[level-membership-for-cardiovascular-category]24
Epigenetics in Cardiac Rhythm Diseases
Definition of Epigenetics
Genetic mutations are well-recognized causes of heritable phenotypic shifts. The completion of the Human Genome Project and technical advances that allow for high-throughput sequencing have advanced the exploration for genetic signatures that associate with and, perhaps, cause disease states. However, it is very possible that many disease states result from environmental influences that affect the expression or repression of different genes without altering the underlying DNA sequence. By definition, an epigenetic phenomenon is a heritable alteration in phenotype without an underlying DNA mutation. Epigenetic phenomena regulate gene expression profiles by controlling genes in a binary manner such that genes are actively expressed (i.e., “on”) or repressed (i.e., “off”). In addition, true epigenetic changes that occur as a result of a stimulus should be heritable and stable, even in the subsequent absence of that stimulus. Although the concept of epigenetics seems simple, it differs distinctly from the classic notion that only mutations at the DNA level can cause heritable phenotypic shifts (Figure 24-1). Clinicians and scientists are exploring epigenetic mechanisms and their contribution to the development of disease states, because these phenomena provide a rational mechanistic explanation for how environmental factors can alter phenotypes in a heritable manner in the absence of DNA mutations.
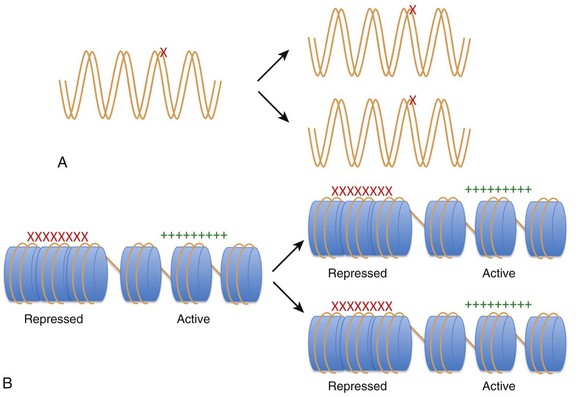
Figure 24-1 Heritability of phenotypic changes at the level of DNA (A) and by epigenetic mechanisms (B). A, The genetic code is defined by the DNA template (orange lines). In this model, DNA mutations (red X) can alter the expression of genes and the resultant phenotype. DNA replication results in mutations being passed from a mother cell to her progeny. B, The process by which epigenetic mechanisms can regulate gene expression profiles in a heritable manner without mutating the underlying DNA. The expression of genes can be regulated by interactions between nucleosome components (blue cylinders) and the DNA (orange strand) such that certain genes are repressed (red X) and certain genes are active (green +). The DNA and the nucleosome components are both heritable so that the active and repressed gene patterns are similar in the mother cells and daughter progeny.
Historical Experiments That Illustrate the Salient Features of Epigenetics
Early work in Drosophila species development revealed that the homeotic genes define segment identity and the position of appendages along the body axes. Thus, developmental biologists became interested in mechanisms to explain why specific homeotic genes are expressed in one body segment and not in a neighboring segment. By using genetic approaches, it was demonstrated that the spatial and temporal expression of the homeotic genes was enhanced by the Trithorax group of genes and repressed by the Polycomb group of genes in a segment-specific manner.1 However, the manner in which the Trithorax and Polycomb groups of proteins controlled homeotic gene expression was not initially understood. Investigators using yeast and Tetrahymena discovered that enzymes that modify histones (the packaging structure for DNA) could silence specific genes.2–4 Biochemistry work revealed that the histone-modifying enzymes that repress gene expression contain a SET domain that catalyzes histone methylation. The importance of histone methylation in Drosophila species development became apparent when the Polycomb protein complex was shown to have a SET domain–containing enzyme capable of methylating H3K9, a repressive epigenetic mark.5,6 Similarly, the Trithorax proteins TRX and Ash1 were found to contain SET domains and capable of methylating histones with a mark that facilitates active gene expression.7–9 Taken together, these studies reveal that histone modifications are well-conserved phenomena that are critical for regulating gene expression profiles during development.
The creation of Dolly the sheep also revealed important features of epigenetic mechanisms. Wilmut et al.10 sought to clone a sheep by taking the nucleus from a differentiated cell (mammary gland cell) and placing that nucleus into an unfertilized oocyte (developing egg cell). Although they were eventually successful, this process of nuclear transfer was extremely inefficient. This work revealed that although the mammary gland cell DNA contains the blueprint necessary to develop an entire sheep, it was not easy to revert a differentiated nucleus back to a pluripotent state. It became apparent that although the blueprint of DNA is the same in an embryonic stem (ES) cell and a differentiated cell, nonmutation-based changes have occurred to the DNA in the differentiated cell that prevent the simple reversion or manipulation of a cell’s identity. This study supports the idea that, as cells differentiate, they accumulate stable and heritable changes in their nucleus that restrict gene expression patterns and define identity. These changes are, by definition, epigenetic.
Basis of Epigenetic Phenomena
The study of epigenetic phenomena presupposes that genes can be marked as transcriptionally “on” or “off” by modifications that are independent of the primary nucleotide coding sequence. The DNA in each cell is composed of approximately 3 × 109 nucleotide bases that could presumably stretch to 2 m in length. To facilitate efficient packaging, DNA is wrapped in a threadlike fashion around protein spools called histones. The histone octamer consists of two molecules of each histone: H2A, H2B, H3, and H4.11–13 The histone octamer and the 146 bp of DNA looped around it forms a structure called a nucleosome, the functional unit of chromatin. In addition to serving as packaging facilitators, nucleosomes are amenable to modifications and dynamic remodeling, which makes them an active participant in many chromosomal processes including transcription, replication, DNA repair, kinetochore and centromere construction, and telomere maintenance.
The regulation of gene expression at the chromatin level occurs by: (1) covalent histone tail modifications; (2) adenosine triphosphate (ATP)-dependent chromatin remodeling; and (3) DNA methylation (Figure 24-2).14 This chapter focuses on covalent histone tail modifications and ATP-dependent chromatin remodeling. The amino terminal tails of histones protrude and are subjected to various covalent posttranslational modifications (e.g., methylation, acetylation, sumoylation, phosphorylation).11 These histone tail modifications regulate whether the neighboring DNA is transcriptionally active (euchromatin) or repressed (heterochromatin). Activating histone modifications attract ATP-dependent nucleosome remodeling factors that can reposition or eject histones, thus promoting transcription or repression of the neighboring DNA. Thus, complex interplay between covalent histone modifications, ATP-dependent chromatin remodeling enzymes, and the transcriptional machinery (i.e., DNA binding factors, transcription initiation, elongation complexes) determines whether genes are actively expressed or repressed.
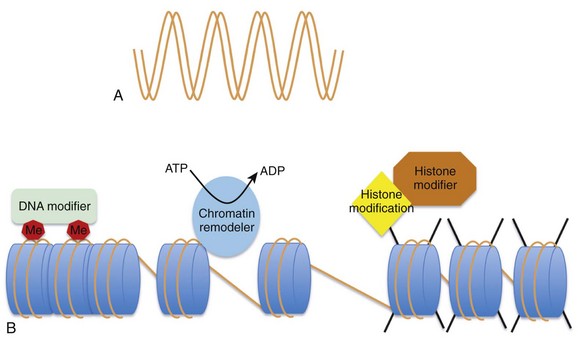
Figure 24-2 A, DNA provides the template that encodes all potential genes. Not all genes are expressed in all cells. Chromatin provides a way of partitioning this genetic information into active and repressed components. The three mechanisms whereby chromatin can modify the expression or repression of genes includes DNA methylation (Me), chromatin remodelers (blue oval), and histone modifications (e.g., phosphorylation, methylation, acetylation) imparted by histone modifiers. (Adapted from Chang CP, Bruneau BG: Epigenetics and cardiovascular development. Annu Rev Physiol 74:41–68, 2012.)
Histone Tail Modifications
From an epigenetic standpoint, the two most important tail modifications are acetylation and methylation.15 Histone tail acetylation is associated with transcriptionally active chromatin. In contrast, histone tail methylation can be associated with either euchromatin or heterochromatin, depending upon the extent of methylation (mono- [me], di- [me2], or tri- [me3] methylation) and the specific tail residues that are modified. For example, lysine (K) residues available for methylation include K4, K9, K27, and K36 of histone H3, and K20 of histone H4. Methylation of H3K9 and H3K27 is associated with gene repression gene expression,16 whereas dimethyl (H3K4me2) and trimethyl (H3K4me3) marks at H3K4 are associated with actively expressed genes.17 As shown in Figure 24-3,18 the extent, specific lysine residue, and location of different histone methylation marks around a gene provide a signature for the transcriptional state of that gene. For example, H3K27me3 marks (black line) are located around the 5′ regulatory region of silenced genes. In contrast, H3K4me3 marks are highly enriched at the 5′ transcription start site of actively expressed genes. In contrast, H3K4me3 marks are not as enriched around poised and silenced genes.
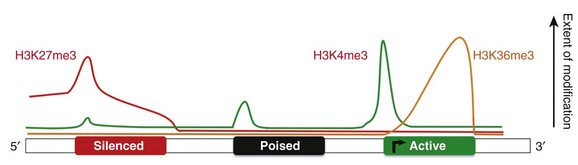
Figure 24-3 Epigenetic signatures differ around genes that are repressed (red rectangle), poised (black rectangle), and active (green rectangle). H3K27me3 marks (red line) are enriched at the 5′ region of silenced genes H3K4me3 marks (green line) are enriched at actively expressed genes. Thus active genes show enrichment of H3K4me3 marks and a paucity of H3K27me3 marks at the 5′ region. H3K36me3 (yellow line) marks are enriched at active genes. This illustrates the histone code hypothesis, which suggests that the epigenetic signature of individual genes, determines whether that gene is actively expressed or repressed. (Adapted from Lee BM, Mahadevan LC: Stability of histone modifications across mammalian genomes: implications for “epigenetic” marking. J Cell Biochem 108:22–34, 2009.)
Histone methylation marks and acetylation marks are imparted and removed by enzymes that are part of protein complexes. Histone acetyltransferases, acetylate histones, and histone deacetylases reverse this acetylation process. Similarly, histone methyltransferase (HMT) complexes impart histone methylation marks, and histone demethylases remove histone methylation marks. HMT complexes often consist of a SET domain–containing HMT enzyme that is capable of adding methyl groups to specific lysine residues. The mammalian homologues to the yeast Set1 and the Drosophila Trithorax proteins are the mixed lineage leukemia family of proteins, now known as KMTA-D. These proteins contain a SET domain and form HMT protein complexes. In some cases, these HMT and acetylase complexes are directed to specific sites of DNA by their interaction with transcription factors (Figure 24-4).19
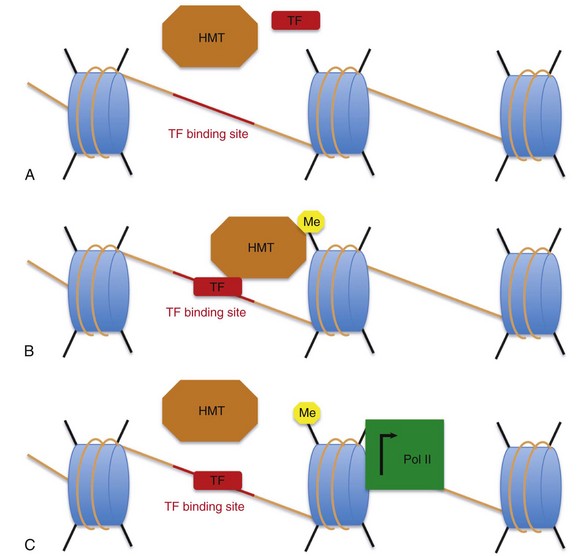
Figure 24-4 How are certain histones marked with activating methylation marks? A, Transcription factors (red rectangle), histone methyltransferase (HMT) complexes (orange octagon), DNA (orange line), histones (blue cylinders), and the histone tails (black lines) are present in the cell. Upon activation, a transcription factor (TF) binds to the DNA at defined binding sites (red line). B, These transcription factors interact with histone methyltransferase complexes (orange hexagon), which impart activating methylation marks on neighboring histones (yellow hexagon). As shown in (C), these active histone methylation marks then interact with the transcription machinery (green square) to facilitate transcription (black arrow).
ATP-Dependent Chromatin Remodeling Complexes
Chromatin-modifying enzymes use the energy derived from ATP to alter the chromatin structure in a manner that permits (euchromatin) or represses gene expression (heterochromatin) by restructuring, mobilizing, and ejecting nucleosomes. There are four families of SWI-like, ATP-dependent chromatin remodeling complexes: the switching defective/sucrose nonfermenting (SWI/SNF); imitation switch (ISWI); chromodomain, helicase, DNA-binding (CHD); and inositol requiring 80 complexes (INO80).20 These chromatin-remodeling complexes share a well-conserved SWI-like ATPase catalytic domain in combination with a unique functional domain. The ATPase domain uses ATP hydrolysis to remodel nucleosomes and render the neighboring DNA accessible or repressed.
Epigenetics in Induced Pluripotent Stem Cells and Direct Reprogramming
Cells accumulate epigenetic marks that restrict their cell fates during development. Pluripotent ES cells have an epigenome that permits the expression and repression of all genes that are necessary for development and differentiation. During development, ES cells respond to external cues and begin to differentiate. During differentiation, ES cells accumulate stable and heritable epigenetic marks that restrict their fate and the fate of their daughter cells. Nuclear transfer experiments demonstrated that removing a nucleus from fully a differentiated cell, transferring that nucleus into a denucleated zygote, and developing a pluripotent stem is a very inefficient process, partly because of an inability to reset the epigenetic marks that are present in the differentiated cell nucleus.10 Despite the stability of these epigenetic marks, investigators have more recently discovered that they can revert differentiated fibroblasts into induced pluripotent stem cells (iPSCs) by introducing a cocktail of transcription factors that revert the epigenome to a more pluripotent state.21,22 Thus, although epigenetic stability is important for establishing cell identity during development in vivo, the ability to revert differentiated cells to pluripotent cells by manipulating the epigenome in vitro has garnered much excitement.
Manipulating the epigenome to develop iPSCs has significant clinical relevance. iPSCs can serve as a source of pluripotent cells for regenerative medicine, and they provide a method of studying genetic-based disease states in vitro. For example, Yazawa et al. used fibroblasts from patients with Timothy syndrome to generate iPSCs and differentiate them into cardiac myocytes in vitro.23 Using this model, they studied the electrical and calcium-handling properties of myocytes that possess the Timothy syndrome mutation in vitro. Although the forced expression of a cocktail of transcription factors reverts differentiated cells to pluripotent iPSCs that share many features of ES cells, recent work has demonstrated that iPSCs have incomplete epigenetic reprogramming.24 To determine the importance of epigenetic mechanisms on cellular reprogramming, Onder et al. investigated how different chromatin-modifying enzymes affect cellular reprogramming.25 They demonstrated that inhibition of components of PRC1 and PRC2 reduces reprogramming efficiency. In comparison, inhibition of SUV39H1, YY1, and the H3K79 methyltransferase DOT1L enhanced reprogramming.
To harness the ability of transcription factors to reprogram cells in a clinically relevant manner, Ieda et al.26 demonstrated that the combination of three developmental cardiac transcription factors (Gata4, Mef2c, and Tbx5) could be used to directly reprogram dermal and postnatal cardiac fibroblasts into differentiated beating cardiaclike cells without first developing iPSCs. They showed that direct reprogramming of fibroblasts resulted in the stable shift of the epigenetic signature of several cardiac genes—Actn2, Ryr2, and Tnnt2.
Heart Muscle Development
SWI/SNF Complexes in Heart Development
Many components of the BAF complex are necessary for the development of the heart chambers and cardiac muscle. Brg1, an adenosine triphosphatase (ATPase)-containing protein that is a member of the BAF complex has been well studied in myocardial development and provides an example of the importance of chromatin-remodeling factors in heart development. Brg1 is necessary for early cardiomyocyte cell proliferation as well as the differentiation into mature cardiac myocytes. Murine models with cardiac-specific deletion of Brg1 have a thinned myocardium and absent interventricular septums.27 Work by Hang et al.27 revealed that Brg1 is necessary for the effective expression of Bmp10 and the repression of p57kip2, a cyclin dependent kinase inhibitor, such that the deletion of Brg1 results in a lack of Bmp10 and ectopic expression of p57kip2 that represses cell cycle progression.
In addition to regulating cell cycle progression, Brg1 is also necessary for the differentiation of cardiac myocytes. Beta-myosin heavy chain (β-MHC) is the primary MHC isoform expressed in fetal myocytes whereas alpha-myosin heavy chain (α-MHC) is the primary MHC expressed in adult myocytes. Brg1 plays an important role in MHC isoform switching. In the absence of Brg1, embryonic myocytes prematurely switch from β-MHC to α-MHC. Thus, Brg1 is important in the normal progression of differentiation from fetal to adult cardiac myocytes.27
Brg1 has also been used to demonstrate the importance of the relationship between cardiac transcription factors and epigenetic complexes. Brg1+/– mice have variable cardiac phenotypes including ventricular septal defects (VSDs), patent foramen ovales (PFO), conduction abnormalities, and cardiac dilatation.27 Brg1 has been shown to interact with the key cardiac transcription factors Tbx5, Nkx2-5, and Gata4 and potentiate transcription factor–mediated transcription.28 Furthermore, the combination of Brg1+/– haploinsufficiency with NKX2-5 or Tbx20 heterozygous mice results in the development of severe lethal cardiac defects. These studies suggest that the effective development of cardiac myocytes requires an interaction between transcription factors and epigenetic complexes.
Ezh2 in Cardiac Development
Polycomb repressive complexes (PRCs) play a role in repressing gene expression profiles. Ezh2 in the is a key histone methyltransferase of PRC2 complex which trimethylates histone 3 at lysine 27 (H3K27). H3K27 methylation marks are associated with transcriptionally repressed chromatin. As cardiac myocytes develop, they undergo an increase in H3K27me3 levels.29 The deletion of Ezh2 during cardiac development in murine models has resulted in disparate results. He et al.30 deleted a floxed Ezh2 allele in early cardiac development in mice using an NKX2.5:Cre driver. These mice demonstrated lethal congenital heart malformations including compact myocardial hypoplasia, hypertrabeculation, and ventricular septal defects. Deleting Ezh2 with Tnnt:Cre, which comes on after NKX2-5:Cre, attenuated the phenotype observed with NKX2-5:Cre, suggesting that the establishment of H3K27me3 epigenetic marks occurs during a finite window of time during cardiac development.30 In a different study, Delgado-Olguin et al. reported that deletion of Ezh2 using the NKX2.5:Cre mice resulted in mild cardiac hypertrophy. However, using a Mef2cAHF:Cre driver that is expressed in cardiac progenitors of the anterior heart field, they demonstrated that the absence of an Ezh2 results in attenuation of the observed increase that accompanies normal H3K27me3 marks development and an inability to repress Six1 gene expression. The inappropriate expression of Six1 resulted in massive right ventricular hypertrophy and mild pulmonary stenosis in adult mice.29
Epigenetics in Disease States
Diabetes and Epigenetics
It is well established that patients with type I and type 2 diabetes have a substantially increased risk of developing cardiovascular complications.31,32 Thus, investigators have explored the effect that tight glucose control has on diabetic complications. Basic science and human clinical trials have demonstrated that a transient exposure to hyperglycemia results in a higher incidence of diabetic vascular disease despite a return to normal glucose levels.33 This “legacy effect” is referred to as metabolic memory. Because epigenetic mechanisms can induce stable changes in gene expression that persist despite the subsequent absence of the inciting stimulus, investigators have explored epigenetic mechanisms to explain the legacy effect.34 In corroboration with these clinical observations, in vitro work on human aortic endothelial cells has demonstrated that transient exposure to hyperglycemia results in sustained activation of NF-κB, a proinflammatory transcriptional regulator.35 A hyperglycemic exposure recruits epigenetic machinery to the promoter region of the p65 subunit of NF-κB. The epigenetic machinery facilitates a decrease in H3K9me repressive marks, an increase in H3K4me activating marks, and a sustained transcriptional increase in the p65 subunit of NF-κB. The increase in p65 results in the persistent expression of a proinflammatory gene expression profile. This in vitro work demonstrates how environmental factors (e.g., hyperglycemia) can be translated into a stable shift in disease-defining gene expression profiles by epigenetic mechanisms.
Cardiac Hypertrophy and Cardiac Failure
The development of cardiac hypertrophy and cardiac failure is a result of a stimulus-induced shift in the cardiac phenotype. Kaneda et al. investigated which shifts in histone tail modifications accompany rat and human models of heart failure.36 By using genome-wide approaches, they demonstrated that there is a significant shift in the profile of H3K4me3 (activating) and H3K9me3 (repressive) marks in Dahl salt–sensitive rats with heart failure. Next, using human heart failure samples, they confirmed that human heart failure is also associated with a significant shift in genome-wide H3K4me3 and H3K9me3 profiles. They found that the activating H3K4me3 marks are associated with many important canonical signaling pathways (e.g., calcium signaling, nitric oxide signaling, cAMP-mediated signaling, G-protein–coupled receptor signaling, actin signaling) that are important in heart failure. Taken together, these results suggest that there is an association between H3K4me3 marks and H3K9me3 marks and the alteration in gene expression profiles that accompanies cardiac disease states.
To investigate the mechanistic role of histone-modifying proteins in the development of cardiac hypertrophy, Zhang et al. looked at the importance of Jmjd2a in the development of cardiac hypertrophy.37 Jmjd2a is a member of the JmjC domain–containing family JMJD2 of histone demethylases. The JMJD2 family of proteins demethylate H3K9me3 and H3K36me3. H3K9me3 is associated with heterochromatin; thus, Jmjd2a acts as an activator by removing repressive epigenetic marks. By using murine models of pressure overload–induced hypertrophy, Zhang et al. demonstrated that Jmjd2a deletion attenuates the development of cardiac hypertrophy. They also showed that cardiac-specific overexpression of Jmjd2a resulted in an exaggerated hypertrophic response.
Studies in cardiac development revealed that Brg1 is critical for the switch from β-MHC to α-MHC. Interestingly, the development of cardiac hypertrophy is known to be accompanied by a switch from the adult α-MHC isoform to the fetal β-MHC isoform. Human studies reveal that Brg1 is upregulated in cardiac tissue from patients with hypertrophic cardiomyopathy. The degree of Brg1 expression correlates with the extent of the hypertrophic phenotype. Brg1 is normally downregulated in adult heart tissue; however, pressure overload–induced hypertrophy in murine models results in re-expression of Brg1. Deletion of Brg1 in murine models attenuates the hypertrophic response, blunts the development of fibrosis, and blocks the switch in MHC isoforms.27
Electromechanical Function and Arrhythmias
Although epigenetic mechanisms are important for defining a cell’s identity during development, it remains unclear whether an inability to maintain the epigenome once a cell is fully differentiated can be a causative mechanism of disease. To test this hypothesis, Stein et al. deleted PTIP, a protein that is part of a histone methyltransferase complex that enriches at actively expressed genes, in fully differentiated murine adult myocytes using an inducible Cre driver.38 These studies revealed that deleting a component of the histone methyltransferase complex in a fully differentiated myocyte results in an inability to maintain H3K4me3 marks. This epigenetic instability resulted in a disease-like phenotype that included significant attenuation in Kcnip2 gene expression, a decrease in the transient outward potassium current, an increase in calcium transients, and a propensity to develop premature ventricular beats. This work demonstrated that it is necessary to maintain epigenetic profiles in fully differentiated tissues, and that an inability to maintain these marks over time can be a mechanistic cause of disease.
References
1. Ringrose, L, Paro, R. Polycomb/trithorax response elements and epigenetic memory of cell identity. Development. 2007; 134:223–232.
2. Nakayama, J, Rice, JC, Strahl, BD, et al. Role of histone h3 lysine 9 methylation in epigenetic control of heterochromatin assembly. Science. 2001; 292:110–113.
3. Rea, S, Eisenhaber, F, O’Carroll, D, et al. Regulation of chromatin structure by site-specific histone h3 methyltransferases. Nature. 2000; 406:593–599.
4. Strahl, BD, Ohba, R, Cook, RG, et al. Methylation of histone h3 at lysine 4 is highly conserved and correlates with transcriptionally active nuclei in tetrahymena. Proc Natl Acad Sci U S A. 1999; 96:14967–14972.
5. Czermin, B, Melfi, R, McCabe, D, et al. Drosophila enhancer of zeste/esc complexes have a histone h3 methyltransferase activity that marks chromosomal polycomb sites. Cell. 2002; 111:185–196.
6. Muller, J, Hart, CM, Francis, NJ, et al. Histone methyltransferase activity of a Drosophila polycomb group repressor complex. Cell. 2002; 111:197–208.
7. Beisel, C, Imhof, A, Greene, J, et al. Histone methylation by the drosophila epigenetic transcriptional regulator ash1. Nature. 2002; 419:857–862.
8. Milne, TA, Briggs, SD, Brock, HW, et al. Mll targets set domain methyltransferase activity to hox gene promoters. Mol Cell. 2002; 10:1107–1117.
9. Nakamura, T, Mori, T, Tada, S, et al. All-1 is a histone methyltransferase that assembles a supercomplex of proteins involved in transcriptional regulation. Mol Cell. 2002; 10:1119–1128.
10. Campbell, KH, McWhir, J, Ritchie, WA, et al. Sheep cloned by nuclear transfer from a cultured cell line. Nature. 1996; 380:64–66.
11. Luger, K, Rechsteiner, TJ, Flaus, AJ, et al. Characterization of nucleosome core particles containing histone proteins made in bacteria. J Mol Biol. 1997; 272:301–311.
12. Germond, JE, Hirt, B, Oudet, P, et al. Folding of the DNA double helix in chromatin-like structures from simian virus 40. Proc Natl Acad Sci U S A. 1975; 72:1843–1847.
13. Olins, AL, Olins, DE. Spheroid chromatin units (V bodies). Science. 1974; 183:330–332.
14. Chang, CP, Bruneau, BG. Epigenetics and cardiovascular development. Annu Rev Physiol. 2012; 74:41–68.
15. Kouzarides, T. Chromatin modifications and their function. Cell. 2007; 128:693–705.
16. Litt, MD, Simpson, M, Gaszner, M, et al. Correlation between histone lysine methylation and developmental changes at the chicken beta-globin locus. Science. 2001; 293:2453–2455.
17. Santos-Rosa, H, Schneider, R, Bannister, AJ, et al. Active genes are tri-methylated at k4 of histone h3. Nature. 2002; 419:407–411.
18. Lee, BM, Mahadevan, LC. Stability of histone modifications across mammalian genomes: Implications for “epigenetic” marking. J Cell Biochem. 2009; 108:22–34.
19. Patel, SR, Kim, D, Levitan, I, et al. The brct-domain containing protein ptip links pax2 to a histone h3, lysine 4 methyltransferase complex. Dev Cell. 2007; 13:580–592.
20. Saha, A, Wittmeyer, J, Cairns, BR. Chromatin remodelling: The industrial revolution of DNA around histones. Nat Rev Mol Cell Biol. 2006; 7:437–447.
21. Takahashi, K, Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006; 126:663–676.
22. Park, IH, Zhao, R, West, JA, et al. Reprogramming of human somatic cells to pluripotency with defined factors. Nature. 2008; 451:141–146.
23. Yazawa, M, Hsueh, B, Jia, X, et al. Using induced pluripotent stem cells to investigate cardiac phenotypes in timothy syndrome. Nature. 2011; 471:230–234.
24. Lister, R, Pelizzola, M, Kida, YS, et al. Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature. 2011; 471:68–73.
25. Onder, TT, Kara, N, Cherry, A, et al. Chromatin-modifying enzymes as modulators of reprogramming. Nature. 2012; 483:598–602.
26. Ieda, M, Fu, JD, Delgado-Olguin, P, et al. Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell. 2010; 142:375–386.
27. Hang, CT, Yang, J, Han, P, et al. Chromatin regulation by brg1 underlies heart muscle development and disease. Nature. 2010; 466:62–67.
28. Lickert, H, Takeuchi, JK, Von Both, I, et al. Baf60c is essential for function of baf chromatin remodelling complexes in heart development. Nature. 2004; 432:107–112.
29. Delgado-Olguin, P, Huang, Y, Li, X, et al. Epigenetic repression of cardiac progenitor gene expression by ezh2 is required for postnatal cardiac homeostasis. Nat Genet. 2012; 44:343–347.
30. He, A, Ma, Q, Cao, J, et al. Polycomb repressive complex 2 regulates normal development of the mouse heart. Circ Res. 2012; 110:406–415.
31. Retinopathy and nephropathy in patients with type 1 diabetes four years after a trial of intensive therapy. The diabetes control and complications trial/epidemiology of diabetes interventions and complications research group. N Engl J Med. 2000; 342:381–389.
32. Writing Team for the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Research Group. Sustained effect of intensive treatment of type 1 diabetes mellitus on development and progression of diabetic nephropathy: The epidemiology of diabetes interventions and complications (EDIC) study. JAMA. 2003; 290:2159–2167.
33. Nathan, DM, Cleary, PA, Backlund, JY, et al. Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N Engl J Med. 2005; 353:2643–2653.
34. Siebel, AL, Fernandez, AZ, El-Osta, A. Glycemic memory associated epigenetic changes. Biochem Pharmacol. 2010; 80:1853–1859.
35. Brasacchio, D, Okabe, J, Tikellis, C, et al. Hyperglycemia induces a dynamic cooperativity of histone methylase and demethylase enzymes associated with gene-activating epigenetic marks that coexist on the lysine tail. Diabetes. 2009; 58:1229–1236.
36. Kaneda, R, Takada, S, Yamashita, Y, et al. Genome-wide histone methylation profile for heart failure. Genes Cells. 2009; 14:69–77.
37. Zhang, QJ, Chen, HZ, Wang, L, et al. The histone trimethyllysine demethylase jmjd2a promotes cardiac hypertrophy in response to hypertrophic stimuli in mice. J Clin Invest. 2011; 121:2447–2456.
38. Stein, AB, Jones, TA, Herron, TJ, et al. Loss of h3k4 methylation destabilizes gene expression patterns and physiological functions in adult murine cardiomyocytes. J Clin Invest. 2011; 121:2641–2650.
[/level-membership-for-cardiovascular-category][not-level-membership-for-cardiovascular-category]24
Epigenetics in Cardiac Rhythm Diseases
Definition of Epigenetics
Genetic mutations are well-recognized causes of heritable phenotypic shifts. The completion of the Human Genome Project and technical advances that allow for high-throughput sequencing have advanced the exploration for genetic signatures that associate with and, perhaps, cause disease states. However, it is very possible that many disease states result from environmental influences that affect the expression or repression of different genes without altering the underlying DNA sequence. By definition, an epigenetic phenomenon is a heritable alteration in phenotype without an underlying DNA mutation. Epigenetic phenomena regulate gene expression profiles by controlling genes in a binary manner such that genes are actively expressed (i.e., “on”) or repressed (i.e., “off”). In addition, true epigenetic changes that occur as a result of a stimulus should be heritable and stable, even in the subsequent absence of that stimulus. Although the concept of epigenetics seems simple, it differs distinctly from the classic notion that only mutations at the DNA level can cause heritable phenotypic shifts (Figure 24-1). Clinicians and scientists are exploring epigenetic mechanisms and their contribution to the development of disease states, because these phenomena provide a rational mechanistic explanation for how environmental factors can alter phenotypes in a heritable manner in the absence of DNA mutations.
Figure 24-1 Heritability of phenotypic changes at the level of DNA (A) and by epigenetic mechanisms (B). A, The genetic code is defined by the DNA template (orange lines). In this model, DNA mutations (red X) can alter the expression of genes and the resultant phenotype. DNA replication results in mutations being passed from a mother cell to her progeny. B, The process by which epigenetic mechanisms can regulate gene expression profiles in a heritable manner without mutating the underlying DNA. The expression of genes can be regulated by interactions between nucleosome components (blue cylinders) and the DNA (orange strand) such that certain genes are repressed (red X) and certain genes are active (green +). The DNA and the nucleosome components are both heritable so that the active and repressed gene patterns are similar in the mother cells and daughter progeny.
Historical Experiments That Illustrate the Salient Features of Epigenetics
Early work in Drosophila species development revealed that the homeotic genes define segment identity and the position of appendages along the body axes. Thus, developmental biologists became interested in mechanisms to explain why specific homeotic genes are expressed in one body segment and not in a neighboring segment. By using genetic approaches, it was demonstrated that the spatial and temporal expression of the homeotic genes was enhanced by the Trithorax group of genes and repressed by the Polycomb group of genes in a segment-specific manner.1 However, the manner in which the Trithorax and Polycomb groups of proteins controlled homeotic gene expression was not initially understood. Investigators using yeast and Tetrahymena discovered that enzymes that modify histones (the packaging structure for DNA) could silence specific genes.2–4 Biochemistry work revealed that the histone-modifying enzymes that repress gene expression contain a SET domain that catalyzes histone methylation. The importance of histone methylation in Drosophila species development became apparent when the Polycomb protein complex was shown to have a SET domain–containing enzyme capable of methylating H3K9, a repressive epigenetic mark.5,6 Similarly, the Trithorax proteins TRX and Ash1 were found to contain SET domains and capable of methylating histones with a mark that facilitates active gene expression.7–9 Taken together, these studies reveal that histone modifications are well-conserved phenomena that are critical for regulating gene expression profiles during development.
The creation of Dolly the sheep also revealed important features of epigenetic mechanisms. Wilmut et al.10 sought to clone a sheep by taking the nucleus from a differentiated cell (mammary gland cell) and placing that nucleus into an unfertilized oocyte (developing egg cell). Although they were eventually successful, this process of nuclear transfer was extremely inefficient. This work revealed that although the mammary gland cell DNA contains the blueprint necessary to develop an entire sheep, it was not easy to revert a differentiated nucleus back to a pluripotent state. It became apparent that although the blueprint of DNA is the same in an embryonic stem (ES) cell and a differentiated cell, nonmutation-based changes have occurred to the DNA in the differentiated cell that prevent the simple reversion or manipulation of a cell’s identity. This study supports the idea that, as cells differentiate, they accumulate stable and heritable changes in their nucleus that restrict gene expression patterns and define identity. These changes are, by definition, epigenetic.
Basis of Epigenetic Phenomena
The study of epigenetic phenomena presupposes that genes can be marked as transcriptionally “on” or “off” by modifications that are independent of the primary nucleotide coding sequence. The DNA in each cell is composed of approximately 3 × 109 nucleotide bases that could presumably stretch to 2 m in length. To facilitate efficient packaging, DNA is wrapped in a threadlike fashion around protein spools called histones. The histone octamer consists of two molecules of each histone: H2A, H2B, H3, and H4.11–13 The histone octamer and the 146 bp of DNA looped around it forms a structure called a nucleosome, the functional unit of chromatin. In addition to serving as packaging facilitators, nucleosomes are amenable to modifications and dynamic remodeling, which makes them an active participant in many chromosomal processes including transcription, replication, DNA repair, kinetochore and centromere construction, and telomere maintenance.
The regulation of gene expression at the chromatin level occurs by: (1) covalent histone tail modifications; (2) adenosine triphosphate (ATP)-dependent chromatin remodeling; and (3) DNA methylation (Figure 24-2).14 This chapter focuses on covalent histone tail modifications and ATP-dependent chromatin remodeling. The amino terminal tails of histones protrude and are subjected to various covalent posttranslational modifications (e.g., methylation, acetylation, sumoylation, phosphorylation).11 These histone tail modifications regulate whether the neighboring DNA is transcriptionally active (euchromatin) or repressed (heterochromatin). Activating histone modifications attract ATP-dependent nucleosome remodeling factors that can reposition or eject histones, thus promoting transcription or repression of the neighboring DNA. Thus, complex interplay between covalent histone modifications, ATP-dependent chromatin remodeling enzymes, and the transcriptional machinery (i.e., DNA binding factors, transcription initiation, elongation complexes) determines whether genes are actively expressed or repressed.
Figure 24-2 A, DNA provides the template that encodes all potential genes. Not all genes are expressed in all cells. Chromatin provides a way of partitioning this genetic information into active and repressed components. The three mechanisms whereby chromatin can modify the expression or repression of genes includes DNA methylation (Me), chromatin remodelers (blue oval), and histone modifications (e.g., phosphorylation, methylation, acetylation) imparted by histone modifiers. (Adapted from Chang CP, Bruneau BG: Epigenetics and cardiovascular development. Annu Rev Physiol 74:41–68, 2012.)
Histone Tail Modifications
From an epigenetic standpoint, the two most important tail modifications are acetylation and methylation.15 Histone tail acetylation is associated with transcriptionally active chromatin. In contrast, histone tail methylation can be associated with either euchromatin or heterochromatin, depending upon the extent of methylation (mono- [me], di- [me2], or tri- [me3] methylation) and the specific tail residues that are modified. For example, lysine (K) residues available for methylation include K4, K9, K27, and K36 of histone H3, and K20 of histone H4. Methylation of H3K9 and H3K27 is associated with gene repression gene expression,16 whereas dimethyl (H3K4me2) and trimethyl (H3K4me3) marks at H3K4 are associated with actively expressed genes.17 As shown in Figure 24-3,18 the extent, specific lysine residue, and location of different histone methylation marks around a gene provide a signature for the transcriptional state of that gene. For example, H3K27me3 marks (black line) are located around the 5′ regulatory region of silenced genes. In contrast, H3K4me3 marks are highly enriched at the 5′ transcription start site of actively expressed genes. In contrast, H3K4me3 marks are not as enriched around poised and silenced genes.
Figure 24-3 Epigenetic signatures differ around genes that are repressed (red rectangle), poised (black rectangle), and active (green rectangle). H3K27me3 marks (red line) are enriched at the 5′ region of silenced genes H3K4me3 marks (green line) are enriched at actively expressed genes. Thus active genes show enrichment of H3K4me3 marks and a paucity of H3K27me3 marks at the 5′ region. H3K36me3 (yellow line) marks are enriched at active genes. This illustrates the histone code hypothesis, which suggests that the epigenetic signature of individual genes, determines whether that gene is actively expressed or repressed. (Adapted from Lee BM, Mahadevan LC: Stability of histone modifications across mammalian genomes: implications for “epigenetic” marking. J Cell Biochem 108:22–34, 2009.)
[/not-level-membership-for-cardiovascular-category]
