CHAPTER 8 Endocrinologic disorders
Endocrine assessment
1. Critical illness initiates the stress response.
2. The stress response increases the metabolic rate.
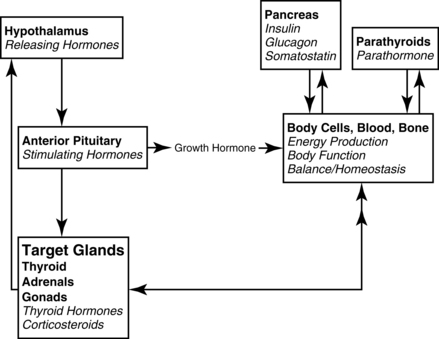
3. The hypothalamic-pituitary axis (Figure 8-1) regulates the metabolic rate. The thyroid and adrenal glands are extremely stressed by the stimulus of critical illness to maintain the increased metabolic rate, along with meeting the energy demands at the cellular level. Hypofunction of the thyroid and adrenals requires assessment of not only the primary glands, but also the hypothalamus (produces releasing factors) and anterior pituitary (produces stimulating hormones.) The hypothalamic-pituitary-target organ feedback loop must be fully intact to maintain normal metabolism.
4. The stress response markedly increases endogenous glucocorticoids, which increase blood glucose. The pancreas may not be able to produce sufficient insulin to manage the glucose level, resulting in hyperglycemia. Insulin may be required to manage hyperglycemia until the stress of illness resolves. People with diabetes mellitus are always challenged with hyperglycemia, and with additional illness, can experience the crisis states of diabetic ketoacidosis (DKA) and hyperosmolar hyperglycemic syndrome (HHS).
5. Under prolonged extreme stress, both the adrenal glands and thyroid may be unable to sustain hormone production to support the stress level. Supplemental glucocorticoids (corticosteroids) and thyroid hormones may be needed.
6. The assessment of the critically ill patient should focus on the signs of failure of the endocrine system to support the stress response. Signs of hypofunction are explained the sections on hyperglycemia, adrenal crisis, and myxedema coma. Patients with adequate function of the pancreas, thyroid, and adrenal glands under normal conditions may not be able to maintain balance when exposed to the stress of critical illness. Those with underlying hypofunction are more likely to experience crises.
7. Hyperthermia and cardiac symptoms can make diagnosis of thyroid storm difficult, as the crisis mimics other cardiac crises and infection. Thyroid storm results from underlying Graves’ disease/hyperthyroidism; not a complication of critical illness.
8. ![]() Assessment of the causes of unusual fluid and electrolyte imbalances should include evaluation of posterior pituitary function, in addition to screening for renal dysfunction. Posterior pituitary dysfunction may result in abnormal levels of antidiuretic hormone (ADH) and may be a complication of critical illness, or result from hypothalamic or pituitary disease. Diabetes insipidus (DI ) produces dehydration and Syndrome of Inappropriate ADH (SIADH) produces hyponatremia, which can reach critical states if not properly addressed. Nephrogenic DI results from failure of the kidneys to respond to ADH. The elderly are at higher risk of complications with DI as a result of age-related changes to the thirst mechanism and renal function.
Assessment of the causes of unusual fluid and electrolyte imbalances should include evaluation of posterior pituitary function, in addition to screening for renal dysfunction. Posterior pituitary dysfunction may result in abnormal levels of antidiuretic hormone (ADH) and may be a complication of critical illness, or result from hypothalamic or pituitary disease. Diabetes insipidus (DI ) produces dehydration and Syndrome of Inappropriate ADH (SIADH) produces hyponatremia, which can reach critical states if not properly addressed. Nephrogenic DI results from failure of the kidneys to respond to ADH. The elderly are at higher risk of complications with DI as a result of age-related changes to the thirst mechanism and renal function.
9.  Medication noncompliance for management of existing endocrine disease must be assessed. People with lower income are at higher risk of crisis due to inability to purchase medications and teenagers may not practice meticulous management; particularly those with diabetes mellitus. Elders may not take medications appropriately due to lack of understanding or mis-dosing due to deteriorating short-term memory.
Medication noncompliance for management of existing endocrine disease must be assessed. People with lower income are at higher risk of crisis due to inability to purchase medications and teenagers may not practice meticulous management; particularly those with diabetes mellitus. Elders may not take medications appropriately due to lack of understanding or mis-dosing due to deteriorating short-term memory.
Acute adrenal insufficiency (adrenal crisis)
Pathophysiology
1. Stimulate the pituitary to secrete stimulating hormones (adrenocorticotropic hormone [corticotropin] [ACTH]), which in turn
2. Stimulates the adrenal glands to secrete glucocorticoids (cortisol) and mineralocorticoids (aldosterone), which in turn
3. Facilitates cellular functions throughout the body (metabolism), which in turn
4. Creates signals back to the hypothalamus from the cells to increase or decrease releasing hormones (see Figure 8-1).
If primary adrenal insufficiency is the cause of the crisis, the adrenal glands are the root cause of the problem. Addison disease manifests when the entire adrenal cortex is destroyed, which stops the production of glucocorticoids (cortisol) and mineralocorticoids (aldosterone). Glucocorticoids are essential hormones produced by the adrenal cortex that help maintain vascular tone and cardiac contractility, facilitate wound healing, and support immunity. Cortisol deficiency intensifies the clinical effects of hypovolemia by promoting a decrease in vascular tone, which is partially related to unopposed endothelial production of nitric oxide, and a decreased vascular response to the catecholamine hormones epinephrine and norepinephrine. Relative hypoglycemia may be present, as the breakdown of stored glycogen is not possible without cortisol. Mineralocorticoid hormones are primary regulators of fluid and electrolyte balance, and when unavailable, patients experience hyponatremia, hypovolemia, hyperkalemia, and metabolic acidosis. Large amounts of sodium and water are excreted in the urine. Severe hypotension, shock, and eventually death may occur without intravenous adrenocortical hormone and fluid replacement. In patients with chronic primary adrenocortical insufficiency or Addison disease, acute crises may be prevented by tripling hormone replacement doses during periods of stress.
8-1 RESEARCH BRIEF
The results were dismal in terms of mortality reduction because there was little difference from placebo. Of note, however, was that the group receiving hydrocortisone spent less time in shock by the prior mentioned definition.
From Sprung CL, Annane D, Keh D, et al: Hydrocortisone therapy for patients in septic shock. N Engl J Med 358(2):111–124, 2008.
Endocrine assessment adrenals
Goal of system assessment:
To evaluate for severe hypotension, refractory to volume and vasopressor administration
History and risk factors
• Extreme emotional or physiologic stress, which increases the need for adrenocortical “stress” hormones to mediate the stress response
• Patients previously receiving glucocorticoids (steroids) who may be abruptly withdrawn from steroids or are not given sufficient steroids to manage additional stress
• Adrenalectomy, hypophysectomy, sepsis, human immunodeficiency (HIV) disease
• Other medications: beta adrenergic blockers, diuretics, angiotensin-converting enzyme (ACE) inhibitors, angiotensin release blockers (ARBs), nitrates, aspirin, and other platelet inhibitors
Observation and vital signs: primary (first-degree) and secondary (second-degree) insufficiency
• Refractory, severe hypotension resulting from vascular collapse
• Acute abdomen assessment findings: abdominal pain, distention
• Hyperpyrexia, with temperatures often reaching in excess of 105°F
• Cyanosis, confusion; may be comatose
• Relative hypoglycemia: tremors, diaphoresis, tachycardia, tachypnea
Observation and vital signs: primary (first-degree) insufficiency only
• Prominent nausea and vomiting
• Signs of dehydration: poor skin turgor, sunken, soft eyeballs, weight loss
• Bronze hue to the skin secondary to excess production of ACTH
• Hyperkalemia, which may be associated with metabolic acidosis: peaked T waves, widening QRS complex, prolonged PR interval, flattened-to-absent P wave
Screening labwork
For suspected acute adrenal crisis
• Random plasma cortisol level: Drawn prior to initiating hydrocortisone replacement but must be interpreted with caution in critically ill patients, because greater than 90% of circulating cortisol is protein bound. When patients are hypoproteinemic, with serum albumin less than 2.5 g/dl, low values may be gleaned from all cortisol testing in patients who have normal adrenal function. A random plasma cortisol level of greater than 25 mcg/dl excludes both primary and secondary adrenal insufficiency.
• Free cortisol level: Done in the setting of hypoproteinemia to better determine the cortisol level. An abnormal test may require a complete endocrinology assessment when the patient stabilizes.
For noncritical adrenocortical insufficiency
• Corticotropin (ACTH) stimulation test: The goal is to differentiate primary from secondary adrenocortical insufficiency or to assess if the adrenal cortex is capable of producing cortisol. Testing of the hypothalamic-pituitary-adrenal axis using this test can differentiate primary from secondary insufficiency. Baseline plasma cortisol level is drawn immediately prior to ACTH administration.
• Adrenal insufficiency is diagnosed when:
| Test | Purpose | Abnormal Findings |
|---|---|---|
| Noninvasive | ||
| Chest radiograph | Assess for heart size and presence of opportunistic infections (primary) | The chest radiogram may be normal but often reveals a small heart. Stigmata of earlier infection or current evidence of tuberculosis (TB) or fungal infection may be present when this is the cause of Addison disease. |
| Computed tomography (CT) scan of abdomen | Assess abdominal organs, size, presence of blood (primary) | Abdominal CT scan may be normal but may show bilateral enlargement of the adrenal glands in patients with Addison disease because of TB, fungal infections, adrenal hemorrhage, or infiltrating diseases involving the adrenal glands. In Addison disease due to TB or histoplasmosis, evidence of calcification involving both adrenal glands may be present. In idiopathic autoimmune Addison disease, the adrenal glands usually are atrophic. |
| Blood Studies | ||
| Complete blood count (CBC) Hemoglobin (Hgb) Hematocrit (Hct) RBC count (RBCs) WBC count (WBCs) |
Assess for anemia, inflammation and infection (primary and secondary) | CBC count may reveal a normocytic normochromic anemia, which, upon initial presentation, may be masked by dehydration and hemoconcentration. Relative lymphocytosis and eosinophilia may be present. |
| Electrolytes Potassium (K+) Sodium (Na+) |
Assess for abnormalities of aldosterone (primary) | Elevation in K+ may cause dysrhythmias; decrease of Na+ may indicate fluid retention and/or concomitant heart failure. |
| Serum glucose | Assess for relative hypoglycemia (primary and secondary) | Hypoglycemia may be present in fasted patients, or it may occur spontaneously. It is caused by the increased peripheral utilization of glucose and increased insulin sensitivity. It is more prominent in children and in patients with secondary adrenocortical insufficiency. |
| Thyroid-stimulating hormone | Assess for thyroid dysfunction (primary and secondary) | Increased thyroid-stimulating hormone, with or without low thyroxine, with or without associated thyroid autoantibodies, and with or without symptoms of hypothyroidism, may occur in patients with Addison disease and in patients with secondary adrenocortical insufficiency due to isolated ACTH deficiency. These findings may be reversible with cortisol replacement. |
Collaborative management
DIAGNOSIS AND MANAGEMENT OF CORTICOSTEROID INSUFFICIENCY IN CRITICALLY ILL PATIENTS
| Recommendations for the diagnosis and management of corticosteroid insufficiency in critically ill adult patients: consensus statements from an international task force by the American College of Critical Care Medicine. | |
| In 2008, an interdisciplinary, multispecialty task force of experts in critical care medicine was convened from the membership of the Society of Critical Care Medicine and the European Society of Intensive Care Medicine. In addition, international experts in endocrinology were invited to participate. The goal was to develop a strategic tool for defining and treating critical illness acute adrenal insufficiency. | |
| Treatment | Rationale |
| Moderate dose of hydrocortisone (200–300 mg/day) for critically ill patients with septic shock. | Six randomized control trials demonstrate significant and greater shock reversal in patients who received hydrocortisone although no difference in mortality. |
| Moderate dose of hydrocortisone in the management of severe early ARDS (PF ratio <200) instituted before day 14. | Five randomized studies evaluated moderate hydrocortisone administration in ARDS from various origins. Consistent improvement was reported in the PF ratio, inflammatory markers were reduced, and both ventilator days and ICU length of stay were reduced. |
| In patients with septic shock, intravenous hydrocortisone should be given in a dose of 200 mg/day in four divided doses or as a bolus of 100 mg followed by a continuous infusion at 10 mg/hr (240 mg/day).The optimal initial dosing regimen in patients with early severe ARDS is 1 mg/kg/day methylprednisolone as a continuous infusion. | Multiple clinical trials, both randomized and not, as well as prospective and retrospective of patients in severe sepsis and ARDS. |
| Glucocorticoid (GC) treatment should be tapered slowly and not stopped abruptly. | Abruptly stopping hydrocortisone will likely result in a rebound of proinflammatory mediators, with recurrence of the features of shock (and tissue injury). |
| Treatment with dexamethasone has previously been suggested in patients with septic shock until an ACTH stimulation test is performed, this approach can no longer be endorsed | Physiologic and pathologic understanding that dexamethasone leads to immediate and prolonged suppression of ACTH. |
From Marik PE, Pastores SM, Annane D, et al: Crit Care Med 36(6):1937–1949, 2008.
Care priorities
Use vasopressors if intravascular volume replacement fails to effectively increase BP (see Appendix 6). Response to catecholamine infusions (epinephrine, norepinephrine, dopamine) is reduced in adrenal insufficiency; higher-than-normal doses may be needed to manage refractory hypotension.
• Administer 100 mg of hydrocortisone in 100 ml of normal saline solution by continuous IV infusion at a rate of 12 ml/hr. Infusion may be initiated with 100 mg of hydrocortisone as an IV bolus.
• A continuous infusion method maintains plasma cortisol levels effectively if the stress level is steady or constant, particularly in patients who rapidly metabolize the drug. Rapid metabolizers have a greater likelihood of having low plasma cortisol levels between IV boluses.
• An alternative method of hydrocortisone administration is 50-75 mg IV every 4-6 hours for 5 days.
• Improvement in BP and other vital signs should be evident within 4 to 6 hours of hydrocortisone infusion. If not, the diagnosis of adrenal insufficiency is questionable.
• After 2 to 3 days, the stress hydrocortisone dose should be reduced to 100 to 150 mg, infused over a 24-hour period regardless of the patient’s status. In addition to helping with adrenal recovery, lower doses may help abate gastrointestinal (GI) bleeding.
• As the patient improves and the clinical situation allows, the hydrocortisone infusion can be gradually tapered over the following 4 to 5 days to daily replacement doses of approximately 3 mg/hr (72 to 75 mg over 24 hours) and eventually to daily oral replacement doses when oral intake is possible.
• If the patient receives at least 100 mg of hydrocortisone in 24 hours, no mineralocorticoid replacement is necessary, because the mineralocorticoid activity of hydrocortisone in this dosage is sufficient.
• As the hydrocortisone dose continues to be weaned, mineralocorticoid replacement should begin in doses equivalent to the daily adrenal gland aldosterone output of 0.05 to 0.1 mg daily or every other day.
4. Maintain normal blood glucose level:
If patient is initially hypoglycemic, 50% dextrose may be needed to correct hypoglycemia. When hydrocortisone or other cortisol replacement is initiated, hyperglycemia may result. An insulin infusion may be needed to control the blood glucose (see Hyperglycemia,p 711).
CARE PLANS FOR ADRENAL INSUFFICIENCY
![]()
Fluid and electrolyte management
1. Monitor vital signs and hemodynamic measurements every 15 minutes until stabilized for 1 hour. Consult physician or midlevel practitioner promptly for deterioration in vital signs or hemodynamics.
2. Administer IV fluids to replace fluid volume. Initially, rapid fluid replacement is essential.
3. Maintain accurate input and output (I&O) record. Weigh patient daily.
4. Monitor for electrolyte imbalance. Imbalances associated with adrenal insufficiency include the following:
5. Monitor ECG continuously; observe for potassium-related changes. Increased ventricular irritability may signal hypokalemia. (See Fluid and Electrolyte Disturbances, Hypokalemia, p 52.)
6. Monitor laboratory results. With appropriate treatment, serum sodium levels should rise to normal and serum potassium levels should fall to normal. Prevent rapid correction or overcorrection of hyponatremia. Serum sodium levels should not be allowed to increase greater than 12 mEq/L during the first 24 hours of treatment because of the risk of neurologic damage. (See Fluid and Electrolyte Disturbances, Hyponatremia, p 46, or Syndrome of Inappropriate ADH, p 734.)
7. Assess mental and respiratory status at frequent intervals. Institute safety measures as indicated. Reorient and reassure patient as needed.
8. Encourage oral fluid intake as patient’s condition stabilizes. Add sodium-rich foods (see Box 8-1) as tolerated. Begin oral glucocorticoid replacement therapy as prescribed.
9. Consult physician or midlevel practitioner if signs and symptoms of fluid and/or electrolyte imbalance persist or worsen.
Box 8-1 PATIENT AND FAMILY EDUCATION CONCERNING GLUCOCORTICOID AND MINERALOCORTICOID REPLACEMENT
Glucocorticoids (e.g., cortisone, acetate, prednisone)
• Take medication in a diurnal pattern to mimic normal secretion (i.e., two thirds of dose in the morning and one third of dose in the afternoon).
• Take steroids with food to decrease gastric irritation.
• Weigh self regularly, and report to physician gains of greater than 2 lb/wk.
• Avoid exposure to infection, and be alert to indicators of infection (e.g., fever, nausea, diarrhea, malaise).
• Contact physician promptly during periods of physical or emotional stress; dosages will require adjustment at these times.
• Indicators of overreplacement: weight gain (moon face, truncal obesity); edema, thin, fragile skin (striae, easy bruising); slow wound healing; chronic fatigue; emotional lability
• Indicators of underreplacement: weight loss, hyperpigmentation, skin creases, anorexia, nausea, abdominal discomfort, chronic fatigue, depression, irritability
Mineralocorticoids (e.g., fludrocortisone, desoxycorticosterone acetate)
• As prescribed, modify diet with liberal amounts of sodium (see Box 1-4), protein, and carbohydrates.
• Weigh self regularly, and report to physician sudden gains or losses greater than 2 lb/wk.
• Contact physician promptly during periods of physical or emotional stress; dosages will require adjustment at these times.
• Indicators of overreplacement: edema, muscle weakness, hypertension
• Indicators of underreplacement: excessive urination, weight loss, decreased skin turgor
 related to potential for acute regulatory dysfunction (cortisol and aldosterone deficiency) secondary to increased psychological, emotional, or physical stressors with increased hormonal demand and inadequate adrenal reserves
related to potential for acute regulatory dysfunction (cortisol and aldosterone deficiency) secondary to increased psychological, emotional, or physical stressors with increased hormonal demand and inadequate adrenal reserves![]()
Immune Status; Infection Status; Energy Conservation
1. Monitor and report signs of increasing crisis: urinary output increased from usual amount, changes in LOC, orthostatic hypotension, nausea, vomiting, and tachycardia.
2. Provide a quiet, nonstressful environment. Adjust lighting to meet needs of individual activities, avoiding direct light in the eyes. Control noise when possible. Prevent unnecessary interruptions, and allow for rest periods. Limit the number of visitors and the length of time they spend with patient. Speak softly and reassuringly to patient.
3. Monitor for and manage hyperthermia using tepid baths, antipyretics, and cooling blankets.
4. Maintain a cool environmental temperature. Maintain strict environmental asepsis, and monitor patient carefully for signs of infection. Avoid exposing patient to staff members or visitors who have colds or infections.
![]() Fluid Monitoring; Environmental Management; Infection Protection
Fluid Monitoring; Environmental Management; Infection Protection
Deficient knowledge: illness care
 related to prevention of adrenal crisis in patients with chronic adrenal insufficiency or those undergoing steroid therapy
related to prevention of adrenal crisis in patients with chronic adrenal insufficiency or those undergoing steroid therapy![]()
Knowledge: Disease Process; Knowledge: Energy Conservation; Knowledge: Medication
1. Teach patient about prescribed medications, including purpose, dosage, route of administration, and potential side effects (Box 8-1). Medication administration should mimic normal diurnal pattern of plasma cortisol levels (e.g., two thirds in the morning and one third in late afternoon).
2. Provide dietary instruction: dietary sodium and potassium may need to be adjusted on the basis of the patient’s clinical condition and drug therapy (see discussions of sodium and potassium in Fluid and Electrolyte Disturbances, p. 37).
3. Explain the importance of controlling stress, both emotional and physiologic, which increases adrenal demand. Teach patient to seek medical intervention during times of increased stress (e.g., fever, infection), inasmuch as medication dosages may need to be increased.
4. Teach indicators of overreplacement and underreplacement of steroids, which require prompt medical attention (see Box 8-1).
5. Stress the importance of never abruptly discontinuing use of any steroid preparation. Use must be tapered to avoid precipitation of crisis.
6. Remind patient of the importance of continued medical follow-up.
7. Explain the procedure for obtaining a medical-alert bracelet or card.
Diabetes insipidus
Pathophysiology
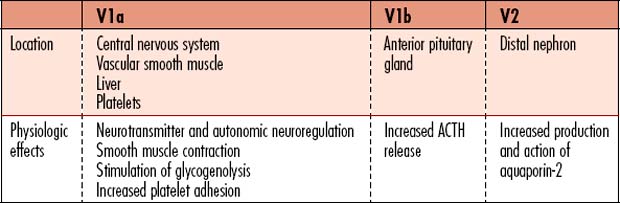
Diabetes insipidus (DI) is a metabolic disorder that affects total body free water regulation, resulting in an abnormally high output of extremely dilute urine, increased fluid intake, and constant thirst. The volume of hypotonic urine excreted is 3-20L/day. Vasopressin (antidiuretic hormone [ADH]) is a key component in the regulation of fluid and electrolyte balance, through direct effects on renal water regulation. Vasopressin is produced in the hypothalamus, is stored in the posterior pituitary gland, and exerts action in the kidneys for water regulation. Three subtypes of receptors respond to the effects of vasopressin (Table 8-1).
• Rapid changes of plasma osmolality: Rapid increases in plasma osmolality result in an abnormal increase in ADH/vasopressin release.
• Drinking fluids: Oral fluid consumption rapidly suppresses the release of ADH, through afferent pathways originating in the oropharynx.
• Pregnancy: The osmotic threshold for ADH release is lowered in pregnancy.
• Aging:  Plasma vasopressin concentrations increase with age, together with enhanced ADH responses to osmotic stimulation. Age-related changes in ADH production can result in blunting of the thirst response, decreased fluid intake, impaired ability to excrete a free water load, and reduced ability of the kidneys to concentrate urine. These changes predispose the elderly to both hypernatremia and hyponatremia.
Plasma vasopressin concentrations increase with age, together with enhanced ADH responses to osmotic stimulation. Age-related changes in ADH production can result in blunting of the thirst response, decreased fluid intake, impaired ability to excrete a free water load, and reduced ability of the kidneys to concentrate urine. These changes predispose the elderly to both hypernatremia and hyponatremia.
• Central, hypothalamic or pituitary DI (neurogenic DI) is the most common type and is caused by lack of vasopressin (ADH) production by a diseased or destroyed posterior pituitary gland. Lack of ADH results in massive diuresis, because ADH normally prompts the kidney to concentrate the urine. Approximately 50% of central DI is idiopathic, as diagnostic testing does not reveal a cause. Central DI is usually permanent, but the signs and symptoms (i.e., thirst, drinking fluids, and urination) are controlled by daily use of synthetic vasopressin.
• Nephrogenic DI (NDI) is caused by inability of the kidneys to respond to normal amounts of ADH, resulting from a variety of drugs or kidney diseases including genetic predisposition. The collecting tubules have decreased permeability to water caused by decreased response to vasopressin by the nephrons. NDI does not improve with synthetic vasopressin and may not improve when probable causes are managed. Familial NDI requires lifelong management. Treatments partially relieve the signs and symptoms. Medications, including lithium, amphotericin B, and demeclocycline can induce NDI. Hypercalcemia can sometimes prompt NDI.
• Gestational or gestogenic DI results from a lack of vasopressin that develops during the third trimester of pregnancy if the pregnant woman’s thirst center is abnormal, causing a blunted thirst response, and/or the placenta destroys vasopressin too rapidly. The placenta may increase the action of vasopressinase, the enzyme that breaks down vasopressin. The condition is controlled using synthetic vasopressin until the DI resolves. Vasopressin can generally be discontinued 4 to 6 weeks after delivery. Signs and symptoms of DI will recur with subsequent pregnancies.
• Dipsogenic DI or primary polydipsia results from vasopressin suppression caused by excessive fluid intake. Primary polydipsia is most often caused by an abnormality in the thirst center of the brain. Unquenchable thirst results in water intoxication. Dipsogenic DI is differentiated from central (pituitary) DI using the water deprivation test. There is no cure for dipsogenic DI at present, but symptoms can be safely relieved. Psychogenic polydipsia is another subtype due to psychosomatic causes that has no treatment that is recognized as consistently effective.
In normal individuals, a more concentrated circulating volume stimulates ADH release through activation of osmoreceptors that monitor serum osmolality. ADH is also released as part of the renin-angiotensin-aldosterone mechanism as a result of hypotension sensed by the juxtomedullary apparatus located outside the glomerulus of the kidney. A 5% to 10% decrease in arterial BP is necessary to increase circulating vasopressin concentrations. Progressive hypotension in healthy individuals results in an exponential increase in plasma ADH via baroreceptor stimulation, while osmoregulated ADH release in response to dehydration is more linear. If the hypothalamus is damaged, production of ADH may not be possible and both the ability to regulate circulating volume and vascular tone may be affected.
Endocrine assessment: diabetes insipidus
Goal of assessment
The clinical presentation of DI is dependent on the overall health of the patient and the primary cause. Evaluate for degree of dehydration and its effects on overall hemodynamics, heart rate/rhythm, and mental status. Generally, DI is recognized and managed prior to resulting in serious complications. Practitioners working with brain dead organ donors must be particularly alert to the occurrence of DI. Severe, unmanaged or under managed dehydration in cases of DI may cause hemoconcentration, which may predispose the patient to thrombosis. Dehydration and electrolyte imbalance must be prevented or managed immediately to avoid possible organ damage.
Observation
• Polyuria with dilute urine; 3-20 L/day of urine may be excreted, with specific gravity of 1.000 to 1.005.
• History may include nocturia (getting up frequently at night to urinate) and/or enuresis (bed wetting).
• Dehydration: Poor skin turgor, dry mucous membranes, sunken eyes, slow capillary refill
• Altered mental status: Serum hyperosmolality and hypernatremia affect consciousness and behavior. Changes may also be related to the underlying disease.
Screening labwork
| Test | Purpose | Abnormal Findings |
|---|---|---|
| Urine osmolality | Assesses for decreased concentration or dilute urine | Decreased to <200 mOsm/kg; may be higher if volume depletion is present |
| Urine specific gravity | Assesses for dilute urine | Specific gravity: <1.005 Normal is 1.010–1.025 |
| Serum osmolality | Assesses for concentrated blood/hemoconcentration | Increased to >290 mOsm/kg |
| Serum sodium | Monitors for hypernatremia | Increased to >147 mEq/L |
| Plasma ADH level (vasopressin level) | Assesses if vasopressin is elevated or decreased | Central DI: Decreased Nephrogenic DI: Normal or increased Gestational DI: Decreased Dipsogenic DI: Decreased |
| Water deprivation test (Miller-Moses Test) Dehydration should prompt the kidneys to concentrate urine. |
To distinguish between the types of DI. Assesses for changes in weight, serum and urine osmolality, and specific gravity when fluid intake is prohibited. | Differentiates psychogenic polydipsia from DI. Central DI and NDI are unaffected by this test. |
| ADH (Vasopressin) test ADH administration will correct the problem if ADH was lacking. |
Assesses if the kidneys begin to concentrate urine when ADH is administered. Distinguishes NDI from other types of DI. | Corrects central/neurogenic DI, wherein ADH is lacking. NDI is unaffected by ADH administration, since the problem is unrelated to lack of ADH. |
| Brain or Pituitary magnetic resonance imaging (MRI) | MRI scan used to identify pituitary lesions that may have caused the DI. | If the patient has the “bright spot” or hyperintense emission from the posterior pituitary gland, the patient likely has primary polydipsia. If the “bright spot” is small or absent, the patient likely has central DI. |
Collaborative management
Care priorities
1. Rehydrate if dehydration and/or hypovolemia are present
• Place at least two large-bore IV lines, or have a central line inserted.
• Administer hypotonic IV fluids (5% Dextrose solution, 0.45% saline.). Hyperglycemia and volume overload should be avoided.
• Avoid overly aggressive correction of hypernatremia. Do not decrease sodium level by more than 5mEq/L per hour or 12 mEq/L within 24 hours.
• Patient may be allowed to drink fluids, with the volume accurately recorded
• Monitor urine output judiciously. Total water deficit may be estimated by assuming body water composes approximately 60% of total body weight in kg.
• Monitor continuous ECG for tachycardia and dysrhythmias.
• Evaluate basic ABCs: airway, breathing, and circulation if the patient becomes hypotensive.
2. Administer exogenous adh (vasopressin)
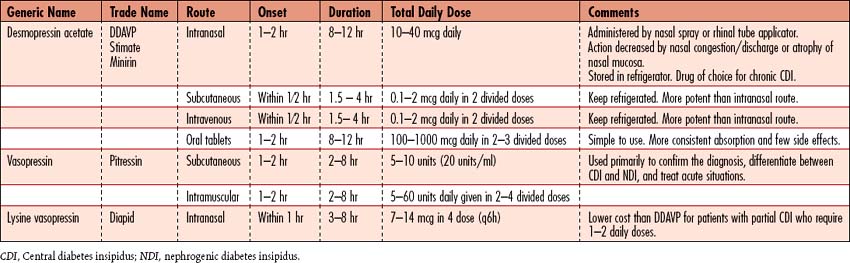
• Use of desmopressin acetate (DDAVP) is popular because it produces fewer side effects (Table 8-2).
• Several preparations are available, and dosage is adjusted to patient response to DI management. Vasopressin’s potential vasoconstrictive effects occur rarely with appropriate dosing for management of DI. Excessive dosing of ADH may cause hypertension and cardiac symptoms; other side effects include abdominal cramping and increased peristalsis.
5. Manage ndi (adh insensitive) with pharmacotherapy
• Thiazide diuretics (e.g., hydrochlorothiazide [HCTZ]) in combination with a low-sodium diet are the major form of therapy for NDI, to reduce the loss of free water in the urine.
• Chlorpropamide stimulates the release of ADH and facilitates the renal response to ADH.
• Amiloride hydrochloride (a potassium-sparing diuretic) is the medication of choice for the treatment of lithium-induced NDI.
• Nonsteroidal anti-inflammatory drugs (NSAIDs) such as indomethacin have been used as adjunctive therapy in NDI.
CARE PLANS FOR DIABETES INSIPIDUS
related to diuresis secondary to ADH deficiency or altered ADH action
![]()
Fluid Balance, Electrolyte and Acid-Base Balance, Hydration
1. Monitor vital signs every 15 minutes until patient is stable for 1 hour. Monitor CVP, MAP, and, if hemodynamic monitoring was in place, pulmonary artery pressure (PAP), and pulmonary capillary wedge pressure (PCWP), if ordered. Consult physician or midlevel practitioner for the following: HR greater than 140 bpm or BP less than 90/60 or decreased 20 mm Hg or greater, or MAP decreased 10 mm Hg or greater from baseline, CVP less than 2 mm Hg, and PAWP less than 6 mm Hg. Manage judiciously in all patients, including brain-dead organ donors.
2.  Monitor hydration status: mucous membranes, pulse rate and quality, and BP. Excessive water intake may result in fluid overload, particularly in elders and children.
Monitor hydration status: mucous membranes, pulse rate and quality, and BP. Excessive water intake may result in fluid overload, particularly in elders and children.
3. Administer hypotonic solutions (e.g., D5W, D50.25, or 0.45 NaCl) for intracellular rehydration. Usually, fluids are administered as follows: 1 ml IV fluid for each 1 ml of urine output. In patients with brain injury, moderate diuresis may be permitted to avoid the need for administering osmotic diuretics. Hypernatremia, if present, must be corrected slowly (at a rate no greater than 0.5 mEq/L/hr or 12 mEq/L/day) to prevent cerebral edema, seizures, permanent neurologic damage, or death.
4. Administer vasopressin (DDAVP) as ordered. Observe for and document effects. Also be alert to side effects of therapy: hypertension, cardiac ischemia, and hyponatremia.
5. Weigh patient daily, at the same time and using the same scale and garments to prevent error. Consult physician or midlevel practitioner for weight loss greater than 1 kg/day.
6. Observe for indications of dehydration (e.g., poor skin turgor, delayed capillary refill, weak/thready pulse, dry mucous membranes, hypotension).
7.  Monitor for fluid overload, which can occur as a result of rapid infusion of fluid or excessive fluid intake in patients with heart failure: jugular vein distention, dyspnea, crackles (rales), and CVP greater than 12 mm Hg.
Monitor for fluid overload, which can occur as a result of rapid infusion of fluid or excessive fluid intake in patients with heart failure: jugular vein distention, dyspnea, crackles (rales), and CVP greater than 12 mm Hg.
8. If urinary catheter has been removed, observe for resolution of nocturia (waking up at night to urinate) and enuresis (bed wetting) as treatment progresses.
1. Monitor laboratory studies, observing for an appropriate response to treatment, including a decrease in serum sodium, increase in serum and urine osmolality, and increase in urine specific gravity.
2. Monitor urine specific gravity hourly to evaluate response to therapy. Patients may be allowed to develop hypotonic polyuria between doses of vasopressin to demonstrate persistence of DI when transient DI is suspected.
3. Report lack of improvement or deterioration to the physician or midlevel practitioner. Urine output greater than 200 ml/hr for 2 consecutive hours, or 500 ml/hrs in the presence of risk factors should be reported.
4. Instruct patients with permanent DI to wear a medical-alert bracelet labeled with DI. Immediate family members should be familiar with the patient’s current treatment plan in case they are contacted in an emergency.
Disturbed sensory perception (visual and auditory)
resulting from hyperosmolality, dehydration, or hypernatremia
![]()
1. Monitor neurologic status frequently. Notify physician or midlevel practitioner of deterioration.
2. Keep bed in lowest position with side rails raised, if patient is confused.
3. Consider nasogastric tube with suction for comatose or brain-dead organ donor patients to decrease likelihood of aspiration.
4. Elevate head of the bed (HOB) to 30 degrees to minimize the risk of aspiration.
![]()
Infection Severity, Immune Status
1. For patients who have undergone transphenoidal hypophysectomy, inspect nasal packing often for frank bleeding or evidence of CSF leak. If glucose is detected in clear nasal drainage (tested using a glucose reagent stick), CSF is leaking, which indicates a flaw in cranial bone integrity. (See Care of the Patient After Intracranial Surgery, p 638.)
2. Elevate the HOB to minimize the chance of bacterial migration into the brain if CSF leak is suspected. Consult physician or midlevel practitioner promptly.
3. Monitor for infection, including elevated temperature, nuchal rigidity, and altered LOC.
4. Monitor for increased WBC count, which initially may reflect dehydration or the stress response.
5. Since patient is at higher risk for bacterial infection, invasive lines should be managed carefully to avoid bloodstream infection (BSI). Central lines should be removed as soon as possible.
6. To prevent injury and contamination of operative site, patients should not brush their teeth until instructed to do so by physician. Provide sponge-tipped applicators for oral hygiene.
![]() Incision Site Care; Infection Protection; Neurologic Monitoring
Incision Site Care; Infection Protection; Neurologic Monitoring
Deficient knowledge: illness care
![]()
Knowledge: Illness Care; Knowledge: Medication; Knowledge: Treatment Regimen
1. Teach patient appropriate administration of exogenous vasopressin and its side effects.
2. Explain exogenous hormone replacement if the anterior pituitary gland was damaged or removed during surgery. If patient is also experiencing anterior pituitary dysfunction (panhypopituitarism), teach the indicators of hormone replacement excess or deficiency.
3. Demonstrate the method for accurate measurement of urine specific gravity and the importance of keeping accurate records of test results.
4. Teach when to seek medical attention, including signs of dehydration (hypernatremia) and water intoxication (hyponatremia).
5. Explain the importance of obtaining a medical-alert bracelet and identification (ID) card.
6. Stress the importance of continued medical follow-up.
7. For patients with permanent need for hormone replacement, explain the method for obtaining a medical-alert bracelet and ID card outlining diagnosis and appropriate treatment in the event of an emergency.
![]() Teaching: Disease Process; Teaching: Prescribed Medication; Emotional Support
Teaching: Disease Process; Teaching: Prescribed Medication; Emotional Support
Additional nursing diagnoses
If patient has developed DI following a transphenoidal hypophysectomy, see Decreased intracranial adaptive capacity, Ineffective breathing pattern, Risk for infection, and Pain in Care of the Patient After Intracranial Surgery, p. 638; also see Hypernatremia and Hyponatremia in Fluid and Electrolyte Disturbances, p 37.)
Hyperglycemia
Pathophysiology
Patients in the Van den Berghe study who received tight glycemic control for new-onset hyperglycemia realized a much greater improvement in outcomes than did patients with diabetes mellitus, a particularly difficult concept for the medical community to accept. A subset of those with “new-onset” hyperglycemia were found to be undiagnosed diabetics, which prompted a recommendation that all patients undergo glycohemoglobin screening (hemoglobin A1c) performed on hospital admission. Assessment of glycemic control prior to hospitalization helps care providers anticipate whether patients will need insulin following hospitalization. Those with diabetes mellitus may be discharged on oral hypoglycemic agents or insulin. “Stress responders” without diabetes generally do not require insulin following hospitalization, because the stress of their illness or procedure resolves.
8-3 RESEARCH BRIEF
From Van den Berghe G, et al: Intensive insulin therapy in critically ill patients. N Engl J Med 345(19):1359–1367, 2001.
Physicians create insulin titration regimens, because no one method of insulin dosing has been universally accepted. Unlike the frequent titration of medications used to manage hypotension or hypertension, most dosage adjustments of IV insulin infusions are done hourly, because a continuous reading of glucose level is not possible. Dose responses are more difficult to assess. Few protocols individualize dosing based on insulin sensitivity or resistance. Individualization increases the complexity of care if nurses must perform mathematical calculations to make dosing adjustments.
Diabetic ketoacidosis
Pathophysiology
DKA is a life-threatening complication of diabetes mellitus characterized by hyperglycemic crisis, ketosis, acidosis, hypovolemic shock due to dehydration, and electrolyte imbalance involving potassium. Progressive hyperglycemia occurs due to inadequate circulating insulin, preventing cellular uptake of glucose and resulting in a state of starvation at the cellular level. Starvation prompts glucagon secretion from the pancreas and release of other stress hormones including catecholamines, cortisol, and growth hormone (GH), which facilitate glycogenolysis and gluconeogenesis, further raising plasma glucose. Proteolysis and lipolysis ensue, forming free fatty acids, which are converted to ketoacids (acetoacetate, beta-hydroxybutyrate, and acetone), due to lack of intracellular glucose required for normal metabolic conversion of the acids.
Hyperglycemic hyperosmolar syndrome
Pathophysiology
Historically, HHS and DKA were described as distinct syndromes, but one third of patients exhibit findings of both conditions. HHS and DKA may be at opposite ends of a range of decompensated diabetes, differing in time of onset, degree of dehydration, and severity of  ketosis. HHS occurs most commonly in older people with type 2 diabetes, but with the recent obesity epidemic, occasionally obese children and teenagers with both diagnosed and undiagnosed type 2 diabetes manifest HHS. The cascade of events in HHS begins with osmotic diuresis. Glycosuria impairs the ability of the kidney to concentrate urine, which exacerbates the water loss. Normally, the kidneys eliminate glucose above a certain threshold and prevent a subsequent rise in blood glucose level. In HHS, the decreased intravascular volume or possible underlying renal disease decreases the glomerular filtration rate (GFR), causing the glucose level to increase. More water is lost than sodium, resulting in hyperosmolarity. Insulin is present, but not in adequate amounts to decrease blood glucose levels, and with type 2 diabetes, significant insulin resistance is present. DKA and HHS are compared in Table 8-3.
ketosis. HHS occurs most commonly in older people with type 2 diabetes, but with the recent obesity epidemic, occasionally obese children and teenagers with both diagnosed and undiagnosed type 2 diabetes manifest HHS. The cascade of events in HHS begins with osmotic diuresis. Glycosuria impairs the ability of the kidney to concentrate urine, which exacerbates the water loss. Normally, the kidneys eliminate glucose above a certain threshold and prevent a subsequent rise in blood glucose level. In HHS, the decreased intravascular volume or possible underlying renal disease decreases the glomerular filtration rate (GFR), causing the glucose level to increase. More water is lost than sodium, resulting in hyperosmolarity. Insulin is present, but not in adequate amounts to decrease blood glucose levels, and with type 2 diabetes, significant insulin resistance is present. DKA and HHS are compared in Table 8-3.
Table 8-3 COMPARISON OF DIABETIC KETOACIDOSIS (DKA) AND HYPERGLYCEMIC HYPEROSMOLAR SYNDROME (HHS)
| Criterion | DKA | HHS |
|---|---|---|
 Diabetes type Diabetes type |
Type 1 | Type 2; rarely, Type 1 |
| Typical age group | More common in young children and adolescents than adults | 57-69 yrs, with average age 60 yrs |
| Signs and symptoms | Polyuria, polydipsia, polyphagia, weakness, orthostatic hypotension, lethargy, changes in LOC, fatigue, nausea, vomiting, abdominal pain | Same as DKA, but slower onset Also, very commonly, neurologic symptoms predominate |
| Physical assessment | Dry and flushed skin, poor skin turgor, dry mucous membranes, decreased BP, tachycardia, altered LOC (irritability, lethargy, coma), Kussmaul respirations, fruity odor to the breath | Same as DKA, but no Kussmaul respirations or fruity odor to the breath; instead, occurrence of tachypnea with shallow respirations |
| History and risk factors | Recent stressors such as surgery, trauma, infection, MI; insufficient exogenous insulin; undiagnosed type 1 diabetes mellitus | Undiagnosed type 2 diabetes mellitus; recent stressors such as surgery, trauma, pancreatitis, MI, infection; high-calorie enteral or parenteral feedings in a compromised patient; use of diabetogenic drugs (e.g., phenytoin, thiazide diuretics, thyroid preparations, mannitol, corticosteroids, sympathomimetics) |
| Monitoring parameters | ECG: Dysrhythmias associated with hyperkalemia: peaked T waves, widened QRS complex, prolonged PR interval, flattened or absent P wave. Hypokalemia (K+ <3 mEq/L), which may produce depressed ST segments, flat or inverted T waves, or increased ventricular dysrhythmias | ECG evidence of hypokalemia as listed with DKA Hemodynamic measurements: CVP >3 mm Hg below patient’s baseline; PADP and PAWP >4 mm Hg below patient’s baseline |
| Diagnostic tests | Serum glucose: Greater than 250 mg/dl | Greater than 600 mg/dl |
| Serum ketones: Large presence | Usually absent to mild presence due to dehydration | |
| Urine glucose: Positive | Positive | |
| Urine acetone: “Large” | Usually Negative | |
| Serum osmolality: Greater than 290 mOsm/L | Greater than 320 mOsm/L | |
| Bicarbonate: Less than 15 mEq/L | Greater than 15 mEq/L | |
| Serum pH: <7.2 | Normal or mildly acidotic (pH < 7.4) | |
| Anion Gap: Elevated greater than 13 | Normal | |
| Serum potassium: normal or elevated >5.0 mEq/L initially and then decreased | Normal or <3.5 mEq/L | |
| Serum sodium: elevated, normal, or low | Elevated, normal, or low | |
| Serum Hct: elevated because of osmotic diuresis with hemoconcentration | Elevated because of hemoconcentration | |
| BUN: elevated >20 mg/dl | Elevated | |
| Serum creatinine: >1.5 mg/dl | Elevated | |
| Serum phosphorus, magnesium, chloride: decreased | Elevated | |
| WBC: elevated, even in the absence of infection | Normal unless infection present | |
| Onset | A few days | Days to weeks |
| Mortality | 1-10% | 14%–58% because of age group and complications such as stroke, thrombosis, renal failure |
BP, Blood pressure; BUN, blood urea nitrogen; CVP, central venous pressure; DKA, diabetic ketoacidosis; ECG, electrocardiogram; Hct, hematocrit; HHS, hyperglycemic hyperosmolar syndrome; LOC, level of consciousness; MI, myocardial infarction; PADP, pulmonary artery diastolic pressure; PAWP, pulmonary artery wedge pressure; WBC, white blood cell count.
Primary causes of HHS include infections, noncompliance with a diabetes management regimen, undiagnosed diabetes, medications, substance abuse, and coexisting diseases. Infections are the leading cause (57% of patients); pneumonia (often gram negative) is the most common, followed by UTI and sepsis. Lack of compliance with diabetic medications or other aspects of diabetes management may be a frequent cause (21%). Undiagnosed diabetes prompts a failure to recognize early symptoms of the complications of unmanaged hyperglycemia. Acute  coronary syndrome (MI), stroke, pulmonary embolus, and mesenteric thrombosis have caused HHS. In urban populations, at least one study revealed the three leading causes to be lack of compliance with medications, drinking alcohol, and use of cocaine. Chronic use of steroids and gastroenteritis are commonly associated with HHS in children.
coronary syndrome (MI), stroke, pulmonary embolus, and mesenteric thrombosis have caused HHS. In urban populations, at least one study revealed the three leading causes to be lack of compliance with medications, drinking alcohol, and use of cocaine. Chronic use of steroids and gastroenteritis are commonly associated with HHS in children.
 Unlike DKA, wherein acidosis produces severe symptoms, HHS develops slowly, and frequently symptoms are nonspecific. Polyuria and polydipsia occur but may be ignored. Neurologic deficits may be mistaken for senility. Similarity of symptoms to other disease processes in older adults may delay diagnosis and treatment, allowing the process to progress.
Unlike DKA, wherein acidosis produces severe symptoms, HHS develops slowly, and frequently symptoms are nonspecific. Polyuria and polydipsia occur but may be ignored. Neurologic deficits may be mistaken for senility. Similarity of symptoms to other disease processes in older adults may delay diagnosis and treatment, allowing the process to progress.Metabolic assessment: hyperglycemia
History and risk factors
Hyperglycemia manifests more commonly in hospitalized patients with diabetes mellitus or impaired glucose tolerance than in normal patients with an exaggerated response to stress. Obese patients are more likely to have insulin resistance associated with metabolic syndrome, impaired tolerance for glucose, or undiagnosed type 2 diabetes mellitus. Type 2 diabetic patients generally manifest HHS when hyperglycemia is ineffectively managed. Rarely, a type 2 patient will manifest DKA. The majority of patients with DKA have type 1 diabetes mellitus. The type 1 patient will die without adequate insulin administration.
Recent stressors that prompt dka in patients with diabetes mellitus include:
• Infection (20% to 55%): May be overestimated because DKA may prompt leukocytosis and vasodilation, which mimic sepsis.
• Inadequate insulin/noncompliance (15% to 40% ): Teenagers may be at higher risk for noncompliance; all illnesses increase stress, which increases the need for insulin. Type 1 patients are totally reliant on administration of exogenous insulin to control hyperglycemia, because without functional beta islet cells in the pancreas, they have no ability to produce insulin.
• Undiagnosed diabetes (10% to 25%): Onset of type 1 diabetes is generally preceded by a significant illness—often a viral infection or childhood disease.
• Other medical illness (10% to 15%): Pneumonia, urinary tract infection, ischemic bowel, pregnancy, hypothyroidism, pancreatitis, pulmonary embolism, surgery, and new medications (notably corticosteroids, sympathomimetics, alpha and beta blockers, fluoroquinolone antibiotics [Levaquin], and diuretics)
• Cardiovascular disease (3% to 10%): Significant cardiovascular disease may be the result of diabetes mellitus, and subsequently, unstable patients may experience variable stress levels making control of hyperglycemia difficult. Vascular events such as MI, cerebrovascular accident (CVA), or ischemic bowel may precipitate or worsen DKA.
• Cause unknown (5% to 35%): Any physiologically stressing illness or event has the potential to cause the condition. Certain women are more likely to go into DKA at the time of menstruation. Severe emotional stress is associated with onset of DKA.
Recent stressors that prompt hhs in patients with diabetes mellitus:
•  Diabetes: First symptomatic presentation of undiagnosed hyperglycemia; poor control of hyperglycemia (noncompliance, inadequate resources, abuse or neglect of patient by caregivers or self-inflicted, accidental omission of medication, or ingestion of excessive carbohydrates)
Diabetes: First symptomatic presentation of undiagnosed hyperglycemia; poor control of hyperglycemia (noncompliance, inadequate resources, abuse or neglect of patient by caregivers or self-inflicted, accidental omission of medication, or ingestion of excessive carbohydrates)
Observation
• Hyperglycemia alone may not cause overt physical assessment changes.
• Skin should be examined for lesions, rashes, cellulitis, and other signs of possible infection.
• If patient presents to the emergency department, observe for signs of recent alcohol consumption.
• With DKA: Kussmaul respirations (rapid, deep) are present to exhale CO2 as a compensatory response to relieve metabolic acidosis; may appear fatigued, with or without diaphoresis from Kussmaul breathing
 Altered LOC (confusion, disorientation, agitation) is more common with older adults; strokelike symptoms may be present; coma is present in about 10% of patients presenting with HHS; seizures are present in 25% of HHS patients.
Altered LOC (confusion, disorientation, agitation) is more common with older adults; strokelike symptoms may be present; coma is present in about 10% of patients presenting with HHS; seizures are present in 25% of HHS patients.Auscultation
Screening labwork
• POC capillary blood glucose (bedside glucose monitoring)
• If POC glucose is elevated, a plasma glucose sample should be drawn.
Diagnostic Tests for Hyperglycemia and Hyperglycemic Emergencies
| Test | Purpose | Abnormal Findings |
|---|---|---|
| Hemoglobin A1c (HbA1c) or glycosylated hemoglobin Performing this test more frequently than every 6–8 weeks does not yield useful information about blood glucose control |
Assesses for control of blood glucose for the 6 to 8 weeks preceding the test. Recommended screening for all hospitalized patients so poorly controlled blood glucose readings can be addressed immediately to avoid development of DKA (diabetic ketoacidosis) or HHS (hyperglycemia hyperosmolar syndrome) | HbA1c >7 reflects poor control. AACE and ADA guidelines have varied; stricter guidelines recommend patients with HbA1c >6 have inadequate control. If a patient with diabetes mellitus who has been managing at home presents with an elevated value, home management and medications should be adjusted. If a patient without diabetes has an elevated value, the patient should undergo a full evaluation for presence of undiagnosed diabetes. |
| Fasting blood glucose (FBG) Test is performed in the morning after fasting all night and prior to consuming breakfast |
Evaluates the effectiveness of basal insulin dosage by assessing for presence of hyperglycemia or hypoglycemia; used for daily screening of blood glucose control during hospitalization | <40 mg/dl: Severe hypoglycemia 41–69 mg/dl: Hypoglycemia 70–110mg/dl: Normoglycemia 111–125mg/dl: Borderline hyperglycemia 126–180mg/dl: Hyperglycemia 181–220mg/dl: Significant hyperglycemia >220 mg/dl: Possible impending DKA or HHNS if glucose is not managed |
| Mealtime blood glucose Generally, a point-of-care (POC) reading is done either 15 to 30 minutes prior to a meal, or as the meal begins. |
Assesses blood glucose control with existing hyperglycemia management program | If reading is <140 mg/dl (2009 AACE and ADA recommendation), no mealtime, short acting, subcutaneous insulin is needed. The recommendations have not been universally accepted. Thresholds for supplemental insulin vary with each hospital and sometimes, each physician. |
| Postprandial blood glucose May be done 1 or 2 hours following meals using serum glucose or point of care capillary glucose readings |
Evaluates ability of glucose to normalize following a meal. Readings may be done 1 or 2 hours following the meal. | >180 mg/dl: If glucose is >180 at 1 hour following a meal, the patient is unable to produce enough insulin, or has not received enough mealtime insulin, or may be insulin resistant, or may require initiation of mealtime insulin. >140 mg/dl: If glucose is >140 at 2 hours following a meal, the patient is unable to produce enough insulin or may be insulin resistant. |
| Oral glucose tolerance test (OGTT) Following at least 8 hours of fasting, oral glucose is consumed by the patient to determine how quickly it is cleared from the blood. |
Used to test for diabetes, insulin resistance, and reactive hypoglycemia. Fasting blood glucose (FBG) is used at the beginning of the test. Additional readings are done 2 hours later. Fasting readings are compared to 2 hours post glucose ingestion to determine extent of glucose intolerance. | FBG >126 mg/dl with 2-hour reading >200 mg/dl: Confirms diagnosis of diabetes mellitus (DM) FBG 111–125 mg/dl with 2-hour reading >140 mg/dl: Patient has impaired glucose tolerance (IGT) FBG 111–125mg/dl with 2-hour reading <140 mg/dl: Patient has impaired fasting glucose (IFT) |
| Arterial blood gas analysis (ABG) Done promptly, following confirmation of hyperglycemia to assess for DKA and HHS. |
Assess for abnormal gas exchange or compensation for metabolic derangements in patients in hyperglycemic crisis; profound acidosis can indicate DKA is present, since HHS typically presents with minimal to mild acidosis unless prolonged, severe hypovolemic shock is present. | pH changes: With DKA, may be 6.8–7.2; acidosis results from ketosis or lactic acidosis Carbon dioxide: With DKA, decreased CO2 reflects tachypnea and Kussmaul respirations Hypoxemia: With DKA or HHS, PaO2 <80 mm Hg may indicate pneumonia precipitated the crisis Oxygen saturation: If pneumonia or heart failure is present, SaO2 may be <92% Bicarbonate: HHS: HCO3− 15-22 mEq/L; DKA: HCO3– may be <15 mEq/L Base deficit: HHS <−2; with DKA, <−10 |
| Complete blood count (CBC) | Evaluates for presence of infection | Increased WBC count: >11,000/mm3 is seen with bacterial pneumonias, urinary tract infections and other infections. |
| Sputum Gram stain, culture and sensitivity | Screens for pneumonia, a common underlying cause of hyperglycemic crisis; identifies infecting organism | Gram stain positive: Indicates organism is present; Culture: Identifies organism Sensitivity: Reflects effectiveness of drugs on identified organism. |
| Blood culture and sensitivity | Screens for sepsis, a common underlying cause of hyperglycemic crisis Identifies whether an organism has become systemic |
Secondary bacteremia: a frequent finding; patients with bacteremia are at higher risk for developing respiratory failure. |
| Blood chemistry | Screens for electrolyte imbalances; potassium imbalances may create potentially dangerous dysrhythmias | DKA and HHS: Hypernatremia is present: BUN, creatinine, and K+ may be elevated, normal, or low. Anion gap DKA: >13 Anion gap HHS: 10–12 |
| Plasma osmolality | Screens for elevated osmolality associated with severe hyperglycemia | Osmolality is increased more with HHS than with DKA. DKA: 290–320 mmol/L HHS: >320 mmol/L |
| Urine ketones | Screens for the presence of ketones to confirm diagnosis of DKA; HHS does not cause ketonuria. |
DKA: Ketones strongly positive HHS: Ketones negative, or mildly positive |
| 12-Lead ECG | Used to rule out myocardial infarction as the cause of HHS or DKA | Tall, peaked T waves if ↑K+ is present prior to management of hyperglycemia, hypovolemia and acidosis; VPCs/ventricular irritability is seen with ↓K+ seen as insulin normalizes glucose. |
| Chest radiograph | Screens for pneumonia and acute respiratory distress syndrome, which may prompt DKA and HHS | “Fluffy whiteness” may not initially be present due to dehydration, but may appear as patient is rehydrated revealing pneumonia or ARDS |
| Computed tomography (CT) brain scan | Screens for ischemic and hemorrhagic stroke, which may prompt DKA and HHS | Generally not done until patient has had at least 1 hour of rehydration and insulin therapy to see if symptoms resolve spontaneously. |
Collaborative management
1. Oral hypoglycemic agents are not recommended for use in acutely ill or unstable hospitalized patients because the response to therapy is unpredictable. If further glucose control is needed, the use of insulin superimposed on oral hypoglycemic agents may prompt episodes of hypoglycemia.
2. Guidelines should be evidence based and parallel the recommendations of the recognized expert organizations (ADA and AACE).
3. Protocols/order sets must be “user-friendly” and clearly written with minimal abbreviations and strive to keep mathematical calculations to a minimum.
4. A system should be in place to identify patients who need insulin or adjustment in an existing insulin regimen.
5. Variations in nutritional requirements/nutritional support should be identified, recognized, and included in the planning of any insulin dosing regimen for patients with varying levels of stability as they move through the hospital.
6. Requirements for safe insulin administration, including availability of point of care testing, IV pumps that can deliver volumes less than 1 ml accurately, and staffing with competent nurses must be considered prior to implementation of a glycemic control program.
7. Expert nurse consultants and/or certified diabetes educators who can provide diabetes patient education should be available to both patients and staff nurses.
8. An interdisciplinary team should be formed to address the following questions about the hospital’s hyperglycemia management practices:
Care priorities
1. Resuscitate patients with severe dehydration in hypovolemic shock:
• Evaluate basic ABCs: airway, breathing, and circulation.
• Intubate and ventilate patients with hypoxemia/decreasing oxygen saturation.
• Place at least two large-bore IV lines, or have a central line inserted.
• Monitor continuous ECG, pulse oximetry, and frequent, if not constant, BP.
• Apply oxygen, or set oxygen appropriately if mechanical ventilation is used.
• Insert a urinary catheter and monitor urine output judiciously.
• Consider inserting a nasogastric tube if high risk for aspiration.
2. Provide aggressive rehydration to replace fluid loss from polyuria:
• HHS: Large amounts of isotonic IV fluids (normal saline, sometimes greater than 9 L) may be required to rehydrate the patient. Half normal saline is sometimes used. Fluids should not include dextrose. Osmotic diuresis causes a 100 to 200 ml/kg fluid loss. At least 1 L of saline should be administered during the first hour of therapy. Hemodynamic or CVP monitoring may be needed to provide aggressive rehydration, as large amounts of fluids may not be well tolerated in older adults. The remaining 8 L should ideally be infused within the next 24 hours. Highly viscous blood is prone to thrombosis. Patients are at risk of developing thrombotic complications before, during, and after a severe hyperglycemic crisis.
• DKA: Large amounts of nondextrose, isotonic IV fluids are needed. Half normal saline is sometimes used. From 1 to 2 L may be needed over the first 60 to 90 minutes. Approximately 4 L should be infused over the first 5 hours, and then the patient should be reevaluated for need for further hydration. Older patients or those with comorbidities that complicate therapy may require hemodynamic or CVP monitoring to guide fluid replacement. Rehydration improves perfusion, which improves oxygen delivery and thus helps to reestablish aerobic metabolism. As cellular metabolism normalizes, lactic and ketotic acidosis resolve.
6. Identify and manage the precipitating cause:
As hyperglycemia is managed, further efforts should be under way to identify the cause of the hyperglycemic crisis. A robust listing of risk factors and probable causes was included earlier, with infections and cardiac and vascular occlusive events as the most likely precipitating events for the hyperglycemic crisis. If the pH and anion gap fail to improve with hydration and insulin, other causes of shock and acidosis should be evaluated and managed accordingly.
CARE PLANS FOR HYPERGLYCEMIA
Glucose, risk for unstable blood
![]()
Blood Glucose Level, Hydration
• Monitor blood glucose levels as ordered or according to protocol.
• Facilitate patient having HbA1c measured to assess for glycemic control prior to hospitalization. If patient has been transfused, HbA1c is no longer a reliable measure of blood glucose control, as patient is circulating blood that includes another person’s glycohemoglobin.
• Assess for signs and symptoms of hyperglycemia including polyuria, polyphagia, blurred vision, headache, change in LOC, weakness, and lethargy.
• Monitor for urine ketones if patient has type 1 diabetes, is a ketosis-prone type 2 diabetic, or manifests severe hyperglycemia.
• Monitor ABGs in severely hyperglycemic patients to assess if acidosis is present.
• Identify possible causes of hyperglycemia and work with physicians to construct an individualized management plan.
• Evaluate hydration status if patient has been hyperglycemic with polyuria due to osmotic diuresis.
• Encourage oral noncaloric fluid/water intake.
• Monitor for potassium imbalance, with awareness that hyperglycemic patients can experience wide variation in potassium when IV insulin therapy is used to control hyperglycemia. As glucose normalizes, hypokalemia may be present and should be managed with careful potassium replacement.
• Instruct patient and significant others on how to prevent, recognize, and manage hyperglycemia.
• Encourage patient to participate in POC testing to assist in refining testing techniques if needed.
• Discuss the need to count and control ingested carbohydrates to provide the best opportunity for glycemic control. Explain the difference in simple (bad) and complex (good) carbohydrates and how they are metabolized.
• Ensure patient understands the need for adherence to the prescribed diet and exercise regimen.
• Assess whether patient has the financial means to procure the proper food and medications to control hyperglycemia following discharge from the hospital.
1. Identify patients at increased risk for hypoglycemia.
2. Monitor blood glucose levels carefully as ordered, especially if insulin is used to manage hyperglycemia.
3. Assess for signs and symptoms of hypoglycemia, including changes in personality, irritability, shakiness or tremors, sweating, nervousness, palpitations, tachycardia, nausea, headache, dizziness, weakness, faintness, blurred vision, difficulty concentrating, confusion, coma, or seizures.
4. Maintain IV access for more precise management of hypoglycemia using IV 50% dextrose, rather than glucagon for instances of severe hypoglycemia that render patient unable to take glucose tablets, juice, or milk.
5. Ensure patient is given IV fluids containing 5% dextrose when glucose approaches 250 mg/dl if receiving an IV insulin infusion for management of severe hyperglycemia.
6. Keep patient NPO or on a no-calorie liquid diet while receiving an insulin infusion. If patients receive meals while insulin is infusing, additional mealtime subcutaneous insulin should be given, rather than adjusting IV insulin to cover glucose increases resulting from meals. If the IV insulin infusion is titrated upward throughout the day for meals, when the patient stops eating meals at night, the probability of hypoglycemia is high.
7. Instruct patient and significant others regarding the signs, symptoms, and management of hypoglycemia.
8. Collaborate with patient and care team members to make changes in insulin regimen if hypoglycemic episodes occur more than occasionally.
related to hyperglycemia-induced dehydration and osmotic diuresis
![]()
Fluid Balance, Electrolyte and Acid-Base Balance, Hydration
1. Monitor vital signs every 15 minutes until patient is stable for 1 hour. Monitor CVP, MAP, and possibly PAP and PCWP, if ordered. Consult physician or midlevel practitioner for the following: HR greater than 140 bpm or BP less than 90/60 or decreased 20 mm Hg or greater, or MAP decreased 10 mm Hg or greater from baseline, CVP less than 4 mm Hg, and PAWP less than 6 mm Hg.
2. Monitor hydration status: mucous membranes, pulse rate and quality, and BP.
3. Monitor I&O. Decreased urine output may indicate inadequate fluid volume or impending renal failure. Consult physician for urine output less than 0.5 ml/kg/hr for 2 consecutive hours.
4.  Replace volume with IV fluids. Monitor for fluid overload, which can occur as a result of rapid infusion of fluids: jugular vein distention, dyspnea, crackles (rales), and CVP greater than 6 mm Hg.
Replace volume with IV fluids. Monitor for fluid overload, which can occur as a result of rapid infusion of fluids: jugular vein distention, dyspnea, crackles (rales), and CVP greater than 6 mm Hg.
1. Monitor for abnormal electrolyte levels as ordered.
2. Note an increase in anion gap (greater than 14 mEq/L), signaling increased production or decreased excretion of acids. Anion gap should decrease steadily with successful treatment of DKA.
3. Monitor for symptomatic cardiac dysrhythmias. (See Fluid and Electrolyte Disturbances, p. 37.)
4. Observe for clinical signs of electrolyte imbalance associated with DKA and its treatment:
![]()
Infection Severity, Immune Status
1. Monitor for signs of infection. Fever may be suppressed secondary to acidosis. Monitor for increased WBC count, which initially may reflect dehydration or the stress response.
2. Since patient is at higher risk for bacterial infection, invasive lines should be managed carefully to avoid bloodstream infection (BSI). Central lines should be removed as soon as possible.
3. Manage urinary catheters meticulously to prevent UTI.
4. Maintain skin integrity. Assess for areas of decreased sensation.
 related to hyperosmolality, dehydration, or hypoglycemia
related to hyperosmolality, dehydration, or hypoglycemia
![]()
1. Monitor neurologic status frequently. Notify physician or midlevel practitioner of deterioration.
2. Keep bed in lowest position with side rails raised, if patient is confused.
3. Consider nasogastric tube with suction for comatose patients to decrease likelihood of aspiration.
4. Elevate HOB to 30 degrees to minimize the risk of aspiration.
5. Monitor blood glucose hourly while on insulin infusion. Consult physician or midlevel practitioner if blood glucose drops faster than 100 mg/dl/hr or if it drops to less than 250 mg/dl. Obtain prescription for glucose-containing IV solution to prevent hypoglycemia and allow the continued administration of insulin necessary to correct acidosis.
 related to new-onset diabetes or misunderstanding of the causes and prevention of DKA or HHS
related to new-onset diabetes or misunderstanding of the causes and prevention of DKA or HHS
![]()
Knowledge: Illness Care; Knowledge: Medications; Health-seeking behavior
1. Consider referral to diabetes educator for new-onset diabetes or if patient has not managed condition well in the past. Provide instructions simply, incorporating patient teaching into patient care routines.
2. Explain the relationship of DKA or HHS to illness and stress. Emphasize importance of adhering to the diabetes regimen, including meal planning, medication, exercise, and monitoring.
3. Review illness guidelines for individuals with diabetes (i.e., need for increased fluid and insulin with illness) with patient and significant others.
4. Provide hospital and community resources for diabetes education and support.
5. Provide address and websites for the American Diabetes Association (ADA): American Diabetes Association, Inc., 18 East 48th Street, New York, NY 10017; www.ada.org and www.diabetes.org
Additional nursing diagnoses
See also nursing diagnoses and interventions in Hyperkalemia, Hypokalemia, and Hypovolemia in Fluid and Electrolyte Disturbances, p 37; Alterations in Consciousness, p 24; Prolonged Immobility (p. 149); and Emotional and Spiritual Support of the Patient and Significant Others (p. 200).
Myxedema coma
Pathophysiology
Individuals who are acutely or critically ill may have an extreme disruption of the normal hypothalamic–anterior pituitary–thyroid axis, particularly related to the nocturnal surge that is normally seen with thyrotropin. These patients will have a low T3 level even after the TSH is restored to normal and are commonly referred to as presenting with low T3 syndrome. Patients with poor heart function or more intense inflammatory reaction show more pronounced downregulation of the thyroid system. During sepsis, the pituitary gland is activated via blood-borne proinflammatory cytokines and through a complex interaction between the autonomic nervous system and the immune cells. Sepsis elicits a pattern of pituitary hormone dysfunction which may cause a significant decrease in the secretion of TSH.
1. Autoimmune: The most frequent cause of acquired hypothyroidism is autoimmune thyroiditis (Hashimoto thyroiditis). The body recognizes the thyroid antigens as foreign, and a chronic immune reaction ensues, resulting in lymphocytic infiltration of the gland and progressive destruction of functional thyroid tissue. Most affected individuals have circulating antibodies to thyroid tissue.
2. Postpartum thyroiditis: 10% of postpartum women develop lymphocytic thyroiditis 2 to 10 months after delivery. The frequency may be 5% in women with type 1 diabetes mellitus. The condition may only last 2 to 4 months but usually requires treatment with levothyroxine; however, postpartum patients with lymphocytic thyroiditis are at increased risk of permanent hypothyroidism. The hypothyroid state may be preceded by a short thyrotoxic state.
3. Subacute granulomatous thyroiditis: Inflammatory conditions or viral syndromes may be associated with transient hyperthyroidism followed by transient hypothyroidism. This presentation is linked to fever, malaise, and a painful and tender gland.
Tertiary hypothyroidism is related to hypothalamic dysfunction and is diagnosed by the release of thyrotropin-releasing hormone (TRH). Other causes are listed in Box 8-2.
Because all metabolically active cells require thyroid hormone, the effects of hormone deficiency vary. Systemic effects are due to either derangements in metabolic processes or direct effects by myxedematous infiltration in the tissues. The patient’s presentation may vary from asymptomatic to, rarely, coma with multisystem organ failure (myxedema coma).  Hypothyroidism is eight times more likely to occur in women than in men, and it frequently presents in the later years of life; older women are the most likely candidates to present with myxedema.
Hypothyroidism is eight times more likely to occur in women than in men, and it frequently presents in the later years of life; older women are the most likely candidates to present with myxedema.
Endocrine assessment: myxedema coma
 A change in mental status may be the most compelling sign to assist in making the diagnosis.
A change in mental status may be the most compelling sign to assist in making the diagnosis.Observation
Cardiac: Nonspecific ECG changes, prolonged conduction times, prolonged QT syndrome. Cardiac enlargement, possible effusion. If hypotensive, may be refractory to volume and vasopressors until thyroid hormone given.
Respiratory: Respiratory depression from reduced hypoxic drive and decreased ventilatory response and increasing CO2. The increase in CO2 is a major factor in the induction of coma. May also have edema of the conducting airways.
Gastrointestinal: Anorexia, nausea, abdominal pain, and constipation. Quiet abdomen, ileus, and megacolon not uncommon.
Hypothyroidism
• Possible presence of obesity and/or weight gain from fluid retention
• The skin may be dry, cool, and coarse, and the hair may be thin, coarse, and brittle. The tongue may be enlarged (macroglossia), and the reflexes may be slowed. Periorbital edema may be noted.
• There may be a surgical scar or a goiter when evaluating the neck. Nodules may be palpated.
• The patient may have muscle weakness, memory and mental impairment, and constipation.
• Polysaccharide substances may be deposited beneath the skin (myxedema), which may prompt hypovolemia.
 A change in mental status may be the most important sign. It is commonly noted first to be lethargy, then stupor, and ultimately coma.
A change in mental status may be the most important sign. It is commonly noted first to be lethargy, then stupor, and ultimately coma.Screening labwork
Blood studies may reveal the presence of thyroid dysfunction. Expected abnormalities include (see also Figure 8-2):
Common Diagnostic Tests for Hypothyroid Crisis: Myxedema Coma
| Test | Purpose | Abnormal Findings |
|---|---|---|
| Blood Studies | ||
| Thyroid-stimulating hormone (TSH) Standard normal: 0.4–4.5 mIU/L Revised normal: 0.4–2.5 mIU/L TSH >2.5 mIU/L from the NAHAMES III study indicated hypothyroidism. |
Measures TSH output from the anterior pituitary. Completes a negative feedback loop with the thyroid. When thyroid hormone level decreases, TSH level should increase. | Elevated unless hypothyroidism is longstanding or severe. When TSH is higher than 2.5 mIU/L in the presence of clinical symptoms, the diagnosis of hypothyroidism will be considered positive until proved otherwise. Always beneficial to also evaluate T4 at the same time. If TSH is higher than 4.5, the diagnosis of hypothyroidism is considered positive. |
| Thyroperoxidase antibodies | Assesses for thyroid antibodies | Positive test signals chronic autoimmune thyroiditis. |
| Free T4 or free thyroxine index (FTI) Normal: 60–170 nmol/L |
Measures the level of primary thyroid hormone May be unreliable in the face of critical illness |
Decreased. When levels are below normal, diagnosis of hypothyroidism is made. If the TSH is high, diagnosis would be a primary hypothyroidism, If TSH is normal or low and T4 is low, the problem is in the hypothalamic- pituitary response to elevated circulating thyroid levels (secondary hypothyroidism). However if the T3 is also low, this may signify euthyroid sick syndrome. |
| T3 (triiodothyronine) Normal: 0.8–2.7 nmol/L |
Measures the more metabolically active form of the thyroid hormone. | Controversial regarding value of treatment of low levels, since it has a higher frequency of adverse cardiac events and is generally reserved for patients who are not improving clinically on LT4. |
| Thyroid-binding globulin (TBG) | To measure the level of the protein that binds with circulating thyroid hormones. Abnormal T4 or T3 measurements are often due to binding protein abnormalities rather than abnormal thyroid function. | Total T4 or T3 must be evaluated with a measure of thyroid hormone binding such as T3 resin uptake or assay of thyroid-binding globulin. These methods are known as free T4 or free T3 even though they do not measure free hormone directly. |
| Electrolytes Potassium (K+) Magnesium (Mg2+) Calcium (Ca2+) Sodium (Na+) |
Assess for possible abnormalities | Frequently, abnormalities of calcium are related to parathyroid disorders exist concurrently with thyroid dysfunction. |
| Radiology/Imaging | ||
| Thyroid scan with radioactive iodine uptake123| or 99mTc pertechnetate | To identify thyroid nodules | Not beneficial in hypothyroidism as uptake of radioactive iodine may not occur |
| Thyroid scan 131I and radioactive iodine uptake | To identify thyroid nodules | In primary hypothyroidism, will be less than 10% in a 24-hour period. In secondary hypothyroidism, uptake increases with administration of exogenous TSH. |
| Chest radiograph | Assess size of heart, and presence of pericardial or pleural effusion | Cardiac enlargement or fluid around the heart is common with myxedema |
| Ultrasound | Assess size and presence of nodules and goiter | Abnormal thyroid is unusual in Hashimoto disease. |
| Fine needle biopsy | Evaluate suspicious nodes | Cancerous cells indicate thyroid cancer |
Collaborative management
Care priorities
• Perform emergent endotracheal intubation and provide mechanical ventilation support: Required to relieve or prevent profound hypoxemia and CO2 narcosis detected by SpO2 and ETCO2 monitoring. Monitoring of arterial blood gas is done periodically to validate SpO2, ETCO2 and to monitor pH.
• Treat hypothermia: Although the hypothermia will be addressed ultimately by the institution of thyroid hormone, it may take several days. Extreme caution must be taken when re-warming (only passive techniques should be used) in order to avoid uncompensated vasodilation and profound hypotension.
•  Treat hypotension: Prior to institution of even passive re-warming, careful volume resuscitation should occur. Administer of IV isotonic fluids (normal saline and lactated Ringer’s solution). Hypotonic solutions, such as 5% dextrose in water (D5W), are contraindicated because they decrease serum Na+ levels further. These patients respond poorly to vasopressors because of alteration in sympathetic response. Consider IV vasopressin or IV corticosteroids.
Treat hypotension: Prior to institution of even passive re-warming, careful volume resuscitation should occur. Administer of IV isotonic fluids (normal saline and lactated Ringer’s solution). Hypotonic solutions, such as 5% dextrose in water (D5W), are contraindicated because they decrease serum Na+ levels further. These patients respond poorly to vasopressors because of alteration in sympathetic response. Consider IV vasopressin or IV corticosteroids.
2. Administer IV thyroid hormone as soon as possible:
8-4 RESEARCH BRIEF
From Grozinsky-Glasberg S: Thyroxine-triiodothyronine combination therapy versus thyroxine monotherapy for clinical hypothyroidism: meta-analysis of randomized controlled trials. J Clin Endocrinol Metab 91(7):2592–2599, 2006.
 Rapid IV administration of thyroid hormone should be done with careful monitoring, as this may cause hyperadrenalism. Concomitant administration of IV hydrocortisone helps prevent adrenal problems but may cause hypoadrenalism if not carefully monitored.
Rapid IV administration of thyroid hormone should be done with careful monitoring, as this may cause hyperadrenalism. Concomitant administration of IV hydrocortisone helps prevent adrenal problems but may cause hypoadrenalism if not carefully monitored.6. Administer stool softeners:
To minimize constipation owing to decreased gastric secretions and peristalsis
CARE PLANS FOR MYXEDEMA COMA
Ineffective protection (myxedema coma)
related to inadequate response to treatment of hypothyroidism or stressors such as infection
![]()
1. Monitor vital signs frequently and note bradycardia, hypotension, or decrease in RR. Report systolic BP greater than 90 mm Hg, HR less than 60 bpm, or RR less than 12 breaths/min.
2. Monitor patient for hypoxia. Report significant findings to physician.
3. Monitor serum electrolytes and glucose levels. Note decreasing Na+ (less than 137 mEq/L) and glucose (less than 60 mg/dl).
4. Administer IV thyroid replacement hormones with IV hydrocortisone and IV glucose to treat hypoglycemia.
5. Monitor for heart failure: jugular vein distention, crackles (rales), shortness of breath (SOB), peripheral edema, weakening peripheral pulses, and hypotension. Notify physician of any significant findings.
6. Keep an oral airway and manual resuscitator at the bedside in the event of seizure, coma, or the need for ventilatory assistance.
![]() Vital Signs Monitoring; Respiratory Monitoring; Shock Prevention; Cardiac Care: Acute
Vital Signs Monitoring; Respiratory Monitoring; Shock Prevention; Cardiac Care: Acute
![]()
Respiratory Status: Ventilation; Vital Signs
1. Assess rate, depth, and quality of breath sounds. Monitor for inadequate ventilation: changes in respiratory rate or pattern, falling SpO2, pallor, or cyanosis. Report findings to physician or midlevel practitioner promptly, including presence of adventitious sounds (e.g., from developing pleural effusion) or decreasing or crowing sounds (e.g., from swollen tongue or glottis).
2. Measure SpO2 intermittently or continuously in patients with decreased ventilatory drive.
3. Teach patient coughing, deep breathing, and use of incentive spirometer. For respiratory distress, assist physician with intubation or tracheostomy and maintenance of mechanical ventilatory assistance. Suction upper airway as needed.
related to compromised regulatory mechanisms occurring with associated adrenal insufficiency
![]()
1. Monitor I&O hourly for evidence of decreasing output.
2. Weigh patient at the same time every day, using the same scale. Monitor for the following indicators of heart failure: jugular vein distention, crackles (rales), SOB, dependent edema of extremities, and decreased pulse amplitude. Report significant findings to physician.
 related to weakness and fatigue secondary to slowed metabolism and decreased cardiac output caused by pericardial effusions, atherosclerosis, and decreased adrenergic stimulation
related to weakness and fatigue secondary to slowed metabolism and decreased cardiac output caused by pericardial effusions, atherosclerosis, and decreased adrenergic stimulation![]()
Activity Tolerance; Endurance; Energy Conservation
1. Monitor vital signs frequently for hypotension, slow pulse, dysrhythmias, complaints of chest pain or discomfort, decreasing urine output, and changes in mentation. Promptly report significant changes to physician.
2. Balance activity with adequate rest to decrease workload of the heart.
3. Administer IV isotonic solutions such as normal saline to help prevent hypotension.
related to compromised immunologic status secondary to alterations in adrenal function
![]()
1. Monitor for infection: fever, erythema, swelling, or discharge from wounds or IV sites; urinary frequency, urgency, or dysuria; cloudy or malodorous urine; presence of adventitious sounds on auscultation of lung fields; and changes in color, consistency, and amount of sputum. Minimize risk of UTI by providing care of catheters.
2. Provide care to maintain skin integrity and prevent pressure ulcers.
3.  Advise visitors who have contracted or been exposed to a communicable disease not to enter patient’s room without appropriate infection control precautions.
Advise visitors who have contracted or been exposed to a communicable disease not to enter patient’s room without appropriate infection control precautions.
 related to inadequate dietary intake of roughage and fluids, bed rest, and/or decreased peristalsis secondary to slowed metabolism
related to inadequate dietary intake of roughage and fluids, bed rest, and/or decreased peristalsis secondary to slowed metabolismWithin 48 to 72 hours of admission, patient resumes normal pattern of bowel elimination.
![]()
1. Monitor bowel function; report problems to the physician. Note decreasing bowel sounds, distention, and increased abdominal girth (may indicate ileus or an obstruction).
2. Encourage patient to maintain a diet with adequate roughage and fluids. Ensure that fluid intake in persons without underlying cardiac or renal disease is at least 2 to 3 L/day.
Deficient knowledge: illness care
related to management of hypothyroidism
![]()
1. Provide teaching about medications including drug name, purpose, dosage, schedule, precautions, drug-drug and food-drug interactions, and potential side effects. Remind patient that thioamides, iodides, and lithium are contraindicated because they decrease thyroid activity. Be sure patient is aware that thyroid replacement medications are to be taken for life.
2. Review dietary requirements and restrictions, which may change as hormone replacement therapy takes effect.
3. Explain expected changes with hormone replacement therapy: increased energy level, weight loss, and decreased peripheral edema. Neuromuscular problems should improve.
4. Stress the importance of continued, frequent medical follow-up.
5. Discuss importance of avoiding physical and emotional stress, and ways for patient to maximize coping mechanisms for dealing with stress.
6. Review signs and symptoms that require medical attention: fever or other symptoms of upper respiratory, urinary, or oral infections and signs and symptoms of hyperthyroidism, which may result from excessive hormone replacement.
Syndrome of inappropriate antidiuretic hormone
Pathophysiology
ADH (vasopressin) is produced in the hypothalamus and stored in the posterior pituitary and regulates free water volume in the kidney. Hyponatremia resulting from chronic SIADH is not always caused by reduced water excretion or volume overload. Plasma ADH level may not be high and measurement is often not helpful in establishing the diagnosis. Findings may reflect dilute (hypo-osmolar) plasma and hyponatremia with a normal circulating blood volume. Morbidity and mortality of hyponatremia associated with SIAD stem from cerebral edema and abnormal nerve function. Values of serum sodium 100 mEq/L or less are life-threatening. Four patterns of abnormal vasopressin secretion have been identified in Table 8-4. The disorders and medications associated with SIAD are listed in Table 8-5.
Table 8-4 SYNDROME OF INAPPROPRIATE ANTIDIURESIS (SIAD): FOUR TYPES
| Characteristics | Prevalence | |
|---|---|---|
| Type A | Large fluctuations in plasma ADH concentration independent of osmolality | 35% |
| Type B | Subnormal osmotic threshold for ADH release Osmoregulation is set around a subnormal threshold |
30% |
| Type C | ADH is not suppressed when plasma osmolality is low Normal response to osmotic changes |
∼25% |
| Type D | Normal osmoregulated ADH release Unable to excrete excess body water. |
<10% |
Table 8-5 MEDICATIONS AND DISEASES ASSOCIATED WITH SIADH AND HYPONATREMIA
| Cancer Related | Pulmonary Disorders |
| Carcinoma (bronchus, duodenum, pancreas, bladder, ureter, prostate) Carcinoid bronchial adenoma Ewing sarcoma Lymphoma, leukemia Mesothelioma Thymoma |
Aspergillosis Cystic fibrosis Empyema Pneumothorax Pneumonia Tuberculosis |
| Brain/Nervous System | Medications and Recreational Drugs |
| Alcohol withdrawal syndrome Brain abscess or tumor Cavernous sinus thrombosis Cerebellar and cerebral atrophy Cerebral hemorrhage/stroke Guillain-Barré syndrome Head injury, neurosurgery Hydrocephalus Meningitis, encephalitis Peripheral neuropathy Shy-Drager syndrome Seizures Subdural hematoma |
ACE inhibitors Alkylating agents and Vinca alkaloids Angiotensin II receptor antagonists (ARBs) Anticonvulsants Carbamazepine Carboplatin Chlorpropamide Cisplatin Clofibrate Cyclophosphamide DDAVP Dopamine antagonists Ifosfamide MAO inhibitors MDMA (“ecstasy”) NSAIDs Opiates Selective serotonin reuptake inhibitors (SSRIs) Sodium valproate Sulphonylureas Thiazides & Loop diuretics Tricyclic antidepressants Venlafaxine Vinblastine Vincristine |
| Other | |
| Abdominal surgery Hyperglycemia Idiopathic Porphyria Psychosis |
|
Hyponatremia resulting in SIAD is often drug induced, reflecting either direct stimulation of ADH/VP release from the hypothalamus, indirect stimulation of ADH action on the hypothalamus, or abnormal resetting of the hypothalamic osmotic threshold that governs release of ADH. The prevalence of hyponatremia in patients taking high-dose dopamine antagonists is greater than 25% and is associated with more than one class of these drugs. Hyponatremia secondary to antidepressants is common, occurring with most selective serotonin reuptake inhibitors (SSRIs). Patients receiving concurrent diuretic therapy are at high risk, indicating that hypovolemia contributes to the hyponatremia. Loop diuretics promote both sodium and water loss. Anticonvulsants commonly cause hyponatremia, which prompts SIAD. Patients treated with carbamazepine have a 5% to 40% incidence of SIAD.
Endocrine assessment: syndrome of inappropriate antidiuresis
Vital signs
• Findings vary significantly, depending on the degree of hyponatremia and fluid overload.
• Hyponatremia alone may have no affect on vital signs if the patient is able to achieve brain and CNS compensation.
• Tachycardia and tachypnea are present with fluid overload.
• Pulse may be irregular if hypokalemia has caused dysrhythmias.
• Mild elevation in BP is possible.
• Hypotension may be present with heart failure.
• Weight gain; slightly elevated BP
• CVP less than 6 mm Hg in the absence of underlying cardiac or pulmonary disease
Observation
• Patients may be asymptomatic or have mild symptoms if hyponatremia is less severe and develops slowly.
• Lesser symptoms reflect adaptation of the brain and CNS.
• Oliguria with concentrated urine; urine output less than 0.5 ml/kg/hr with specific gravity greater than 1.030 in the presence of adequate fluid intake
• Headache, difficulty concentrating, impaired memory, altered taste sensation
•  Electrolyte imbalance: Generalized weakness, possible exhaustion, nausea and vomiting, impaired vision, and leg cramps may be present from hyponatremia or hypocalcemia; patients may become unable to get out of bed; dysrhythmias result from hypokalemia.
Electrolyte imbalance: Generalized weakness, possible exhaustion, nausea and vomiting, impaired vision, and leg cramps may be present from hyponatremia or hypocalcemia; patients may become unable to get out of bed; dysrhythmias result from hypokalemia.
•  Altered mental status: Serum hypo-osmolality and hyponatremia affect consciousness and behavior. Changes may also be related the underlying disease.
Altered mental status: Serum hypo-osmolality and hyponatremia affect consciousness and behavior. Changes may also be related the underlying disease.
 Heart sounds may reflect heart failure if fluid overload exceeds the ability of the pumping action of the heart. Only a subset of these patients has fluid overload.
Heart sounds may reflect heart failure if fluid overload exceeds the ability of the pumping action of the heart. Only a subset of these patients has fluid overload.Screening labwork
• Biochemical panel: To assess for hyponatremia and altered concentrations of other electrolytes associated with hemodilution. Decreased blood urea nitrogen (BUN) with a urinary sodium level greater than 40 mmol/L in patients with hyponatremia suggests SIADH but is not diagnostic.
• Plasma and urine osmolality: Assesses for plasma hypo-osmolality in the presence of urine hyperosmolality
• Urine sodium level: Increased
• Urinalysis/specific gravity: Assesses for highly concentrated urine
| Test | Purpose | Abnormal Findings |
|---|---|---|
| Urine osmolality | Assesses for increased concentration of urine | Must exceed 100 mOsm/kg water when the plasma osmolality is low Normal: 300–1090 mOsm/kg |
| Urine specific gravity | Assesses for concentrated urine. | Specific gravity: >1.030 Normal: 1.010–1.025 |
| Urine sodium | Assesses for excessive loss of sodium in the urine | Increased to >20 mEq/L; increases to >60 mEq/L are common |
| Serum osmolality | Assesses for dilute blood/hemodilution | Decreased to <275 mOsm/kg Normal: 275–300 mOsm/kg |
| Serum sodium | Monitors for hyponatremia | Decreased to <130 mEq/L Normal: 137–147 mEq/L |
| Serum arginine vasopressin (VP) level (ADH level) See Table 8-4. |
Assesses whether vasopressin is elevated or decreased in relation to serum osmolality *VP/ADH level may be done with the water load test to provide specific information about VP secretion in response to the water load. |
Type A: Wide fluctuations Type B: Normal or increased Type C: Normal or increased Type D: Normal or increased *Not recommended for routine screening; urinary osmolality >100 mOsm per kilogram of water indicates excessive VP. |
| Water load test The test is not required for diagnosis; can be useful in the management of chronic or recurrent hyponatremia |
Abnormal ADH/VP secretion resulting in hyponatremia is diagnosed by assessing excretion of a standard water load over 4 hours. Urine output, urine and plasma osmolality are measured hourly. Plasma sodium is measured 2 hours following test completion, and the next morning, along with plasma osmolality. | Excretion of water or urine output may be reduced to 30% to 40% of the ingested load in the presence of VP production. Normal urine output is 78% to 82% of the ingested water load during the 4-hour test. In SIADH, low plasma osmolality is present with highly concentrated urine, with decreased plasma sodium indicating abnormal secretion of ADH/VP. |
| Urinary aquaporin-2 (AQP-2) Test provides limited information; role in diagnosis remains unclear |
May be useful in the differentiation of SIADH from other causes of hyponatraemia | There is a positive correlation between plasma VP concentration and urinary excretion of AQP-2, but urinary AQP-2 cannot clearly diagnose hyponatremic states associated with significant VP/ADH production. SIAD and chronic hypovolemia may generate similar plasma VP concentrations and similar urine AQP-2 levels. |
Collaborative management
Care priorities
1. Correct acute, symptomatic hyponatremia.
• Key considerations: The patient’s symptoms, severity, and duration of hyponatremia. Electrolytes should be measured every 4 hours. The approximate sodium deficit can be estimated using the following formula:
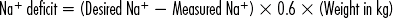
• Treatment goal: Increase serum sodium level by 1 to 2 mEq/L/hr using an IV 3% saline infusion; aim for resolution of major neurologic symptoms (i.e., seizures) and then decrease the correction rate. An increase in serum sodium levels of less than 10 mEq/L reduces the symptoms while preventing complications. The approximate volume of 3% saline needed:
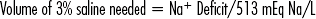
• Rate of sodium correction: The rate of correction should not exceed 0.5 mEq/L/hr. Correction during the first 24 hours of treatment should be an increase of 8 to 12 mEq/L and no greater than 18 to 25 mEq/L during the first 48 hours, regardless of the severity of hyponatremia:
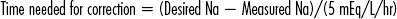
• Hypertonic (3%) saline infusion: The best method for calculating the initial infusion rate remains controversial. The rate of infusion may be calculated:
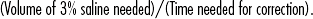
• If calculations are not used: 3% saline may be infused at a rate of 1 to 2 ml/kg of body weight/hr. The serum sodium level will increased by 1 to 2 mmol/L/hr. The rate may be doubled (2 to 4 ml/kg/hr) for a short time in patients with coma or seizures; half the rate (0.5 ml/kg/hr) is used if symptoms are mild.
• Diuretics: Consider concomitant use of IV furosemide (1 mg/kg); experts disagree on use. Some recommend avoiding or using it exclusively for those with fluid overload/extracellular-fluid volume expansion. Once renal function normalizes, correct K+ level.
2. Manage chronic (or uncertain duration) hyponatremia.
• Occurrence out of hospital: Most patients have chronic hyponatremia with few symptoms. Severe symptoms seen in marathon runners, users of the street drug 3,4-MDMA (ecstasy), and those who drink excessive amounts of water usually indicate acute hyponatremia and require rapid correction.
• Symptomatic hyponatremia of unknown duration: A correction of 8 mEq/L in 24 hours or 18 mEq/L in 48 hours is recommended; Serum sodium levels may be measured every 2 to 3 hours to avoid overcorrection.
• Asymptomatic patients with chronic hyponatremia: Fluid restriction is used to correct the sodium level slowly while avoiding osmotic demyelination. Maximum fluid intake tolerated depends on the oral osmotic load, so adequate intake of protein and salt should be encouraged. Oral intake of urea (30 grams daily) is effective but poorly tolerated.
• Demeclocycline: A dose of 300 to 600 mg twice daily may decrease urinary osmolality while increasing serum sodium levels; effects are variable and treatment may prompt nephrotoxicity.
3. Vasopressin-receptor antagonist therapy
• Conivaptan (Vaprisol): A vasopressin-receptor antagonist approved by the Food and Drug Administration in 2005 for the treatment of euvolemic hyponatremia and in 2007 for the treatment of hypervolemic hyponatremia. Intravenous conivaptan is used to treat hospitalized patients with symptomatic moderate-to-severe hyponatremia without seizures, delirium, or coma, who are candidates for hypertonic saline.
• Tolvaptan: Oral vasopressin receptor–selective V2 receptor antagonist used to increase serum sodium in SIAD patients. The appropriate role for vasopressin antagonist therapy continues to be studied and evaluated. These agents cause dry mouth and thirst, which prompts water intake, thus slowing the rise in serum sodium levels. Use of this agent in practice requires close monitoring of serum sodium levels.
• Hemodynamic monitoring: May be useful in guiding fluid management strategies and efficacy and may help recognize the patient’s response to the effects of nonselective vasopressin receptor blockade if shock symptoms occur. Distributive shock has not been reported, but is a possibility with use of these agents. Euvolemic patients may be at higher risk of hypotension.
5. Differentiate siad from cerebral salt wasting:
SIAD is often difficult to distinguish from cerebral salt wasting, a condition associated with patients with disease, an event or trauma of the CNS. Cerebral salt wasting is consistently associated with extracellular fluid volume depletion, but clinical assessment of volume status is difficult. Many physicians use saline infusion rather than fluid restriction for patients who have hyponatremia with subarachnoid hemorrhage, because of the risks associated with volume depletion in these patients. (See Cerebral Aneurysms and Subarachnoid Hemorrhage, Table 7-1, p 631.)
6. Monitor for signs of osmotic demyelination:
Several case reports have revealed this rare event resulting from too rapid correction of hyponatremia may be managed with IV vasopressin administration, which lowers the sodium level prompting improvement in neurological symptoms.
7. Manage postoperative hyponatremia:
CARE PLANS FOR SYNDROME OF INAPPROPRIATE ANTIDIURESIS
Decreased intracranial adaptive capacity with alteration in neurologic function
related to management of hyponatremia
![]()
Fluid Balance, Electrolyte and Acid-Base Balance (Serum Sodium)
Electrolyte management: hyponatremia
1. Assess LOC, vital signs, hemodynamic measurements, and I&O hourly; weigh patient daily. Monitor for decreased LOC; elevated BP, CVP, and PAWP; urine output less than 0.5 ml/kg/hr; and weight gain.
2. Monitor for changes in neurologic/neuromuscular symptoms of hyponatremia: lethargy, coma, seizures, headache, confusion, and weakness. Sodium levels of less than 120 mEq/L can cause life-threatening symptoms.
3. Obtain laboratory specimens to assess sodium levels (e.g., serum and urine sodium, serum and urine osmolality, urine specific gravity). Monitor for decreased serum sodium and plasma osmolality, urine osmolality that is disproportionately elevated compared to plasma osmolality, and increased urine sodium. Consult physician or designee for significant findings.
4. Administer hypertonic sodium chloride (3% saline infusion) as prescribed. Rate of administration is usually based on serial serum sodium levels. To minimize the risk of too-rapid correction of hyponatremia, ensure that laboratory specimens are drawn on time. Serum sodium should not be allowed to increase greater than 12 mEq/L in 24 hours because of the risk of neurologic damage (osmotic demyelination), particularly if the hyponatremia is chronic rather than acute. Monitor for indications of fluid overload (e.g., crackles, elevated CVP or PCWP, edema) as appropriate. Consult physician or designee promptly for significant findings.
5. Institute seizure precautions. These include padded side rails, supplemental oxygen, bite block, and oral airway at the bedside. Side rails should remain up when a staff member is not present.
6. Restrict fluids if ordered. Explain treatment to patient and significant others. Do not keep water or ice chips at the bedside. Give IV solutions with an infusion pump.
7. Elevate HOB no greater than 20 degrees to promote venous return and thus reduce ADH release. Decreased venous return is a stimulus to the release of ADH.
8. Administer demeclocycline, conivaptan, tolvaptan, and furosemide as prescribed; carefully observe and document patient’s response.
9. Provide care calmly and gently to minimize discomfort, which increases ADH release.
![]() Fluid/Electrolyte Management; Neurologic Monitoring; Seizure Precautions
Fluid/Electrolyte Management; Neurologic Monitoring; Seizure Precautions
Thyrotoxicosis crisis (thyroid storm)
Pathophysiology
Thyroid storm is a medical emergency caused by uncontrolled hyperthyroidism. Patients occasionally present with cardiovascular collapse and shock. Hyperthyroidism or thyrotoxicosis is a condition of increased circulating thyroid hormone. The crisis results from a surge of thyroid hormones into the bloodstream, which results in profound stimulation of the sympathetic nervous system, with marked increases in body metabolism. The hypothalamus, anterior pituitary, and thyroid normally work together to balance the level of circulating thyroid hormone. Hyperthyroidism may be caused by an increased synthesis and secretion of thyroid hormones (thyroxine [T4] and triiodothyronine [T3]) from the thyroid or from increased secretion of TSH from the anterior pituitary, possibly by an increase in TRH from the hypothalamus or by autonomous thyroid hyperfunction. Symptoms of hyperthyroidism can also result from excessive release of thyroid hormone from the thyroid without increased synthesis. Such release is commonly caused by the destructive changes of various types of thyroiditis. Thyrotoxicosis crisis may also follow subtotal thyroidectomy because of manipulation of the gland during surgery. Various clinical syndromes also produce hyperthyroidism, but thyroid storm is most often associated with Graves’ disease, also known as diffuse toxic goiter. Causes of acute hyperthyroid states are listed in Box 8-3. Not all conditions listed are associated with thyroid storm.
Abnormal laboratory analysis of thyroid function provides a relatively definitive diagnosis. Initial serum measurements should include a free T4 and TSH, but concerns regarding thyroidal dysfunction should be referred to an endocrinology specialist for a more thorough and evaluative diagnostic panel. Calcium regulation may also be affected by thyroid disease if there is a problem with the level of thyrocalcitonin, a third thyroid hormone that is stimulated by increased calcium levels to help lower the calcium level. Recent diagnostic tests have isolated a long-acting thyroid stimulator, suggesting the disease is an autoimmune response. An acute decrease in thyroxine-binding globulin (inactivating or binding thyroid hormone) facilitates high levels of free and metabolically active hormone. The increased circulating levels of active thyroid hormone increase the response of beta adrenergic receptors (sympathetic stimulation increase) and also increase the system responsiveness to catecholamines.
Endocrine assessment: hyperthyroidism
History and risk factors
A patient with a history of hyperthyroidism resulting from Graves’ disease, thyroid nodules, or toxic goiter who has undergone a recent stressful experience including severe infection, trauma, major surgery, thromboembolism, DKA, or preeclampsia, pregnancy, labor, and/or delivery. Complaints include night sweats, unexplained weight loss, rapid gastric turnover with increased bowel movements or diarrhea, feelings of impending doom, rapid heart rate, and sleep disturbances. (See SIRS, Sepsis, and MODS, p. 924, Hyperglycemia, p. 711, High-Risk Obstetrics, p. 882.)
Observation
• Confused, possibly psychotic, disoriented person with CNS irritability
• Hyperreflexia and fine tremor may be present.
• Coma, heart failure, and generalized muscle weakness may be present.
• Patients with Graves’ disease (only in this form of thyrotoxicosis) have significant immune system components that may express as soft tissue swelling around the eye orbit, causing the protrusion of the eyes (exopthalmos), stare, and/or lid lag.
• Males often have gynecomastia. Many patients have fine hair and thin skin.
• Dependent lower extremity edema, alteration in appetite, change in vision, and fertility or menstrual dysfunctions are possible.
Palpation
• The thyroid gland in Graves’ disease will usually be enlarged, soft, and symmetrical, although occasionally it may be firm and irregular.
•  Older patients may not have enlarged thyroid glands, which may lead to dismissal of the diagnosis.
Older patients may not have enlarged thyroid glands, which may lead to dismissal of the diagnosis.
• Palpation is not the definitive method to evaluate thyroid dysfunction.
Screening labwork
• TSH level: Any type or cause of hyperthyroid disease (except when the cause is secondary excessive TSH production) will create a suppression of TSH. The development of TSH sensitive testing has made the diagnosis of thyroid dysfunction much simpler.
• T4 level: Unstable patients who have been diagnosed previously and/or are treated for thyroid disorders require a serum thyroxine (T4), which will be significantly more valuable in diagnosis. See Figure 8-1 for diagnostic principles.
| Blood Studies | ||
| Test | Purpose | Abnormal Findings |
| Thyroid-stimulating hormone (TSH) TSH is produced by the anterior pituitary gland in response to decreased T4 level. The hypothalamus, anterior pituitary gland and thyroid are connected in an “axis” of function. Normal/standard: 0.4 and 4.5 mIU/L |
To assess if TSH level is normal. TSH is decreased when T4 level is increased. If T4 is constantly increased, the TSH level is suppressed (low or decreased) by the high level of T4. Normally, TSH release is needed to stimulate the thyroid to produce additional T4. | When TSH is low in a patient with symptoms of thyroid storm, the diagnosis of hyperthyroidism will be considered positive until proven otherwise. If TSH is not prompting the increase in T4, the thyroid has started generating T4 abnormally. It is always beneficial to also evaluate T4 at the same time. If the TSH is low while the T4 is high, the patient is diagnosed with primary hyperthyroidism. |
| Free T4 (thyroxine) Normal: 60–170 nmol/L |
Measures the primary thyroid hormone | When levels are above normal, diagnosis of hyperthyroidism is made. If TSH and T4 are both high, the problem is in the hypothalamic- pituitary response to elevated circulating thyroid levels (secondary hyperthyroidism). |
| T3 (triiodothyronine) Normal: 0.8–2.7 nmol/L |
Measures the more metabolically active form of the thyroid hormone. | Controversial |
| Thyroid-binding globulin (TBG) | To measure the level of the protein that binds with circulating thyroid hormones. Abnormal T4 or T3 measurements are often due to binding protein abnormalities rather than abnormal thyroid function. | Total T4 or T3 must be evaluated with a measure of thyroid hormone binding such as T3 resin uptake or assay of thyroid-binding globulin. These methods are known as free T4 or free T3 even though they do not measure free hormone directly. |
| Radioactive iodine uptake | Differentiates the cause of thyroid disorder | May identify “hot” or “cold” nodules |
| Cholesterol analysis | Routine screening in biochemical profile | Hypercholesterolemia |
| Glucose | Routine screening in biochemical profile | Hyperglycemia; patients also have an impaired glucose tolerance test. |
| Electrolytes Potassium (K+) Magnesium (Mg2+) Calcium (Ca2+) Sodium (Na+) |
Assess for possible abnormalities | Frequently, abnormalities of calcium are related to parathyroid disorders that exist concurrently with thyroid dysfunction. |
| Radiology | ||
| Thyroid scan 123I (preferably) or 99mTc pertechnetate |
Assess size of heart, thoracic cage (for fractures), thoracic aorta (for aneurysm), and lungs (pneumonia, pneumothorax) Assists with differential diagnosis of chest pain |
Scan is done to help determine the cause of the hyperthyroidism. The scan may also be useful in assessing the functional status of any palpable thyroid irregularities or nodules associated with a toxic goiter. |
Collaborative management
Care priorities
• Reduce the stress response with beta adrenergic–blocking agents (e.g., esmolol, metoprolol, propranolol): Control tachycardia, manage atrial fibrillation if present, anxiety, heat intolerance, and tremor. Does not decrease the metabolic rate and has a mild effect on global oxygen consumption. DO NOT ASSUME improvement in the thyroid disorder itself, only its symptoms. Calcium channel blockers may be used if beta blockers are contraindicated.
• Oxygen: Used per nasal cannula or mask as needed to maintain SPO2 greater than 90%.
• Control fever: Antipyretic medications (i.e., acetaminophen) or hypothermia blanket is used. Aspirin is contraindicated, as it worsens thyroid crisis.
• Rehydrate using intravenous fluids: Fever and rapid metabolism can lead to dehydration.
• Correct electrolyte imbalance: Various imbalances may occur. Replace electrolytes as appropriate.
• Glucocorticoids: Administered to help inhibit conversion of thyroxine (T4) to T3 and prevent adrenal insufficiency.
• Minimize anxiety with mild tranquilizers: Also promote rest.
2. Treat the thyroid with antithyroid agents (thioamides):
• Propylthiouracil (PTU) has been the first line of treatment and is more effective in thyroid storm than methimazole. Can be used in pregnant patients. If the patient is unable to swallow, the medication is given using a nasogastric tube. Iodides (potassium iodide or Lugol’s solution) are given several hours after PTU to avoid a buildup of hormones stored in the thyroid gland.
• Methimazole may also be used. Both thioamides may cause leukopenia, rash, urticaria, fever, arthralgias, and, rarely, agranulocytosis. Methimazole may have a lower rate of these effects. Agranulocytosis (complete lack of granulocytes) causes fever and sore throat. If agranulocytosis is confirmed by complete blood count (CBC), the thioamide should be stopped.
• Iodides (SSKI): Sodium or potassium iodide drops may be given to help inhibit the release of thyroid hormone or to help abate accumulation of hormones stored in the thyroid. May stain the teeth.
4. Perform a subtotal thyroidectomy to provide a rapid, effective, curative therapy:
CARE PLANS FOR THYROID STORM
![]()
Immune Hypersensitivity Response
1. Measure and report rectal or core temperature greater than 38.3°C (101°F): often the first sign of impending thyroid storm.
2. Monitor vital signs hourly for evidence of hypotension and increasing tachycardia and fever.
3. Monitor patient for signs of congestive heart failure, which occurs as an effect of thyroid storm: jugular vein distention, crackles (rales), decreased amplitude of peripheral pulses, peripheral edema, and hypotension. Immediately report any significant findings to physician, and prepare to transfer patient to ICU if they are noted. Maternal and fetal monitoring is initiated on pregnant patients.
4. Provide a cool, calm, protected environment to minimize emotional stress if possible. Reassure patient, and explain all procedures. Limit the number of visitors.
5. Ensure good hand washing and meticulous aseptic technique for dressing changes and all procedures. Advise visitors who have contracted or been exposed to a communicable disease either not to enter patient’s room or to use appropriate infection control precautions.
6. Administer acetaminophen to decrease temperature.
7. Provide cool sponge baths or apply ice packs to patient’s axilla and groin areas to decrease fever. If high temperature continues, obtain a prescription for a hypothermia blanket.
8. Administer PTU as prescribed to prevent further synthesis and release of thyroid hormones.
9. Administer beta blockers as prescribed to block sympathetic nervous system (SNS) effects.
10. Administer IV fluids as prescribed to provide adequate hydration and prevent vascular collapse. Fluid volume deficit may occur because of increased fluid excretion by the kidneys or excessive diaphoresis. Carefully monitor I&O hourly to prevent fluid overload or inadequate fluid replacement. Decreasing output with normal specific gravity may indicate decreased cardiac output, whereas decreasing output with increased specific gravity can signal dehydration.
11. Administer iodides as prescribed, 1 hour after administering PTU.
12. Administer small doses of insulin as prescribed to control hyperglycemia. Hyperglycemia can occur as an effect of thyroid storm because of the hypermetabolic state.
13. Administer prescribed supplemental oxygen to support increased metabolism.
Impaired swallowing (or risk for same)
related to edema or laryngeal nerve damage resulting from surgical procedure
![]()
Aspiration Prevention, Swallowing Status
1. Monitor respiratory status for signs of edema (i.e., dyspnea, choking, inspiratory stridor, inability to swallow). Assess patient’s voice. Slight hoarseness is normal after surgery. Persistent hoarseness indicates laryngeal nerve damage. If bilateral nerve damage is present, upper airway obstruction can occur. Report findings to physician promptly.
2. Elevate HOB 30 to 45 degrees to minimize edema and incisional stress. Support patient’s head with flat or cervical pillows so that it is in a neutral position.
3. Keep tracheostomy set and oxygen equipment at the bedside at all times. Suction upper airway as needed, using gentle suction to avoid stimulating laryngospasm.
4. To minimize pain and anxiety and enhance patient’s ability to swallow, administer analgesics promptly and as prescribed.
![]() Management; Nutrition Therapy; Respiratory Monitoring; Swallowing Therapy
Management; Nutrition Therapy; Respiratory Monitoring; Swallowing Therapy
![]()
Anxiety Level, Anxiety Self-Control
1. Assess for anxiety; administer short-acting sedatives (e.g., lorazepam) as prescribed.
2. Provide a quiet, stress-free environment away from loud noises or excessive activity.
3. Limit number of visitors and the amount of time they spend with patient. Advise significant others to avoid discussing stressful topics and to refrain from arguing with the patient.
4. Administer beta blockers as prescribed to reduce anxiety, tachycardia, and heat intolerance.
5. Reassure patient that anxiety is related to the disease and that treatment decreases severity.
6. Inform significant others that the patient’s agitated behavior should not be taken personally.
Imbalanced nutrition: less than body requirements
related to hypermetabolic state and/or inadequate nutrient absorption
![]()
1. Provide foods high in calories, protein, carbohydrates, and vitamins.
2. Administer vitamin supplements as prescribed, and explain their importance to patient.
3. Administer prescribed antidiarrheal medications, which increase absorption of nutrients from the GI tract.
4. Weigh patient daily, and report significant losses to physician.
![]() Weight Management; Weight Gain Assistance; Nutritional Counseling
Weight Management; Weight Gain Assistance; Nutritional Counseling
related to accelerated metabolism
Within 48 hours of hospital admission, patient relates the attainment of sufficient rest and sleep.
Impaired tissue integrity of the cornea
related to dryness that can occur with exophthalmos in persons with Graves disease
Within 24 hours of admission, patient’s corneas are moist and intact.
![]()
Tissue Integrity: Skin and Mucous Membranes
1. Teach patient to wear dark glasses to protect the cornea.
2. Administer lubricating eye drops as prescribed to supplement lubrication and decrease SNS stimulation, which can cause lid retraction.
3. If appropriate, apply eye shields or tape the eyes shut at bedtime.
4. Administer thioamides as prescribed to maintain normal metabolic state and halt progression of exophthalmos.
Deficient knowledge: medications
![]()
Knowledge: Medications; Knowledge: Illness Care
1. Explain importance of taking antithyroid medications daily, as prescribed.
2. Teach indicators of hypothyroidism (e.g., early fatigue, weight gain, anorexia, constipation, menstrual irregularities, muscle cramps, lethargy, inability to concentrate, hair loss, cold intolerance, and hoarseness), which may occur from excessive antithyroid medication, and the signs and symptoms that necessitate medical attention, including cold intolerance, fatigue, lethargy, and peripheral or periorbital edema.
3. Teach side effects of thioamides and symptoms that require medical attention: appearance of a rash, fever, or pharyngitis, which can occur in the presence of agranulocytosis and require prompt medical intervention.
4. Discuss signs of worsening hyperthyroidism including high body temperature, palpitations, rapid HR, irritability, anxiety, and feelings of restlessness or panic.
5. Explain importance of continued and frequent medical follow-up.
6. Indicators that require medical attention: fever, rash, or sore throat (side effects of thioamides), and symptoms of hypothyroidism or worsening hyperthyroidism.
7. For patients receiving radioactive iodine, explain the importance of not holding children to the chest for 72 hours following therapy, because children are more susceptible to the effects of radiation. Explain that there is negligible risk for adults.
8. Stress the importance of avoiding physical and emotional stress early in the recuperative stage and maximizing coping mechanisms for dealing with stress.
Additional nursing diagnoses
See Compromised Family Coping, in Oncologic Emergencies, p. 893; Nutritional Support, p. 117; Alterations in Consciousness, p. 24; Emotional and Spiritual Support of the Patient and Significant Others, p. 200; Dysrhythmias and Conduction Disturbances, p. 492; Heart Failure, p. 421.
AACE Thyroid Task Force. American Association of Clinical Endocrinologists: Medical guidelines for clinical practice for the evaluation and treatment of hyperthyroidism and hypothyroidism(2006 amended version). Endocr Pract. 2002;8(6):457–469. http://www.aace.com/pub/pdf/guidelines/hypo_hyper.pdf.
Aghar A, Thornton E, et al. Posterior pituitary dysfunction after traumatic brain injury. J Clin -Endocrinol Metab. 2004;89:5987–5992.
Albright TN, Zimmerman MA, Selzman CH. Vasopressin in the cardiac surgery intensive care unit. Am J Crit Care. 2002;11:326–330.
Ball SG, Bayliss PH. Normal and abnormal physiology of the hypothalamus-posterior pituitary (including DI and SIADH). Chapter 2, Endotext.org downloaded. http://www.endotext.org/neuroendo/neuroendo2/neuroendoframe2.htm, June 15, 2009.
Bornstein SR. Predisposing factors for adrenal insufficiency. N Engl J Med. 2009;360:2328–2339.
Cathie K, Levin M, Faust SN. Drug use in acute meningococcal disease. Educ Pract. 2009;93:151–158.
Cohen J, Ward G, Prins J, et al. Variability of cortisol assays can confound the diagnosis of adrenal insufficiency in the critically ill population. Intens Care Med. 2006;32:1901–1905.
Daley MR, Seam N, Luboshitzky R. the CORTICUS Study Group, Corticosteroids for septic shock. N Engl J Med. 2008;358:2068–2071.
Dunser MW, Hasibeder WR. Sympathetic overstimulation during critical illness, adverse effects of adrenergic stress. J Intens Care Med. 2009;24:293–316.
Ellison DH, Berl T. The syndrome of inappropriate antidiuresis. N Engl J Med. 2007;356:2064–2072.
Eskes S, Wiersinga WM. Amiodarone and thyroid. Clin Endocr Metab. 2009;23(6):735–751.
Grozinsky-Glasberg S, Fraser A, Nahshoni E. Thyroxine-triiodothyronine combination therapy versus thyroxine monotherapy for clinical hypothyroidism: meta-analysis of randomized -controlled trials. J Clin Endocrinol Metab. 2006;91(7):2592–2599.
Hamrahain AH, Osenis TS, Arafah BM. Measurements of serum free cortisol in critically ill patients. N Engl J Med. 2004;350:1629.
Hollowell JG, Staehling NW, Flanders WD, et al. Serum TSH, T(4), and thyroid antibodies in the United States population (1988 to 1994): National Health and Nutrition Examination Survey (NHANES III). J Clin Endocrinol Metab. 2002;87(2):489–499. http://www.endotext.org/-neuroendo/neuroendo2/neuroendoframe2.htm.
Krinsley JS. Effect of an intensive glucose management protocol on the mortality of critically ill adult patients. Mayo Clin Proc. 2004;79:992–1000.
Ladenson P, Kim M. Thyroid. In: Goldman L, Ausiello DA, editors. Cecil Medicine. ed 23. Philadelphia: Saunders Elsevier; 2007:1698–1712.
Langouche L, Van den Berghe G. The dynamic neuroendocrine response to critical illness. Endocrinol Metab Clin North Am. 2006;35(4):777–791. ix
Lazar HL, McDonnell M, Chipkin SR, et al. The Society of Thoracic Surgeons practice guideline series: blood glucose management during adult cardiac surgery. Ann Thorac Surg. 2009;87(2):663–669.
LeBeau SO, Mandel SJ. Thyroid disorders during pregnancy. Endocrinol Metab Clin North Am. 2006;35(1):117–136. vii
Lee CR, Watkins M, Patterson JH, et al. Vasopressin, a new target for the treatment of heart failure. Am Heart J. 2003;146:9–18.
Marik PE. Critical illness-related corticosteroid insufficiency. Chest. 2009;135:181–193.
Marik PE, Pastores SM, Annane D, et al. Recommendations for the diagnosis and management of corticosteroid insufficiency in critically ill adult patients: consensus statements from an international task force by the American College of Critical Care Medicine. Crit Care Med. 2008;36(6):1937–1949.
Moghissi ES, Korytkowski MT, Dinardo N, et al. American Association of Clinical Endocrinologists and American Diabetes Association consensus on inpatient diabetes control. Endocr Pract. 2009;15(4):1–16.
Moliach ME. Anterior Pituitary. In: Goldman L, Ausiello DA, editors. Cecil Medicine. 23rd ed. Philadelphia, PA: Saunders Elsevier; 2007:1674–1691.
Moliach ME. Neuroendocrinology and the neuroendocrine system. In: Goldman L, Ausiello DA, editors. Cecil Medicine. ed 23. Philadelphia, PA: Saunders Elsevier; 2007:1664–1674.
Navak B, Hodak SP. Hyperthyroidism. Endocrinol Metab Clin N Am. 2007;36(3):617–656.
O’Connor KJ, Wood KE, Lord K. Intensive management of organ donors to maximize transplantation. Crit Care Nurse. 2006;26:94–100.
Osburne RC, Cook CB, Stockton L, et al. Improving hyperglycemia management in the intensive care unit preliminary report of a nurse-driven quality improvement project using a redesigned insulin infusion algorithm. Diabetes Educator. 2006;32(3):394–403.
Reid JR, Wheeler SF. Hyperthyroidism, diagnosis and treatment. Am Fam Phys. 2005;2:623–630.
Schrier RW, Gross P, Gheorghiade M, et al. Tolvaptan, a selective oral vasopressin V2-receptor -antagonist, for hyponatremia. N Engl J Med. 2006;355:2099–2112.
Sprung CL, Annane D, Keh D, et al. Hydrocortisone therapy for patients with septic shock. N Engl J Med. 2008;358:111–124.
The NICE SUGAR Study Investigators. Intensive vs conventional glucose control in critically ill patients. N Engl J Med. 2009;360(13):1283–1297.
Wartofsky L. Myxedema coma. Endocrinol Metab Clin North Am. 2006;35(4):687–698. vii-viii
Wartofsky L, Dickey RA. Controversy in clinical endocrinology, The evidence for a narrower -thyrotropin reference range is compelling. J Clin Endocrinol Metab. 2005;90:5483–5488.
Wilson M, Weinreb J, Hoo GWS. Intensive insulin therapy in critical care: a review of 12 protocols. Diab Care. 2007;30(4):1005–1011.
