Esophageal Atresia and Tracheoesophageal Fistula Malformations
Esophageal atresia (EA) and tracheoesophageal fistula (TEF) anomalies present the pediatric surgeon with a unique and complex congenital disease, which tests both the diagnostic and technical skill of the surgeon. Most pediatric surgeons consider the surgical correction of these malformations to be the height of neonatal surgical care. In 1959, Dr Willis Potts wrote, ‘To anastomose the ends of an infant’s esophagus, the surgeon must be as delicate and precise as a skilled watchmaker. No other operation offers a greater opportunity for pure technical artistry.’1 While this statement still remains true, improvements in anesthetic and neonatal intensive care have made repair of these anomalies and their postoperative management much more routine so that a good outcome can be achieved in most cases. Technical advances, including the application of the minimally invasive approach, have also decreased the morbidity from these operations.
The first report of EA was by Durston in 1670,2 who found a blind upper pouch in one of a pair of thoracopagus conjoined twins, but the initial classic description was by Thomas Gibson in 1697.3 However, it was not until 1939 when a baby with EA/TEF survived following successful staged repairs described separately by Leven and Ladd.4,5 In 1940, Haight described the first survival following primary anastomosis.6 By the mid-1980s, most neonatal centers were performing primary repair and reporting successful outcomes in up to 90%.7–10
Embryology
The embryology of the foregut is still subject to controversy.11 What is known, however, is that during the fourth week of gestation the foregut starts to differentiate into a ventral respiratory part and a dorsal esophageal part. The laryngotracheal diverticulum then invaginates ventrally into the mesenchyme. The traditional theory postulates that the ventral respiratory system separates from the esophagus by the formation of lateral tracheoesophageal folds that fuse in the midline and create the tracheoesophageal septum. At 6 to 7 weeks of gestation, the separation between trachea and esophagus is complete. Incomplete fusion of the folds results in a defective tracheoesophageal septum and abnormal connection between the trachea and esophagus.
This theory of longitudinal tracheoesophageal folds merging to form a septum has been challenged.12,13 In chick embryo studies, these folds could not be demonstrated. Instead, cranial and caudal folds were found in the region of tracheoesophageal separation. According to this theory, EA/TEF would then be due to an imbalance in the growth of these folds. Furthermore, rat studies suggest that EA/TEF results from disturbances in either epithelial proliferation or apoptosis.14
More recent studies show that ectopic expression of sonic hedgehog occurs in the tissues between the notochord and the gut. Knockout mice models have helped elucidate the functions of different genes in the development of the foregut aberrations such as EA/TEF.15 Also, the relationship between BMP4 (bone morphogenic protein) and Nog, the gene encoding noggin (which is a BMP antagonist), may also have an impact on the development of TEF.16–18
Epidemiology
The birth incidence of EA/TEF varies between 1 in 2500 to 3000 live births.19–21 There is a slight male preponderance of 1.26 : 1. There is no evidence for a link between EA/TEF and maternal age when chromosomal cases are excluded.22 The risk for a second child with EA/TEF among parents of one affected child is 0.5–2%, increasing to 20% when more than one child is affected. The empirical risk of an affected child born to an affected person is 3–4%.23 The relative risk for EA/TEF in twins is 2.56 when compared with singletons.24 The concordance rate in twins is low, but the risk among twins of the same gender is high.25
Environmental factors that have been implicated include the use of methimazole in early pregnancy, prolonged use of contraceptive pills, progesterone and estrogen exposure, maternal diabetes, and thalidomide exposure.26–30 EA is occasionally seen in the fetal alcohol syndrome and in maternal phenylketonuria.31,32
Chromosomal anomalies are found in 6–10% of the patients.33–35 The total number of trisomy 18 cases exceeds the total number of trisomy 21 cases. As the incidence of trisomy 18 is higher, it would seem to indicate that trisomy 18 is a greater risk for EA development. Three separate genes have been associated with EA/TEF: MYCN haploinsufficiency in Feingold syndrome, CHD7 in CHARGE syndrome, and SOX2 in the anophthalmia–esophageal–genital (AEG) syndrome.34–37
EA may occasionally be part of the Opitz G/BB syndrome, Fanconi anemia, oculo-auriculo-vertebral syndrome, Bartsocas–Papas syndrome, or Frijns syndrome.38
Associated Anomalies
The factor or factors responsible for the early disturbance in organogenesis causing EA may affect other organs or systems that are developing at the same time. EA can be divided clinically into isolated EA and syndromic EA, occurring at roughly the same rate.38
The most frequent associated malformations encountered in syndromic EA are:
Vertebral anomalies are confined mainly to the thoracic region. An earlier claim that the presence of 13 pairs of ribs is a good indicator of long-gap EA has not been substantiated.39
Nonrandom associations have been documented as well. Two of these are the VACTERL association (vertebral, anorectal, cardiac, tracheo-esophageal, renal, and limb abnormalities) and the CHARGE association (coloboma, heart defects, atresia of the choanae, developmental retardation, genital hypoplasia, and ear deformities). In 1973, VACTERL was originally described as VATER, an acronym made up of vertebral defects, anal atresia, tracheo-esophageal fistula with EA, and radial dysplasia.40 It was later extended with the C for cardiac anomalies and the L for limb anomalies. In a cohort of 463 patients with EA, 107 (23%) had at least two additional VACTERL defects.41 Seventeen of these patients had a chromosomal defect or a syndrome without a known genetic defect. Interestingly, as many as 70% of the remaining 90 patients had additional defects other than VACTERL anomalies.
Classification
EA and TEF present in many forms and various classification systems have been used to describe them. It is clear that EA should be thought of as a spectrum of anomalies (Fig. 27-1).42 The original classification system was devised by Vogt in 1929.43 Ladd put forth his own classification in 19455 and Gross revised this in 1953.44 These classifications tend to be confusing, as the same subclasses are named differently. For clarity, it seems much better to give descriptive names to the major subtypes.
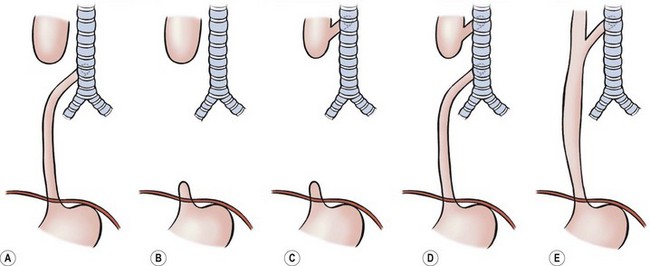
FIGURE 27-1 Classification of EA and/or TEF: (A) esophageal atresia with distal tracheoesophageal fistula: Vogt IIIb, Ladd III, Gross C; (B) esophageal atresia without fistula: Vogt II, Ladd I, Gross A; (C) esophageal atresia with proximal fistula: Vogt IIIa, Ladd II, Gross B; (D) esophageal atresia with proximal and distal fistulas: Vogt IIIc, Ladd V, Gross D; (E) tracheoesophageal fistula (H-type) without atresia: Vogt IV, Gross E.
Esophageal Atresia with Distal Fistula (Gross Type C)
This is the most common subtype, accounting for about 85% of EA anomalies.45 The very dilated proximal esophagus has a thickened wall and descends into the superior mediastinum usually to the third or fourth thoracic vertebrae. The distal esophagus is slender and has a thin wall. It enters the trachea posteriorly either at the level of the carina or 1–2 cm higher. The distance between the esophageal ends varies from very small to quite wide. Very rarely, the distal fistula may be occluded, leading to the misdiagnosis of EA without distal fistula.46
Pure Esophageal Atresia without TEF (Gross Type A)
Pure EA has an incidence of about 7%. The proximal and distal esophagus end blindly in the posterior mediastinum. The proximal end is dilated and has a thickened wall as in the more common EA/TEF. If there is no concomitant proximal fistula, the upper esophagus ends at the level of the azygos vein. The distal esophagus is short and often suspended by a fibrotic band. The distance between the two segments is considerable, usually precluding immediate anastomosis.
H-type Fistula without Esophageal Atresia (Gross Type E)
H-type TEF without atresia is usually discussed together with EA because it may be part of the VACTERL association. It occurs with an incidence of about 4%. The fistula starts from the membranous trachea and runs caudad to enter the esophagus. Normally it is short, although the diameter may be variable. The fistula is usually situated at the thoracic aperture or higher in the neck.47
Esophageal Atresia with Proximal Fistula (Gross Type B)
The association of a proximal fistula in a patient with pure EA is generally thought to be about 2%, but may be higher than is generally appreciated. In a recent series of 13 children without distal fistula, a proximal fistula was found in seven.48 An upper esophageal fistula is usually not found at the end of the pouch. This fistula is similar to the H-type starting proximally on the trachea and ending distally in the dilated proximal esophagus. Usually, there is only one proximal fistula, but two or three have been described.49 The fistula is usually located at the thoracic aperture or higher in the neck. Although limited in length, its diameter may vary from tiny to large. If not diagnosed preoperatively, it may be suspected during operative repair when bubbles are seen when opening the proximal esophagus.
Esophageal Atresia with Proximal and Distal Fistulas (Gross Type D)
The incidence of EA with proximal and distal fistulas is thought to be less than 1%. EA with one distal fistula and two proximal fistulas has also been described.49 Also reported is a near-complete membranous obstruction of the esophagus in conjunction with a single TEF at the level of the membrane, communicating with both parts of the esophagus.50
Diagnosis
Antenatal Diagnosis
The prenatal diagnosis of EA/TEF relies, in principle, on two nonspecific signs: polyhydramnios and an absent or small stomach bubble. Polyhydramnios is associated with a wide range of fetal abnormalities and is nonspecific. Similarly, the ultrasonographic (US) absence of a stomach bubble may point to a variety of fetal anomalies. The combination of a small stomach together with a dilated cervical esophagus (the pouch sign) has been confirmed to be diagnostic for pure EA in a number of patients.51–53 Nevertheless, it is encountered in only a few patients. Current ultrasound technology does not allow for the certain diagnosis of EA/TEF. Therefore, definitive counseling of the parents should be guarded.54 The application of 3D power Doppler imaging seems promising, both antenatally and postnatally. Aortic arch anomalies, for example, have been diagnosed using this modality.55,56
Magnetic resonance imaging (MRI) has been used to identify other fetal thoracic lesions, and may be beneficial in patients deemed to be at risk on prenatal ultrasound. Sensitivity in various studies is between 60–100% and the diagnosis is made by the lack of visualization of the thoracic esophagus.57–59
Postnatal Diagnosis
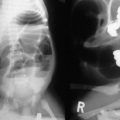
As EA prevents the passage of saliva down the esophagus, saliva accumulates in the proximal esophagus and mouth, and feeding should be withheld until esophageal continuity is confirmed. This is best done with a stiff 10 French catheter inserted either through the nose or mouth. A chest film is then obtained with downward pressure on the tube. With EA, the tip of the tube is found to be slightly curled in the blind upper pouch around T2–T4 (Fig. 27-2). This technique not only identifies the atresia but gives some clue to the length of the upper pouch. Often, the dilated upper esophageal pouch is visualized by air within it. Air in the stomach signifies the presence of a distal TEF. If the tip of the catheter passes beyond the level of the carina, then the diagnosis of EA should be questioned. Esophageal stenosis, tracheal rings, and iatrogenic perforation of the esophagus can be confused with EA.60,61 If there is any question about the diagnosis, a small amount of contrast can be dripped into the upper pouch, but this needs to be done with fluoroscopy and under direction of the surgeon to assure that the contrast is not aspirated.
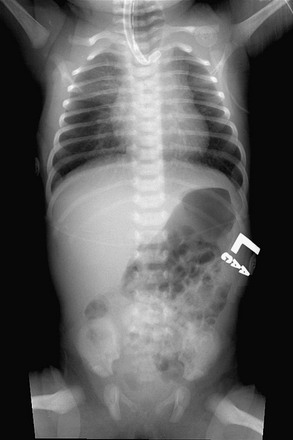
FIGURE 27-2 This plain radiograph depicts the classic features seen in an infant with EA and TEF. A nasoesophageal tube is seen in the upper pouch and has kinked a little at the end of the pouch. There is air in the stomach and bowel, which signifies the presence of a distal TEF.
Radiographs may reveal associated anomalies such as vertebral and rib anomalies, or other problems such as duodenal atresia (Fig. 27-3). The absence of air in the stomach points to EA without distal fistula (see Fig. 27-3A). Mediastinal ultrasound has been suggested as a helpful adjunct in the diagnosis of pure EA.62
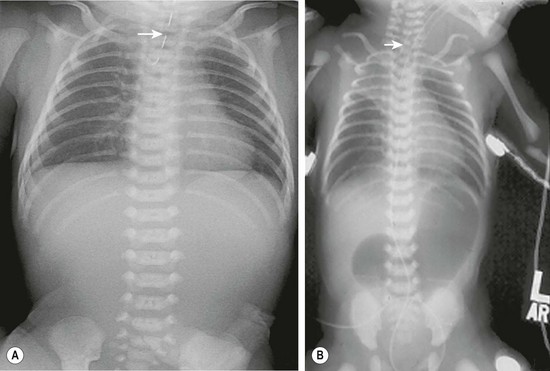
FIGURE 27-3 These two radiographs depict less common presentations of EA. (A) Isolated EA. The nasoesophageal tube (arrow) is seen in the proximal pouch. There is no air in the gastrointestinal tract. (B) This patient has EA with distal TEF and duodenal atresia and has been endotracheally intubated. A nasoesophageal catheter (arrow) sits in the upper esophageal pouch. An endotracheal tube is also seen. The stomach and bulbous duodenum are distended with air but no air is seen distal to the duodenum.
The length of the esophageal gap is usually not known preoperatively. Absence of air in the stomach has been linked with a long gap, but has also been described in association with a distal fistula occluded with mucus.46 Even in true long-gap EA (atresia without distal fistula), the gap length varies. In newborns with isolated EA, the first procedure is generally a gastrostomy, which allows for enteral feeding and also allows for assessment of the length and location of the lower pouch. It can be identified radiologically, either with metal bougies, a small gastroscope, or with a contrast injected through the gastrosomy.63 This can be done at the time of the initial gastrostomy or more routinely seven to ten days later. If bougies or a telescope are introduced into the distal esophagus, the amount of pressure on these instruments will affect the measurement of the gap between the two esophageal segments and may under- or over-estimate the gap length. In one report by an experienced surgeon, operative management was linked to the measured gap length: less than two vertebrae, then primary anastomosis; two to six vertebrae, then delayed primary anastomosis; more than six vertebrae, then esophageal replacement.64 In the era of thoracoscopy, an initial thoracic exploration can be considered if the upper pouch appears fairly long. If the lower pouch is identified and seen to be of adequate length, a primary repair can be attempted. If not, then a gastrostomy can be placed and delayed repair planned.65
In EA/TEF, a longer gap between the two esophageal ends should be expected when the distal fistula is found at the carina.66 Combined with a short upper pouch, this can mean a long gap exists between the two esophageal segments and may not be amenable to an initial primary repair. Unfortunately this may not fully be appreciated until the time of exploration. However since it is usually necessary to ligate the fistula in the early postnatal period, the gap length can be assessed at that time. A thoracoscopic approach in this scenario allows for minimal morbidity if the decision is to ligate the fistula only without reconstruction. Bronchoscopy can also be performed prior to exploration, not only to assess the site of the distal fistula, but to also look for an upper pouch fistula. Echocardiography should be performed prior to operation as it may reveal cardiac and/or aortic arch anomalies. A right descending aorta, which occurs in about 2.5% of the cases, may make a left-sided thoracic approach preferable.67 Renal ultrasound and spine radiographs should be obtained as well. Because EA may be part of a syndrome, consultation by a geneticist is recommended at some point.
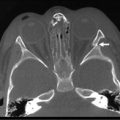
There is little doubt that better preoperative imaging of the neck and chest allows for better preoperative planning. At present, the two best imaging modalities are CT and MRI. Although MRI would certainly be preferable for its absence of radiation exposure, it requires general anesthesia. On the other hand, MRI is better for diagnosing cardiac and aortic arch anomalies.68 CT has also been performed in children with EA/TEF (Fig. 27-4).69–72
Management
Preoperative
Once the diagnosis of EA has been established, the baby, if not delivered in a maternal/fetal/neonatal center, should be transferred to a pediatric surgical center. A 10 French Replogle tube is placed in the upper esophagus and set to continuous suction.73 The child is positioned head-up and on his or her side. Intravenous access is important, and the vital signs are monitored.
If the child is in respiratory distress, endotracheal intubation and ventilation may be needed. Forceful ventilation will over distend the stomach, possibly causing diaphragmatic splinting and even gastric rupture.74,75 Gentle low-pressure ventilation is therefore essential. In these cases, some feel high-frequency ventilation is advantageous. However, on occasion, emergency ligation of the fistula may be needed as a life-saving maneuver.64,74–76 After ligation of the fistula, a delayed primary repair can be performed when the infant is more stable. Some recommend not postponing longer than seven to 14 days because recanalization of the fistula can occur.64,77,78 In these patients, we have performed thoracoscopic fistula ligation followed by repair two to six weeks later. We have not seen recanalization in our experience. If the infant is extremely unstable, then an emergency gastrostomy to decompress the stomach may be the best option. However this can result in a significant loss of tidal volume and may create respiratory issues as well.
Operative Repair
Esophageal Atresia with Distal Fistula
The operation is performed with the patient under general anesthesia and with adequate venous access. An arterial line is occasionally beneficial depending on the baby’s clinical status. Generally, a pulse oximeter and end tidal CO2 monitor are adequate. The operation can be performed through a thoracotomy or using a thoracoscopic approach. The side of entrance into the chest is opposite the turn of the aortic arch: right for a left descending aorta, left for a right descending aorta. If a right-sided aortic arch is not detected until the operation has begun, change to the left side is appropriate if the thoracoscopic approach was initially chosen. If a thoracotomy has been performed, an anastomosis from the right chest should be attempted, but there is a higher morbidity in this setting.67
Preoperative Bronchoscopy
The value of routine preoperative rigid bronchoscopy is much debated because the incidence of a simultaneous proximal and distal fistula is less than 1%.63 With the availability of small-diameter flexible fiberscopes, tracheobronchoscopy can now be performed after intubation through the endotracheal tube.79,80 Forceful ventilation must be avoided, not only to avoid lung damage,81 but also to prevent gastric distention and gastric perforation with insufflation through the distal fistula. Bronchoscopy may reveal abnormalities such as a second proximal fistula, a laryngotracheoesophageal cleft, tracheal stenosis, or a tracheal bronchus to the right upper lobe.82–84 The entrance of the distal fistula is usually well seen (Fig. 27-5). Its distance to the carina provides a clue as to the gap between the esophageal segments: the closer the fistula is to the carina, the longer the distance. An indication of the severity of tracheomalacia is only possible when the child is breathing spontaneously. The problem with bronchoscopy is that it may prolong the procedure and the infant can decompensate prior to ligation of the fistula.85–88 The surgeon and the anesthesiologist should discuss the advantages/disadvantages in each individual case. Following intubation, it may be helpful to position the endotracheal tube distal to the fistula, assuming the fistula is not at the carina.
Repair via Thoracotomy
The patient’s arm is positioned over the head (Fig. 27-6). Suction is removed from the Replogle tube, but the tube is left in place so that it can be advanced during the operation to aid in identifying the proximal pouch.
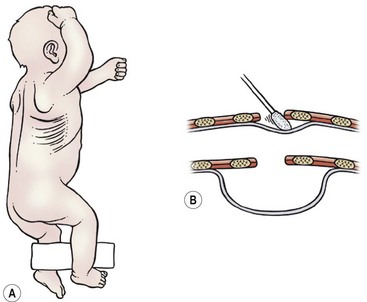
FIGURE 27-6 (A) The infant is positioned for a right thoracotomy. The ipsilateral arm is positioned over the head of the patient. A 4–5 cm incision is made 1 cm below the tip of the scapula. (B) This schematic depicts a peanut or sterile cotton swab being used to gently push the pleura away from the chest wall.
A slightly curved 4–5 cm long incision is made 1 cm below the inferior tip of the scapula. With the use of a muscle-sparing approach, the auscultatory triangle is opened and the muscles are retracted (i.e., the latissimus dorsi posteriorly and the serratus anterior anteriorly).89,90 If the serratus muscle needs to be transected, this should be done as low as possible to preserve the long thoracic nerve. The fourth or fifth intercostal space is then entered.
An extrapleural approach has been suggested to protect the pleural space in case of an anastomotic leak, but there is no evidence that it is better than a transpleural one.91,92 However an extrapleural approach does aid in exposure as it is easier to retract the lung when it is incased in the pleura. With the extrapleural approach, the pleura is gently pushed away from the endothoracic fascia, first in the middle of the incision so that an infant rib spreader can be inserted and opened (see Fig. 27-6B). With the rib spreader opened even farther, the pleura is carefully pushed away posteriorly until the posterior mediastinum is exposed.
The distal fistula may start from the trachea directly underneath the azygos vein, in which case the azygos vein is transected between 3-0 or 4-0 absorbable ligatures or simply cauterized and divided (Fig. 27-7). If the distal fistula originates more cephalad on the trachea, the vein can be left intact. Recently, a relationship between azygos vein transection and anastomotic leak has been suggested.93,94
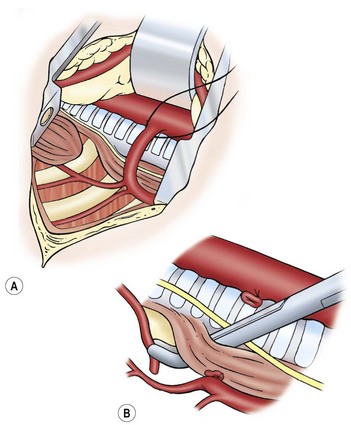
FIGURE 27-7 (A) Ligation of the azygos vein. (B) The distal fistula is being mobilized from its insertion in the trachea.
The distal esophagus is easily found because it distends with each inspiration and the vagus nerve is intimately attached. Once identified, the distal segment should be followed proximally to locate where the fistula enters the trachea. The fistula should be dissected and mobilized close to the trachea, which will spare as many vagal nerve branches as possible (see Fig. 27-7B). The fistula can be encircled with a vessel loop or suture to aid in exposure. There are several ways to ligate the fistula on the tracheal side. Ligation in continuity can result in a higher recanalization rate.95,96 We prefer to divide the fistula sharply after placing traction sutures at each end. A series of 5-0 PDS (Ethicon, Inc, Sommerville NJ) sutures are then used to close the tracheal side taking care not to compromise the tracheal lumen (Fig. 27-8). Another option is to apply a single 5mm clip across the fistula where it connects to the membranous trachea. We have found this technique to be simple and efficient, and results in a smaller pouch remnant on the posterior tracheal wall. However, there are anecdotal reports of clip migration using this technique. The tracheal closure can be checked by irrigating with warm water and applying a higher ventilation pressure to assess for an air leak.
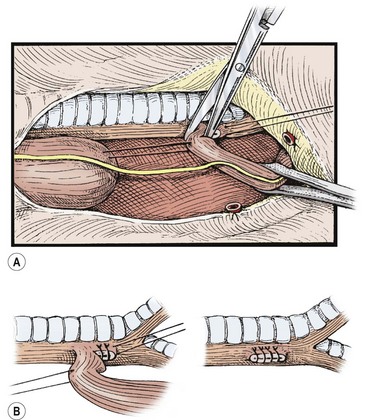
FIGURE 27-8 The distal esophagus is identified, looped, and carefully dissected up to its junction with the trachea, meticulously sparing the segmental vessels from the aorta. (A) Traction sutures may be placed for gentle handling of the segments. The fistula is divided close to the trachea without narrowing its lumen. (B) The tracheal end of the tracheoesophageal fistula is closed with continuous or interrupted sutures. Adjacent tissue, if available, is tacked over the closure. (From Holder TM, Manning PB. Esophageal atresia and tracheoesophageal fistula. Surg Rounds 1991;14:492–502.)
Attention is then turned to the proximal esophagus which can be identified by asking the anesthesiologist to push on the Replogle tube. A traction suture, taking a good bite of the muscular wall, is placed in the most distal part of the proximal pouch. Using the traction suture, the proximal pouch can be freed posteriorly and laterally by blunt dissection. Anteriorly, however, the pouch may be adherent to the membranous trachea. Usually, it can be dissected sharply, staying on the esophageal side to avoid entrance into the membranos trachea. Extensive dissection is not warranted, unless there is a long gap, because the dissection can damage the tracheal or esophageal walls and may interfere with innervation to the upper esophagus.97 Extensive dissection of the proximal pouch searching for a proximal fistula should not be performed routinely because the incidence of a proximal fistula in combination with a distal one is only about 1%. If a proximal fistula is present, it is usually of the H-type. If missed at birth and diagnosed after repair of the EA, it can usually be repaired through the neck at a later date. If the trachea is entered during the dissection of the proximal pouch, one should be wary that a proximal fistula may have been opened as well. There should not be a common wall between the trachea and esophagus.
After mobilization of the proximal pouch, the tip is now amputated so that the lumen and mucosa become visible. An end-to-end anastomosis is performed with 5-0 absorbable sutures starting in the middle of the back wall of each esophageal segment (Fig. 27-9). It is important to include both the mucosa and the muscular wall with each suture. The sutures in the back wall of the anastomosis are tied intralumenally. Then, the front part of the anastomosis is performed with sutures tied on the outside. Before finishing the anastomosis, an 8 French or 10 French tube is passed into the stomach. This protects from inadvertent closure of the lumen and for gastric decompression. Others feel the placement of an intralumenal tube or stent causes an increased risk of stricture or leak, and choose not to use it.
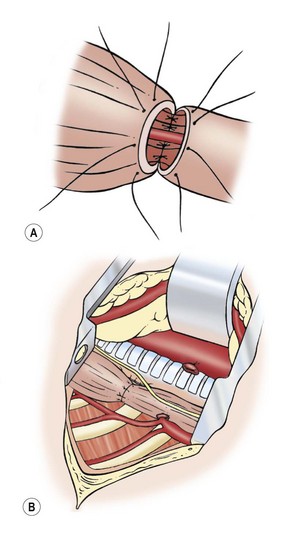
FIGURE 27-9 Esophageal anastomosis. (A) The back wall of the anastomosis has been sutured from the inside, and a small nasogastric tube has been passed through the anastomosis. The front part of the anastomosis is being sutured with knots on the outside. (B) Completed anastomosis.
The use of a chest drain is also optional.98,99 We use one until a contrast study is obtained on day 4 or 5. The chest incision is then closed in layers. The ribs should be approximated with one 3-0 absorbable suture. This suture should be tied gently so that the intercostal space is not obliterated. If a muscle-sparing approach was used, the muscles are allowed to fall back into their normal position and the skin is closed with a 5-0 absorbable subcuticular suture.
Thoracoscopic Repair
The first successful repair was reported in 2000 and the first series reported in 2002.100,101 Since then, there have been several retrospective reports describing experience with the thoracoscopic approach.102–105 During this early experience, attempts were made to obtain single lung ventilation by intubating the left mainstem bronchus. However, this proved time consuming and often unsuccessful so now the endotracheal tube is usually left in the trachea just above the carina and right lung collapse is achieved with CO2 insufflation alone.106 Others have described using the oscillating ventilator to effect lung collapse and negate the adverse effects of prolonged hypercarbia.107
Positioning.
Once the endotracheal tube is secure, the patient is placed in a modified prone position with the right side elevated approximately 30° (Fig. 27-10). The patient is placed near the edge of the table so that the handles of the instruments do not collide with the table. (If there is a right-sided arch, then a left-sided approach is used.) This positioning gives the surgeon access to the area between the anterior and posterior axillary lines for port placement while allowing gravity to retract the lung away from the posterior mediastinum. This arrangement allows excellent exposure of the fistula and esophageal segments without the need for an extra instrument or lung retractor. The assistant should not be placed on the opposite side of the table as this will place him/her at a complete paradox with the telescope. The scrub nurse can be on either side of the baby depending on the room layout. Because of the fine manipulation necessary, the surgeon and the assistant should position themselves so that they are in the most ergonomic and comfortable position.
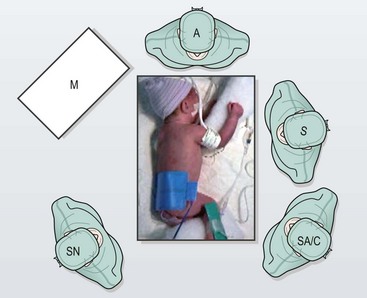
FIGURE 27-10 This infant is positioned more prone than lateral for right thoracoscopic repair. The surgeon (S) stands on the left side of the operating table when the aortic arch turns to the left. The surgeon, operative field, and screen are in-line. The assistant and camera holder (SA/C) is situated to the left of the surgeon when the surgeon is right-handed. The scrub nurse (SN) stands to the right of the operating table. M, monitor; A, anesthesiologist. (From Holcomb GW, Rothenberg SS, Georgeson KE. Atlas of Pediatric Laparoscopy and Thoracoscopy. Elsevier; 2009.)
Port placement is extremely important because of the small chest cavity and the intricate nature of the dissection and reconstruction. Usually three ports are satisfactory, but a fourth one can be used if needed (Fig. 27-11).
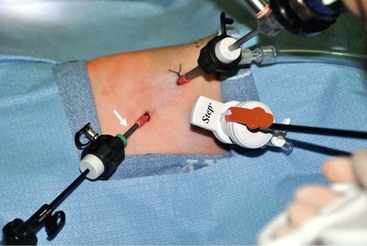
FIGURE 27-11 Optimal port positions for a thoracoscopic repair are seen. The ports are inserted in triangulation to allow the working instruments to meet at 90°. The camera port is inserted just below and posterior to the tip of the scapula. It is also the site for insufflation. One working port (Step, in photograph) is inserted in the midaxillary line in the axilla. The second working port (arrow) is introduced in the posterior axillary line. (From Holcomb GW, Rothenberg SS, Georgeson KE. Atlas of Pediatric Laparoscopy and Thoracoscopy. Elsevier; 2009.)
The initial port (3 mm or 4 mm) is placed in the fifth intercostal space behind the tip of the scapula. This is the telescope and camera port and allows excellent visualization of the posterior mediastinum. An angled telescope (30° or 45°) is essential. The two instrument ports are then introduced. The first is in the midaxillary line one or two interspaces above the telescope port in the axilla. This cephalad port is 5 mm for the clip applier and needle driver/suture. The lower port is 3 mm and is located in the posterior axillary line two interspaces below the telescope port. Ideally, these ports are positioned so that the instrument tips will approximate a 90° angle at the level of the fistula which will facilitate performing the anastomosis.
With the vein divided, the lower esophageal segment is identified and followed proximally to the fistula. Because of the magnification afforded by the thoracoscopic approach, it is easy to visualize exactly where the distal fistula enters the back wall of the trachea (Fig. 27-12). A 5 mm endoscopic clip can then be applied safely. Care should be taken to avoid the vagus nerve. A single clip is usually sufficient. The fistula is then divided with scissors. As the distal segment can retract, making it difficult to visualize, it may be preferable to wait until the upper pouch is mobilized before completely dividing the fistula. The tracheal opening can also be suture ligated, but this requires delicate suturing at a time when an air leak from the opened fistula may be causing respiratory compromise.
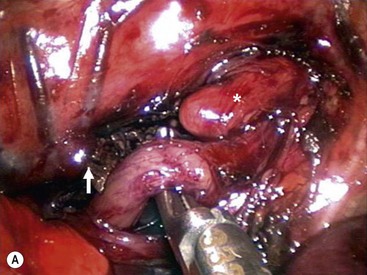
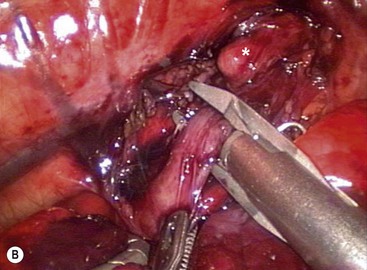
FIGURE 27-12 (A) After ligation and division of the azygos vein (arrow), the tracheoesophageal fistula has been mobilized and is being encircled with an angled dissecting instrument. The upper esophageal pouch (asterisk) was mobilized before ligation and division of the fistula. (B) The tracheoesophageal fistula is being ligated with a 5 mm endoscopic clip applier. Two clips are usually applied and the fistula is divided distal to the second clip. The upper esophageal pouch is marked with an asterisk. (From Holcomb GW III, Rothenberg SS, Georgeson KE. Atlas of Pediatric Laparoscopy and Thoracoscopy. Elsevier; 2009.)
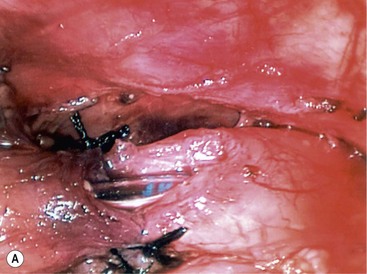
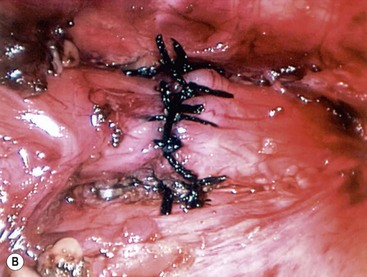
Once adequate mobilization is achieved, the distal tip of the pouch is resected, exposing the mucosa. With the two esophageal segments mobilized, the anastomosis is performed using a 4-0 or 5-0 suture on a small tapered needle. The sutures are placed one at a time in an interrupted fashion, back wall first, and then the front after the nasogastric tube is advanced, as described for the open procedure (Fig. 27-13). Once the anastomosis is complete, a chest drain is introduced through the lower port site and its tip is positioned near the anastomosis. The other ports are removed and the sites are closed with absorbable suture.
Esophageal Atresia without Distal Fistula
Initial Treatment
A gasless abdomen is the signature of EA without distal fistula. Very rarely, a blocked distal fistula is responsible for the lack of air of the abdomen.46 One should be aware that the incidence of a proximal fistula, in the absence of a distal fistula, is relatively high.
Esophageal Reconstruction
The timing of the esophageal reconstruction is debatable and often depends on the gap length. A period of a few weeks to three months has been used.64,108–112 During this waiting period, the proximal esophagus is emptied by continuous suction on a transnasally placed 10 French Replogle tube. If the child has persistent respiratory problems, a proximal fistula should again be considered. Cervical esophagostomy should be avoided as this will likely jeopardize the ability to perform a primary esophago-esophagostomy in the future. Home care during the waiting period has been advocated, but most of the reported patients have required a long time in the hospital before they were sent home.111,112
At the time of definite repair, several techniques to lengthen the native esophagus have been described, including esophageal myotomy or extensive mobilization of the proximal and distal esophagus (see Box 27-1). There is little doubt that all these maneuvers damage the esophagus and that the long-term results may be less than optimal. If a delayed primary anastomosis with or without lengthening is not feasible, an alternative procedure should be considered, such as a gastric pull-up or a jejunal, ileal, or colonic interposition. An alternative option is to attempt esophageal growth and lengthening by placing trans-thoracic traction sutures, but the results are variable.140,141
Thoracoscopy is ideal to evaluate the gap length to be bridged (Fig. 27-14). Moreover, the ends can be dissected and mobilized as well. An anastomosis can also be performed thoracoscopically. In one series, all long gaps were successfully closed.142 Also, a thoracoscopic Foker procedure can be performed.
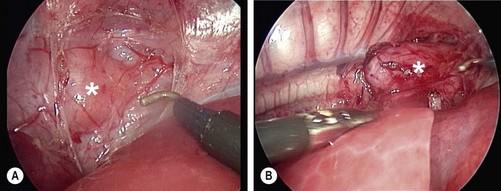
FIGURE 27-14 Thoracoscopic view of esophageal atresia without fistula in a 3-month old neonate. (A) The short distal esophageal segment (asterisk) is seen in the inferior portion of the mediastinum. (B) The proximal pouch (asterisk) reaches to the azygos vein, which has been coagulated and transected.
Postoperative Management
Mechanical ventilation with muscle relaxation for five days has been advocated after an anastomosis is performed under considerable tension.143–145 Evidence for the effectiveness of this approach is lacking.146 A chest drain is optional and feedings can be started early through the nasogastric tube if there is no evidence of a leak or concern about significant gastroesophageal reflux (GER).
Complications
Anastomotic Leaks
An overall leak rate of 3.5–17% has been reported by several authors.9,147,148 Major leaks which require active intervention occur much less frequently—3.5% in the first study and 4.5% in the more recent one, which confirms the statement that most leaks will close spontaneously. In the largest thoracoscopic report, the leak rate was 7.6%,103 but this was an multi-institutional study and early in each institution’s experience. Published reports often include all types of EA and do not always mention whether the leak was detected at routine esophagography or how it was identified.
Anastomotic Stricture
As with anastomotic leaks, no uniform definition is used for an anastomotic stricture. It has been defined as a narrowing of more than 50% of the lumen149 or as a narrowing detected on a contrast study, or at esophagoscopy in combination with symptoms.150 Reported incidences range from 17–60%.150–152 Four of 104 patients (3.8%) undergoing thoracoscopic repair developed a stricture, which was defined on the initial esophagogram.103 Routine dilatation is not indicated because many patients never require dilatation. Anastomotic tension, anastomotic leakage, and GER have been implicated as risk factors.153–155 Strictures usually respond well to dilation. Resection of the stricture is rarely required. Balloon dilatation seems superior, but there is no hard evidence.
Recurrent Tracheoesophageal Fistula
The reported incidence of a recurrent TEF varies between 3–15%. In one study, the 10% incidence in the period 1986 to 1995 had dropped to 5% in the period 1996 to 2005.7 In the large multi-institution study of thoracoscopic repairs, the incidence was 1.9%.103 The etiology of a recurrent fistula is almost certainly secondary to an anastomotic leak.
A recurrent fistula is suspected when the child starts to cough during feeding, has apneic or cyanotic episodes, or has repeat respiratory infections. The diagnosis can sometimes be made by an esophagram using a water-soluble contrast medium (Fig. 27-15) but bronchoscopy is usually needed to confirm the diagnosis.
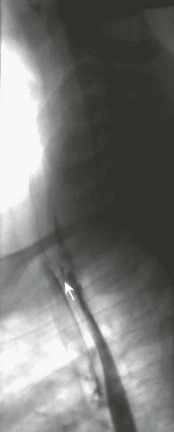
Treatment of a recurrent TEF can be difficult. Thus, attempts at prevention have been described. Interposition of a biosynthetic patch between the tracheal closure and esophageal anastomosis has been described for this purpose (Fig. 27-16).156 Native tissue such as pleura, pericardium, or intercostal muscle can also be used.
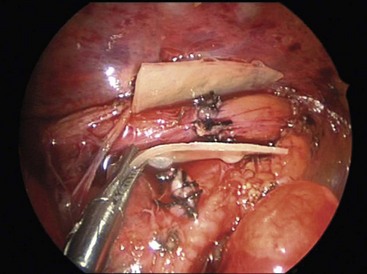
FIGURE 27-16 After thoracoscopic repair of esophageal atresia and distal fistula in this infant, Surgisis was interposed between the tracheal closure and esophageal anastomosis to help prevent a recurrent TEF. (From St. Peter SD, Calkins CM, Holcomb GW III. The use of biosynthetic mesh to separate the anastomoses during the thoracoscopic repair of esophageal atresia and tracheoesophageal fistula. J Laparoendosc Adv Surg Tech 2007;17:380–2. Reprinted with permission.)
Once developed, attempts at endoscopic management with cautery, fibrin glue, and small intestine submucosa have been described (Fig. 27-17).157–159 The results are mixed with success rates reported between 20–80%.
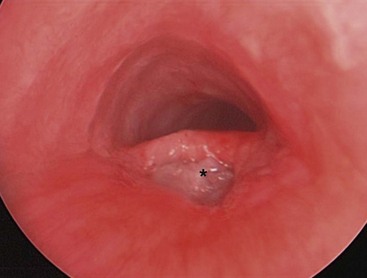
Tracheomalacia
Tracheomalacia is a generalized or localized weakness of the trachea that allows the anterior and posterior walls to come together during expiration or coughing.15 The area of collapse is usually in the region of the fistula.160 The cartilage of the rings is softened, and the length of the transverse muscle is increased. As a result, the airway collapses during expiration, which produces expiratory stridor varying from a hoarse barking cough to acute life-threatening episodes of cyanosis or apnea.161 In EA without fistula, tracheomalacia does not seem to occur very often.162
Treatment is required in severely symptomatic infants. The therapy of choice is aortopexy.163–168 The principle behind this operation is that if ascending aorta and arch are suspended against the posterior surface of the sternum, the anterior wall of the trachea, which is loosely attached to the aorta, is suspended as well. This anterior aortic suspension opens the tracheal lumen. Aortopexy is classically performed through a left anterolateral thoracotomy or median sternotomy, but can also be performed through a low cervical incision and partial sternal split,169 or thoracoscopically either from the left170,171 or from the right.172 It is best to check the effect of the suspension by simultaneous tracheoscopy.
Disordered Peristalsis/Gastroesophageal Reflux/Esophageal Cancer
Disturbed motility of the esophagus in EA patients has been recognized for a long time.173–176 Symptoms included dysphagia, episodes of foreign body impaction, heartburn, vomiting, and various respiratory disorders.177–179 The oral phase of swallowing is normal, but the pharyngeal and esophageal phases are abnormal in all patients, both on video-fluoroscopy180,181 and with manometry.182–187
Whether the cause of these disturbed motility problems is congenital or acquired has been a long-standing debate. Whatever the cause, it brings with it several problems, not the least of which is GER. While GER tends to improve during the first year of life in most infants, this is not the case in infants with EA. The incidence of significant reflux in patients with EA has been stated to be up to 50%, and about half of these will require antireflux surgery.103,188–190 A considerable number of patients with EA have complaints in adult life pointing toward GER as the etiology including Barrett’s esophagitis.189 Early normal pH values in the esophagus do not exclude significant reflux on follow-up.190 Antireflux medication, including gastric acid suppression, is successful in only about half of the cases.191 Antireflux operations in patients with repaired EA have a higher failure rate than in non-EA patients,192 but there seems to be no other alternative. While some surgeons have advocated partial wraps because of concerns over dysmotility,193 there is little evidence to support this practice. The question remains whether adults with repaired EA as an infant should be screened as there may be a higher incidence of squamous cell carcinoma.194
Vocal Cord Dysfunction
Another complication which is becoming more recognized is injury to the recurrent laryngeal nerves during EA repair. In a retrospective review of 150 patients undergoing repair of EA and associated anomalies, five patients (3%) were found to have vocal fold paralysis on subsequent evaluation.195 Bilateral paralysis was found in three patients and two patients had unilateral paralysis. In this study, the etiology of the paralysis was difficult to assess. However, the authors recommend preoperative laryngoscopy or bronchoscopy to identify infants with congenital vocal fold paralysis prior to operative repair. This is especially true for patients requiring revision surgery. An older review found a 12% incidence of vocal cord injury following EA repair.196
Respiratory Morbidity
Respiratory morbidity after EA repair is high.197,198 In a series of 334 patients aged 1 to 37, just under half were subsequently hospitalized with a respiratory illness.197 Two-thirds of the admissions were before 5 years of age. Also, a high proportion of the adult patients reported respiratory symptoms. These symptoms can be attributed to tracheomalacia and GER. Aortopexy is effective in preventing further life-threatening spells, but does not prevent the increased susceptibility to respiratory infections.199
Thoracotomy-Related Morbidity
Thoracotomy, especially in the newborn, can lead to significant morbidity, such as winged scapula, elevation or fixation of the shoulder, asymmetry of the chest wall, rib fusion, scoliosis, and breast and pectoral muscle maldevelopment.200–207 Moreover, chronic pain after thoracic surgery is a serious problem, at least in adults, and has been reported in more than 50% of patients.208,209 These negative consequences of a thoracotomy can be alleviated by using the thoracoscopic approach.
H-Type Fistula
The incidence of H-type TEF is about 4%.14,15 Isolated TEF is associated with the same anomalies as seen in EA, although at lower incidences.210 The fistula runs from the trachea downward to the esophagus, typically intramurally, and is short (see Fig. 27-1). In a series of 20 children, the fistula was at C5–C6 in two, C6–C7 in three, C7–T1 in eight, T1–T2 in three, and T2–T3 in one.47 Rarely, there is a second fistula.211
Respiratory symptoms, especially choking, often occur immediately after birth with feeding. Sometimes, there are unexplained cyanotic spells. Symptoms subside when the child is fed by a nasogastric tube. Often the abdomen is distended with air, and flatulence may be present. Older children can present with recurrent pneumonia. Symptoms can often be traced back to the neonatal period. On occasion, symptoms develop later in life.212,213
It can be difficult to diagnose an H-type fistula even if there is a high degree of suspicion. A water-soluble, low osmolar contrast study seems to be the best initial investigation and demonstrates the fistula in many cases (Fig. 27-18). Bronchoscopy (usually rigid) is another diagnostic modality, but even then, the fistula can be missed.214 CT esophagography is another option but has the disadvantage of radiation exposure.215
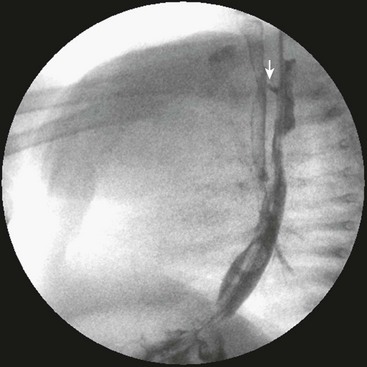
FIGURE 27-18 Imaging for an H-type TEF. A contrast swallow is performed using a low osmolar, water-soluble medium. The fistula (arrow) is clearly seen on the lateral chest film. The fistula is at the level of the thoracic inlet. Contrast material entered the trachea and bronchial tree.
A cervical approach can be used in most (80%) cases.216 With the use of a small feeding tube or guide wire placed through the fistula at tracheoscopy and pulled out of the esophagus, most lower H-type fistulas can be pulled up and approached through the neck as well.217,218 The classic approach is through a small low right cervical incision (Fig. 27-19). The sternal head of the sternocleidomastoid muscle may need transection. The esophagus is easily identified and separated from the trachea, taking care not to injure the recurrent laryngeal nerve. The left recurrent laryngeal nerve is vulnerable as well. The proximal esophagus, as well as the fistula, is encircled with separate vessel loops and so is the distal esophagus. Traction sutures are placed at the upper and lower ends of the fistula, which is transected close to the esophagus. The trachea is closed longitudinally and the esophagus transversely with interrupted absorbable sutures. To eliminate a recurrent fistula, the sternal head of the sternocleidomastoid muscle can be interposed between the cut ends of the fistula.
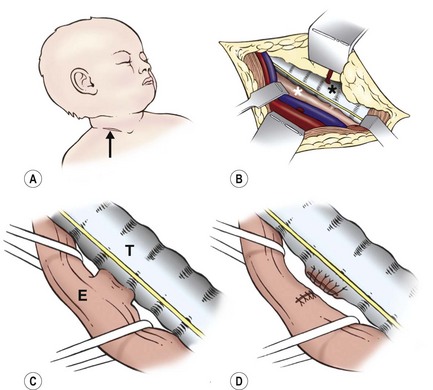
FIGURE 27-19 Cervical approach used to repair an H-fistula. (A) Low right transverse cervical incision (arrow). (B) The esophagus (white asterisk) is approached medial to the carotid artery and jugular vein. Care is taken not to injure the recurrent laryngeal nerve on either side. The trachea is marked with a black asterisk. (C) The esophagus (E) is encircled both above and below the fistula. The fistula will be transected close to the trachea (T). (D) The tracheal opening is closed vertically, and the esophageal defect is closed transversely.
It is evident that less trauma is inflicted when a thoracotomy can be avoided. The availability of thoracoscopy obviates the need for a thoracotomy, thus making a cervical approach for the extremely low H-fistulas less imperative. Several reports of thoracoscopically repaired H-fistulas have been described.219–221 Whether a fistula should be approached from the neck or the chest depends on the location of the fistula based on preoperative imaging, and the surgeon and patient preference.
Laryngeal and Laryngotracheoesophageal Cleft
Laryngeal and laryngotracheoesophageal clefts are rare congenital anomalies with an incidence of approximately 1 in 10,000 to 20,000 live births.222 The cleft consists of a midline communication of the larynx, trachea, and rarely bronchus with the pharynx and upper esophagus. Its embryology is poorly understood. Originally, it was thought to be the consequence of a failure of the tracheoesophageal septum to fuse in the caudocranial direction. However, as previously mentioned, this theory of tracheoesophageal separation by fusion of a tracheoesophageal septum has been challenged.11–18
Many congenital malformations can coexist, including congenital heart disease, gastrointestinal and genitourinary tract abnormalities, and midline defects.223 Familial occurrence has been described.224 Clefts may be part of the Opitz–Frias syndrome and the Pallister–Hall syndrome.225 TEFs have been found in 20–37% of these patients. Several classifications have been described (Fig. 27-20) and can be seen in Table 27-1.226–228

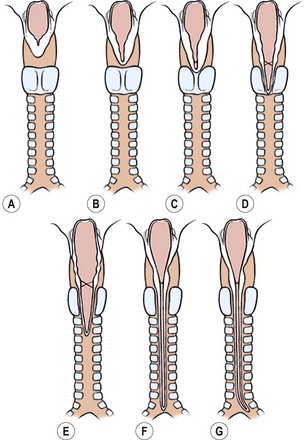
FIGURE 27-20 This schematic depicts the classification of laryngotracheoesophageal clefts described by Myer and colleagues.228 The laryngoesophageal clefts are seen in the top row, and the laryngotracheoesophageal clefts are seen on the bottom row. (A) Normal anatomy. (B) Interarytenoid cleft (LI). (C) Partial cricoid cleft (LII). (D) Complete cricoid cleft (LIII). (E) Cleft extends into trachea (LTEI). (F) Cleft extends into the carina (LTEII). (G) Cleft extends into the bronchus (LTEIII, proposed).
Contrast radiographs may be diagnostic, although it can be difficult to differentiate between overflow and aspiration. Tracheobronchoscopy is diagnostic (Fig. 27-21). The diagnosis can be missed, especially in less severe cases. A high degree of suspicion is often required to make the diagnosis. Often pressure applied to the scope while sitting on the arytenoids will open the cleft.
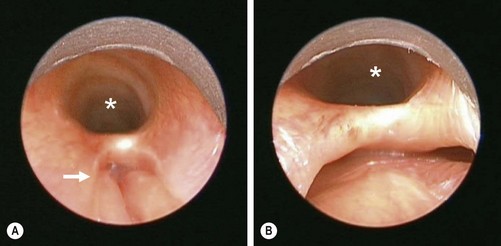
FIGURE 27-21 This newborn began to have respiratory distress shortly after birth. Clinically, it was felt that the baby might have a tracheoesophageal fistula without esophageal atresia as a small nasogastric tube was able to be passed into the stomach. The bronchoscopic examination is shown. In both figures, the trachea is identified with the asterisk. (A) The laryngotracheoesophageal cleft is visualized. The esophagus is marked with the arrow. (B) The distal end of the laryngotracheoesophageal cleft is easily seen with the trachea anteriorly (asterisk) and the esophagus posteriorly. According to the classification of laryngotracheoesophageal clefts described by Meyer, this would be an LTE1 (see Fig. 27-20E).
Using the Myer classification system,228 LI may need no treatment other than antireflux therapy. If still symptomatic, these patients may need an open or endoscopic repair. LII, LIII, and LTEI are best managed through anterior laryngotomy. LTEII and LTEIII appear best treated by a combined cervical and thoracic approach.229 A lateral cervical approach provides suboptimal access.230 An anterior approach under cardiopulmonary bypass has also been used,231,232 and may be the best option in some cases.
This is a rare anomaly, and reported series are small. Tracheal instability, tracheomalacia, and GER with recurrent aspiration have been noted. The anastomotic leak rate is high, and hospitalization is often prolonged.
References
1. Potts, WJ. The Surgeon and the Child. W. B. Saunders Co; 1959.
2. Durston, W. A Narrative of a Monstrous Birth in Plymouth, Oct. 22, 1670; together with the Anatomical Observations, taken thereupon by William Durston Doctor in Physick, and communicated to Dr. Tim. Clerk. Philos Trans (London). 1670; 5:2096–2098.
3. Gibson, T. Anatomy of Humane bodies Epitomized, 5th ed. London: printed by TW for Awnsham and John Churchill, at the Black Swan in Pater-Noster-Row, and sold by Timothy Childe, at the White-Hart, the West end of St. Paul’s Church Yard; 1697.
4. Levin, NL. Congenital atresia of the esophagus with tracheo-esophageal fistula. J Thorac Cardiovasc Surg. 1941; 10:648–657.
5. Ladd, WE. The surgical treatment of esophageal atresia and tracheoesophageal fistulas. N Engl J Med. 1944; 230:625–637.
6. Haight, C, Towsley, HA. Congenital atresia of the esophagus with tracheoesophageal fistula: Extrapleural ligation of fistula and end-to-end anastomosis. Surg Gynecol Obstet. 1943; 76:672–688.
7. Lilja, HE, Wester, T. Outcome in neonates with esophageal atresia treated over the last 20 years. Pediatr Surg Int. 2008; 24:531–536.
8. Spitz, L, Kiely, EM, Morecroft, JA, et al. Oesophageal atresia: At-risk groups for the 1990s. J Pediatr Surg. 1994; 29:723–725.
9. Tonz, M, Kohli, S, Kaiser, G. Oesophageal atresia: What has changed in the last 3 decades? Pediatr Surg Int. 2004; 20:768–772.
10. Orford, J, Cass, DT, Glasson, MJ. Advances in the treatment of oesophageal atresia over three decades: The 1970s and the 1990s. Pediatr Surg Int. 2004; 20:402–407.
11. Felix, JF, Keijzer, R, van Dooren, MF, et al. Genetics and developmental biology of oesophageal atresia and tracheo-oesophageal fistula: Lessons from mice relevant for paediatric surgeons. Pediatr Surg Int. 2004; 20:731–736.
12. Zaw-Tun, HA. The tracheo-esophageal septum—fact or fantasy? Origin and development of the respiratory primordium and esophagus. Acta Anat (Basel). 1982; 114:1–21.
13. Kluth, D, Fiegel, H. The embryology of the foregut. Semin Pediatr Surg. 2003; 12:3–9.
14. Qi, BQ, Beasley, SW. Stages of normal tracheo-bronchial development in rat embryos: Resolution of a controversy. Dev Growth Differ. 2000; 42:145–153.
15. Felix, JF, Keijzer, R, van Dooren, MF, et al. Genetics and developmental biology of oesophageal atresia and tracheo-oesophageal fistula: Lessons from mice relevant for paediatric surgeons. Pediatr Surg Int. 2004; 20:731–736.
16. Que, J Choi, M, Zeil, JW, et al. Morphogenesis of the trachea and esophagus; current players and new roles for Noggin and BMPS. Differentiation. 2006; 74:422–437.
17. Que, J, Okinbo, T, Goldenring, JR, et al. Multiple dose-dependent roles for Sox2 in pathway and differentiation of the anterior foregut endoderm. Development. 2009; 134:2521–2531.
18. Jacobs, J, Ku, WY, Que, J. Genetic and cellular mechanisms regulating anterior foregut and esophageal development. Dev Biol. 2012; 1:54–64.
19. Depaepe, A, Dolk, H, Lechat, MF. The epidemiology of tracheo-oesophageal fistula and oesophageal atresia in Europe. EUROCAT Working Group. Arch Dis Child. 1993; 68:743–748.
20. Robert, E, Mutchinick, O, Mastroiacovo, P, et al. An international collaborative study of the epidemiology of esophageal atresia or stenosis. Reprod Toxicol. 1993; 7:405–421.
21. Harris, J, Kallen, B, Robert, E. Descriptive epidemiology of alimentary tract atresia. Teratology. 1995; 52:15–29.
22. Shaw-Smith, C. Oesophageal atresia, tracheo-oesophageal fistula, and the VACTERL association: Review of genetics and epidemiology. J Med Genet. 2006; 43:545–554.
23. Pletcher, BA, Friedes, JS, Breg, WR, et al. Familial occurrence of esophageal atresia with and without tracheoesophageal fistula: Report of two unusual kindreds. Am J Med Genet. 1991; 39:380–384.
24. Mastroiacovo, P, Castilla, EE, Arpino, C, et al. Congenital malformations in twins: An international study. Am J Med Genet. 1999; 83:117–124.
25. Robert, E, Mutchinick, O, Mastroiacovo, P, et al. An international collaborative study of the epidemiology of esophageal atresia or stenosis. Reprod Toxicol. 1993; 7:405–421.
26. Clementi, M, Di Gianantonio, E, Pelo, E, et al. Methimazole embryopathy: Delineation of the phenotype. Am J Med Genet. 1999; 83:43–46.
27. Ramirez, A, Espinosa de los Monteros, A, Parra, A, et al. Esophageal atresia and tracheoesophageal fistula in two infants born to hyperthyroid women receiving methimazole (Tapazol) during pregnancy. Am J Med Genet. 1992; 44:200–202.
28. Szendrey, T, Danyi, G, Czeizel, A. Etiological study on isolated esophageal atresia. Hum Genet. 1985; 70:51–58.
29. Nora, AH, Nora, JJ. A syndrome of multiple congenital anomalies associated with teratogenic exposure. Arch Environ Health. 1975; 30:17–21.
30. Chen, H, Goei, GS, Hertzler, JH, et al. Family studies in congenital esophageal atresia with and without tracheoesophageal fistula. In: Epstein CJ, Curry CJR, Packman S, eds. Risks, Communication, and Decision Making in Genetic Counseling. New York: Alan R. Liss for the National Foundation March-of-Dimes, 1979.
31. Martinez-Frias, ML, Rodriguez-Pinilla, E. Tracheoesophageal and anal atresia in prenatal children exposed to a high dose of alcohol. Am J Med Genet. 1991; 40:128.
32. Lipson, A, Beuhler, B, Bartley, J, et al. Maternal hyperphenylalaninemia fetal effects. J Pediatr. 1984; 104:216–220.
33. Depaepe, A, Dolk, H, Lechat, MF. The epidemiology of tracheo-oesophageal fistula and oesophageal atresia in Europe. EUROCAT Working Group. Arch Dis Child. 1993; 68:743–748.
34. Torfs, CP, Curry, CJ, Bateson, TF. Population-based study of tracheoesophageal fistula and esophageal atresia. Teratology. 1995; 52:220–232.
35. van Bokhoven, H, Celli, J, van Reeuwijk, J, et al. MYCN haploinsufficiency is associated with reduced brain size and intestinal atresias in Feingold syndrome. Nat Genet. 2005; 37:465–467.
36. Vissers, LE, van Ravenswaaij, CM, Admiraal, R, et al. Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat Genet. 2004; 36:955–957.
37. FitzPatrick DR, Magee A, Friedler Z, et al. Mutations in SOX2 cause Rogers syndrome (anophthalmia, tracheoesophageal fistula, and genitourinary anomalies). Presented before the American Society of Human Genetics annual meeting, 2004(session 54), p 190.
38. Genevieve, D, de Pontual, L, Amiel, J, et al. An overview of isolated and syndromic oesophageal atresia. Clin Genet. 2007; 71:392–399.
39. Kulkarni, B, Rao, RS, Oak, S, et al. 13 pairs of ribs—a predictor of long gap atresia in tracheoesophageal fistula. J Pediatr Surg. 1997; 32:1453–1454.
40. Quan, L, Smith, DW. The VATER association: Vertebral defects, Anal atresia, T-E fistula with esophageal atresia, Radial and Renal dysplasia: A spectrum of associated defects. J Pediatr. 1973; 82:104–107.
41. de Jong, EM, Felix, JF, Deurloo, JA, et al. Non–VACTERL-type anomalies are frequent in patients with esophageal atresia/tracheo-esophageal fistula and full or partial VACTERL association. Birth Defects Res A Clin Mol Teratol. 2008; 82:92–97.
42. Kluth, D. Atlas of esophageal atresia. J Pediatr Surg. 1976; 11:901–919.
43. Vogt, EC. Congenital esophageal atresia. Am J Roentgenol. 1929; 22:463–465.
44. Gross, RE. The Surgery of Infancy and Childhood. Philadelphia: WB Saunders; 1953.
45. Harmon, C, Coran, GC. Congenital anomalies of the esophagus. In: Grosfeld J, O’Neill JA, Coran AG, eds. Pediatric Surgery. 6th ed. Philadelphia: Mosby; 2006:1051–1081.
46. Goh, DW, Brereton, RJ, Spitz, L. Esophageal atresia with obstructed tracheoesophageal fistula and gasless abdomen. J Pediatr Surg. 1991; 26:160–162.
47. Laffan, EE, Daneman, A, Ein, SH, et al. Tracheoesophageal fistula without esophageal atresia: Are pull-back tube esophagograms needed for diagnosis? Pediatr Radiol. 2006; 36:1141–1147.
48. Bax, KN, Roskott, AM, van der Zee, DC. Esophageal atresia without distal tracheoesophageal fistula: High incidence of proximal fistula. J Pediatr Surg. 2008; 43:522–525.
49. Kane, T, Atri, P, Potoka, DA. Triple fistula: Management of a double tracheoesophageal fistula with a third H-type proximal fistula. J Pediatr Surg. 2007; 42:e1–e3.
50. Touloukian, RJ. Membranous esophageal obstruction simulating atresia with a double tracheoesophageal fistula in a neonate. J Thorac Cardiovasc Surg. 1973; 65:191–194.
51. Centini, G, Rosignoli, L, Kenanidis, A, et al. Prenatal diagnosis of esophageal atresia with the pouch sign. Ultrasound Obstet Gynecol. 2003; 21:494–497.
52. Has, R, Gunay, S. Upper neck pouch sign in prenatal diagnosis of esophageal atresia. Arch Gynecol Obstet. 2004; 270:56–58.
53. Kalache, KD, Chaoui, R, Mau, H, et al. The upper neck pouch sign: A prenatal sonographic marker for esophageal atresia. Ultrasound Obstet Gynecol. 1998; 11:138–140.
54. Houben, CH, Curry, JI. Current status of prenatal diagnosis, operative management and outcome of esophageal atresia/tracheoesophageal fistula. Prenat Diagn. 2008; 28:667–675.
55. Chaoui, R, Schneider, MB, Kalache, KD. Right aortic arch with vascular ring and aberrant left subclavian artery: Prenatal diagnosis assisted by three-dimensional power Doppler ultrasound. Ultrasound Obstet Gynecol. 2003; 22:661–663.
56. Sivaprakasam, MC, Vettukattil, JJ. 3-D echocardiographic imaging of double aortic arch. Eur J Echocardiogr. 2006; 7:476–477.
57. Levine, D, Barnewolt, CE, Mehta, TS, et al. Fetal thoracic abnormalities: MR imaging. Radiology. 2003; 228:379–388.
58. Matsuoka, S, Takeuchi, K, Yamanaka, Y, et al. Comparison of magnetic resonance imaging and ultrasonography in the prenatal diagnosis of congenital thoracic abnormalities. Fetal Diagn Ther. 2003; 18:447–453.
59. Langer, JC, Hussain, H, Khan, A, et al. Prenatal diagnosis of esophageal atresia using sonography and magnetic resonance imaging. J Pediatr Surg. 2001; 36:804–807.
60. Eklof, O, Lohr, G, Okmian, L. Submucosal perforation of the esophagus in the neonate. Acta Radiol Diagn (Stockh). 1969; 8:187–192.
61. Sapin, E, Gumpert, L, Bonnard, A, et al. Iatrogenic pharyngoesophageal perforation in premature infants. Eur J Pediatr Surg. 2000; 10:83–87.
62. Gassner, I, Geley, TE. Sonographic evaluation of oesophageal atresia and tracheoesophageal fistula. Pediatr Radiol. 2005; 35:159–164.
63. Gross, ER, Reichstein, A, Gander, JW, et al. The role of fiberoptic endoscopy in the evaluation and management of long gap esophageal atresia. Pediatr Surg Int. 2010; 45:1223–1227.
64. Spitz, L. Esophageal atresia: Lessons I have learned in a 40-year experience. J Pediatr Surg. 2006; 41:1635–1640.
65. Rothenberg, SS. Thoracoscopic repair of tracheoesophageal fistula. Semin Pediatr Surg. 2005; 14:2–7.
66. Gupta, DK, Arora, M, Srinivas, M. Azygos vein anomaly: The best predictor of a long gap in esophageal atresia and tracheoesophageal fistula. Pediatr Surg Int. 2001; 17:101–103.
67. Babu, R, Pierro, A, Spitz, L, et al. The management of oesophageal atresia in neonates with right-sided aortic arch. J Pediatr Surg. 2000; 35:56–58.
68. Cantinotti, M, Hedge, S, Bell, A, et al. Diagnostic role of magnetic resonance imaging in identifying aortic arch anomalies. Congent Heart Dis. 2008; 3:117–123.
69. Tam, PK, Chan, FL, Saing, H. Diagnosis and evaluation of esophageal atresia by direct sagittal CT. Pediatr Radiol. 1987; 17:68–70.
70. Ratan, SK, Varshney, A, Mullick, S, et al. Evaluation of neonates with esophageal atresia using chest CT scan. Pediatr Surg Int. 2004; 20:757–761.
71. Ou, P, Seror, E, Layouss, W, et al. Definitive diagnosis and surgical planning of H-type tracheoesophageal fistula in a critically ill neonate: First experience using air distension of the esophagus during high-resolution computed tomography acquisition. J Thorac Cardiovasc Surg. 2007; 133:1116–1117.
72. Islam, S, Cavanaugh, E, Honeke, R, et al. Diagnosis of a proximal tracheoesophageal fistula using three-dimensional CT scan: A case report. J Pediatr Surg. 2004; 39:100–102.
73. Replogle, RL. Esophageal atresia: Plastic sump catheter for drainage of the proximal pouch. Surgery. 1963; 54:296–297.
74. Holcomb, GW, 3rd. Survival after gastrointestinal perforation from esophageal atresia and tracheoesophageal fistula. J Pediatr Surg. 1993; 28:1532–1535.
75. Maoate, K, Myers, NA, Beasley, SW. Gastric perforation in infants with esophageal atresia and distal tracheo-oesophageal fistula. Pediatr Surg Int. 1999; 15:24–27.
76. Beasley, SW, Myers, NA, Auldist, AW. Management of the premature infant with esophageal atresia and hyaline membrane disease. J Pediatr Surg. 1992; 27:23–25.
77. Holmes, S, Keily, EM, Spitz, L. Tracheo-oesophageal atresia and the respiratory distress syndrome. Pediatr Surg Int. 1987; 2:16–18.
78. Malone, PS, Kiely, EM, Brain, AJ, et al. Tracheo-oesophageal fistula and pre-operative mechanical ventilation. Aust N Z J Surg. 1990; 60:525–527.
79. Atzori, P, Iacobelli, BD, Bottero, S, et al. Preoperative tracheobronchoscopy in newborns with esophageal atresia: Does it matter? J Pediatr Surg. 2006; 41:1054–1057.
80. Usui, N, Kamata, S, Ishikawa, S, et al. Anomalies of the tracheobronchial tree in patients with esophageal atresia. J Pediatr Surg. 1996; 31:258–262.
81. Iannoli, ED, Litman, RS. Tension pneumothorax during flexible fiberoptic bronchoscopy in a newborn. Anesth Analg. 2002; 94:512–513.
82. Holzki, J. Bronchoscopic findings and treatment in congenital tracheoesophageal fistula. Paediatr Anaesth. 1992; 2:297–303.
83. Johnson, AM, Rodgers, BM, Alford, B, et al. Esophageal atresia with double fistula: The missed anomaly. Ann Thorac Surg. 1984; 38:195–200.
84. Katsura, S, Shono, T, Yamanouchi, T, et al. Esophageal atresia with double tracheoesophageal fistula—a case report and review of the literature. Eur J Pediatr Surg. 2005; 15:354–357.
85. Bikhazi, G, Davis, PJ. Anesthesia for neonates and premature infants. In: Motoyama E, ed. Anesthesia for Infants and Children. St. Louis: Mosby; 1990:450–453.
86. Salem, MR, Wong, AY, Lin, YH, et al. Prevention of gastric distention during anesthesia for newborns with tracheoesophageal fistulas. Anesthesiology. 1973; 38:82–83.
87. Schwartz, N, Eisenkraft, JB. Positioning the endotracheal tube in an infant with tracheoesophageal fistula. Anesthesiology. 1988; 69:289–290.
88. Alabbad, SI, Shaw, K, Puligandla, PS, et al. The pitfalls of endotracheal intubation beyond the fistula in babies with type C esophageal atresia. Semin Pediatr Surg. 2009; 18(2):116–118.
89. Soucy, P, Bass, J, Evans, M. The muscle-sparing thoracotomy in infants and children. J Pediatr Surg. 1990; 27:1257–1258.
90. Rothenberg, SS, Pokorny, WJ. Experience with a total muscle sparing approach for thoracotomies in neonates, infants and children. J Ped Surg. 1992; 27:1157–1160.
91. McKinnon, LJ, Kosloske, AM. Prediction and prevention of anastomotic complications of esophageal atresia and tracheoesophageal fistula. J Pediatr Surg. 1990; 25:778–781.
92. Bishop, PJ, Klein, MD, Philippart, AI, et al. Transpleural repair of esophageal atresia without a primary gastrostomy: 240 patients treated between 1951 and 1983. J Pediatr Surg. 1985; 20:823–828.
93. Sharma, S, Sinha, SK, Rawat, JD, et al. Azygos vein preservation in primary repair of esophageal atresia with tracheoesophageal fistula. Pediatr Surg Int. 2007; 23:1215–1218.
94. Upadhyaya, VD, Gangopadhyaya, AN, Gopal, SC, et al. Is ligation of azygos vein necessary in primary repair of tracheoesophageal fistula with esophageal atresia? Eur J Pediatr Surg. 2007; 17:236–240.
95. Touloukian, RJ, Pickett, LK, Spackman, T, et al. Repair of esophageal atresia by end-to-side anastomosis and ligation of the tracheoesophageal fistula: A critical review of 18 cases. J Pediatr Surg. 1974; 9:305–310.
96. Poenaru, D, Laberge, JM, Neilson, IR, et al. A more than 25-year experience with end-to-end versus end-to-side repair for esophageal atresia. J Pediatr Surg. 1991; 26:472–477.
97. Davies, MR. Anatomy of the extrinsic motor nerve supply to mobilized segments of the oesophagus disrupted by dissection during repair of oesophageal atresia with distal fistula. Br J Surg. 1996; 83:1268–1270.
98. McCallion, WA, Hannon, RJ, Boston, VE. Prophylactic extrapleural chest drainage following repair of esophageal atresia: Is it necessary? J Pediatr Surg. 1992; 27:561.
99. Kay, S, Shaw, K. Revisiting the role of routine retropleural drainage after repair of esophageal atresia with distal tracheoesophageal fistula. J Pediatr Surg. 1999; 34:1082–1085.
100. Rothenberg, SS. Thoracoscopic repair of tracheoesophageal fistula in a newborn. Ped Endosurg Innovative Tech. 2000; 289–294.
101. Rothenberg, SS. Thoracoscopic repair of tracheoesophageal fistula in newborns. J Pediatr Surg. 2002; 37:869–872.
102. Bax, KM, van Der Zee, DC. Feasibility of thoracoscopic repair of esophageal atresia with distal fistula. J Pediatr Surg. 2002; 37:192–196.
103. Holcomb, GW, 3rd., Rothenberg, SS, Bax, KM, et al. Thoracoscopic repair of esophageal atresia and tracheoesophageal fistula: A multi-institutional analysis. Ann Surg. 2005; 242:422–430.
104. van der Zee, DC, Bax, NM. Thoracoscopic repair of esophageal atresia with distal fistula. Surg Endosc. 2003; 17:1065–1067.
105. Bax, N, van der Zee, DC, et al. The thoracoscopic approach to esophageal atresia with distal fistula. In: Bax N, Georgeson KE, Rothenberg SS, eds. Endoscopic Surgery in Infants and Children. Heidelberg: Springer; 2008:199–205.
106. Rothenberg, SS. Thoracoscopic repair of esophageal atresia and tracheoesophageal fistula in neonates: Evolution of a technique. J Laparoendosc Adv Surg Tech A. 2012; 195–199.
107. Mortellaro, VE, Fike, FB, Adibe, OO, et al. The use of high-frequency oscillating ventilation to facilitate stability during neonatal thoracoscopic operations. J Laparoendosc Adv Surg Tech. 2011; A 21:877–879.
108. Puri, P, Blake, N, O’Donnell, B, et al. Delayed primary anastomosis following spontaneous growth of esophageal segments in esophageal atresia. J Pediatr Surg. 1981; 16:180–183.
109. Puri, P, Ninan, GK, Blake, NS, et al. Delayed primary anastomosis for esophageal atresia: 18 months’ to 11 years’ follow-up. J Pediatr Surg. 1992; 27:1127–1130.
110. Spitz, L, Kiely, E, Brereton, RJ, et al. Management of esophageal atresia. World J Surg. 1993; 17:296–300.
111. Aziz, D, Schiller, D, Gerstle, JT, et al. Can ‘long-gap’ esophageal atresia be safely managed at home while awaiting anastomosis? J Pediatr Surg. 2003; 38:705–708.
112. Hollands, CM, Lankau, CA, Jr., Burnweit, CA. Preoperative home care for esophageal atresia—a survey. J Pediatr Surg. 2000; 35:279–282.
113. Howard, R, Myers, NA. Esophageal atresia: A technique for elongating the upper pouch. Surgery. 1965; 58:725–727.
114. Mahour, GH, Woolley, MM, Gwinn, JL. Elongation of the upper pouch and delayed anatomic reconstruction in esophageal atresia. J Pediatr Surg. 1974; 9:373–383.
115. Hays, DM, Woolley, MM, Snyder, WH, Jr. Changing techniques in the management of esophageal atresia. Arch Surg. 1966; 92:611–616.
116. Hendren, WH, Hale, JR. Electromagnetic bougienage to lengthen esophageal segments in congenital esophageal atresia. N Engl J Med. 1975; 293:428–432.
117. Livaditis, A, Radberg, L, Odensjo, G. Esophageal end-to-end anastomosis: Reduction of anastomotic tension by circular myotomy. Scand J Thorac Cardiovasc Surg. 1972; 6:206–214.
118. Kate, JT. Method of suturing operations for congenital oesophageal atresia. Arch Chir Neerland. 1952; 4:43–47.
119. Gough, MH. Esophageal atresia—use of an anterior flap in the difficult anastomosis. J Pediatr Surg. 1980; 15:310–311.
120. Kimura, K, Soper, RT. Multistaged extrathoracic esophageal elongation for long gap esophageal atresia. J Pediatr Surg. 1994; 29:566–568.
121. Martinez-Ferro, M, et al. Thoracoscopic repair of esophageal atresia without fistula. In: Bax N, Georgeson KE, Rothenberg SS, eds. Endoscopic surgery in infants and children. Heidelberg: Springer; 2008:207–219.
122. Davison, P, Poenaru, D, Kamal, I. Esophageal atresia: Primary repair of a rare long gap variant involving distal pouch mobilization. J Pediatr Surg. 1999; 34:1881–1883.
123. Lessin, MS, Wesselhoeft, CW, Luks, FI, et al. Primary repair of long-gap esophageal atresia by mobilization of the distal esophagus. Eur J Pediatr Surg. 1999; 9:369–372.
124. Lai, JY, Sheu, JC, Chang, PY, et al. Experience with distal circular myotomy for long-gap esophageal atresia. J Pediatr Surg. 1996; 31:1503–1508.
125. Giacomoni, MA, Tresoldi, M, Zamana, C, et al. Circular myotomy of the distal esophageal stump for long gap esophageal atresia. J Pediatr Surg. 2001; 36:855–857.
126. Rehbein, F, Schweder, N. Reconstruction of the esophagus without colon transplantation in cases of atresia. J Pediatr Surg. 1971; 6:746–752.
127. Okmian, L, Booss, D, Ekelund, L. An endoscopic technique for Rehbein’s silver olive method. Z Kinderchir. 1975; 16:212–215.
128. Vogel, AM, Yang, EY, Fishman, SJ. Hydrostatic stretch-induced growth facilitating primary anastomosis in long-gap esophageal atresia. J Pediatr Surg. 2006; 41:1170–1172.
129. Schaerli, A. Esophageal reconstruction in very long atresia by elongation of the lesser curvature. Pediatr Surg Int. 1992; 7:101–105.
130. Rao, KL, Menon, P, Samujh, R, et al. Fundal tube esophagoplasty for esophageal reconstruction in atresia. J Pediatr Surg. 2003; 38:1723–1725.
131. Waterston, D. Colonic replacement of the esophagus (intrathoracic). Surg Clin North Am. 1994; 44.
132. Anderson, KD, Randolph, JG. The gastric tube for esophageal replacement in children. J Thorac Cardiovasc Surg. 1973; 66:333–342.
133. Heimlich, HJ, Winfield, JM. The use of a gastric tube to replace or by-pass the esophagus. Surgery. 1955; 37:549–559.
134. Spitz, L, Kiely, E, Pierro, A. Gastric transposition in children—a 21-year experience. J Pediatr Surg. 2004; 39:276–281.
135. Ure, BM, Jesch, NK, Sumpelmann, R, et al. Laparoscopically assisted gastric pull-up for long gap esophageal atresia. J Pediatr Surg. 2003; 38:1661–1662.
136. Bax, NM, van der Zee, DC. Jejunal pedicle grafts for reconstruction of the esophagus in children. J Pediatr Surg. 2007; 42:363–369.
137. Ring, WS, Varco, RL, L’Heureux, PR, et al. Esophageal replacement with jejunum in children: An 18 to 33 year follow-up. J Thorac Cardiovasc Surg. 1982; 83:918–927.
138. Saitua, F, Madrid, A, Capdeville, F, et al. Pharyngoesophageal reconstruction by free jejunal graft and microvascular anastomosis in a 10-year-old girl. J Pediatr Surg. 2004; 39:e10–e12.
139. Bax, NM, Van Renterghem, KM. Ileal pedicle grafting for esophageal replacement in children. Pediatr Surg Int. 2005; 21:369–372.
140. Foker, JE, Linden, BC, Boyle, EM, Jr., Marquardt, C. Development of a true primary repair for the full spectrum of esophageal atresia. Ann Surg. 1997; 226:533–541.
141. Foker, JE, Kendall Krosch, TC, Catton, K, et al. A flexible approach to achieve a true primary repair for all infants with esophageal atresia. Semin Pediatr Surg. 2009; 18:23–29.
142. van der Zee, DC, Vieirra-Travassos, D, Kramer, WL, et al. Thoracoscopic elongation of the esophagus in long gap esophageal atresia. J Pediatr Surg. 2007; 42:1785–1788.
143. Spitz, L, Kiely, E, Brereton, RJ, et al. Management of esophageal atresia. World J Surg. 1993; 17:296–300.
144. MacKinlay, G, Burlles, R. Oesophageal atresia: Paralysis and ventilation management of the wide gap. Pediatr Surg Int. 1987; 2:10–12.
145. Lyall, P, Bao-Quan, Q, Beasley, S. The effect of neck flexion on oesophageal tension in the pig and its relevance to repaired oesophageal atresia. Pediatr Surg Int. 2001; 17:193–195.
146. Beasley, SW. Does postoperative ventilation have an effect on the integrity of the anastomosis in repaired oesophageal atresia? J Paediatr Child Health. 1999; 35:120–122.
147. Chittmittrapap, S, Spitz, L, Kiely, EM, et al. Anastomotic leakage following surgery for esophageal atresia. J Pediatr Surg. 1992; 27:29–32.
148. Yanchar, NL, Gordon, R, Cooper, M, et al. Significance of the clinical course and early upper gastrointestinal studies in predicting complications associated with repair of esophageal atresia. J Pediatr Surg. 2001; 36:815–822.
149. Said, M, Mekki, M, Golli, M, et al. Balloon dilatation of anastomotic strictures secondary to surgical repair of oesophageal atresia. Br J Radiol. 2003; 76:26–31.
150. Spitz, L, et al. Oesophageal atresia and tracheoesophageal fistula. In: Freeman NV, ed. Surgery of the Newborn. New York: Churchill Livingstone; 1994:353–373.
151. Chittmittrapap, S, Spitz, L, Kiely, EM, et al. Anastomotic stricture following repair of esophageal atresia. J Pediatr Surg. 1990; 25:508–511.
152. Koivusalo, A, Turunen, P, Rintala, RJ, et al. Is routine dilatation after repair of esophageal atresia with distal fistula better than dilatation when symptoms arise? Comparison of results of two European pediatric surgical centers. J Pediatr Surg. 2004; 39:1643–1647.
153. Ashcraft, KW, Goodwin, C, Amoury, RA, et al. Early recognition and aggressive treatment of gastroesophageal reflux following repair of esophageal atresia. J Pediatr Surg. 1977; 12:317–321.
154. Fonkalsrud, EW. Gastroesophageal fundoplication for reflux following repair of esophageal atresia: Experience with nine patients. Arch Surg. 1979; 114:48–51.
155. Pieretti, R, Shandling, B, Stephens, CA. Resistant esophageal stenosis associated with reflux after repair of esophageal atresia: A therapeutic approach. J Pediatr Surg. 1974; 9:355–357.
156. St. Peter, SD, Calkins, CM, Holcomb, GW, III. The use of biosynthetic mesh to separate the anastomoses during the thoracoscopic repair of esophageal atresia and tracheoesophageal fistula. J Laparoendosc Adv Surg Tech. 2007; 17:380–382.
157. Richter, GT, Ryckman, F, Brown, RL, et al. Endoscopic management of recurrent tracheoesophageal fistula. J Pediatr Surg. 2008; 43:238–245.
158. Keckler, SJ, St. Peter, SD, Calkins, CM, et al. Occlusion of a recurrent tracheoesophageal fistula. J Pediatr Surg. 2008; 18:465–468.
159. Meier, JD, Sulman, CG, Almond, PS, et al. Endoscopic management of recurrent congenital tracheoesophageal fistula: A review of techniques and result. Int J Pediatr Otorhinolaryngol. 2007; 71:691–697.
160. Wailoo, MP, Emery, JL. The trachea in children with tracheooesophageal fistula. Histopathology. 1979; 3:329–338.
161. Cohen, B, Glasson, M. Tracheomalacia in association with congenital tracheoesophageal fistula. Surgery. 1978; 79:504–508.
162. Rideout, DT, Hayashi, AH, Gillis, DA, et al. The absence of clinically significant tracheomalacia in patients having esophageal atresia without tracheoesophageal fistula. J Pediatr Surg. 1991; 26:1303–1305.
163. Delius, RE, Wheatley, MJ, Coran, AG. Etiology and management of respiratory complications after repair of esophageal atresia with tracheoesophageal fistula. Surgery. 1992; 112:527–532.
164. Gross, RE, Neuhauser, EBD. Compression of the trachea by an anomalous innominate artery: An operation for its relief. Am J Dis Child. 1948; 75:570–574.
165. Schwartz, MZ, Filler, RM. Tracheal compression as a cause of apnea following repair of tracheoesophageal fistula: Treatment by aortopexy. J Pediatr Surg. 1980; 15:842–848.
166. Applebaum, H, Woolley, MM. Pericardial flap aortopexy for tracheomalacia. J Pediatr Surg. 1990; 25:30–32.
167. Koyluoglu, G, Gunay, I, Ceran, C, et al. Pericardial flap aortopexy: An easy and safe technique in the treatment of tracheomalacia. J Cardiovasc Surg (Torino). 2002; 43:295–297.
168. Blair, GK, Cohen, R, Filler, RM. Treatment of tracheomalacia: Eight years’ experience. J Pediatr Surg. 1986; 21:781–785.
169. Vaishnav, A, MacKinnon, AE. New cervical approach for tracheopexy. Br J Surg. 1986; 73:441–442.
170. DeCou, JM, Parsons, DS, Gauderer, MWL. Thoracoscopic aortopexy for severe tracheomalacia. Pediatr Endosurg Innov Tech. 2001; 5:205–208.
171. Bax, N, van der Zee, DC, et al. Aortosternopexy for tracheomalacia. In: Bax N, Georgeson KE, Rothenberg SS, eds. Endoscopic Surgery in Infants and Children. Heidelberg: Springer; 2008:157–162.
172. Kane, T, Nadler, EP, Potoka, DA. Thoracoscopic aortopexy for vascular compression of the trachea: Approach from the right. J Laparoendosc Adv Surg Tech A. 2008; 18:313–316.
173. Kirkpatrick, JA, Cresson, SL. Pilling GP 4th. The motor activity of the esophagus in association with esophageal atresia and tracheoesophageal fistula. Am J Roentgenol Radium Ther Nucl Med. 1961; 86:884–887.
174. Lind, JF, Blanchard, RJ, Guyda, H. Esophageal motility in tracheoesophageal fistula and esophageal atresia. Surg Gynecol Obstet. 1966; 123:557–564.
175. Shermeta, DW, Whitington, PF, Seto, DS, et al. Lower esophageal sphincter dysfunction in esophageal atresia: Nocturnal regurgitation and aspiration pneumonia. J Pediatr Surg. 1977; 12:871–876.
176. Tovar, JA, Diez Pardo, JA, Murcia, J, et al. Ambulatory 24-hour manometric and pH metric evidence of permanent impairment of clearance capacity in patients with esophageal atresia. J Pediatr Surg. 1995; 30:1224–1231.
177. Deurloo, JA, Ekkelkamp, S, Bartelsman, JF, et al. Gastroesophageal reflux: Prevalence in adults older than 28 years after correction of esophageal atresia. Ann Surg. 2003; 238:686–689.
178. Zigman, A, Yazbeck, S. Esophageal foreign body obstruction after esophageal atresia repair. J Pediatr Surg. 2002; 37:776–778.
179. Hormann, M, Pokieser, P, Scharitzer, M, et al. Videofluoroscopy of deglutition in children after repair of esophageal atresia. Acta Radiol. 2002; 43:507–510.
180. Dutta, HK, Rajani, M, Bhatnagar, V. Cineradiographic evaluation of postoperative patients with esophageal atresia and tracheoesophageal fistula. Pediatr Surg Int. 2000; 16:322–325.
181. Montgomery, M, Witt, H, Kuylenstierna, R, et al. Swallowing disorders after esophageal atresia evaluated with videomanometry. J Pediatr Surg. 1998; 33:1219–1223.
182. Dutta, HK, Grover, VP, Dwivedi, SN, et al. Manometric evaluation of postoperative patients of esophageal atresia and tracheo-esophageal fistula. Eur J Pediatr Surg. 2001; 11:371–376.
183. Kawahara, H, Kubota, A, Hasegawa, T, et al. Lack of distal esophageal contractions is a key determinant of gastroesophageal reflux disease after repair of esophageal atresia. J Pediatr Surg. 2007; 42:2017–2021.
184. Shono, T, Suita, S, Arima, T, et al. Motility function of the esophagus before primary anastomosis in esophageal atresia. J Pediatr Surg. 1993; 28:673–676.
185. Shono, T, Suita, S. Motility studies of the esophagus in a case of esophageal atresia before primary anastomosis and in experimental models. Eur J Pediatr Surg. 1997; 7:138–142.
186. Cheng, W, Poon, KH, Lui, VC, et al. Esophageal atresia and achalasia-like esophageal dysmotility. J Pediatr Surg. 2004; 39:1581–1583.
187. Tibboel, D, Pattenier, JW, Van Krutgen, RJ, et al. Prospective evaluation of postoperative morbidity in patients with esophageal atresia. Pediatr Surg Int. 1988; 4:252–255.
188. Koch, A, Rohr, S, Plaschkes, J, et al. Incidence of gastroesophageal reflux following repair of esophageal atresia. Progr Pediatr Surg. 1986; 19:103–113.
189. Koivusalo, A, Pakarinen, MP, Rintala, RJ. The cumulative incidence of significant gastroesophageal reflux in patients with oesophageal atresia with a distal fistula—a systematic clinical, pH-metric, and endoscopic follow-up study. J Pediatr Surg. 2007; 42:370–374.
190. Koivusalo, A, Pakarinen, M, Rintala, RJ, et al. Does postoperative pH monitoring predict complicated gastroesophageal reflux in patients with esophageal atresia? Pediatr Surg Int. 2004; 20:670–674.
191. Van Biervliet, S, Van Winckel, M, Robberecht, E, et al. High-dose omeprazole in esophagitis with stenosis after surgical treatment of esophageal atresia. J Pediatr Surg. 2001; 36:1416–1418.
192. Wheatley, MJ, Coran, AG, Wesley, JR. Efficacy of the Nissen fundoplication in the management of gastroesophageal reflux following esophageal atresia repair. J Pediatr Surg. 1993; 28:53–55.
193. Snyder, CL, Ramachandran, V, Kennedy, AP, et al. Efficacy of partial wrap fundoplication for gastroesophageal reflux after repair of esophageal atresia. J Pediatr Surg. 1997; 32:1089–1092.
194. Sistonen, SJ, Koivusalo, A, Lindahl, H, et al. Cancer after repair of esophageal atresia: Population-based long-term follow-up. J Pediatr Surg. 2008; 43:602–605.
195. Mortellaro, VE, Pettiford, JN, St. Peter, SD, et al. Incidence, diagnosis and outcomes of vocal fold immobility after esophageal atresia (EA) and/or tracheoesophageal fistula (TEF) repair. Eur J Pediatr Surg. 2011; 21:386–388.
196. Robertson, JR, Birck, HG. Laryngeal problems following infant esophageal surgery. Laryngoscope. 1976; 86:965–970.
197. Chetcuti, P, Phelan, PD. Respiratory morbidity after repair of esophageal atresia and tracheoesophageal fistula. Arch Dis Child. 1993; 68:167–170.
198. Malmström, K, Lohi, J, Lindahl, H, et al. Longitudinal follow-up of bronchial inflammation, respiratory symptoms, and pulmonary function in adolescents after repair of esophageal atresia with tracheoesophageal fistula. J Pediatr Surg. 2008; 153:396–401.
199. Vazquez-Jimenez, JF, Sachweh, JS, Liakopoulos, OJ, et al. Aortopexy in severe tracheal instability: Short-term and long-term outcome in 29 infants and children. Ann Thorac Surg. 2001; 72:1898–1901.
200. Durning, RP, Scoles, PV, Fox, OD. Scoliosis after thoracotomy in tracheoesophageal fistula patients: A follow-up study. J Bone Joint Surg Am. 1980; 62:1156–1159.
201. Jaureguizar, E, Vazquez, J, Murcia, J, et al. Morbid musculoskeletal sequelae of thoracotomy for tracheoesophageal fistula. J Pediatr Surg. 1985; 20:511–514.
202. Cherup, LL, Siewers, RD, Futrell, JW. Breast and pectoral muscle maldevelopment after anterolateral and posterolateral thoracotomies in children. Ann Thorac Surg. 1986; 41:492–497.
203. Westfelt, JN, Nordwall, A. Thoracotomy and scoliosis. Spine. 1991; 16:1124–1125.
204. Chetcuti, P, Dickens, DR, Phelan, PD. Spinal deformity in patients born with oesophageal atresia and tracheo-oesophageal fistula. Arch Dis Child. 1989; 64:1427–1430.
205. Chetcuti, P, Myers, NA, Phelan, PD, et al. Chest wall deformity in patients with repaired esophageal atresia. J Pediatr Surg. 1989; 24:244–247.
206. Emmel, M, Ulbach, P, Herse, B, et al. Neurogenic lesions after posterolateral thoracotomy in young children. Thorac Cardiovasc Surg. 1996; 44:86–91.
207. Schier, F, Korn, S, Michel, E. Experiences of a parent support group with the long-term consequences of esophageal atresia. J Pediatr Surg. 2001; 36:605–610.
208. Perttunen, K, Tasmuth, T, Kalso, E. Chronic pain after thoracic surgery: A follow-up study. Acta Anaesthesiol Scand. 1999; 43:563–567.
209. Rogers, ML, Duffy, JP. Surgical aspects of chronic post-thoracotomy pain. Eur J Cardiothorac Surg. 2000; 18:711–716.
210. Haller, JO, Berdon, WE, Levin, TL, et al. Tracheoesophageal fistula (H-type) in neonates with imperforate anus and the VATER association. Pediatr Radiol. 2004; 34:83–85.
211. Fordham, LA. Imaging of the esophagus in children. Radiol Clin North Am. 2005; 43:283–302.
212. Azoulay, D, Regnard, JF, Magdeleinat, P, et al. Congenital respiratory-esophageal fistula in the adult: Report of nine cases and review of the literature. J Thorac Cardiovasc Surg. 1992; 104:381–384.
213. Garand, SA, Kareti, LR, Dumont, TM, et al. Thoracoscopic repair of tracheoesophageal fistula in a septuagenarian. Ann Thorac Surg. 2006; 81:1899–1901.
214. Crabbe, DC. Isolated tracheo-oesophageal fistula. Paediatr Respir Rev. 2003; 4:74–78.
215. Nagata, K, Kamio, Y, Ichikawa, T, et al. Congenital tracheoesophageal fistula successfully diagnosed by CT esophagography. World J Gastroenterol. 2006; 12:1476–1478.
216. LaSalle, AJ, Andrassy, RJ, Ver Steeg, K, et al. Congenital tracheoesophageal fistula without esophageal atresia. J Thorac Cardiovasc Surg. 1979; 78:583–588.
217. Garcia, NM, Thompson, JW, Shaul, DB. Definitive localization of isolated tracheoesophageal fistula using bronchoscopy and esophagoscopy for guide wire placement. J Pediatr Surg. 1998; 33:1645–1647.
218. Ko, BA, Frederic, R, DiTirro, PA, et al. Simplified access for division of the low cervical/high thoracic H-type tracheoesophageal fistula. J Pediatr Surg. 2000; 35:1621–1622.
219. Rothenberg, SS. Experience with thoracoscopic tracheal surgery in infants and children. J Laparoendosc Adv Surg Tech A. 2009; 19:671–674.
220. Allal, H, Montes-Tapia, F, Andina, G, et al. Thoracoscopic repair of H-type tracheoesophageal fistula in the newborn: A technical case report. J Pediatr Surg. 2004; 39:1568–1570.
221. Aziz, GA, Schier, F. Thoracoscopic ligation of a tracheoesophageal H-type fistula in a newborn. J Pediatr Surg. 2005; 40:e35–e36.
222. Roth, B, Rose, KG, Benz-Bohm, G, et al. Laryngo-tracheo-oesophageal cleft: Clinical features, diagnosis and therapy. Eur J Pediatr. 1983; 140:41–46.
223. Walner, DL, Stern, Y, Collins, M, et al. Does the presence of a tracheoesophageal fistula predict the outcome of laryngeal cleft repair? Arch Otolaryngol Head Neck Surg. 1999; 125:782–784.
224. Phelan, PD, Stocks, JG, Williams, HE, et al. Familial occurrence of congenital laryngeal clefts. Arch Dis Child. 1973; 48:275–278.
225. Tyler, DC. Laryngeal cleft: Report of eight patients and a review of the literature. Am J Med Genet. 1985; 21:61–75.
226. Pettersson, G. Inhibited separation of larynx and the upper part of trachea from oesophagus in a newborn: Report of a case successfully operated upon. Acta Chir Scand. 1955; 110:250–254.
227. Ryan, DP, Muehrcke, DD, Doody, DP, et al. Laryngotracheoesophageal cleft (type IV): Management and repair of lesions beyond the carina. J Pediatr Surg. 1991; 26:962–969.
228. Myer, CM, 3rd., Cotton, RT, Holmes, DK, et al. Laryngeal and laryngotracheoesophageal clefts: Role of early surgical repair. Ann Otol Rhinol Laryngol. 1990; 99:98–104.
229. Donahoe, PK, Gee, PE. Complete laryngotracheoesophageal cleft: Management and repair. J Pediatr Surg. 1984; 19:143–148.
230. Mathur, NN, Peek, GJ, Bailey, CM, et al. Strategies for managing Type IV laryngotracheoesophageal clefts at Great Ormond Street Hospital for Children. Int J Pediatr Otorhinolaryngol. 2006; 70:1901–1910.
231. Moukheiber, AK, Camboulives, J, Guys, JM, et al. Repair of a type IV laryngotracheoesophageal cleft with cardiopulmonary bypass. Ann Otol Rhinol Laryngol. 2002; 111:1076–1080.
232. Geiduschek, JM, Inglis, AF, Jr., O’Rourke, PP, et al. Repair of a laryngotracheoesophageal cleft in an infant by means of extracorporeal membrane oxygenation. Ann Otol Rhinol Laryngol. 1993; 102:827–833.