Erythrodermas, Immunodeficiency, and Metabolic Disorders
Moise L. Levy
Erythrodermas
The term ‘erythroderma’ is used in dermatology to describe a skin eruption characterized by diffuse erythema, usually in association with scaling. Infantile erythroderma is caused by or associated with a large number of disorders (Box 18.1). The differential diagnosis includes inflammatory, infectious, inherited, and immunologic diseases, many of which have a hereditary basis. Some of these diseases are potentially life-threatening, and erythroderma itself can cause serious medical complications, such as electrolyte imbalance, sepsis, and temperature instability resulting from heat loss. It is therefore important for the physician to accurately diagnose and treat the problem.
Inflammatory diseases
Atopic dermatitis
Severe generalized atopic dermatitis is unusual in neonates, though may often be seen in infancy. Because atopic dermatitis is such a common problem, it is the most common cause of acquired erythroderma in infants (see Chapter 15). Classic infantile atopic dermatitis involves the scalp, cheeks, and extensor surfaces of the extremities and may not appear until the infant is several months of age.1 When the distribution is generalized and the onset is early, diagnosis can be more difficult.
The presence of pruritus, an almost invariable feature of this condition, is not always apparent in neonates and young infants. There is often a family history of atopy. Typically, the diaper region is spared, even in cases of widespread atopic dermatitis, as a result of the moist, occlusive environment of diapered skin. In contrast to infants with severe metabolic or immunologic disease, infants with atopic dermatitis usually grow normally and thrive, assuming the disease is recognized and treated promptly. Severe and long-standing disease, however, can be a cause of failure to thrive. Other clinical features such as repeated pneumonia, viral infections such as HSV or molluscum, and skeletal abnormalities may suggest the autosomal dominant hyper-IgE syndrome (HIES) or the autosomal recessive DOCK8 deficiency syndrome.2,3 Atopic dermatitis generally responds rapidly to appropriate therapy with topical anti-inflammatory agents and emollients. Skin biopsy in atopic dermatitis demonstrates acanthosis (thickening of the epidermis) and varying degrees of spongiosis (epidermal edema), as well as lymphohistiocytic inflammatory infiltrates, often with scattered eosinophils and plasma cells.4
Seborrheic dermatitis
Seborrheic dermatitis is a common problem during the neonatal period and is generally easily recognized (see also Chapter 15). Typically, there is scaling and erythema involving seborrheic areas such as the scalp and body folds. The yellow, greasy, scalp scale may encompass the entire forehead, including the eyebrows, and erythema and maceration can involve body folds such as the retroauricular areas, neck, axillae, and groin. Occasionally, a more diffuse pattern of seborrheic dermatitis can occur, which must be distinguished from atopic dermatitis, neonatal candidiasis, psoriasis, and other causes of infantile erythroderma (Box 18.1 and Fig. 18.1).
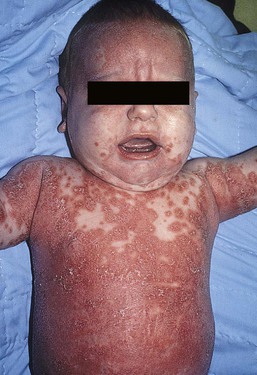
The distribution of the dermatitis is more helpful than any other criterion in differentiating between atopic and seborrheic dermatitis, but it can be difficult and sometimes impossible to differentiate the two conditions accurately early in their course. Although the scalp can be red and scaly in both conditions, seborrheic dermatitis tends to involve the groin and other body folds, which are generally spared in atopic dermatitis. As treatment for both conditions in infancy is similar, from a practical standpoint accurate differentiation can be an academic exercise. However, the course of this disease differs: seborrheic dermatitis usually resolves over several months, whereas atopic dermatitis often persists for several years. Skin biopsy findings in seborrheic dermatitis are similar to those in atopic dermatitis. There is mild acanthosis, spongiosis, and a mild lymphohistiocytic inflammatory infiltrate; parakeratotic scale may be present.
If clinical features suggest widespread seborrheic dermatitis in an infant who is otherwise well and thriving, and the skin readily clears after the application of low- to mid-potency topical corticosteroids without chronic rebound when therapy is tapered, the diagnosis of seborrheic dermatitis is probably accurate. Otherwise, alternative diagnoses should be considered. Severe seborrheic dermatitis in a child who is not thriving can suggest an immunodeficiency or Netherton syndrome.
Psoriasis
Less than 1% of all cases of psoriasis are said to occur in children less than 1 year of age. Infantile psoriasis can be difficult to diagnose because of its clinical similarity to both seborrheic dermatitis and atopic dermatitis. Infantile psoriasis can look like that seen in older individuals, with discrete oval erythematous plaques with white scale involving the trunk, extremities, and face. Psoriatic plaques in infants may have less hyperkeratosis than usually seen in adults. Facial involvement is more common in the infant, and the scalp, palms, and soles may have diffuse erythema and scaling. A periumbilical distribution may be helpful in distinguishing psoriasis from either seborrheic or atopic dermatitis. In contrast to atopic dermatitis, psoriasis in young infants often involves the diaper area because it develops in areas of injured skin (the Koebner phenomenon), e.g. after a prior irritant or Candida diaper dermatitis (see Chapter 16).5 Pustular psoriasis, either in a diffuse distribution or limited to the palms and soles, may be seen rarely. A positive family history for psoriasis is helpful. Some neonatal or infantile cases are HLA-B17 positive.6
Rarely, infantile psoriasis is generalized, a presentation that has been reported in young infants, and can even be present at birth. Erythroderma can evolve into and even alternate with pustulosis. Infantile generalized pustular psoriasis can be associated with lytic bone lesions,7 and be complicated by the acute respiratory distress syndrome (pulmonary capillary leak syndrome) that is also described in adults with acute generalized pustular psoriasis (B. Krafchik pers. comm.).8 Skin biopsy can be helpful in differentiating causes of neonatal erythroderma and in some cases, is diagnostic.4 Biopsy usually shows psoriasiform hyperplasia with elongated rete ridges and parakeratotic scale, often containing neutrophils. Occasionally, the diagnostic finding of a spongiform micropustule or microabscess in the upper epidermis is seen. Skin biopsies of erythrodermic psoriasis are often indistinguishable from those of any chronic dermatitis, lacking the classic features, and it may take several biopsies and close observation over time to confirm the diagnosis.
Localized psoriasis may be treated with emollients and low-potency topical corticosteroids, but often clears only partially or recurs. Cases of infantile psoriasis may prove to be mild and occasionally even clear completely as the child gets older.9 The prognosis of generalized erythrodermic or pustular psoriasis in infancy is more guarded, and treatment usually requires systemic retinoid therapy, as well as supportive care.10
Drug exanthem
Erythroderma due to medications is fortunately rare, though cases in pediatric patients, including infants and neonates have been described.13 Severe reactions such as Stevens–Johnson syndrome or the drug reaction with eosinophilia and systemic symptoms (DRESS) represent particular challenges for clinicians.6 The approach to patients, regardless of age, requires a high index of suspicion and a broad understanding of other causes of erythroderma.14
Acute generalized exanthematous pustulosis
Acute generalized exanthematous pustulosis (AGEP) presents acutely with pustules overlying diffuse erythema after exposure to an offending medication, mercury exposure or viral illness. Antibiotics reported to cause AGEP in infants include amoxicillin and amoxicillin-clavulanic acid. AGEP has also been reported in infants with no antibiotic exposure, suggesting an infectious trigger in these cases.15 Subcorneal pustules are seen by skin biopsy (see Chapter 20).16
Boric acid poisoning
Boric acid poisoning is now very rare, but was seen in the past as a result of the frequent use of boric acid-containing powders and lotions for the treatment of diaper dermatitis. It presents with a maculopapular eruption that can evolve into a generalized erythroderma, the appearance of which has been likened to a boiled lobster. A report of this in an adult after ingestion of a boric acid-containing pesticide, has been published.17 A positive Nikolsky sign and desquamation are additional features. Like staphylococcal scalded skin syndrome, the condition may be accentuated in periorificial and intertriginous areas. Alopecia may also develop. Affected infants are usually ill, with fever, irritability, vomiting, and diarrhea, which can progress to shock and even death.
Diffuse cutaneous mastocytosis
The various forms of cutaneous mastocytosis are discussed fully in Chapter 28. Only the rare diffuse cutaneous form of the disease is associated with neonatal erythroderma.18 The affected infant usually has generalized thickening of the skin, which can be subtle. The thickening is due to infiltration of the dermis by mast cells. Because these mast cells release histamine and other vasoactive substances, they cause the skin to be very reactive, with a tendency to develop erythema, flushing, and wheals. Urtication with minor trauma (Darier’s sign), and blisters, which develop either spontaneously or superimposed upon wheals, may be seen. The absence of scale and the presence of the above findings differentiate mastocytosis from other causes of erythroderma. C-kit mutations are seen in most patients with systemic mast cell disease.18
Skin biopsy is diagnostic revealing a dense, band-like infiltrate of mast cells in the upper dermis, which can be confirmed with Giemsa stain.
Infectious diseases
Staphylococcal scalded skin syndrome
Staphylococcal scalded skin syndrome (SSSS) is an uncommon cause of neonatal erythroderma (see Chapter 12). It is characterized by the abrupt onset of diffuse erythema, which rapidly evolves to erosive desquamation involving most skin surfaces. Although there is often periorificial accentuation, giving a dry, chapped appearance, the staphylococcal toxin requires keratinizing epithelium and therefore spares mucous membrane surfaces. The toxins act at a subcorneal location due to effects on desmoglein 1.6,13 Epidermal sloughing, occurring with minor trauma, helps distinguish SSSS from the other causes of infantile erythroderma, with the exception of toxic epidermal necrolysis (TEN)19 (see Chapter 12) and boric acid poisoning (see above). Rarely, a widespread form of staphylococcal pustulosis has been observed acutely in otherwise healthy infants. The pustules develop on an erythematous macular base and are small and superficial. They subsequently desquamate, thereby mimicking a true erythroderma.
Candidiasis
Candidal infection can cause neonatal erythroderma in two clinical settings: intrauterine acquisition with the development of congenital candidiasis, and postnatal onset in very premature infants. Congenital candidiasis typically presents either at birth or within the first few days of life with generalized erythema, vesicles, pustules, papules, and scaling. The pustules can be subtle at first, with erythroderma predominating. The palms and soles are often involved, which may be a helpful clue to diagnosis. The condition can occur in either term or preterm infants. Candida albicans can also cause a diffuse burn-like erythema within the first 2 weeks of life in premature infants (see Fig. 14.5A); 20 Also diffuse scaling and erythema, most pronounced over the back may be seen.21 The diagnosis is made by examination of skin scrapings (KOH preparation) and/or surface culture for Candida, or by skin biopsy. The latter will show fungal elements within the epidermis and/or dermis, mixed inflammatory infiltrates, and occasional areas of necrosis and hemorrhage.
The risk of extracutaneous disease and the prognosis depend on the gestational age of the infant. In term infants, the prognosis is excellent, and topical anti-yeast therapies are usually curative. In infants weighing less than 1500 g with either congenital or acquired generalized cutaneous candidiasis, there is a significant risk of disseminated disease, and parenteral antifungal agents are recommended. Skin biopsies of affected areas in such infants may be used to predict the ultimate dissemination of disease. In one series, the finding of subcorneal invasion of Candida on skin biopsy was associated with a 69% risk for disseminated disease.21 Both conditions are discussed in more detail in Chapter 14.
Herpes simplex
Although most cases of herpes simplex (HSV) infection have characteristic vesicular lesions localized on the presenting part, the so-called intrauterine variant of HSV can present at birth with either isolated or diffuse erythema, and scaling or crusted erosions on an erythematous base (see Chapter 13). It may be difficult to recognize clinically because vesicles may not be present.22 In fact, 44% of neonates with cutaneous disease do not have vesicles or bullae.22 This type of widespread involvement is generally associated with very severe neurologic disease. Multinucleated giant cells should be demonstrable on Tzanck smears of vesicular lesions. A skin biopsy, scrapings for direct fluorescent antibody staining, and viral cultures will help confirm the diagnosis.
Syphilis
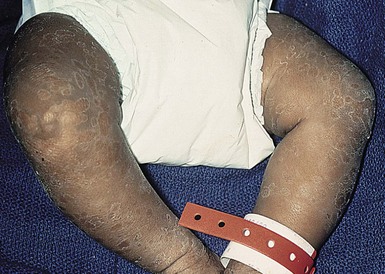
Congenital syphilis may cause diffuse erythema and scaling (see Chapter 12). This presentation is most typically seen in infants 6–8 weeks of age, in whom exposure to syphilis occurred either very late in pregnancy or at the time of delivery.23 Superficial erosions or bullae over the hands or feet of a newborn, together with a diffuse scaling dermatitis (Fig. 18.2), should alert the practitioner to the possibility of syphilis. Infiltrated mucosal papules and plaques (condyloma lata) may be seen in a perianal location and are similar to the mucous patches seen on other mucous membrane sites in older patients with secondary syphilis. Periosteal changes of long bones, such as the clavicles, as well as hepatosplenomegaly, are additional features. Appropriate serologies are generally diagnostic, and darkfield examination of mucous membrane lesions should reveal spirochetes.
Genodermatoses
Ichthyosis
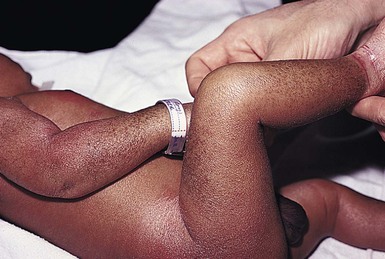
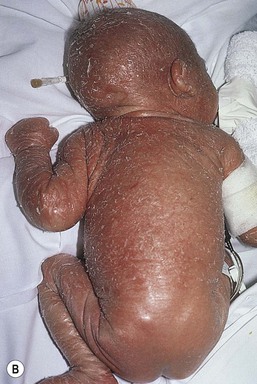
The ichthyoses are a group of genetic disorders characterized by generalized skin scaling; generalized erythroderma is a common presentation for some types of ichthyosis (see Chapter 19). Many infants with this presentation have the autosomal recessive congenital ichthyosis (ARCI) phenotype,24 which is notable for either diffuse erythematous appearance of the skin and overlying fine white scale or thicker plate-like scale (Fig. 18.3). Ectropion can occur. Testing for TGM1, ALOXE3, ALOX12B, or ICHTHYIN genes can be done for individual cases (see Chapter 19). Infants with autosomal dominant bullous ichthyosis, epidermolytic ichthyosis (EI), typically present with diffusely erythematous skin and mild hyperkeratosis, often in association with areas of denuded skin. The lack of mucous membrane involvement in EI helps distinguish it from epidermolysis bullosa. Over subsequent weeks and months, the blistering subsides and is replaced by varying degrees of an ichthyosiform erythroderma. Ultimately, marked hyperkeratosis is evident diffusely, with accentuation on flexural surfaces. The characteristic histopathologic findings of epidermal cytolysis of the upper spinous and granular layers help confirm the diagnosis of EI. Consideration of screening for mutations of keratins 1 and/or 10 is needed for confirmation.
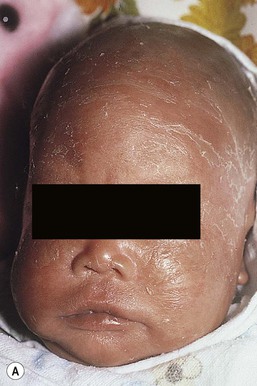
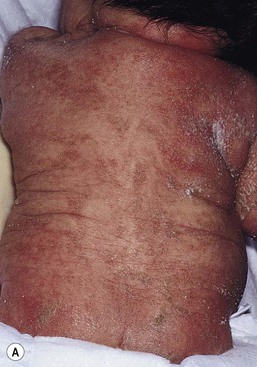
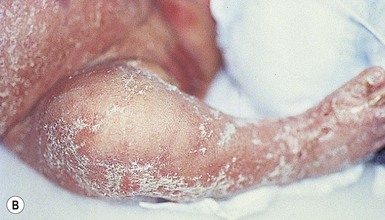
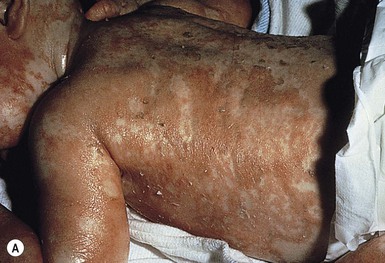
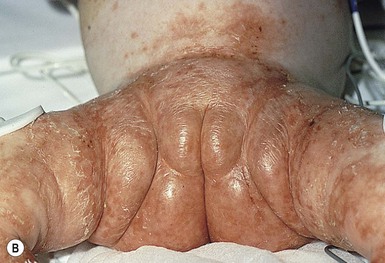
The ichthyosis most likely to be confused with other causes of erythroderma is Netherton syndrome, a rare disorder caused by mutations in SPINK5, which encodes a serine protease inhibitor, LEKTI.25 A publication has outlined specific mutations in SPINK5 and the use of such data for prenatal diagnosis.26 Some laboratories are able to stain tissue specimens for this protein.4 Netherton syndrome is marked by severe diffuse erythroderma, scaling, and varying degrees of alopecia, including sparse eyebrows (Fig. 18.4).27,28 Affected infants often fail to thrive as a result of the extreme metabolic demands presented by their skin disease, and they can also develop hypernatremic dehydration. Most have a markedly elevated IgE level. The diagnosis of Netherton syndrome is often delayed because of the late presentation of the diagnostic hair shaft abnormality, trichorrhexis invaginata (bamboo hair). Such hairs, when examined by routine light microscopy, will appear to have telescoped into themselves along the length of the shaft (Fig. 18.5). Small bulbous areas of thickening at the site of the telescoping correspond to the areas of increased fragility and ultimate breakage of affected hairs. Plucking of eyebrow hairs and evaluation of multiple areas of the scalp over time may be required to visualize the characteristic hair changes. Later in the course, patients may continue with generalized, scaling erythroderma or show a distinctive skin finding, ichthyosis linearis circumflexa. This is an erythematous scaling eruption with polycyclic and/or serpiginous morphology and elevated borders.

Sjögren–Larsson syndrome is due to a deficiency of fatty aldehyde dehydrogenase (FALDH) and can present with varying degrees of erythroderma.29,30 Most cases are due to mutations in the ALDH3A2 gene, which codes for FALDH, though other factors play a role in some cases.31 A collodion membrane at birth is unusual. Affected infants may have a phenotype consistent with ARCI. Nonprogressive spasticity and mental retardation become apparent during the early years of life. After the first year, many affected patients have distinctive glistening dots seen on careful retinal examination.
Chondrodysplasia punctata (Conradi–Hünermann syndrome) presents with either diffuse erythroderma or bands of erythema, and a patterned ichthyosis occurring along Blaschko’s lines (Fig. 18.6).32–34 Alopecia may be seen. These areas of ichthyosis typically resolve and may be replaced by a follicular atrophoderma. This X-linked dominant syndrome is also marked by skeletal defects (dwarfism), cataracts, and other features. Plain radiographs at the time of birth may show stippling of the epiphyseal areas of bones. It is due to a defect in emopamil binding protein (3β-hydroxysteroid-Δ8, Δ7-isomerase).
Infantile erythroderma can also be seen in keratosis–ichthyosis–deafness (KID) syndrome,35,36 and neutral lipid storage disease with ichthyosis (Chanarin–Dorfman syndrome).37,38 A report of erythroderma as a presenting sign of Menkes disease has been published.39
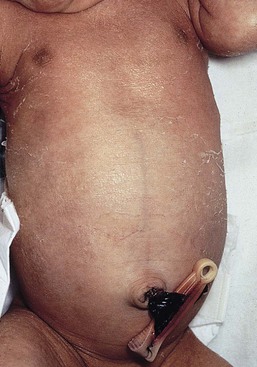
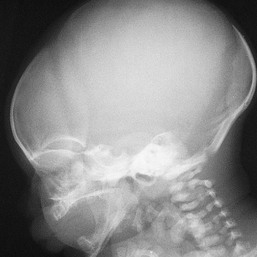
Occasionally, males affected with X-linked hypohidrotic ectodermal dysplasia may present at birth with a mild diffuse erythroderma and fine superficial scaling (Fig. 18.7).40 Such infants have the typical facial features of ED and sparse hair, lashes, and eyebrows. Periorbital hyperpigmentation and fine wrinkling can be seen at birth, and lateral plain films of the skull will demonstrate no or few tooth buds (Fig. 18.8). Some of these patients have been reported to have primary immunodeficiency as well.41,42
Immunologic diseases
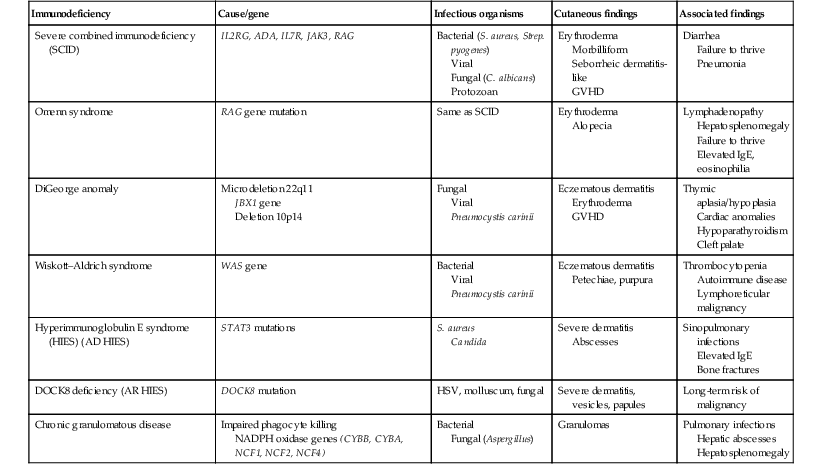
Several immunologic diseases marked by immunodeficiency may produce similar initial clinical signs, with an eczematous dermatitis, diarrhea and failure to thrive. Children with a persistent eczematous eruption accompanied by failure to thrive may warrant an immunologic evaluation. The following discussion encompasses immunodeficiency syndromes with either erythroderma or other cutaneous manifestations in the neonatal period. These are outlined in Table 18.1.
TABLE 18.1
Immunodeficiency syndromes with cutaneous manifestations in the neonatal period
| Immunodeficiency | Cause/gene | Infectious organisms | Cutaneous findings | Associated findings |
| Severe combined immunodeficiency (SCID) | IL2RG, ADA, IL7R, JAK3, RAG | Bacterial (S. aureus, Strep. pyogenes) Viral Fungal (C. albicans) Protozoan |
Erythroderma Morbilliform Seborrheic dermatitis-like GVHD |
Diarrhea Failure to thrive Pneumonia |
| Omenn syndrome | RAG gene mutation | Same as SCID | Erythroderma Alopecia |
Lymphadenopathy Hepatosplenomegaly Failure to thrive Elevated IgE, eosinophilia |
| DiGeorge anomaly | Microdeletion 22q11 JBX1 gene Deletion 10p14 |
Fungal Viral Pneumocystis carinii |
Eczematous dermatitis Erythroderma GVHD |
Thymic aplasia/hypoplasia Cardiac anomalies Hypoparathyroidism Cleft palate |
| Wiskott–Aldrich syndrome | WAS gene | Bacterial Viral Pneumocystis carinii |
Eczematous dermatitis Petechiae, purpura |
Thrombocytopenia Autoimmune disease Lymphoreticular malignancy |
| Hyperimmunoglobulin E syndrome (HIES) (AD HIES) | STAT3 mutations | S. aureus Candida |
Severe dermatitis Abscesses |
Sinopulmonary infections Elevated IgE Bone fractures |
| DOCK8 deficiency (AR HIES) | DOCK8 mutation | HSV, molluscum, fungal | Severe dermatitis, vesicles, papules | Long-term risk of malignancy |
| Chronic granulomatous disease | Impaired phagocyte killing NADPH oxidase genes (CYBB, CYBA, NCF1, NCF2, NCF4) |
Bacterial Fungal (Aspergillus) |
Granulomas | Pulmonary infections Hepatic abscesses Hepatosplenomegaly |
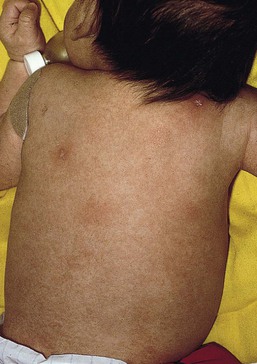
Erythroderma and failure to thrive
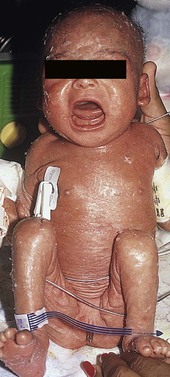
In 1988, Glover and colleagues43 reported a group of five infants with erythroderma, diarrhea, and failure to thrive. None had a yeast opsonization defect (as described in so-called ‘Leiner disease’), however, a variety of other immunologic abnormalities, including elevated IgE levels and hypogammaglobulinemia, were found. Some patients were subsequently diagnosed as having Netherton or Omenn syndromes. This paper established the need to consider the diagnosis of immunodeficiency when evaluating erythrodermic infants, and established the multiple etiologies of what had formerly been called Leiner disease.
Infants in this category have a clinical phenotype of (1) noncongenital or acquired erythroderma (Fig. 18.9); (2) diarrhea; and (3) failure to thrive. Infants with these findings need thorough investigations searching for the underlying cause of their disorder. Most of the diseases listed in Box 18.1 need to be considered, especially immunodeficiencies, Netherton syndrome, Omenn syndrome, and eosinophilic gastroenteritis (see below). Baseline immune studies of such infants should include chest radiograph, full blood count, quantitative immunoglobulins, and specific measures of T-cell function. More detailed testing should be pursued as indicated.
The prognosis and treatment of this condition are entirely dependent on the specific diagnosis. The associated diarrhea and failure to thrive must be treated aggressively with adequate nutritional support up to and including parenteral hyperalimentation, as indicated.
Severe combined immunodeficiency
Severe combined immunodeficiency (SCID) is a heterogeneous group of inherited disorders with similar clinical manifestations and immunologic deficiencies, with a profound deficiency of T lymphocytes and defects in both cellular and humoral immunity. Subtypes are classified by the abnormal development of other lymphocyte lineages, predominantly B lymphocytes and natural killer (NK) cells. Most cases are inherited in an autosomal recessive manner, although approximately 40% are X-linked recessive. About 20% of SCID cases are caused by adenosine deaminase (ADA) deficiency.44 Several other genetic defects account for the balance of causes of SCID.45
The immunologic deficiency results in an increased susceptibility to bacterial, viral, fungal and protozoan infections. Recurrent infections, diarrhea, and failure to thrive are evident by 3–6 months of age.46 Among the most common mucocutaneous infections are those due to Candida albicans, Staphylococcus aureus, and Streptococcus pyogenes. In addition to infectious cutaneous manifestations, all reported forms of SCID can present with a diffuse skin involvement, either erythroderma (Fig. 18.10), morbilliform, or seborrheic dermatitis-like eruptions. Patients with SCID may develop graft-versus-host disease from nonirradiated blood products or engraftment of maternal lymphocytes (Fig. 18.11). Cutaneous manifestations vary from a morbilliform rash or exfoliative dermatitis in acute GVHD, to a lichenoid or sclerodermoid rash in chronic GVHD. Early extracutaneous infections may also include viral-induced chronic diarrhea, otitis media, and pneumonia due to bacteria, viruses or Pseudomonas carinii.
The prognosis of SCID is poor and, without intervention, most patients die from overwhelming infection by 1 year of age. The importance of early diagnosis cannot be overemphasized. Some states screen newborns for SCID on a routine basis.45,47 Screening for T-cell receptor excision circles (TRECs) has been useful as a screen for SCID when neonates are found to have unusually low lymphocyte counts. TRECs are biomarkers for new and naive T cells.45 Follow-up testing for more specific genetic causes of SCID can be done based upon this test which can be done by routine newborn blood screening.48 Management includes protective isolation and vigorous treatment of infections. All blood products should be irradiated to prevent GVHD. Hematopoietic stem cell transplantation is the treatment of choice. Enzyme replacement with ADA coupled to polyethylene glycol (PEG-ADA) has helped several infants with ADA deficiency, and ADA is a target for gene therapy protocols. However, enzyme replacement alone may not be sufficient.44,49
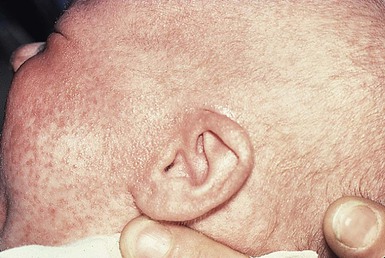
Omenn syndrome
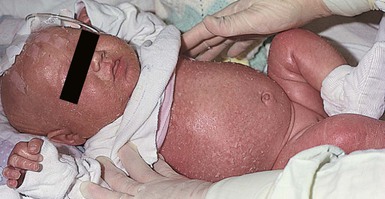
Omenn syndrome is a rare autosomal recessive form of SCID with reticuloendothelial cell proliferation caused by recombinase-activating gene (RAG1 or RAG2) deficiency in most patients. This syndrome was originally described as familial reticuloendotheliosis with eosinophilia. This T-cell deficient state is marked by abnormal histiocytic cells and extreme elevations of eosinophils in affected tissues and in the peripheral blood. During early infancy these patients develop a generalized exfoliative erythroderma (Fig. 18.12), lymphadenopathy, hepatosplenomegaly, recurrent infections, and failure to thrive.50 Diffuse alopecia may be seen as well. Although the condition is primarily one of T-cell dysregulation, both humoral and cellular immune defects are seen.51 Abnormal antibody production and elevated IgE levels occur. There is usually a marked leukocytosis with eosinophilia, anemia, hypogammaglobulinemia, and depressed T-cell mediated immunity. As noted above, RAG1 or RAG2 gene mutations are seen in most cases of Omenn syndrome. These result in faulty T- and B-cell development. Other gene mutations exist in this syndrome.51 The disorder is often difficult to distinguish from GVHD by skin biopsy.4 The only known effective treatment is bone marrow transplantation.
Digeorge anomaly
The DiGeorge anomaly (DGA) is one of a group of disorders that have in common a chromosomal deletion resulting in monosomy 22q11, known as the DiGeorge syndrome chromosome region, or DGCR. A small fraction of patients with clinical features of DGA have deletions of chromosome 10p14.52 Autosomal dominant, autosomal recessive and X-linked inheritance have all been reported. All forms are associated with a T-cell defect and normal humoral immunity. The compromise in T-cell production is a result of thymic hypoplasia or aplasia, which is in turn part of a larger developmental anomaly of the third and fourth pharyngeal pouches. In addition to the thymus defect, conotruncal cardiac anomalies, hypoparathyroidism, dysmorphism, and cleft palate are prominent features. Characteristic facial features include a short philtrum, low-set ears, and hypertelorism.53 Neonatal tetany may occur with hypocalcemia due to aplastic parathyroid glands. Also mapping to 22q11 is the velocardiofacial syndrome which shares many of these features as well as mental disorders. Many such cases are due to mutations in the TBX1 gene. These disorders are part of a spectrum.54
Infants with DiGeorge syndrome can have a maculopapular or eczematous dermatitis that may become generalized.55,56 A group of patients with incomplete DiGeorge syndrome and diffuse eczematous dermatitis have been studied by histopathology. Findings included dyskeratosis of keratinocytes, satellite cell necrosis, and parakeratosis with neutrophils.57 Many patients have recurrent mucocutaneous candidal infections as neonates, as well as increased susceptibility to viral, Pneumocystis carinii and fungal infections. GVHD may occur in patients who are given nonirradiated blood products. Noninfectious granulomas have also been described. Patients with DGA may have erythroderma, similar to that seen in Omenn syndrome, which occurs in association with a dramatic oligoclonal expansion of a few founding T-cells, often of an exclusively memory type.58 No live vaccines should be given and blood products should be irradiated. Patients with dramatically low levels of T-cells should be considered for an HLA-identical bone marrow transplant or a thymus transplant.58
Graft-versus-host disease
Graft-versus-host disease (GVHD) is caused by the interaction between immunocompetent lymphoid cells and immunodeficient host cells.59 Nearly all cases in neonates and young infants are caused by severe T-cell immunodeficiency states (such as severe combined immunodeficiency) with maternal engraftment either in utero or at the time of delivery.60 Clinically, affected infants typically present with scaling and erythema that often begin on the scalp and face and moves downward. Fine erythematous papules may also occur. The findings may be patchy or diffuse, in some cases progressing to frank erythroderma (Fig. 18.10).60 The most extreme cutaneous manifestation of GVHD is severe, generalized desquamation, a finding that may rarely be evident at the time of delivery. Diffuse alopecia is a common finding and often involves the eyebrows as well as scalp hair. Although milder forms of GVHD sometimes respond (at least partially) to emollients or topical corticosteroids, recurrences are the rule. If unrecognized and untreated, GVHD progresses and may affect a variety of organ systems.
The diagnosis should be suspected in any young infant with erythroderma and frequent infections, chronic diarrhea, and/or failure to thrive. If present, a family history of prior early infant deaths is helpful because many forms of immunodeficiency are familial. Skin biopsy can be very useful in confirming the diagnosis. The histopathologic changes of GVHD are usually graded by severity (from I to IV).61 Most authors agree that minimum criteria for the histopathologic diagnosis of GVHD include the presence of epidermal lymphocytes, dyskeratosis, and satellite cell necrosis. The latter refers to the finding of a lymphocyte apposed to an eosinophilic keratinocyte (dyskeratotic epidermal cell) within the epidermis. Similar changes may also result from the conditioning therapy utilized for some patients before bone marrow transplantation, as well as from the effect of certain viruses.62 In some cases of GVHD caused by maternal engraftment, a spongiotic dermatitis may predominate.60,63 Multiple skin biopsies obtained over days or weeks may be necessary if the diagnosis is strongly suspected, but without confirmatory histopathology.4 Immunophenotyping of skin biopsies has been used to complement the characteristic findings on routine histopathology described previously.64 It should be emphasized, however, that immunophenotyping alone should not be considered diagnostic of GVHD, and that the lack of such features should not discount the presence of compatible clinical and histopathologic findings of this condition.
Because the diagnosis of GVHD in the absence of a known organ or bone marrow transplant implies a severe immunodeficiency, a complete evaluation of the immune system should be undertaken. Although lymphopenia is characteristic of severe T-cell or severe combined immunodeficiency, the lymphocyte count in the blood may be normal or even elevated because of the presence of circulating maternal lymphocytes. Eosinophilia is often present. Although small numbers of circulating maternal cells are considered a normal finding in the first few weeks of life, the presence of large numbers of maternal lymphocytes is highly suggestive of an underlying immunodeficiency, and in the setting of erythroderma, strongly suggests the diagnosis of GVHD. Maternal engraftment can be documented by performing analysis for chimerism to demonstrate maternal T lymphocytes.6
The differential diagnosis is generally confined to other more common conditions, such as seborrheic dermatitis, infantile atopic dermatitis, or the unusual viral or drug eruptions.
The course of this condition can be variable and depends on the degree of organ involvement, as well as the severity of the immunodeficiency. In most instances, the skin can be treated with bland emollients, as well as low- or mid-potency topical corticosteroid preparations. More severe reactions, particularly those with systemic manifestations, may require systemic corticosteroid therapy, cytotoxic drugs, or monoclonal antibodies.59 For subacute or chronic skin disease, in the absence of systemic involvement, phototherapy, either PUVA or narrowband UVB, has proven useful both as a primary therapy and as a means to decrease or discontinue altogether the use of systemic therapies.65,66 Such therapy is usually reserved for older patients.
The skin manifestations of GVHD occurring after transplantation, or rarely resulting from transfusions, have been well-characterized and are varied. They include an acute phase with morbilliform erythema, papular dermatitis, diffuse erythroderma, or in severe cases, diffuse bullae or frank necrosis, such as are seen in toxic epidermal necrolysis. Often, such skin changes begin on the head and neck and extend in a caudal fashion. The palms and soles are predominantly involved. Chronic changes (more than 100 days post-transplantation) may include oral mucous membrane changes, nail dystrophy, and localized or diffuse lichenoid (flat-topped) papules. Extracutaneous features of the disease are primarily gastrointestinal. Hepatitis may be found, as well as varying degrees of diarrhea. Skin biopsies of representative lesions will generally reveal features of GVHD, as described above.
Wiskott–aldrich syndrome
Wiskott–Aldrich syndrome (WAS) is a rare X-linked recessive disorder characterized by recurrent pyogenic infections, bleeding due to thrombocytopenia, and recalcitrant eczematous dermatitis. The gene for WAS has been mapped to chromosome Xp1128 and encodes for WAS protein (WASP), a cytoplasmic protein constitutively expressed in all hematopoietic stem cell-derived lineages.67
The most consistent finding in WAS is thrombocytopenia with small platelets. Because it is present from birth, initial presenting signs in the majority of patients are related complications such as petechiae, purpura, epistaxis, gastrointestinal bleeding, or intracranial hemorrhage.68 Recurrent bacterial infections begin in infancy and include otitis media, pneumonia, infectious diarrhea, sinusitis, meningitis, and septicemia. There is also increased susceptibility to viruses and Pneumocystis carinii. There is also an increased risk of autoimmune disease and malignancy, mainly lymphoreticular malignancies.52 Laboratory examination may also reveal characteristic elevated IgA and IgE and decreased IgM.67
Dermatitis usually develops during the first few months of life and meets the criteria for atopic dermatitis (see Fig. 15.16). The face, scalp, and flexural areas are usually the most severely involved, although involvement can be widespread. Of patients with WAS, 80–100% will have eczematous skin disease, which can be widespread and difficult to control.69 Excoriated areas often have associated petechiae or purpura, and secondary bacterial infection of eczematous lesions is common, as are eczema herpeticum and molluscum.52 Other infections of the skin may be present independently, and may include impetigo, cellulitis, and abscesses.
Treatment is largely symptomatic and includes antibiotics and intravenous immunoglobulin in selected cases. Splenectomy may be used to treat thrombocytopenia, although there is an increased risk of post-splenectomy sepsis. Platelet transfusion is reserved for cases of life-threatening hemorrhage.52 Blood products should be irradiated. At present, bone marrow transplant or cord blood stem cell transplant is the only curative therapy for WAS.68
Hyperimmunoglobulin E syndrome
Hyperimmunoglobulin E (hyper-IgE) syndrome (HIES), formerly also known as Job syndrome, consists of a severe dermatitis with recurrent abscess formation and recurrent sinopulmonary infections associated with markedly elevated serum IgE levels.70 Inheritance follows an autosomal dominant pattern with variable penetrance. STAT3 mutations are the cause of the majority of autosomal dominant HIES.3
The severe dermatitis may be present from birth or early childhood, and as in atopic dermatitis, the rash is typically pruritic and often lichenified, although its distribution may be atypical for true atopic dermatitis (see Fig. 15.17). A distinct papulopustular eruption of the face and scalp has been reported in the first year of life (see Fig. 10.28).71 A history of allergic diseases, absence of bone or joint abnormalities, or deep-seated abscesses suggests atopic dermatitis rather than HIES.72
Cutaneous infections are frequent and start in infancy. They may take the form of crusted plaques, pustules, furuncles, cellulitis, lymphangitis, or abscesses. The abscesses may be erythematous and tender, or may be fluctuant masses which are neither hot nor tender and are not associated with systemic symptoms (‘cold abscesses’). They are filled with pus that grows Staphylococcus aureus on culture. This diagnosis should be considered in children with recurrent abscesses complicating chronic eczema. Skin biopsy reveals an eosinophilic infiltrate similar to that seen in eosinophilic pustular folliculitis. Infection with nonbacterial pathogens may also occur, especially Candida infections of the mouth, nails, and skin.73
In addition to skin infections, sinopulmonary and bone infections are common. S. aureus is also the most common organism for these infections. Skeletal abnormalities, including hyperextensible joints, are seen in hyper-IgE syndrome.73 Coarse facial features manifested by a prominent forehead and a broad nasal bridge are typically not seen at birth but appear later in childhood. Patients with viral infections such as warts, molluscum, or severe/recurrent HSV with atopic dermatitis should be considered for DOCK8 deficiency. Such patients also have an increased risk for malignancies such as squamous cell carcinoma and lymphoma.2,3
The management of hyper-IgE syndrome includes prophylactic antibiotics to prevent S. aureus infections. The eczematous dermatitis may require treatment with topical corticosteroids to reduce inflammation and antihistamines to control pruritus. Incision and drainage may be required for abscesses.74
Chronic granulomatous disease
Chronic granulomatous disease (CGD) represents a group of genetic disorders in which impaired intracellular microbial killing by phagocytes leads to recurrent bacterial and fungal infections and granuloma formation. Defects in the nicotinamide dinucleotide phosphate (NADPH) oxidase complex result in failure to generate superoxide radicals during the respiratory burst, leading to an inability of phagocytic lymphocytes to kill intracellular bacteria and fungi, especially Aspergillus spp. CGD can be caused by mutations in any of the five structural genes of NADPH oxidase. The most common type is X-linked recessive, and the remainder are autosomal recessive.75 Of genetic mutations, 70% are of the CYBB gene resulting in the X-linked form of CGD.76
The earliest lesions are usually staphylococcal infections of the skin around the nose and mouth, which may be present at birth. Skin abscesses, usually caused by S. aureus, occur in 42% of patients. Purulent inflammatory reactions may occur at sites of minor cutaneous trauma or sites of regional lymph node drainage with suppurative lymphadenitis. There are also reports of cutaneous granulomas containing the typical pigmented macrophages also seen in visceral granulomas.77
The extracutaneous organs most frequently involved are the lymph nodes, lungs, and liver. Bronchopneumonia is the most prevalent infection, with abscess formation and empyema as frequent complications. These patients have hepatosplenomegaly, and granulomas of the liver and spleen are common. Hepatic abscesses are usually caused by S. aureus and may require surgical drainage.
In the past, the screening test for CGD is the nitroblue tetrazolium (NBT) reduction assay. The dihydrorhodamine (DHR) test has become the more frequently used testing method. This measures NADPH oxidase activity by flow cytometry.76 Management includes antibacterial and antifungal prophylaxis and treatment. Patients with all forms of CGD have shown clinical improvement after administration of subcutaneous recombinant IFN-γ.78 Allogeneic hematopoietic stem cell transplantation is curative.
Eosinophilic gastroenteritis
Eosinophilic gastroenteritis was first reported by Waldman and colleagues79,80 who called the condition allergic gastroenteropathy. Cutaneous features included edema, particularly over the face, as well as generalized atopic dermatitis. The extracutaneous manifestations are striking and include growth retardation, extreme hypoalbuminemia, hypogammaglobulinemia, anemia, and eosinophilia, as well as mild gastrointestinal symptoms consisting of intermittent diarrhea or vomiting after the ingestion of certain foods, and excessive loss of protein into the gastrointestinal tract.81 Asthma and allergic rhinitis may also be present. Diagnosis is confirmed with intestinal biopsies, which reveal mucosal eosinophilia. The disease is now subclassified into protein-sensitive and idiopathic forms.82 The protein-sensitive form is more common, responds to dietary restriction of cow’s milk or soy protein, and ultimately resolves with time. The idiopathic form requires steroid therapy to control symptoms. The dermatitis improves rapidly and dramatically with resolution of the other symptoms when the dermatitis is aggressively treated.
Metabolic disorders
Disorders of metabolism (Table 18.2) often present during the neonatal period and may exhibit skin manifestations. Rarely, metabolic diseases are associated with erythroderma, either shortly after birth, or later resulting from subsequent therapeutic dietary restrictions. Although cutaneous manifestations may be seen with such diseases, many metabolic disorders manifest with feeding or neurological abnormalities, or through biochemical abnormalities alone.83,84 Lysosomal storage diseases may present with coarse facial features as well as organomegaly.
TABLE 18.2
Classification of inherited metabolic disorders
| Organic acid disorders | Fatty acid disorders | Amino acid disorders | Others |
| BKT | MCAD | PKU | BIOT |
| GA 1 | VLCAD | MSUD | GALT |
| HMG | LCHAD | HCY | CF |
| MCD | CUD | CIT | LSD |
| IVA | ASA | ||
| MUT | |||
| 3MCC | |||
| Cb1 A, B | |||
| PROP |
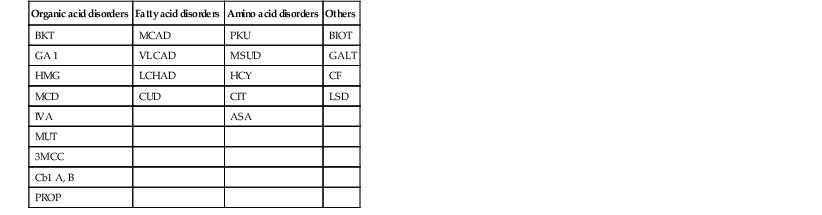
BKT, b-ketothiolase deficiency; GA-1, glutaric acidemia, type 1; HMG, 3-OH 3-CH3 glutaric aciduria; MCD, multiple carboxylase deficiency; IVA, isovaleric acidemia; MUT, methylmalonic acidemia (mutase deficiency); 3MCC, 3-methylcrotonyl-CoA carboxylase deficiency; Cb1 A, B, methylmalonic acidemia (cobalamin 1 A, B); PROP, propionic acidemia; MCAD, medium-chain acyl-CoA dehydrogenase deficiency; VLCAD, very long-chain acyl-CoA dehydrogenase deficiency; LCHAD, long-chain L-3-OH acyl-CoA dehydrogenase deficiency; CUD, carnitine uptake defect; PKU, phenylketonuria; MSUD, maple syrup urine disease; HCY, homocystinuria; CIT, citrullinemia; ASA, argininosuccinic-CoA lyase deficiency (argininosuccinic aciduria); BIOT, biotinidase deficiency; GALT, classic galactosemia; CF, cystic fibrosis; LSD, lysosomal storage disease.
Modified from Seashore MR, Seashore CJ. Newborn screening and the pediatric practioner. Semin Perinatol 2005; 29:182–8.
Many metabolic conditions have been well-described over the last several decades and are now associated with documented molecular bases. Due to the similarity of skin findings seen with this group of disorders, the term acrodermatitis dysmetabolica has been suggested.85 This describes diffuse and/or periorificial scale and erythema.
Organic acid disorders
Methylmalonic acidemia
Methylmalonic acidemia (MMA) is an inborn error of metabolism inherited in an autosomal recessive manner.86 It includes a group of diseases caused by a defect in the metabolism of branched-chain amino acids, which results in the accumulation of methylmalonic acid. Although some cases of methylmalonic acidemia, especially those caused by a cobalamin F and cobalamin C type of defect, have presented with a dermatitis similar to that seen in acrodermatitis enteropathica,86 more commonly the dermatitis begins after the institution of dietary restrictions.87 In either case, the appearance is similar and in a primarily periorificial location. There are erythema and ulceration in the corners of the mouth and genital areas. A more diffuse dermatitis resembling SSSS has also been described. Extracutaneous manifestations can include poor feeding, vomiting, hypotonia, and acidosis, often leading to coma and death.
Assays for serum amino acid and metabolic analysis of cultured skin fibroblasts from affected patients confirm the diagnosis. Urinary organic acids should also be examined for elevations of methylmalonate and homocystine. Enzyme analysis in white cells or fibroblasts or mutation analysis can be used for confirmation of this diagnosis.88 Skin biopsy shows vacuolar dermatitis with dyskeratotic keratinocytes, mild psoriasiform changes, and epidermal pallor as seen in acrodermatitis enteropathica. There is a lymphocytic perivascular infiltrate within the dermis, as well as areas of orthokeratosis and parakeratosis, with spongiosis of the epidermis. The differential diagnosis of this disease includes other metabolic and nutritional deficiency states, such as acrodermatitis enteropathica, other aminoacidurias, and biotinidase deficiency. Management consists of dietary restrictions of branched-chain amino acids, specifically isoleucine and valine, and, in those cases marked by cobalamin deficiency, supplementation with cobalamin. The prognosis of MMA is guarded, with many patients remaining severely impaired neurologically in spite of aggressive dietary support.
Fatty acid disorders
Essential fatty acid deficiency
Essential fatty acid (EFA) deficiency was seen more frequently before 1975, in patients on parenteral hyperalimentation, before the need for EFA supplementation was recognized. It is now an extremely unusual condition though reports continue to exist in this setting.89 It presents with a diffuse fine desquamation and mild or even absent erythema. The condition sometimes occurs in patients with severe fat malabsorption and may be one of the causes of the dermatitis of cystic fibrosis.
Amino acid disorders
Maple syrup urine disease and other inborn errors of metabolism
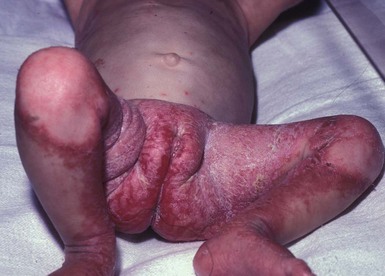
In maple syrup urine disease (MSUD),90–92 diffuse exfoliative erythroderma has been well described. MSUD is another inborn error of metabolism in which the metabolism of branched-chain amino acids is defective. There is an abnormality in the degradation of the branched-chain amino acids causing diagnostic elevations of isoleucine, leucine, and valine in the serum, as well as in urine and cultured tissue fibroblasts. Again, enzyme analysis of WBC or fibroblasts or mutation analysis will confirm this diagnosis.88 An erythematous scaling eruption that becomes erosive begins in a primarily periorificial distribution within days after initiating dietary therapy. The eruption is similar to that seen in acrodermatitis enteropathica. It can generalize, however. Such infants may also present with poor feeding, vomiting, lethargy, and seizures. Death may occur if the disorder is not promptly recognized. The skin disease may be caused by low isoleucine levels due to the dietary restrictions required for the disease. A similar, albeit milder dermatitis has been induced in infants fed diets deficient in isoleucine,93 and similar eruptions have also been noted in citrullinemia,94 carbamoyl phosphate synthetase deficiency, and argininosuccinicaciduria.95 The cause of the dermatitis in each of these cases is also presumed to be caused by an abnormality of branched-chain amino acids, such as isoleucine. Histopathology of skin biopsies may show a very superficial perivascular lymphohistiocytic infiltrate with erosion of the outer epidermis, again as may be seen in skin biopsies from patients with other inborn errors of metabolism. The differential diagnosis includes other organic acidemias in which periorificial dermatitis may occur after initiation of dietary therapy, such as propionic acidemia and methylmalonic acidemia (Fig. 18.13). Treatment requires diligent attention to dietary restrictions. The diet must be liberalized so that sufficient branched-chain amino acids are delivered to raise plasma concentrations above the subnormal range.
Cystic fibrosis
Infants with cystic fibrosis (CF) can develop widespread, scaly erythematous lesions as a manifestation of global malnutrition during the first 3 or 4 months of life, and these are occasionally the initial presentation of CF.96–99 The dermatitis is variable. A diffuse erythematous papular dermatitis, diffuse desquamating erythema, a distinctly periorificial erythema and scaling or, commonly, a generalized desquamative erythroderma may be seen (Fig. 18.14). The dermatitis does not respond completely to treatment with topical corticosteroids or antifungals. In contrast to infants affected with classic acrodermatitis enteropathica, infants with cystic fibrosis typically lack paronychial involvement. Affected infants often have depressions of zinc levels, increased liver transaminases, and normal or slightly depressed levels of alkaline phosphatase. The dermatitis of CF occasionally clears with zinc therapy, but does so more reliably with appropriate enzyme replacement and nutritional supplements. The etiology of the dermatitis is likely multifactorial with data suggesting zinc, essential fatty acid, and protein deficiencies.99
Testing for metabolic diseases
With the abundance of diagnostic testing for metabolic diseases, many are now currently included in mandated state newborn screening programs. Newborn screening began with testing for phenylketonuria (PKU) in 1940 and – in addition to hypothyroidism – might include only galactosemia routinely. Testing availability varies by locality. The Secretary of the US Department of Health and Human Services Advisory Committee on Heritable Disorders in Newborns and Children has suggested a uniform screening panel of 31 core disorders.100 The importance of such screening is intervention at a time in advance of the often devastating consequences of these disorders. Many of these disorders will present during the first week of life with metabolic acidosis and/or hyperammonemia, as well as altered neurologic status or emesis. Such findings are nonspecific and may be initially diagnosed as sepsis. Severe mental status alteration, however, should be a clue to a primary metabolic disease. Extreme elevations in the anion gap will suggest organic acidemias, such as propionic acidemia, methylmalonic academia, isovaleric acidemia, or multiple carboxylase deficiency. Elevations in serum ammonia levels may be a sign of the organic acid disorders, as well as urea cycle abnormalities, mitochondrial diseases, or disorders of fatty acid β-oxidation. Table 18.3 lists diseases to be considered when a clinical or laboratory finding is present, along with suggested testing.
TABLE 18.3
Clinical clues to metabolic disorders
| Clinical sign/Lab finding | Differential diagnosis | Suggested testing |
| Metabolic acidosis without lactic acidosis or ketonuria | Pyroglutamic aciduria | Urine organic acids Molecular testing |
| Metabolic acidosis without lactic acidosis, with ketonuria | Organic acidemias | Urine organic acids Plasma acylcarnitine profile Molecular testing |
| Lactic acidosis with ketonuria | Pyruvate carboxylase deficiency (severe) Glycogen storage disease I |
Urine organic acids Plasma amino acids Molecular testing |
| Lactic acidosis, without ketonuria | Pyruvate carboxylase deficiency (mild) Pyruvate dehydrogenase deficiency Mitochondrial dysfunction Hypoxia or hypoperfusion |
Urine organic acid analysis Molecular testing |
| Hyperammonemia, moderate–severe | Urea cycle disorders Organic acidemias Transient hyperammonemia of newborn HHH syndrome Lysinuric protein intolerance |
Plasma amino acids Plasma acylcarnitine profile Urine organic acids Molecular testing |
| Hyperammonemia, mild–moderate | Organic acidemias Severe pyruvate carboxylase deficiency Fatty acid oxidation disorders Carnitine disorders Non-fasting sample |
Urine organic acids Plasma amino acids Plasma acylcarnitine profile Plasma carnitine, free and total Molecular testing |
| Hypoglycemia | Fatty acid oxidation disorders Carnitine disorders Glycogen storage diseases Hyperinsulinism |
Urine organic acids Plasma acylcarnitine profile Plasma carnitine, free and total Molecular testing |
| Seizures | Non-ketotic hyperglycinemia Sulfite oxidase deficiency Molybdenum cofactor deficiency Pyridoxal phosphate sensitive encephalopathy Serine deficiency disorder GLUT-1 deficiency |
Plasma amino acids CSF amino acids Urine sulfite (dipstick) Blood and CSF glucose Molecular testing |
| Liver disease | Galactosemia Disorders of bile acid synthesis |
Galactose-1-phosphate (RBC) GALT enzyme assay (RBC) Molecular testing |
| Cataracts | Galactosemia, mitochondrial dysfunction | Galactose and galactose-1-phosphate. GALT enzyme assay, urine organic acids Molecular testing |
| Neutropenia | Barth syndrome, glycogen storage disease 1 b, organic acidemias, abnormalities of folate and B12 metabolism | Urine organic acids, plasma total homocysteine, plasma amino acids Molecular testing |
| Cardiomyopathy, arrhythmia | Long chain fatty acid oxidation disorders, carnitine disorders, Barth syndrome, mitochondrial dysfunction | Plasma acylcarnitine profile Plasma carnitine, free and total Urine organic acids Molecular testing |
Many of these disorders now have well-described molecular diagnoses. Most are inherited in either autosomal recessive or X-linked recessive fashion. Basic screening tests include plasma amino acid levels and urine organic acids. Screening for acylcarnitine levels can provide clues to both organic acidurias and fatty acid oxidation disorders, such as medium-chain acyl-CoA dehydrogenase deficiency (MCAD). Serum amino acid studies will diagnose disorders such as maple syrup urine disease and homocystinuria. More specific testing is required for disorders such as biotinidase deficiency, galactosemia, cystic fibrosis, peroxisomal, or lysosomal storage diseases. As noted above, specific enzyme analysis and confirmatory testing is available for most biochemical defects.88 Clinicians caring for infants should be aware of the often broad and nonspecific presentation of metabolic disorders and work closely with genetic/metabolic consultants. The goal of prompt diagnosis and treatment is to attempt to avoid the severe long-term sequelae of neurologic deficits or death.
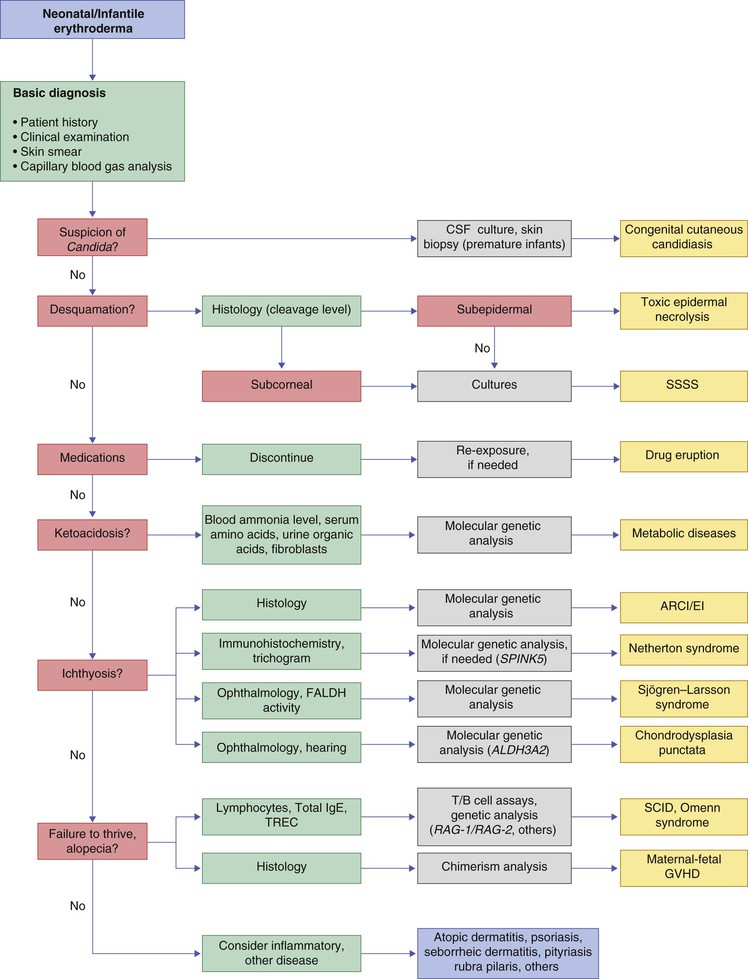
Evaluation and management of the red scaly baby
The history and physical examination may provide important diagnostic clues to the etiology of erythroderma. Specific parameters that may be of value in determining an underlying cause include: congenital onset, skin induration, and the presence of large scaling plaques, alopecia with or without hair dysplasia, evolution, response to corticosteroid therapy, presence of infections, and failure to thrive.6,13,101 If the infant appears to have atopic or seborrheic dermatitis, then appropriate therapy can be instituted. If there is no response to therapy, or if the infant appears to be systemically ill, fails to thrive, or shows other evidence of a more generalized disease, a more comprehensive evaluation should be undertaken (Boxes 18.1, 18.2 and Fig. 18.15). Laboratory tests useful in evaluating erythroderma in neonates and infants are outlined in Box 18.2. The selection of which tests to perform depends on which disease(s) are most suspected. Appropriate smears and cultures for fungal, bacterial, or viral disease should be performed if infection is suspected. A chest radiograph may be useful to evaluate the thymic shadow, which may be absent in neonates with SCID. A CBC showing lymphopenia in a neonate can suggest a primary immunodeficiency. IgG levels, if obtained during the first months of life, are reflective of maternal values. These may, however, be useful in infants over 6 months of age. Liver function tests may be indicated in primary or secondary nutritional disease, such as cystic fibrosis. In the latter, one would expect elevations of the transaminases and severely decreased serum albumin. Serum amino and urine organic acids are necessary to screen for suspected cases of primary metabolic diseases, such as the aminoacidurias or biotin deficiencies. A biotinidase level can be obtained if biotin deficiency is suspected. Skin biopsy can be useful for direct histopathologic examination of representative lesions, and fibroblast culture can help in definitive diagnosis of several metabolic diseases.4 Lastly, specific molecular testing for genodermatoses, immunodeficiencies, or metabolic diseases is available as discussed above. Consultation with appropriate specialists may be necessary to decide which of these tests are best pursued.
When an infant presents with erythroderma, immediate attention to fluid and electrolytes is paramount. For example, infants with ichthyosis can develop life-threatening hypernatremic dehydration. Infectious complications, primarily bacterial or fungal, must also be considered. These infants need a warm, humid environment to minimize their metabolic demands. Topical therapy consisting of bland emollients such as petrolatum or Aquaphor® is helpful in minimizing transepidermal water loss and may decrease potential infectious complications.102
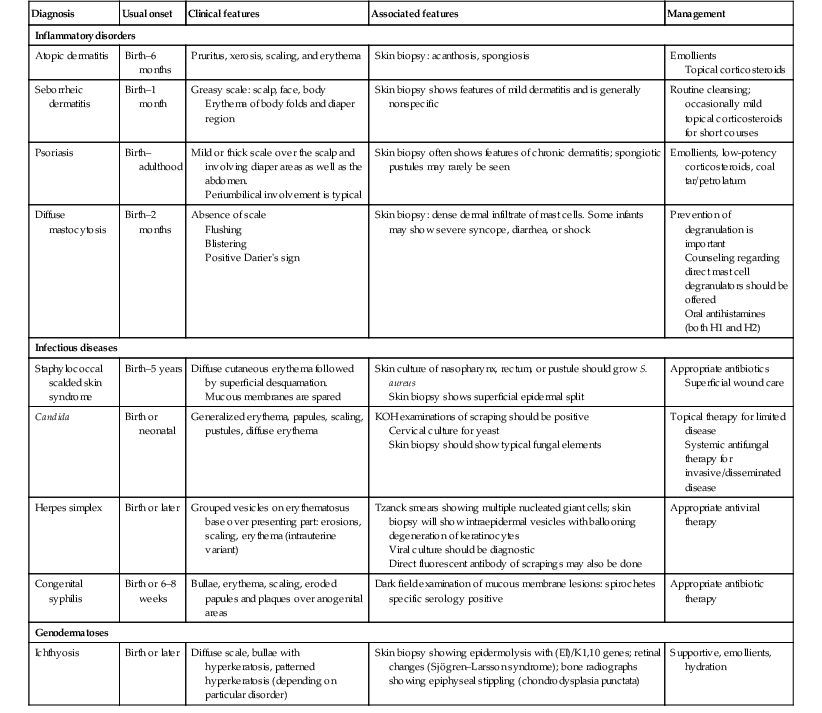
A summary of the evaluation and management of many of the disorders is found in Table 18.4.
TABLE 18.4
Evaluation and management of disorders
| Diagnosis | Usual onset | Clinical features | Associated features | Management |
| Inflammatory disorders | ||||
| Atopic dermatitis | Birth–6 months | Pruritus, xerosis, scaling, and erythema | Skin biopsy: acanthosis, spongiosis | Emollients Topical corticosteroids |
| Seborrheic dermatitis | Birth–1 month | Greasy scale: scalp, face, body Erythema of body folds and diaper region |
Skin biopsy shows features of mild dermatitis and is generally nonspecific | Routine cleansing; occasionally mild topical corticosteroids for short courses |
| Psoriasis | Birth–adulthood | Mild or thick scale over the scalp and involving diaper areas as well as the abdomen. Periumbilical involvement is typical |
Skin biopsy often shows features of chronic dermatitis; spongiotic pustules may rarely be seen | Emollients, low-potency corticosteroids, coal tar/petrolatum |
| Diffuse mastocytosis | Birth–2 months | Absence of scale Flushing Blistering Positive Darier’s sign |
Skin biopsy: dense dermal infiltrate of mast cells. Some infants may show severe syncope, diarrhea, or shock | Prevention of degranulation is important Counseling regarding direct mast cell degranulators should be offered Oral antihistamines (both H1 and H2) |
| Infectious diseases | ||||
| Staphylococcal scalded skin syndrome | Birth–5 years | Diffuse cutaneous erythema followed by superficial desquamation. Mucous membranes are spared |
Skin culture of nasopharynx, rectum, or pustule should grow S. aureus Skin biopsy shows superficial epidermal split |
Appropriate antibiotics Superficial wound care |
| Candida | Birth or neonatal | Generalized erythema, papules, scaling, pustules, diffuse erythema | KOH examinations of scraping should be positive Cervical culture for yeast Skin biopsy should show typical fungal elements |
Topical therapy for limited disease Systemic antifungal therapy for invasive/disseminated disease |
| Herpes simplex | Birth or later | Grouped vesicles on erythematosus base over presenting part: erosions, scaling, erythema (intrauterine variant) | Tzanck smears showing multiple nucleated giant cells; skin biopsy will show intraepidermal vesicles with ballooning degeneration of keratinocytes Viral culture should be diagnostic Direct fluorescent antibody of scrapings may also be done |
Appropriate antiviral therapy |
| Congenital syphilis | Birth or 6–8 weeks | Bullae, erythema, scaling, eroded papules and plaques over anogenital areas | Dark field examination of mucous membrane lesions: spirochetes specific serology positive | Appropriate antibiotic therapy |
| Genodermatoses | ||||
| Ichthyosis | Birth or later | Diffuse scale, bullae with hyperkeratosis, patterned hyperkeratosis (depending on particular disorder) | Skin biopsy showing epidermolysis with (EI)/K1,10 genes; retinal changes (Sjögren–Larsson syndrome); bone radiographs showing epiphyseal stippling (chondrodysplasia punctata) | Supportive, emollients, hydration |
Access the full reference list at ExpertConsult.com 
