2 EPITHELIAL GLANDS
Development of epithelial glands
Most glands develop as epithelial outgrowths into the underlying connective tissue (Figure 2-1). Exocrine glands remain connected to the surface of the epithelium by an excretory duct that transports the secretory product to the outside. Endocrine glands lack an excretory duct, and their product is released into the blood circulation.
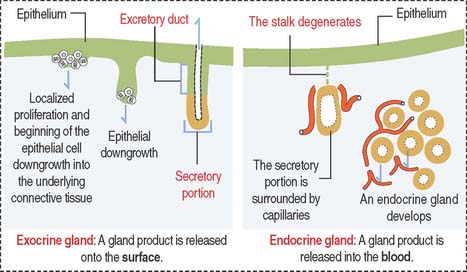
Endocrine glands are surrounded by fenestrated capillaries and commonly store the secretions they synthesize and release after stimulation by chemical or electrical signals. Exocrine and endocrine glands can be found together (for example, in the pancreas), as separate structures in endocrine organs (thyroid, parathyroid), or as single cells (enteroendocrine cells). Endocrine glands will be studied later in Chapter 18, Neuroendocrine System, and Chapter 19, Endocrine System.
Classification of epithelial glands
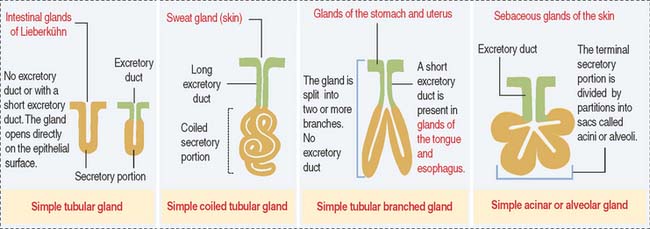
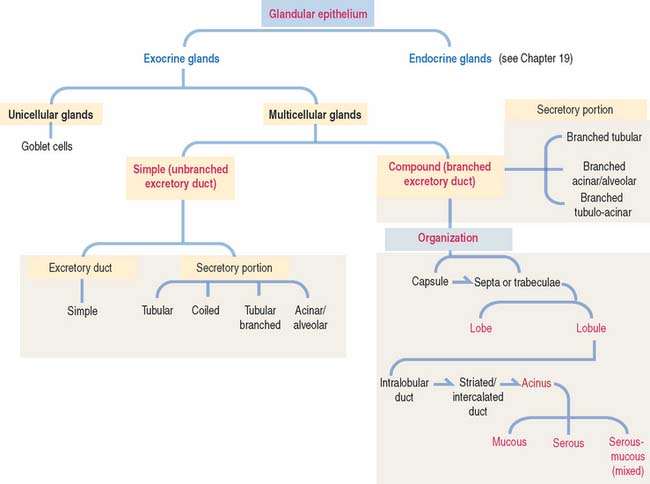
Glands are classified according to the type of excretory duct into simple and branched (also called compound) glands. The gland can be simple (Figure 2-2) when the excretory duct is unbranched. The gland can be branched when the excretory duct subdivides (Figure 2-3).
The secretory portion can be unicellular or multicellular
An exocrine gland has two components: a secretory portion and an excretory duct. The secretory portion of a gland may be composed of one cell type (unicellular, for example, goblet cells in the respiratory epithelium and intestine) or many cells (multicellular).
According to the shape of the secretory portion (see Figures 2-2 and 2-3), glands can be tubular, coiled, or alveolar (Latin alveolus, small hollow sac; plural alveoli), also called acinar (Latin acinus, grape; plural acini).
Shape of the secretory portion
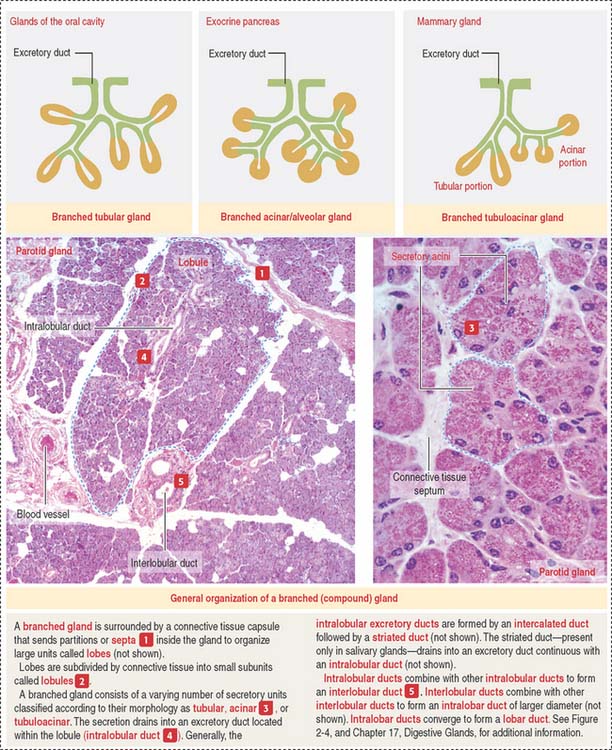
A branched gland (Figure 2-4) is surrounded by a capsule. Septa or trabeculae extend from the capsule into the glandular tissue. Large septa divide the gland into a number of lobes. Branches from the septa separating adjacent lobes divide the lobes into smaller compartments called lobules.
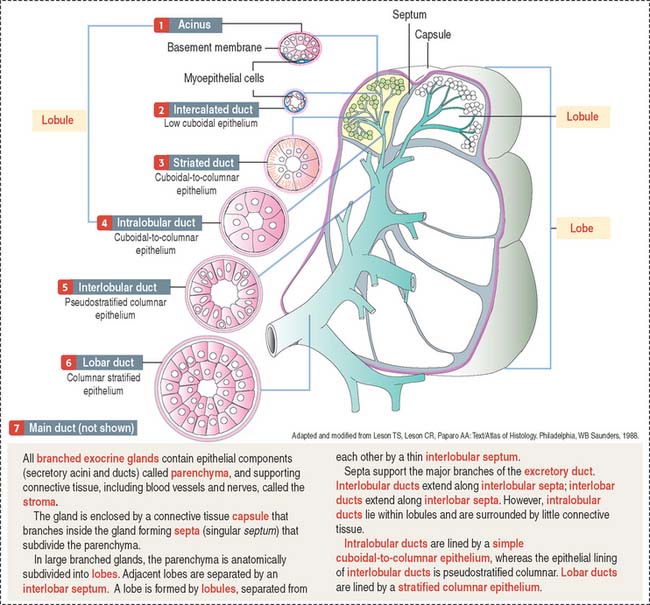
During development, a main excretory duct gives rise to branches that lie either between (interlobar) or within lobes (intralobar). Small branches derived from each of these ducts generate small subdivisions that constitute the lobule of a gland. These branches can be found first between lobules (interlobular) and within lobules (intralobular). Additional details are presented in Chapter 17, Digestive Glands.
Types of secretion
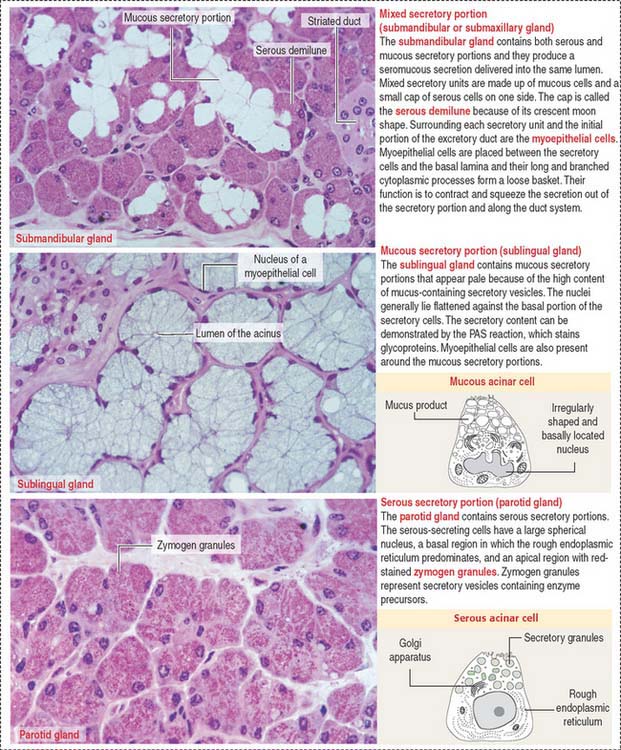
Based on the type of secretion, exocrine glands can be classified as mucous glands, when their products are rich in glycoproteins and water; serous glands, with secretions enriched with proteins and water; and mixed glands, which contain both mucous and serous cells (Figure 2-5).
CYTOMEMBRANES
Plasma membrane
A review of major concepts of cytomembranes and organelles and their clinical relevance is presented in this chapter. Epithelial glands are a convenient topic for this integration. We initiate the review by addressing the structural and biochemical characteristics of the plasma membrane. Additional information related to plasma membrane–mediated cell signaling is presented in Chapter 3, Cell Signaling.
The plasma membrane determines the structural and functional boundaries of a cell. Intracellular membranes, called cytomembranes, separate diverse cellular processes into compartments known as organelles. The nucleus, mitochondria, peroxisomes, and lysosomes are membrane-bound organelles; lipids and glycogen are not membrane-bound and are known as inclusions.
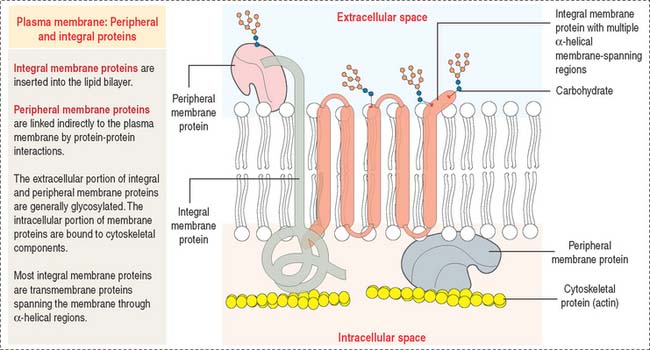
The plasma membrane consists of both lipids and proteins. The phospholipid bilayer is the fundamental structure of the membrane and forms a bilayer barrier between two aqueous compartments: the extracellular and intracellular compartments. Proteins are embedded within the phospholipid bilayer and carry out specific functions of the plasma membrane such as cell-cell recognition and selective transport of molecules (see Box 2-A).
Box 2-A Lipid rafts
Phospholipid bilayer
The four major phospholipids of plasma membranes are phosphatidylcholine, phosphatidylethanolamine, phosphatidylserine, and sphingomyelin (Figure 2-7). They represent more than one half the lipid of most membranes. A fifth phospholipid, phosphatidylinositol, is localized to the inner leaflet of the plasma membrane.
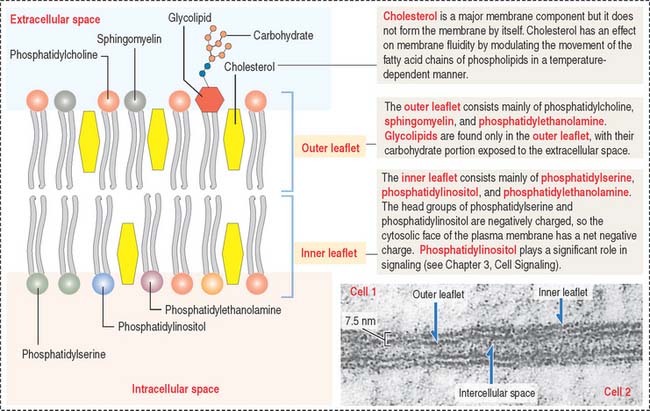
Two general aspects of the phospholipid bilayer are important to remember:
Membrane proteins
Most plasma membranes consist of about 50% lipid and 50% protein (Figure 2-8). The carbohydrate component of glycolipids and glycoproteins represents 5% to 10% of the membrane mass. The surface of a plasma membrane is coated by a glycocalyx (see Box 2-B).
Box 2-B Glycocalyx
Restrictions in the mobility of membrane proteins are responsible for the polarized nature of epithelial cells, divided into distinct apical and basolateral domains that differ in protein composition and function. Tight junctions between adjacent epithelial cells (discussed in Chapter 1, Epithelium) not only seal the space between cells but also serve as barriers to the diffusion of proteins and lipids between the apical and basolateral domains.
Portions of integral membrane proteins are inserted into the lipid bilayer. They can only be released by solubilization using detergents. Detergents are chemical agents that contain both hydrophobic and hydrophilic groups. The hydrophobic domains of the detergent penetrate the membrane lipids and bind to the membrane-inserted hydrophobic portion of the protein. The hydrophilic domains combine with the protein, forming aqueous-soluble detergent-protein complexes.
Freeze-fracture: Differences between a surface and a face
Figure 2-9 indicates the nomenclature for the identification of surfaces and faces in electron micrographs of freeze-fracture preparations.
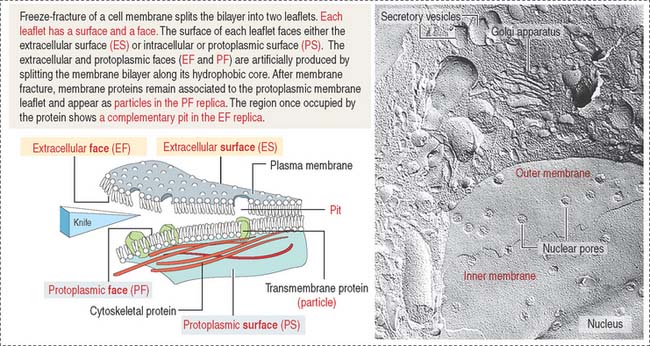
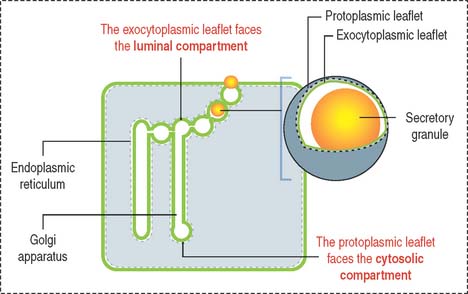
The face of the membrane leaflet looking to the extracellular space (the exocytoplasmic leaflet in the illustration) is labeled EF, for extracellular face. Similarly, the face of the leaflet facing the protoplasmic space (identified as a protoplasmic leaflet) is PF, for protoplasmic face.
Transporter and channel proteins
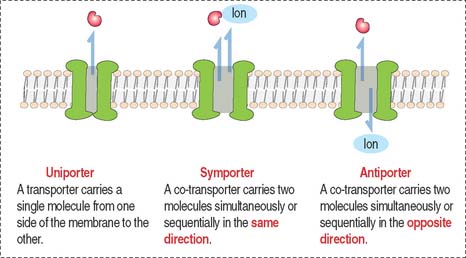
Other biological molecules (such as glucose, charged molecules, and small ions—H+, Na+, K+, and Cl−) are unable to dissolve in the hydrophobic interior of the phospholipid bilayer. They require the help of specific transport proteins (Figure 2-10) and channel proteins, which facilitate the diffusion of most biological molecules.
Endoplasmic reticulum
The endoplasmic reticulum is an interconnected network of membrane-bound channels within the cytoplasm, part of the cytomembrane system and distinct from the plasma membrane.
The smooth endoplasmic reticulum lacks ribosomes and is generally in proximity to deposits of glycogen and lipids in the cytoplasm. The smooth endoplasmic reticulum has an important role in detoxification reactions required for the conversion of harmful lipid-soluble or water-insoluble substances into water-soluble compounds more convenient for discharge by the kidneys. It also participates in steroidogenesis (see Chapter 19, Endocrine System).
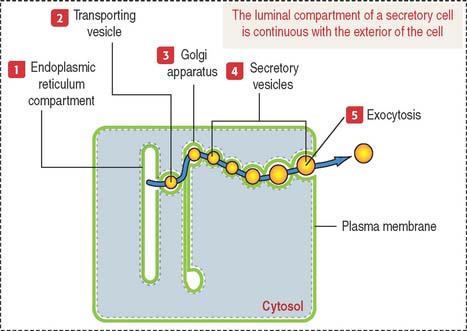
Products released into the luminal compartment of the endoplasmic reticulum are transported to the Golgi apparatus by a transporting vesicle and eventually to the exterior of the cell by exocytosis. One can visualize the sequence in which the lumen of the cytomembrane system is interconnected and remains as such in an imaginary stage; you can visualize that the luminal compartment of a secretory cell is continuous with the exterior of the cell (Figure 2-11). The surrounding space is the cytosolic compartment in which soluble proteins, cytoskeletal components, and organelles are present.
Now, let us visualize the membrane of each component of the cytomembrane system as consisting of two leaflets (Figure 2-12):
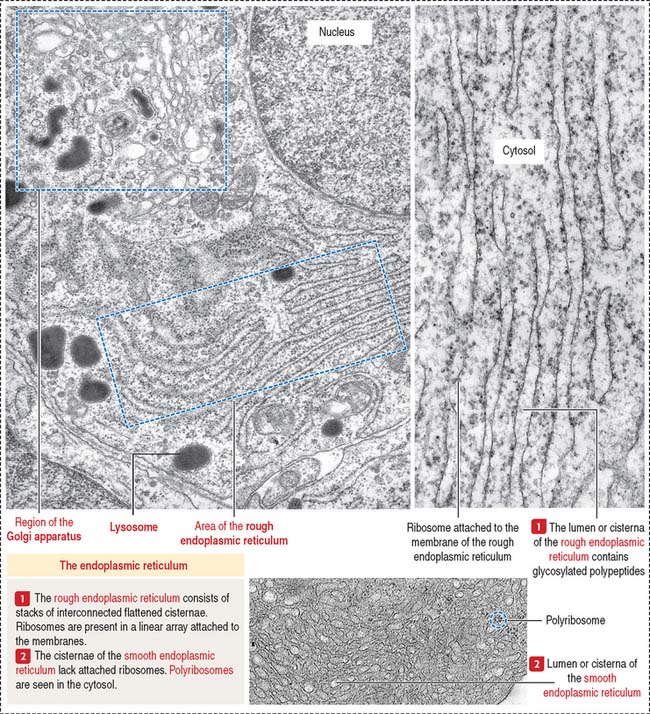
Rough endoplasmic reticulum
The rough endoplasmic reticulum is involved in the synthesis of proteins, carried out by their attached ribosomes (Figure 2-13). In contrast, the membranes of the smooth endoplasmic reticulum lack attached ribosomes (see Figure 2-13). Most proteins exit the rough endoplasmic reticulum in vesicles transported to the cis portion of the Golgi apparatus (see Figures 2-16 and 2-17). Other proteins are retained by the rough endoplasmic reticulum to participate in the initial steps of protein synthesis (see Figure 2-15). The retained proteins contain the targeting sequence Lys-Asp-Glu-Leu (KDEL) at the C-terminal. A lack of the KDEL sequence marks proteins for transport to the Golgi apparatus.
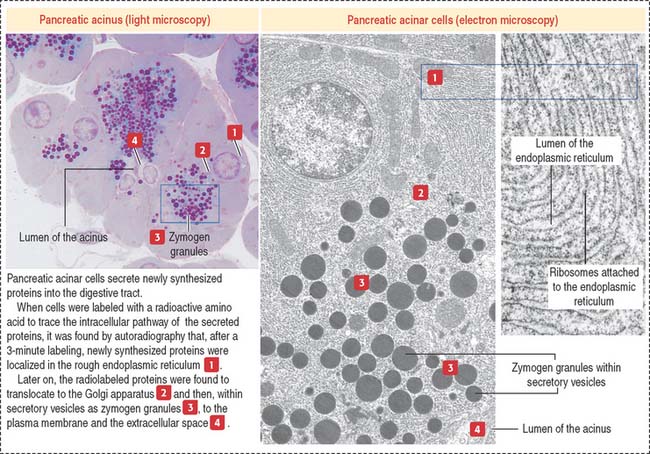
Protein synthesis and sorting
The role of the endoplasmic reticulum in protein synthesis and sorting was demonstrated by incubating pancreatic acinar cells in a medium containing radiolabeled amino acids and localizing radiolabeled proteins by autoradiography. The secretory pathway taken by secretory proteins includes the following sequence: rough endoplasmic reticulum, to Golgi apparatus, to secretory vesicles, to the extracellular space or lumen (Figure 2-14). Plasma membrane and lysosomal proteins also follow the sequence of rough endoplasmic reticulum to Golgi apparatus but are retained within the cell.
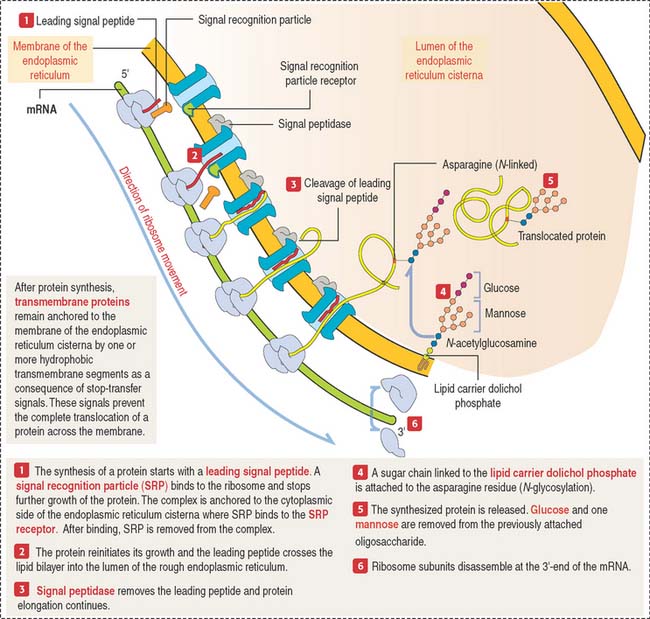
The mechanism by which secretory proteins are directed to the endoplasmic reticulum is explained by the signal hypothesis (Figure 2-15).
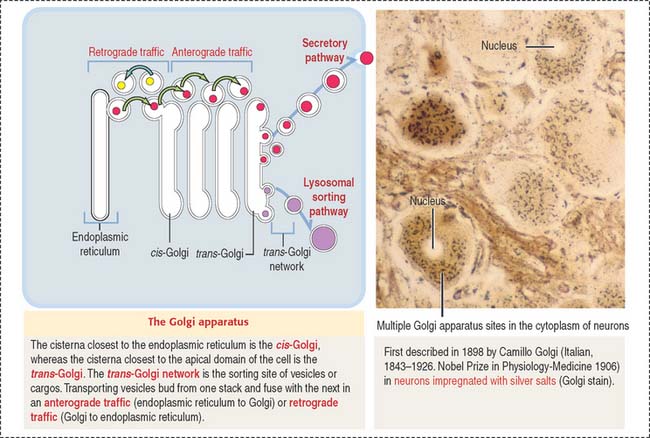
GOLGI APPARATUS
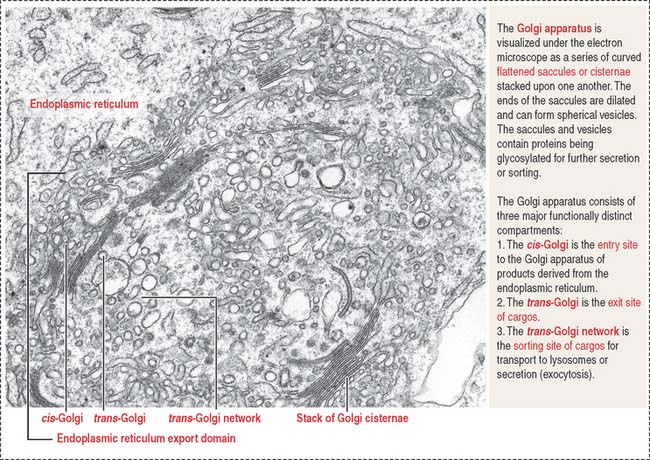
The Golgi apparatus consists of a cluster of flattened stacks of sacs called cisternae (Figures 2-16 and 2-17). Each Golgi stack has two distinct faces: an entry, or cis, face and an exit, or trans, face. The cis-Golgi is adjacent to the endoplasmic reticulum; the trans-Golgi points toward the plasma membrane or the nucleus.
The Golgi apparatus undergoes a permanent turnover. It disassembles during mitosis/meiosis and reassembles in interphase.
Functions of the Golgi apparatus
Once processed, cargos bud off from the Golgi apparatus and are either sorted to the secretory of lysosomal sorting pathway (anterograde traffic) or back to the endoplasmic reticulum (retrograde traffic) (see Figure 2-16).
Cargo sorting occurs along microtubules or actin filaments with the help of motor proteins. The presence of specific lipids domains in the membrane of a vesicle cargo recruit coating proteins and tethering factors to sort the cargo in the direction of an acceptor membrane site. Essentially, the sorting and transport of cargos depend on specialized coats preparing the cargo to be moved along the cytoskeleton by molecular motor proteins. Tethering factors (rod-shaped proteins) attach the cargo to the cytoskeleton. When the vesicle cargo reaches an acceptor membrane, it fuses with the help of fusion proteins.
Exocytosis or secretory pathway and endocytic pathway
These two events depend on distinctive proteins coating the cytosolic side of the membrane of the transport vesicle that becomes a coated vesicle. The coat helps the recruitment of molecules for transport. Before fusion with the acceptor membrane, the vesicle sheds its coat, allowing the membranes to interact directly and fuse.
Transport vesicles are coated by the protein clathrin. Clathrin-coated vesicles are seen in the exocytosis/secretory and endocytosis pathway. In the endocytosis pathway (Figure 2-18), vesicles start at the plasma membrane as clathrin-coated pits. Clathrin molecules assemble in a basket-like arrangement on the cytosolic face of the plasma membrane and the pit shape changes into a vesicle.
Sorting of clathrin-coated vesicles and COP-coated vesicles
A continual process of budding and fusion of transport vesicles mobilizes products from the endoplasmic reticulum to the Golgi apparatus (anterograde traffic), between membranous stacks of the Golgi apparatus, and from the Golgi apparatus to the endoplasmic reticulum (retrograde traffic) (see Figure 2-16).
The vesicular transport mechanism involves two types of coated vesicles (Figure 2-19):
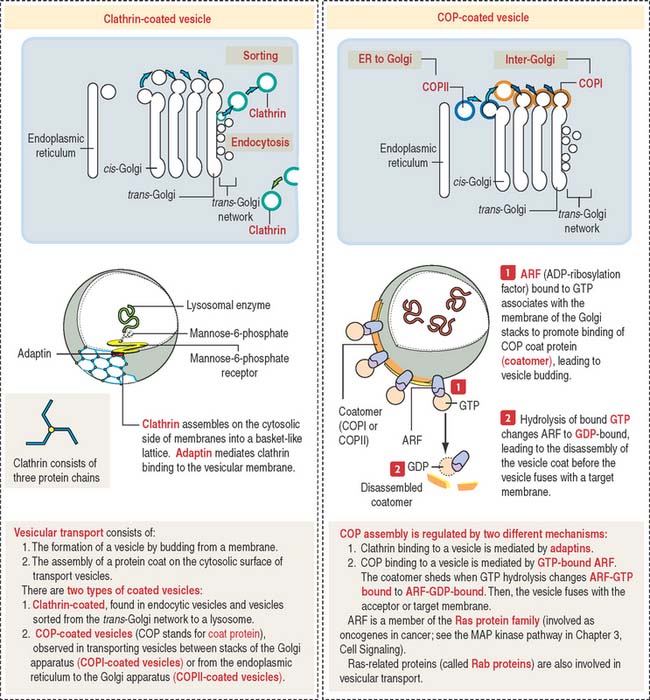
A guanosine triphosphate (GTP)-binding protein called ARF (for adenosine diphosphate [ADP]-ribosylation factor), is required for the assembly of COPI and COPII molecules to form a protein coat called a coatomer on the cytosolic side of a transporting vesicle. When GTP is converted to guanosine diphosphate (GDP) by hydrolysis, the coatomer dissociates from the vesicle just before the vesicle fuses with a target membrane. ARF is related to Ras proteins, a group of oncogene proteins also regulated by the alternate binding of GTP and GDP (see the MAP kinase pathway in Chapter 3, Cell Signaling).
Vesicle fusion to a target membrane requires NSF and SNARE proteins
The fusion of a transporting vesicle to a target membrane (Figure 2-20) requires the recognition of the specific target membrane so the vesicle and target membranes can fuse to deliver the transported cargo.
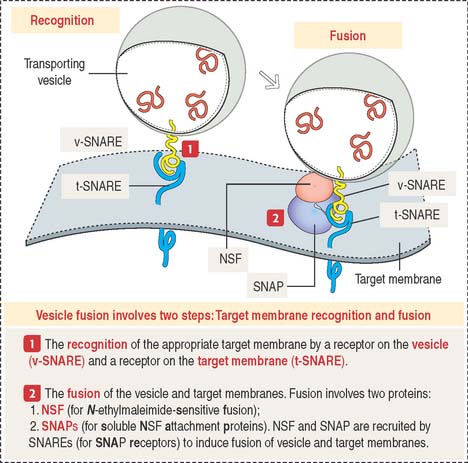
Lysosomal sorting pathway: significance of M6P and its receptor
Lysosomal hydrolases are synthesized in the endoplasmic reticulum, transported to the cis-Golgi, and finally sorted to lysosomes. This sorting mechanism involves two important steps (Figure 2-21):
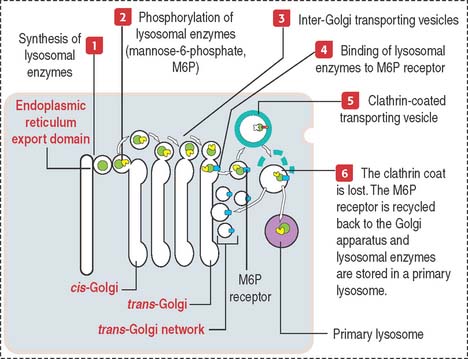
By this mechanism, M6P-containing lysosomal enzymes are separated from other glycoproteins in vesicles with the M6P receptor. After being transported to a clathrin-coated transporting vesicle, lysosomal enzymes dissociate from the M6P receptor and become surrounded by a membrane to form a primary lysosome. Membranes containing free M6P receptor are returned to the Golgi apparatus for recycling.
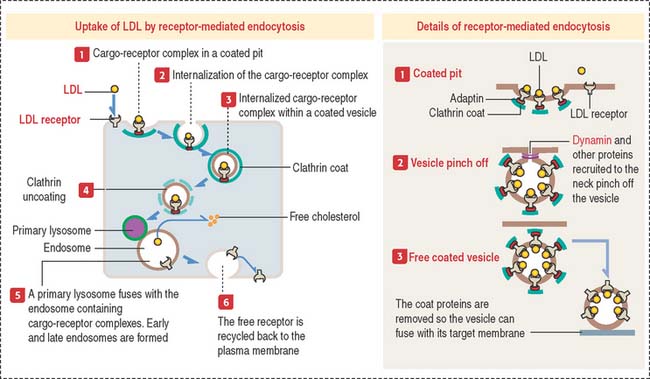
Receptor-mediated endocytosis: Uptake of cholesterol
The internalization of a ligand (such as LDL, transferrin, polypeptide hormones, or growth factors) by a cell requires a specific membrane receptor. The LDL receptor–LDL complex is internalized by receptor-mediated endocytosis. We have seen that this process involves the assembly of the protein clathrin on the cytosolic side of the plasma membrane, which forms a coated pit (see Figure 2-16).
The LDL receptor, in turn, is continuously recycled back to the plasma membrane to be used again. The LDL receptor can recycle every 10 minutes and can make several hundred cycles in its 20-hour life span.
Lysosomes and intracellular digestion
Two types of lysosomes are recognized: primary lysosomes (Figure 2-22), defined as the primary storage site of lysosomal hydrolases, and secondary lysosomes, regarded as lysosomes engaged in a catalytic process.
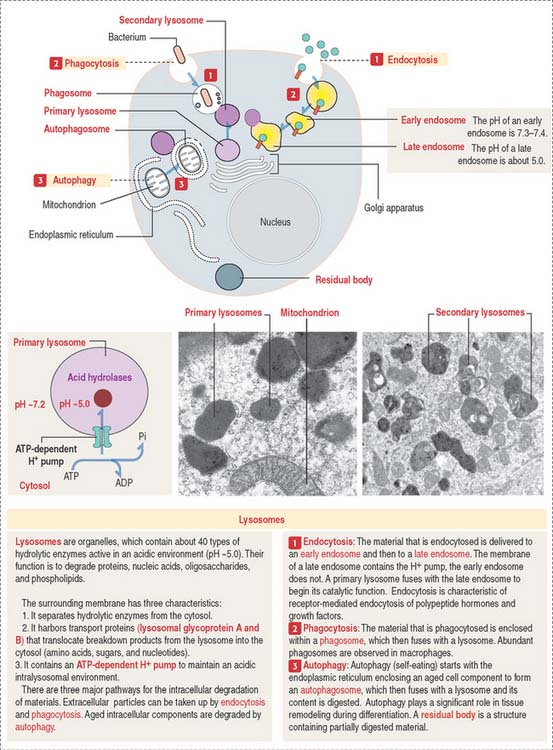
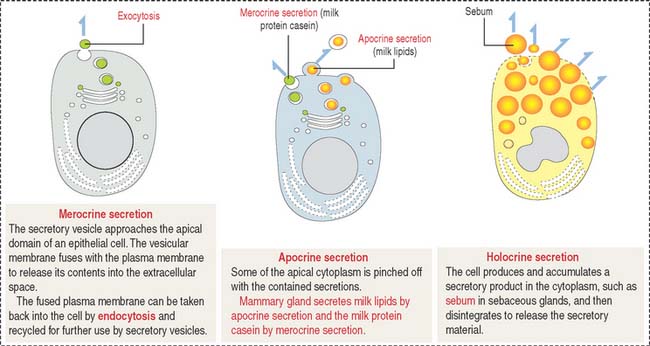
As discussed, the plasma membrane can internalize extracellular particles and fluids using vesicles resulting from the invagination of the membrane by a process called endocytosis. Endocytosis has two important goals: to bring material into the cell, and to recycle the plasma membrane. The reverse process, called exocytosis, is the transport outside the cell of products processed or synthesized by the cell.
Most cells take in fluid by pinocytosis but phagocytosis is the function of specialized cells, including macrophages. We study them in Chapter 4, Connective Tissue (macrophages), Chapter 6, Blood and Hematopoiesis (white blood cells), and Chapter 10, Immune-Lymphatic System (macrophages and antigen-presenting cells). Phagocytic cells scavenge cell remnants during apoptosis and aging blood cells in spleen.
The lysosome is a membrane-bound organelle that, in addition to hydrolytic enzymes, has membrane-bound transporters that allow digested products, such as amino acids, sugars, and nucleotides, to reach the cytosol for reuse or excretion. The lysosomal membrane also contains an ATP-dependent pump that provides H+ into the lysosome to maintain an acidic environment (see Figure 2-22).
We review the lysosomal sorting pathway (see Figure 2-21) to highlight important steps: (1) Lysosomal enzymes and lysosomal membrane proteins are synthesized in the endoplasmic reticulum and transported through the Golgi apparatus to the trans-Golgi network; (2) an important event in the cis-Golgi is the tagging of lysosomal enzymes with a specific phosphorylated sugar group, M6P, that is recognized in the trans-Golgi by the corresponding receptor, M6P receptor; and (3) tagging enables enzymes to be sorted and packaged into transport vesicles that leave the trans-Golgi network toward the lysosomes.
The different paths of various materials to lysosomes are described in Figure 2-22. Note some important terminology: phagosomes fuse with lysosomes and endocytic vesicles fuse with an endosome before delivery to a lysosome. Also, autophagy signifies the degradation of disposable components of the cell itself with the help of an autophagosome (see Box 2-C).
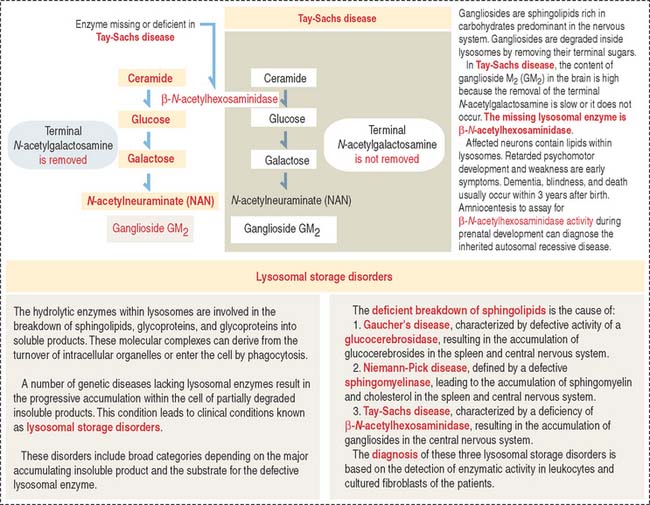
Clinical significance: Lysosomal storage disorders
Lysosomal storage disorders or diseases are caused by the progressive accumulation of cell membrane components within cells because of a hereditary deficiency of enzymes required for their breakdown. An example is Tay-Sachs disease. Additional details on the mechanism leading to lysosomal storage diseases are presented in Figure 2-23.
MITOCHONDRIA
A mitochondrion consists of an outer mitochondrial membrane and an inner mitochondrial membrane creating an intermembrane space between them (Figure 2-24). The inner mitochondrial membrane surrounds a large compartment called the matrix. The matrix is partitioned by infoldings of the inner mitochondrial membrane known as cristae. Cristae amplify the inner mitochondrial membrane on which ATP synthesis takes place.
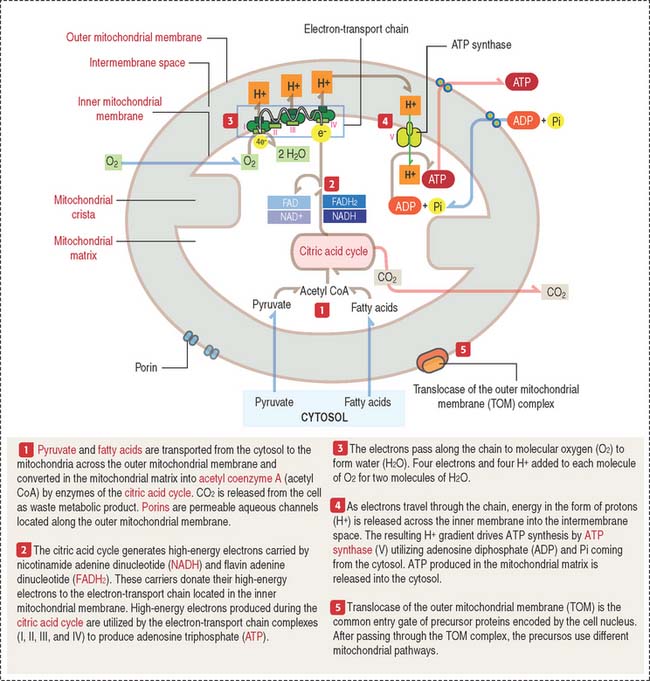
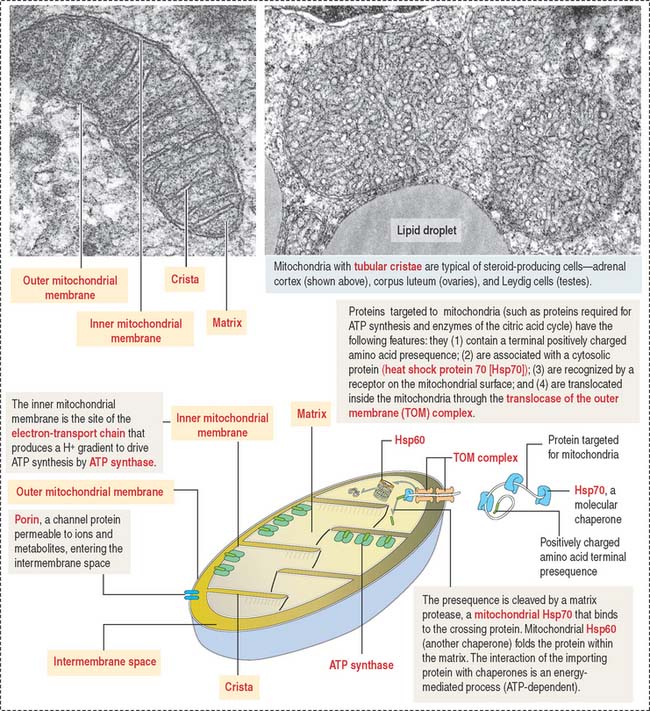
Mitochondria contain DNA and RNA, including ribosomes to synthesize some of their own proteins in the matrix. Only 1% of mitochondrial proteins are encoded by mitochondrial DNA. Most of mitochondrial proteins are encoded by nuclear genes, synthesized in cytosol ribosomes and imported into mitochondria by targeting signals that are recognized by the translocase of the outer mitochondrial membrane complex (TOM) on the outer mitochondrial membrane. TOM is the most common entry route of imported mitochondrial proteins. Targeting polypeptide signals and chaperones (Hsp60 and Hsp70) enable proteins to reach the matrix (Figure 2-25).
The inner mitochondrial membrane is the site of electron-transport and proton (H+) pumping and contains the ATP synthase. Most of the proteins embedded in the inner mitochondrial membrane are components of the electron-transport chain, involved in oxidative phosphorylation.
Mitochondria participate in apoptosis, steroidogenesis, and thermogenesis
Mitochondria participate in three additional functions: programmed cell death or apoptosis, stereidogenesis (production of steroid hormones), and thermogenesis. Enzyme proteins transported to the matrix must cross the outer and inner mitochondrial membranes.
Concerning apoptosis, mitochondria contain procaspases-2, -3, and -9 (precursors of proteolytic enzymes), apoptosis initiation factor (AIF), and cytochrome c. The release of these proteins in the cytosol initiates apoptosis. We come back to mitochondria and apoptosis in Chapter 3, Cell Signaling.
Concerning steroidogenesis, mitochondrial membranes contain enzymes involved in the synthesis of the steroids aldosterone, cortisol, and androgens. We discuss the participation of mitochondria in steroid production in Chapter 19, Endocrine System, and Chapter 20, Spermatogenesis.
Concerning thermogenesis, most of the energy from oxidation is dissipated as heat rather than converted to ATP. Uncoupling proteins (UCPs), members of the superfamily of mitochondrial anion-carrier proteins present in the mitochondrial inner membrane, mediate the regulated discharge of H+ (called proton leak), resulting in the release of heat. Proton leak across the mitochondrial inner membrane is mediated by UCP–1. UCP-1 is present in the mitochondrial inner membrane of brown adipocytes. Its role is to mediate regulated thermogenesis in response to cold exposure (see section on adipose tissue in Chapter 4, Connective Tissue).
Clinical significance: Mitochondrial inheritance
Histologic preparations of muscle biopsies of individuals with MERRF display a peripheral red-stained material corresponding to aggregates of abnormal mitochondria, giving a ragged appearance to red muscle fibers. MERRF is caused by a point mutation in a mitochondrial DNA gene encoding tRNA for lysine. An abnormal tRNA causes a deficiency in the synthesis of proteins required for electron transport and ATP production.
Three maternally inherited mitochondrial diseases affect males more severely than females:
PEROXISOMES
Peroxisomes are single membrane-bound structures (Figure 2-26). They are assembled from proteins synthesized on free ribosomes in the cytosol and then imported into peroxisomes. Peroxisomes contain about 32 different proteins collectively called peroxins. Many of the peroxisomal metabolic pathways are directed toward the production of hydrogen peroxide and its breakdown by catalase.
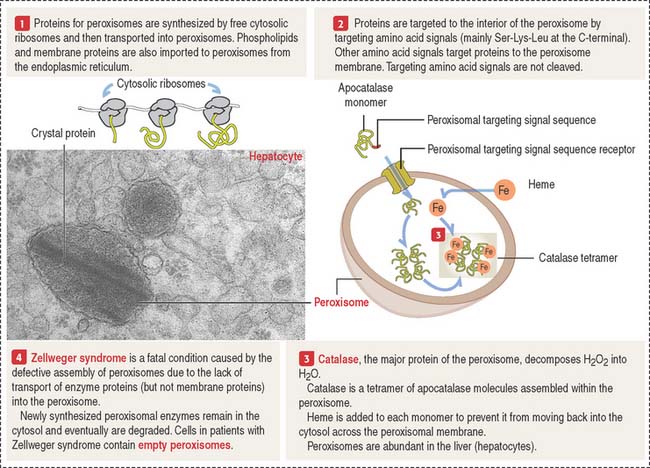
Clinical significance: Zellweger syndrome
Zellweger syndrome (see Figure 2-26) is a rare, congenital disease, fatal within the first year of life. It belongs to the group of leukodystrophies, affecting myelin formation in axons of the brain. Zellweger syndrome is characterized by a reduction or absence of peroxisomes in hepatocytes, and cells of the kidney and brain. Multiple peroxisomal proteins, peroxins, fail to be imported into peroxisomes.
Epithelial Glands
Cytomembranes and the plasma membrane. Intracellular membranes, called cytomembranes, separate diverse cellular processes into compartments. Cytomembranes are components of the endoplasmic reticulum and Golgi apparatus. The nucleus, mitochondria, lysosomes, and peroxisomes are bound by cytomembranes and are called organelles. The nucleus and mitochondria are surrounded by a double membrane; lysosomes and peroxisomes are surrounded by a single membrane. Lipids and glycogen are not membrane-bound and are called inclusions.
Receptor-mediated endocytosis of a ligand requires a plasma membrane receptor. The ligand-receptor complex is internalized by the process of receptor-mediated endocytosis. This process involves: (1) the formation of a clathrin-coated pit (to concentrate ligand-receptor complexes in a small surface area); (2) the invagination of the coated pit to form a coated vesicle; (3) the pinching off of the coated vesicle from the plasma membrane; (4) transport of the vesicle to an endosome; (5) removal of the clathrin coat before fusion of the vesicle with the endosome; and (6) recycling back of the receptor-containing vesicle to the plasma membrane.