CHAPTER 74 EPIDEMIOLOGY AND GENETICS OF MULTIPLE SCLEROSIS
Multiple sclerosis is a chronic inflammatory disease of the central nervous system (CNS) that usually begins in early adulthood and is characterized by demyelination, gliosis, a varying degree of axonal pathology, and episodic or progressive neurological disability. More than 1 million people worldwide and at least 350,000 individuals in the United States alone are affected with multiple sclerosis, which is second only to trauma as a cause of acquired disability in young adults in most white populations.1,2
DISEASE HETEROGENEITY
Inflammatory events are considered to initiate and drive the disease process during early stages. The myelin damage and axonal injury that account for the permanent neurological deficit seen during later phases of multiple sclerosis probably result from a complex sequence of events, including processes intrinsic to the CNS, such as increased vulnerability to tissue injury and/or poor repair, which might progress independently of immune factors. Multiple sclerosis is therefore not solely a disease of the immune system; CNS-specific components, although largely overlooked in their potential pathogenetic role, are presumably equally important for its pathogenesis.3
POPULATION PREVALENCE
The disease prevalence varies between 60 and 200 per 100,000 in North America and northern Europe and generally follows a north-to-south decreasing gradient on the northern hemisphere and the opposite on the southern hemisphere, with very low rates or virtual absence of the disease near the equator (Fig. 74-1).
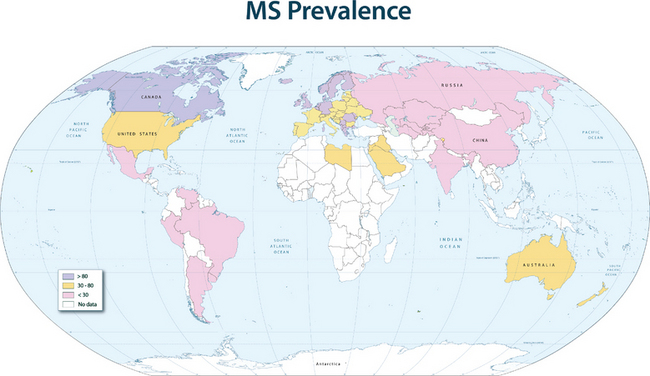
This geographical distribution can be attributed to both environmental factors and genetic effects. For many years, an infectious etiology of multiple sclerosis has been suspected, because it is consistent with a number of epidemiological observations and with immunopathological characteristics of the disease. Migration studies showed that individuals who migrate from high-risk to low-risk areas after the age of 15 tend to retain their risk of multiple sclerosis, whereas individuals who migrate from high-risk to low-risk areas before the age of 15 acquire a lower risk; this indicates that childhood exposure to an environmental factor increases disease susceptibility. Supportive data for an infectious agent also come from reports of endemic clusters of multiple sclerosis. After the British occupation of the Faroe Islands, off the coast of Denmark, where no cases of multiple sclerosis had been reported before, several islanders developed the disease between 1940 and the end of World War II, and the affected areas were found to be locations of British troop encampments after 1940. Other examples of multiple sclerosis epidemics have been described in northern America and Europe. These observations suggest that an environmental factor is relevant for the initiation of the disease process, and in the established disease, infections are additionally known to be capable of triggering exacerbations.
The idea that genetic factors may have a role in multiple sclerosis was first raised in the 1890s with the identification of familial aggregation. Further milestones in establishing the concept of genetic susceptibility to multiple sclerosis were the appreciation of ethnic risks in the 1920s, comprehensive studies on the geographical distribution of the disease in the 1950s, and the description of the MHC associations in the 1970s. It took, however, a century until the first systematic genetic epidemiological analyses on familial aggregation were initiated. These studies formed the groundwork for the understanding of multiple sclerosis as a genetically determined disease.
FAMILIAL AGGREGATION
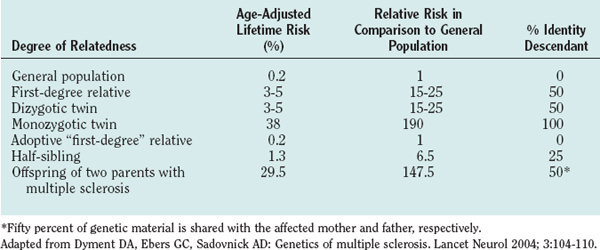
Familial aggregation studies, including research with twins, siblings, and adoptees, demonstrated that the risk of developing multiple sclerosis increases with the degree of relatedness between individuals.4 For example, monozygotic twins of patients with multiple sclerosis have a risk more than 100 times higher than that of the general population of developing the disease, and full siblings have a lifetime risk approximately 20 times higher than that of the general population (Table 74-1). Although these recurrence risk values are considerably lower than the ones for mendelian-dominant disorders such as Huntington’s disease (approximately 5000-fold increased risk for siblings), they are similar to the risks that have been reported for other complex polygenic diseases, such as systemic lupus erythematosus or type I diabetes mellitus (approximately 20- to 30-fold increased risk for siblings).
Twin Studies
Twin studies consistently showed that the concordance rate (whereby both twins develop the disease) is approximately 25% to 30% for monozygotic twins and 2% to 5% for dizygotic twins.5 The 10-fold increased risk for monozygotic twins indicates a strong genetic component, whereas the fact that the concordance rate is not nearly 100% is usually taken as an argument for the impact of nongenetic, environmental factors. Studies on animal models of systemic lupus erythematosus, however, demonstrated that the rate of disease expression can be influenced by the number of disease-associated genes under identical environmental influences.6 A simple concordance rate in monozygotic twins might therefore not optimally reflect, and might underestimate, the contribution of genetic factors to a complex genetic trait disease such as multiple sclerosis.
Adoption and Half-Sibling Studies
An elegant approach to dissecting the effect of genetic sharing versus a shared family environment for the development of multiple sclerosis are epidemiological studies of adoptees (no genetic sharing) and half-siblings in comparison with full siblings (25% and 50% genetic sharing, respectively). Adopted relatives, although raised from infancy with the patients who have multiple sclerosis, do not develop multiple sclerosis more frequently than would be expected for the general population.4 Half-siblings raised apart (different environments) or together (same environment) with patients who have multiple sclerosis have similar risks of developing the disease, and the recurrence risk for half-siblings is significantly lower than that for full siblings raised in the same family (1.32% versus 3.46%).
SUSCEPTIBILITY GENES
Numerous searches have been performed and are currently ongoing in order to identify single candidate genes or a set of genes that are relevant for the disease. The strongest and most consistent evidence exists for the area of the MHC, the human leukocyte antigen (HLA), on chromosome 6p21. The total genetic susceptibility attributed to the HLA locus in multiple sclerosis is estimated to be between 15% and 50%.7
In an alternative approach to a traditional case-control design that has been applied to investigate allelic variants of single candidate genes, whole-genome screens of affected relatives have been used to assess linkage of polymorphic markers spread throughout the genome. Linkage studies have been successfully applied in genetic diseases that follow a mendelian single-gene trait with a high penetrance of the allelic variant and a clear clinical phenotype, such as those in Huntington’s disease or in muscular dystrophy.
Because no major single risk locus could be identified by whole-genome linkage studies, a large number of common allelic variants, each with only subtle but important variations, might synergistically lead to the major genetic risk that is associated with the occurrence of the disease. For example, the overall number of susceptibility loci that have been identified in the most comprehensive genomic screen so far is 411.8 Because of the low resolution and the weak signals that can be achieved by traditional linkage studies in polygenic diseases, the identification of such common variants requires different approaches. To date, the MHC class II region remains the only area of the whole genome clearly associated with multiple sclerosis, although the precise genes within this area that confer disease susceptibility are not yet known.
THE HUMAN LEUKOCYTE ANTIGEN GENE COMPLEX
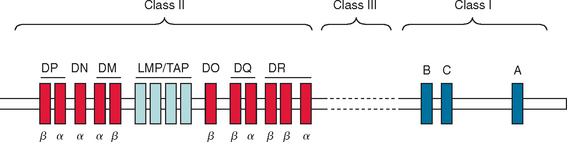
The MHC region on chromosome 6p21 encompasses 4.5 megabases and encodes approximately 200 genes. Aside from genes for histones and transfer RNA, most of the encoded molecules play an important role in the development, maturation, and regulation of the T cell repertoire and other immunological processes. The MHC regions harbors separate clusters of MHC class I, class II, and class III genes (Fig. 74-2). The classic class I genes are HLA-A, HLA-B, and HLA-C. These genes encode the heavy (α) chain of MHC class I molecules. The class II region contains genes for the α and β chains of the antigen-presenting MHC class II molecules HLA-DR, HLA-DP, and HLA-DQ. The class III region encodes structurally and functionally diverse proteins such as complement components, tumor necrosis factors α and β, and heat shock proteins (HSP70). Other interesting candidates within this region include the gene for myelin oligodendrocyte glycoprotein, a minor structural but immunogenic component of the CNS myelin and so called class I–like genes such as the MHC class I polypeptide-related sequence gene family that regulates the activation and inhibition of natural killer cells.
The most consistent association of multiple sclerosis was found for HLA-DR and HLA-DQ genes. In particular, the HLA-DR15 haplotype (DRB1*1501, DRB5*0101, DQA1*0102, DQB1*0602) showed a strong association with the disease among white persons in northern and central Europe, North America, and Australia, and even a dose effect of HLA-DR15 could be identified in homozygous patients with multiple sclerosis. Because DRB1 and DQB1 genes are in strong linkage disequilibrium, it has been difficult to distinguish the relative contribution from each allelic variant to the susceptibility haplotype. Both additive and independent effects have been reported for the DQA1*0102/DQB1*0602 locus. More recent investigations, however, indicated a primary role of the DRB1*1501 locus independent of DQB1*0602 at least in a white population.9
The HLA-DR15 association was comparably weak or lacking in other ethnic groups. Mediterranean populations with a distinct genetic background, such as Sardinians, showed a stronger association with HLA-DR4, and no apparent association with any HLA haplotype could be identified in a southern Chinese population. Much less information is available for risks conferred by class I alleles, although HLA-A3 and HLA-B7 are the first MHC genes that were found to be associated with multiple sclerosis.10 Their association with multiple sclerosis, however, appears to be much lower than that of the HLA-DR and HLA-DQ alleles. In addition to these risk genes, certain MHC class I and class II allelic variants have been identified as exerting protective properties. Table 74-2 summarizes the reports on risk/protective alleles for multiple sclerosis within the MHC region.
TABLE 74-2 Selected Studies on the Associations of MHC Class I and Class II Alleles with Multiple Sclerosis
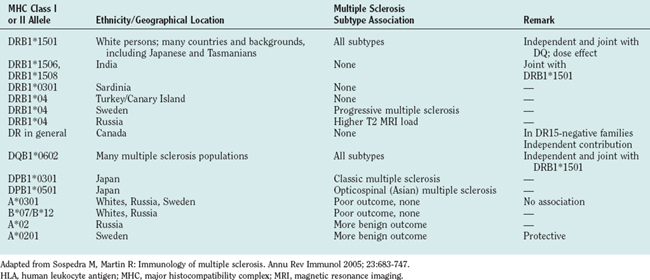
Only limited data are available for the association of clinical features with the HLA-DR/HLA-DQ risk alleles in multiple sclerosis patients. It has been reported that patients expressing the DR15 haplotype have an earlier disease onset, more often have relapsing-remitting multiple sclerosis, are female, and have optic neuritis or spinal involvement as an initial event. DR4-positive patients are described to have a worse clinical outcome or progressive course. However, as mentioned previously, the HLA association studies are heterogeneous with regard to sample size, methodology, ethnic background, and clinical findings. In older studies, the exact MHC class II gene has not even been determined by molecular typing techniques.
FUTURE DIRECTIONS
Dyment DA, Ebers GC, Sadovnick AD. Genetics of multiple sclerosis. Lancet Neurol. 2004;3:104-110.
Hafler DA, De Jager PL. Applying a new generation of genetic maps to understand human inflammatory disease. Natl Rev Immunol. 2005;5:83-91.
Sospedra M, Martin R. Immunology of multiple sclerosis. Annu Rev Immunol. 2005;23:683-747.
1 McFarlin DE, McFarland HF. Multiple sclerosis (first of two parts). N Engl J Med. 1982;307:1183-1188.
2 McFarlin DE, McFarland HF. Multiple sclerosis (second of two parts). N Engl J Med. 1982;307:1246-1251.
3 Sospedra M, Martin R. Immunology of multiple sclerosis. Annu Rev Immunol. 2005;23:683-747.
4 Ebers GC, Sadovnick AD, Risch NJ. A genetic basis for familial aggregation in multiple sclerosis. Canadian Collaborative Study Group. Nature. 1995;377:150-151.
5 Sadovnick AD, Armstrong H, Rice GP, et al. A population-based study of multiple sclerosis in twins: update. Ann Neurol. 1993;33:281-285.
6 Wakeland EK, Liu K, Graham RR, et al. Delineating the genetic basis of systemic lupus erythematosus. Immunity. 2001;15:397-408.
7 Haines JL, Terwedow HA, Burgess K, et al. Linkage of the MHC to familial multiple sclerosis suggests genetic heterogeneity. The Multiple Sclerosis Genetics Group. Hum Mol Genet. 1998;7:1229-1234.
8 GAMES; Transatlantic Multiple Sclerosis Genetics Cooperative. A meta-analysis of whole genome linkage screens in multiple sclerosis. J Neuroimmunol. 2003;143:39-46.
9 Oksenberg JR, Barcellos LF, Cree BA, et al. Mapping multiple sclerosis susceptibility to the HLA-DR locus in African Americans. Am J Hum Genet. 2004;74:160-167.
10 Jersild C, Ammitzboll T, Clausen J, et al. Association between HL-A antigens and measles antibody in multiple sclerosis. Lancet. 1973;1:151-152.






