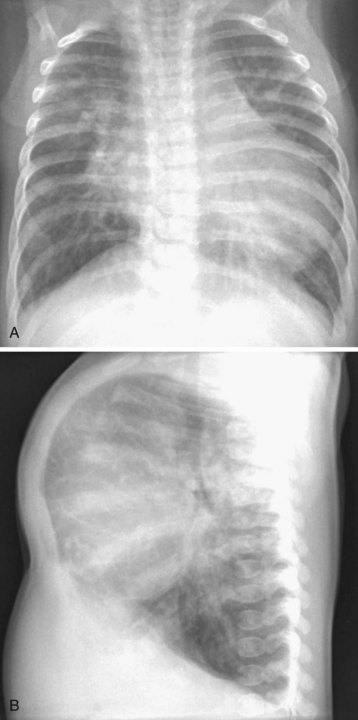
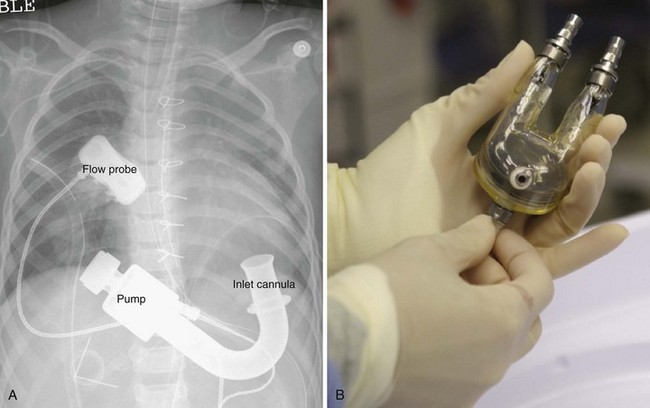
Most children with DCM present with signs and symptoms of congestive heart failure (e.g., tachypnea, tachycardia, gallop rhythm, diminished pulses, hepatomegaly). The chest radiograph typically demonstrates cardiomegaly, pulmonary vascular congestion, and in some cases, atelectasis (Fig. 14-7). The ECG may identify the likely cause of the cardiac dysfunction in those with cardiomyopathy due to rhythm disorders or anomalous origin of the left coronary artery from the pulmonary root (ALCAPA). The echocardiogram can confirm the diagnosis by demonstrating a dilated left ventricle with decreased systolic function (Video 14-6, A and B) .57 Therapy in the acute setting is supportive and aimed at stabilization. Management may include afterload reduction, inotropic support, and mechanical ventilation. Unlike children with HCM, those with DCM have a volume-loaded, poorly contractile ventricle. Gentle diuresis is beneficial. The infusion of large fluid boluses may be poorly tolerated and result in hemodynamic decompensation and cardiovascular collapse. The outcomes of children with dilated cardiomyopathy vary. For most, recovery of left ventricular systolic function occurs, but others eventually require cardiac transplantation.58 In a subset of children with severe disease, mechanical circulatory support may be necessary as a bridge to recovery or cardiac transplantation (Fig. 14-8) (see Chapter 19).59–61
.57 Therapy in the acute setting is supportive and aimed at stabilization. Management may include afterload reduction, inotropic support, and mechanical ventilation. Unlike children with HCM, those with DCM have a volume-loaded, poorly contractile ventricle. Gentle diuresis is beneficial. The infusion of large fluid boluses may be poorly tolerated and result in hemodynamic decompensation and cardiovascular collapse. The outcomes of children with dilated cardiomyopathy vary. For most, recovery of left ventricular systolic function occurs, but others eventually require cardiac transplantation.58 In a subset of children with severe disease, mechanical circulatory support may be necessary as a bridge to recovery or cardiac transplantation (Fig. 14-8) (see Chapter 19).59–61
Restrictive cardiomyopathy (RCM) is the least common of the major types of cardiomyopathies (5%) and portends a poor prognosis when it manifests during childhood.62–67 The disorder is characterized by diastolic dysfunction related to a marked increase in myocardial stiffness resulting in impaired ventricular filling. Most cases are thought to be idiopathic. Presenting symptoms are nonspecific and primarily respiratory. Occasionally, the diagnosis is made after a syncopal or sudden near-death event. The physical examination may demonstrate hepatomegaly, peripheral edema, and ascites.
The echocardiographic hallmark of RCM is that of severe atrial dilation and normal- to small-sized ventricles (Video 14-7) . The marked diastolic dysfunction leads to increased end-diastolic pressures, left atrial hypertension, and secondary pulmonary hypertension. Children with RCM are prone to thromboembolic complications, and anticoagulation therapy is frequently recommended. This is an important consideration during perioperative care because adjustments in the anticoagulation regimen may be necessary. Atrial and ventricular tachyarrhythmias may also occur. Optimal medical treatment is controversial because no specific agents or strategies have been shown to significantly alter outcomes.67 Similar to children with HCM, diuretics often cause a decrease in the needed preload with detrimental effects on hemodynamics. Inotropic agents are not indicated because systolic function is preserved and the arrhythmogenic properties of inotropic drugs can induce a terminal event. In many centers, cardiac transplantation has been effectively used.68,69
. The marked diastolic dysfunction leads to increased end-diastolic pressures, left atrial hypertension, and secondary pulmonary hypertension. Children with RCM are prone to thromboembolic complications, and anticoagulation therapy is frequently recommended. This is an important consideration during perioperative care because adjustments in the anticoagulation regimen may be necessary. Atrial and ventricular tachyarrhythmias may also occur. Optimal medical treatment is controversial because no specific agents or strategies have been shown to significantly alter outcomes.67 Similar to children with HCM, diuretics often cause a decrease in the needed preload with detrimental effects on hemodynamics. Inotropic agents are not indicated because systolic function is preserved and the arrhythmogenic properties of inotropic drugs can induce a terminal event. In many centers, cardiac transplantation has been effectively used.68,69
Myocarditis
Myocarditis is defined as inflammation of the myocardium, often associated with necrosis and myocyte degeneration. In the United States, it is most often caused by a viral infection. Over the past 20 years, the spectrum of viral pathogens causing myocarditis has changed, such that adenovirus, enteroviruses (e.g., coxsackievirus B), and parvovirus have become the most frequent causes of fulminant disease.70–72
The overall incidence of myocarditis is unknown because it is frequently underdiagnosed and unrecognized as a nonspecific viral syndrome. A large, 10-year, population-based study on cardiomyopathy found an annual incidence of 1.24 cases per 100,000 children younger than 10 years of age; only a fraction of cases represented those with myocarditis.73 The diagnosis is made using clinical history, physical examination, and imaging modalities. Myocarditis is highly suspected when a child presents with new-onset congestive heart failure or ventricular arrhythmias without evidence of structural heart disease. The ECG typically demonstrates low-voltage QRS complexes with tachycardia, which sometimes is ventricular in origin. Chest radiography often shows cardiomegaly with pulmonary vascular congestion (Fig. 14-9). Echocardiography displays ventricular dilation with decreased systolic function, similar to DCM, and it is useful in the exclusion of alternative diagnoses, such as pericardial effusion or anomalous origin of a coronary artery. Myocarditis is a clinical diagnosis because definitive confirmation requires the analysis of tissue obtained through myocardial biopsy in the catheterization laboratory or the operating room (rarely performed).74
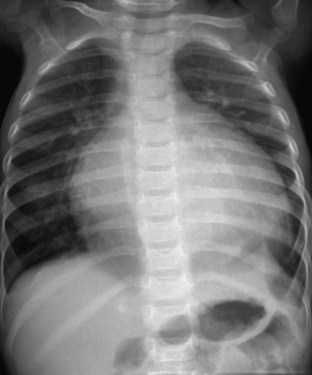
Many children with myocarditis have subclinical or mild clinical disease, whereas others progress to overt heart failure or arrhythmias, or both. Among children with heart failure, approximately one third will regain full ventricular function, one third will recover but continue to demonstrate impaired systolic function, and one third will require cardiac transplantation.75,76 A subset of children, not all of whom initially manifest severe symptoms in the acute period, will progress to have DCM.
Although no specific therapies have been identified to directly treat the myocardial injury, a variety of strategies have been employed.77–80 The current paradigm includes diuresis and afterload reduction to improve myocardial performance without placing a large burden on an already failing heart.81 Rhythm disturbances are treated appropriately. Therapy with immune modulation or suppression with intravenous immunoglobulin is the standard of care at many centers.81–83 Mechanical circulatory support may be required in fulminant disease.84–89
Rheumatic Fever and Rheumatic Heart Disease
Acute rheumatic fever and rheumatic heart disease are leading causes of acquired cardiac disease in developing countries and still occur, albeit infrequently, in developed countries.90 In the United States, the availability of antibiotic therapy for streptococcal pharyngitis has markedly reduced the incidence of this disease, but sporadic cases still occur.91 In children, the peak incidence occurs between 5 and 14 years of age.
The clinical diagnosis of rheumatic fever is based on the modified Jones criteria.92 Major criteria include carditis, polyarthritis, chorea, subcutaneous nodules, and erythema marginatum. Without a history of rheumatic fever or echocardiographic evidence of typical valvular involvement, the diagnosis in children requires evidence of a prior streptococcal infection along with two major criteria or one major and two minor criteria. Fever and arthritis are common symptoms. The polyarthritis has a migratory pattern, typically affecting large joints. Cardiac involvement or carditis occurs in almost 50% of children with their first attack of rheumatic fever. Rheumatic heart disease represents a sequela of the acute process, and it most frequently affects the mitral and aortic valves.
Primary prevention of rheumatic fever and rheumatic heart disease begins with prompt recognition and appropriate treatment of the initial streptococcal infection.93,94 Secondary prevention, with ongoing therapy in individuals with a known history of rheumatic fever, has been extremely effective in preventing recurrent attacks. Although there is debate regarding the optimal regimen, intramuscular injections of benzathine penicillin every 3 to 4 weeks appear to be most efficacious.95
In a subset of children with severe cardiac involvement, elective or emergent surgery may be required.96 Valvular disease, rather than global myocarditis, is often the cause of congestive symptoms; medical management therefore has limited efficacy. Valve repair is preferred to replacement.
Infective Endocarditis
Causes and Treatment
Children with structural or acquired heart disease are at risk for infective endocarditis.97,98 The risk to a great extent is based on the nature of the cardiac condition. The infection results from deposition of bacteria or other pathogens on tissues in areas of abnormal or turbulent blood flow. The diagnosis of endocarditis is made clinically by applying the modified Duke criteria (Table 14-2).99,100 Major criteria include demonstration of microorganisms and evidence of pathologic lesions. The presentation of the disease may be acute or subacute. New or changing heart murmurs may indicate the development of regurgitation or obstruction on an affected valve. Among the physical findings may be signs of systemic embolization (i.e., minor criteria). Splinter hemorrhages (i.e., linear streaks under the nail beds), Janeway lesions (i.e., painless macules on the hands or feet), Osler nodes (i.e., small, painful nodules on the fingers), and Roth spots (i.e., retinal hemorrhages with clear centers) may also be present. Inflammatory markers, such as erythrocyte sedimentation rate and C-reactive protein, are typically increased, albeit nonspecific. Microscopic hematuria, as a manifestation of renal involvement, is frequently seen.
TABLE 14-2 Modified Duke Criteria for Infective Endocarditis
Typical microorganism consistent with IE from two separate blood cultures:
Viridians streptococcus, Streptococcus bovis, HACEK† group of microorganisms, Staphylococcus aureus, or community-acquired enterococci without a primary focus
Microorganism consistent with IE from persistently positive blood cultures
Single positive blood culture for Coxiella burnetii or antiphase I IgG antibody titer greater than 1 : 800
Positive echocardiogram for IE:
Oscillating intracardiac mass on valve or supporting structures, in the path of regurgitant jets, or on implanted material in the absence of an alternative anatomic explanation, or abscess
New partial dehiscence of prosthetic valve
New valvular regurgitation (worsening of or change in preexisting murmur not sufficient)
Predisposition: predisposing cardiac condition or intravenous drug use
Vascular phenomena: major arterial emboli, septic pulmonary infarctions, mycotic aneurysm, intracranial hemorrhage, conjunctival hemorrhages, and Janeway lesions
Immunologic problems: glomerulonephritis, Osler nodes, Roth spots, and rheumatoid factor
Microbiologic evidence: positive blood culture that does not meet a major criterion (above) or serologic evidence of active infection with organism consistent with IE
*Duke Clinical Criteria for Infective Endocarditis requires two major criteria or one major and three minor criteria or five minor criteria to establish the diagnosis.
†For definition, see http://en.wikipedia.org/wiki/HACEK_endocarditis
Modified from Li JS, Sexton DJ, Mick N, et al. Proposed modifications to the Duke criteria for the diagnosis of infective endocarditis. Clin Infect Dis 2000;30:633-8.
Acute bacterial endocarditis is most commonly caused by Staphylococcus aureus.101,102 The clinical presentation includes high fevers, chills, myalgias, fatigue, and lethargy. Some children present in a critically ill state or in shock. Both left- and right-sided endocarditis can occur in children with CHD.103 Children with indwelling venous catheters have an expanded spectrum of pathogens known to cause acute endocarditis, including coagulase-negative staphylococcal species or other nonbacterial organisms.
Initial evaluation for bacterial endocarditis includes serial blood cultures drawn from separate sites before initiation of antimicrobial therapy. The temporal frequency of cultures depends on the clinical scenario and stability of the child. In up to 20% of children with evidence of endocarditis, a pathogen cannot be isolated (i.e., negative culture), requiring empirical treatment throughout. Transthoracic echocardiography is routinely performed to evaluate evidence of vegetations or other abnormalities.104 Although visualization of a vegetation establishes the diagnosis, a negative study does not exclude the diagnosis. Depending on how strongly the diagnosis is suspected, further imaging, including transesophageal echocardiography, may be necessary (Video 14-8, A and B) .105 These imaging modalities are also valuable during follow-up.
.105 These imaging modalities are also valuable during follow-up.
Parenteral antibiotics are initiated after blood cultures are collected. Broad-spectrum agents are used initially, and after a pathogen has been identified, the antibiotic regimen is narrowed.97 Daily blood cultures are obtained until three consecutive cultures remain sterile. A prolonged course of therapy (e.g., 4 to 6 weeks) is required in all children. This can be facilitated by placement of a peripherally inserted central catheter (PICC) line. Home therapy for endocarditis may be feasible in some patients, but it depends on many factors, including clinical status, initial response to antibiotics, sensitivity of organism to antimicrobial therapy, and ability of infrastructure to support outpatient treatment of a serious infection (e.g., parental or family member’s ability, home health care provider).
A high level of suspicion for endocarditis must be maintained when evaluating a child with known heart disease and with persistent bacteremia (or fungemia) or a fever of unknown origin. The same holds true for any child with foreign material in the heart or vascular tissue, such as indwelling central venous catheters, pacemakers or defibrillators, and closure devices.106
Endocarditis Prophylaxis
The risk for developing endocarditis from transient bacteremia is extremely low in children with normal intracardiac anatomy, however, certain cardiac conditions are predisposed to acquiring endocarditis. The American Heart Association guidelines do not recommend antibiotic prophylaxis based exclusively on an increased lifetime risk of endocarditis, but they propose that it should be restricted to those at greatest risk for an adverse outcome resulting from endocarditis. Children in this category include those with prosthetic cardiac valves, a history of endocarditis, certain congenital heart defects, after specific interventions, and cardiac transplant recipients with valvular disease (Table 14-3).107
TABLE 14-3 Cardiac Conditions Associated with the Greatest Risk of Adverse Outcomes from Endocarditis
• Previous infective endocarditis
• Congenital heart disease (CHD)*
• Cardiac transplantation recipients who develop cardiac valvulopathy
*Except for the conditions listed, antibiotic prophylaxis is no longer recommended for other forms of CHD.
†Prophylaxis is recommended because endothelialization of prosthetic material occurs within 6 months after the procedure.
Modified with permission from Wilson W, Taubert KA, Gewitz M, et al., editors. Prevention of infective endocarditis: guidelines from the American Heart Association. A guideline from the American Heart Association Rheumatic Fever, Endocarditis, and Kawasaki Disease Committee, Council on Cardiovascular Disease in the Young, and the Council on Clinical Cardiology, Council on Cardiovascular Surgery and Anesthesia, and the Quality of Care and Outcomes Research Interdisciplinary Working Group. Circulation 2007;116:1736-54.
Transient bacteremia may occur during dental procedures that involve the gingival tissues or the periapical region of teeth or perforation of the oral mucosa. Although several respiratory tract procedures are associated with transient bacteremia, no definitive data demonstrate a cause-and-effect relationship between these procedures and endocarditis. Caution may be warranted for children at high risk undergoing invasive procedures of the respiratory tract that involve incision or biopsy of the mucosa. In contrast to previous guidelines, routine prophylactic administration of antibiotics solely to prevent endocarditis is not recommended for those undergoing genitourinary or gastrointestinal tract procedures. However, for specific clinical scenarios, antibiotic prophylaxis may be considered in these children. Routine endoscopy or transesophageal echocardiography does not merit routine antibiotic administration.108 Prophylaxis is not considered necessary for cardiac catheterization; and although many practitioners routinely administer antibiotics during transcatheter placement of devices, there is insufficient evidence to support this practice.109
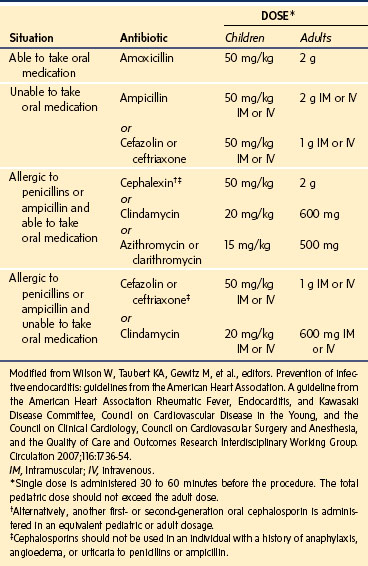
The guidelines recommend the administration of antibiotic prophylaxis 30 to 60 minutes before the procedure to achieve adequate tissue levels of antibiotics before bacteremia occurs (Table 14-4).107 The standard prophylactic regimen for children is for oral amoxicillin. For the child who is allergic to penicillin or ampicillin, oral alternatives include cephalexin, clindamycin, azithromycin, or clarithromycin. In children who are unable to ingest oral medications, alternative antibiotics include ampicillin, cefazolin, and ceftriaxone by an intravenous or intramuscular route. If the child is allergic to penicillin or ampicillin and unable to swallow oral medications, cefazolin, ceftriaxone, or clindamycin may be used.
TABLE 14-4 American Heart Association Guidelines for the Prevention of Infective Endocarditis: Antibiotic Regimens
Although many health care providers have adopted the updated recommendations, some have been hesitant to alter their practice regarding endocarditis prophylaxis, particularly when caring for patients with CHD undergoing gastrointestinal or genitourinary procedures.110 Only the American Dental Association has adopted the new guidelines for endocarditis prophylaxis; other associations such as the gastrointestinal and genitourinary groups remain uncommitted to the new guideline.
Kawasaki Disease
Kawasaki disease (i.e., mucocutaneous lymph node syndrome) represents a fairly common and potentially fatal form of systemic vasculitis of unknown origin.111–115 It is seen predominantly in infants and young children. The disease can affect the coronary arteries resulting in dilation and aneurysm formation.116,117
The diagnosis relies on clinical features. To meet criteria, a child must have persistent fevers and at least four of the following findings118,119:

 Peripheral extremity changes (e.g., erythema, desquamation, edema of the hands or feet)
Peripheral extremity changes (e.g., erythema, desquamation, edema of the hands or feet)
 Bilateral, nonexudative conjunctivitis
Bilateral, nonexudative conjunctivitis
 Cervical lymphadenopathy (often unilateral)
Cervical lymphadenopathy (often unilateral)
 Oral changes (i.e., strawberry tongue; red, dry, or cracked lips)
Oral changes (i.e., strawberry tongue; red, dry, or cracked lips)
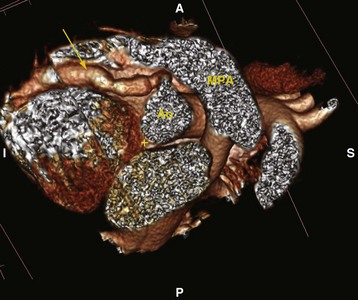
Intravenous gamma globulin (IVIG) and high-dose aspirin are recommended during the acute phase of the disease. The incidence of coronary artery aneurysms is significantly reduced if IVIG is administered within the first 10 days of the illness.119 The presence of coronary artery aneurysms is considered diagnostic for Kawasaki disease (Fig. 14-10). In children with coronary artery aneurysms, low-dose aspirin therapy is administered, in some cases in combination with anticoagulants or antiplatelet drugs.120 Myocardial ischemia and infarction, although uncommon, are important potential complications.117,121 Anesthetic care in these children requires careful consideration regarding myocardial oxygen demand and supply; on rare occasions, coronary revascularization may be necessary.
Cardiac Tumors
Because cardiac tumors are rare in children, the natural history and optimal treatment strategies are often determined from limited case series.122–124 Atrial myxomas represent more than 90% of cardiac tumors in adults, but in children, they tend to be rhabdomyomas or fibromas.125 Less common types include hemangiomas, myxomas (Video 14-9) , Purkinje cell tumors, and teratomas. In adults, most tumors are found in the left atrium, but cardiac tumors in children occur in all four cardiac chambers. Malignant primary tumors are rare, and data on their outcomes are limited. Other nonprimary cardiac tumors, such as neuroblastoma, can invade vascular structures and extend into the heart.
, Purkinje cell tumors, and teratomas. In adults, most tumors are found in the left atrium, but cardiac tumors in children occur in all four cardiac chambers. Malignant primary tumors are rare, and data on their outcomes are limited. Other nonprimary cardiac tumors, such as neuroblastoma, can invade vascular structures and extend into the heart.
Rhabdomyomas are the most common primary cardiac tumors in children. They often involve the ventricular septum and left ventricle and are multiple in most cases.126 Although they are considered benign, children may present with cardiomegaly, congestive heart failure, arrhythmias, or sudden death. The significance of a rhabdomyoma is determined largely by its size and any obstruction it may cause. Many tumors regress over time or completely resolve; surgery is not indicated unless symptoms are present.127,128 Many children with cardiac rhabdomyomas have associated tuberous sclerosis.129,130
Cardiac fibromas are the second most common type of pediatric primary cardiac tumors.131 They are typically single and involve the ventricular free wall. In a subset of fibromas, the tumor can invade the conduction system.132 Surgery or cardiac transplantation may be required.133,134 The tumors can be very large, and complete surgical resection may result in severely depressed cardiac function. Partial resections have been found to result in an arrest in growth with good outcomes while sparing cardiac function.128
The primary concerns in the perioperative care of children with cardiac tumors are the impact of the mass on hemodynamics and the associated abnormalities of cardiac rhythm.135
Heart Failure in Children
Definition and Pathophysiology
Heart failure has become a major field of interest and investigation in pediatric cardiology and the subject of various publications,136 scientific meetings, and several textbooks.137,138 Pediatric heart failure results from markedly different causes from those reported in adults.139 The cellular basis of heart failure, compensatory mechanisms, and therapeutic advances represent areas of interest in children.136–138,140,141 The following discussion highlights key concepts as they relate to anesthetic practice.
Heart failure is considered to be a pump failure and a circulatory failure involving neurohumoral aspects of the circulation.142 Several conditions may ultimately compromise the ability to generate an adequate cardiac output to meet the systemic circulatory demands. This disease state does not necessarily imply impairment of ventricular systolic function, but diastolic heart failure is an increasingly recognized clinical entity.
Etiology and Clinical Features
During the first year of life, most cases of heart failure are caused by structural heart disease. Other causes include cardiomyopathies due to inborn errors of metabolism or acute events such as myocarditis. In infants with heart failure, tachypnea, dyspnea, tachycardia, feeding difficulties, and failure to thrive are prominent symptoms.143 Physical examination reveals grunting respirations, rales, intercostal retractions, a gallop rhythm, and hepatosplenomegaly. Frequently, a mitral regurgitant murmur is present.
Treatment Strategies
Therapy is tailored to the cause of the cardiac dysfunction and may include supportive care, mechanical ventilation, inotropic support, afterload reduction, prostaglandin E1 therapy to maintain pulmonary or systemic blood flow, maneuvers to balance the systemic and pulmonary circulations, catheter-based interventions, or surgery.144–148 Maintaining organ perfusion is the main goal of therapy for acute heart failure. Pharmacologic agents include inotropes (used on a very-short-term basis, if necessary) and inodilators. Although the administration of digoxin was the mainstay of chronic therapy in the past, this is no longer the case. Favored agents for use in children include diuretics, angiotensin-converting enzyme inhibitors, and β-blockade.149,150 Newer agents that have received increasing attention in the management of pediatric heart failure include nesiritide (a recombinant form of human B-type natriuretic peptide)151–153 and carvedilol (a third-generation β-blocker).154–156
Anesthetic Considerations
Anesthesia for children with heart failure can be quite challenging. The severity of the condition and degree of baseline decompensation can influence the likelihood of an untoward event and the potential for hemodynamic instability and a bad outcome. Several publications have addressed the risks associated with anesthesia in this setting.157–159 It is important to first reexamine the risk–benefit ratio in these patients before going forward with the planned procedure. In most surgical settings, tracheal intubation and mechanical ventilation are indicated. The need for invasive monitoring should be based on the clinical situation, anticipated nature of the procedure, and impact on hemodynamic state.
Syndromes, Associations, and Systemic Disorders: Cardiovascular Disease and Anesthetic Implications
Chromosomal Syndromes
Trisomy 21
Trisomy 21 (i.e., Down syndrome) is the most frequent chromosomal anomaly, occurring with a frequency of 1 case per 800 live births. The incidence increases sharply with advanced maternal age. The syndrome results from trisomy 21 in most children, but it may occur from a balanced or unbalanced chromosome translocation or mosaicism. The phenotypes are indistinguishable. Affected children are typically smaller than normal for age. Craniofacial features include microbrachycephaly, short neck, oblique palpebral fissures, epicanthal folds, Brushfield spots, small and low-set ears, macroglossia, and microdontia with fused teeth. Mandibular hypoplasia and a broad flat nose are typical. A narrow nasopharynx with hypertrophic lymphatic tissue (e.g., tonsils, adenoids) in combination with generalized hypotonia frequently leads to sleep apnea. Other conditions include mental retardation, cervical spine disorders with vertebral and ligamentous instability (i.e., subluxation risk), thyroid disease, leukemia, obesity, subglottic stenosis,160 and gastrointestinal problems.
Airway issues include the potential for upper airway obstruction due to a large tongue, postextubation stridor,161 and cervical spine injury.162–164 Vascular access can be challenging. Subjectively, children have small and abnormal radial vessel sizes,164a vascular hyperreactivity, and fragile tissue consistency, and they may suffer from more complications after arterial cannulation.
Cardiovascular defects occur in 40% to 50% of children with Down syndrome, and it has been recommended that they all should undergo screening for CHD in early infancy.165 The most common lesions include AV septal defects (Video 14-10) , VDSs, TOF, and PDA. Bradycardia under anesthesia occurs commonly, although the mechanism is poorly understood.166,167 Pulmonary hypertension may result from the cardiac pathology or from chronic hypoxemia due to upper airway obstruction (i.e., obstructive sleep apnea) and should be considered in their management.168 Reduced nitric oxide bioavailability has been reported in patients with Down syndrome, leading to endothelial cell dysfunction169 and possibly explaining the observed increased pulmonary vascular reactivity.
, VDSs, TOF, and PDA. Bradycardia under anesthesia occurs commonly, although the mechanism is poorly understood.166,167 Pulmonary hypertension may result from the cardiac pathology or from chronic hypoxemia due to upper airway obstruction (i.e., obstructive sleep apnea) and should be considered in their management.168 Reduced nitric oxide bioavailability has been reported in patients with Down syndrome, leading to endothelial cell dysfunction169 and possibly explaining the observed increased pulmonary vascular reactivity.
Trisomy 18
Trisomy 18 (i.e., Edwards syndrome) is recognized as the second most common chromosomal trisomy (incidence of 1 case per 3500 live births). Most children exhibit microcephaly, delayed psychomotor development, and mental retardation.170 Characteristic craniofacial features include micrognathia or retrognathia, microstomia, malformed ears, and microphthalmia.171 These abnormalities can affect airway management.172,173 Skeletal anomalies include clenched fingers and severe growth retardation. Neurologic problems include developmental delay, hypotonia, and central nervous system malformations. The high mortality rate for children with trisomy 18 is related to cardiac and renal problems, feeding difficulties, sepsis, and apnea caused by neurologic abnormalities.
Cardiovascular disease is present in most children with trisomy 18 and consists primarily of VSDs and polyvalvular disease.174,175 Implications for anesthesia care include the high incidence of congestive heart failure and aspiration pneumonia.173 These children may require interventions to address associated gastrointestinal or genitourinary anomalies.
Trisomy 13
Trisomy 13 (i.e., Patau syndrome) is an uncommon autosomal trisomy with an incidence that ranges from 1 case per 5000 to 12,000 live births. Major features include cleft lip and palate, holoprosencephaly, polydactyly, rocker-bottom feet, microphthalmia, microcephaly, and severe mental retardation.176,177 Almost all children have associated cardiovascular defects that include PDA, septal defects, valve abnormalities, and dextrocardia.175 The overall prognosis for these children is extremely poor.
Turner Syndrome
Turner syndrome is a genetic disorder characterized by partial or complete X chromosome monosomy.178 The estimated incidence is 1 case per 5000 liveborn female infants. A high degree of spontaneous abortion is seen among affected fetuses. Features of this syndrome include webbed neck, low-set ears, multiple pigmented nevi and micrognathia, lymphedema, short stature, and ovarian failure.179 Systemic manifestations include cardiac defects (notably aortic coarctation and bicuspid aortic valve), hypertension, hypercholesterolemia, renal anomalies (up to 33%), liver disease, and inflammatory bowel disease. Obesity is common in older children, as is a high incidence of endocrine abnormalities such as hypothyroidism and diabetes.178,180
Gene Deletion Syndromes
Williams Syndrome
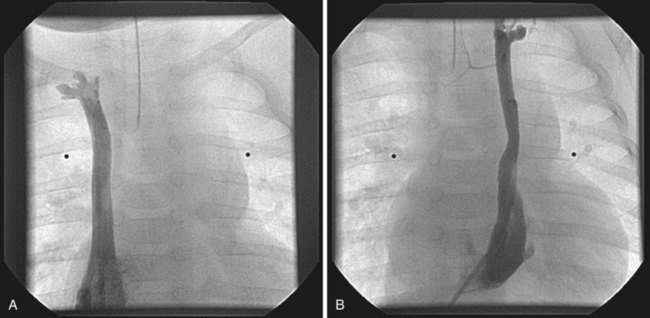
Williams syndrome is a congenital disorder with an incidence of 1/20,000 live births. In most cases, the long arm of chromosome 7 is deleted, altering the elastin gene.181,182 The absence of this gene is detected by fluorescence in situ hybridization (FISH). Features of Williams syndrome include characteristic elfin facies, outgoing personality, endocrine abnormalities (including hypercalcemia and hypothyroidism), mental retardation, growth deficiency, and altered neurodevelopment. Cardiovascular abnormalities can include valvar and supravalvular aortic stenosis (Fig. 14-11) and aortic coarctation.183,184 This arteriopathy can also involve the origin of the coronary arteries or other vessels. Diffuse narrowing of the abdominal aorta may be associated with renal artery stenosis.
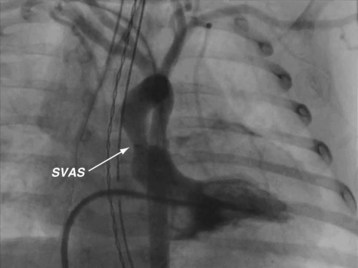
FIGURE 14-11 The angiogram displays the classic supravalvar aortic stenosis (SVAS, arrow) in a child with Williams syndrome.
Several reports have described unanticipated events during anesthesia for these children.185–187 Two conditions can increase anesthetic morbidity and the potential for mortality: coronary artery stenosis leading to myocardial ischemia and severe biventricular outflow tract obstruction. A thorough cardiac evaluation of all children is advisable because the spectrum of disease and the potential devastating implications in affected individuals varies.188 Children occasionally may require further evaluation before undergoing anesthetic care. Even asymptomatic children and those without evidence of clinical cardiovascular disease may be at risk for morbidity and death in the perioperative period.187 Extreme vigilance and particular attention to signs of myocardial ischemia is warranted, as is a plan of action in the event of acute decompensation. This syndrome is one of the leading causes of cardiac arrest in the Pediatric Perioperative Cardiac Arrest (POCA) registry.189
Children with Williams syndrome may exhibit some degree of muscular weakness, and the cautious use and application of muscle relaxants has been recommended.188 Associated neurodevelopmental delay, attention-deficit disorder, and autistic behavior often requires adequate premedication. Subclinical hypothyroidism has a high prevalence among these children.190 Renal manifestations include renovascular hypertension, reduced function, and hypercalcemia-induced nephrocalcinosis.
Chromosome 22q11.2 Deletion Syndrome: DiGeorge and Velocardiofacial Syndrome
The 22q11.2 deletion syndrome, with an estimated incidence of approximately 1 case per 3000 live births, encompasses DiGeorge, conotruncal face, and velocardiofacial syndromes. The syndrome is also known as CATCH 22, a mnemonic for cardiac defects, abnormal facies, thymic hypoplasia, cleft palate, and hypocalcemia, all of which are commonly present.191 Cardiac malformations, speech delay, and immunodeficiency are the most common features of the chromosome 22q deletion syndromes. Because no single feature is overwhelmingly associated with the deletion, the diagnosis should be considered for any child with a conotruncal anomaly, neonatal hypocalcemia, or any of the less common features when seen in association with dysmorphic facial features.
Cardiac malformations are often described as conotruncal anomalies; however, outflow tract problems are also frequently seen.192 The remainder of the cardiac defects encompasses an enormous spectrum of pathologies. Only a few children have a normal cardiovascular system. As a consequence of thymic hypoplasia, children typically have diminished T-cell numbers and function. Their immunodeficiency requires the use of irradiated blood products and strict aseptic precautions during vascular access. Neurodevelopmental features include primarily speech delay and attention-deficit disorders. Psychiatric disorders are well described in these individuals.193
Single-Gene Defects
Noonan Syndrome
Noonan syndrome, an autosomal dominant syndrome, occurs with a frequency of 1 case per 1000 to 2500 live births. Dysmorphic features include neck webbing, low-set ears, chest deformities, hypertelorism, and short stature.194 The diagnosis of Noonan syndrome depends primarily on clinical features.195 In neonates, the facial features may be less apparent; however, generalized edema and excess nuchal folds may be present as in Turner syndrome. The facial features are more difficult to detect in later adolescence and adulthood.
The disorder is associated with a high incidence of CHD (about 50%), and pulmonary valve dysplasia or stenosis is the most common feature.196 HCM may develop during the first few years of life (10% to 20%).197 Clinical problems may also include developmental delay and bleeding diathesis.198
Marfan Syndrome
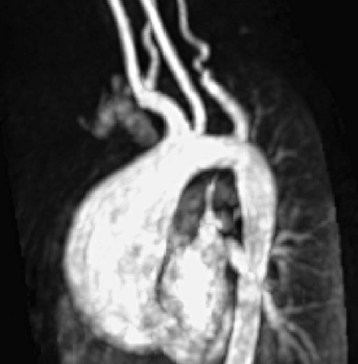
Marfan syndrome is a multisystem disorder with variable expression resulting from a mutation in the fibrillin gene, a connective tissue protein, located on chromosome 15.199 Clinical manifestations typically involve the cardiovascular, skeletal, and ocular systems.200,201 Cardiovascular pathology includes mitral valve prolapse and regurgitation, ascending aortic dilation (Fig. 14-12), and main pulmonary artery dilatation. The risk of aortic dissection rises considerably with increasing aortic size but may occur at any point in the course of the disease.202 Cardiac arrhythmias may be related to valvular heart disease, cardiomyopathy, or congestive heart failure.
β-Blocker therapy and aggressive blood pressure control has been the standard of care in children with aortic root dilation203 and should be continued perioperatively. Aortic root replacement in Marfan syndrome has been associated with a greater risk of repeat dissection and recurrent aneurysm compared with other children who have undergone similar interventions.204 It is wise to maintain hemodynamics near baseline values in the perioperative period. After aortic root surgery some individuals may require chronic anticoagulation therapy. Preoperative hospitalization may be necessary to adjust the anticoagulation regimen in anticipation of surgery in these children. In emergency cases, administration of coagulation factors and other blood products may be required. In addition to vascular pathology, children with Marfan syndrome have a predisposition for ventricular dilation and abnormal systolic function.205,206
Several factors can result in pulmonary disease in these children.207 Chest wall deformities and progressive scoliosis can contribute to restrictive lung disease. The fibrillin defect may affect lung development and homeostasis, impairing pulmonary function. Development of a pneumothorax is relatively common.
Associations
Vater or VACTERL Association
VACTERL association is an acronym given to describe a series of nonrandom anomalies that include vertebral, anal, cardiovascular, tracheoesophageal, renal, and limb defects.208,209 Up to three fourths of children with VACTERL association have CHD. The most common lesions include VSDs, ASDs, and TOF. Complex pathology such as truncus arteriosus and transposition of the great arteries occur less frequently.
Vertebral and tracheal anomalies can complicate airway management and regional anesthesia. Approximately 70% of children with VACTERL have vertebral anomalies, usually consisting of hypoplastic vertebrae or hemivertebra and predisposing children to scoliosis. Anal atresia or imperforate anus is reported in about 55% of cases. These anomalies often require surgery in the first days of life. Esophageal atresia with tracheoesophageal fistula occurs in a large number of affected infants. Low birth weight (less than 1500 g) and associated cardiac pathology have been identified to be independent predictors of mortality in infants undergoing surgery for esophageal atresia or tracheoesophageal fistula (see Chapter 36). The presence of a ductal-dependent cardiac lesion further increases perioperative morbidity and mortality.210 Limb defects occur in most children, potentially affecting vascular access and monitor placement. Renal defects occur in about 50% of children.
CHARGE Association
CHARGE association is characterized by congenital anomalies that include coloboma, heart defects, choanal atresia, retardation of growth and development, genitourinary problems, and ear abnormalities. The association is estimated to occur at a rate of 1 case per 10,000 to 12,000 live births. The cause is unknown although many theories have been suggested.211,212
Specific genetic abnormalities have also been identified in some individuals.213,214 Cardiac defects occur in as many as 50% to 70% of children and commonly include conotruncal and aortic arch anomalies.215 Delayed growth and development usually results from cardiac disease, nutritional problems, and/or growth hormone deficiency. Most children have some degree of cognitive impairment. Anesthetic implications, in addition to those related to the cardiac defects, focus on the airway216; a retrospective review of 50 cases reported upper airway abnormalities in 56% of children apart from choanal atresia and cleft lip and palate.217
Other Disorders
Tuberous sclerosis is a rare genetic disease with an autosomal dominant inheritance pattern and an incidence of approximately 1 case per 25,000 to 30,000 births.218 In a relatively large number of children, it can be attributed to spontaneous mutations. This systemic disease primarily manifests as cutaneous and neurologic symptoms, but cardiac and renal lesions are frequent findings.
The presence of upper airway nodular tumors, fibromas, or papillomas in affected children may interfere with airway management. Developmental delay, autism, attention-deficit disorder, and aggressive behavior are common. Brain tumors and renal tumors (60% to 80%) may produce significant comorbidities. Cardiac pathology includes cardiac rhabdomyoma in 60% of children and coexisting CHD in 33% of cases.219–221 Cardiac abnormalities with obstruction to flow, heart failure, arrhythmias, conduction defects, or preexcitation may affect the selection of anesthetic agents. Preoperative evaluation in most cases should include an ECG to assess arrhythmia, conduction defects, or preexcitation.219 Blood pressure and renal function should also be assessed. Anticonvulsants should be optimized and continued until the morning of surgery. Baseline medical treatment should be resumed as soon as possible because seizures are the most common postoperative complication.222 A child with mental retardation may require premedication with oral midazolam or ketamine, or both, to facilitate parental separation.
Selected Vascular Anomalies and Their Implications for Anesthesia
Aberrant Subclavian Arteries
An aberrant or anomalous subclavian artery usually arises from the descending aorta as a separate vessel distal to the usual last subclavian artery in a posterior location. In a left aortic arch, this arrangement is as follows. The first branch is the right carotid artery, followed by the left carotid artery and the left subclavian artery. The aberrant right subclavian artery, rather than arising proximally from the innominate artery as the first arch branch, originates distal to the last (left) subclavian artery as the fourth branch and courses behind the esophagus toward the right arm. This variant is one of the most common aortic arch anomalies. It occurs in 0.4% to 2% of the general population and may or may not be associated with CHD.223 This anomaly has a high incidence among children with Down syndrome and is associated with VSDs, TOF, and other lesions. In a right aortic arch, the anomalous left subclavian artery originates distal to the origin of the right subclavian artery (Fig. 14-13). This anomaly may be seen in the context of conotruncal malformations. The diagnosis of an aberrant subclavian artery is made by most currently available imaging modalities.
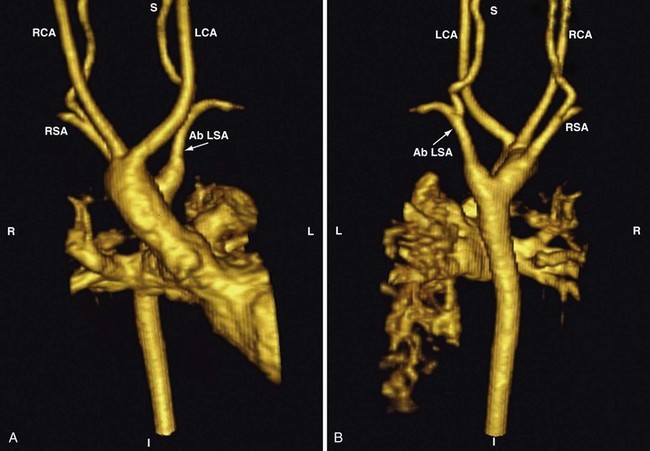
This anomaly has several implications:
 The presence of an aberrant subclavian artery may influence the location of placement of a systemic-to-pulmonary artery shunt.
The presence of an aberrant subclavian artery may influence the location of placement of a systemic-to-pulmonary artery shunt.
 This anomaly should be considered in the selection of a site for arterial line placement if transesophageal echocardiography is planned during surgery. The aberrant vessel may be compressed along its retroesophageal course by the imaging probe, resulting in inaccurate recordings.224 Regardless of the site of arterial line placement, it may be wise to monitor the arm supplied by the anomalous vessel by pulse oximetry or other methods during esophageal instrumentation.
This anomaly should be considered in the selection of a site for arterial line placement if transesophageal echocardiography is planned during surgery. The aberrant vessel may be compressed along its retroesophageal course by the imaging probe, resulting in inaccurate recordings.224 Regardless of the site of arterial line placement, it may be wise to monitor the arm supplied by the anomalous vessel by pulse oximetry or other methods during esophageal instrumentation.
 An aberrant subclavian artery sometimes is part of a vascular ring.
An aberrant subclavian artery sometimes is part of a vascular ring.

Persistent Left Superior Vena Cava to the Coronary Sinus
A persistent left superior vena cava (LSVC) is a form of anomalous systemic venous drainage identified in 4.4% of children with CHD, most frequently those with septal defects.225 It represents a venous remnant that typically involutes during development. If it persists, it remains patent and drains into the right atrium through an enlarged coronary sinus. Bilateral superior vena cavae may be present (Fig. 14-14), or the right superior vena cava may be absent. Bilateral superior vena cavae may communicate through an innominate or bridging vein. This anomaly has several implications:
 In the absence of an innominate vein, a catheter placed in the left arm or left internal jugular vein and advanced into the central circulation may rest within the coronary sinus, a potentially undesirable location in a small infant. On chest radiography, an unusual course is identified as the catheter courses along the left aspect of the mediastinum and can be mistaken for intracarotid, intrapleural, or mediastinal locations.
In the absence of an innominate vein, a catheter placed in the left arm or left internal jugular vein and advanced into the central circulation may rest within the coronary sinus, a potentially undesirable location in a small infant. On chest radiography, an unusual course is identified as the catheter courses along the left aspect of the mediastinum and can be mistaken for intracarotid, intrapleural, or mediastinal locations.






Evaluation of the Child with a Cardiac Murmur
The finding of an incidental murmur during the perioperative period may result in significant distress to the child or family; may trigger additional diagnostic studies, including a cardiology consultation; and has the potential to delay the scheduled procedure when identified preoperatively. Although cardiac auscultation is a challenging skill that takes many years of practice to master,226 it is important for the anesthesiologist who routinely cares for children to recognize the main physical findings that may distinguish an innocent cardiac murmur from a pathologic one. Knowledge of several core concepts and red flags can help avoid overlooking potentially important diagnoses.
Although a complete discussion of cardiac murmur evaluation is beyond the scope of this chapter, it is pertinent to review a few key concepts related to the distinction between innocent and pathologic murmurs.227,228 The basic systematic approach when assessing a heart murmur is the same as when evaluating any child’s cardiovascular system. Auscultation with the diaphragm and bell of the stethoscope in the positions of the four primary cardiac valves should occur with the child and environment as quiet as possible. Innocent murmurs of infancy and childhood include pulmonary flow murmur, a Still murmur, physiologic pulmonary branch stenosis, venous hum, and carotid bruit. Innocent murmurs are usually of low intensity (grades I and II of VI) and are associated with a normal cardiovascular examination (e.g., normal precordial activity, first and second heart sounds, peripheral pulses, capillary refill). Innocent murmurs, such as those associated with peripheral pulmonary branch stenosis, right ventricular outflow murmurs, and Still murmurs, tend to be soft, systolic ejection type, and not holosystolic in duration. Physiologic murmurs often resolve by changing the child’s hemodynamic state with maneuvers such as lying down or sitting up or with temporal changes such as resolution of fever and improvement in anemia. Diastolic or continuous murmurs are typically abnormal, with the exception of a venous hum. This murmur is thought to be related to turbulent flow of systemic venous return in the jugular veins and superior vena cava and is best heard at the base of the neck. Murmurs accompanied by a palpable thrill are always pathologic.
When there is doubt regarding the benign or pathologic nature of a murmur, consultation with a pediatric cardiologist is indicated. A chest radiograph and ECG, although thought by some to add minimal value in the initial diagnostic assessment of a cardiac murmur229,230 and to not be cost-effective,231 can be helpful when considering if further consultation is indicated.
Basic Interpretation of the Electrocardiogram in Children
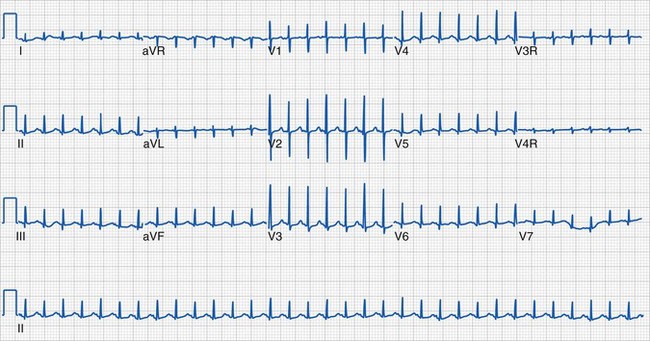
Although the characteristic features of a normal ECG in infants and children were described many decades ago, it is surprising that it continues to be one of the most often misinterpreted screening tests in pediatric medicine.232 This is largely because of the developmental changes that occur in the normal individual as he or she progresses from the neonatal period through childhood, adolescence, and adulthood.233 Normal values for children of various ages have been established.234 Knowledge of normal configurations and values for various ages of children is essential for accurate interpretation.233–236 Immediately after birth, there is a predominance of right ventricular forces represented by tall R waves in the right precordial leads (V1 and V2) (Fig. 14-15). Over the first several years, the typical electrocardiographic changes to a more familiar left heart–dominant configuration with larger S waves in the right precordial leads and a gradual RS progression with tall R waves in the left precordial leads (V5 and V6) (Fig. 14-16). The predominance of right heart forces and the need to evaluate for dextrocardia are the primary reasons that pediatric ECGs should include the V3R and V4R leads, which are not routinely obtained in adult studies. These electrodes are placed in the corresponding V3 and V4 locations over the right precordium.
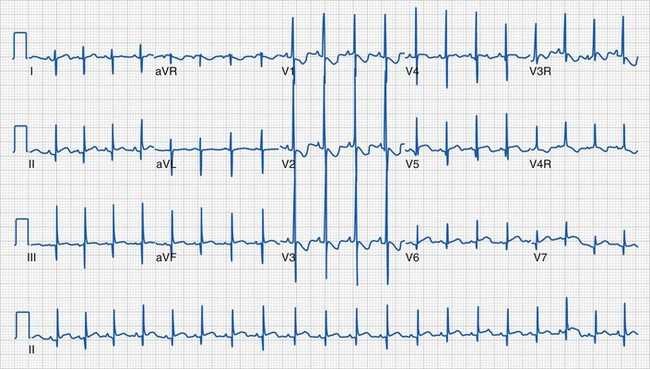
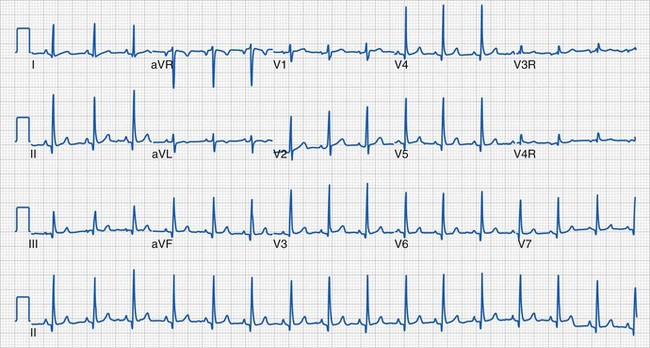
Clinical information of relevance in the interpretation of an ECG includes the child’s age, gender, suspected or documented diagnosis, and indications for the examination. Several requirements are essential for accurate interpretation, including appropriate skin preparation, electrode placement, and an artifact-free recording. The approach to the pediatric ECG should be systematic and organized. Determination of the rate and rhythm, with evaluation of the P-wave vector and the relationship between each P wave and QRS complex, is the first step. It is important to consider the influences of age, autonomic nervous system, level of physical activity, medications, pain, and temperature on the child’s heart rate. The P wave should be upright or positive in leads I and aVF, indicating that the sinus node is the pacemaker of the heart (i.e., sinus rhythm) (see Fig. 14-16). Normally, the P wave should precede the QRS complex. Next, the QRS electrical axis should be determined. The QRS frontal plane axis is determined by identifying the most isoelectric lead, which is perpendicular to the direction of ventricular depolarization. Alternatively, the direction of depolarization in leads I and aVF can be examined to roughly estimate the axis. As with all other components of the evaluation, the physiologic changes that occur with growth are responsible for the change in normal values for the QRS axis based on age. Regardless of age, QRS axes that lie in the northwest quadrant (between 180 and 270 degrees with an S-wave–dominant pattern in leads I and aVF) are always abnormal and merit further investigation. This is a frequent finding in children with AV septal defects. Evaluating the T wave, or repolarization axis, is also important, because a difference of greater than 90 degrees between the QRS and T-wave axes can represent strain on the ventricle, a potential finding in ventricular hypertrophy (see Fig. 14-5).
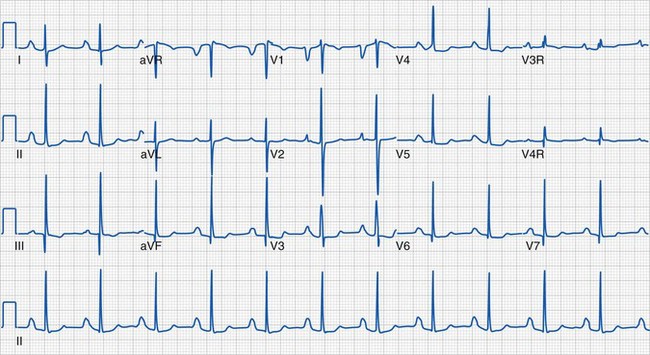
After the evaluation of rhythm and axes is complete, each component of the cardiac cycle as reflected by the ECG should be examined. The P wave represents atrial systole; and its morphology, with particular interest in leads II and V1, can demonstrate right atrial (P-wave amplitude greater than 2.5 mm or 3.0 mm based on age) or left atrial (P-wave duration greater than 100 to 120 msec based on age) enlargement (Fig. 14-17). The PR interval represents the time required for passage of an impulse from the sinoatrial node until ventricular depolarization and is largely composed of the AV nodal delay. A prolonged PR interval, which is age specific, indicates first-degree AV block. A short PR interval should prompt evaluation of the QRS duration for signs of preexcitation (i.e., Wolff-Parkinson-White syndrome) (Fig. 14-18), although a short PR interval may also reflect a low right atrial pacemaker.
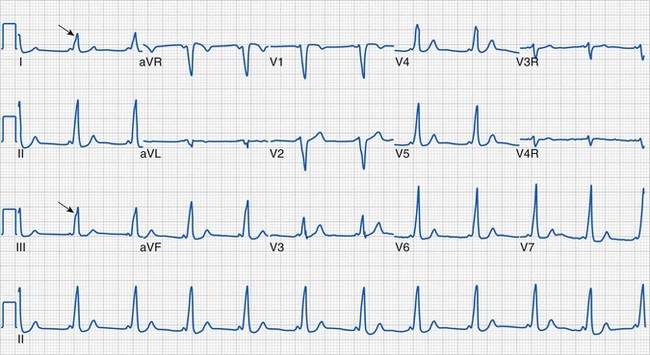
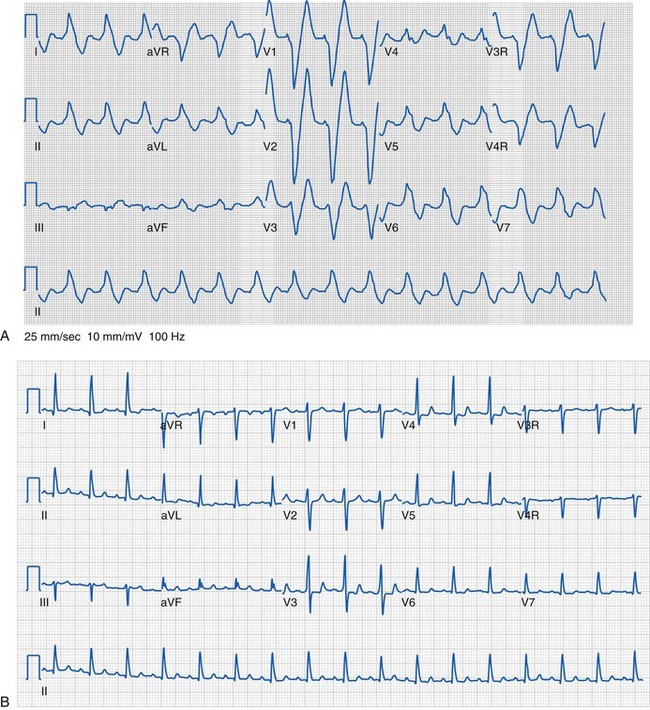
In addition to the QRS duration, the components of the QRS complex should be examined. Q waves are often present in the lateral and inferior leads and in lead aVR, but they should be narrow (less than 40 msec) and shallow (age dependent but usually less than 5 mm deep). Deep or wide Q waves suggest myocardial ischemia and require further evaluation. An uncommon but crucial finding occurs in infants with ALCAPA. Classically, the ECG in this lesion demonstrates deep, wide Q waves in leads I and aVL with ST-segment and T-wave changes in the anterior distribution (V2 to V4) consistent with compromised myocardial blood flow (Fig. 14-19). The QRS amplitudes are also important in assessing left and right ventricular hypertrophy. Conditions associated with increased QRS voltages that likely require echocardiographic assessment include HCM, left ventricular noncompaction, and Pompe disease (see Fig. 14-6).

Although a detailed organized approach to interpretation of a pediatric ECG is necessary, there are occasions when particular conditions or circumstances cause global electrocardiographic changes that must be quickly recognized. One such case that may occur in the operating room is related to the electrocardiographic changes associated with hyperkalemia. As the potassium level increases, the T-wave amplitude increases. This is followed by widening of the QRS duration (Fig. 14-20) due to an intraventricular conduction delay and by AV block and arrhythmias, including ventricular tachycardia and fibrillation. Other electrolyte disturbances may result in characteristic changes on the ECG:
 Hypokalemia: decreased T-wave amplitude, ST-segment depression, and the presence of U waves
Hypokalemia: decreased T-wave amplitude, ST-segment depression, and the presence of U waves
 Hypercalcemia: shortening of the QT interval, sinus rate slowing, and sinoatrial block
Hypercalcemia: shortening of the QT interval, sinus rate slowing, and sinoatrial block


Essentials of Cardiac Rhythm Interpretation and Acute Arrhythmia Management in Children
1. Operating room, bedside, or transport monitors and strip recordings facilitate the recognition of rhythm disorders, but in most cases, they are inadequate for definitive diagnosis. A 15-lead surface ECG and rhythm strip should be obtained for all children when feasible.
2. Clinicians caring for children should have a basic knowledge of cardiac rhythm interpretation. Although a comprehensive discussion of arrhythmia interpretation is beyond the scope of this chapter, a brief overview of the characteristic features of normal and abnormal cardiac rhythms in the pediatric age group is presented in the following section.
3. The need for acute therapy for a rhythm disturbance should be based primarily on the nature of the disorder, urgency of the situation, and the likelihood that this abnormality would or would not be tolerated beyond the immediate short-term period. The guidelines established by the American Heart Association for Pediatric Advanced Life Support should be followed in all patients.237 In otherwise healthy children and in contrast to ventricular arrhythmias, supraventricular tachyarrhythmias are rarely life-threatening.
4. The degree of comfort in the characterization and management of pediatric cardiac arrhythmias is likely to be quite variable among anesthesia care providers. For arrhythmias caused by respiratory compromise, electrolyte imbalance, or metabolic derangements, consultation with a pediatric cardiologist is probably not required. This is also the case for variants or benign rhythm disturbances such as sinus arrhythmia, low atrial rhythms, or occasional premature atrial beats. Consultation is appropriate for most children with known structural or acquired cardiovascular pathology, in those with a history of a cardiac rhythm disorder under the care of a cardiologist, and in most of those with acute arrhythmias, particularly when initiation of antiarrhythmic drug therapy is contemplated.
5. Information that may be helpful to a consultant includes pertinent details regarding the child’s history, clinical diagnosis, nature of the procedure/intervention, relevant laboratory values, description or characterization of the rhythm abnormality, associated hemodynamic parameters, circumstances surrounding the event (including the presence or absence of an intracardiac catheter), review of the pharmacologic agents administered (including anesthetic agents), and other therapies if applicable. The specialist should assist in the characterization of the rhythm disorder, advise about whether further evaluation is indicated, make recommendations for treatment, and facilitate diagnostic/therapeutic interventions as necessary.
Basic Rhythms
Sinus Rhythm
Sinus rhythm is characterized by a P wave that precedes every QRS, a QRS that follows every P wave, and an upright P wave in leads I and aVF (see Fig. 14-16).
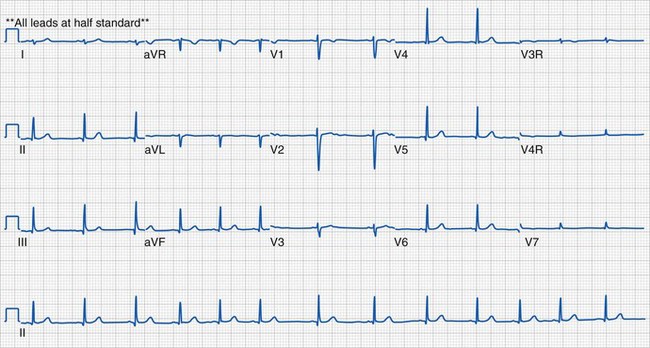
Sinus Arrhythmia
Sinus arrhythmia represents cyclic changes in the heart rate during breathing. This is a normal finding in healthy children (Fig. 14-21).
Sinus Bradycardia
Sinus bradycardia is characterized by sinus rhythm with heart rates below normal for age (see Fig. 14-5). Slow heart rates can be observed during sleep or at times of high vagal tone. When there is significant sinus bradycardia, a slow junctional escape rhythm or a slow atrial rhythm originating from an ectopic focus may be present. Certain forms of CHD may be prone to slow heart rhythms (i.e., heterotaxy syndromes).
In the intraoperative setting, particularly on induction of anesthesia, with laryngoscopy, endotracheal intubation, or tracheal suctioning, sinus bradycardia may occur. Sinus bradycardia may also result from drug administration (i.e., opioids) or increased parasympathetic tone. This type of sinus bradycardia rarely poses significant hemodynamic compromise and, if necessary, can be easily treated with removal of the stimulus, administration of a vagolytic agent (e.g., pancuronium), or with chronotropic drugs such as atropine or epinephrine. Sinus bradycardia may also result from hypoxemia, hypothermia, acidosis, electrolyte imbalance, or increased intracranial pressure. Bradycardia related to hypoxemia should be treated promptly with the administration of supplemental oxygen and appropriate airway management (see Chapter 39). The approach to other secondary forms of sinus bradycardia should focus on addressing the underlying cause. For worrisome slow heart rates, particularly in small infants, or for clinical evidence of compromised hemodynamics, pharmacologic therapy (i.e., epinephrine, atropine, or isoproterenol infusion) or temporary pacing should be considered.
Sinus Tachycardia
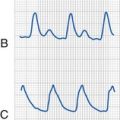
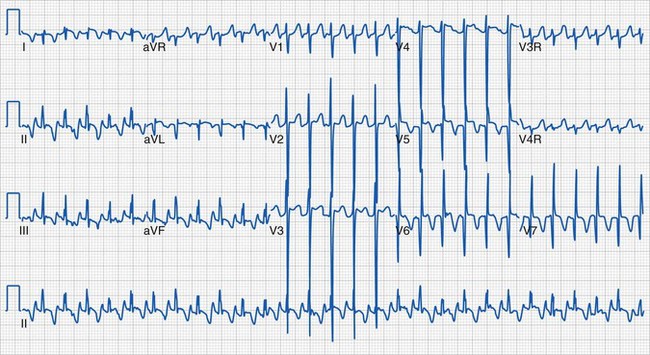
During sinus tachycardia, sinus rhythm occurs at a heart rate above normal for age (Fig. 14-22). In the perioperative setting this is often the result of surgical stimulation, stress, pain, hypovolemia, anemia, fever, medications (i.e., inotropic agents), malignant hyperthermia, or a high catecholamine state. Treatment is directed at the underlying cause. Prolonged periods of sinus tachycardia may impair diastolic filling time, limit ventricular preload, and compromise cardiac output. Children at risk for hemodynamic decompensation include those with significant degrees of ventricular hypertrophy or diastolic dysfunction, aortic stenosis, Williams syndrome, and HCM.
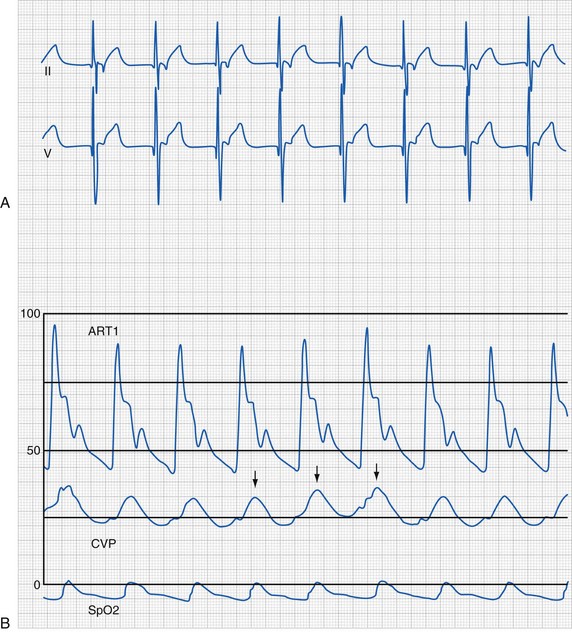
Junctional Rhythm
A junctional rhythm is characterized by QRS complexes of morphology identical to that of sinus rhythm without preceding P waves. This rhythm is slower than the expected sinus rate. When this rhythm completely takes over the pacemaker activity of the heart, retrograde P waves and AV dissociation may be seen. Junctional rhythm during cardiac surgery is frequently the result of manipulation or dissection near the right atrium. The central venous pressure contour typically demonstrates prominent v waves (i.e., right atrial pressure wave at the end of systole) due to the loss of AV synchrony (Fig. 14-23). The lack of atrial contribution to ventricular filling may result in decreases in the systemic arterial blood pressure.
Conduction Disorders
Atrioventricular Block
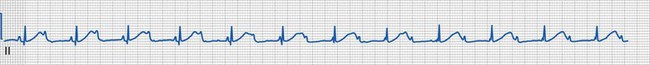
Third-Degree Atrioventricular Block
Third-degree (complete) AV block is characterized by total failure of conduction of atrial impulses to the ventricle. It can be congenital or acquired. There is complete AV dissociation, with more atrial than ventricular contractions, and the ventricular rate is usually slow and regular (Fig. 14-24). Temporary pacing may be indicated in the acute setting.
Cardiac Arrhythmias
Supraventricular Arrhythmias
Supraventricular Tachycardia
Supraventricular tachycardia (SVT) is the most common significant arrhythmia in infants and children.238,239 It is characterized by a regular tachyarrhythmia (tachycardia heart rate is age dependent but typically more than 230 beats/min in children) with a narrow or usual complex QRS morphology. Supraventricular tachycardia can occur in structurally normal hearts and in various forms of CHD. Usual complex implies that the QRS morphology in tachycardia is similar to that in normal sinus rhythm (Fig. 14-25). Occasionally, widening of the QRS in SVT may result from bundle branch block or related to the tachycardia mechanism (i.e., SVT with aberrancy). A wide QRS complex may make the distinction between supraventricular and ventricular tachycardia difficult.
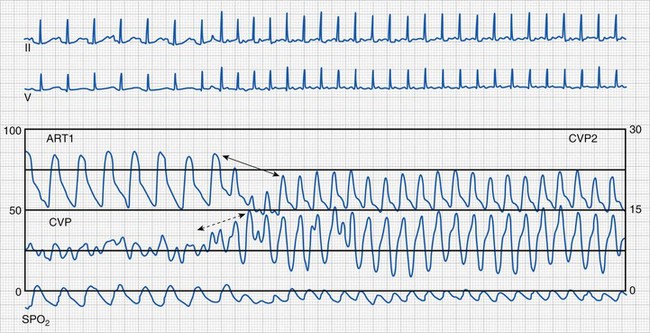
 Hemodynamic stability should be determined. Synchronized direct current cardioversion (0.5 to 1.0 J/kg) should be performed for hemodynamic instability.237
Hemodynamic stability should be determined. Synchronized direct current cardioversion (0.5 to 1.0 J/kg) should be performed for hemodynamic instability.237
 Antiarrhythmic therapy is based primarily on the clinical condition and suspected tachycardia mechanism. Vagal maneuvers may be considered but should not delay treatment. Adenosine is the drug of choice in the acute setting for diagnosis and termination of most supraventricular tachycardias.240 β-Blockers are most often used for chronic therapy.
Antiarrhythmic therapy is based primarily on the clinical condition and suspected tachycardia mechanism. Vagal maneuvers may be considered but should not delay treatment. Adenosine is the drug of choice in the acute setting for diagnosis and termination of most supraventricular tachycardias.240 β-Blockers are most often used for chronic therapy.

 In addition to pharmacologic therapy, atrial pacing or cardioversion may be required.
In addition to pharmacologic therapy, atrial pacing or cardioversion may be required.
Ventricular Arrhythmias
Ventricular Tachycardia
Ventricular tachycardia (VT) is relatively uncommon in children. It is defined as three or more consecutive ventricular beats occurring at a rate greater than 120 beats/min (Fig. 14-26). The QRS morphology in VT is different from that in sinus rhythm, and the QRS duration is typically prolonged for age. ECG features that support this diagnosis include AV dissociation, intermittent fusion (i.e., QRS complex of intermediate morphology between two other distinct QRS morphologies), QRS morphology of VT similar to that of single PVCs, and tachycardia rate in children usually below 250 beats/min.
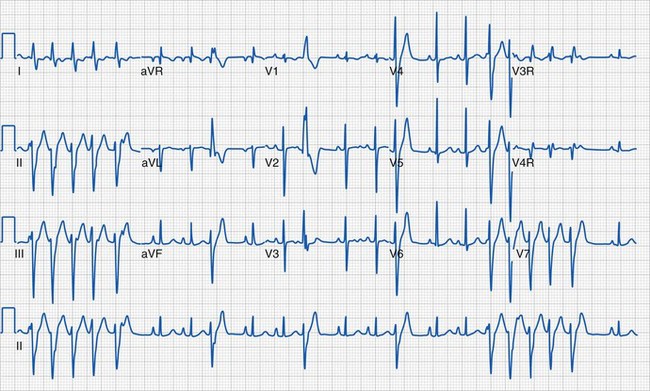
Acute onset of VT in pediatric patients may be caused by hypoxia, acidosis, electrolyte imbalance, or metabolic problems. Ventricular tachycardia may also occur in the context of depressed ventricular function, halothane anesthesia with or without hypercarbia, poor hemodynamics, prior surgical interventions, cardiomyopathies, myocardial tumors, acute injury (e.g., inflammation, trauma), and prolonged QT syndromes.241,242
Long QT Syndrome
Long QT syndrome (LQTS) (Fig. 14-27) is an electrical cardiac disturbance that can predispose children to arrhythmias that include torsades de pointes ventricular tachycardia (Fig. 14-28), ventricular fibrillation, and bradyarrhythmias, and any of these can result in syncope, cardiac arrest, or sudden death.243 It occurs with an incidence of 1 case per 2500 births; congenital and acquired forms have been described. The congenital varieties are likely the result of genetic defects in the cardiac ion channels responsible for maintaining electrical homeostasis.244 Thirteen genotypes have been described on five chromosomes. The cLQT1-3 (Romano-Ward) cases account for 90% of the children. The Romano-Ward syndrome has an incidence of 1 case per 10,000 births and an autosomal dominant pattern of inheritance. The Jervill Lange-Nielsen syndrome has an incidence of 1 case per 1,000,000 births, an autosomal recessive pattern of inheritance, and an association with deafness. Diagnostic criteria proposed for the long QT syndrome include electrocardiographic findings, clinical history (e.g., deafness, syncope), and family history.245 A frequent feature is prolongation of the QTc on the resting ECG.
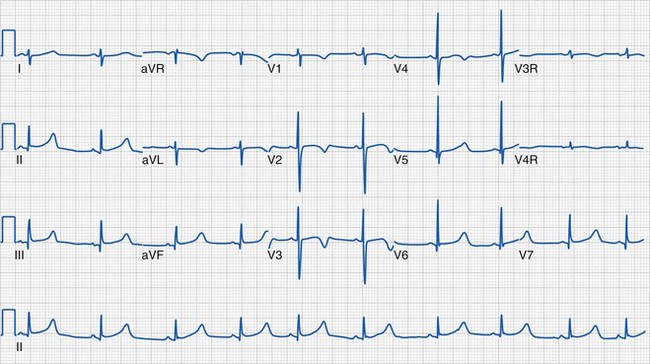
FIGURE 14-27 For a patient with long QT syndrome, the electrocardiogram demonstrates prolongation of the QT interval.
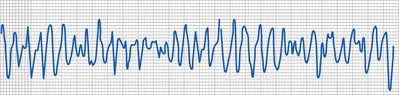
An important consideration in the care of children with cLQTS is ensuring adequate β-adrenergic blockade preoperatively and minimizing adrenergic stimulation.243 Since the introduction of β-adrenergic blockade in patients with cLQTS in 1975, the mortality rate has decreased more than 10-fold. The risk for developing torsades de pointes from the many drugs known to trigger it is almost unpredictable; these drugs have been divided into several groups that are listed and updated online (www.qtdrugs.org).
In a retrospective study of children with cLQTS, three adverse events were reported during emergence from anesthesia immediately after administration of ondansetron and anticholinesterase medications; one was described as torsades de pointes.244 These arrhythmias resolved quickly with intravenous β-blockers, lidocaine, or the administration of both agents. This report suggests that children with cLQTS are at risk for arrhythmias during periods of enhanced sympathetic activity (i.e., during emergence), particularly in the presence of drugs that prolong the QT interval. Conditions (e.g., hypothermia) and drugs (www.qtdrugs.org) that are known to prolong the QT interval should not be combined if possible. Despite the fact that most intravenous and inhalational medications routinely administered during anesthesia prolong the QT interval, adverse events are rare. This may be attributed in part to the need for a second abnormality beyond a prolonged QT interval to be present to trigger arrhythmias (i.e., increased dispersion of repolarization). Fortunately, most anesthetics do not increase the dispersion of repolarization.
Prolongation of the QT interval and genesis of torsades de pointes occur more commonly in the presence of several conditions and drugs: electrolyte derangements (e.g., hypokalemia, hypocalcemia, hypomagnesemia), combination drug therapies (e.g., antibiotics, antiarrhythmic agents, class III antiarrhythmics such as amiodarone and procainamide), antipsychotic drugs, cisapride, neurologic or endocrine abnormalities (e.g., hypothyroidism), 5-HT3 receptor–blocking drugs (except palonosetron), neostigmine, stress (including induction of and emergence from anesthesia and laryngoscopy), female sex, bradycardia, and coronary artery disease. Although many anesthetics prolong the QT interval, few affect the dispersion of repolarization (as in the case of sevoflurane),246 and the risk of torsades de pointes during general anesthesia in children is rare.
Management of torsades de pointes includes the following considerations:
1. Although some atypical forms of supraventricular tachyarrhythmias may mimic VT, a wide QRS tachycardia should always be considered to be of ventricular origin.
2. The initial approach in the setting of an acute ventricular rhythm disturbance consists of prompt evaluation of clinical status and hemodynamic stability. Sustained ventricular arrhythmias are poorly tolerated and require immediate attention. In the unstable child, cardiopulmonary resuscitation should be instituted while preparing for cardioversion. Expert consultation is advisable when advanced drug therapy is contemplated. Potential pharmacologic interventions include lidocaine, amiodarone, and procainamide.247 The latter two should not routinely be administered during torsades de pointes due to QT prolongation.
3. Magnesium sulfate is considered the first-line treatment of torsades de pointes. Procainamide and amiodarone are contraindicated due to prolongation of the QT interval. Isoproterenol and overdrive pacing may be effective for bradycardia. Electrical cardioversion (1 to 2 J/kg) should be performed only if the arrhythmia is refractory to pharmacologic treatment.
4. cLQTS should be treated with β-blockade (not overdrive pacing); some forms of the cLQTS may require the implantation of a cardioverter-defibrillator.
Ventricular Fibrillation
Management of VF includes the following considerations:
1. This is a lethal arrhythmia if untreated.
2. Immediate defibrillation (initial dose of 2 J/kg) is the definitive therapy. Cardiopulmonary resuscitation, beginning with chest compressions, should be immediately resumed and continued for 2 minutes. If defibrillation is unsuccessful, the energy dose should be doubled (4 J/kg) and repeated. Pediatric paddles (2.2 cm in diameter) are recommended for children weighing less than 10 kg. Adult paddles (8 to 9 mm in diameter) are suggested for children weighing more than 10 kg to reduce impedance and maximize current flow.
3. Adequate airway control and chest compressions should be rapidly instituted while preparing for defibrillation or between shocks if several defibrillation attempts are needed. Resuscitative drugs and amiodarone should be considered without delaying defibrillation.
Pacemaker Therapy in the Pediatric Age Group
Pacemaker Nomenclature
Pacemaker nomenclature follows the guidelines of the North American Society of Pacing and Electrophysiology and the British Pacing and Electrophysiology Group248 (Table 14-5):
 First letter: chambers paced (A = atrium, V = ventricle, D = dual or both, O = none)
First letter: chambers paced (A = atrium, V = ventricle, D = dual or both, O = none)
 Second letter: chambers sensed (A = atrium, V = ventricle, D = dual or both, O = none)
Second letter: chambers sensed (A = atrium, V = ventricle, D = dual or both, O = none)

 Fourth letter: rate modulation (R = rate modulation, O = none)
Fourth letter: rate modulation (R = rate modulation, O = none)
 Fifth letter: multisite pacing (A = atrium, V = ventricle, D = dual or both, O = none)
Fifth letter: multisite pacing (A = atrium, V = ventricle, D = dual or both, O = none)
Permanent Cardiac Pacing
Indications
The most recent guidelines for permanent pacing in children, adolescents, and patients with CHD were published in 2008.249 Indications include symptomatic sinus bradycardia, bradycardia-tachycardia syndromes, congenital third-degree AV block, and advanced second- or third-degree AV block.249,250
Perioperative Considerations
Preoperative evaluation of children with implanted pacemakers should include device interrogation.251 Familiarity with unit type, settings, date of and indications for implantation, device location, and underlying rhythm is highly recommended. If records are not available and there is no identification card providing details about the unit implanted, a radiopaque marker on a chest radiograph may assist in the identification of the device. Major pacemaker manufacturers can also be contacted at any time because they maintain computerized records of all implanted devices. Results of a recent 15-lead ECG should be reviewed if available.
Reprogramming may be required before the planned procedure to avoid potential problems with pacemaker malfunction related to electrocautery. This represents one of the most common potential sources of electromagnetic interference in children with implanted cardiac devices. Recommendations for the perioperative management of these children include the use of bipolar cautery versus a unipolar configuration if possible, avoidance of cauterization near the generator, and positioning of the indifferent plate for electrocautery away from the pacemaker so that the device is not between the electrocautery electrodes.252 Devices such as the harmonic scalpel and battery-operated, hot wire, handheld cautery units do not interfere with implanted cardiac devices. Rate-responsive features should be deactivated in most cases.
Chronotropic drugs and alternate pacing modalities should be readily available in the event of pacemaker malfunction and compromising underlying rate. Although insertion of a transvenous pacing system has been advised in children with complete AV block undergoing pacemaker implantation, a 10-year review indicated no benefit to routine preoperative temporary pacing.253 Capture thresholds can be affected by pharmacologic agents, and this should be considered if pacing is required in the child receiving antiarrhythmic drug therapy.
Perioperative conditions may also influence pacing thresholds. A magnet should be accessible to allow asynchronous pacing if required.254,255 Most generators respond to magnet application by pacing at a fixed rate asynchronously (i.e., AOO, VOO, or DOO). A potential problem is that the specific magnet rate, as determined by the manufacturer for the particular device, may be different from the desirable or optimal pacing rate. The use of a magnet should not be considered a substitute for preoperative pacemaker interrogation/programming. In addition to perioperative electrocardiographic monitoring, additional modalities that confirm pulse generation during pacing (e.g., esophageal stethoscope for assessment of heart sounds, pulse oximetry, invasive arterial blood pressure monitoring) are strongly encouraged. The device should be tested and reprogrammed after the procedure is completed.
Transcutaneous Pacing
Several devices that combine defibrillation and cardioversion capabilities with external pacing features are available. Emergency transthoracic pacing may be considered as a temporizing measure for children with symptomatic bradycardia,256 but this has not been effective in the treatment of asystole in children.257 Pacing electrode size should be selected according to patient size (e.g., smaller adhesive pads for weight less than 15 kg). Device settings typically include pacing rate and power output. Sedation may be necessary to tolerate soft tissue discomfort. Prolonged periods of transcutaneous pacing may result in local cutaneous injury. In addition to monitoring for pacemaker capture by ECG, ongoing clinical assessment of the adequacy of cardiac output should be undertaken.
Implantable Cardioverter-Defibrillators
The primary goal of an implantable cardioverter-defibrillator (ICD) is the prevention of sudden death. Children with long QT syndrome, HCM, history of near-death events, arrhythmogenic right ventricular dysplasia, and operated CHD with a history of malignant arrhythmias may be considered suitable candidates for device implantation.250,258,259 The capabilities of these units include pacing and defibrillation. It may be feasible to terminate tachyarrhythmias by pacing.
Over the past several years, more children have been considered for device implantation.249,259–263 This implies an increasing number of children with these devices who may require anesthetic care. The primary issues associated with intraoperative management of these devices relate to patient monitoring, managing issues with potential for electromagnetic interference, and performing emergent defibrillation, cardioversion, or heart rate support.252 Perioperative consultation with a cardiologist or electrophysiologist is essential. These devices should be interrogated and likely require programming before and at the conclusion of the planned procedure.
Diagnostic Modalities in Pediatric Cardiology
Chest Radiography
The standard posteroanterior and lateral chest radiographs provide clues to a child’s underlying cardiovascular anatomy; however, plain radiographs are an insensitive screening tool for cardiac disease.264 Children with numerous types of significant CHD may have initially normal-appearing radiographs; alternatively, an infant with a poor inspiratory effort and the presence of a large thymus may give the appearance of cardiomegaly and have normal intracardiac anatomy (Fig. 14-29).
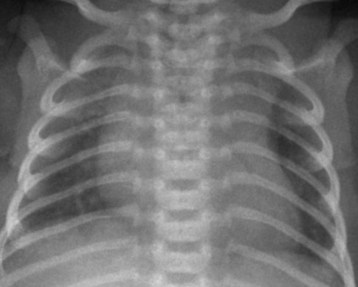
Barium Esophagram
The current applications and uses of barium esophagram (swallow) studies in the diagnosis of CHD are limited. This modality has been largely replaced by MRI and chest tomography.265–267 In some cases, a barium esophagram is used as an initial screening tool when there is concern about the presence of a vascular ring.268,269 A double aortic arch represents one of the most common types of vascular rings in young children, but other vascular anomalies such as a right aortic arch with an aberrant left subclavian artery and left-sided ligamentum arteriosus may also cause symptoms. The indentation pattern in the barium column is consistent with the specific vascular anomaly (Fig. 14-30).
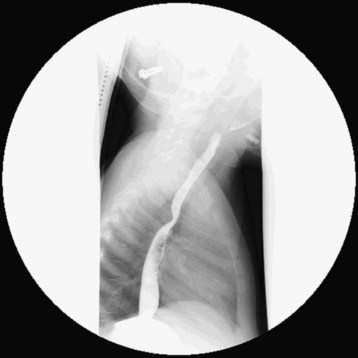
Echocardiography
Echocardiography is the primary diagnostic modality for the initial evaluation and serial assessment in most types of pediatric heart disease.270,271 Using a variety of transducers, ultrasound is used to acquire real-time images of cardiovascular structures. Various echocardiographic modalities are available, including transthoracic,272 transesophageal,273–276 fetal,277,278 epicardial,279 intracardiac imaging,280 and intravascular ultrasound.281 Each plays an important role in the diagnostic evaluation and management of children with suspected or confirmed cardiovascular disease.
A standard transthoracic study consists of a two-dimensional examination, M-mode imaging, and Doppler evaluation (i.e., color flow, pulsed-wave, or continuous-wave modalities). Two-dimensional imaging provides structural assessment of the heart and adjacent vasculature. Cross-sectional images are obtained from several windows that allow excellent anatomic detail in multiple planes (Video 14-11) . In most cases, this is adequate for a detailed segmental evaluation of the cardiac anatomy as described earlier. M-mode echocardiography allows one-dimensional imaging of the heart with excellent temporal resolution (Fig. 14-31). It is known as an ice pick view of the heart in real time and is primarily used in the assessment of ventricular dimensions and function.
. In most cases, this is adequate for a detailed segmental evaluation of the cardiac anatomy as described earlier. M-mode echocardiography allows one-dimensional imaging of the heart with excellent temporal resolution (Fig. 14-31). It is known as an ice pick view of the heart in real time and is primarily used in the assessment of ventricular dimensions and function.
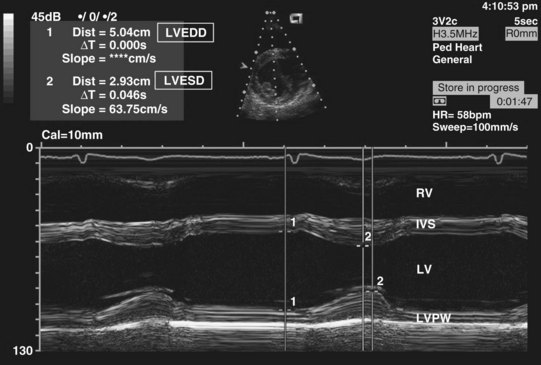
 .
.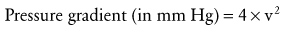
The applications of three-dimensional echocardiography in CHD continue to be investigated.282–284 This approach can provide clear and useful volumetric assessments when the images are adequate. A significant advantage of this modality is that it is able to display cardiovascular structures and their interrelationships in detail, in many cases facilitating the understanding of pathologic conditions over two-dimensional imaging. Three-dimensional echocardiography may also be useful when interventions are planned.
Interpretation of an Echocardiographic Report
Assessment of Ventricular Function
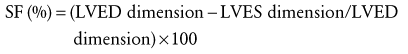
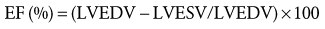
Although these functional indices are routinely and easily obtained, they have significant limitations. Estimation of ejection fraction is based on geometric assumptions for the elliptical left ventricle, and this may not be applicable to a systemic right ventricle or other types of ventricular geometries.285 This accounts for an escalating interest in alternative approaches that may provide more sensitive and comprehensive information regarding ventricular performance, even in the absence of clinical disease. These techniques include the myocardial performance index (MPI), also known as the Tei index, which combines systolic and diastolic intervals to assess global ventricular function286–288; Doppler tissue imaging (DTI), which is used to evaluate intramural myocardial velocities289; and strain and strain rate imaging to quantitate the rate of segmental myocardial deformation.290 Although values in normal children have been established for these imaging modalities and alterations in the presence of pathologic conditions have been described,291,292 additional studies documenting their clinical applications in specific types of cardiovascular pathology are needed.
 . For example, if a peak regurgitant velocity of 3 m/sec is recorded across the tricuspid valve using the simplified Bernoulli equation, the pressure gradient or difference between the right atrial and right ventricular systolic pressures can be estimated to be 4 × 32 = 36 mm Hg. If a normal right atrial pressure is assumed (4 to 6 mm Hg), it would predict a right ventricular systolic pressure of approximately 40 mm Hg. Similarly, if the peak or maximal flow velocity across a ventricular septal defect is measured at 4.5 msec, it predicts a pressure gradient of 4 × 4.52 = 81 mm Hg between the ventricles, implying that the defect is pressure restrictive and the right ventricular and pulmonary artery systolic pressures are relatively low.
. For example, if a peak regurgitant velocity of 3 m/sec is recorded across the tricuspid valve using the simplified Bernoulli equation, the pressure gradient or difference between the right atrial and right ventricular systolic pressures can be estimated to be 4 × 32 = 36 mm Hg. If a normal right atrial pressure is assumed (4 to 6 mm Hg), it would predict a right ventricular systolic pressure of approximately 40 mm Hg. Similarly, if the peak or maximal flow velocity across a ventricular septal defect is measured at 4.5 msec, it predicts a pressure gradient of 4 × 4.52 = 81 mm Hg between the ventricles, implying that the defect is pressure restrictive and the right ventricular and pulmonary artery systolic pressures are relatively low.Evaluation of Gradients
Estimation of a peak instantaneous gradient is the most clinically useful method for quantifying the severity of obstructions across semilunar valves and outflow tracts. It is derived by application of the simplified Bernoulli equation. When obtained across the pulmonary valve, these estimates tend to have a better correlation with catheterization peak-to-peak gradients than those measured across the aortic valve, for which mean gradients (obtained by automated integration of the velocities under a spectral Doppler tracing) have been shown to have a greater correlation.293 The mean gradient rather than a peak gradient determined by Doppler echocardiography is considered a better indicator of the severity of the obstruction across AV valves and other low-flow venous pathways.
Magnetic Resonance Imaging
Cardiovascular MRI-angiography has emerged as a complementary technology to other imaging modalities (Videos 14-14 and 14-15). Benefits have been reported in the assessment of complex pathology,294–300 delineation of systemic and pulmonary vascular anomalies,299,300 evaluation of global and regional ventricular function,301 assessment of myocardial viability,302 and characterization of pulmonary blood supply in children with structural alterations of the pulmonary vascular tree.303 Additional applications that may further expand the utility of cardiovascular MRI include the quantification of left-to-right shunts304–306 and measurement of blood oxygen saturation.307 MRI is beneficial for guiding interventions in pediatric heart disease.308–310
Benefits have been reported in the assessment of complex pathology,294–300 delineation of systemic and pulmonary vascular anomalies,299,300 evaluation of global and regional ventricular function,301 assessment of myocardial viability,302 and characterization of pulmonary blood supply in children with structural alterations of the pulmonary vascular tree.303 Additional applications that may further expand the utility of cardiovascular MRI include the quantification of left-to-right shunts304–306 and measurement of blood oxygen saturation.307 MRI is beneficial for guiding interventions in pediatric heart disease.308–310
An additional limitation of MRI is the need for patient immobility during long examinations for optimal image quality. For small children, this requirement usually necessitates the use of deep sedation or general anesthesia.311–313 For infants with complex and often cyanotic CHD, specialists are often asked to provide care during the procedures. The severity of the cardiovascular disease and the need for breath holding may add to the challenges presented to the anesthesia care provider in a remote location.314,315 The lengthy nature of the studies and the significant time requirements to perform postprocessing of the images cause MRI to be much more time intensive for the interpreting physician than other noninvasive imaging modalities.
Computed Tomography
Cardiac CT, with or without electrocardiographic gating, has become an option among the various cardiovascular imaging modalities (Fig. 14-32).294,298,316,317 The major advantage of CT over MRI is the very rapid scan times, and for most children, sedation is minimal or unnecessary. A significant drawback of CT is the large radiation burden, although typically estimated to be similar to or slightly greater than a diagnostic cardiac catheterization, and the likely need for iodinated contrast agents with their concomitant risks.
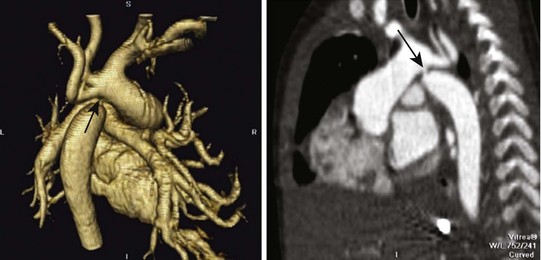
Cardiac Catheterization and Angiography
Cardiac catheterization involves the invasive measurement of intracardiac and vascular pressures and blood oxygen saturation coupled with angiography to assess cardiac anatomy and hemodynamics (Figs. 14-33 and 14-34). Before the era of two-dimensional echocardiography, cardiac catheterization was frequently used for diagnostic purposes. With the advances in noninvasive imaging, diagnostic procedures represent a relatively small proportion of these studies. Current indications for cardiac catheterization at most centers include the assessment of physiologic parameters such as pressure and resistance data, anatomic definition when other diagnostic modalities are inadequate, need for electrophysiologic testing or treatment, and when interventions are anticipated.
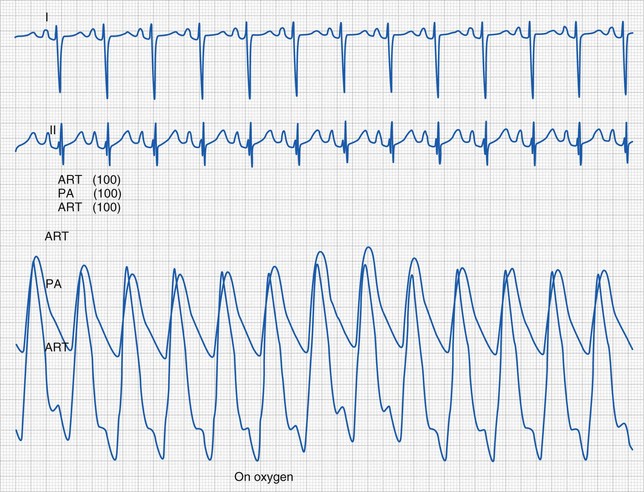
Most catheterizations are performed for interventions, including endomyocardial biopsies; angioplasties and stenting of stenotic vessels, dilation of valves, and conduits; and occlusion techniques for native defects, fistulous connections, and surgically created defects no longer considered necessary (Fig. 14-35, Video 14-16)  (see Chapter 20). In some cases, such as critically ill neonates with complex heart disease, catheter-based interventions such as balloon atrial septostomy and other procedures can be lifesaving.
(see Chapter 20). In some cases, such as critically ill neonates with complex heart disease, catheter-based interventions such as balloon atrial septostomy and other procedures can be lifesaving.
In most children, access to the central circulation is accomplished percutaneously through a femoral approach. Most examinations involve hemodynamic evaluation with recording of pressure data through catheters positioned at various sites of interest. Oxygen saturation data are obtained by reflectance oximetry or blood gas measurement from various cardiac chambers and vessels. In contrast to the oxygen saturation calculations derived from a blood gas analysis, reflectance oximetry provides measured values. This allows determination of oxygen content (i.e., total amount of hemoglobin in the blood) and, when combined with values of oxygen consumption, assessment of blood flows and other calculations (e.g., shunts).318 Additional data that may be obtained include pressure gradients, cardiac output, and parameters for deriving vascular resistances and valve areas.
Fluoroscopy and cineangiography are essential components of most cardiac catheterization studies. Of the two, cineangiography accounts for most of the radiation exposure as images are recorded during the injection of contrast material, typically at 15 or 30 frames/sec.319 Most angiograms are obtained during biplane imaging by positioning the equipment to obtain optimal views allowing for delineation of the pathology in question (i.e., axial angiography) (Video 14-17, A and B) .320,321
.320,321
Although cardiac catheterization has evolved over the years, providing an improved margin of safety, it remains an invasive procedure involving several risks. They include excessive blood loss, vascular complications,322,323 infection, arrhythmias, vascular or cardiac perforation,324 systemic air embolization, myocardial ischemia, and those associated with the administration of contrast agents. These complications are more likely in infants and small children.325 Interventional catheterizations, by the nature of the procedures, are associated with a greater rate of complications and greater potential for morbidity and mortality. However, as transcatheter interventions become safer and more effective, an increasing number of children may obviate the need for surgery, often undergoing procedures on an outpatient basis.326 Evolving approaches in this field include percutaneous implantation of valves,327 strategies that combine cardiac catheterization and surgical intervention (hybrid procedures),328–330 and catheter-based interventions during fetal life.331
 ) to systemic (
) to systemic (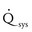 ) blood flow ratio. In the equation, Ssysa
) blood flow ratio. In the equation, Ssysa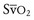 is the mixed venous O2 saturation, Spulmv
is the mixed venous O2 saturation, Spulmv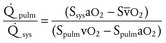
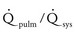 ratio that exceeds 3 to 1 is considered a significant shunt, although smaller ratios may be associated with considerable symptoms.
ratio that exceeds 3 to 1 is considered a significant shunt, although smaller ratios may be associated with considerable symptoms.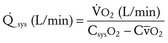
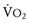 is the oxygen consumption (assumed or measured), Csys
is the oxygen consumption (assumed or measured), Csys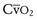 is the mixed venous O2 content. The O2 content = O2 saturation × (1.36 × 10 × hemoglobin concentration).
is the mixed venous O2 content. The O2 content = O2 saturation × (1.36 × 10 × hemoglobin concentration).

Perioperative Considerations for Children with Cardiovascular Disease
General Issues
Anesthesia for children with heart disease332 is challenged by the following factors:
 The remarkable spectrum of disease
The remarkable spectrum of disease
 The wide range of congenital lesions and their underlying physiologic consequences
The wide range of congenital lesions and their underlying physiologic consequences
 The numerous interventional and surgical options in CHD (Table 14-6), in addition to their hemodynamic implications
The numerous interventional and surgical options in CHD (Table 14-6), in addition to their hemodynamic implications

| Procedure | Description | Goal or Result |
|---|---|---|
| Arterial switch (Jatene) operation | Arterial trunks transected above the level of the semilunar valves, relocated to their appropriate respective ventricles, coronary arteries reimplanted into the neoaortic root | Establishes the normal ventricular-arterial connection (right ventricle to pulmonary artery and left ventricle to aorta) in transposition of the great arteries |
| Atrioventriculoseptal defect (atrioventricular canal) repair | Patch closure of atrial and ventricular communications, reconstruction of atrioventricular valves, closure of cleft in left-sided atrioventricular valve | Eliminates the intracardiac shunt |
| Blalock-Taussig shunt | Subclavian artery to pulmonary artery communication; modified implies placement of a graft | Allows or increases pulmonary blood flow |
| Central shunt, Waterston shunt, Pott shunt | Creation of communication between systemic and pulmonary circulations | Allows or increases pulmonary blood flow |
| Closure of septal defects | Patch or primary closure of communications at the atrial or ventricular levels | Eliminates the intracardiac shunt |
| Coarctation repair | Relief of aortic arch obstruction (various approaches) | Establishes patency across the aortic arch |
| Damus-Kaye-Stansel procedure | End-to-side anastomosis of main pulmonary artery onto the aorta; necessitates reestablishing pulmonary blood flow through an alternative route (graft from a systemic artery into the pulmonary artery or a right ventricular to pulmonary artery conduit) | Allows unobstructed systemic outflow in the context of single ventricle associated with obstruction to aortic flow or other settings |
| Division or ligation of a patent ductus arteriosus | Obliteration of the ductus arteriosus | Eliminates shunting at the level of the great arteries |
| Fontan procedure | Connection that directs inferior vena cava blood into the pulmonary circulation | Separates the pulmonary and systemic circulations in patients with single ventricle physiology; usually the final step in the single-ventricle palliation pathway |
| Glenn anastomosis (cavopulmonary connection) | Superior vena cava to pulmonary artery direct anastomosis (bidirectional implies flow from superior vena cava into both pulmonary arteries) | Provides pulmonary blood flow while unloading the single ventricle; may be the first or intermediate step in the single-ventricle palliation pathway |
| Konno-Rastan procedure (aortoventriculoplasty) | Enlargement of the left ventricular outflow tract and aortic annulus; defect created in the ventricular septum to enlarge the outflow tract repaired with a large patch | Alleviates subvalvar and valvar aortic obstruction; when the aortic root is replaced by an autologous pulmonary root, it is referred to as a Ross-Konno procedure. Alternatively, cryopreserved homograft tissue may be used in the form of an extended aortic root replacement. |
| Norwood procedure (stage I palliation) | Involves aortic reconstruction, an atrial septectomy, and placement of a systemic-to-pulmonary artery shunt | Addresses systemic outflow tract obstruction by allowing the right ventricle to eject into a reconstructed aorta. Atrial septectomy provides unobstructed drainage of the pulmonary venous return into the right atrium. The systemic-to-pulmonary artery shunt supplies the pulmonary blood flow. |
| Pulmonary artery banding | Constrictive band placed around the main pulmonary artery | Limits excessive pulmonary blood flow |
| Rastelli operation | Creation of an intracardiac tunnel that allows left ventricular output into the aorta while closing a ventricular septal defect and placement of a right ventricular conduit to pulmonary artery | Allows the left ventricle to eject solely into the aorta, abolishes intracardiac shunting at the ventricular level, and provides unobstructed pulmonary blood flow. The procedure results in separation of the pulmonary and systemic circulations. |
| Sano modification of the Norwood procedure | Placement of graft between the right ventricle and main pulmonary artery as an alternative to a modified Blalock-Taussig shunt in the Norwood operation | Provides pulmonary blood flow |
| Senning or Mustard procedure (atrial switch) | Intraatrial baffle procedure | Allows pulmonary venous blood to be rerouted through the tricuspid valve into the right ventricle (as the systemic chamber that ejects into the aorta). Systemic venous return is channeled across the mitral valve into the left ventricle, which pumps into the main pulmonary artery. |
| Tetralogy of Fallot repair | Closure of ventricular septal defect and relief of right ventricular outflow tract obstruction | Eliminates intracardiac shunting at the ventricular level (cyanosis) and addresses right ventricular outflow tract obstruction (often at several levels) |
| Truncus arteriosus repair | Closure of the ventricular septal defect and establishment of right ventricular to pulmonary artery continuity (usually with a homograft) | Abolishes intracardiac shunting and restores the normal connection between the ventricles and great arteries |
| Valvectomy | Valve excision | Relieves valvar obstruction |
| Valvotomy | Opening of stenotic valve | Relieves valvar obstruction |
| Valve replacement | Placement of bioprosthetic or mechanical valve | Addresses valvar pathology (obstruction and regurgitation) |
| Valvuloplasty | Valve repair | Relieves valvar regurgitation and stenosis |
To optimally care for these children, the following objectives should be met:
 Familiarity with the cardiovascular defects
Familiarity with the cardiovascular defects
 Understanding of the physiologic abnormalities and available therapies
Understanding of the physiologic abnormalities and available therapies
 Recognition of compensatory mechanisms, signs of limited reserve, and potential perioperative risks333
Recognition of compensatory mechanisms, signs of limited reserve, and potential perioperative risks333

This combination of daunting challenges and difficult objectives can be intimidating even for the most experienced clinician. When caring for children with cardiovascular disease, an interdisciplinary approach is recommended, allowing for the formulation and execution of optimal individualized management plans. If available, consultation with the child’s cardiologist or primary care physician should include inquiries about the details of the child’s disease, overall clinical status, past and current medical treatment, prior catheterization or surgical interventions, and presence of residual pathology. The interaction between members of the perioperative team should allow an exchange of information, discussion of concerns, and recommendations that may facilitate patient care and the development of comprehensive care plans.334 This is particularly important in the management of children with complex disease.
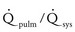 exceeding 3 to 1, outflow tract gradients greater than 50 mm Hg, pulmonary hypertension (i.e., mean pulmonary artery pressure above 30 mm Hg), increased pulmonary vascular resistance index (more than 2 Woods units/m2), or polycythemia (i.e., hematocrit greater than 60%). Several clinical states may place children at significant risk for severe cardiopulmonary decompensation during anesthesia and surgery: recent congestive heart failure, uncontrolled arrhythmias, severe ventricular dysfunction, unexplained syncope, substantial exercise intolerance, or any condition associated with significant functional cardiac or pulmonary impairment. For some children, a planned admission to the intensive care unit (with or without a tracheal tube) should be discussed with the parents, child, and whole care team.
exceeding 3 to 1, outflow tract gradients greater than 50 mm Hg, pulmonary hypertension (i.e., mean pulmonary artery pressure above 30 mm Hg), increased pulmonary vascular resistance index (more than 2 Woods units/m2), or polycythemia (i.e., hematocrit greater than 60%). Several clinical states may place children at significant risk for severe cardiopulmonary decompensation during anesthesia and surgery: recent congestive heart failure, uncontrolled arrhythmias, severe ventricular dysfunction, unexplained syncope, substantial exercise intolerance, or any condition associated with significant functional cardiac or pulmonary impairment. For some children, a planned admission to the intensive care unit (with or without a tracheal tube) should be discussed with the parents, child, and whole care team.Clinical Condition and Status of Prior Repair
Children with CHD may present for anesthesia care before or after palliation or for definitive procedures. Corrective procedures are those that result in a normal life expectancy and full cardiovascular reserve.335 Children undergoing these interventions usually require no further medical or surgical treatment. In the strict sense, only a few procedures fulfill these criteria: ligation, division, or occlusion of a PDA and closure of an isolated secundum ASD. Other interventions or surgical procedures may result in repair or correction but not necessarily in normal hemodynamics or life expectancy. The clinician should assume some limitation in cardiovascular reserve, a need for close follow-up, further medical management, and potential or additional surgical therapies. In other cases, as in children with palliated CHD, the circulation may still be abnormal. These individuals have been reported to be at greater risk for adverse perioperative events.336–338 Published data from the Pediatric Perioperative Cardiac Arrest (POCA) Registry examined anesthesia-related cardiac arrests in children with congenital and acquired heart disease.339 Compared with other children, cardiac arrests were found to occur more frequently in children with heart disease. Causes were primarily cardiovascular in nature. These events occurred more frequently in the general operating room, usually during the surgical maintenance phase. The most common anatomic substrate in this setting was that of a single ventricle, particularly those early in the palliation pathway. The overall mortality rate for children with heart disease was greater than those without heart disease, with the greatest mortality rate occurring in children with aortic stenosis and cardiomyopathy.
The effects of previous procedures on the heart and other systems require careful consideration. Problems may remain or develop after surgical intervention include residual shunts, valvar stenoses or outflow tract obstruction, valvar regurgitation, pulmonary hypertension, arrhythmias, and ventricular dysfunction. Children who require a detailed appraisal of perioperative risks include those with residual significant pathology, suspected or known pulmonary hypertension, or single-ventricle physiology and those after conduit placement or cardiac transplantation (see Chapters 15, 16, and 21).
Bai W, Voepel-Lewis T, Malviya S. Hemodynamic changes in children with Down syndrome during and following inhalation induction of anesthesia with sevoflurane. J Clin Anesth. 2010;22:592–597.
Cordina RL, Celermajer DS. Chronic cyanosis and vascular function: implications for patients with cyanotic congenital heart disease. Cardiol Young. 2010;20:242–253.
Lang RM, Mor-Avi V, Sugeng L, et al. Three-dimensional echocardiography: the benefits of the additional dimension. J Am Coll Cardiol. 2006;48:2053–2069.
Murphy T, Smith J, Ranger M, et al. General anesthesia for children with severe heart failure. Pediatr Cardiol. 2011;32:139–144.
Pharis CS, Conway J, Warren AE, et al. The impact of 2007 infective endocarditis prophylaxis guidelines on the practice of congenital heart disease specialists. Am Heart J. 2011;161:123–129.
1 Hoffman JI. Incidence of congenital heart disease: I. Postnatal incidence. Pediatr Cardiol. 1995;16:103–113.
2 Patel MS, Kogon BE. Care of the adult congenital heart disease patient in the United States: a summary of the current system. Pediatr Cardiol. 2010;31:511–514.
3 Seal R. Adult congenital heart disease. Paediatr Anaesth. 2011;21:615–622.
4 Warnes CA, Williams RG, Bashore TM, et al. ACC/AHA 2008 guidelines for the management of adults with congenital heart disease: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Develop Guidelines on the Management of Adults with Congenital Heart Disease). Developed in collaboration with the American Society of Echocardiography, Heart Rhythm Society, International Society for Adult Congenital Heart Disease, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons. J Am Coll Cardiol. 2008;52:e143–e263.
5 Hoffman JI, Kaplan S. The incidence of congenital heart disease. J Am Coll Cardiol. 2002;39:1890–1900.
6 Tutar E, Ekici F, Atalay S, Nacar N. The prevalence of bicuspid aortic valve in newborns by echocardiographic screening. Am Heart J. 2005;150:513–515.
7 Grabitz RG, Joffres MR, Collins-Nakai RL. Congenital heart disease: incidence in the first year of life. The Alberta Heritage Pediatric Cardiology Program. Am J Epidemiol. 1988;128:381–388.
8 Ooshima A, Fukushige J, Ueda K. Incidence of structural cardiac disorders in neonates: an evaluation by color Doppler echocardiography and the results of a 1-year follow-up. Cardiology. 1995;86:402–406.
9 Roguin N, Du ZD, Barak M, et al. High prevalence of muscular ventricular septal defect in neonates. J Am Coll Cardiol. 1995;26:1545–1548.
10 Rosenthal GL, Wilson PD, Permutt T, et al. Birth weight and cardiovascular malformations: a population-based study. The Baltimore-Washington Infant Study. Am J Epidemiol. 1991;133:1273–1281.
11 Sands AJ, Casey FA, Craig BG, et al. Incidence and risk factors for ventricular septal defect in “low risk” neonates. Arch Dis Child Fetal Neonatal Ed. 1999;81:F61–F63.
12 Botto LD, Correa A, Erickson JD. Racial and temporal variations in the prevalence of heart defects. Pediatrics. 2001;107:E32.
13 Loffredo CA. Epidemiology of cardiovascular malformations: prevalence and risk factors. Am J Med Genet. 2000;97:319–325.
14 Anderson RH, Becker AE, Freedom RM, et al. Sequential segmental analysis of congenital heart disease. Pediatr Cardiol. 1984;5:281–287.
15 Shinebourne EA, Macartney FJ, Anderson RH. Sequential chamber localization: logical approach to diagnosis in congenital heart disease. Br Heart J. 1976;38:327–340.
16 Tynan MJ, Becker AE, Macartney FJ, et al. Nomenclature and classification of congenital heart disease. Br Heart J. 1979;41:544–553.
17 Van Mierop LH. Diagnostic code for congenital heart disease. Pediatr Cardiol. 1984;5:331–362.
18 Van Praagh R. The segmental approach to diagnosis in congenital heart disease. The Cardiovascular System. Birth Defects. 1972;8:4–23.
19 Van Praagh R. The segmental approach clarified. Cardiovasc Intervent Radiol. 1984;7:320–325.
20 Van Praagh R, Santini F, Geva T. Segmental situs in congenital heart disease: a fundamental concept. G Ital Cardiol. 1990;20:246–253.
21 Van Praagh R. The importance of segmental situs in the diagnosis of congenital heart disease. Semin Roentgenol. 1985;20:254–271.
22 Hagler DJ. Echocardiographic segmental approach to complex congenital heart disease in the neonate. Echocardiography. 1991;8:467–475.
23 Greeley WJ, Galli KK. Cardiac surgery: anesthetic considerations and postoperative management. In: Bissonnette B, Dalens BJ, eds. Pediatric anesthesia: principles and practice. New York: McGrawHill; 2002:1164–1185.
24 Mahajan A, Marijic J. Hemodynamic management. In: Andropoulos DB, Stayer SA, Russell IA, eds. Anesthesia for congenital heart disease. Malden, Mass: Blackwell Publishing; 2005:225–402.
25 Edwards JE. Classification of congenital heart disease in the adult. Cardiovasc Clin. 1979;10:1–26.
26 Haselby KA, Moorthy SS. Noncardiac surgery in the patient with congenital heart disease. Semin Pediatr Surg. 1992;1:65–73.
27 Sauvage LR, Mansfield PB, Stamm SJ. Physiologic classification of congenital heart disease. AORN J. 1973;18:61–83.
28 Van Praagh R, Plett JA, Van Praagh S. Single ventricle: pathology, embryology, terminology and classification. Herz. 1979;4:113–150.
29 Torres AJ, DiLiberti J, Pearl RH, et al. Noncardiac surgery in children with hypoplastic left heart syndrome. J Pediatr Surg. 2002;37:1399–1403.
30 Walker SG, Stuth EA. Single-ventricle physiology: perioperative implications. Semin Pediatr Surg. 2004;13:188–202.
31 Bilgic A, Ozbarlas N, Ozkutlu S, et al. Cardiomyopathies in children: clinical, epidemiological and prognostic evaluation. Jpn Heart J. 1990;31:789–797.
32 Towbin JA. Pediatric myocardial disease. Pediatr Clin North Am. 1999;46:289–312.
33 Bozic I, Fabijanic D, Carevic V, et al. Echocardiography in the diagnosis and management of isolated left ventricular noncompaction: case reports and review of the literature. J Clin Ultrasound. 2006;34:416–421.
34 Conraads V, Paelinck B, Vorlat A, et al. Isolated noncompaction of the left ventricle: a rare indication for transplantation. J Heart Lung Transplant. 2001;20:904–907.
35 Pignatelli RH, McMahon CJ, Dreyer WJ, et al. Clinical characterization of left ventricular noncompaction in children: a relatively common form of cardiomyopathy. Circulation. 2003;108:2672–2678.
36 Sen-Chowdhry S, Lowe MD, Sporton SC, et al. Arrhythmogenic right ventricular cardiomyopathy: clinical presentation, diagnosis, and management. Am J Med. 2004;117:685–695.
37 Lipshultz SE. Exposure to anthracyclines during childhood causes cardiac injury. Semin Oncol. 2006;33:S8–14.
38 Bryant RM. Hypertrophic cardiomyopathy in children. Cardiol Rev. 1999;7:92–100.
39 Lipshultz SE, Sleeper LA, Towbin JA, et al. The incidence of pediatric cardiomyopathy in two regions of the United States. N Engl J Med. 2003;348:1647–1655.
40 Berger S, Dhala A, Dearani JA. State-of-the-art management of hypertrophic cardiomyopathy in children. Cardiol Young. 2009;19(Suppl 2):66–73.
41 Maron BJ. Hypertrophic cardiomyopathy in childhood. Pediatr Clin North Am. 2004;51:1305–1346.
42 Bashyam MD, Savithri GR, Kumar MS, et al. Molecular genetics of familial hypertrophic cardiomyopathy (FHC). J Hum Genet. 2003;48:55–64.
43 Towbin JA. Molecular genetic aspects of cardiomyopathy. Biochem Med Metab Biol. 1993;49:285–320.
44 Decker JA, Rossano JW, Smith EO, et al. Risk factors and mode of death in isolated hypertrophic cardiomyopathy in children. J Am Coll Cardiol. 2009;54:250–254.
45 Ostman-Smith I, Wettrell G, Keeton B, et al. Echocardiographic and electrocardiographic identification of those children with hypertrophic cardiomyopathy who should be considered at high-risk of dying suddenly. Cardiol Young. 2005;15:632–642.
46 Pelliccia A, Corrado D, Bjornstad HH, et al. Recommendations for participation in competitive sport and leisure-time physical activity in individuals with cardiomyopathies, myocarditis and pericarditis. Eur J Cardiovasc Prev Rehabil. 2006;13:876–885.
47 Seggewiss H, Rigopoulos A. Management of hypertrophic cardiomyopathy in children. Paediatr Drugs. 2003;5:663–672.
48 Ciszewski A, Bilinska ZT, Lubiszewska B, et al. Dilated cardiomyopathy in children: clinical course and prognosis. Pediatr Cardiol. 1994;15:121–126.
49 Gagliardi MG. Dilated cardiomyopathy in children. Acta Paediatr Suppl. 2006;95:14–16.
50 Hsu DT, Canter CE. Dilated cardiomyopathy and heart failure in children. Heart Fail Clin. 2010;6:415–432.
51 Keesler MJ, Fisher SD, Lipshultz SE. Cardiac manifestations of HIV infection in infants and children. Ann N Y Acad Sci. 2001;946:169–178.
52 Towbin JA, Solaro RJ. Genetics of dilated cardiomyopathy: more genes that kill. J Am Coll Cardiol. 2004;44:2041–2043.
53 Towbin JA, Lowe AM, Colan SD, et al. Incidence, causes, and outcomes of dilated cardiomyopathy in children. JAMA. 2006;296:1867–1876.
54 Dodo H, Gow RM, Hamilton RM, et al. Chaotic atrial rhythm in children. Am Heart J. 1995;129:990–995.
55 Friedman RA, Moak JP, Garson AJ. Clinical course of idiopathic dilated cardiomyopathy in children. J Am Coll Cardiol. 1991;18:152–156.
56 Salim MA, Case CL, Gillette PC. Chaotic atrial tachycardia in children. Am Heart J. 1995;129:831–833.
57 Chen SC. Echocardiography of dilated cardiomyopathy in children. Echocardiography. 1991;8:207–217.
58 Tsirka AE, Trinkaus K, Chen SC, et al. Improved outcomes of pediatric dilated cardiomyopathy with utilization of heart transplantation. J Am Coll Cardiol. 2004;44:391–397.
59 Kim JJ, Denfield SW, McKenzie ED, et al. Mechanical circulatory support as a bridge to combined dual organ transplantation in children. J Heart Lung Transplant. 2006;25:1480–1482.
60 Minich LL, Tani LY, Hawkins JA, et al. Intra-aortic balloon pumping in children with dilated cardiomyopathy as a bridge to transplantation. J Heart Lung Transplant. 2001;20:750–754.
61 Morrow WR. Cardiomyopathy and heart transplantation in children. Curr Opin Cardiol. 2000;15:216–223.
62 Denfield SW, Rosenthal G, Gajarski RJ, et al. Restrictive cardiomyopathies in childhood: etiologies and natural history. Tex Heart Inst J. 1997;24:38–44.
63 Denfield SW. Sudden death in children with restrictive cardiomyopathy. Card Electrophysiol Rev. 2002;6:163–167.
64 Lewis AB. Clinical profile and outcome of restrictive cardiomyopathy in children. Am Heart J. 1992;123:1589–1593.
65 Rivenes SM, Kearney DL, Smith EO, et al. Sudden death and cardiovascular collapse in children with restrictive cardiomyopathy. Circulation. 2000;102:876–882.
66 Russo LM, Webber SA. Idiopathic restrictive cardiomyopathy in children. Heart. 2005;91:1199–1202.
67 Weller RJ, Weintraub R, Addonizio LJ, et al. Outcome of idiopathic restrictive cardiomyopathy in children. Am J Cardiol. 2002;90:501–506.
68 Bograd AJ, Mital S, Schwarzenberger JC, et al. Twenty-year experience with heart transplantation for infants and children with restrictive cardiomyopathy: 1986-2006. Am J Transplant. 2008;8:201–207.
69 Fenton MJ, Chubb H, McMahon AM, et al. Heart and heart-lung transplantation for idiopathic restrictive cardiomyopathy in children. Heart. 2006;92:85–89.
70 Kuhl U, Schultheiss HP. Myocarditis in children. Heart Fail Clin. 2010;6:483–496.
71 Shekerdemian L, Bohn D. Acute viral myocarditis: epidemiology and pathophysiology. Pediatr Crit Care Med. 2006;7:S2–S7.
72 Levine MC, Klugman D, Teach SJ. Update on myocarditis in children. Curr Opin Pediatr. 2010;22:278–283.
73 Nugent AW, Daubeney PE, Chondros P, et al. The epidemiology of childhood cardiomyopathy in Australia. N Engl J Med. 2003;348:1639–1646.
74 Webber SA, Boyle GJ, Jaffe R, et al. Role of right ventricular endomyocardial biopsy in infants and children with suspected or possible myocarditis. Br Heart J. 1994;72:360–363.
75 Amabile N, Fraisse A, Bouvenot J, et al. Outcome of acute fulminant myocarditis in children. Heart. 2006;92:1269–1273.
76 English RF, Janosky JE, Ettedgui JA, et al. Outcomes for children with acute myocarditis. Cardiol Young. 2004;14:488–493.
77 Bohn D. Acute viral myocarditis in children: guidelines. Pediatr Crit Care Med. 2006;7:S1.
78 Vashist S, Singh GK. Acute myocarditis in children: current concepts and management. Curr Treat Options Cardiovasc Med.. 2009;11:383–391.
79 Schwartz SM, Wessel DL. Medical cardiovascular support in acute viral myocarditis in children. Pediatr Crit Care Med. 2006;7:S12–S16.
80 Wheeler DS, Kooy NW. A formidable challenge: the diagnosis and treatment of viral myocarditis in children. Crit Care Clin. 2003;19:365–391.
81 Drucker NA, Colan SD, Lewis AB, et al. Gamma-globulin treatment of acute myocarditis in the pediatric population. Circulation. 1994;89:252–257.
82 Gagliardi MG, Bevilacqua M, Bassano C, et al. Long-term follow-up of children with myocarditis treated by immunosuppression and of children with dilated cardiomyopathy. Heart. 2004;90:1167–1171.
83 Robinson JL, Hartling L, Crumley E, et al. A systematic review of intravenous gamma globulin for therapy of acute myocarditis. BMC Cardiovasc Disord. 2005;5:12.
84 Bohn D, Macrae D, Chang AC. Acute viral myocarditis: mechanical circulatory support. Pediatr Crit Care Med. 2006;7:S21–S24.
85 Duncan BW, Bohn DJ, Atz AM, et al. Mechanical circulatory support for the treatment of children with acute fulminant myocarditis. J Thorac Cardiovasc Surg. 2001;122:440–448.
86 Grinda JM, Chevalier P, D’Attellis N, et al. Fulminant myocarditis in adults and children: biventricular assist device for recovery. Eur J Cardiothorac Surg. 2004;26:1169–1173.
87 Reiss N, El-Banayosy A, Arusoglu L, et al. Acute fulminant myocarditis in children and adolescents: the role of mechanical circulatory assist. ASAIO J. 2006;52:211–214.
88 Wu ET, Huang SC, Chen YS, et al. Children with fulminant myocarditis rescued with extracorporeal membrane oxygenation. Heart. 2006;92:1325–1326.
89 Wilmot I, Morales DL, Price JF, et al. Effectiveness of mechanical circulatory support in children with acute fulminant and persistent myocarditis. J Card Fail. 2011;17:487–494.
90 Seckeler MD, Hoke TR. The worldwide epidemiology of acute rheumatic fever and rheumatic heart disease. Clin Epidemiol. 2011;3:67–84.
91 Gordis L. The virtual disappearance of rheumatic fever in the United States: lessons in the rise and fall of disease. T. Duckett Jones Memorial Lecture. Circulation. 1985;72:1155–1162.
92 Guidelines for the diagnosis of rheumatic fever: Jones criteria, 1992 update. Special Writing Group of the Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease of the Council on Cardiovascular Disease in the Young of the American Heart Association. JAMA. 1992;268:2069–2073.
93 Dajani AS, Bisno AL, Chung KJ, et al. Prevention of rheumatic fever: a statement for health professionals by the Committee on Rheumatic Fever, Endocarditis and Kawasaki Disease of the Council on Cardiovascular Disease in the Young, the American Heart Association. Pediatr Infect Dis J. 1989;8:263–266.
94 Dajani A, Taubert K, Ferrieri P, et al. Treatment of acute streptococcal pharyngitis and prevention of rheumatic fever: a statement for health professionals. Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease of the Council on Cardiovascular Disease in the Young, the American Heart Association. Pediatrics. 1995;96:758–764.
95 Cilliers AM. Rheumatic fever and its management. BMJ. 2006;333:1153–1156.
96 Hillman ND, Tani LY, Veasy LG, et al. Current status of surgery for rheumatic carditis in children. Ann Thorac Surg. 2004;78:1403–1408.
97 Baddour LM, Wilson WR, Bayer AS, et al. Infective endocarditis: diagnosis, antimicrobial therapy, and management of complications. A statement for healthcare professionals from the Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease, Council on Cardiovascular Disease in the Young, and the Councils on Clinical Cardiology, Stroke, and Cardiovascular Surgery and Anesthesia, American Heart Association: endorsed by the Infectious Diseases Society of America. Circulation. 2005;111:e394–e434.
98 Knirsch W, Nadal D. Infective endocarditis in congenital heart disease. Eur J Pediatr. 2011;170:1111–1127.
99 Durack DT, Lukes AS, Bright DK. New criteria for diagnosis of infective endocarditis: utilization of specific echocardiographic findings. Duke Endocarditis Service. Am J Med. 1994;96:200–209.
100 Li JS, Sexton DJ, Mick N, et al. Proposed modifications to the Duke criteria for the diagnosis of infective endocarditis. Clin Infect Dis. 2000;30:633–638.
101 Saiman L, Prince A, Gersony WM. Pediatric infective endocarditis in the modern era. J Pediatr. 1993;122:847–853.
102 Martin JM, Neches WH, Wald ER. Infective endocarditis: 35 years of experience at a children’s hospital. Clin Infect Dis. 1997;24:669–675.
103 Ferrieri P, Gewitz MH, Gerber MA, et al. Unique features of infective endocarditis in childhood. Circulation. 2002;105:2115–2126.
104 Penk JS, Webb CL, Shulman ST, et al. Echocardiography in pediatric infective endocarditis. Pediatr Infect Dis J. 2011;30:1109–1111.
105 Humpl T, McCrindle BW, Smallhorn JF. The relative roles of transthoracic compared with transesophageal echocardiography in children with suspected infective endocarditis. J Am Coll Cardiol. 2003;41:2068–2071.
106 Morris CD, Reller MD, Menashe VD. Thirty-year incidence of infective endocarditis after surgery for congenital heart defect. JAMA. 1998;279:599–603.
107 Wilson W, Taubert KA, Gewitz M, et al. Prevention of infective endocarditis: guidelines from the American Heart Association. A guideline from the American Heart Association Rheumatic Fever, Endocarditis, and Kawasaki Disease Committee, Council on Cardiovascular Disease in the Young, and the Council on Clinical Cardiology, Council on Cardiovascular Surgery and Anesthesia, and the Quality of Care and Outcomes Research Interdisciplinary Working Group. editors. Circulation. 2007;116:1736–1754.
108 Steckelberg JM, Khandheria BK, Anhalt JP, et al. Prospective evaluation of the risk of bacteremia associated with transesophageal echocardiography. Circulation. 1991;84:177–180.
109 Banai S, Selitser V, Keren A, et al. Prospective study of bacteremia after cardiac catheterization. Am J Cardiol. 2003;92:1004–1007.
110 Pharis CS, Conway J, Warren AE, et al. The impact of 2007 infective endocarditis prophylaxis guidelines on the practice of congenital heart disease specialists. Am Heart J. 2011;161:123–129.
111 Kawasaki T, Kosaki F, Okawa S, et al. A new infantile acute febrile mucocutaneous lymph node syndrome (MLNS) prevailing in Japan. Pediatrics. 1974;54:271–276.
112 Newburger JW, Fulton DR. Kawasaki disease. Curr Opin Pediatr. 2004;16:508–514.
113 Falcini F. Kawasaki disease. Curr Opin Rheumatol. 2006;18:33–38.
114 Baker AL, Newburger JW. Cardiology patient pages. Kawasaki disease. Circulation. 2008;118:e110–e112.
115 Yeung RS. Kawasaki disease: update on pathogenesis. Curr Opin Rheumatol. 2010;22:551–560.
116 Fukazawa R. Long-term prognosis of Kawasaki disease: increased cardiovascular risk? Curr Opin Pediatr. 2010;22:587–592.
117 Levy DM, Silverman ED, Massicotte MP, et al. Long-term outcomes in patients with giant aneurysms secondary to Kawasaki disease. J Rheumatol. 2005;32:928–934.
118 AHA Scientific Statement. Diagnostic guidelines for Kawasaki disease. Council on Cardiovascular Disease in the Young; Committee on Rheumatic Fever, Endocarditis and Kawasaki Disease; American Heart Association. Circulation. 2001;103:335–336.
119 Newburger JW, Takahashi M, Gerber MA, et al. Diagnosis, treatment, and long-term management of Kawasaki disease: a statement for health professionals from the Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease, Council on Cardiovascular Disease in the Young, American Heart Association. Pediatrics. 2004;114:1708–1733.
120 Rowley AH, Shulman ST. Recent advances in the understanding and management of Kawasaki disease. Curr Infect Dis Rep. 2010;12:96–102.
121 Gordon JB, Kahn AM, Burns JC. When children with Kawasaki disease grow up: myocardial and vascular complications in adulthood. J Am Coll Cardiol. 2009;54:1911–1920.
122 Freedom RM, Lee KJ, MacDonald C, et al. Selected aspects of cardiac tumors in infancy and childhood. Pediatr Cardiol. 2000;21:299–316.
123 Isaacs HJ. Fetal and neonatal cardiac tumors. Pediatr Cardiol. 2004;25:252–273.
124 Wang JN, Yao CT, Chen JS, et al. Cardiac tumors in infants and children. Acta Paediatr Taiwan. 2003;44:215–219.
125 Becker AE. Primary heart tumors in the pediatric age group: a review of salient pathologic features relevant for clinicians. Pediatr Cardiol. 2000;21:317–323.
126 Sallee D, Spector ML, Van Heeckeren DW, et al. Primary pediatric cardiac tumors: a 17 year experience. Cardiol Young. 1999;9:155–162.
127 Ross RD, Farooki ZQ. Medical versus surgical management of intracardiac rhabdomyomas. Am J Cardiol. 1991;68:836.
128 Stiller B, Hetzer R, Meyer R, et al. Primary cardiac tumours: when is surgery necessary? Eur J Cardiothorac Surg. 2001;20:1002–1006.
129 Jozwiak S, Kotulska K, Kasprzyk-Obara J, et al. Clinical and genotype studies of cardiac tumors in 154 patients with tuberous sclerosis complex. Pediatrics. 2006;118:e1146–e1151.
130 Tworetzky W, McElhinney DB, Margossian R, et al. Association between cardiac tumors and tuberous sclerosis in the fetus and neonate. Am J Cardiol. 2003;15:487–489.
131 Burke AP, Rosado-de-Christenson M, Templeton PA, et al. Cardiac fibroma: clinicopathologic correlates and surgical treatment. J Thorac Cardiovasc Surg. 1994;108:862–870.
132 Kusano KF, Ohe T. Cardiac tumors that cause arrhythmias. Card Electrophysiol Rev. 2002;6:174–177.
133 Padalino MA, Basso C, Milanesi O, et al. Surgically treated primary cardiac tumors in early infancy and childhood. J Thorac Cardiovasc Surg. 2005;129:1358–1363.
134 Gowdamarajan A, Michler RE. Therapy for primary cardiac tumors: is there a role for heart transplantation? Curr Opin Cardiol. 2000;15:121–125.
135 Kussman BD, Devavaram P, Hansen DD, et al. Anesthetic implications of primary cardiac tumors in infants and children. J Cardiothorac Vasc Anesth. 2002;16:582–586.
136 Madriago E, Silberbach M. Heart failure in infants and children. Pediatr Rev. 2010;31:4–12.
137 Chang AC, Towbin JA. Heart failure in children and young adults: from molecular mechanisms to medical and surgical strategies. Philadelphia: Saunders; 2006.
138 Shaddy RE, Wernovsky G. Pediatric heart failure. Boca Raton, Fla.: Taylor & Francis Group; 2005.
139 Webster G, Zhang J, Rosenthal D. Comparison of the epidemiology and comorbidities of heart failure in the pediatric and adult populations: a retrospective, cross-sectional study. BMC Cardiovasc Disord. 2006;6:23–30.
140 Auslender M. Pathophysiology of pediatric heart failure. Prog Pediatr Cardiol. 2000;11:175–184.
141 Ohuchi H, Takasugi H, Ohashi H, et al. Stratification of pediatric heart failure on the basis of neurohormonal and cardiac autonomic nervous activities in patients with congenital heart disease. Circulation. 2003;108:2368–2376.
142 Packer M. How should physicians view heart failure? The philosophical and physiological evolution of three conceptual models of the disease. Am J Cardiol. 1993;71:3C–11C.
143 Ross RD, Bollinger RO, Pinsky WW. Grading the severity of congestive heart failure in infants. Pediatr Cardiol. 1992;13:72–75.
144 Hsu DT, Pearson GD. Heart failure in children: part I. History, etiology, and pathophysiology. Circ Heart Fail. 2009;2:63–70.
145 Hsu DT, Pearson GD. Heart failure in children: part II. Diagnosis, treatment, and future directions. Circ Heart Fail. 2009;2:490–498.
146 Clark B, Jr. Treatment of heart failure in infants and children. Heart Dis. 2000;2:354–361.
147 Momma K. ACE inhibitors in pediatric patients with heart failure. Paediatr Drugs. 2006;8:55–69.
148 Wernovsky G, Hoffman TM. Pediatric heart failure management: solving the puzzle. Crit Care Med. 2001;29:S212–S213.
149 Gazit AZ, Oren PP. Pharmaceutical management of decompensated heart failure syndrome in children: current state of the art and a new approach. Curr Treat Options Cardiovasc Med. 2009;11:403–409.
150 Shaddy RE. Beta-adrenergic blockers in the treatment of pediatric heart failure. Prog Pediatr Cardiol. 2000;12:113–118.
151 Baden HP. Does nesiritide offer something new in the management of pediatric heart failure? Pediatr Crit Care Med. 2005;6:613–614.
152 Feingold B, Law YM. Nesiritide use in pediatric patients with congestive heart failure. J Heart Lung Transplant. 2004;23:1455–1459.
153 Jefferies JL, Denfield SW, Price JF, et al. A prospective evaluation of nesiritide in the treatment of pediatric heart failure. Pediatr Cardiol. 2006;27:402–407.
154 Bruns LA, Chrisant MK, Lamour JM, et al. Carvedilol as therapy in pediatric heart failure: an initial multicenter experience. J Pediatr. 2001;138:505–511.
155 Greenway SC, Benson LN. The use of carvedilol in pediatric heart failure. Cardiovasc Hematol Disord Drug Targets. 2006;6:35–42.
156 Shaddy RE, Curtin EL, Sower B, et al. The pediatric randomized carvedilol trial in children with chronic heart failure: rationale and design. Am Heart J. 2002;144:383–389.
157 Murphy TW, Smith JH, Ranger MR, et al. General anesthesia for children with severe heart failure. Pediatr Cardiol. 2011;32:139–144.
158 Groban L, Butterworth J. Perioperative management of chronic heart failure. Anesth Analg. 103, 2006. 557–555
159 Rosenthal DN, Hammer GB. Cardiomyopathy and heart failure in children: anesthetic implications. Paediatr Anaesth. 2011;21:577–584.
160 Shapiro NL, Huang RY, Sangwan S, et al. Tracheal stenosis and congenital heart disease in patients with Down syndrome: diagnostic approach and surgical options. Int J Pediatr Otorhinolaryngol. 2000;54:137–142.
161 Shott SR. Down syndrome: analysis of airway size and a guide for appropriate intubation. Laryngoscope. 2000;110:585–592.
162 Hata T, Todd MM. Cervical spine considerations when anesthetizing patients with Down syndrome. Anesthesiology. 2005;102:680–685.
163 Lin YC. Cervical spine disease and Down syndrome in pediatric anesthesia. Anesth Clin North Am. 1998;16:911–923.
164 Litman RS, Zerngast BA, Perkins FM. Preoperative evaluation of the cervical spine in children with trisomy-21: results of a questionnaire study. Paediatr Anaesth. 1995;5:355–361.
164a Lo RN, Leung MP, Lau KC, Yeung CY. Abnormal radial artery in Down’s syndrome. Arch Dis Child. 1986;61:885–890.
165 Wells GL, Barker SE, Finley SC, et al. Congenital heart disease in infants with Down’s syndrome. South Med J. 1994;87:724–727.
166 Bai W, Voepel-Lewis T, Malviya S. Hemodynamic changes in children with Down syndrome during and following inhalation induction of anesthesia with sevoflurane. J Clin Anesth. 2010;22:592–597.
167 Kraemer FW, Stricker PA, Gurnaney HG, et al. Bradycardia during induction of anesthesia with sevoflurane in children with Down syndrome. Anesth Analg. 2010;111:1259–1263.
168 Clapp S, Perry BL, Farooki ZQ, et al. Down’s syndrome, complete atrioventricular canal, and pulmonary vascular obstructive disease. J Thorac Cardiovasc Surg. 1990;100:115–121.
169 Cappelli-Bigazzi M, Santoro G, Battaglia C, et al. Endothelial cell function in patients with Down’s syndrome. Am J Cardiol. 2004;94:392–395.
170 Goc B, Walencka Z, Wloch A, et al. Trisomy 18 in neonates: prenatal diagnosis, clinical features, therapeutic dilemmas and outcome. J Appl Genet. 2006;47:165–170.
171 Pont SJ, Robbins JM, Bird TM, et al. Congenital malformations among liveborn infants with trisomies 18 and 13. Am J Med Genet A. 2006;140:1749–1756.
172 Courreges P, Nieuviarts R, Lecoutre D. Anaesthetic management for Edward’s syndrome. Paediatr Anaesth. 2003;13:267–269.
173 Miller C, Mayhew JF. Edward’s syndrome (trisomy 18). Paediatr Anaesth. 1998;8:441–442.
174 Chen CP. Congenital heart defects associated with fetal trisomy 18. Prenat Diagn. 2006;26:483–485.
175 Musewe NN, Alexander DJ, Teshima I, et al. Echocardiographic evaluation of the spectrum of cardiac anomalies associated with trisomy 13 and trisomy 18. J Am Coll Cardiol. 1990;15:673–677.
176 Baty BJ, Blackburn BL, Carey JC. Natural history of trisomy 18 and trisomy 13: I. Growth, physical assessment, medical histories, survival, and recurrence risk. Am J Med Genet. 1994;49:175–188.
177 Rios A, Furdon SA, Adams D, et al. Recognizing the clinical features of trisomy 13 syndrome. Adv Neonatal Care. 2004;4:332–343.
178 Gravholt CH. Epidemiological, endocrine and metabolic features in Turner syndrome. Arq Bras Endocrinol Metabol. 2005;49:145–156.
179 Doswell BH, Visootsak J, Brady AN, et al. Turner syndrome: an update and review for the primary pediatrician. Clin Pediatr. 2006;45:301–313.
180 Gravholt CH. Clinical practice in Turner syndrome. Nat Clin Pract Endocrinol Metab. 2005;1:41–52.
181 Ewart AK, Morris CA, Atkinson D, et al. Hemizygosity at the elastin locus in a developmental disorder, Williams syndrome. Nat Genet. 1993;5:11–16.
182 Morris CA, Mervis CB. Williams syndrome and related disorders. Annu Rev Genomics Hum Genet. 2000;1:461–484.
183 Bruno E, Rossi N, Thuer O, et al. Cardiovascular findings, and clinical course, in patients with Williams syndrome. Cardiol Young. 2003;13:532–536.
184 Zalzstein E, Moes CA, Musewe NN, et al. Spectrum of cardiovascular anomalies in Williams-Beuren syndrome. Pediatr Cardiol. 1991;12:219–223.
185 Bragg K, Fedel GM, DiProsperis A. Cardiac arrest under anesthesia in a pediatric patient with Williams syndrome: a case report. AANA J. 2005;73:287–293.
186 Horowitz PE, Akhtar S, Wulff JA, et al. Coronary artery disease and anesthesia-related death in children with Williams syndrome. J Cardiothorac Vasc Anesth. 2002;16:739–741.
187 Burch TM, McGowan FXJ, Kussman BD, et al. Congenital supravalvular aortic stenosis and sudden death associated with anesthesia: what’s the mystery? Anesth Analg. 2008;107:1848–1854.
188 Medley J, Russo P, Tobias JD. Perioperative care of the patient with Williams syndrome. Paediatr Anaesth. 2005;15:243–247.
189 Ramamoorthy C, Haberkern CM, Bhananker SM, et al. Anesthesia-related cardiac arrest in children with heart disease: data from the Pediatric Perioeraptve Cardiac Arrest (POCA) registry. Anesth Analg. 2010;110:1376–1382.
190 Cambiaso P, Orazi C, Digilio MC, et al. Thyroid morphology and subclinical hypothyroidism in children and adolescents with Williams syndrome. J Pediatr. 2007;150:62–65.
191 Hall JG. CATCH 22. J Med Genet. 1993;30:801–802.
192 Soares G, Alvares S, Rocha C, et al. Congenital heart defects and chromosomal anomalies including 22q11 microdeletion (CATCH 22). Rev Port Cardiol. 2005;24:349–371.
193 Eliez S, Palacio-Espasa F, Spira A, et al. Young children with velo-cardio-facial syndrome (CATCH-22): psychological and language phenotypes. Eur Child Adolesc Psychiatry. 2000;9:109–114.
194 Noonan JA. Noonan syndrome revisited. J Pediatr. 1999;135:667–668.
195 Noonan J. Noonan syndrome: then and now. Cardiol Young. 1999;9:545–546.
196 Marino B, Digilio MC, Toscano A, et al. Congenital heart diseases in children with Noonan syndrome: an expanded cardiac spectrum with high prevalence of atrioventricular canal. J Pediatr. 1999;135:703–706.
197 Nishikawa T, Ishiyama S, Shimojo T, et al. Hypertrophic cardiomyopathy in Noonan syndrome. Acta Paediatr Jpn. 1996;39:91–98.
198 Staudt JM, Van der Horst CM, Peters M, et al. Bleeding diathesis in Noonan syndrome. Scand J Plast Reconstr Surg Hand Surg. 2005;4:247–248.
199 Mizuguchi T, Matsumoto N. Recent progress in genetics of Marfan syndrome and Marfan-associated disorders. J Hum Genet. 2007;52:1–12.
200 De Paepe A, Devereux RB, Dietz HC, et al. Revised diagnostic criteria for the Marfan syndrome. Am J Med Genet. 1996;62:417–426.
201 Pyeritz RE. The Marfan syndrome. Annu Rev Med. 2000;51:481–510.
202 Sharkey AM. Cardiovascular management of Marfan syndrome in the young. Curr Treat Options Cardiovasc Med. 2006;8:396–402.
203 Shores J, Berger KR, Murphy EA, et al. Progression of aortic dilatation and the benefit of long-term beta-adrenergic blockade in Marfan’s syndrome. N Engl J Med. 1994;330:1335–1341.
204 Detter C, Mair H, Klein HG, et al. Long-term prognosis of surgically-treated aortic aneurysms and dissections in patients with and without Marfan syndrome. Eur J Cardiothorac Surg. 1998;13:416–423.
205 Das BB, Taylor AL, Yetman AT. Left ventricular diastolic dysfunction in children and young adults with Marfan syndrome. Pediatr Cardiol. 2006;27:256–258.
206 Savolainen A, Nisula L, Keto P, et al. Left ventricular function in children with the Marfan syndrome. Eur Heart J. 1994;15:625–630.
207 Wood JR, Bellamy D, Child AH, et al. Pulmonary disease in patients with Marfan syndrome. Thorax. 1984;39:780–784.
208 Corsello G, Maresi E, Corrao AM, et al. VATER/VACTERL association: clinical variability and expanding phenotype including laryngeal stenosis. Am J Med Genet. 1992;44:813–815.
209 Rittler M, Paz JE, Castilla EE. VACTERL association, epidemiologic definition and delineation. Am J Med Genet. 1996;63:529–536.
210 Diaz LK, Akpek EA, Dinavahi R, et al. Tracheoesophageal fistula and associated congenital heart disease: implications for anesthetic management and survival. Paediatr Anaesth. 2005;15:862–869.
211 Thomas IT, Frias JL. The heart in selected congenital malformations: a lesson in pathogenetic relationships. Ann Clin Lab Sci. 1987;17:207–210.
212 Van Meter TD, Weaver DD. Oculo-auriculo-vertebral spectrum and the CHARGE association: clinical evidence for a common pathogenetic mechanism. Clin Dysmorphol. 1996;5:187–196.
213 Vissers LE, van Ravenswaaij CM, Admiraal R, et al. Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat Genet. 2004;36:955–957.
214 Blake KD, Davenport SL, Hall BD, et al. CHARGE association: an update and review for the primary pediatrician. Clin Pediatr (Phila). 1998;37:159–173.
215 Wyse RK, al-Mahdawi S, Burn J, et al. Congenital heart disease in CHARGE association. Pediatr Cardiol. 1993;14:75–81.
216 Hoshi T, Matsumiya N, Satsumae T, et al. [A case of the CHARGE association with failed tracheal intubation]. Masui. 1998;47:487–489.
217 Stack CG, Wyse RK. Incidence and management of airway problems in the CHARGE Association. Anaesthesia. 1991;46:582–585.
218 Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. N Engl J Med. 2006;355:1345–1356.
219 Shenkman Z, Rockoff MA, Eldredge EA, et al. Anaesthetic management of children with tuberous sclerosis. Paediatr Anaesth. 2002;12:700–704.
220 Watson GH. Cardiac rhabdomyomas in tuberous sclerosis. Ann NY Acad Sci. 1991;615:50–57.
221 Webb DW, Thomas RD, Osborne JP. Cardiac rhabdomyomas and their association with tuberous sclerosis. Arch Dis Child. 1993;68:367–370.
222 Septer S, Thompson ES, Willemsen-Dunlap A. Anesthesia concerns for children with tuberous sclerosis. AANA J. 2006;74:219–225.
223 Freed K, Low VH. The aberrant subclavian artery. AJR Am J Roentgenol. 1997;168:481–484.
224 Bensky AS, O’Brien JJ, Hammon JW. Transesophageal echo probe compression of an aberrant right subclavian artery. J Am Soc Echocardiogr. 1995;8:964–966.
225 Tak T, Crouch E, Drake GB. Persistent left superior vena cava: incidence, significance and clinical correlates. Int J Cardiol. 2002;82:91–93.
226 Pelech AN. The physiology of cardiac auscultation. Pediatr Clin North Am. 2004;51:1515–1535.
227 Frommelt MA. Differential diagnosis and approach to a heart murmur in term infants. Pediatr Clin North Am. 2004;51:1023–1032.
228 Rosenthal A. How to distinguish between innocent and pathologic murmurs in childhood. Pediatr Clin North Am. 1984;31:1229–1240.
229 Danford DA. Effective use of the consultant, laboratory testing, and echocardiography for the pediatric patient with heart murmur. Pediatr Ann. 2000;29:482–488.
230 Newburger JW, Rosenthal A, Williams RG, et al. Noninvasive tests in the initial evaluation of heart murmurs in children. N Engl J Med. 1983;308:61–64.
231 Yi MS, Kimball TR, Tsevat J, et al. Evaluation of heart murmurs in children: cost-effectiveness and practical implications. J Pediatr. 2002;141:504–511.
232 Wathen JE, Rewers AB, Yetman AT, et al. Accuracy of ECG interpretation in the pediatric emergency department. Ann Emerg Med. 2005;46:507–511.
233 Sharieff GQ, Rao SO. The pediatric ECG. Emerg Med Clin North Am. 2006;24:195–208.
234 Davignon A, Rautaharju PM, Boisselle E, et al. Normal ECG standards for infants and children. Pediatr Cardiol. 1979;1:123–152.
235 Dickinson DF. The normal ECG in childhood and adolescence. Heart. 2005;91:1626–1630.
236 Schwartz PJ, Garson AJ, Paul T, et al. Guidelines for the interpretation of the neonatal electrocardiogram: a task force of the European Society of Cardiology. Eur Heart J. 2002;23:1329–1344.
237 American Heart Association guidelines for cardiopulmonary resuscitation (CPR) and emergency cardiovascular care part 12: pediatric advanced life support. Circulation. 2005;112(Suppl IV):167–187.
238 Balaji S. Supraventricular tachycardia in children. Curr Treat Options Cardiovasc Med. 2000;2:521–528.
239 Chun TU, Van Hare GF. Advances in the approach to treatment of supraventricular tachycardia in the pediatric population. Curr Cardiol Rep. 2004;6:322–326.
240 Dixon J, Foster K, Wyllie J, et al. Guidelines and adenosine dosing in supraventricular tachycardia. Arch Dis Child. 2005;90:1190–1191.
241 Khairy P, Dore A, Talajic M, et al. Arrhythmias in adult congenital heart disease. Expert Rev Cardiovasc Ther. 2006;4:83–95.
242 Meldon SW, Brady WJ, Berger S, et al. Pediatric ventricular tachycardia: a review with three illustrative cases. Pediatr Emerg Care. 1994;10:294–300.
243 Bar-Cohen Y, Silka MJ. Congenital long QT syndrome: diagnosis and management in pediatric patients. Curr Treat Options Cardiovasc Med. 2006;8:387–395.
244 Roden DM, Lazzara R, Rosen M, et al. Multiple mechanisms in the long-QT syndrome: current knowledge, gaps, and future directions. The SADS Foundation Task Force on LQTS. Circulation. 1996;94:1996–2012.
245 Schwartz PJ, Moss AJ, Vincent GM, et al. Diagnostic criteria for the long QT syndrome: an update. Circulation. 1993;88:782–784.
246 Whyte SD, Sanatani S, Lim J, Booker PD. A comparison of the effect on dispersion of repolarization of age-adjusted MAC values of sevoflurane in children. Anesth Analg. 2007;104:277–282.
247 Nathan AT, Berkowitz DH, Montenegro LM, et al. Implications of anesthesia in children with long QT syndrome. Anesth Analg. 2011;112:1163–1168.
248 Bernstein AD, Daubert JC, Fletcher RD, et al. The revised NASPE/BPEG generic code for antibradycardia, adaptive-rate, and multisite pacing. Pacing Clin Electrophysiol. 2002;25:260–264.
249 Epstein AE, DiMarco JP, Ellenbogen KA, et al. ACC/AHA/HRS 2008 guidelines for device-based therapy of cardiac rhythm abnormalities: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the ACC/AHA/NASPE 2002 Guideline Update for Implantation of Cardiac Pacemakers and Antiarrhythmia Devices) developed in collaboration with the American Association for Thoracic Surgery and Society of Thoracic Surgeons. J Am Coll Cardiol. 2008;51:e1–62.
250 Silka MJ, Bar-Cohen Y. Pacemakers and implantable cardioverter-defibrillators in pediatric patients. Heart Rhythm. 2006;3:1360–1366.
251 Navaratnam M, Dubin A. Pediatric pacemakers and ICDs: how to optimize perioperative care. Paediatr Anaesth. 2011;21:512–521.
252 Practice advisory for the perioperative management of patients with cardiac rhythm management devices: pacemakers and implantable cardioverter-defibrillators. A report by the American Society of Anesthesiologists Task Force on Perioperative Management of Patients with Cardiac Rhythm Management Devices. Anesthesiology. 2005;103:186–198.
253 Bennie RE, Dierdorf SF, Hubbard JE. Perioperative management of children with third degree heart block undergoing pacemaker placement: a ten year review. Paediatr Anaesth. 1997;7:301–304.
254 Bloomfield P, Bowler GM. Anaesthetic management of the patient with a permanent pacemaker. Anaesthesia. 1989;44:42–46.
255 Bourke ME. The patient with a pacemaker or related device. Can J Anaesth. 1996;43:R24–R41.
256 Beland MJ, Hesslein PS, Finlay CD, et al. Noninvasive transcutaneous cardiac pacing in children. Pacing Clin Electrophysiol. 1987;10:1262–1270.
257 Quan L, Graves JR, Kinder DR, et al. Transcutaneous cardiac pacing in the treatment of out-of-hospital pediatric cardiac arrests. Ann Emerg Med. 1992;21:905–909.
258 Kron J, Oliver RP, Norsted S, et al. The automatic implantable cardioverter-defibrillator in young patients. J Am Coll Cardiol. 1990;16:896–902.
259 Alexander ME, Cecchin F, Walsh EP, et al. Implications of implantable cardioverter defibrillator therapy in congenital heart disease and pediatrics. J Cardiovasc Electrophysiol. 2004;15:72–76.
260 Eicken A, Kolb C, Lange S, et al. Implantable cardioverter defibrillator (ICD) in children. Int J Cardiol. 2006;107:30–35.
261 Dubin AM, Berul CI, Bevilacqua LM, et al. The use of implantable cardioverter-defibrillators in pediatric patients awaiting heart transplantation. J Card Fail. 2003;9:375–379.
262 Kaski JP, Tome Esteban MT, Lowe MD, et al. Outcomes following implantable cardioverter-defibrillator therapy in children with hypertrophic cardiomyopathy. Heart. 2006;92:832–835.
263 Stephenson EA, Batra AS, Knilans TK, et al. A multicenter experience with novel implantable cardioverter defibrillator configurations in the pediatric and congenital heart disease population. J Cardiovasc Electrophysiol. 2006;17:41–46.
264 Laya BF, Goske MJ, Morrison S, et al. The accuracy of chest radiographs in the detection of congenital heart disease and in the diagnosis of specific congenital cardiac lesions. Pediatr Radiol. 2006;36:677–681.
265 Beekman RP, Hazekamp MG, Sobotka MA, et al. A new diagnostic approach to vascular rings and pulmonary slings: the role of MRI. Magn Reson Imaging. 1998;16:137–145.
266 Hernanz-Schulman M. Vascular rings: a practical approach to imaging diagnosis. Pediatr Radiol. 2005;35:961–979.
267 Shah RK, Mora BN, Bacha E, et al. The presentation and management of vascular rings: an otolaryngology perspective. Int J Pediatr Otorhinolaryngol. 2007;71:57–62.
268 Burch M, Balaji S, Deanfield JE, et al. Investigation of vascular compression of the trachea: the complementary roles of barium swallow and echocardiography. Arch Dis Child. 1993;68:171–176.
269 Woods RK, Sharp RJ, Holcomb GW, 3rd., et al. Vascular anomalies and tracheoesophageal compression: a single institution’s 25-year experience. Ann Thorac Surg. 2001;72:434–438. discussion 438-9
270 Mertens L, Friedberg MK. The gold standard for noninvasive imaging in congenital heart disease: echocardiography. Curr Opin Cardiol. 2009;24:119–124.
271 Lopez L. Advances in echocardiography. Curr Opin Pediatr. 2009;21:579–584.
272 Lai WW, Geva T, Shirali GS, et al. Guidelines and standards for performance of a pediatric echocardiogram: a report from the Task Force of the Pediatric Council of the American Society of Echocardiography. J Am Soc Echocardiogr. 19, 2006. 1413–1410
273 Ayres NA, Miller-Hance W, Fyfe DA, et al. Indications and guidelines for performance of transesophageal echocardiography in the patient with pediatric acquired or congenital heart disease: report from the task force of the Pediatric Council of the American Society of Echocardiography. J Am Soc Echocardiogr. 2005;18:91–98.
274 D’souza MG, Thys DM. Transesophageal echocardiography. Curr Opin Anaesthesiol. 2000;13:21–25.
275 Muhiudeen Russell IA, Miller-Hance WC, Silverman NH. Intraoperative transesophageal echocardiography for pediatric patients with congenital heart disease. Anesth Analg. 1998;5:1058–1076.
276 Russell IA, Rouine-Rapp K, Stratmann G, et al. Congenital heart disease in the adult: a review with internet-accessible transesophageal echocardiographic images. Anesth Analg. 2006;102:694–723.
277 Germanakis I, Sifakis S. The impact of fetal echocardiography on the prevalence of liveborn congenital heart disease. Pediatr Cardiol. 2006;27:465–472.
278 Smrcek JM, Berg C, Geipel A, et al. Detection rate of early fetal echocardiography and in utero development of congenital heart defects. J Ultrasound Med. 2006;25:187–196.
279 Ungerleider RM, Greeley WJ, Sheikh KH, et al. Routine use of intraoperative epicardial echocardiography and Doppler color flow imaging to guide and evaluate repair of congenital heart lesions: a prospective study. J Thorac Cardiovasc Surg. 1990;100:297–309.
280 Rigatelli G. Expanding the use of intracardiac echocardiography in congenital heart disease catheter-based interventions. J Am Soc Echocardiogr. 2005;18:1230–1231.
281 Pedra SR, Pedra CA, Abizaid AA, et al. Intracoronary ultrasound assessment late after the arterial switch operation for transposition of the great arteries. J Am Coll Cardiol. 2005;45:2061–2068.
282 Baker GH, Shirali G, Ringewald JM, et al. Usefulness of live three-dimensional transesophageal echocardiography in a congenital heart disease center. Am J Cardiol. 2009;103:1025–1028.
283 Hung J, Lang R, Flachskampf F, et al. 3D Echocardiography: a review of the current status and future directions. J Am Soc Echocardiogr. 2007;20:213–233.
284 Lang RM, Mor-Avi V, Sugeng L, et al. Three-dimensional echocardiography: the benefits of the additional dimension. J Am Coll Cardiol. 2006;48:2053–2069.
285 Friedberg MK, Rosenthal DN. New developments in echocardiographic methods to assess right ventricular function in congenital heart disease. Curr Opin Cardiol. 2005;20:84–88.
286 Cui W, Roberson DA. Left ventricular Tei index in children: comparison of tissue Doppler imaging, pulsed wave Doppler, and M-mode echocardiography normal values. J Am Soc Echocardiogr. 2006;19:1438–1445.
287 Ikemba CM, Su JT, Stayer SA, et al. Myocardial performance index with sevoflurane-pancuronium versus fentanyl-midazolam-pancuronium in infants with a functional single ventricle. Anesthesiology. 2004;101:1298–1305.
288 Prakash A, Printz BF, Lamour JM, et al. Myocardial performance index in pediatric patients after cardiac transplantation. J Am Soc Echocardiogr. 2004;17:439–442.
289 Nikitin NP, Witte KK. Application of tissue Doppler imaging in cardiology. Cardiology. 2004;101:170–184.
290 Pislaru C, Abraham TP, Belohlavek M. Strain and strain rate echocardiography. Curr Opin Cardiol. 2002;17:443–454.
291 McMahon CJ, Nagueh SF, Eapen RS, et al. Echocardiographic predictors of adverse clinical events in children with dilated cardiomyopathy: a prospective clinical study. Heart. 2004;90:908–915.
292 McMahon CJ, Pignatelli RH, Nagueh SF, et al. Left ventricular noncompaction cardiomyopathy in children: characterization of clinical status using tissue Doppler-derived indices of left ventricular diastolic relaxation. Heart. 2007;93:676–681.
293 Gjertsson P, Caidahl K, Svensson G, et al. Important pressure recovery in patients with aortic stenosis and high Doppler gradients. Am J Cardiol. 2001;88:139–144.
294 Boxt LM. Magnetic resonance and computed tomographic evaluation of congenital heart disease. J Magn Reson Imaging. 2004;19:827–847.
295 Dorfman AL, Geva T. Magnetic resonance imaging evaluation of congenital heart disease: conotruncal anomalies. J Cardiovasc Magn Reson. 2006;8:645–659.
296 Fenchel M, Greil GF, Martirosian P, et al. Three-dimensional morphological magnetic resonance imaging in infants and children with congenital heart disease. Pediatr Radiol. 2006;36:1265–1272.
297 Gutierrez FR, Siegel MJ, Fallah JH, et al. Magnetic resonance imaging of cyanotic and noncyanotic congenital heart disease. Magn Reson Imaging Clin North Am. 2002;10:209–235.
298 Samyn MM. A review of the complementary information available with cardiac magnetic resonance imaging and multislice computed tomography (CT) during the study of congenital heart disease. Int J Cardiovasc Imaging. 2004;20:569–578.
299 Fogel MA, Hubbard AM, Fellows KE, et al. MRI for physiology and function in congenital heart disease: functional assessment of the heart preoperatively and postoperatively. Semin Roentgenol. 1998;33:239–251.
300 Johnson TR, Goldmuntz E, McDonald-McGinn DM, et al. Cardiac magnetic resonance imaging for accurate diagnosis of aortic arch anomalies in patients with 22q11.2 deletion. Am J Cardiol. 2005;96:1726–1730.
301 Fogel MA. Assessment of cardiac function by magnetic resonance imaging. Pediatr Cardiol. 2000;21:59–69.
302 Prakash A, Powell AJ, Krishnamurthy R, et al. Magnetic resonance imaging evaluation of myocardial perfusion and viability in congenital and acquired pediatric heart disease. Am J Cardiol. 2004;93:657–661.
303 Powell AJ, Chung T, Landzberg MJ, et al. Accuracy of MRI evaluation of pulmonary blood supply in patients with complex pulmonary stenosis or atresia. Int J Card Imaging. 2000;16:169–174.
304 Beerbaum P, Korperich H, Gieseke J, et al. Rapid left-to-right shunt quantification in children by phase-contrast magnetic resonance imaging combined with sensitivity encoding (SENSE). Circulation. 2003;108:1355–1361.
305 Hundley WG, Li HF, Lange RA, et al. Assessment of left-to-right intracardiac shunting by velocity-encoded, phase-difference magnetic resonance imaging: a comparison with oximetric and indicator dilution techniques. Circulation. 1995;91:2955–2960.
306 Powell AJ, Geva T. Blood flow measurement by magnetic resonance imaging in congenital heart disease. Pediatr Cardiol. 2000;21:47–58.
307 Nield LE, Qi XL, Valsangiacomo ER, et al. In vivo MRI measurement of blood oxygen saturation in children with congenital heart disease. Pediatr Radiol. 2005;35:179–185.
308 Geva T, Marshall AC. Magnetic resonance imaging-guided catheter interventions in congenital heart disease. Circulation. 2006;113:1051–1052.
309 Moore P. MRI-guided congenital cardiac catheterization and intervention: the future? Catheter Cardiovasc Interv. 2005;66:1–8.
310 Rickers C, Seethamraju RT, Jerosch-Herold M, et al. Magnetic resonance imaging guided cardiovascular interventions in congenital heart diseases. J Interv Cardiol. 2003;16:143–147.
311 Allen JG, Sury MR. Sedation of children undergoing magnetic resonance imaging. Br J Anaesth. 2006;97:898–899. author reply 899
312 Dalal PG, Murray D, Cox T, et al. Sedation and anesthesia protocols used for magnetic resonance imaging studies in infants: provider and pharmacologic considerations. Anesth Analg. 2006;103:863–868.
313 Sarikouch S, Schaeffler R, Korperich H, et al. Cardiovascular magnetic resonance imaging for intensive care infants: safe and effective? Pediatr Cardiol. 2009;30:146–152.
314 Gooden CK. Anesthesia for magnetic resonance imaging. Curr Opin Anaesthesiol. 2004;17:339–342.
315 Osborn IP. Magnetic resonance imaging anesthesia: new challenges and techniques. Curr Opin Anaesthesiol. 2002;15:443–448.
316 Han BK, Lindberg J, Grant K, et al. Accuracy and safety of high pitch computed tomography imaging in young children with complex congenital heart disease. Am J Cardiol. 2011;107:1541–1546.
317 Chandran A, Fricker FJ, Schowengerdt KO, et al. An institutional review of the value of computed tomographic angiography in the diagnosis of congenital cardiac malformations. Cardiol Young. 2005;15:47–51.
318 Freed MD, Miettinen OS, Nadas AS. Oximetric detection of intracardiac left-to-right shunts. Br Heart J. 1979;42:690–694.
319 Boothroyd A, McDonald E, Moores BM, et al. Radiation exposure to children during cardiac catheterization. Br J Radiol. 1997;70:180–185.
320 Fellows KE, Keane JF, Freed MD. Angled views in cineangiocardiography of congenital heart disease. Circulation. 1977;56:485–490.
321 Soto B, Coghlan CH, Bargeron LM. Present status of axially angled angiocardiography. Cardiovasc Intervent Radiol. 1984;7:156–165.
322 Bennett D, Marcus R, Stokes M. Incidents and complications during pediatric cardiac catheterization. Paediatr Anaesth. 2005;15:1083–1088.
323 Bulbul ZR, Galal MO, Mahmoud E, et al. Arterial complications following cardiac catheterization in children less than 10 kg. Asian Cardiovasc Thorac Ann. 2002;10:129–132.
324 Pophal SG, Sigfusson G, Booth KL, et al. Complications of endomyocardial biopsy in children. J Am Coll Cardiol. 1999;34:2105–2110.
325 Rhodes JF, Asnes JD, Blaufox AD, et al. Impact of low body weight on frequency of pediatric cardiac catheterization complications. Am J Cardiol. 2000;86:1275–1278.
326 Arpagaus M, Gray D, Zierler B. Cardiac catheterization in children as outpatients: potential, eligibility, safety and costs. Cardiol Young. 2003;13:275–283.
327 Feinstein JA, Kim N, Reddy VM, et al. Percutaneous pulmonary valve placement in a 10-month-old patient using a hand crafted stent-mounted porcine valve. Catheter Cardiovasc Interv. 2006;67:644–649.
328 Bacha EA, Cao QL, Galantowicz ME, et al. Multicenter experience with perventricular device closure of muscular ventricular septal defects. Pediatr Cardiol. 2005;26:169–175.
329 Galantowicz M, Cheatham JP. Lessons learned from the development of a new hybrid strategy for the management of hypoplastic left heart syndrome. Pediatr Cardiol. 2005;26:190–199.
330 Hjortdal VE, Redington AN, de Leval MR, et al. Hybrid approaches to complex congenital cardiac surgery. Eur J Cardiothorac Surg. 2002;22:885–890.
331 Tworetzky W, Marshall AC. Fetal interventions for cardiac defects. Pediatr Clin North Am. 2004;51:1503–1513.
332 Andropoulos DB, Stayer SA, Russell IA, eds. Anesthesia for congenital heart disease. Malden, Mass.: Blackwell Publishing, 2005.
333 Cordina RL, Celermajer DS. Chronic cyanosis and vascular function: implications for patients with cyanotic congenital heart disease. Cardiol Young. 2010;20:242–253.
334 Jaquiss RD, Tweddell JS. The neonate with congenital heart disease: what the cardiac surgeon needs to know from the neonatologist and the cardiologist. Clin Perinatol. 2005;32:947–961.
335 Greeley WJ, Kern FH, Schulman SR. Congenital heart disease. In: Estafanous FG, Barash PG, Reves JG, eds. Cardiac anesthesia: principles and clinical practice. 2nd ed. Philadelphia: Lippincott Williams & Wilkins; 2001:585–615.
336 Baum VC, Perloff JK. Anesthetic implications of adults with congenital heart disease. Anesth Analg. 1993;76:1342–1358.
337 Baum VC, Barton DM, Gutgesell HP. Influence of congenital heart disease on mortality after noncardiac surgery in hospitalized children. Pediatrics. 2000;105:332–335.
338 Warner MA, Lunn RJ, O’Leary PW, et al. Outcomes of noncardiac surgical procedures in children and adults with congenital heart disease. Mayo Perioperative Outcomes Group. MayoClin Proc. 1998;73:728–734.
339 Ramamoorthy C, Haberkern CM, Bhananker SM, et al. Anesthesia-related cardiac arrest in children with heart disease: data from the Pediatric Perioperative Cardiac Arrest (POCA) registry. Anesth Analg. 2010;110:1376–1382.