[level-membership-for-pulmolory-and-respiratory-category]
Chapter 49 Eosinophilic Lung Disease
The Eosinophil Leukocyte and Eosinophilic Pneumonia
Diagnosis of Eosinophilic Pneumonia
The diagnosis of eosinophilic pneumonia relies on both characteristic clinical-imaging features and the demonstration of alveolar eosinophilia and/or peripheral blood eosinophilia (Box 49-1). Bronchoalveolar lavage (BAL) is a noninvasive alternative to lung biopsy in this setting. The percentage of eosinophils in BAL fluid is less than 2% in normal control subjects, and a differential cell count for eosinophils of 2% to 25% may be found in nonspecific conditions. Therefore, a cutoff value of 25% eosinophils or more in BAL fluid, and preferably 40% or more, is recommended for the diagnosis of eosinophilic pneumonia. The presence of marked eosinophilia in BAL fluid obviates the need for lung biopsy in this disorder, especially when the eosinophils are the predominant cell population in BAL fluid (macrophages excepted). Markedly elevated peripheral blood eosinophilia (greater than 1000/µL and preferably 1500/µL) together with typical clinical radiologic features are highly suggestive of the diagnosis of eosinophilic pneumonia, and BAL may not be always mandatory in such patients, although other disorders may be associated with pulmonary infiltrates and peripheral eosinophilia (e.g., bacterial pneumonia, parasitic pneumonia, infiltrates related to lymphoma). Peripheral blood eosinophilia may be absent at presentation, especially in IAEP and in patients receiving corticosteroid treatment.
Box 49-1
Diagnosis of Eosinophilic Pneumonias
Eosinophilic pneumonia may be separated into that of undetermined origin, which usually may be included within well-individualized syndromes, and that with a definite cause (mainly infection and drug reaction) (Box 49-2). Potential causes must be thoroughly investigated, because identification of a cause may lead to effective therapeutic measures.
Eosinophilic Lung Diseases of Known Origin
Allergic Bronchopulmonary Aspergillosis
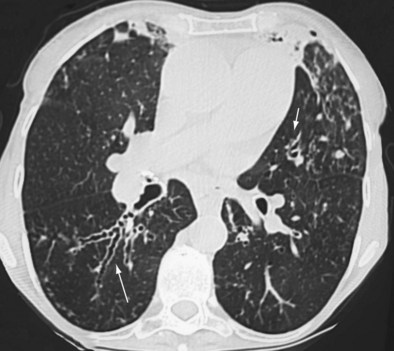
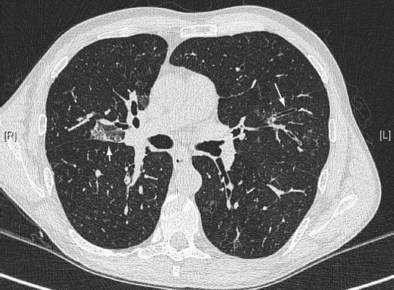
Early ABPA is characterized by fever, expectoration of mucous plugs, peripheral blood eosinophilia with counts higher than 1000/µL, and pulmonary infiltrates caused by eosinophilic pneumonia, or segmental or lobar atelectasis caused by mucous plugging. Chronic ABPA is characterized by asthma, eosinophilia, and bronchopulmonary manifestations including bronchiectasis (Figures 49-1 and 49-2 and Box 49-3). The presence of bronchiectasis on computed tomography (CT) images in a patient with asthma is therefore highly suggestive of ABPA; however, typical proximal bronchiectasis may be absent, and such cases are designated ABPA-seropositive. Centrilobular nodules and mucoid impaction on CT scan (typically characterized by a V-shaped lesion, with the vertex pointing toward the hilum) also are highly suggestive of ABPA. The expectoration of mucous plugs, the presence of Aspergillus in sputum, and late skin reactivity to Aspergillus antigen also are common at this stage; however, Aspergillus is not reliably identified in sputum or BAL fluid samples of patients with ABPA, and positive cultures are not required for diagnosis. The finding of Aspergillus organisms may reflect colonization and thus is not specific for ABPA. Total serum IgE levels of at least 1000 IU/mL constitute a hallmark of ABPA. Peripheral blood eosinophilia is common but is not a required diagnostic criterion.
Box 49-3
Minimal Essential Diagnostic Criteria for Allergic Bronchopulmonary Aspergillosis (ABPA) in Patients With Asthma
Patients With Asthma and Central Bronchiectasis
Patients With Cystic Fibrosis
1. Clinical deterioration (increased cough, wheezing, exercise intolerance, increased sputum, decrease in pulmonary function)
2. Immediate cutaneous reactivity to Aspergillus or presence of IgE–A. fumigatus
3. Total serum IgE concentration <417 kU/L (1000 ng/mL)
4. Precipitating antibodies to A. fumigatus or serum IgG–A. fumigatus
5. Abnormalities on chest radiograph (infiltrates, mucous plugging, or a change from earlier films)
Iatrogenic Eosinophilic Pneumonias Secondary to Drugs, Toxic Agents, and Radiation Therapy
Drugs taken in the weeks preceding clinical onset of eosinophilic pneumonia must be thoroughly investigated, including illicit drugs (cocaine or heroin). Eosinophilic pneumonia has been reported in association with many drugs, but causality has been confidently established for fewer than 20 specific agents (Box 49-4).
Eosinophilic Lung Disease of Unknown Origin
Idiopathic Chronic Eosinophilic Pneumonia
Imaging features of ICEP (Figure 49-3) characteristically consist of alveolar opacities, with ill-defined margins and variable density, ranging from ground glass to consolidation. Migration of the infiltrates highly suggestive of the diagnosis occurs in approximately a quarter of the cases; however, it also may occur in cryptogenic organizing pneumonia. A peripheral predominance of the lesions (described as the classic pattern of “photographic negative of pulmonary edema”) is seen in approximately one fourth of patients. On high-resolution computed tomography (HRCT), the opacities are almost always bilateral and predominate in the upper lobes, with coexisting peripheral ground glass and consolidation opacities. In addition, septal line thickening, bandlike opacities parallel to the chest, or mediastinal lymph node enlargement may be seen. Small pleural effusions are present in only 10% of cases at HRCT.
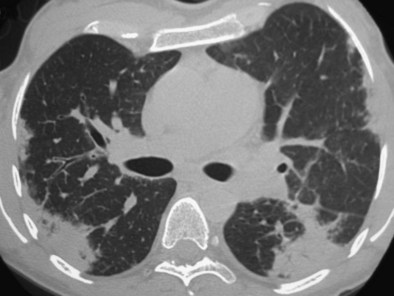
High-level peripheral blood eosinophilia is the key to the diagnosis (mean blood eosinophil count of approximately 5500/µL) (Box 49-5). Alveolar eosinophilia, with BAL fluid differential cell count usually more than 40%, is a hallmark of ICEP. Because the BAL eosinophil cell count drops within days after institution of corticosteroid treatment, assessment for alveolar eosinophilia should be undertaken before any corticosteroid intake. Sputum eosinophilia also may be present; however, its diagnostic value has not been evaluated. C-reactive protein levels are elevated. Total blood IgE level is increased in approximately half the cases.
Box 49-5
Diagnostic Criteria for Idiopathic Chronic Eosinophilic Pneumonia
• Diffuse pulmonary alveolar consolidation with air bronchogram and/or ground glass opacities on chest imaging, especially with peripheral predominance
• Eosinophilia on BAL fluid differential cell count: ≥40% (or peripheral blood eosinophilia, with counts ≥1.0 × 109 cells/L)
• Respiratory symptoms present for at least 2 to 4 weeks
• Absence of other known causes of eosinophilic lung disease (especially exposure to any of various drugs known to induce pulmonary eosinophilia)
Idiopathic Acute Eosinophilic Pneumonia
IAEP differs from ICEP not only in its acute onset (less than 1 month) and severity but also in the absence of relapse after recovery. This acute pneumonia develops in previously healthy people, with possible respiratory failure, and may be misdiagnosed as infectious pneumonia or acute respiratory distress syndrome (ARDS). Patients with IAEP often are admitted to the intensive care unit. Blood eosinophilia, often lacking at presentation, contrasts with frank alveolar eosinophilia in BAL fluid. Current diagnostic criteria are listed in Box 49-6.
Box 49-6
Diagnostic Criteria for Idiopathic Acute Eosinophilic Pneumonia
• Acute onset with febrile respiratory manifestations (present for ≤1 month, and especially ≤7 days before medical examination)
• Bilateral diffuse infiltrates on imaging
• PaO2 on room air ≤60 mm Hg, or PaO2/FIO2 ≤300 mm Hg, or oxygen saturation on room air <90%
• Lung eosinophilia, with ≥25% eosinophils on BAL fluid differential cell count (OR eosinophilic pneumonia at lung biopsy, if done)
• Absence of determined cause of acute eosinophilic pneumonia, including infection or exposure to drugs known to induce pulmonary eosinophilia, although patient history may include recent onset of tobacco smoking or exposure to inhaled dusts
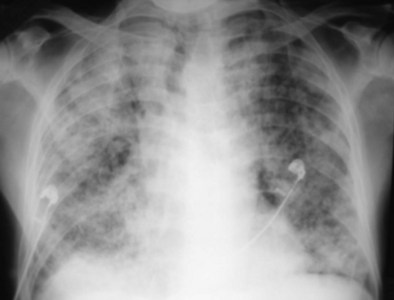
IAEP manifests with the acute onset of cough, dyspnea, fever, and chest pain, sometimes with abdominal complaints or myalgias. Tachypnea, tachycardia, and crackles are present on examination. The chest radiograph shows bilateral infiltrates (see Figure 49-4), with mixed alveolar interstitial and opacities, especially Kerley lines. Chest CT mainly shows ground glass opacities and air space consolidation, together with poorly defined nodules, interlobular septal thickening, and bilateral pleural effusions (in two thirds of patients)—an imaging pattern very distinct from that of ICEP that may evoke the diagnosis of IAEP.
Churg-Strauss Syndrome
CSS (see also Chapter 59) is included in the group of small-vessel vasculitides and is defined as an eosinophil-rich and granulomatous inflammation involving the respiratory tract and necrotizing vasculitis affecting small to medium-sized vessels; it is associated with asthma and eosinophilia. All of the pathologic lesions—initially described from autopsied cases—are seldom found in a single biopsy specimen, and abnormalities often are limited to an eosinophilic perivascular infiltration of the tissues characteristic of the early (prevasculitic) phase of the disease. CSS is one of the pulmonary vasculitides associated with antineutrophil cytoplasmic antibodies (ANCA), together with granulomatosis with polyangiitis (Wegener) and microscopic polyangiitis, with ANCA found only in about 40% of cases of CSS.
The chest radiograph may remain normal in appearance throughout the course of the disease. Lung opacities, present in more than half of patients, correspond to eosinophilic pneumonia and consist mainly of ill-defined pulmonary infiltrates, sometimes migratory and transient. Pleural eosinophilic exudate, present in approximately one fourth of patients, must be distinguished from the transudate associated with cardiac failure resulting from cardiac eosinophilic involvement with severe cardiomyopathy. HRCT mainly shows areas of ground glass attenuation or air space consolidation, with peripheral predominance or random distribution (Figure 49-5). Centrilobular nodules, bronchial wall thickening or dilatation, interlobular septal thickening, and hilar or mediastinal lymphadenopathy are less common. These abnormalities are nonspecific and resemble those seen in ICEP, although the subpleural distribution of consolidation is less pronounced, centrilobular nodules within ground glass opacities are more frequent, and traction bronchiectases are less frequent in CSS than in ICEP. Pulmonary cavitary lesions are exceptional.
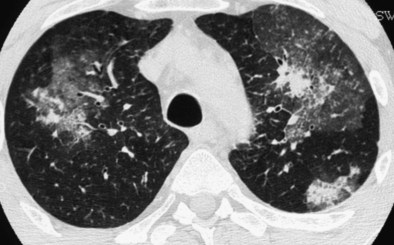
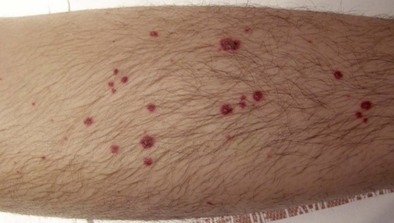
Extrapulmonary manifestations of CSS usually include asthenia, weight loss, fever, arthralgias, and/or myalgias. Neurologic involvement typically consists of mononeuritis multiplex or asymmetric polyneuropathy. Cardiac involvement manifesting as eosinophilic myocarditis (or, more rarely, coronary arteritis) often is of insidious onset and asymptomatic and may lead to dilated cardiomyopathy, although marked clinical improvement may follow corticosteroid treatment. Pericardial effusion is common, but tamponade is rare. Endomyocardial fibrosis is rare, in contrast with the idiopathic hypereosinophilic syndrome. Myocardial involvement is a common finding in systematic studies by echocardiography or magnetic resonance imaging (MRI), and its frequency may have been heretofore underestimated. Digestive tract involvement usually manifests as isolated abdominal pain or diarrhea, but intestinal vasculitis (with ulcerations, perforations, or hemorrhage) and cholecystitis may occur. Cutaneous lesions, seen in approximately half of the patients, consist mainly of palpable purpura (Figure 49-6) on the extremities, subcutaneous nodules, erythematous rashes, and urticaria. Renal involvement, present in a fourth of the cases, usually is mild.
Currently used diagnostic criteria for CSS are shown in Box 49-7. ANCA also may be considered a major diagnostic criterion when present. Conversely, absence of ANCA does not exclude the diagnosis of CSS. A histopathologic diagnosis of CSS may be obtained by examination of skin, nerve, or muscle biopsy specimens. Lung biopsy and transbronchial biopsy are not helpful. The diagnosis of CSS may be difficult, especially at an early stage, with mild or limited manifestations corresponding to “formes frustes” of CSS. These may consist of cases in which the disease has been partly controlled by corticosteroids given for asthma.
Box 49-7
Practical Diagnostic Criteria of Churg-Strauss Syndrome
2. Peripheral blood eosinophilia ≥1500/µL and/or alveolar eosinophilia ≥25%
3. Extrapulmonary clinical manifestations of disease (other than rhinosinusitis), with at least one of the following:
ANCA are found in only approximately 40% of patients and consist mainly of perinuclear ANCA (p-ANCA), with a specificity for myeloperoxidase on ELISA, whereas other ANCA (especially cytoplasmic ANCA [c-ANCA] with proteinase-3 specificity) are very rare in CSS. Of interest, two phenotypes of CSS can be distinguished on the basis of these antibodies (Table 49-1): Patients with ANCA have a vasculitic phenotype (with more frequent vasculitis on biopsy, renal involvement, peripheral neuropathy, and purpura), whereas patients without ANCA have more frequent cardiac and pulmonary involvement. Therefore, patients with CSS seen in respiratory medicine departments or chest clinics are at high risk for cardiac involvement, which can be subclinical (with especially no chest pain) and should be systematically evaluated with measurement of serum troponin, electrocardiogram, echocardiography, and possibly MRI of the heart, which can identify impaired cardiac function and myocardial involvement. Serum measurement of eotaxin-3 and of CCL17/thymus and activation–related chemokine may reflect disease activity and merits further study as a potential biomarker of disease monitoring.
| Feature | Vasculitic Phenotype | Tissue Disease Phenotype |
|---|---|---|
| Respective frequency | ~40% | ~60% |
| ANCA | Present (mostly p-ANCA with antimyeloperoxidase specificity) | Absent |
| Predominant clinical and histopathologic features | Glomerular renal disease Peripheral neuropathy Purpura Biopsy-proven vasculitis |
Cardiac involvement (eosinophilic myocarditis) Eosinophilic pneumonia Fever |
ANCA, antineutrophil cytoplasmic antibodies; p-ANCA, perinuclear ANCA.
Controversies and Pitfalls
• The diagnosis of parasitic diseases in patients with eosinophilic pneumonia may be difficult and requires appropriate serologic tests and repeated search for parasites in the feces. Such diseases must be considered in any patient who lived in or visited an endemic area.
• Eosinophilic pneumonia has been reported in association with a variety of drugs, but causality has been confidently established for fewer than 20 specific agents.
• The dose and duration of treatment of ICEP with corticosteroids have not been established. Disease control must be balanced with treatment side effects.
• “Idiopathic” acute eosinophilic pneumonia may be misdiagnosed as severe infectious pneumonia.
• Established diagnostic criteria for CSS currently are lacking. ANCA are found in about 40% of patients, and true vasculitis is a feature in only a minority of cases.
• Asthma is a common denominator in most patients with ICEP and CSS.
• The respiratory manifestations of the lymphocytic and myeloid variants of the HESs have not been described in detail.
Agarwal R. Allergic bronchopulmonary aspergillosis. Chest. 2009;135:805–826.
Cottin V, Khouatra C, Dubost R, et al. Persistent airflow obstruction in asthma of patients with Churg-Strauss syndrome and long-term follow-up. Allergy. 2009;64:589–595.
Dennert RM, van Paassen P, Schalla S, et al. Cardiac involvement in Churg-Strauss syndrome. Arthritis Rheum. 2010;62:627–634.
Kunst H, Mack D, Kon OM, et al. Parasitic infections of the lung: a guide for the respiratory physician. Thorax. 2011;66:528–536.
Marchand E, Cordier JF. Idiopathic chronic eosinophilic pneumonia. Orphanet J Rare Dis. 2006;1:11.
Marchand E, Etienne-Mastroïanni B, Chanez P, et al. Groupe d’Etudes et de Recherche sur les Maladies Orphelines Pulmonaires: Idiopathic chronic eosinophilic pneumonia and asthma: how do they influence each other? Eur Respir J. 2003;22:8–13.
Nair P, Pizzichini MM, Kjarsgaard M, et al. Mepolizumab for prednisone-dependent asthma with sputum eosinophilia. N Engl J Med. 2009;360:985–993.
Pagnoux C, Guilpain P, Guillevin L. Churg-Strauss syndrome. Curr Opin Rheumatol. 2009;19:25–32.
Patterson K, Strek ME. Allergic bronchopulmonary aspergillosis. Proc Am Thorac Soc. 2010;7:237–244.
Sablé-Fourtassou R, Cohen P, Mahr A, et al. Antineutrophil cytoplasmic antibodies and the Churg-Strauss syndrome. Ann Intern Med. 2005;143:632–638.
Walsh ER, August A. Eosinophils and allergic airway disease: there is more to the story. Trends Immunol. 2010;31:39–44.
[/level-membership-for-pulmolory-and-respiratory-category][not-level-membership-for-pulmolory-and-respiratory-category]
Chapter 49 Eosinophilic Lung Disease
The Eosinophil Leukocyte and Eosinophilic Pneumonia
Diagnosis of Eosinophilic Pneumonia
The diagnosis of eosinophilic pneumonia relies on both characteristic clinical-imaging features and the demonstration of alveolar eosinophilia and/or peripheral blood eosinophilia (Box 49-1). Bronchoalveolar lavage (BAL) is a noninvasive alternative to lung biopsy in this setting. The percentage of eosinophils in BAL fluid is less than 2% in normal control subjects, and a differential cell count for eosinophils of 2% to 25% may be found in nonspecific conditions. Therefore, a cutoff value of 25% eosinophils or more in BAL fluid, and preferably 40% or more, is recommended for the diagnosis of eosinophilic pneumonia. The presence of marked eosinophilia in BAL fluid obviates the need for lung biopsy in this disorder, especially when the eosinophils are the predominant cell population in BAL fluid (macrophages excepted). Markedly elevated peripheral blood eosinophilia (greater than 1000/µL and preferably 1500/µL) together with typical clinical radiologic features are highly suggestive of the diagnosis of eosinophilic pneumonia, and BAL may not be always mandatory in such patients, although other disorders may be associated with pulmonary infiltrates and peripheral eosinophilia (e.g., bacterial pneumonia, parasitic pneumonia, infiltrates related to lymphoma). Peripheral blood eosinophilia may be absent at presentation, especially in IAEP and in patients receiving corticosteroid treatment.
Box 49-1
Diagnosis of Eosinophilic Pneumonias
Eosinophilic pneumonia may be separated into that of undetermined origin, which usually may be included within well-individualized syndromes, and that with a definite cause (mainly infection and drug reaction) (Box 49-2). Potential causes must be thoroughly investigated, because identification of a cause may lead to effective therapeutic measures.
Eosinophilic Lung Diseases of Known Origin
Allergic Bronchopulmonary Aspergillosis
Early ABPA is characterized by fever, expectoration of mucous plugs, peripheral blood eosinophilia with counts higher than 1000/µL, and pulmonary infiltrates caused by eosinophilic pneumonia, or segmental or lobar atelectasis caused by mucous plugging. Chronic ABPA is characterized by asthma, eosinophilia, and bronchopulmonary manifestations including bronchiectasis (Figures 49-1 and 49-2 and Box 49-3). The presence of bronchiectasis on computed tomography (CT) images in a patient with asthma is therefore highly suggestive of ABPA; however, typical proximal bronchiectasis may be absent, and such cases are designated ABPA-seropositive. Centrilobular nodules and mucoid impaction on CT scan (typically characterized by a V-shaped lesion, with the vertex pointing toward the hilum) also are highly suggestive of ABPA. The expectoration of mucous plugs, the presence of Aspergillus in sputum, and late skin reactivity to Aspergillus antigen also are common at this stage; however, Aspergillus is not reliably identified in sputum or BAL fluid samples of patients with ABPA, and positive cultures are not required for diagnosis. The finding of Aspergillus organisms may reflect colonization and thus is not specific for ABPA. Total serum IgE levels of at least 1000 IU/mL constitute a hallmark of ABPA. Peripheral blood eosinophilia is common but is not a required diagnostic criterion.
[/not-level-membership-for-pulmolory-and-respiratory-category]
