[level-membership-for-pathology-category]
Enteropathies Associated with Chronic Diarrhea and Malabsorption in Childhood
Pierre Russo
Introduction
The goal of this chapter is to outline the pathologic features of the major intestinal disorders of infancy and early childhood, with an emphasis on congenital disorders that result in chronic diarrhea and malabsorption, and to illustrate their appearance in small intestinal biopsy and resection specimens (Box 9.1). Other major categories of disorders that cause chronic diarrhea, such as infections, immunodeficiencies (primary and secondary), gluten-sensitive enteropathy and other food allergies, and motility and pancreatic disorders are discussed in other chapters of this book.
Chronic diarrhea occurring in the neonatal period presents particularly difficult diagnostic and therapeutic challenges. Intractable diarrhea of infancy is a term initially coined by Avery and colleagues1 to refer to chronic diarrhea in neonates, most of which remained undiagnosed and was associated with a high mortality rate. Since these initial reports, more precise identification of disorders that cause intractable diarrhea of infancy has led to the use of prolonged parenteral nutrition, immunosuppression, and even bowel transplantation to try to improve survival, emphasizing the need for timely and accurate diagnosis. Investigation of some of these disorders has also led to significant advances in understanding of gastrointestinal (GI) and immunologic functions. For instance, investigation of microvillous inclusion disease has helped identify genes responsible for intracellular vesicular transport. The discovery that mutations in the gene that codes for FOXP3 causes the immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome, as well as its animal homolog in the Scurfy mouse, has led to recognition of the critical role played by that gene in the control of the human immune response, as well as in autoimmunity and immune tolerance.
Biopsy Sampling and Indications In Children
In most pediatric GI practices, intestinal biopsy specimens are most frequently obtained from the duodenum via forceps during endoscopic examination, during which time samples are also obtained from the esophagus and stomach (esophagogastroduodenoscopy, or EGD). Endoscopic forceps biopsies have largely replaced Crosby suction biopsies because of the greater ease of the procedure, greater patient comfort, avoidance of radiation exposure, direct visualization of the GI tract, and the ability to perform multiple biopsies at several sites. In addition to being submitted for routine histology, biopsy samples may be snap-frozen (for disaccharidase analysis) or submitted for electron microscopy (for confirmation of microvillous inclusion disease). Biopsies of both the proximal and distal duodenum are recommended, including endoscopically normal mucosa, because many disorders that affect the duodenum have a focal distribution. For example, pathologic lesions in children with gluten-sensitive enteropathy may be patchy, and villous atrophy may coexist with otherwise normal mucosa.2 Furthermore, villous atrophy may be limited to, or most severe in, the duodenal bulb at the time of clinical presentation.2,3
Since pediatric EGD evolved into a routine outpatient procedure, the indications for its use have changed. For instance, at The Children’s Hospital of Philadelphia, the “first-time” EGD rate increased 12-fold in the 20 years between 1985 and 2005, with isolated abdominal pain replacing GI bleeding as the most frequent indication.4 Much of this increase was driven by the dramatic increase in food allergy–related disorders, such as eosinophilic esophagitis; by the increased prevalence of celiac disease and its clinically atypical forms, for which intestinal biopsy is the gold standard for establishing a diagnosis; and by the routine use of EGD in addition to colonoscopy for evaluation of children with suspected inflammatory bowel disease. In a 2012 study, the most frequent indications for EGD and colonoscopies in children younger than 1 year of age were diarrhea, failure to thrive, reflux, and rectal bleeding.5 Histologic abnormalities were detected in two thirds of cases, and only 2% of the mucosal biopsies were considered insufficient. Sampling of endoscopically normal-appearing mucosa is recommended because it may help assess the “background” features of the mucosa and because histologic examination may reveal clinically relevant findings previously unsuspected by the endoscopist (e.g., granulomas).6,7 One pediatric study found that duodenal biopsy performed routinely for indications such as gastroesophageal reflux, vomiting, abdominal pain, anemia, and evaluation of Crohn’s disease yielded pathologic findings in approximately 17% of cases.7
Some entities, such as congenital transport disorders, are associated with normal intestinal biopsy findings, whereas others, such as autoimmune enteropathy (AIE) or celiac disease, exhibit various degrees of villous atrophy either with or without inflammation. In contrast, a few disorders, such as abetalipoproteinemia or microvillous inclusion disease, usually reveal characteristic findings in intestinal biopsies (Table 9.1).
Table 9.1
Diagnostic Findings on Small Intestinal Biopsies
| Intestinal Biopsy | Differential Diagnosis |
| Normal |

Intestinal Development in Children
Villi appear in the duodenum during the eighth week after fertilization, and crypts of Lieberkühn are seen during the ninth week. They spread caudally in the gut, affecting the distal ileum by the 14th week of gestation. The definitive histologic features of the duodenum are established by the 14th week after fertilization, and the histology closely resembles that of a newborn by 20 weeks’ gestation. The transition from pyloric to duodenal mucosa is gradual in some individuals. Duodenal-like villi may be found in the distal pylorus, and pyloric-like epithelium may be found in the duodenum. Enteroendocrine and goblet cells appear differentiate before 14 weeks’ gestation under the control of transcription factors such as Math1.8,9 The ratio of villus height to crypt depth in the duodenum of a newborn is similar to that in an adult. Newborns usually lack plasma cells in the first week of life and gradually acquire them during the first month. At that time, immunoglobulin M (IgM)-containing plasma cells predominate, but by 3 months of life, immunoglobulin A (IgA) plasma cells predominate. Normal numbers of plasma cells and normal ratios of IgA, IgM, and IgG antibodies are attained by the first year of life.10
Congenital Disorders of Intestinal Digestion, Absorption, and Transport
Specific gene defects associated with various disorders of substrate transport have been characterized (Table 9.2). Genetic studies are instrumental in providing a picture of enterocyte function at the molecular level. Except for disorders associated with fat processing, small intestinal biopsies in these cases are typically normal or only very slightly abnormal. Normal-appearing small bowel mucosa from a patient with prolonged diarrhea, especially a young infant, should alert the clinician to these entities. Only those disorders with significant histologic findings or with confounding clinical features are discussed here.
Table 9.2
Molecular Basis of Disorders of Digestion, Absorption, and Transport
| Disease | Gene | Location | Function |
| Disaccharidase Deficiency | |||
| Congenital lactase deficiency | LCT | 2q21 | Lactase-phlorizin hydrolase activity |
| Sucrase-isomaltase deficiency | SI (EC 3.2.1.48) | 3q25-q26 | Isomaltase-sucrase activity |
| Maltase-glucoamylase deficiency | MGAM | 7q34 | Maltase-glucoamylase activity |
| Ion and Nutrient Transport Defects | |||
| Glucose-galactose malabsorption | SLC5A1 (SGLT1) | 22q13.1 | Na+/glucose contransporter |
| Fructose malabsorption | SLC2A5 (GLUT5) | 1p36 | Fructose transporter |
| Fanconi-Bickel syndrome | SLC2A2 (GLUT2) | 3q26 | Basolateral glucose transporter |
| Cystic fibrosis | CFTR | 7q31.2 | cAMP-dependent Cl− channel |
| Acrodermatitis enteropathica | SLC39A4 (ZIP4) | 8q24.3 | Zn2+ transporter |
| Congenital chloride diarrhea | SLC26A3 (DRA) | 7q22-q31.1 | Cl−/base exchanger |
| Congenital sodium diarrhea | SPINT2 | 19q13.1 | Serine-protease inhibitor |
| Congenital bile acid diarrhea | SLC10A2 (ASBT) | 13q3 | Ileal Na+/bile salt cotransporter |
| Lysinuric protein intolerance | SLC7A7 | 14q11 | Hydrolyzes endo-/exopeptidases amino acid basolateral transport |
| Pancreatic Insufficiency | |||
| Enterokinase deficiency | TMPRSS15 (PRSS7) | 21q21 | Proenterokinase |
| Trypsinogen deficiency | PRSS1 | 7q35 | Trypsinogen synthesis |
| Pancreatic lipase deficiency | PNLIP | 10q26.1 | Hydrolyzes triglycerides to fatty acids |
| Lipid Trafficking | |||
| Abetalipoproteinemia | MTTP | 4q22 | Transfer lipids to apolipoprotein |
| Hypobetalipoproteinema | APOB | 2p24 | Apolipoprotein that forms chylomicrons |
| Chylomicron retention disease | SAR1B | 5q31.1 | Intracellular chylomicron trafficking |
cAMP, Cyclic adenosine monophosphate.
From Canani RB, Terrin G, Cardillo G, et al. Congenital diarrheal disorders: improved understanding of gene defects is leading to advances in intestinal physiology and clinical management. J Pediatr Gastroenterol Nutr. 2010;50:362.
Carbohydrate Malabsorption and Disaccharidase Deficiencies
Clinically, congenital disaccharidase deficiency and carbohydrate malabsorption is an osmotic type of diarrhea caused by unabsorbed solute in the ileum. The normal rapid transit of the GI tract in children results in a more severe form of diarrhea than in adults. Histologically, the appearance of the small intestine in these cases is usually unremarkable. Therefore, the diagnosis is usually confined by determination of disacchariadase activity in homogenates of small bowel biopsy specimens or by breath testing.
Congenital disaccharidase and transporter deficiencies are rare; these disorders are much more likely to be secondary, resulting from diffuse mucosal damage due to infectious gastroenteritis, gluten-sensitive enteropathy, or other food allergies. Accelerated crypt shedding, which occurs in mucosal injury, may outpace expression of brush border enzymes and transporter proteins, leading to worsening of the diarrhea and malabsorption. This may be one reason why tacrolimus, which reduces the rate of T cell–driven crypt cell proliferation and shedding, has been found to be effective in conditions such as “tufting” enteropathy and AIE.11,12
Pathogenesis
Carbohydrate absorption begins with the breakdown of complex carbohydrates by salivary and gastric enzymes into oligosaccharides, which are then hydrolyzed to monosaccharides by specific disaccharidases located at the enterocyte brush border. There are also facilitative hexose transporters located at the basolateral membrane. The latter have been cloned and functionally characterized in several different tissues: GLUT1, erythrocyte; GLUT2, hepatocyte; GLUT3, brain; GLUT4, muscle and fat; and GLUT5, small intestine.
There are four major disaccharidase enzymes: lactase-phlorizin hydrolase (or lactase-glycosyl ceramidase), sucrase-isomaltase, maltase-glucoamylase, and trehalase.11 Lactase-phlorizin hydrolase degrades lactose, the main carbohydrate in milk. Sucrase-isomaltase is responsible for the degradation of dietary sucrose. Maltase-glucoamylase hydrolyses starch into small polymers. Trehalose occurs mainly in mushrooms. Trehalase deficiency is extremely rare. It has been described only in older children and adults. The expression of these enzymes on the intestinal brush border appears to follow a time-dependent sequence. Lactase-phlorizin hydrolase, which is highly expressed at birth, typically declines in early life in most individuals (although it persists through adult life in whites). Sucrase-isomaltase, which is undetectable at birth, reaches adult levels within the first few months of life. These changes coincide with a switch from milk to a more solid diet.
Lactose malabsorption in children can be caused by primary, genetically determined deficiency of lactase-phlorizin hydrolase, which follows a variable clinical course, or by a secondary lactase deficiency resulting from mucosal injury, as in infectious diarrhea, celiac disease, or Crohn’s disease.12 Congenital sucrose-isomaltase deficiency typically becomes clinically apparent when the child begins to consume fruits and juices.13 Glucose-galactose malabsorption is a rare autosomal recessive disorder that is caused by a mutation in the gene on chromosome 22 that encodes the sodium/glucose cotransporter, solute carrier family 5, member 1 (SLC5A1, also known as SGLT1), which can result in severe chronic diarrhea.14 Congenital fructose malabsorption is caused by a mutation in the gene for the hexose transporter isoform, GLUT5, which is located on chromosome 1 and designated SLC2A5.15,16
Lipid Trafficking Disorders
Fat absorption by enterocytes begins with emulsification and solubilization of cholesterol in the intestinal lumen by biliary lipids and salts. Most clinical disorders of fat malabsorption result from severe liver disease, pancreatic disease (e.g., cystic fibrosis), or extensive ileal resection (as in Crohn’s disease), with loss of the enterohepatic circulation of bile acids. Intestinal biopsies play a limited role in the diagnosis of these disorders. However, primary disorders involving abnormalities of fat transport within enterocytes, although uncommon, may result in a characteristic vacuolization of enterocytes in intestinal biopsy specimens.
Abetalipoproteinemia, hypolipoproteinemia, and chylomicron retention disease (Anderson disease) share many characteristics, including fat malabsorption, low levels of serum lipids, failure to thrive in childhood, neurologic and visual problems resulting from malabsorption of fat-soluble vitamins, and accumulation of lipid droplets in the enterocytes. These conditions are associated with disorders of apolipoproteins, which reside on the surface of chylomicrons. Apoliproproteins native to the intestine are apolipoprotein A-I (apo A-I), apo A-IV, and apo B, which has two forms, apo B-100 and apo B-48, both encoded by the same gene located on chromosome 2.17 Localization of apolipoproteins in the Golgi apparatus and along the microvilli of enterocytes has been demonstrated by immuno-electron microscopy.18
Abetalipoproteinemia
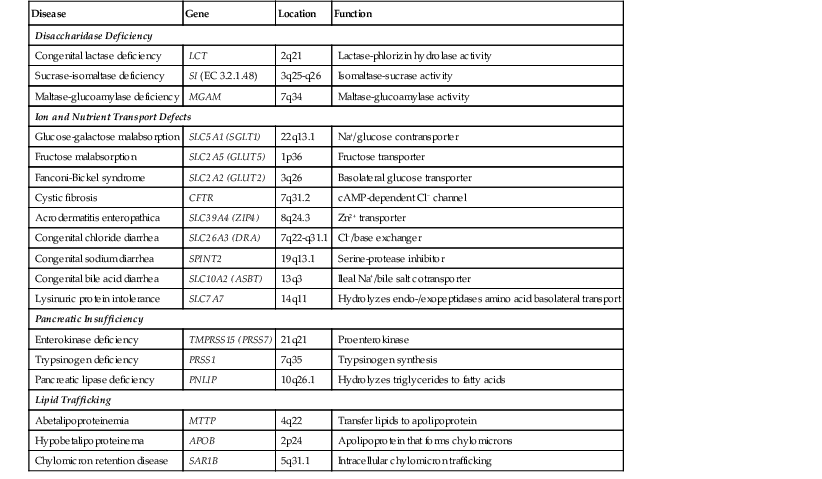
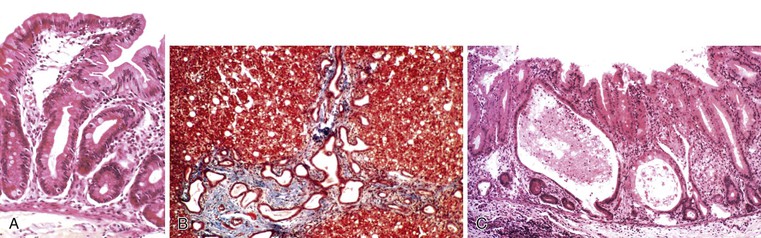
Abetalipoproteinemia is an autosomal recessive disorder characterized by absence of apo B-containing lipoproteins. The molecular basis is a mutation of MTTP, the gene that codes for microsomal triglyceride transfer protein, which is located on chromosome 4q22 (see Table 9.2).19 MTTP is responsible for the assembly of lipoprotein particles and for the proper folding of apo B, which prevents its premature degradation.20 Because of the mutation, fatty acids within intestinal cells cannot be exported as chylomicrons. Patients develop diarrhea and fat malabsorption usually within the first few months of life, with acanthocytosis; deficiencies in fat-soluble vitamins then result in retinitis pigmentosa and neurologic symptoms. However, there is clinical heterogeneity because signs and symptoms may occasionally manifest in older individuals. Serum levels of cholesterol and triglycerides are typically low and, most importantly, do not rise after a fatty meal. Small bowel biopsies typically reveal preserved villi (Fig. 9.1, A), but characteristic multivacuolated, fat-filled enterocytes may be seen in specimens from fasting patients, which on electron microscopy are irregular in size and generally not membrane bound (see Fig. 9.1, B). In addition, lipid does not accumulate in the extracellular space. Hepatic biopsies typically reveal steatosis, with numerous non-membrane-bound lipid droplets within hepatocyte cytoplasm.21 Fibrosis evolving to cirrhosis has been reported in some patients.22
Hypobetalipoproteinemia
Hypobetalipoproteinemia is an autosomal dominant disorder caused by a mutation in the APOB gene located on chromosome 2, which leads to the development of a truncated apo B protein.23 Homozygous patients have a clinical and histologic phenotype that is essentially indistinguishable from abetalipoproteinemia, whereas heterozygous patients have only a mild disease.
Chylomicron Retention Disease
Anderson disease is similar to abetalipoproteinemia in its GI manifestations and impact on growth, although acanthocytosis is usually absent and neurologic and ocular abnormalities are much less severe. However, in contrast to abetalipoproteinemia, serum fasting triglyceride levels are typically normal, and hypocholesterolemia is less marked. The causative gene, SAR1B, codes for a guanosine triphosphatase (GTPase) that is associated with coat protein carriers involved in transport from the endoplasmic reticulum to the Golgi apparatus, particularly transport of chylomicrons and low-density lipoproteins.24 The pathologic features in small bowel biopsy specimens are essentially indistinguishable from those of abetalipoproteinemia (see Fig. 9.1, C).
Minor degrees of enterocyte vacuolization (e.g., resulting from a recent feed) are common in intestinal biopsy specimens in infants. In these cases, vacuolization is not as marked or as diffused as in abetalipoproteinemia or chylomicron storage disease. In addition, lipid droplets are present in the intercellular spaces and lacteals after feeding but are absent from these spaces in disorders that cause impaired lipid transport. By contrast, lipid-containing macrophages are present in the lamina propria in several storage disorders in which digestive symptoms can occasionally be significant and in which an intestinal biopsy is obtained in the course of a workup for failure to thrive (see Metabolic Diseases).
Amino Acid Transport Disorders
Disorders of amino acid transport rarely cause prominent GI manifestations, except for lysinuric protein intolerance (LPI), which results from mutations in the SLCA7 gene. This gene codes for the light chain subunit of the heterodimer amino acid transporter at the basolateral membrane of intestinal and renal epithelial cells.25 Clinically, LPI manifests with failure to thrive, vomiting, and diarrhea. Hepatosplenomegaly, hematologic anomalies, and neurologic involvement including hyperammonemic coma are recurrent clinical features. The urea cycle intermediates ornithine and arginine fail to exit from intestinal epithelium in LPI, resulting in a urea cycle dysfunction with hyperammonemia and alterations of mental status. Two major complications, pulmonary alveolar proteinosis and renal disease, are increasingly observed in patients with LPI.26,27 The condition is recognized by the presence of markedly elevated urinary levels of lysine and other dibasic amino acids; DNA testing may be required for confirmation of the diagnosis. One 5-year-old boy who was reported to have chronic diarrhea and a “flat gut” on small intestinal biopsy was unsuccessfully treated with a gluten-free diet.28 Other complications associated with this disorder include systemic lupus erythematosus, hemophagocytic lymphohistiocytosis,29 and sudden infant death.30
Electrolytes and Trace Elements
The biology of intestinal ion transport is a complex field. Disorders range from rare selective deficiencies to multisystem diseases such as cystic fibrosis. Defects that manifest primarily with severe diarrhea in infancy include congenital chloride diarrhea and congenital sodium diarrhea. Intestinal biopsy findings in these cases have been reported to be normal or to show only mild partial villus atrophy.31 Congenital chloride diarrhea is caused by mutations in the gene for solute-linked carrier family 26, member 3 (SLC26A3). It manifests as a life-threatening congenital diarrhea. Long-term manifestations include renal disease, spermatoceles, and male subfertility.32 Congenital sodium diarrhea results in hyponatremia and metabolic acidosis. The syndromic form of this disorder has been linked to mutations in SPINT2 and is also characterized by cloanal and anal atresia, hypertelorism, double kidney, cleft palate, and digital anomalies.13
Acrodermatitis enteropathica is an autosomal recessive disorder that is caused by a defect in intestinal absorption of zinc. Mutations in a candidate gene, ZIP4 (SLC39A4), result in defective epithelial transport in the intestine and kidney.13 There is a characteristic clinical syndrome of diarrhea combined with acral and orificial skin lesions. Rodin and Goldman found pancreatic islet hyperplasia, absence of the thymus and germinal centers, and plasmacytosis of the spleen and lymph nodes in autopsy studies.33 Inclusion bodies in Paneth cells have been reported in patients with this disorder.34,35 A decrease in villus height is observed in animal models of zinc deficiency and is corrected by zinc supplementation.36 Interference with chylomicron development and function has been noted.37 Severe zinc deficiency is also seen in other disorders, such as the Cronkhite-Canada syndrome.38
Menkes disease is an X-linked disorder that results from a defect in intestinal absorption of copper. Affected patients present with characteristic hair shaft anomalies (pili torti), cerebral degeneration, hypopigmentation, abnormal bones, vomiting and diarrhea, and, occasionally, protein-losing enteropathy (PLE). The disorder maps to Xq12-q13 and is caused by mutations in the ATP7A gene, which encodes a copper-transporting P1B-type adenosine triphosphatase that is part of a family of membrane proteins responsible for cation transport across membranes.39,40
Vitamins
Congenital vitamin B12 transport disorders include congenital intrinsic factor deficiency, selective absence of intrinsic factor without any gastric anomaly, congenital pancreatic insufficiency of various causes, and congenital selective vitamin B12 malabsorption—the latter an autosomal recessive disorder characterized by megaloblastic anemia, hepatosplenomegaly, vomiting, diarrhea and proteinuria. These disorders are caused by mutations in the gene CUBN, which encodes an ileal cell surface receptor protein known as cubilin.41 None of these disorders results in any characteristic histologic changes on intestinal biopsies.
A rare congenital disorder of intracellular vitamin B12 metabolism belonging to the cobalamin C (CblC) family has also been described. Patients present with diarrhea, renal failure, and systemic thromboemboli in association with a striking gastric atrophy.42 PLE responsive to hydroxycobalamin treatment has been reported in one patient with this disorder,43 presumably on the basis of the gastric anomaly.
Bile Acids
Heubi and colleagues44 described a severe type of refractory diarrhea caused by a primary disorder of bile acid absorption; intestinal biopsy findings were normal. A defect in the gene coding for an ileal Na+-dependent bile acid transporter, SLC10A2, has been found in this disorder.45 Bile acid–related diarrheal illnesses are much more commonly secondary to chronic pancreatic insufficiency46 or to loss of ileal surface, as occurs with short gut syndrome resulting from necrotizing enterocolitis47 or with extensive ileal resection in Crohn’s disease.48,49
Congenital Defects of Intestinal Epithelial Differentiation
Microvillus Inclusion Disease
Clinical Features
Initially described by Davidson et al in 197850 and subsequently recognized worldwide, microvillus inclusion disease (MVID) is an autosomal recessive disease characterized by refractory secretory diarrhea usually within the first week of life, although late-onset symptoms may manifest in the first few months of life.51 Investigation of clusters of cases within the Navajo population identified mutations in the gene MYO5B.52 Homozygosity mapping of an extended Turkish kindred allowed Muller and colleagues to locate the gene locus to chromosome 18q21.53 MYO5B codes for myosin Vb, part of a family of proteins responsible for actin-dependent organelle transport and regulation of endosome recycling. Mutations in the MYO5A gene cause Griscelli syndrome, an immunodeficiency disorder also characterized by abnormal transfer of melanin granules to keratinocytes.53
Pathology
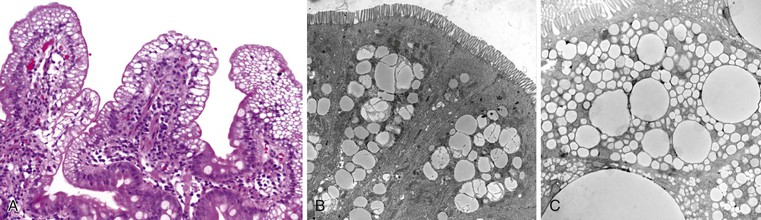
Small bowel biopsy specimens in MVID are usually characterized by severe villus atrophy, mild or moderate crypt hyperplasia, and a variable degree of inflammation in the lamina propria, but without crypt destruction (Fig. 9.2, A). The diagnosis may be strongly suspected on paraffin-embedded sections by the absence of a distinct brush border using the periodic acid–Schiff (PAS) stain, and by the presence of PAS-positive diastase-resistant densities in the apex of enterocytes (see Fig. 9.2, B and C). Similar observations are seen on immunohistochemical staining for alkaline phosphatase and anti-CD10. More recently, similar results using antibodies directed against anti-Rab11a, a small GTPase protein on the surface of recycling endosomes, has provided further evidence that MVID is a disorder of apical plasma membrane recycling.54 The pathognomonic ultrastructural features include absent or small stubby microvilli, vesicular structures located toward the apex of enterocytes containing microvilli, and granules containing dense amorphous material (Fig. 9.3). Microvillus inclusions have also been reported in the colon, gallbladder, and renal tubular epithelium in these patients.55 A variety of ultrastructural features have also been noted in patients with this disorder, and finding the “typical” inclusions may require a prolonged search.56
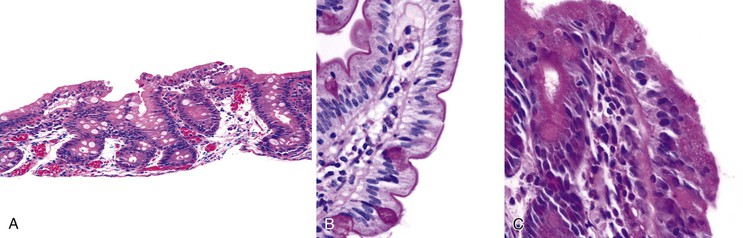
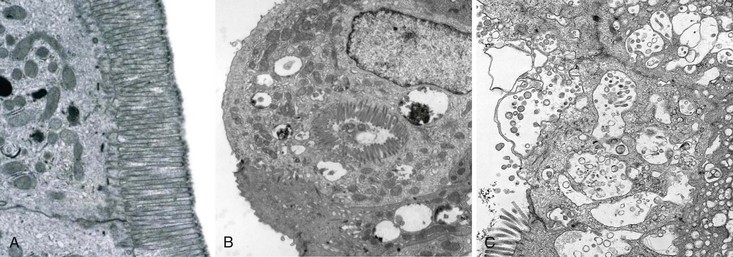
Atypical inclusions include the presence of numerous small electron-lucent vesicles and accumulation of osmiophilic vermiform structures.57 Cases with these features have been previously reported as “MVID with atypical features” or as “microvillous dystrophy.”58,59 Genetic analysis helps in defining whether the ultrastructural (and sometimes clinical) variability represent different entities or variations in the spectrum of manifestations of a single disorder, especially given the enormous prognostic and therapeutic implications.
Treatment
Patients with MVID are dependent on total parenteral nutrition (TPN). Medical therapy in these patients is generally ineffective, and improved survival may require small bowel transplantation.60 An intriguing instance of apparent resolution of the disease with improvement of the mucosal features in a patient on long-term TPN has been reported.61
Congenital Tufting Enteropathy (Epithelial Dysplasia)
Clinical Features
Congenital tufting enteropathy (CTE) was first described by Reifen and colleagues62 in patients presenting in the neonatal period with watery diarrhea. The prenatal history is usually uneventful. The disease appears to be inherited in an autosomal recessive fashion, as suggested by the finding of other affected siblings and frequent parental consanguinity. The incidence is estimated at 1 : 50,000 to 1 : 100,000 live births in Europe, and it is more frequent in patients of Arabic origin.63 Cases of tufting enteropathy have been described in association with other malformations, including facial dysmorphism, micrognathia and anal imperforation,64 and choanal atresia and hair abnormalities.65 Superficial punctate keratitis and conjunctival erosions have been observed in about 50% of patients.66 In addition, an association has been reported between CTE and autoimmune phenomena, including autoimmune anemia, thrombocytopenia,67 and chronic arthritis.68 Using single nucleotide polymorphism (SNP) analysis in an affected kindred, Sivagnanam and co-workers69 identified the epithelial cell adhesion molecule gene (EPCAM), located on chromosome 2p21, as the causative gene. EpCAM belongs to a family of cell adhesion receptors, is associated with tight junction proteins, and is responsible for cell–cell interaction by recruiting actin filaments to sites of contact.70
Pathology
The histologic hallmarks of CTE are severe villus atrophy with the formation of “tufts” of rounded, teardrop-shaped enterocytes that appear to shed into the lumen. There may be a mild increase in lamina propria inflammatory cells, but intraepithelial lymphocytes do not appear to be significantly increased. The brush border is normal by PAS staining, and electron microscopic findings are nonspecific. Dilated crypts have also been described. Epithelial abnormalities typically vary over time and may be subtle or absent in the first biopsy (Fig. 9.4). Abnormalities in immunohistochemical staining for basement membrane laminin and heparan sulfate proteoglycans,55 ultrastructural irregularities of desmosomes, and abnormal distribution of α2β1 integrin71 have been observed in patients with this disorder. Immunohistochemistry of intestinal tissue reveals decreased expression of EpCAM, which may be helpful in diagnosis.72 Intestinal features resembling CTE have been observed in a knockout mouse model for transcription factor Elf3.73
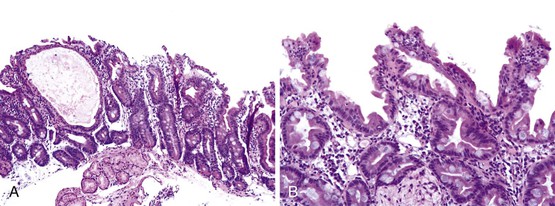
Treatment
Dietary manipulations, antibiotic therapy, and immunosuppressive therapy have proved ineffective in these patients. Long-term TPN and small bowel transplantation are currently the major treatment modalities for these patients. However, the disease may have a variable clinical course, with some patients reported to have been weaned off TPN.74 A report of a patient with similar phenotypic and biopsy features who was found to harbor a mutation in SPINT2, which is associated with congenital sodium diarrhea, suggests that the CTE phenotype may overlap that of other congenital forms of diarrhea.75
A likely related disorder resulting from the absence of intestinal epithelial α6β4 integrin has also been described. The salient features are severe watery diarrhea within the first 2 weeks of life in association with pyloric atresia, with extensive sloughing of epithelium observed in biopsies.76 Diarrhea has also been observed in cases of pyloric atresia–junctional epidermolysis bullosa, in which mutations in the genes that code for laminin-5 and α6β4 integrin have been described.77
Enteroendocrine Cell Dysgenesis (Enteric Anendocrinosis)
Clinical Features
Enteroendocrine cell dysgenesis (ECD) is an autosomal recessive disorder associated with congenital absence of enteroendocrine cells in the small and large bowel secondary to mutations in the NEUROG3 gene, located on chromosome 10q21.3. This gene is required for differentiation of epithelial cells to an endocrine phenotype.78 Affected patients exhibit a congenital diarrheal syndrome with profound malabsorption of all nutrients from birth. Patients also typically develop insulin-dependent diabetes, some during infancy and others later in childhood.78,79 Pancreatic exocrine function is normal. Neurogenin-3 null mice lack enteroendocrine cells80 and are also severely diabetic, because neurogenin-3 is required for pancreatic endocrine development.81
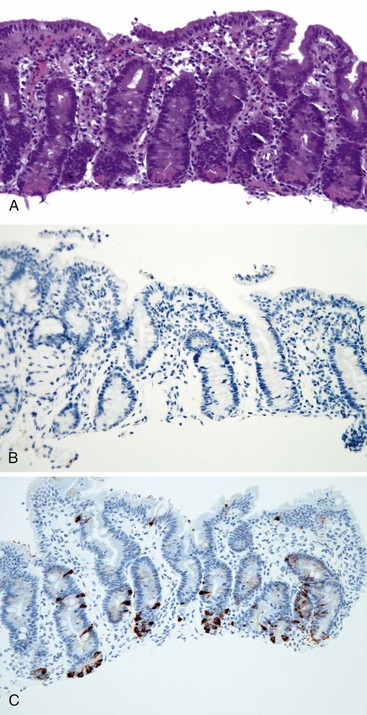
Pathology
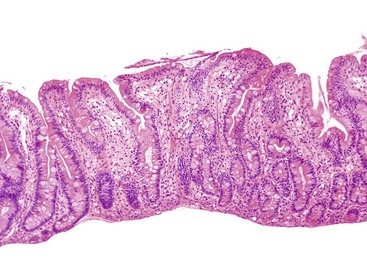
Intestinal biopsies may be normal or may reveal villous atrophy, the severity of the changes perhaps related to different mutations in the NEUROG3 gene79 (Fig. 9.5). The brush border is normal, and goblet and Paneth cells are present. Inflammatory cells in the lamina propria do not appear significantly increased, and there is no crypt-destructive process. Electron microscopy confirms the presence of microvilli and the absence of microvillus inclusions.82 The characteristic absence of enteroendocrine cells in the small bowel and colon can be confirmed by immunohistochemistry for chromogranin A (see Fig. 9.5, B). Patients with autoimmune polyglandular syndrome I may also have absent or markedly reduced enteroendocrine cells, which may be transient.83 In addition, loss of enteroendocrine cells may be observed in patients with autoimmune enteropathy, along with loss of goblet and Paneth cells. Treatment consists of parenteral feeding with limited oral feeding.
Syndromatic Diarrhea (Trichohepatoenteric Syndrome)
Clinical Features
Girault and associates84 described a group of patients with dysmorphic features consisting of a prominent forehead, broad nose, hypertelorism, and wooly, easily removable abnormal hair (trichorrhexis nodosa), in association with intractable diarrhea and immunodeficiency, which they termed syndromatic intractable diarrhea because of the constellation of extraintestinal manifestations. Most of the patients reported were of Middle Eastern origin, and their histories included parental consanguinity and similarly affected siblings. The immunodeficiency was characterized by impaired T-cell and antibody responses despite normal immunoglobulin levels. Infants with similar clinical features had been previously reported as having “tricho-hepato-enteric syndrome” (THE)85,86; in these patients, thin, sparse hair (trichomalacia) was seen in association with hypertelorism and chronic diarrhea, and there was a history of premature delivery with intrauterine growth retardation. In addition, the patients described by Verloes and colleagues86 presented with neonatal hemochromatosis, characterized by liver failure, cirrhosis, and multivisceral iron deposition. The phenotypic similarity between THE and the cases described by Girault led Fabre and colleagues to suggest that these manifestations represent a spectrum of the same disorder.87 This appeared to be confirmed when mutations in TTC37, described in cases of THE,88 were also observed in patients with the features of “syndromic diarrhea.”89 TTC37 maps to chromosome 5q14.3-5q21.2 and codes for a tetratricopeptide repeat protein, thespin, whose function is uncharacterized.88
Pathology
Intestinal biopsies have revealed variable degrees of villous atrophy without a conspicuous increase in inflammatory cells and without other specific features. Serially obtained biopsies suggest that villous atrophy improves with time and that the inflammatory infiltrate is not consistent. The hair is described as woolly with microscopic features of trichorrhexis nodosa. Hepatic involvement is inconsistent. Hartley and co-workers also observed platelet abnormalities consisting of reduced α-granules, abnormal lipid inclusions, and an abnormal canalicular system.88
Affected children require long-term parenteral nutrition, though some may be weaned to full enteral feeding eventually.
Disorders of Immunomodulation
Autoimmune Enteropathy
Clinical Features
Autoimmune enteropathy (AIE) is the disorder that most frequently leads to infantile intractable diarrhea.90 Most cases occur in infancy or within the first year of life. The clinical picture in infants is usually dominated by secretory diarrhea, dermatitis, and various endocrine disorders, of which diabetes mellitus is the most frequent. These patients also suffer from numerous complications, mainly infectious, related to loss of a functional gut barrier, central lines, underlying immunodeficiency, and immunosuppressive therapy. Features include a strong male preponderance, a family history of other affected siblings, frequent extraintestinal involvement, and the presence of various circulating autoantibodies.91 However, this entity has also been reported in older children, in girls, and even in adults, in whom it may be responsible for a proportion of cases referred to as “refractory sprue.” In adults, the clinical picture resembles celiac disease unresponsive to gluten withdrawal.92–99 Corazza and colleagues reported on four adult women with refractory celiac disease, none of whom had had malabsorptive symptoms in childhood, which confirms the possibility of adult onset.94 Akram and associates92 reported a series of 15 patients (7 female), aged 42 to 67 years, in whom celiac disease was excluded by lack of response to a gluten-free diet or by absence of the human leukocyte antigen (HLA) genotypes with celiac disease susceptibility. All patients had protracted diarrhea, weight loss, and malnutrition.
Pathogenesis
The strongest and most commonly recognized association of AIE is with the IPEX syndrome, which was initially described by Powell and colleagues.100 Extraintestinal diseases in these patients include insulin-dependent diabetes, thyroiditis, membranous glomerulopathy, and interstitial nephritis.101 This syndrome has been related to mutations in the FOXP3 gene (Xp11.23).102 Humans bearing this mutation share a phenotype very similar to that of the Scurfy mouse, showing an absence of naturally occurring CD4+ CD25+ regulatory T cells (Tregs). Mutations in the FOXP3 gene or its promoter region impair suppression of the inflammatory response by regulatory T cells, such that inflammation triggered by a relatively trivial viral infection can result in an extensive autoimmune cascade.103,104 However, approximately 50% of patients, including females, with otherwise typical clinical manifestations of IPEX have no detectable mutations of FOXP3.105 The term IPEX-like has been suggested for these cases.106
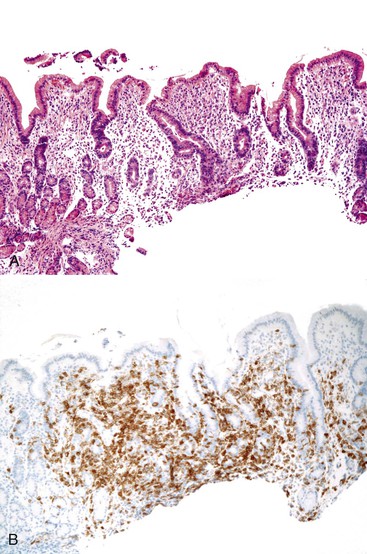
One of the hallmarks of this entity, noted in the initial reports of AIE, is the presence of anti-enterocyte antibodies, which can be detected by indirect immunofluorescence using the patient’s serum on frozen sections of normal bowel. Positive fluorescence usually results in a linear pattern along the apex and basolateral border of the enterocyte100–101,107–111 (Fig. 9.6, D). The antibodies are predominantly IgG and have been described as complement-fixing,107 although IgM and IgA have also been described.109,111 Antibodies reacting against mucus or goblet cells have also been described, and intestinal biopsies in these cases have shown a marked depletion of goblet cells.112,113
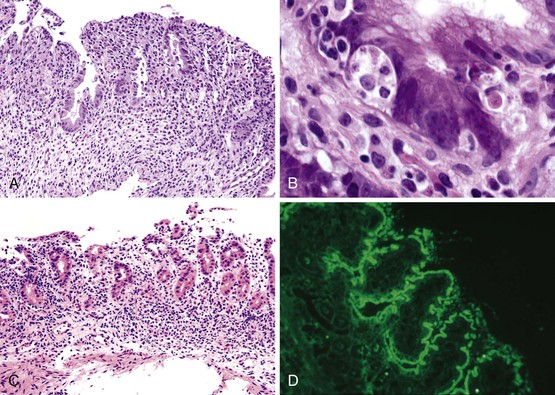
The target of the anti-enterocyte antibodies is still a matter of investigation, although a 75-kilodalton (kDa) antigen reactive with autoantibody in the sera of patients with the IPEX syndrome has been reported.114 This protein is expressed in the epithelial cells of the small bowel, colon, and kidney and may play a role in protein–protein interactions in enterocytes. More than one antigen is likely to be involved. More recently, a 95-kDa antigen identified as villin has been reported in IPEX patients.115
The role of these antibodies in the diagnosis of AIE is debatable. They have been reported to occur after the onset of disease and disappear before restoration of mucosal integrity110; therefore, they may represent an epiphenomenon without playing a causative role in mucosal damage. Similar anti-enterocyte antibodies have been described in adult AIDS patients without digestive symptoms.116 The specificity of anti–goblet cell antibodies has also been questioned. Anti–goblet cell antibodies have been detected in patients with chronic inflammatory bowel disease (Crohn’s and ulcerative colitis) and their first degree relatives117 and in a series of treated and untreated patients with celiac disease and controls.118 Finally, the usefulness of these antibodies for the diagnosis of AIE in young infants is also limited, given that there is little IgG production during the first 3 months of life and that IgG detected in infants is mostly maternal in origin.119
Pathology
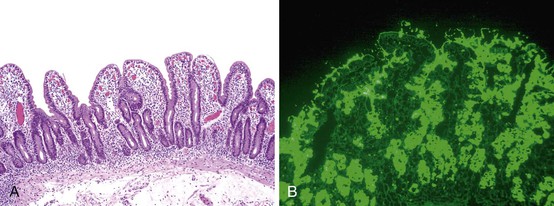
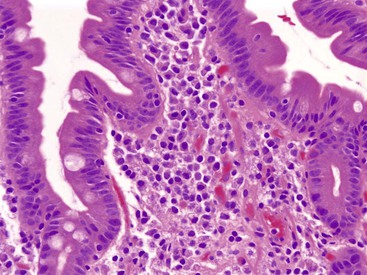
The entire GI tract is often involved in AIE. The histopathologic findings in duodenal and small bowel biopsies are typically characterized by severe villus atrophy, crypt hyperplasia, and a mixed inflammatory infiltrate of the lamina propria. Marked inflammatory destruction of intestinal crypts, with extensive apoptosis, is often observed and is similar to that seen in intestinal graft-versus-host disease, suggesting an abnormal immune-mediated attack against intestinal epithelium101 (see Fig. 9.6). However, there appears to be marked variation in the histologic features, with some cases showing milder degrees of intestinal damage. In contrast to gluten-sensitive enteropathy, a condition that also reveals flat villi, intraepithelial lymphocytes in AIE tend to be relatively few in number. A concomitant crypt-destructive colitis and gastritis are present in the majority of cases as well.
The differential diagnosis of AIE includes, in addition to celiac disease and graft-versus-host disease, Crohn’s disease and enterocolitis associated with various immunodeficiency states. A high index of suspicion for AIE should be present when there is a severe inflammatory crypt-destructive process with villous atrophy and increased apoptosis in the bases of the crypts, loss of goblet and Paneth cells, or an intestinal inflammatory process in a patient with other autoimmune phenomena (Fig. 9.7). Testing for circulating anti-enterocyte antibodies is useful in older children and adults but may be of more limited use in young infants. Immunostaining for the FOXP3 gene product applied to formalin-fixed paraffin-embedded GI biopsies may be useful to screen for the IPEX syndrome in young patients with severe diarrhea.120
Treatment
Mortality in cases of AIE has been reported to be high, as much as one third of reported cases, but was more frequent in older reports. Diet changes and steroids alone have been largely ineffective, and immunosuppressants appear to be necessary in most cases. Bone marrow transplantation may be curative. Immunosuppression is required in older children and adults.92 A good response to sirolimus has been reported in IPEX-like cases.106 Bone marrow transplantation appears to offer the only cure for IPEX.121
Major differentiating features for various etiologies of severe diarrhea in early infancy are presented in Table 9.3.
Table 9.3
Differentiating Features of Severe Diarrhea of Early Infancy
| Presentation | First 2 Weeks | After 1 Month | ||
| Microvillous Inclusion Disease | Tufting Enteropathy | Enteroendocrine Cell Dysgenesis | Autoimmune Enteropathy | |
| Gene defect | MYO5B (18q21) | EPCAM (2p21) | NEUROG3 (10q21.3) | FOXP3 (Xp11.23) in IPEX syndrome |
| Extraintestinal disease | Low GGT cholestasis after bowel transplantation | Dysmorphism; autoimmune diseases (arthritis) | Insulin-dependent diabetes | Polyendocrinopathies |
| Anti-enterocyte antibodies | No | No | No | Yes |
| Villous atrophy | Yes | Variable | Variable | Variable |
| Surface epithelium | Absent brush border | Tufting and desquamation | Normal | Normal or atrophic |
| Lamina propria inflammation | Minimal | Variable | Minimal | Usually increased |
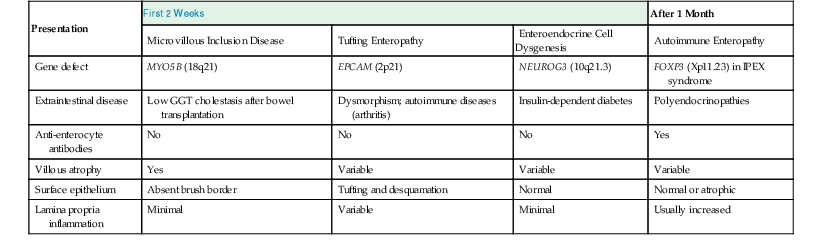
GGT, γ-Glutamyl transferase.
Autoimmune Polyendocrine Syndrome (APS 1)
Autoimmune polyendocrine syndrome (APS 1), also referred to as autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED), is an autosomal recessive disorder with heterogeneous clinical manifestations. It is caused by mutations in the AIRE gene, which codes for a transcription factor primarily expressed in medullary thymic epithelial cells, where negative selection is thought to occur.122 The type I form is characterized by various endocrinopathies of an autoimmune nature, often beginning in childhood or early teenage years; by chronic mucocutaneous candidiasis due to a T-cell defect starting soon after birth; and by dystrophy of ectodermal tissues.123 Malabsorption occurs in as much as 25% of patients and appears to result from destruction of intestinal endocrine cells.124 A specific deficiency of cholecystokinin-producing enteroendocrine cells has been reported in one patient, with seeming reappearance of the cells when the diarrhea abated.83 Small bowel biopsy specimens show mild changes or can even be normal, in contrast to the crypt-destructive inflammation usually seen in AIE.
Necrotizing Enterocolitis
Clinical Features
Neonatal necrotizing enterocolitis (NEC) is the most serious type of acquired GI disorder of neonates and the most common cause of intestinal perforation and acquired short gut syndrome among patients in neonatal intensive care units.125 The severity of the disease and its attendant complications is inversely proportional to the gestational age, reaching an incidence of 10% and a mortality rate of 26% among very-low-birth-weight (<1500 g) infants.126 However, approximately 10% of cases occur in infants who are almost full term but have a variety of risk factors such as cyanotic heart disease, volvulus, aganglionosis, or gastroschisis.
Early signs and symptoms of NEC may be subtle and nonspecific; they may include apnea, bradycardia, and lethargy, suggesting sepsis. More specific GI signs include abdominal distention, absent bowel sounds, and vomiting and diarrhea; radiographic findings include ileus and pneumatosis intestinalis. More advanced disease is characterized by perforation in a severely ill child.127
Pathogenesis
The pathogenesis of NEC is incompletely understood but is believed to be multifactorial, resulting from a combination of genetic predisposition, intestinal immaturity with inadequate motility, a heightened and inappropriate inflammatory response, abnormal microbial colonization, and an imbalance in microvascular tone.125 Ischemic damage in NEC has been postulated to be due, in part, to circulatory alterations that divert blood away from abdominal organs to the heart and brain during episodes of hypoxia (the “diving reflex”). Intramural vessels are hypothesized to be the primary target of a combination of ischemia and inflammation, mediated by the release of vasoconstrictor agents such as peptide endothelin-1 and free radicals such as nitric oxide.126 Because NEC usually occurs after the beginning of enteral feeding, enteral alimentation has also been proposed as a contributing factor. Human milk appears to reduce the risk for NEC.127 A number of different bacterial pathogens have also been associated with NEC, as well as various cytokines. Endotoxin, platelet-activating factor, and tumor necrosis factor-α have also induced ischemic damage in experimental models of NEC.128
Pathology
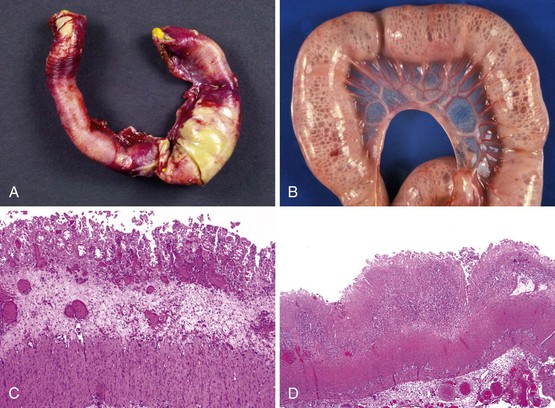
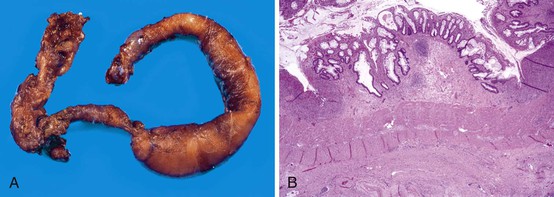
The ileocecal region, a vascular “watershed,” is most frequently involved, although 25% of pediatric patients may have involvement of only the ileum or colon. Disease extending from the ligament of Treitz to the rectum (pan-NEC) may be seen in fatal cases. Continuous and discontinuous involvement occurs in an equal proportion of children. The affected bowel is typically distended, with a grayish or purple color (if hemorrhage is present). The wall is thin and fragile, with grossly discernable gas bubbles in cases of pneumatosis (Fig. 9.8). Ischemic hemorrhagic necrosis is the predominant histologic manifestation; it may be limited to the mucosa, or it may involve the entire thickness of the bowel wall. Increasing degrees of inflammatory cell infiltrates occur with progression of disease and appear to be a response to both necrosis and bacterial proliferation. In contrast to infectious colitis, crypt microabscesses are uncommon in patients with NEC.129 Although mesenteric thromboembolism, most frequently resulting from umbilical artery catheterization in neonates, can also cause necrosis of the bowel, occlusion of large vessels is uncommon in patients with NEC; however, it may be noted in autopsy cases, where it is believed to be a secondary phenomenon. Intestinal pneumatosis develops as a result of fermentation of intraluminal contents due to bacterial overgrowth. Healing changes include reepithelialization, granulation tissue formation, and fibrosis, which can be associated with stenosis and strictures in infants who survive (Fig. 9.9).
Treatment
Medical therapy includes stopping enteral feeds, administration of antibiotics, intravenous fluids, and parenteral nutrition along with circulatory support. Surgery is indicated for patients with intestinal perforation, for those with progressive clinical deterioration despite optimal medical management, and for resection of strictures and stenoses in convalescing patients. The goal of surgery is to resect only grossly necrotic tissue and, thus, to preserve as much bowel as possible. Obtaining intraoperative frozen sections for assessment of viability of surgical margins is neither indicated nor useful.
Lymphangiectasia
Clinical Features
Intestinal lymphangiectasia, which may be primary or secondary, results in PLE (Box 9.2). Primary lymphangiectasia is a rare disorder that is usually diagnosed in children younger than 3 years of age who present with diarrhea, PLE, hypoalbuminemia, hypogammaglobulinemia, and lymphopenia, all of which contribute to the development of secondary immunodeficiency. Malabsorption may cause deficiencies in fat-soluble vitamins. Primary disorders often result from congenital obstruction to lymph flow or abnormal lymphatic structure; they may be essentially limited to intestinal lymphatics, as in primary lymphangiectasia,130,131 or they may involve multiple organs, as in hereditary lymphedema (Milroy disease), which is associated with mutations in the gene that encodes for vascular endothelial growth factor receptor factor 3 (VEGFR3).132–134 Other disorders may be associated with characteristic congenital anomalies, such as the Turner, Noonan, and Hennekam syndromes.135,136 Intestinal lymphangiectasia has also been reported in premature infants.137 It has been postulated that congenital hypoplasia of lymphatics may also cause PLE in neonates.138
Secondary lymphangiectasia may be central, resulting from cardiac disease, or local, caused by obstruction of lymphatics due to a variety of potential conditions. In children, secondary immunodeficiency resulting from extensive protein loss in the stools has been reported with intestinal malrotation and cavernous hemangioma of the jejunum.139 Cardiac causes of lymphangiectasia with PLE usually involve obstruction to systemic venous return or surgical procedures that cause increased systemic venous pressure.140,141
PLE occurs in as much as 10% of patients who have undergone the Fontan operation for correction of tricuspid atresia and other right-sided cardiac malformations; it carries a poor prognosis, with a 5-year survival rate of approximately 50%.142 This procedure creates a direct communication between the systemic venous return and the pulmonary arterial system, resulting in chronically sustained increased systemic venous pressure. Associated factors that may contribute to PLE in these patients include inflammatory mediators, decreased enterocyte heparin sulfate synthesis, and chronic mesenteric ischemia.143
Pathology
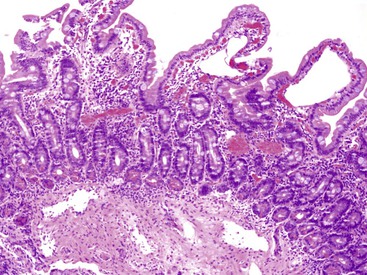
The endoscopic appearance is usually characteristic, with swollen opaque villi, white nodules, or submucosal elevations.144,145 Histologic features include blunted villi with dilated lacteals (Fig. 9.10). The finding of occasional dilated lymphatics in a biopsy specimen from a patient without significant symptoms is not infrequent and is of no diagnostic importance. Conversely, mucosal biopsies may be nondiagnostic when the lesions are focal, and they almost always fail to detect lymphangiectasia located within deeper layers of the bowel wall. Dietary therapy is the cornerstone of treatment and is designed to maintain protein levels.145 Localized forms of intestinal lymphangiectasia may be amenable to surgical resection.146
Metabolic Diseases
Metabolic disorders can result in chronic diarrhea and malabsorption (Box 9.3). Several congenital disorders of glycosylation (CDG) are associated with prominent GI manifestations. These are part of a large group of disorders related to defects in synthesis and attachment of glycans to proteins and lipids, currently comprising approximately 24 entities, most of which have neurologic manifestations. Failure to thrive with diarrhea, PLE, and hepatic abnormalities has been described mainly in phosphomannose isomerase deficiency (MPI-CDG, formerly called CDG type Ib) and in patients with mutations in the gene encoding alpha-1,3-glucosyltransferase (ALG6-CDG, formerly CDG type Ic).147–149 Small bowel biopsy specimens are generally unremarkable or show mild, nonspecific villous atrophy and inflammation unresponsive to gluten or cow’s milk withdrawal. Electron microscopic changes are characterized by distention of the smooth endoplasmic reticulum with lipid-containing inclusions150 or insoluble precipitated protein.151 Absence or marked reduction in the level of enterocyte heparan sulfate may explain the PLE.152,153 Reported hepatic abnormalities range from mild degrees of steatosis to congenital hepatic fibrosis. Congenital hepatic fibrosis, without renal cysts, may be the only feature of MPI-CGD.148 Dilatation of the crypts of the large intestine has also been observed in some cases (Fig. 9.11).154 The diagnosis of CDG can be confirmed by isoelectric focusing of serum transferrin. MPI-CDG can be treated with mannose. Murch and colleagues described three infant boys with massive diarrhea associated with abnormal intestinal glycosaminoglycans and a complete absence of heparan sulfate.152
Almost half of all patients with glycogen storage disease type Ib, according to a report from a consortium of European centers, have protracted diarrhea and a clinical profile similar to that of patients with chronic inflammatory bowel disease. In fact, defects in neutrophil function may underlie the cause of enteritis in these patients.155 Several disorders resulting from deficient lysosomal degradation of glycoconjugates can manifest with severe diarrhea of infancy. Chronic diarrhea can be a feature of mucopolysaccharidoses (MPS), and it has been reported with MPS III (Sanfilippo syndrome) (Fig. 9.12).156 In these cases, nodular infiltration of the bowel wall may be seen endoscopically, although the diarrhea is most likely caused by impairment of the autonomic nervous system. Infiltration of the mucosa by foamy macrophages is characteristic of Wolman disease (Fig. 9.13). Shortening of the microvilli and impairment of disaccharidase activity are features observed in patients with diarrhea in Wolman disease.157 Tangier disease is a high-density lipoprotein (HDL) deficiency syndrome characterized by the accumulation of cholesterol in tissue macrophages throughout the body; it is prevalent in patients with atherosclerosis. It results from mutations in the gene coding for ATP-binding cassette transporter 1 (ABCA1). Endoscopic examination of patients with this disorder reveals characteristic orange-colored nodules in the gut mucosa, with accumulation of fat in macrophages in the lamina propria and smooth muscle cells.158
Glycolipid storage disorders are characterized, in the GI tract, by the presence of lipid deposits in Schwann cells, ganglia, muscle cells, and endothelial cells,159,160 usually without symptoms attributable to the deposits. However, more than half of patients with Fabry disease, an X-linked disorder that results in deficiency of α-galactosidase, experience GI symptoms mimicking irritable bowel syndrome. In this condition, there is accumulation of a lipid, globotriaosylceramide, in the enteric neurons that interferes with normal gut motility and results in delayed gastric emptying, bacterial overgrowth, and subsequent diarrhea and steatorrhea.161 Enzyme replacement therapy with agalsidase alfa has been shown to improve symptoms in these patients.162 Severe protracted diarrhea in association with low cholesterol levels in the first month of life is an important manifestation of some of the more severe forms of peroxisomal disorders, such as infantile Refsum disease.163 The clinical presentation of these disorders should be differentiated from that of lipid transport disorders such as abetalipoproteinemia. Cormier-Daire and colleagues reported on two infants with chronic diarrhea and villous atrophy caused by a respiratory chain complex III deficiency associated with mitochondrial DNA mutations.164 Severe diarrhea has been described as a presenting manifestation in other disorders associated with mitochondrial DNA rearrangements.165
Infantile systemic hyalinosis is a rare, apparently autosomal recessive, disorder characterized by painful joint contractures and skin nodules resulting from widespread deposits of a hyaline substance in the skin, skeletal muscle, GI tract, and endocrine organs. This condition was well documented by Landing and Nadorra,166 although the first description of the disease was probably by Nezelof and associates.167 PLE due to intestinal lymphangiectasia has been described in many of these patients.168,169 On electron microscopy, floccular amorphous material has been observed around blood vessels in the dermis, and this is postulated to interfere with collagen formation.170 The exact biochemical defect is unknown.
Neoplastic Disorders
Neoplasms rarely cause chronic diarrhea in children, especially infants and toddlers, although diarrhea may occasionally be a major presenting manifestation. Some neoplasms cause symptoms by direct infiltration of the bowel wall or by secretion of a hormone or substance that is active in the GI tract. Diffuse infiltration of the bowel wall resulting in severe diarrhea in neonates has been reported in patients with Langerhans cell histiocytosis171,172 and visceral myofibromatosis173 (Fig. 9.14). Neuroblastomas and ganglioneuroblastomas have been shown to cause watery diarrhea, hypokalemia, and achlorhydria in numerous reports.
[/level-membership-for-pathology-category][not-level-membership-for-pathology-category]
Enteropathies Associated with Chronic Diarrhea and Malabsorption in Childhood
Pierre Russo
Introduction
The goal of this chapter is to outline the pathologic features of the major intestinal disorders of infancy and early childhood, with an emphasis on congenital disorders that result in chronic diarrhea and malabsorption, and to illustrate their appearance in small intestinal biopsy and resection specimens (Box 9.1). Other major categories of disorders that cause chronic diarrhea, such as infections, immunodeficiencies (primary and secondary), gluten-sensitive enteropathy and other food allergies, and motility and pancreatic disorders are discussed in other chapters of this book.
Chronic diarrhea occurring in the neonatal period presents particularly difficult diagnostic and therapeutic challenges. Intractable diarrhea of infancy is a term initially coined by Avery and colleagues1 to refer to chronic diarrhea in neonates, most of which remained undiagnosed and was associated with a high mortality rate. Since these initial reports, more precise identification of disorders that cause intractable diarrhea of infancy has led to the use of prolonged parenteral nutrition, immunosuppression, and even bowel transplantation to try to improve survival, emphasizing the need for timely and accurate diagnosis. Investigation of some of these disorders has also led to significant advances in understanding of gastrointestinal (GI) and immunologic functions. For instance, investigation of microvillous inclusion disease has helped identify genes responsible for intracellular vesicular transport. The discovery that mutations in the gene that codes for FOXP3 causes the immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome, as well as its animal homolog in the Scurfy mouse, has led to recognition of the critical role played by that gene in the control of the human immune response, as well as in autoimmunity and immune tolerance.
Biopsy Sampling and Indications In Children
In most pediatric GI practices, intestinal biopsy specimens are most frequently obtained from the duodenum via forceps during endoscopic examination, during which time samples are also obtained from the esophagus and stomach (esophagogastroduodenoscopy, or EGD). Endoscopic forceps biopsies have largely replaced Crosby suction biopsies because of the greater ease of the procedure, greater patient comfort, avoidance of radiation exposure, direct visualization of the GI tract, and the ability to perform multiple biopsies at several sites. In addition to being submitted for routine histology, biopsy samples may be snap-frozen (for disaccharidase analysis) or submitted for electron microscopy (for confirmation of microvillous inclusion disease). Biopsies of both the proximal and distal duodenum are recommended, including endoscopically normal mucosa, because many disorders that affect the duodenum have a focal distribution. For example, pathologic lesions in children with gluten-sensitive enteropathy may be patchy, and villous atrophy may coexist with otherwise normal mucosa.2 Furthermore, villous atrophy may be limited to, or most severe in, the duodenal bulb at the time of clinical presentation.2,3
Since pediatric EGD evolved into a routine outpatient procedure, the indications for its use have changed. For instance, at The Children’s Hospital of Philadelphia, the “first-time” EGD rate increased 12-fold in the 20 years between 1985 and 2005, with isolated abdominal pain replacing GI bleeding as the most frequent indication.4 Much of this increase was driven by the dramatic increase in food allergy–related disorders, such as eosinophilic esophagitis; by the increased prevalence of celiac disease and its clinically atypical forms, for which intestinal biopsy is the gold standard for establishing a diagnosis; and by the routine use of EGD in addition to colonoscopy for evaluation of children with suspected inflammatory bowel disease. In a 2012 study, the most frequent indications for EGD and colonoscopies in children younger than 1 year of age were diarrhea, failure to thrive, reflux, and rectal bleeding.5 Histologic abnormalities were detected in two thirds of cases, and only 2% of the mucosal biopsies were considered insufficient. Sampling of endoscopically normal-appearing mucosa is recommended because it may help assess the “background” features of the mucosa and because histologic examination may reveal clinically relevant findings previously unsuspected by the endoscopist (e.g., granulomas).6,7 One pediatric study found that duodenal biopsy performed routinely for indications such as gastroesophageal reflux, vomiting, abdominal pain, anemia, and evaluation of Crohn’s disease yielded pathologic findings in approximately 17% of cases.7
Some entities, such as congenital transport disorders, are associated with normal intestinal biopsy findings, whereas others, such as autoimmune enteropathy (AIE) or celiac disease, exhibit various degrees of villous atrophy either with or without inflammation. In contrast, a few disorders, such as abetalipoproteinemia or microvillous inclusion disease, usually reveal characteristic findings in intestinal biopsies (Table 9.1).
Table 9.1
Diagnostic Findings on Small Intestinal Biopsies
| Intestinal Biopsy | Differential Diagnosis |
| Normal |
Intestinal Development in Children
Villi appear in the duodenum during the eighth week after fertilization, and crypts of Lieberkühn are seen during the ninth week. They spread caudally in the gut, affecting the distal ileum by the 14th week of gestation. The definitive histologic features of the duodenum are established by the 14th week after fertilization, and the histology closely resembles that of a newborn by 20 weeks’ gestation. The transition from pyloric to duodenal mucosa is gradual in some individuals. Duodenal-like villi may be found in the distal pylorus, and pyloric-like epithelium may be found in the duodenum. Enteroendocrine and goblet cells appear differentiate before 14 weeks’ gestation under the control of transcription factors such as Math1.8,9 The ratio of villus height to crypt depth in the duodenum of a newborn is similar to that in an adult. Newborns usually lack plasma cells in the first week of life and gradually acquire them during the first month. At that time, immunoglobulin M (IgM)-containing plasma cells predominate, but by 3 months of life, immunoglobulin A (IgA) plasma cells predominate. Normal numbers of plasma cells and normal ratios of IgA, IgM, and IgG antibodies are attained by the first year of life.10
Congenital Disorders of Intestinal Digestion, Absorption, and Transport
Specific gene defects associated with various disorders of substrate transport have been characterized (Table 9.2). Genetic studies are instrumental in providing a picture of enterocyte function at the molecular level. Except for disorders associated with fat processing, small intestinal biopsies in these cases are typically normal or only very slightly abnormal. Normal-appearing small bowel mucosa from a patient with prolonged diarrhea, especially a young infant, should alert the clinician to these entities. Only those disorders with significant histologic findings or with confounding clinical features are discussed here.
Table 9.2
Molecular Basis of Disorders of Digestion, Absorption, and Transport
| Disease | Gene | Location | Function |
| Disaccharidase Deficiency | |||
| Congenital lactase deficiency | LCT | 2q21 | Lactase-phlorizin hydrolase activity |
| Sucrase-isomaltase deficiency | SI (EC 3.2.1.48) | 3q25-q26 | Isomaltase-sucrase activity |
| Maltase-glucoamylase deficiency | MGAM | 7q34 | Maltase-glucoamylase activity |
| Ion and Nutrient Transport Defects | |||
| Glucose-galactose malabsorption | SLC5A1 (SGLT1) | 22q13.1 | Na+/glucose contransporter |
| Fructose malabsorption | SLC2A5 (GLUT5) | 1p36 | Fructose transporter |
| Fanconi-Bickel syndrome | SLC2A2 (GLUT2) | 3q26 | Basolateral glucose transporter |
| Cystic fibrosis | CFTR | 7q31.2 | cAMP-dependent Cl− channel |
| Acrodermatitis enteropathica | SLC39A4 (ZIP4) | 8q24.3 | Zn2+ transporter |
| Congenital chloride diarrhea | SLC26A3 (DRA) | 7q22-q31.1 | Cl−/base exchanger |
| Congenital sodium diarrhea | SPINT2 | 19q13.1 | Serine-protease inhibitor |
| Congenital bile acid diarrhea | SLC10A2 (ASBT) | 13q3 | Ileal Na+/bile salt cotransporter |
| Lysinuric protein intolerance | SLC7A7 | 14q11 | Hydrolyzes endo-/exopeptidases amino acid basolateral transport |
| Pancreatic Insufficiency | |||
| Enterokinase deficiency | TMPRSS15 (PRSS7) | 21q21 | Proenterokinase |
| Trypsinogen deficiency | PRSS1 | 7q35 | Trypsinogen synthesis |
| Pancreatic lipase deficiency | PNLIP | 10q26.1 | Hydrolyzes triglycerides to fatty acids |
| Lipid Trafficking | |||
| Abetalipoproteinemia | MTTP | 4q22 | Transfer lipids to apolipoprotein |
| Hypobetalipoproteinema | APOB | 2p24 | Apolipoprotein that forms chylomicrons |
| Chylomicron retention disease | SAR1B | 5q31.1 | Intracellular chylomicron trafficking |
cAMP, Cyclic adenosine monophosphate.
From Canani RB, Terrin G, Cardillo G, et al. Congenital diarrheal disorders: improved understanding of gene defects is leading to advances in intestinal physiology and clinical management. J Pediatr Gastroenterol Nutr. 2010;50:362.
Carbohydrate Malabsorption and Disaccharidase Deficiencies
Clinically, congenital disaccharidase deficiency and carbohydrate malabsorption is an osmotic type of diarrhea caused by unabsorbed solute in the ileum. The normal rapid transit of the GI tract in children results in a more severe form of diarrhea than in adults. Histologically, the appearance of the small intestine in these cases is usually unremarkable. Therefore, the diagnosis is usually confined by determination of disacchariadase activity in homogenates of small bowel biopsy specimens or by breath testing.
Congenital disaccharidase and transporter deficiencies are rare; these disorders are much more likely to be secondary, resulting from diffuse mucosal damage due to infectious gastroenteritis, gluten-sensitive enteropathy, or other food allergies. Accelerated crypt shedding, which occurs in mucosal injury, may outpace expression of brush border enzymes and transporter proteins, leading to worsening of the diarrhea and malabsorption. This may be one reason why tacrolimus, which reduces the rate of T cell–driven crypt cell proliferation and shedding, has been found to be effective in conditions such as “tufting” enteropathy and AIE.11,12
Pathogenesis
Carbohydrate absorption begins with the breakdown of complex carbohydrates by salivary and gastric enzymes into oligosaccharides, which are then hydrolyzed to monosaccharides by specific disaccharidases located at the enterocyte brush border. There are also facilitative hexose transporters located at the basolateral membrane. The latter have been cloned and functionally characterized in several different tissues: GLUT1, erythrocyte; GLUT2, hepatocyte; GLUT3, brain; GLUT4, muscle and fat; and GLUT5, small intestine.
There are four major disaccharidase enzymes: lactase-phlorizin hydrolase (or lactase-glycosyl ceramidase), sucrase-isomaltase, maltase-glucoamylase, and trehalase.11 Lactase-phlorizin hydrolase degrades lactose, the main carbohydrate in milk. Sucrase-isomaltase is responsible for the degradation of dietary sucrose. Maltase-glucoamylase hydrolyses starch into small polymers. Trehalose occurs mainly in mushrooms. Trehalase deficiency is extremely rare. It has been described only in older children and adults. The expression of these enzymes on the intestinal brush border appears to follow a time-dependent sequence. Lactase-phlorizin hydrolase, which is highly expressed at birth, typically declines in early life in most individuals (although it persists through adult life in whites). Sucrase-isomaltase, which is undetectable at birth, reaches adult levels within the first few months of life. These changes coincide with a switch from milk to a more solid diet.
Lactose malabsorption in children can be caused by primary, genetically determined deficiency of lactase-phlorizin hydrolase, which follows a variable clinical course, or by a secondary lactase deficiency resulting from mucosal injury, as in infectious diarrhea, celiac disease, or Crohn’s disease.12 Congenital sucrose-isomaltase deficiency typically becomes clinically apparent when the child begins to consume fruits and juices.13 Glucose-galactose malabsorption is a rare autosomal recessive disorder that is caused by a mutation in the gene on chromosome 22 that encodes the sodium/glucose cotransporter, solute carrier family 5, member 1 (SLC5A1, also known as SGLT1), which can result in severe chronic diarrhea.14 Congenital fructose malabsorption is caused by a mutation in the gene for the hexose transporter isoform, GLUT5, which is located on chromosome 1 and designated SLC2A5.15,16
Lipid Trafficking Disorders
Fat absorption by enterocytes begins with emulsification and solubilization of cholesterol in the intestinal lumen by biliary lipids and salts. Most clinical disorders of fat malabsorption result from severe liver disease, pancreatic disease (e.g., cystic fibrosis), or extensive ileal resection (as in Crohn’s disease), with loss of the enterohepatic circulation of bile acids. Intestinal biopsies play a limited role in the diagnosis of these disorders. However, primary disorders involving abnormalities of fat transport within enterocytes, although uncommon, may result in a characteristic vacuolization of enterocytes in intestinal biopsy specimens.
Abetalipoproteinemia, hypolipoproteinemia, and chylomicron retention disease (Anderson disease) share many characteristics, including fat malabsorption, low levels of serum lipids, failure to thrive in childhood, neurologic and visual problems resulting from malabsorption of fat-soluble vitamins, and accumulation of lipid droplets in the enterocytes. These conditions are associated with disorders of apolipoproteins, which reside on the surface of chylomicrons. Apoliproproteins native to the intestine are apolipoprotein A-I (apo A-I), apo A-IV, and apo B, which has two forms, apo B-100 and apo B-48, both encoded by the same gene located on chromosome 2.17 Localization of apolipoproteins in the Golgi apparatus and along the microvilli of enterocytes has been demonstrated by immuno-electron microscopy.18
Abetalipoproteinemia
Abetalipoproteinemia is an autosomal recessive disorder characterized by absence of apo B-containing lipoproteins. The molecular basis is a mutation of MTTP, the gene that codes for microsomal triglyceride transfer protein, which is located on chromosome 4q22 (see Table 9.2).19 MTTP is responsible for the assembly of lipoprotein particles and for the proper folding of apo B, which prevents its premature degradation.20 Because of the mutation, fatty acids within intestinal cells cannot be exported as chylomicrons. Patients develop diarrhea and fat malabsorption usually within the first few months of life, with acanthocytosis; deficiencies in fat-soluble vitamins then result in retinitis pigmentosa and neurologic symptoms. However, there is clinical heterogeneity because signs and symptoms may occasionally manifest in older individuals. Serum levels of cholesterol and triglycerides are typically low and, most importantly, do not rise after a fatty meal. Small bowel biopsies typically reveal preserved villi (Fig. 9.1, A), but characteristic multivacuolated, fat-filled enterocytes may be seen in specimens from fasting patients, which on electron microscopy are irregular in size and generally not membrane bound (see Fig. 9.1, B). In addition, lipid does not accumulate in the extracellular space. Hepatic biopsies typically reveal steatosis, with numerous non-membrane-bound lipid droplets within hepatocyte cytoplasm.21 Fibrosis evolving to cirrhosis has been reported in some patients.22
Hypobetalipoproteinemia
Hypobetalipoproteinemia is an autosomal dominant disorder caused by a mutation in the APOB gene located on chromosome 2, which leads to the development of a truncated apo B protein.23 Homozygous patients have a clinical and histologic phenotype that is essentially indistinguishable from abetalipoproteinemia, whereas heterozygous patients have only a mild disease.
Chylomicron Retention Disease
Anderson disease is similar to abetalipoproteinemia in its GI manifestations and impact on growth, although acanthocytosis is usually absent and neurologic and ocular abnormalities are much less severe. However, in contrast to abetalipoproteinemia, serum fasting triglyceride levels are typically normal, and hypocholesterolemia is less marked. The causative gene, SAR1B, codes for a guanosine triphosphatase (GTPase) that is associated with coat protein carriers involved in transport from the endoplasmic reticulum to the Golgi apparatus, particularly transport of chylomicrons and low-density lipoproteins.24 The pathologic features in small bowel biopsy specimens are essentially indistinguishable from those of abetalipoproteinemia (see Fig. 9.1, C).
Minor degrees of enterocyte vacuolization (e.g., resulting from a recent feed) are common in intestinal biopsy specimens in infants. In these cases, vacuolization is not as marked or as diffused as in abetalipoproteinemia or chylomicron storage disease. In addition, lipid droplets are present in the intercellular spaces and lacteals after feeding but are absent from these spaces in disorders that cause impaired lipid transport. By contrast, lipid-containing macrophages are present in the lamina propria in several storage disorders in which digestive symptoms can occasionally be significant and in which an intestinal biopsy is obtained in the course of a workup for failure to thrive (see Metabolic Diseases).
Amino Acid Transport Disorders
Disorders of amino acid transport rarely cause prominent GI manifestations, except for lysinuric protein intolerance (LPI), which results from mutations in the SLCA7 gene. This gene codes for the light chain subunit of the heterodimer amino acid transporter at the basolateral membrane of intestinal and renal epithelial cells.25 Clinically, LPI manifests with failure to thrive, vomiting, and diarrhea. Hepatosplenomegaly, hematologic anomalies, and neurologic involvement including hyperammonemic coma are recurrent clinical features. The urea cycle intermediates ornithine and arginine fail to exit from intestinal epithelium in LPI, resulting in a urea cycle dysfunction with hyperammonemia and alterations of mental status. Two major complications, pulmonary alveolar proteinosis and renal disease, are increasingly observed in patients with LPI.26,27 The condition is recognized by the presence of markedly elevated urinary levels of lysine and other dibasic amino acids; DNA testing may be required for confirmation of the diagnosis. One 5-year-old boy who was reported to have chronic diarrhea and a “flat gut” on small intestinal biopsy was unsuccessfully treated with a gluten-free diet.28 Other complications associated with this disorder include systemic lupus erythematosus, hemophagocytic lymphohistiocytosis,29 and sudden infant death.30
Electrolytes and Trace Elements
The biology of intestinal ion transport is a complex field. Disorders range from rare selective deficiencies to multisystem diseases such as cystic fibrosis. Defects that manifest primarily with severe diarrhea in infancy include congenital chloride diarrhea and congenital sodium diarrhea. Intestinal biopsy findings in these cases have been reported to be normal or to show only mild partial villus atrophy.31 Congenital chloride diarrhea is caused by mutations in the gene for solute-linked carrier family 26, member 3 (SLC26A3). It manifests as a life-threatening congenital diarrhea. Long-term manifestations include renal disease, spermatoceles, and male subfertility.32 Congenital sodium diarrhea results in hyponatremia and metabolic acidosis. The syndromic form of this disorder has been linked to mutations in SPINT2 and is also characterized by cloanal and anal atresia, hypertelorism, double kidney, cleft palate, and digital anomalies.13
Acrodermatitis enteropathica is an autosomal recessive disorder that is caused by a defect in intestinal absorption of zinc. Mutations in a candidate gene, ZIP4 (SLC39A4), result in defective epithelial transport in the intestine and kidney.13 There is a characteristic clinical syndrome of diarrhea combined with acral and orificial skin lesions. Rodin and Goldman found pancreatic islet hyperplasia, absence of the thymus and germinal centers, and plasmacytosis of the spleen and lymph nodes in autopsy studies.33 Inclusion bodies in Paneth cells have been reported in patients with this disorder.34,35 A decrease in villus height is observed in animal models of zinc deficiency and is corrected by zinc supplementation.36 Interference with chylomicron development and function has been noted.37 Severe zinc deficiency is also seen in other disorders, such as the Cronkhite-Canada syndrome.38
Menkes disease is an X-linked disorder that results from a defect in intestinal absorption of copper. Affected patients present with characteristic hair shaft anomalies (pili torti), cerebral degeneration, hypopigmentation, abnormal bones, vomiting and diarrhea, and, occasionally, protein-losing enteropathy (PLE). The disorder maps to Xq12-q13 and is caused by mutations in the ATP7A gene, which encodes a copper-transporting P1B-type adenosine triphosphatase that is part of a family of membrane proteins responsible for cation transport across membranes.39,40
Vitamins
Congenital vitamin B12 transport disorders include congenital intrinsic factor deficiency, selective absence of intrinsic factor without any gastric anomaly, congenital pancreatic insufficiency of various causes, and congenital selective vitamin B12 malabsorption—the latter an autosomal recessive disorder characterized by megaloblastic anemia, hepatosplenomegaly, vomiting, diarrhea and proteinuria. These disorders are caused by mutations in the gene CUBN, which encodes an ileal cell surface receptor protein known as cubilin.41 None of these disorders results in any characteristic histologic changes on intestinal biopsies.
A rare congenital disorder of intracellular vitamin B12 metabolism belonging to the cobalamin C (CblC) family has also been described. Patients present with diarrhea, renal failure, and systemic thromboemboli in association with a striking gastric atrophy.42 PLE responsive to hydroxycobalamin treatment has been reported in one patient with this disorder,43 presumably on the basis of the gastric anomaly.
Bile Acids
Heubi and colleagues44 described a severe type of refractory diarrhea caused by a primary disorder of bile acid absorption; intestinal biopsy findings were normal. A defect in the gene coding for an ileal Na+-dependent bile acid transporter, SLC10A2, has been found in this disorder.45 Bile acid–related diarrheal illnesses are much more commonly secondary to chronic pancreatic insufficiency46 or to loss of ileal surface, as occurs with short gut syndrome resulting from necrotizing enterocolitis47 or with extensive ileal resection in Crohn’s disease.48,49
Congenital Defects of Intestinal Epithelial Differentiation
Microvillus Inclusion Disease
Clinical Features
Initially described by Davidson et al in 197850 and subsequently recognized worldwide, microvillus inclusion disease (MVID) is an autosomal recessive disease characterized by refractory secretory diarrhea usually within the first week of life, although late-onset symptoms may manifest in the first few months of life.51 Investigation of clusters of cases within the Navajo population identified mutations in the gene MYO5B.52 Homozygosity mapping of an extended Turkish kindred allowed Muller and colleagues to locate the gene locus to chromosome 18q21.53 MYO5B codes for myosin Vb, part of a family of proteins responsible for actin-dependent organelle transport and regulation of endosome recycling. Mutations in the MYO5A gene cause Griscelli syndrome, an immunodeficiency disorder also characterized by abnormal transfer of melanin granules to keratinocytes.53
Pathology
Small bowel biopsy specimens in MVID are usually characterized by severe villus atrophy, mild or moderate crypt hyperplasia, and a variable degree of inflammation in the lamina propria, but without crypt destruction (Fig. 9.2, A). The diagnosis may be strongly suspected on paraffin-embedded sections by the absence of a distinct brush border using the periodic acid–Schiff (PAS) stain, and by the presence of PAS-positive diastase-resistant densities in the apex of enterocytes (see Fig. 9.2, B and C). Similar observations are seen on immunohistochemical staining for alkaline phosphatase and anti-CD10. More recently, similar results using antibodies directed against anti-Rab11a, a small GTPase protein on the surface of recycling endosomes, has provided further evidence that MVID is a disorder of apical plasma membrane recycling.54 The pathognomonic ultrastructural features include absent or small stubby microvilli, vesicular structures located toward the apex of enterocytes containing microvilli, and granules containing dense amorphous material (Fig. 9.3). Microvillus inclusions have also been reported in the colon, gallbladder, and renal tubular epithelium in these patients.55 A variety of ultrastructural features have also been noted in patients with this disorder, and finding the “typical” inclusions may require a prolonged search.56
Atypical inclusions include the presence of numerous small electron-lucent vesicles and accumulation of osmiophilic vermiform structures.57 Cases with these features have been previously reported as “MVID with atypical features” or as “microvillous dystrophy.”58,59 Genetic analysis helps in defining whether the ultrastructural (and sometimes clinical) variability represent different entities or variations in the spectrum of manifestations of a single disorder, especially given the enormous prognostic and therapeutic implications.
Treatment
Patients with MVID are dependent on total parenteral nutrition (TPN). Medical therapy in these patients is generally ineffective, and improved survival may require small bowel transplantation.60 An intriguing instance of apparent resolution of the disease with improvement of the mucosal features in a patient on long-term TPN has been reported.61
Congenital Tufting Enteropathy (Epithelial Dysplasia)
Clinical Features
Congenital tufting enteropathy (CTE) was first described by Reifen and colleagues62 in patients presenting in the neonatal period with watery diarrhea. The prenatal history is usually uneventful. The disease appears to be inherited in an autosomal recessive fashion, as suggested by the finding of other affected siblings and frequent parental consanguinity. The incidence is estimated at 1 : 50,000 to 1 : 100,000 live births in Europe, and it is more frequent in patients of Arabic origin.63
[/not-level-membership-for-pathology-category]
