2
Epidemiology, genetics and screening for oesophageal and gastric cancer
Definitions
The concentration of disease around the oesophagogastric junction has created differences in opinion with regard to classification. This partly reflects differences in the pathological behaviour of tumours arising at the different sites. In addition, the recent change in the TNM classification1 has described cancers as either oesophageal, including all within 5 cm of the oesophagogastric junction, or gastric. In epidemiology it is important to ensure a clear classification in order to understand differences in incidence and to appreciate aetiological evidence for the observed changes in these cancers.
For the purposes of the following discussion, carcinoma of the oesophagus will include cancers of the thoracic and abdominal oesophagus but will exclude the cervical oesophagus. Oesophagogastric junctional cancers will be considered according to the Siewert and Stein classification:2
• Type I is adenocarcinoma of the distal oesophagus, which usually arises from an area of Barrett’s metaplasia and which may infiltrate the oesophagogastric junction from above.
• Type II is true carcinoma of the cardia arising from the cardiac epithelium or short segments with intestinal metaplasia at the oesophagogastric junction, often referred to as ‘junctional carcinoma’.
• Type III is subcardial gastric carcinoma that infiltrates the oesophagogastric junction and distal oesophagus from below.
Non-cardia gastric cancer will include all cancers of the fundus, body and pyloric antrum.
Epidemiology
Oesophageal cancer
Carcinoma of the oesophagus (ICD code 150) was the eighth commonest cancer in 2008.3 Worldwide there were 481 000 new cases representing 7% of the total cases of cancer. Mortality was high, with 406 000 deaths or 84% of all registered cases. Incidence varies across the world, with the highest risk in the so-called Asian ‘oesophageal cancer belt’, which extends from Northern Iran through Central Asia to North Central China. SCC predominates in these less developed countries, reflecting low socio-economic status and poor diet. Overall, the male to female ratio is 2.1:1, although there are variations. In more developed countries the incidence of SCC has declined, with age-specific rates in white males in the USA at 2.2 per 100 000.4 However, there have been increasing trends in some regions; for example, in Scotland rates are increasing in women and decreasing in men, possibly reflecting the changing patterns of tobacco and alcohol consumption.
Oesophageal and oesophagogastric junctional adenocarcinoma
Adenocarcinoma of the oesophagus and junction accounts for variable proportions of oesophageal cancer across the world, ranging from 0% in parts of China to 10% in Northern Europe to 48% in the UK.4 Adenocarcinoma of the oesophagogastric junction includes cancer located in the distal third of the oesophagus and the cardia of the stomach. Although there is a male predominance there are differences according to organ of origin. For oesophageal tumours the male to female ratio is 2.6:1 and for gastric origin tumours 4:1. Recent data from England show an increase in incidence of lower third oesophageal cancer from 8.1 per 100 000 in 1998 to 10.1 per 100 000 in 2007, although the rate of increase has stabilised since 2002.5 Over the same period there has been a slight decrease in cardia cancer incidence. The peak age group affected is between 50 and 60 years of age.
Gastric cancer
Gastric cancer (ICD code 151) is the fourth most frequent cancer worldwide.3 In 2008 there were 988 000 new cases with 737 000 deaths. This represents 14% of all new cases of malignancy and 10.3% of all cancer deaths. Crude numbers are still increasing in relation to demographic changes of the ageing population throughout the world. The majority of cases occur in less developed countries, where the male to female ratio is 1.8:1. This contrasts with a ratio of 1.6:1 in more developed countries. Highest incidence rates are found in Japan (male 69.2 per 100 000 and female 28.6 per 100 000). Other countries with high incidence include East Asia, Korea, Eastern Europe, and Central and South America. Although distal cancers still predominate in countries with highest incidence, there has been a fall in mid and distal gastric cancer, with a progressive increase in cardia cancer.
Aetiology
Squamous cell carcinoma of the oesophagus
Socio-economic and dietary influences
Areas of highest incidence are those countries of low socio-economic status where poverty and malnutrition predominate. The development of SCC appears to be related to a type of chronic oesophagitis that is different from that found in the West and is often complicated by atrophy and dysplasia (see Chapter 1). It is not usually associated with gastro-oesophageal reflux and is often asymptomatic.
SCC has been associated with ingestion of very hot beverages, a family history of oesophageal cancer, prevalence of oesophagitis among siblings, and a low intake of fresh fruits and wheat flour products.6 Furthermore, riboflavin deficiency and vitamin A and C deficiency7 have been identified as risk factors that are particularly important at a young age. By contrast, vitamin C intake confers a protective benefit; Hu et al.,8 in a case–control study, found that 100 mg of vitamin C per day decreased risk by 39%.
Associated conditions
Achalasia is associated with SCC, but the magnitude of the risk is unclear. Brucher et al.9 report from their single institution series that the risk of developing a carcinoma in long-standing achalasia is increased 140-fold when compared with the general population. The risk appears to relate to retention oesophagitis secondary to stasis and exposure to possible carcinogens in fermenting food residue. There is a lead time of approximately 15–20 years and these cases probably warrant long-term surveillance. Treatment of the achalasia does not seem to reduce the risk.
Tylosis palmarum is a rare inherited autosomal dominant condition in which there is a very high incidence of SCC. Perhaps of greater significance is the finding of the increased risk in low-risk areas for offspring of parents with oesophageal cancer.10 There are numerical and structural chromosomal aberrations in patients with a family history not seen in those without a family history (see below).
Adenocarcinoma of the oesophagus and junctional cancers
Gastro-oesophageal reflux disease (GORD)
Gastro-oesophageal reflux is now the most common symptomatic presentation of all conditions affecting the upper gastrointestinal tract. Estimates suggest that 4–9% of all adults experience daily heartburn and up to 20% experience symptoms on a weekly basis.11 Of these, 60% have no endoscopic abnormality, 30% have oesophagitis and 10% have Barrett’s columnar lined oesophagus. Many are self-treated and do not attend for further investigation, yet 80% with Barrett’s are asymptomatic. The relationship of GORD and oesophageal ACA has been evaluated in case–control studies.12 The individual cancer risk is small because of the high frequency of GORD. Lagergen et al.13 have estimated the risk of developing ACA of the oesophagus by scoring symptoms of heartburn and regurgitation (alone or in combination), timing of symptoms (particularly at night) and frequency of symptoms. Among those with recurrent symptoms of reflux, the odds ratio of developing cancer was 7.7 in comparison with those without symptoms. More frequent, more severe and longer-lasting symptoms of reflux were associated with a much greater risk (odds ratio 44). The risk associated with GORD is related to the development of Barrett’s metaplasia, which is greatest among Caucasian males with a history of alcohol consumption and continuous smoking. Further detailed discussion of the role of Barrett’s in the aetiology of ACA is presented in Chapter 15.
Obesity and dietary factors
In the last 20 years the incidence of junctional cancer has increased in parallel with the epidemic of obesity. There is a three- to sixfold excess risk among overweight individuals.14 Obesity predisposes to hiatus hernia and reflux, and hence contributes mechanically to increase risk. However, data from a number of studies demonstrate an effect independent of reflux. Lindblad et al.15 have reported a 67% increase in the risk of oesophageal ACA in patients with a body mass index (BMI) greater than 25, and this increases with increasing BMI. This effect was noted irrespective of the presence of reflux symptoms.
There appears to be a sex difference in that the effect was only found in women with a BMI greater than 30, whereas in men it was observed in both overweight and obese individuals. Recently this effect in women has been confirmed, with 50% of cases of oesophageal adenocarcinoma in postmenopausal women in the Million Women study being attributed to obesity.16
Evidence is accumulating to support different types of obesity. The distribution of abdominal fat tends to be central and retroperitoneal. This acts as a potent source of growth factors, hormones and regulators of the cell cycle. Such individuals develop the metabolic syndrome, which is linked to raised serum cholesterol and triglycerides, hypertension and hyperglycaemia. In the general population the metabolic syndrome occurs in 10–20%. Power et al.17 have demonstrated that 46% of those with Barrett’s oesophagus and 36% of those with GORD have features of the metabolic syndrome.
The factors released by centrally deposited fat may have an effect on the process of metaplasia transforming to dysplasia. Vaughan et al.18 have examined the potential relationship between a series of biological markers of progression from metaplasia to cancer in obese and overweight patients. There was little evidence of change in the biomarkers in association with increasing obesity. However, abnormalities in the biomarkers were observed in individuals with high anthropometric measures of abdominal fat. The study concluded that increased BMI contributed to reflux and development of metaplasia but it was the ‘male pattern’ of abdominal obesity that was actually associated with malignant transformation.
Helicobacter pylori
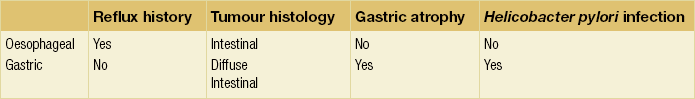
There is accumulating evidence that there may be two distinct types of junctional cancer reflecting the two potential sites of origin. McColl and Going19 have suggested that one is similar to oesophageal cancer and the other gastric cancer. In a series of studies of patients with junctional cancer, they evaluated H. pylori infection from serology, gastric atrophy from pepsinogen I and II ratios, symptoms of reflux and histological subtype of diffuse or intestinal type. They also included biopsies of the distal stomach to document atrophy associated with Helicobacter. Tumours of oesophageal origin are intestinal type with no evidence of gastric atrophy or Helicobacter infection and occur in the context of reflux. By contrast, tumours of gastric origin are diffuse type or intestinal but with evidence of atrophy and Helicobacter infection and without a history of reflux (Table 2.1). Such different characteristics would imply a different carcinogenic process at the two sites and should be considered in prognosis and patient management.
Socio-economic factors
Lifestyle has an effect on the risk for junctional cancers. There is an association with lower socio-economic class but this is not as strong as for SCC. Powell and McConkey20 demonstrated that the increase of ACA of the lower third of the oesophagus and the cardia was mainly in social classes I and II – that is, in professional and managerial occupations. In addition, in a large surgical series, Siewert and Ott21 reported that patients with ACA were more frequently from an educated background, a characteristic not present in the population with SCC. However, the effect of socio-economic class may not be independent as, when adjusted for GORD, BMI and smoking, Jansson et al.22 found the effect to be less apparent.
Gastric cancer
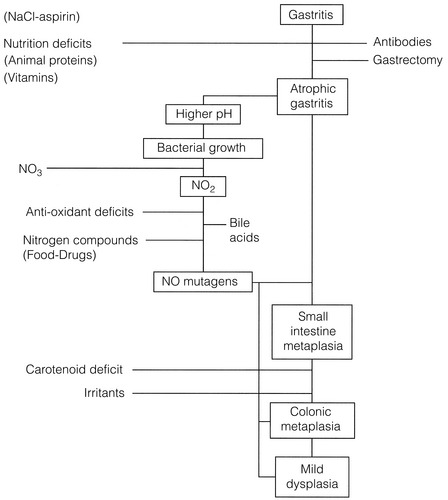
The Correa hypothesis23 (Fig. 2.1) describes the steps in the process of malignant transformation for gastric cancer. It highlights where environmental factors stimulate changes, particularly in the development of intestinal-type gastric cancer (see Chapter 1). These include socio-economic and dietary influences, as well as exposure to carcinogens.
Socio-economic influences
Exposure to potentially carcinogenic agents at an early age is clearly crucial to the risk of developing both precursor lesions and subsequent gastric cancer. Evidence for this risk is available from migrant studies. Japanese migrants to the USA had a far lower rate of intestinal-type cancers than the equivalent population who remained in Japan, indicating environmental or dietary aetiology, whereas the rates of diffuse-type cancer remained the same, suggesting a hereditary component.24
Diet
Salt preservation of food was common during the early years of the 20th century throughout the world; in some landlocked parts of the world this still occurs. In such areas and in those still using salt preservation there have been high rates of gastric cancer. The consumption of salted and pickled fish is high in Japanese and Colombians and correlates with their disease incidence. On the basis that salt induces injury to the gastric mucosa it may act like high carbohydrate intake, as an initiator to allow access for more potent carcinogens. By contrast, the rapid and widespread adoption of refrigerators in the 1950s and 1960s has significantly affected the preservation of fresh foods. The reduction in mortality observed in Japan shows an inverse relationship with the increase in ownership of domestic refrigerators.25
Helicobacter pylori
In 1994 the International Agency for Research on Cancer designated H. pylori to be a type I carcinogen26 for gastric cancer. The initial effect of H. pylori is acute inflammation. Since the infection does not resolve spontaneously, an effect is likely to persist and may proceed to chronic gastritis and associated mucosal atrophy and intestinal metaplasia, dysplasia and eventually cancer. The evidence for its role is from a number of sources. Areas of high cancer incidence have a high rate of H. pylori infection. In a prospective population-based study in Japan, 2.9% of those infected developed gastric cancer compared with none from the uninfected population; 4.7% of those infected who had non-ulcer dyspepsia progressed to cancer.27 In high incidence areas H. pylori infection tends to occur early in life. These early rates of infection are linked to low income, poor education, poor sanitation and overcrowding. There has, however, been a progressive fall in rates of H. pylori serology positivity in longitudinal studies, which have paralled the decline in gastric cancer incidence.
Although the evidence for H. pylori inducing gastric cancer is convincing, not all those infected develop the disease. The risk of malignant transformation appears to be enhanced by bacterial virulence and host factors (see below). Helicobacter pylori with cytotoxin-associated gene A (cagA) appears to be associated with the greatest risk.28 In the West, 60% of H. pylori infections are cagA positive compared with 100% in Japan.29,30 It is likely that H. pylori induces an environment that is susceptible to malignant transformation. It induces tissue monocytes to produce reactive oxygen intermediates, which are potent carcinogens. Infection is associated with a significant reduction in gastric juice ascorbic acid,31 which acts to scavenge and suppress N-nitroso compounds and oxygen free radicals. It also facilitates the proliferation of nitrosating bacteria, which promote the development of N-nitroso compounds.
Prevention of oesophageal and gastric cancer
In oesophageal and gastric cancer primary prevention approaches are currently limited to population education to alter social habits (such as decreasing or stopping tobacco or alcohol consumption) and dietary habits (such as maintaining a diet containing fresh fruit and vegetables with a low or minimal salt intake). In addition, the need to prevent obesity is now well established. The role of H. pylori eradication is important but programmes of eradication should only be considered according to the level of risk for oesophageal or gastric cancer in the population. In populations with a high risk of gastric cancer, eradication is indicated; however, in populations in which oesophageal ACA is common, eradication may have an adverse effect. The overall benefit of these approaches would be greatly enhanced if specific markers of risk could be identified to focus prevention strategies (see Chapter 15).
Secondary prevention depends upon understanding the natural history and detection of premalignant conditions. In SCC there is limited evidence that secondary measures could be effective because of lack of understanding of the histological changes leading to cancer. In oesophageal ACA, surveillance of Barrett’s metaplasia to identify progression to dysplasia is theoretically a positive approach (see Chapter 15). Identification of p53 expression and aneuploidy in biopsies of Barrett’s has been shown to predict the risk of progression.32 In both gastric and oesophageal cancer there is a potential role for chemoprevention. Increasing levels of cyclo-oxygenase-2 (COX-2) are present in the progression of atrophic gastritis to intestinal metaplasia and gastric cancer.33 Smoking, acid and H. pylori are all associated with COX-2 expression. Aspirin and other non-steroidal agents inhibit COX-2 and their use may act as a chemopreventive for gastric cancer. Aspirin also seems to have an effect in Barrett’s metaplasia and in combination with acid suppression may minimise progression to dysplasia. The ASPECT trial in the UK is assessing whether such a strategy can have a secondary preventive effect.34
Genetics of oesophageal and gastric cancer
Oesophageal cancer
Evidence for an inherited type of oesophageal cancer is limited. However, the rare skin condition of tylosis palmaris and familial clustering for Barrett’s cancer raise the possibility of an hereditary risk. The relationship of tylosis palmaris with SCC had been recognised from epidemiological studies. Investigation of a group of families in Liverpool including several generations has identified a specific tylosis oesophageal cancer gene.35 Subsequent studies have detected this gene in 69% of cases of sporadic SCC. Recently, more specific proteins coded for by this gene have been reported to be related to poorly differentiated SCC and potentially predict for those with a poorer prognosis.36
There have been a number of reports of families with Barrett’s metaplasia who have developed adenocarcinoma. In these families the frequency of Barrett’s was more than 20% and the frequency of GORD was approximately 40%. In a case–control study, 24% of those with Barrett’s, oesophageal or junctional ACA had a family history compared to 5% in the control group.37 Multivariate analysis confirmed that family history was an independent risk factor with equal weighting to age, male gender, obesity and alcohol consumption. Further analysis of similar families is required to evaluate possible genetic linkages.
There are very few studies examining polymorphisms in sporadic adenocarcinoma and those that have been done show weak associations with risk of cancer development. In Barrett’s it is likely that there are host polymorphisms, which interact with the environmental factors to promote progression to malignant transformation. The largest body of evidence supports a role for p53 and aneuploidy as markers of risk (see Chapter 15).
Gastric cancer
Hereditary diffuse gastric carcinoma (HDGC)
The first description of a germ-line mutation was in 1998 in three New Zealand Maori families. Mutations in the CDH1 tumour suppressor gene (the E-cadherin gene) have since been described in several families of different ethnic backgrounds. The CDH1 mutation occurs along the gene in these families as opposed to clustering in one site as observed in sporadic cases. In order to develop a common approach for HDGC, the International Gastric Cancer Linkage Consortium defined HDGC as including families with more than two pathologically proven diffuse gastric cancers in individuals under 50 and families with more than three close relatives with pathologically proven diffuse gastric cancer at any age.38
HDGC is an autosomal dominantly inherited syndrome. Carriers of the CDH1 mutation have in excess of 70% lifetime risk of developing diffuse gastric cancer. Female carriers have an additional risk of lobular breast cancer in about 40% of patients. Screening for CDH1 mutations in the research setting has shown its presence in 40% of families with multiple gastric cancers and at least one diffuse gastric cancer in a member under 50. The criteria for potential screening have been extended to include a number of other combinations of presentation (Box 2.1).
The clinical issue for these families is their optimal management. In view of the 70% or more chance of developing diffuse gastric cancer with its attendant poor long-term survival, the options are either prophylactic total gastrectomy or endoscopic surveillance. There have been a number of reports of the pathology of resected stomachs after prophylactic gastrectomy. In clinically, endoscopically ‘normal’ stomachs, supported by biopsy, diffuse multifocal intramucosal disease was identified in all specimens.39,40 Some studies have shown concentration of disease in the distal third, whereas others have more widespread involvement.41 The question arises, however, as to the appropriateness of total gastrectomy in essentially a young population for whom the nutritional sequelae will be lifelong. Furthermore, the morbidity and mortality of such a procedure must be minimal. Therefore the role for endoscopy must be explored. Standard surveillance is limited but the distribution of disease from the pathology studies indicates where biopsies should be concentrated. Advances in endoscopy, which include endoscopic autofluorescence spectroscopy and chromoendoscopy, have the potential to enhance accuracy. A further point is that 20–30% of CDH1 germ-line mutations do not progress to clinically diffuse gastric cancer. There are some data to suggest intramucosal disease may not progress and be of biological rather than clinical importance, analogous to prostatic cancer in elderly men. Thus, counselling of individuals from HDGC families produces very difficult questions, particularly as knowledge is incomplete as to risk, most appropriate management and the role of genetic intervention, as well as the sequelae of life following total gastrectomy.
Hereditary cancer syndromes
The development of molecular genetics has allowed confirmation of primary genetic aetiology for a spectrum of cancers which epidemiology studies had suggested were inherited (Table 2.2). Gastric cancer has been found to be coexistent in these syndromes, further supporting a genetic basis for its development.42 There are differences across the world, consistent with evidence that the gene pool varies within different populations. In patients with familial adenomatous polyposis (FAP) there is an excess of gastric cancer in Japanese families that is not observed in US non-oriental families. Similarly, in the Lynch syndrome, gastric cancer is more common in China and Korea yet rare in Caucasians. Thus, screening surveillance in such populations should be directed accordingly. Unless there is gastric cancer in the family then upper gastrointestinal endoscopy is not routinely required in those with the Lynch syndrome.
Table 2.2
| Syndrome | Main tumours | Associated tumours |
| Lynch syndrome (hereditary non-polyposis colorectal cancer) | Colon carcinoma | Endometrial, gastric, small bowel and urothelial cancer |
| Li–Fraumeni syndrome | Breast cancer, osteosarcoma, brain tumours, soft tissue sarcoma | Gastric and colon cancer, adrenocortical carcinoma, haematological and gynaecological |
| Familial adenomatous polyposis coli | Colon cancer | Gastric cancer, papillary thyroid cancer, desmoid tumours, medulloblastoma and hepatoblastoma |
| Peutz–Jeghers syndrome | Hamartomatous polyps of the small bowel, colon and stomach | Gastrointestinal carcinomas, breast, testicular and ovarian cancers |
| Juvenile polyposis | Hamartomatous polyps of the colon and occasionally stomach and small bowel | Gastrointestinal cancer |
Moderate cancer risk
Worldwide studies have shown that approximately 5–10% of patients with gastric cancer have a family history but without other features to suggest an inherited aetiology. However, it is possible that in this population there is some hereditary predisposition to increased susceptibility to environmental factors such that their risk is increased. Studies have shown increased rates of H. pylori infection with atrophic gastritis and hypochlorhydria in first-degree relatives of gastric cancer patients compared with normal controls. This could of course be purely due to environmental factors. Alternatively, normal variations in the genetic coding sequence of multiple genes (polymorphisms), which are inheritable, may lead to differential inflammatory responses to agents such as H. pylori or tobacco. Thus, the combined effect of inflammation promoting host genetic polymorphisms and different microbiological genotypes such as CagA H. pylori may increase the risk in a particular population. Specific studies including p53 have shown certain polymorphisms to be associated with the production of variant proteins.43 These have been identified more frequently in patients with diffuse gastric cancer than in matched controls. DNA polymorphism in the interleukin-1 gene cluster has been associated with a response to H. pylori infection. It is postulated that the polymorphism increases the production of interleukin-1β, a proinflammatory cytokine, which inhibits gastric acid secretion and hence achlorhydria and gastric atrophy.
Molecular genetics of oesophageal and gastric cancer
The development and progression of oesophageal and gastric cancer has been clearly demonstrated in numerous studies to have a genetic basis. Alterations in tumour suppressor genes and oncogenes have been identified in both cancers. Specifically genes, which have roles in diverse functions such as cell adhesion, signal transduction, differentiation, development, gene transcription or DNA repair, have been demonstrated in both oesophageal and gastric cancer. Figure 2.2 shows some of the changes described in oesophageal cancer arising in Barrett’s metaplasia, and Fig. 2.3 shows the changes in gastric cancer and highlights different mechanisms for the intestinal and diffuse types. Studies of cDNA microarrays for gastric cancer have reported characteristic patterns of gene expression in chronic gastritis, intestinal metaplasia, and intestinal and diffuse gastric cancer. These raise opportunities for identification of molecular markers and gene profiling in cancer progression and for the prediction of prognosis and treatment sensitivity.42,44 Studies of host genetic factors are likely to provide vital information to explain the diverse risks in differing populations. This may require whole genome sequencing studies but these may be limited by the capability of bioinformatics to cope with the associated vast amounts of data.
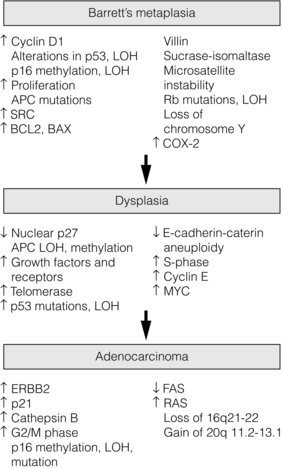
Figure 2.2 Genetic changes described in the progression from Barrett’s metaplasia to oesophageal adenocarcinoma. Reproduced from Lin J, Beer DG. Molecular biology of upper gastrointestinal malignancies. Semin Oncol 2004; 31:476–86. With permission from Elsevier.
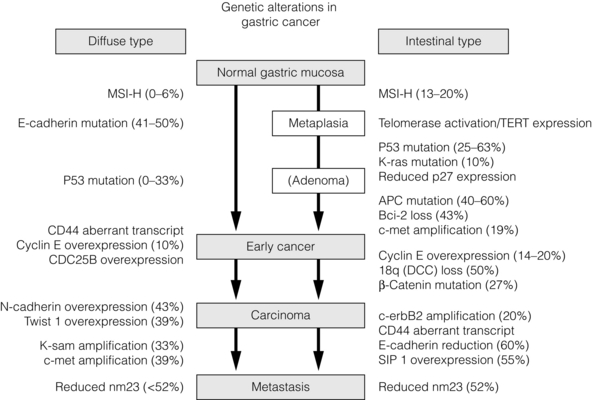
Figure 2.3 Genetic alterations described in gastric cancer. Abbreviations: APC, adenomatous polyposis coli; Bcl-2, B-cell CLL/lymphoma 2; CD44, CD44 antigen; CDC25B, cell division cycle 25B; c-erbB2, v-erb-B2 erythroblastic leukaemia viral oncogene homologue 2; c-met, met proto-oncogene (hepatocyte growth factor receptor); DCC, deleted in colon cancer; K-ras, v-Ki-ras 2 Kirsten rat sarcoma viral oncogene homologue; K-sam, encodes fibroblast growth factor receptor 2; MSI-H, microsatellite instability – high; nm23, non-metastatic cells 1 (protein, NM23, expressed in); p53, tumour protein p53 (Li–Fraumeni syndrome); SIP-1, SMAD-interacting protein 1; TERT, telomerase reverse transcriptase; TWIST 1, twist homologue 1. Reproduced from Keller G, Hofler H, Becker K-F. Molecular medicine of gastric adenocarcinomas. Expert Rev Mol Med 2005; 7:1–13. With permission from Cambridge University Press.
Screening for oesophageal and gastric cancer
Asymptomatic screening
Evaluation of asymptomatic screening for carcinomas of the oesophagus has centred on those parts of China with the highest incidence. The screening test involves swallowing a small deflated balloon, which is then inflated at the lower end of the oesophagus. The balloon surface is covered with a fine mesh; on withdrawal from the oesophagus, this scrapes the mucosa to collect cells. A cytological smear is then made from the scrapings for microscopic examination. Those individuals found to have abnormalities are then subjected to endoscopy and appropriate biopsy. In 132 subjects with early oesophageal cancer detected in this way, 26% had normal radiological appearances.45
The efficiency of this technique has had varying reports. Reviewing data based on 500 000 examinations, Shu46 suggested an accuracy for the differentiation of benign from malignant of 90%. Mass surveys have shown that 73.8% of detected cancers were either in situ or minimally invasive. In a provincial review, Huang47 reported on 17 000 examinations screened during a 1-year period. Abnormalities were found in 68% of the population, with low-grade dysplasia in 37%, high-grade in 26% and in situ cancer in 2%. A group with high-grade dysplasia were followed for up to 8 years. Regression to normal or low-grade change was observed in 40%, 20% remained as high grade, 20% fluctuated between high and low grade, and 20% developed cancer. In the absence of dysplasia, 0.12% developed cancer. Progression from dysplasia to in situ cancer occurred over 3–12 years and from in situ to invasive cancer over 3–7 years. Tumour risk was consistent, with a known distribution of middle-third chronic oesophagitis in 76%. It would seem that the duration of severe dysplasia is the greatest risk for malignant transformation. Follow-up by endoscopy is therefore important and in order to ensure biopsy of the same site vital stains have been used. Huang48 reported that staining with toluidine blue was effective for identifying neoplastic epithelium; 84% of cancers were identified in positively staining areas.
The problem associated with this approach is the management of dysplasia. Oesophageal dysplasia is a dynamic process with both spontaneous regression and progression. Furthermore, even if in situ cancer develops, progress to advanced disease is often prolonged and may be associated with prolonged survival. In one series of 23 untreated patients, 11 developed late-stage disease at a mean of 55 months. In the remainder there was no change for over 6 years and the 5-year survival of the group was 78%.49 Five-year survival needs to be considered with caution as detection of asymptomatic slowly progressive disease introduces lead-time bias and this can falsely give the impression that treatment results for screen-detected cases are better.
As a result an International Union Against Cancer (UICC) recommendation has been to limit oesophageal cancer screening to areas of high risk.50 The aim is to identify the natural history of dysplasia more completely. Common standards are required for the classification of dysplasia to identify those changes with greatest risk. Once the assessment is more reliable, control studies should be developed to determine whether screening intervention could reduce mortality for oesophageal squamous cell cancer.
Gastric cancer
The prominence of gastric cancer as a public health problem in Japan led to the development during the 1960s of a mass screening programme for all men over the age of 40 years. The programme has been based on double-contrast radiology with endoscopic assessment of any abnormalities.51 Members of the public are invited to undergo radiology in mobile units at which seven films are taken after the ingestion of an effervescent contrast agent. Screening is undertaken annually or biannually depending on the area of Japan and the associated risk of disease. Government recommendations set a target of 30% for the annual examination rate. Despite the recognition of gastric cancer as a public health problem, attendance for screening is low. In 1985 over 5 million were examined, representing 13% of the at-risk population. Therein lies one of the problems with any screening programme, namely the cooperation of the public.
Oshima et al.52 compared screened and unscreened populations to determine whether screening was important over and above the other influences on the decrease in mortality. In a case-controlled study they found that the risk of dying from gastric cancer among screened cases was at least 50% less than that for non-screened cases. Other Japanese groups have reported similar results. However, the actual effect on mortality remains to be proven as none of the studies have been randomised or controlled. As a result the UICC recommended that studies should be continued in Japan to resolve the problem, but screening in this way should not be adopted as public health programmes in other parts of the world.50
Symptomatic screening and early detection
Studies have evaluated methods of selecting those potentially at higher risk of having a significant diagnosis. Dyspepsia has been classified as uncomplicated or complicated by alarm symptoms including weight loss, anorexia, vomiting, dysphagia and signs of anaemia or an abdominal mass. Further classification according to age has also been studied as early gastric cancer tends to present approximately 10 years younger than advanced disease.53 Although such studies have increased rates of detection of early gastric cancer to approximately 15–20%, many patients with uncomplicated dyspepsia have undergone normal examinations. In a series of 25 patients under 55 years with gastric cancer, 24 had complicated dyspepsia.54 Furthermore, in a population database of 3293 oesophageal and gastric cancers, 290 were under 50 and 21 (7%) had uncomplicated dyspepsia.55 The simple conclusion of this evidence is to restrict endoscopy for those under 55 years to complicated dyspepsia. However, the alarm symptoms used to define complicated dyspepsia are those of established locally advanced disease with the expected poor prognosis. This has been confirmed in a large case series of open access endoscopy from Newcastle upon Tyne.56 It could be argued that the low index of suspicion for the significance of simple dyspepsia in younger patients had led to a delay in investigation until they developed more significant or alarm symptoms. The failure to diagnose earlier cancers in younger patients may be a result of a failure to initiate investigations until the cancer is advanced and raising the age threshold to 55 for uncomplicated dyspeptics would decrease the rate of diagnosis of upper gastrointestinal cancer. Indeed, the effect of early intervention in unselected dyspepsia not only increases the rate of earlier diagnoses of cancer, but this is also translated into a survival advantage (Fig. 2.4).57
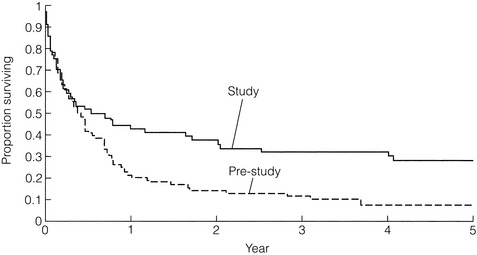
Figure 2.4 Survival after early detection of gastric cancer (study population) compared with historical control population (pre-study population).
A pragmatic approach has been adopted in the UK.58 Urgent specialist referral or endoscopic investigation (within 2 weeks) is indicated for people with dyspepsia of any age when presenting with chronic gastrointestinal bleeding, progressive unintentional weight loss, iron-deficiency anaemia, progressive dysphagia, persistent vomiting, epigastric mass or suspicious barium meal. In addition to these alarm symptoms, similar referral is required for a dyspeptic patient over 55 years with onset of symptoms within the last year and/or continuous symptoms since onset. The advantage of referral within 2 weeks is largely procedural and has only limited support from the literature. For example, gastric cancers limited to the mucosa and submucosa have a doubling time of 1.5–10 years, whereas advanced disease has a doubling time of between 2 months and 1 year.59,60 Reducing symptomatic delay is unlikely to alter outcome for early disease significantly, but may render more advanced disease amenable to resection. In a comparative audit of 2-week referrals (TWRs) with conventional presentations, Radbourne et al.61 have found that although the TWR produced more cancers, the stage of disease was equivalent at diagnosis and survival was comparable between the two groups.
High-risk groups
GORD and Barrett’s oesophagus: A variety of approaches have been assessed for early diagnosis in patients with GORD and at risk of Barrett’s oesophagus. Small-calibre nasal endoscopy in unsedated patients has been evaluated for accuracy of detecting Barrett’s in patients with GORD. It was found to be well tolerated, technically feasible and accurate, despite producing smaller biopsy samples.62
A modification of the Chinese wire mesh covered balloon used to screen for SCC has recently been reported as a non-endoscopic screening test for Barrett’s oesophagus in a population with a history of GORD.63 The device, Cytosponge, has a similar design in which the patient swallows a gelatin capsule attached to a length of string. The capsule contains a compressed mesh, which expands to 3 cm in diameter once the outer gelatin coating dissolves in the proximal stomach. The string is withdrawn and the mesh takes samples of cells from the oesophageal mucosa. The mesh is processed to produce a cytological preparation, which is stained with a marker for Barrett’s oesophagus. Further evaluation is required of this novel technique to determine if this has a role in selecting those with GORD for surveillance.
Helicobacter pylori: The role of H. pylori as a marker for endoscopy has received considerable attention. Both serological estimation and breath tests depending on exhalation of urea have been investigated. Serology has been assessed for concordance with the underlying histological presence of H. pylori. Farinati et al.64 found 82% agreement between a measurable antibody response and histological evidence of H. pylori infection. Urea breath tests are in routine use in Helicobacter eradication programmes for duodenal ulceration. Again, the problem is one of specificity and sensitivity.
Helicobacter pylori seropositivity does not necessarily imply active infection. Equally, seropositivity is a common finding and may not be specific for the at-risk population. It increases with age and to a certain extent parallels gastric atrophy, which is equally an age-related phenomenon and in the majority does not progress to cancer. There is also evidence that seroreversion may occur, with seropositivity frequently seen in early gastric cancer and seronegativity in more advanced disease.65 Whiting et al.66 reported a retrospective analysis of H. pylori seropositivity in cancer patients compared with a group of undiagnosed dyspeptics. Although the cancer patients were significantly more likely to be seropositive, this was very much site related; cardia cancers were not usually seropositive. Thus, any screening programme based on H. pylori serology could miss proximal tumours, which are currently the more common cancers. Further investigation is required and longitudinal studies may resolve the issue of whether patients with H. pylori seropositivity warrant close endoscopic follow-up.
Gastric atrophy and intestinal metaplasia: Those found at endoscopic biopsy to have gastric atrophy and columnar-type gastric intestinal metaplasia may also form a risk group. Whiting et al.67 have followed a group of patients by annual endoscopy who were found to have chronic atrophic gastritis and intestinal metaplasia at diagnostic endoscopy for dyspepsia. This group was reported to have an 11% risk of developing gastric cancer and the authors suggest that such patients should be considered a high-risk group.
References
1. UICC, International Union Against Cancer. TNM classification of malignant tumours, 7th ed. New York: Wiley-Blackwell Press, 2009.
2. Siewert, J.R., Stein, H.J., Classification of adenocarcinoma of the oesophago-gastric junction. Br J Surg 1998; 85:1457–1459. 9823902
3. Globocan 2008. Online. Available at http://globocan.iarc.fr; [accessed 16.11.12].
4. Lambert, R., Hainaut, P., Epidemiology of oesophagogastric cancer. Best Pract Res Clin Gastroenterol 2007; 21:921–945. 18070696
5. Coupland, V.H., Allum, W., Blazeby, J., et al, Incidence and survival of oesophageal and gastric cancer in England between 1998 and 2007, a population-based study. BMC Cancer 2012; 12:11. 22239958
6. Chang-Claude, J.C., Wahrendorf, J., Liang, Q.S., et al, An epidemiological study of precursor lesions of oesophageal cancer among young persons in a high risk population in Huixian. China. Cancer Res 1990; 50:2268–2274. 2317814
7. Esophageal cancer studies in the Caspian littoral of Iran: results of population studies–a prodrome, Joint Iran-International Agency for Research on Cancer Study Group. J Natl Cancer Inst 1977; 59:1127–1138. 561853
8. Hu, J., Nyren, O., Wolk, A., et al, Risk factors for oesophageal cancer in northeast China. Int J Cancer 1994; 57:38–46. 8150539
9. Brucher, B.L., Stein, H.J., Bartels, H., et al, Achalasia and oesophageal cancer: incidence, prevalence and prognosis. World J Surg 2001; 25:745–749. 11376410
10. Li, J.Y., Ershaw, A.G., Chen, Z.J., et al, A case–control study of cancer of the oesophagus and gastric cardia in Linxian. Int J Cancer 1989; 43:755–761. 2714880
11. Cameron, A.J., Epidemiology of columnar-lined oesophagus and adenocarcinoma. Gastroenterol Clin North Am 1997; 26:487–494. 9309399
12. Chow, W.H., Finkle, W.D., McLaughlin, J.K., et al, The relation of gastro-oesophageal reflux disease and its treatment to adenocarcinomas of the oesophagus and gastric cardia. JAMA 1995; 274:474–477. 7629956
13. Lagergen, J., Bergstrom, R., Londgren, A., et al, Symptomatic gastro-oesophageal reflux as a risk factor for oesophageal adenocarcinoma. N Engl J Med 1999; 340:825–831. 10080844
14. Cheng, K.K., Sharp, L., McKinney, P.A., et al, A case–control study of oesophageal adenocarcinoma in women: a preventable disease. Br J Cancer 2000; 83:127–132. 10883680
15. Lindblad, M., Rodriguez, L.A., Lagergen, J., Body mass, tobacco and alcohol and risk of oesophageal, gastric cardia and gastric non-cardia adenocarcinoma among men and women in a nested case control study. Cancer Causes Control 2005; 16:285–294. 15947880
16. Reeves, G.K., Pirie, K., Beral, V., et al, Cancer incidence and mortality in relation to body mass index in the Million Women Study; cohort study. Br Med J 2007; 335:1134–1139. 17986716
17. Power, D.G., Ryan, A.M., Healy, L.A., et al, Barrett’s oesophagus: prevalence of central adiposity, metabolic syndrome and a pro-inflammatory state. Proceedings of Gastrointestinal Cancer Symposium. American Society for Clinical Oncology, 2008:70.
18. Vaughan, T.L., Kristal, A.R., Blount, P.L., et al, Non steroidal anti-inflammatory drug use, body mass index and anthropometry in relation to genetic and flow cytometric abnormalities in Barrett’s oesophagus. Cancer Epidemiol Biomark Prev 2002; 11:745–752. 12163328
19. McColl, K.E.L., Going, J.J., Aetiology and classification of adenocarcinoma of the gastro-oesophageal junction/cardia. Gut 2010; 59:282–284. 20207629
20. Powell, J., McConkey, C.C., The rising trend in oesophageal adenocarcinoma and gastric cardia. Eur J Cancer Prev 1992; 1:265–269. 1467772
21. Siewert, J.R., Ott, K., Are squamous and adenocarcinoma of the oesophagus the same disease? Semin Radiat Oncol 2006; 17:38–44. 17185196
22. Jansson, C., Johansson, A.L., Nyren, O., et al, Socioeconomic factors and risk of oesophageal adenocarcinoma within the European Prospective Investigation into Cancer and Nutrition (EPIC). J Natl Cancer Inst 2006; 98:345–354. 16507831
23. Correa, P., A human model of gastric carcinogenesis. Cancer Res 1988; 48:3554–3560. 3288329
24. Correa, P., Sasano, N., Stemmerman, N., et al, Pathology of gastric carcinoma in Japanese populations: comparisons between Miyagi prefecture, Japan, and Hawaii. J Natl Cancer Inst 1973; 51:1449–1459. 4762929
25. Hirayama, T. Actions suggested by gastric cancer epidemiological studies in Japan. In: Reed P.I., Hill M.J., eds. Gastric carcinogenesis. Amsterdam: Excerpta Medica; 1988:209–228.
26. International Agency for Research on Cancer Working Group on the Evaluation of Carcinogenic Risks to HumansSchistosomes, liver flukes and Helicobacter pylori. Luon: International Agency for Research on Cancer, 1994.
27. Uemara, N., Okamoto, S., Yamamoto, S., et al, Helicobacter pylori infection and the development of gastric cancer. N Engl J Med 2001; 345:784–789. 11556297
28. Tomb, J.F., White, O., Kerlavage, A.R., et al, The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature 1997; 388:539–547. 9252185
29. Vicari, J.J., Peek, R.M., Falk, G.W., et al, The seroprevalence of cagA-positive Helicobaster pylori: strains in the spectrum of gastro-oesophageal reflux disease. Gastroenterology 1998; 115:50–57. 9649458
30. Ito, Y., Azuma, T., Ito, S., et al, Analysis and typing of the vacA gene from cagA-positive strains of Helicobacter pylori isolated in Japan. J Clin Microbiol 1997; 35:1710–1714. 9196179
31. Sobala, G.M., Schorah, C.J., Shires, S. Gastric ascorbic acid concentration and acute Helicobacter pylori infection. Rev Esp Enf Digest. 1990; 78(Suppl. 1):63.
32. Fitzgerald, R.C., Molecular basis of Barrett’s oesophagus and oesophageal adenocarcinoma. Gut 2006; 55:1810–1818. 17124160
33. Ristimaki, A., Houkanen, N., Jankala, H., et al, Expression of cyclo-oxygenase-2 in human gastric carcinoma. Cancer Res 1997; 57:1276–1280. 9102213
34. Jankowski, J., Barr, H., Improving surveillance for Barrett’s oesophagus: AspECT and BOSS trials provide an evidence base. Br Med J 2006; 332:1512. 16793832
35. von Brevern, M., Hollstein, M.C., Risk, J.M., et al, Loss of heterozygosity in sporadic oesophageal tumours in the tylosis oesophageal cancer gene region of chromosome 17q. Oncogene 1998; 17:2101–2105. 9798681
36. Moodley, R., Reddi, A., Chetty, R., et al, Abnormalities of chromosome 17 in oesophageal cancer. J Clin Pathol 2007; 60:990–994. 17046844
37. Chak, A., Lee, T., Kinnard, M.F., et al, Familial aggregation of Barrett’s oesophagus, oesophageal adenocarcinoma, and oesophago-gastric junctional adenocarcinoma in Caucasian adults. Gut 2002; 51:323–328. 12171951
38. Fitzgerald, R.C., Hardwick, R., Huntsman, D., et al, Hereditary diffuse gastric cancer: updated consensus guidelines for clinical management and directions for future research (International Gastric Cancer Linkage Consortium). J Med Genet 2010; 47:436–444. 20591882
39. Huntsman, D.G., Carneiro, F., Lewis, F.R., et al, Early gastric cancer in young asymptomatic carriers of germline E cadherin mutation. N Engl J Med 2001; 344:1904–1909. 11419427
40. Chun, Y.S., Linder, N.M., Smyrk, T.C., et al, Germline E-cadherin germ mutations. Is prophylactic total gastrectomy indicated? Cancer 2001; 92:181–187. 11443625
41. Charlton, A., Blair, V., Shaw, D., et al, Hereditary diffuse gastric cancer: predominance of multiple foci of signet ring cell carcinoma in distal stomach and transitional zone. Gut 2004; 53:814–820. 15138207
42. Keller, G., Hofler, H., Becker, K.F., Molecular mechanisms of gastric adenocarcinoma. Expert Rev Mol Med 2005; 7:1–13. 16274491
43. Hiyama, T., Tanaka, S., Kitadai, Y., et al, p53 codon 72 polymorphism in gastric cancer susceptibility in patients with Helicobacter pylori associated chronic gastritis. Int J Cancer 2002; 100:304–308. 12115545
44. Lin, J., Beer, D.G., Molecular biology of upper gastrointestinal malignancies. Semin Oncol 2004; 31:476–486. 15297940
45. Wang, G.-Q., Endoscopic diagnosis of early oesophageal carcinoma. J R Soc Med 1981; 74:502–503. 7265074
46. Shu, Y.-J., Cytopathology of the oesophagus. Acta Cytol 1983; 27:7–16. 6573834
47. Huang, G.-J. Recognition and treatment of the early lesion. In: Delarae N.C., Wilkins E.W., Wong J., eds. Oesophageal cancer. International trends: general thoracic surgery. 4th ed. St Louis: Mosby; 1988:149–152.
48. Huang, G.J., Early detection and surgical treatment of oesophageal carcinoma. Jpn J Surg 1981; 11:399–405. 7328933
49. Yanjun, M., Li, G., Xianzhil, G., et al, Detection and natural progression of early oesophageal carcinoma – preliminary communication. J R Soc Med 1981; 74:884–886. 7321012
50. Chamberlain, J., Day, N.E., Hakama, M., et al, UICC workshop of the project on evaluation of screening programmes for gastrointestinal cancer. Int J Cancer 1986; 37:329–334. 3949421
51. Hisamichi, S., Screening for gastric cancer. World J Surg 1989; 13:31–37. 2658351
52. Oshima, A., Hirata, N., Ubakata, T., et al, Evaluation of a mass screening programme for stomach cancer with a case–control study design. Int J Cancer 1986; 38:829–834. 3793262
53. Fielding, J.W.L., Ellis, D.J., Jones, B.G., et al, Natural history of ‘early’ gastric cancer: results of a 10-year regional survey. Br Med J 1980; 281:965–967. 7427544
54. Christie, J., Shepherd, N.A., Codling, B.W., et al, Gastric cancer below the age of 55: implications for screening patients with uncomplicated dyspepsia. Gut 1997; 41:513–517. 9391251
55. Salmon, C.A., Park, K.G.M., Rapson, T., et al, Age threshold for endoscopy and risk of missing upper GI malignancy: data from the Scottish Audit of Gastric and Oesophageal Cancer. Aliment Pharmacol Ther. 2006;23(2):229–233. 16393301
56. Bowrey, D.J., Griffin, S.M., Wayman, J., et al, Using alarm symptoms to select dyspeptics for endoscopy which result in patients with curable oesophagogastric cancer being overlooked. Surg Endosc 2006; 20:1725–1728. 17024539
57. Hallissey, M.T., Jewkes, A.J., Allum, W.H., et al, The impact of the dyspepsia study on deaths from gastric cancer vol. 1Nishi M., Sugano H., Takahashi T., eds. International gastric cancer congress. Monduzzi Editore-International Proceedings Division: Bologna, 1995:264.
58. NHS Executive. Referral guidelines for suspected cancer. London: HMSO; 2000.
59. Martin, I.G., Young, S., Sue-Ling, H., et al, Delays in the diagnosis of oesophago-gastric cancer: a consecutive case series. Br Med J 1997; 314:467–470. 9056794
60. Kohli, Y., Kawai, K., Fujita, S., Analytical studies on growth of human gastric cancer. J Clin Gastroenterol 1981; 3:129–133. 6895643
61. Radbourne, D., Walker, G., Joshi, D., et al. The 2 week standard for suspected upper GI cancers: its impact on staging. Gut. 2008; 52:A116.
62. Jobe, B.A., Hunter, J.G., Chang, E.Y., et al, Office-based unsedated small-caliber endoscopy is equivalent to conventional sedated endoscopy in screening and surveillance for Barrett’s esophagus: a randomized and blinded comparison. Am J Gastroenterol 2006; 101:2693–2703. 17227516
63. Kadri, S.R., Lao-Sirieix, P., O’Donovan, M., et al, Acceptability and accuracy of a non-endoscopic screening test for Barett’s oesophagus in primary care: cohort study. Br Med J 2010; 341:c4372–c4380. 20833740
64. Farinati, F., Valiante, F., Germania, B., et al, Prevalence of Helicobacter pylori infection in patients with precancerous changes and gastric cancer. Eur J Cancer Prev 1993; 2:321–326. 8358284
65. Kikuchi, S., Epidemiology of Helicobacter pylori and gastric cancer. Gastric Cancer 2002; 5:6–15. 12021854
66. Whiting, J.L., Hallissey, M.T., Fielding, J.W.L., et al, Screening for gastric cancer by Helicobacter pylori serology: a retrospective study. Br J Surg 1998; 85:408–411. 9529506
67. Whiting, J.L., Sigurdsson, A., Rowlands, D.C., et al, The long term results of endoscopic surveillance of premalignant gastric lesions. Gut 2002; 50:378–381. 11839718

