[level-membership-for-endocrinology-diabetes-and-metabolism-category]5
Endocrine tumours of the pancreas
Introduction
Pancreatic neuroendocrine tumours (pNETs) are the most common endocrine tumours found in the abdomen. They may be broadly classified into two groups: functional tumours, which cause clinical syndromes due to hormone secretion; and non-functional tumours, which may cause symptoms through mass effect or metastatic spread, or may be discovered incidentally. The most common functional pNETs are gastrinomas and insulinomas, but a variety of other rare hormone secreting tumours also occur (Table 5.1). Identification of clinical syndromes and hormone production allows the classification of functional pNETs into specific types. Potentially life-threatening situations caused by hormone overproduction are a major reason to identify and resect these neoplasms. Additionally, pNETs have malignant potential. However, they are less malignant than pancreatic adenocarcinoma and surgical extirpation is beneficial in most instances.
Insulinoma
Endogenous hyperinsulinism was first described in 1927, and was the first syndrome of excessive pancreatic hormone production to be recognised.1 Hyperinsulinaemia and consequent hypoglycaemia is the major cause of morbidity and potential mortality associated with insulinoma. Insulinoma occurs in approximately one person per million population per year (Table 5.1).1 Hyperinsulinaemic hypoglycaemia associated with insulinoma is not well controlled by medical therapy, and surgery has remained the cornerstone of treatment over the past 80 years. Insulinomas are unique among pNETs in that 90% of insulinomas are benign, solitary growths that occur uniformly throughout and almost exclusively within the pancreas, without evidence of local invasion or locoregional lymph node metastases. Tumours may be as small as 6 mm in diameter and are usually less than 2 cm in size, making localisation challenging in many cases.2
Presentation
Excessive and physiologically uncontrolled secretion of insulin by the tumour causes episodes of symptomatic hypoglycaemia and leads patients to seek medical evaluation. Acute neuroglycopenia induces anxiety, dizziness, obtundation, confusion, unconsciousness, personality changes and seizures.2 Symptoms commonly occur during early morning hours, when glucose reserves are low after a period of overnight fasting during which insulin overproduction has continued. Most patients (80%) experience major weight gain as they attempt to treat these symptoms with increased caloric intake. They may first present with symptoms of hypoglycaemia when food intake is then decreased in an attempt to lose weight. A majority (60–75%) of patients are women, and many have undergone extensive psychiatric evaluation before the correct diagnosis is reached. Other patients will have been misdiagnosed with neurological conditions such as seizure disorders, cerebrovascular accidents or transient ischaemic attacks. Potentially life-threatening symptoms may be present for several years prior to diagnosis.2 In a review of 59 patients with insulinoma, the interval from onset of symptoms to time of diagnosis ranged from 1 month to 30 years, with the median time to diagnosis being 2 years.3 Because insulinoma is rare and neuroglycopenic symptoms are relatively non-specific, a high index of suspicion for insulinoma is necessary when other explanations for these symptoms are not evident. The identification of symptomatic patients and the liberal use of simple and precise biochemical tests result in accurate diagnosis of insulinoma prior to life-threatening sequelae.
Diagnosis

The diagnosis of insulinoma is established through a 72-hour supervised fast. The most definitive biochemical result for insulinoma is a plasma insulin concentration above 5 μU/mL in the presence of symptomatic hypoglycaemia.2,4 Factitious hypoglycaemia must be excluded.5
The classic diagnostic triad, proposed by Whipple in 1935 based on his observations in 32 patients, consists of symptoms of hypoglycaemia during a fast, a concomitant blood glucose concentration less than 3 mmol/L and relief of the hypoglycaemic symptoms after glucose administration.1 Factitious hypoglycaemia due to clandestine administration of exogenous insulin or oral hypoglycaemic drugs may also result in this same presentation and can lead to a misdiagnosis of insulinoma.5 Factitious hypoglycaemia classically occurs in patients associated with the medical profession or patients who have relatives with diabetes. The diagnosis of insulinoma must be reached in each patient by performing a 72-hour supervised fast with appropriate biochemical measurements. Urinary sulphonylurea concentrations (to exclude oral hypoglycaemic drugs) should be measured by gas chromatography–mass spectroscopy.
Supervised standard fasting test
The standard fasting test is carried out in a hospital setting and begins with a baseline examination in which memory, calculations and coordination are documented (Fig. 5.1). An intravenous catheter with a heparin lock is then placed, and the patient is allowed to drink only non-caloric beverages. Close observation is necessary. Blood is collected every 6 hours for measurement of serum glucose and immunoreactive insulin concentrations. As the blood glucose level falls below 3 mmol/L, blood samples are collected more frequently (every hour or less) and the patient is observed more closely. When neuroglycopenic symptoms appear, blood is collected immediately for determination of serum insulin, glucose, C-peptide and proinsulin concentrations (Fig. 5.1). Glucose is then administered and the fast is terminated. If a patient remains symptom-free for the entire 72 hours, the test is terminated and the above blood concentrations are measured.
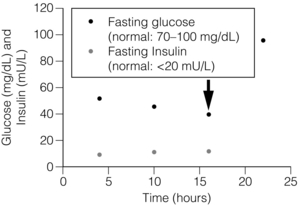
Figure 5.1 Results of supervised fast in a patient with insulinoma. The patient, a 48-year-old woman, experienced acute onset of confusion and blurred vision 16 hours into a supervised fast in the ICU (arrow). Plasma glucose was 40 mg/dL at this time and plasma insulin was 12 μU/mL. Proinsulin measured simultaneously was 87 pmol/L (normal: 3–20 pmol/L). Symptoms rapidly resolved following intravenous administration of dextrose. The most diagnostic biochemical result in a patient with insulinoma is an inappropriately increased plasma insulin concentration above 5 μU/mL at the time of documented hypoglycaemia and symptoms.
Neuroglycopenic symptoms manifest in approximately 60% of patients with insulinomas within 24 hours after fasting begins.4 Approximately 16% of patients with insulinoma develop symptoms when the blood glucose concentration is greater than 2.5 mmol/L.2 The blood glucose concentration eventually decreases below 2.5 mmol/L in approximately 85% of patients with insulinomas during the 72-hour fast (Table 5.2). The most definitive diagnostic biochemical test for insulinoma is an inappropriately increased plasma immunoreactive insulin concentration above 5 μU/mL at the time of documented hypoglycaemia and symptoms.4 The plasma insulin concentration is usually greater than 10 μU/mL.2 Although prolonged maximal stimulation of insulin secretion in normal subjects does not cause the release of the insulin precursor molecule proinsulin, some insulinomas secrete large amounts of uncleaved proinsulin. Patients with high proinsulin-producing tumours may remain euglycaemic and asymptomatic for longer periods during the fast because proinsulin is not biologically active. The proinsulin-like component (PLC) is measured at the time of symptomatic hypoglycaemia and termination of the fast. A value greater than 25% or an increased PLC: total immunoreactive insulin ratio is abnormal and consistent with the diagnosis of insulinoma.2,4 Hypersecretion of endogenous insulin also results in increases of the circulating concentration of C-peptide, a biologically inactive by-product of enzymatic insulin cleavage from the precursor proinsulin molecule. Most patients with insulinomas have C-peptide concentrations greater than 1.7 ng/mL.2
Table 5.2
Standard fasting test results and the differentiation of insulinoma from factitious hypoglycaemia
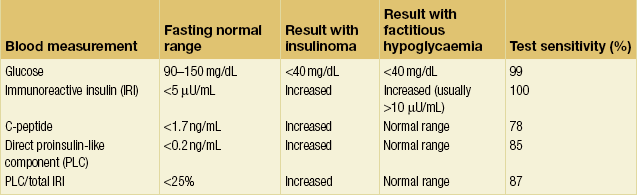
Increased serum concentrations of proinsulin or C-peptide during hypoglycaemia effectively exclude the diagnosis of factitious hypoglycaemia because exogenously administered insulin does not contain these proteins and actually suppresses their production. However, approximately 13–22% of patients with insulinoma do not have increased serum proinsulin or C-peptide concentrations, and a supervised fast prohibiting exogenous insulin administration remains the best test to diagnose insulinoma and conclusively exclude factitious hypoglycaemia. The biochemical parameters measured during the standard fasting test cannot discriminate between patients with MEN1 and patients with sporadic insulinoma.4
Nesidioblastosis
Insulinoma must be distinguished from nesidioblastosis, a condition of islet cell hyperfunction and malregulation that occurs primarily in infants and causes hyperinsulinaemic hypoglycaemia. Age at the time of presentation is the most important distinguishing factor, as nesidioblastosis occurs most commonly in children under the age of 18 months. Approximately half of infants with nesidioblastosis require a spleen-preserving near-total pancreatectomy, in which 95% of the pancreas is removed, because this disorder affects the entire pancreas diffusely.6,7
Adult nesidioblastosis has been reported and was recently described in a small series of adult patients following gastric bypass surgery for morbid obesity.8 However, nesidioblastosis in adults is exceedingly rare. In a review of over 300 patients with hyperinsulinaemic hypoglycaemia evaluated at the Mayo Clinic since 1927, only five adult patients had a reasonably confirmed diagnosis of nesidioblastosis.9 Furthermore, biochemical tests (blood glucose, insulin and C-peptide) do not reliably distinguish hyperinsulinaemic hypoglycaemia caused by an insulinoma from that attributed to nesidioblastosis, and an insulinoma may be present in a patient who has islet cell hyperplasia. Therefore, the diagnosis of nesidioblastosis in the adult must be critically suspect and should not preclude attempts to localise an insulinoma.
Management
Medical management of hypoglycaemia
Medical management aims to prevent hypoglycaemia caused by hyperinsulinism so that symptoms and life-threatening sequelae are avoided. In patients with acute hypoglycaemia, blood glucose concentrations are normalised initially with an intravenous dextrose infusion. To prevent hypoglycaemic episodes during diagnosis, tumour localisation and the preoperative period, euglycaemia is maintained by giving frequent feeds of a high-carbohydrate diet, including a night feed. For patients who continue to become hypoglycaemic between feedings, diazoxide may be added to the treatment regimen at a dose of 400–600 mg orally each day. Diazoxide inhibits insulin release in approximately 50% of patients with insulinoma; however, side-effects of oedema, weight gain and hirsutism occur in 50% of patients, and nausea occurs in over 10%.9 Diazoxide should be discontinued 1 week prior to surgery as it may contribute to intraoperative hypotension. Calcium-channel blockers or phenytoin may also suppress insulin production in some patients. Long-term control of hypoglycaemic symptoms with medical management has generally been ineffective for patients with insulinoma. The surgeon must know the patient’s response to medical management in order to gauge the urgency of surgical intervention.
Octreotide, a synthetic, long-acting analogue of the naturally occurring hormone somatostatin, may be useful for treating symptoms caused by vasoactive intestinal peptide (VIP)-omas and carcinoid tumours but is not generally recommended for insulinomas because its efficacy in inhibiting insulin release is unpredictable.10 The usefulness of radiolabelled octreotide in imaging insulinomas has been equally disappointing. Therefore, long-term medical management of hypoglycaemia in patients with insulinomas generally is reserved for the few patients (< 5%) with unlocalised, unresected tumours after thorough preoperative testing and exploratory laparotomy, and for patients with metastatic, unresectable malignant insulinoma. Patients with malignant insulinomas and refractory hypoglycaemia may even require the placement of implantable glucose pumps for continuous glucose infusion.9
Preoperative tumour localisation

Virtually all insulinomas may be localised by the experienced surgeon through the combination of preoperative modalities and intraoperative ultrasound.11 Blind pancreatic resection is not indicated.
Non-invasive imaging studies: An initial attempt should be made to localise the tumour and identify metastatic disease by using non-invasive tests. Computed tomography (CT) and magnetic resonance imaging (MRI) are both capable of identifying pancreatic tumours as small as 1 cm in diameter (Table 5.3). If CT is elected, a pancreatic protocol study with biphasic contrast injection and fine cuts through the pancreas should be used. Tumours will appear hypervascular on arterial phase images (Fig. 5.2a). The sensitivity of CT for insulinoma is 40–80% in recent series.12,13 MRI may image an islet cell tumour based on increased signal intensity (brightness) on T2-weighted images (Fig. 5.2b). The sensitivity of MRI is similar to that of CT and, as expected, the accuracy of both CT and MRI increases with larger tumour size.14
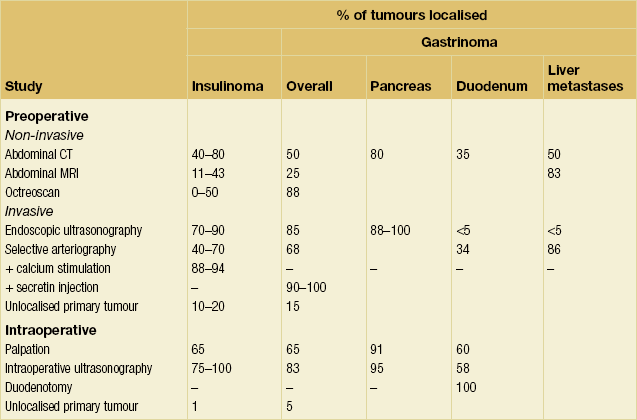
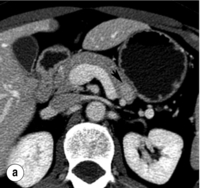
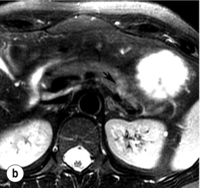
Figure 5.2 A 1.3 × 1.3 cm insulinoma at the junction of the body and tail of pancreas as visualised on preoperative and intraoperative imaging (tumour is identified by arrows). (a) Pancreatic protocol CT demonstrates the hypervascular tumour following administration of intravenous contrast. (b) On T2-weighted MRI images the tumour has increased signal intensity (brightness). (c) Intraoperative ultrasound demonstrates the insulinoma, which is sonolucent compared with the more echodense pancreas.
Invasive localising procedures: Up to 50% of insulinomas are too small to be detected by non-invasive imaging. A variety of more sensitive invasive tests are used to localise these tumours preoperatively. Endoscopic ultrasound (EUS) is safe and highly effective in experienced hands and is the modality of choice for patients whose insulinoma is not localised by CT.15 It has been demonstrated to detect tumours as small as 2–3 mm, well below the limits of detection of CT or MRI,16,17 and it is the most sensitive modality for detection of intrapancreatic tumours.
To perform EUS of the pancreas, the endoscope is passed into the duodenum and a saline-filled balloon is inflated against the intestinal wall. A 5- to 10-MHz transducer is used to generate an image of the pancreas through the intestinal and stomach walls. Tumours as small as 2–3 mm in diameter can be identified at the pancreatic head by moving the transducer through the duodenum at the junction with the pancreas. The endoscope must be passed well into the third portion of the duodenum to adequately visualise the uncinate process. Insulinomas in the pancreatic body and tail are imaged by positioning the transducer in the stomach and scanning through its posterior wall. Insulinomas will appear homogeneously hypoechoic, well circumscribed and round, and are typically easily distinguishable from the surrounding pancreatic parenchyma. Sensitivity of EUS for insulinoma ranges from 70% to 94%.18,19 Rates of detection are highest in the head of the pancreas (83–100%) because the head can be viewed from three angles (from the third portion of the duodenum, through the bulb of the duodenum and through the stomach). They are lower (37–60%) in the body and tail, which can only be viewed through the stomach.20
Despite the tremendous potential and proven benefit of EUS for insulinoma, there are some limitations. First, there may be false positives, which include accessory spleens and intrapancreatic lymph nodes. Further, EUS is limited in assessment of malignancy, identification of pedunculated tumours, and differentiation of large tumours from the pancreatic parenchyma.21
In patients with negative results after non-invasive imaging studies and EUS, calcium arteriography may be useful (Fig. 5.3). This study relies on the functional activity of the insulinoma (i.e. excessive insulin production) and not on the ability to image the tumour (i.e. tumour size). Arteries that perfuse the pancreatic head (gastroduodenal artery and superior mesenteric artery) and the body/tail (splenic artery) are selectively catheterised, and a small amount of calcium gluconate (0.025 mEq Ca2 +/kg body weight) is injected into each artery during sequential runs. A catheter positioned in the right hepatic vein is then used to collect blood for measurement of insulin concentrations. Calcium stimulates a marked increase in insulin secretion from the insulinoma. A greater than twofold increase in the hepatic vein insulin concentration indicates localisation of the tumour to the area of the pancreas being perfused by the injected artery (Fig. 5.3). In this way, calcium provocation may identify the region of the pancreas containing the tumour (head, body or tail). Additionally, injection of contrast may reveal a tumour blush, confirming the location of the insulinoma by imaging the tumour. These combined features are particularly useful in identifying the insulinoma in patients with MEN1 who may have multiple imaged pNETs. The reported sensitivity of calcium stimulation is between 88% and 94% and few false-positive results occur (Table 5.3).22
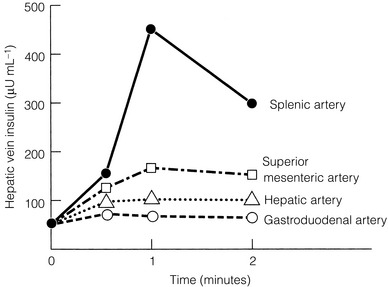
Figure 5.3 Calcium angiogram in a patient with an insulinoma localised to the pancreatic tail. Intra-arterial calcium was selectively injected into splenic, superior mesenteric, hepatic and gastroduodenal arteries in four different runs. Blood samples were taken serially from hepatic vein and insulin concentrations measured at 0, 1 and 2 minutes before and after calcium injection. After injection into the splenic artery there was a rapid marked increase in hepatic vein insulin concentrations at 1 and 2 minutes. This finding localised the insulinoma to the pancreatic tail.
A small proportion of insulinomas will remain unlocalised even after all preoperative studies are obtained. When the diagnosis is certain on the basis of the results of the supervised fast, surgical exploration with careful inspection, palpation and intraoperative ultrasonography (IOUS) of the pancreas is still indicated. Most of these patients (> 90%) will still have an insulinoma identified and removed by the experienced surgeon.1,23 Retrospective reviews have shown that the combination of careful surgical exploration with IOUS will identify almost all insulinomas.20,24
Operative management
Open exploration: In contradistinction to gastrinomas, virtually all insulinomas are located within the pancreas and are uniformly distributed throughout the entire gland.2 Therefore, in the patient with unlocalised insulinoma, the head, body and tail of the pancreas should be sufficiently mobilised to permit evaluation of the entire organ. This requires an extended Kocher manoeuvre, to adequately lift the head of the pancreas out of the retroperitoneum, and division of attachments at the inferior and posterior border of the pancreas, to permit evaluation of the posterior body and tail (Fig. 5.4). The entire pancreatic surface should then be inspected, as an insulinoma may appear as a brownish-red purple mass. Because the head of the pancreas is thick, small tumours that are centrally located may not be easily palpated. The entire pancreatic head must be sufficiently mobilised so that the posterior surface can be adequately examined visually and palpated between the thumb and forefinger. The splenic ligaments may be divided to completely mobilise the spleen out of the retroperitoneum for full examination and palpation of the pancreatic tail.
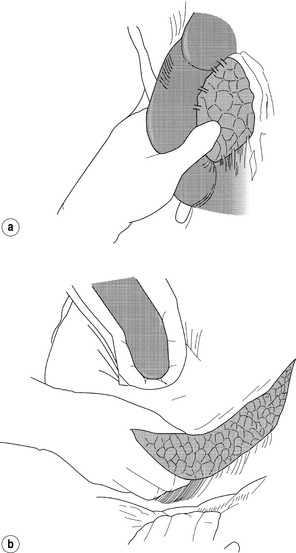
Figure 5.4 Intraoperative manoeuvres to identify insulinoma. (a) Kocher manoeuvre with careful palpation of head of pancreas. (b) Opening gastrocolic ligament, superior retraction of stomach, inferior retraction of transverse colon, and careful palpation of body and tail of pancreas after incision along inferior border.
IOUS is the best intraoperative method to idenitify insulinomas. It is performed by placing the transducer on the surface of the pancreas, which is covered in a pool of saline to maximise image quality. A 10- or 7.5-MHz real-time probe is used, which has a short focal length and high resolution. An insulinoma appears as a sonolucent mass with margins distinct from the uniform, more echodense pancreatic parenchyma (Fig. 5.2c). IOUS can localise an occult insulinoma that has not been identified preoperatively and can identify tumours that are not visible or palpable.25 It is particularly helpful in evaluation of the pancreatic head.2,23 The sensitivity for detecting insulinomas using IOUS is greater than 75%12,24 and approaches 100%2,26 (Table 5.3). A study of 37 consecutive patients showed that IOUS correctly identified 35 (95%). The two tumours that were missed were in the pancreatic tail.11
Resection of insulinoma: Enucleation of the tumour with removal of minmal adjacent pancreatic tissue is the operation of choice for benign insulinomas. However, tumour size, location and surrounding anatomy may dictate that formal pancreatic resection is necessary. It is important to consider the relationship of the tumour to the pancreatic duct by imaging both structures with IOUS prior to tumour excision. Small tumours that are separated from the pancreatic duct and major vessels by normal pancreas can be safely enucleated. If a clear margin of normal pancreatic tissue does not exist between the insulinoma and other structures, then a spleen-preserving distal or subtotal pancreatectomy is advised.11,25 Evidence of malignancy, such as involvement of peripancreatic lymph nodes or tumour invasion, also mandates pancreatic resection. Rarely, pancreatico-duodenectomy is indicated if enucleation cannot be performed safely. Frozen-section confirmation of neuroendocrine tumour is traditionally used as the end-point of surgery.
Insulinoma and MEN1
Approximately 10% of insulinomas occur in the setting of MEN1, and 20% of patients with MEN1 develop insulinomas. Insulinomas in MEN1 may be multiple and may occur simultaneously and diffusely throughout the pancreas. In addition, patients with MEN1 may have multiple non-functioning pNETs. The goal of treatment in this scenario is to ameliorate the hypoglycaemia by eliminating the source of insulin hypersecretion. Difficulty may arise in identifying which tumour produces the excessive insulin. Calcium angiogram is useful to determine if the dominant imaged tumour is responsible for the excessive secretion of insulin. If the insulinoma(s) arises within the body or tail of the pancreas, then a subtotal or distal pancreatectomy is indicated because multiple other islet cell tumours are virtually always present. A tumour that arises in the head of the pancreas is enucleated, if possible, or alternatively resected by pancreatico-duodenectomy. As in the case of sporadic insulinoma, medical management for this condition is reserved for those occasional patients who have failed surgical therapy, those who are poor surgical candidates, or those in whom a single source of hyperinsulinism cannot be found.27
Laparoscopic surgery
Laparoscopic surgery offers many advantages to patients, including shorter hospital stay, reduced pain, smaller incisions and faster recovery. Because insulinomas are typically small, benign and limited to the pancreas, laparoscopic surgery can offer an ideal way to treat these patients. Both enucleation and subtotal pancreatectomy, with or without splenectomy, may be performed laparoscopically.28,29 Good candidates for a minimally invasive approach have benign-appearing tumours that are well localised on preoperative studies. Some recommend that candidates have tumour confined to the body and tail of the pancreas; however, it is also possible to laparoscopically enucleate an insulinoma in the pancreatic head. Even after successful preoperative localisation, tumour location should be confirmed intraoperatively through the use of laparoscopic IOUS. Because palpation is not possible during laparoscopic exploration, the availability of laparoscopic IOUS and an experienced operator is essential. Several published series indicate that laparoscopic pancreatic resection is safe in experienced hands, although operation times are typically longer than for an open procedure.30,31 Complications include pancreatic fistula, and some procedures will require conversion to open surgery.
Outcome
Most patients with insulinoma are cured of hypoglycaemia and return to a fully functional lifestyle with normal long-term survival (Table 5.4). Appropriate localisation of sporadic insulinoma and complete surgical resection results in a cure rate of greater than 95%.2,11 Symptoms resolve postoperatively and the fasting serum concentration of glucose normalises. Although successful resection of the tumour(s) responsible for hyperinsulinism renders most MEN1 patients asymptomatic postoperatively,32 persistent or recurrent hypoglycaemia due to a missed insulinoma or metastatic disease from the original tumour may develop.
Gastrinoma
Each year, approximately 0.1–3 people per million population develop gastrinoma, the second most common functional pNET (Table 5.1).33 The clinical features of this tumour were first described by Zollinger and Ellison in 1955.34 Because of an increased awareness of Zollinger–Ellison syndrome (ZES) and the widespread availability of accurate immunoassays to measure serum concentrations of gastrin, gastrinoma is increasingly diagnosed and treated at an early stage of disease. However, the mean time from symptoms to diagnosis is still as long as 8 years, so improvements in detection are needed.
Patient presentation
Gastrinomas secrete excessive amounts of the hormone gastrin, causing acid hypersecretion, which results in epigastric pain, diarrhoea and oesophagitis. The most common presenting symptoms are those of peptic ulcer disease. Diarrhoea, caused by gastrin-induced hypersecretion and increased bowel motility, is the second most common symptom and may be the only manifestation of ZES in 20% of patients. Oesophagitis with or without stricture occurs with more severe forms of the syndrome. Approximately 20% of patients with ZES will have it as part of MEN1,1 and this syndrome must always be excluded. A significant family history of ulcers, peptic ulceration occurring at a young age, and peptic ulcers in association with hyperparathyroidism or pituitary tumour may all suggest MEN1.
Patients with ZES most frequently present with a solitary ulcer in the proximal duodenum, similar to patients with peptic ulcer disease unrelated to gastrinoma. Therefore, ‘typical’ pattern ulceration should not exclude the diagnosis of ZES. All patients with peptic ulcer disease severe enough to require surgery should be screened preoperatively for gastrinoma by obtaining a fasting serum gastrin concentration. Recurrent ulceration after appropriate medical treatment, or peptic ulceration in multiple locations or unusual locations such as distal duodenum or jejunum, should also raise suspicion of ZES. In addition, patients with peptic ulcer disease in the presence of persistent diarrhoea or in the absence of Helicobacter pylori should be investigated.35
Diagnosis
The evaluation of possible ZES begins by obtaining a fasting serum gastrin concentration (Fig. 5.5). Hypergastrinaemia occurs in almost all patients with ZES and is defined as a serum gastrin concentration > 100 pg/mL.36 Therefore, a normal fasting serum gastrin concentration effectively excludes ZES. Antacid medications like proton-pump inhibitors (PPIs) or H2-receptor antagonists may cause a false-positive increase in serum gastrin concentration and should be withheld for at least 1 week before measurement of the serum gastrin concentration.
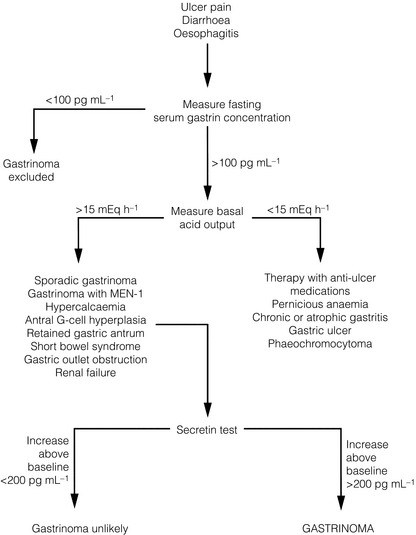
Figure 5.5 Flow diagram for diagnosis and evaluation of patients with suspected Zollinger–Ellison syndrome.
Achlorhydria is a common cause of hypergastrinaemia, and gastric acid secretion is measured to exclude this condition (Fig. 5.5). A basal acid output (BAO) greater than 15 mEq/h (>5 mEq/h in patients who have undergone previous acid-reducing operations) is abnormal and occurs in 98% of patients with ZES. Measurement of gastric pH is a simpler but less accurate indicator of gastric acid hypersecretion. A gastric pH > 3 essentially excludes ZES, whereas a pH ≤ 2 is consistent with ZES.
A markedly increased fasting serum gastrin concentration (> 1000 pg/mL) in the presence of an elevated BAO (> 15 mEq/L) is diagnostic of ZES. However, many patients with ZES have gastric acid hypersecretion and moderately increased fasting serum gastrin concentrations (100–1000 pg/mL). For these patients, the secretin stimulation test is the provocative test of choice.1 The secretin test is carried out after an overnight fast. Secretin, 2 U/kg i.v. injection, is administered, and blood samples are collected immediately before and at 2, 5, 10 and 15 minutes after the injection. A 200 pg/mL increase of gastrin concentration above baseline is diagnostic of ZES. The test sensitivity is not 100%, and approximately 15% of patients with gastrinoma may have a negative secretin test.
Management
Medical control of gastric acid hypersecretion
The management of patients with gastrinoma consists of two phases: control of the symptoms associated with acid hypersecretion and removal of the tumour, which is potentially malignant and life-threatening. The development of H2-receptor antagonists and PPIs has made medical control of gastric acid hypersecretion possible in all patients. Patients with ZES typically require two to five times the usual dose of anti-ulcer medications to keep the BAO < 15 mEq/h. Omeprazole at 20–40 mg p.o. twice a day will usually control acid hypersecretion. Patients who have reflux oesophagitis or who have had prior operations to reduce acid secretion, such as subtotal gastrectomy, should have the acid output maintained at < 5 mEq/h. The intravenous PPI pantoprazole should be administered at the same dose throughout the perioperative period. Once acid hypersecretion is controlled, epigastric discomfort resolves and ulcers heal in virtually all patients.37,38 Because of the availability of effective medical treatment for peptic ulcer disease, total gastrectomy is no longer indicated in patients with gastrinoma.
As medical control of gastric acid hypersecretion has led to fewer ulcer-related complications, there is increased concern about the malignant potential of the gastrinoma. The most important determinant of long-term survival in patients with ZES is the growth of the primary tumour and its metastatic spread. Development of liver metastases is associated with subsequent death from tumour, and surgical resection of the primary gastrinoma can reduce the incidence of liver metastases. Hepatic metastases developed in only 3% of patients with gastrinoma treated by surgical excision of the primary compared with 23% managed without surgery.39 Therefore, the current goal of surgery for ZES has shifted away from controlling gastric acid hypersecretion to aggressive resection of the primary tumour as well as localised metastatic disease. Surgical intervention can also normalise gastrin levels and lessen the requirement for long-term medical therapy. Normalisation of gastrin levels may be an important additional benefit since long-term hypergastrinaemia had been associated with the development of gastric carcinoid tumours.40
The natural history of long-standing ZES in patients in whom the excessive acid secretion is controlled is largely unclear, primarily because effective medical therapy is relatively new. A longitudinal study of 212 patients with ZES and well-controlled acid secretion showed that none of these patients died of acid-related complications.41 Pancreatic (versus duodenal) location of tumour and a tumour diameter > 3 cm were found to be associated with an increased risk of death from gastrinoma, and higher serum gastrin concentrations correlated with more aggressive disease. Liver metastases had a negative impact on survival, as did the development of bone metastases or ectopic Cushing’s syndrome. These results lend further support to early surgical intervention in gastrinoma as well as aggressive surgical resection of limited hepatic metastases.
Preoperative tumour localisation

Somatostatin receptor scintigraphy is the first-line preoperative study of choice for localisation of gastrinoma.42,43
In contradistinction to insulinoma, gastrinoma is malignant in 60–90% of patients.44 Duodenal gastrinomas as small as 2 mm in diameter may have associated regional lymph node metastases.45 At the time of diagnosis, approximately 25–40% of patients have liver metastases.46 Imaging studies must carefully assess the liver, and all patients with ZES should undergo preoperative testing to localise the tumour and to define the extent of disease so that appropriate surgical treatment can be undertaken.
Non-invasive tumour-localising studies: Initial tumour localisation studies should be non-invasive and should adequately assess the liver for metastases. Primary gastrinomas that arise within the pancreas are identified much more readily than those in the duodenum or other extrapancreatic locations (80% vs. 35%). Abdominal CT detects approximately 50% of gastrinomas overall (Fig. 5.6), but sensitivity depends greatly on tumour size, tumour location and the presence of metastases.47 Gastrinomas > 3 cm in diameter are reliably detected by CT, whereas tumours < 1 cm in diameter are rarely detected. Abdominal MRI has a low sensitivity (25%) in localising primary gastrinomas but is excellent for detection of hepatic metastases (Table 5.3). Gastrinoma metastases in the liver appear bright on dynamic T2-weighted MRI images and show a distinct ring with gadolinium enhancement. MRI is especially useful to differentiate gastrinoma metastases within the liver from haemangiomas.
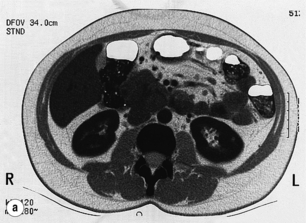
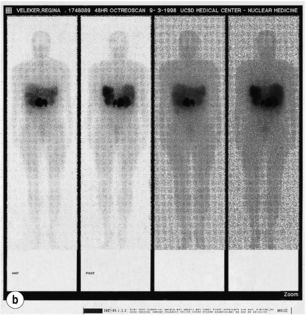
Figure 5.6 CT scan (a) and somatostatin receptor scintigraphy (b) preoperatively identified large gastrinomas in this patient.
The use of somatostatin receptor scintigraphy (SRS) has significantly improved the preoperative localisation of gastrinomas.48 SRS images gastrinomas on the basis of the density of somatostatin type 2 receptors (Fig. 5.6). Because a high proportion of gastrinomas have type 2 receptors, approximately 80% of primary tumours can be identified and the true extent of metastatic disease is delineated more accurately than by CT or MRI.48 SRS is now the non-invasive imaging modality of choice for gastrinomas.
Several prospective studies have evaluated the utility of SRS compared with conventional imaging. In a prospective study of 35 patients, SRS had a greater sensitivity than all other modalities combined (angiography, MRI, CT, ultrasonography).42 The rate of detection correlated closely with tumour size: 30% of gastrinomas < 1.1 cm in diameter and 96% of those > 2 cm were detected. A positive SRS study strongly predicts the presence of tumour, but the variable negative predictive value (33–100%) cautions against excluding a tumour on the basis of a negative study.49 Another prospective study of 146 patients with gastrinoma found a sensitivity of 71%, specificity of 86%, positive predictive value of 85% and a negative predictive value of 52%. These 146 patients underwent 480 SRS studies, with a false-positive localisation rate of 12%. Extra-abdominal false-positive localisation studies were more common than intra-abdominal false-positive scans and were attributed to thyroid, breast or granulomatous lung disease.50 The most common causes of false-positive intra-abdominal SRS scans were accessory spleens, localisation to prior operative sites and renal parapelvic cysts. Only 2.7% of these false-positive studies actually altered management, suggesting the importance of a high awareness of other potential causes for a positive SRS scan.
Invasive tumour-localising modalities: Although non-invasive imaging studies are important to exclude unresectable metastatic disease, these studies may fail to image the primary gastrinoma. Invasive modalities may be useful to localise the primary tumour prior to surgery. As discussed previously, EUS is highly sensitive for detecting intra-pNETs. It has a reported sensitivity of 75–94% for identification of gastrinomas located within the pancreas.19 It has also been reported to precisely identify the location of small lymph node metastases in patients with gastrinoma. Sensitivity for duodenal wall gastrinoma has been disappointing, however (11–50%).51 This represents a significant limitation of EUS, as the majority of gastrinomas are located within the duodenum.
Surgery for tumour eradication
Operative approach: The surgeon should be prepared for hepatic resection if unsuspected liver metastases are identified intraoperatively. An upper abdominal incision that provides adequate exposure for exploration of the entire pancreas, regional lymph nodes and liver is necessary. The abdomen is initially inspected for metastases, with particular attention to possible ectopic sites of tumour such as the ovaries, jejunum and omentum. The entire surface of the liver is then palpated for metastatic lesions. Metastases typically appear tan in colour and feel firm. Deep hepatic metastases may be identified by using IOUS with a 5-MHz transducer. All suspicious hepatic lesions must be either excised or biopsied to exclude malignant gastrinoma. In general, liver metastases that are not identified preoperatively by abdominal MRI or SRS are small and potentially resectable at the time of operation. Similarly, hilar, coeliac and peripancreatic regional lymph nodes are carefully sampled for metastatic disease.
Intraoperative manoeuvres to find the primary gastrinoma:

Duodenotomy has been demonstrated to improve cure rates in surgery for gastrinoma52 and should be performed during every exploration for Zollinger–Ellison syndrome.
Successful intraoperative localisation and resection of tumours may be extremely challenging because gastrinomas only 2 mm in diameter may reside in the wall of the duodenum. There is also a high rate of associated lymph node metastases and even the posibility of primary gastrinomas arising within lymph nodes.53,54 The initial finding of a single involved lymph node may therefore represent a primary tumour or metastatic disease from a very small, unlocalised primary tumour. Preoperative studies, such as SRS, accurately localise the primary gastrinoma and metastases and greatly facilitate the operative management, allowing a surgical approach directed to the area containing the tumour. Intraoperative localisation is still necessary because 20–40% of patients in whom the tumour is not apparent with preoperative studies will still have tumour identified at surgery.
Successful intraoperative tumour identification requires knowledge of where primary gastrinomas arise. The so-called ‘gastrinoma triangle’, bounded by the neck and body of the pancreas medially, the junction of the cystic and common bile ducts superiorly, and the second and third portions of the duodenum inferiorly, contains more than 80% of primary gastrinomas.55 Most gastrinomas arise within the duodenum. The head of the pancreas and duodenum are first exposed by mobilising the hepatic flexure of the colon out of the upper abdomen and dividing the gastrocolic ligament to open the lesser sac. A Kocher manoeuvre is performed to lift the head of the pancreas out of the retroperitoneum. The entire pancreatic surface is carefully examined visually and palpated between the thumb and forefinger. IOUS is very useful for localising intrapancreatic gastrinomas (Table 5.3). The body and tail of the pancreas may be mobilised and similarly examined after dividing the inferior and posterior pancreatic attachments to find the few gastrinomas that may arise in the distal pancreas.
It is increasingly recognised that a high percentage of gastrinomas are located in the duodenal wall.55–58 IOUS is poor at detecting duodenal gastrinomas (Table 5.3), and the surgeon must rely on inspection, palpation and duodenotomy to find these tumours. These gastrinomas are usually very small (< 6 mm) and are difficult to palpate. They are concentrated more proximally in the duodenum, decreasing in density as one moves distally (Fig. 5.7). Duodenotomy is necessary to allow direct inspection of the duodenal mucosa. A review of 143 patients with sporadic ZES who underwent surgical exploration revealed a significantly higher cure rate following duodenotomy, both immediately and in the long term. Duodenotomy was particularly important in the detection of small duodenal tumours, allowing localisation of 90% of tumours < 1 cm versus only 50% discovered on preoperative imaging. Gastrinomas will ultimately be found by an experienced surgeon in > 95% of patients.52,55,59,60 Further, duodenotomy is associated with a higher rate of finding gastrinoma and a higher rate of cure.52
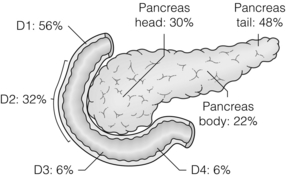
Figure 5.7 Distribution of gastrinomas throughout the duodenum and pancreas. Duodenal gastrinomas are concentrated in the proximal duodenum and become progressively less frequent in the distal duodenum. Pancreatic gastrinomas are most commonly found in the tail of the pancreas.
Approximately 5–24% of gastrinomas are found in extrapancreatic, extraintestinal lymph nodes only, with no identifiable primary pancreatic or duodenal tumour.53,61–65 Whether these represent lymph node primary tumours or metastases from occult pancreatic or intestinal primary tumours is controversial. In one series of patients who underwent exploration for gastrinoma, 10% of patients (13/138) with sporadic ZES met the criteria for lymph node primary gastrinoma in that they achieved long-term cure after resection of a lymph node only. These patients were followed for a mean of 10 years, suggesting that these tumours represent true lymph node primary gastrinoma.53 The discovery that neuroendocrine cells may be found within abdominal lymph nodes offers a possible explanation for the origin of these tumours.54
A prospective study found eight patients to have extrapancreatic, extraduodenal, extralymphatic primary gastrinomas. These tumours were located in the liver (three patients), common bile duct (one), jejunum (one), omentum (one), pylorus (one) and ovary (one).66 A gastrinoma has been reported in the interventricular septum of the heart.67 It is reasonable to conclude the surgical exploration for gastrinoma if careful exploration with duodenotomy does not reveal a primary tumour, all involved lymph nodes have been resected, and other potential ectopic sites have been carefully examined.
Tumour resection: As described for insulinoma, tumour enucleation remains the preferred approach for sporadic gastrinomas. Tumours that arise within the pancreas and that are not near the pancreatic duct or major vessels are safely enucleated. Large pancreatic tumours with vital structures in close proximity must be removed by pancreatic resection. Duodenal gastrinomas may be precisely resected following duodenotomy to localise the tumour. Some normal duodenal wall around the tumour is removed, but as much of the duodenal wall as possible is preserved to allow a non-constricting closure. Special attention is paid to avoid damaging the ampulla of Vater. Regional lymph nodes should be systematically sampled, as lymph node metastases may be inapparent at exploration and will be found in 55% of patients with duodenal tumours. Although most gastrinomas are malignant, performing a more radical pancreatic resection (e.g. pancreatico-duodenectomy) is currently not indicated. Small tumours can be easily enucleated and because of the slow progression of disease, symptomatic relief with medical treatment is easily achieved. However, pancreatico-duodenectomy (Whipple procedure) can be performed with acceptable morbidity and mortality and may be indicated for patients with larger, locally aggressive tumours. Further, the morbidity and mortality of the Whipple procedure are decreasing, making this a potentially acceptable operative procedure for locally advanced gastrinomas and other islet cell tumours.68
The presence of lymph node metastases at the time of operation should not discourage an aggressive surgical approach to remove all gross tumour. Gastrinoma is associated with lymph node involvement in 50–80% of patients and, unlike many other types of cancer, lymph node involvement alone without hepatic or distant metastases does not appear to decrease survival.69,70 Resection of all apparent tumour to eradicate disease increases disease-free survival and may extend overall survival. The development of hepatic and distant metastatic disease occurs in 25–90% of patients and is the most common cause of morbidity and mortality associated with tumour.69,71 Patients should be carefully followed by screening for recurrent disease because if a patient develops an increased serum gastrin concentration in conjunction with a tumour that has been imaged, re-operation should be considered. Approximately one-third of patients with a recurrence can be rendered free of disease.72
Gastrinoma and MEN1
Parathyroidectomy should be performed first in patients with MEN1 who have hyperparathyroidism and ZES, because normalisation of the serum calcium concentration usually results in a marked decrease in serum gastrin, allowing better medical control of the symptoms of ZES.73 Whether patients should undergo abdominal exploration is controversial. Earlier surgical series suggest that resecting gastrinoma does not cure these patients of ZES. Some studies suggest that aggressive surgical approaches may result in normalisation of serum gastrin concentrations in MEN1 patients. However, prospective studies with strict criteria for cure indicate that few, if any, patients with ZES and MEN1 are cured. Preoperative abdominal CT is necessary to identify hepatic metastases and plan surgical resection. SRS is also useful in determining the true extent of disease. In patients with MEN1, 70% of gastrinomas are found within the duodenum, and approximately 50% of patients may have multiple duodenal tumours.74 We advocate performing routine duodenotomy and peripancreatic lymph node sampling during exploration for gastrinoma in a patient with MEN1, as well as enucleation of palpable tumours in the pancreas.
The appropriate extent of surgical resection in patients with ZES and MEN1 is controversial, with only a few studies having enough patients to allow for analysis of surgical outcome. Thompson and colleagues argue for an aggressive surgical approach.75 In a series of 34 patients with ZES and MEN1 who had undergone surgery, 68% remained eugastrinaemic, with a 15-year survival rate of 94%. He recommends performing a distal pancreatectomy (because of the concomitant neuroendocrine tumours in the neck, body and tail of the pancreas in these patients), enucleation of any tumours in the pancreatic head or uncinate process, duodenotomy and exision of any tumours from the first to fourth portions of the duodenum, and a peripancreatic lymph node dissection.75,76 We, however, found that patients with MEN1 and ZES rarely became free of disease despite extensive duodenal exploration, with only 16% of patients free of disease immediately after surgery and only 6% at 5 years. This is in contrast to a surgical cure of approximately 40% of patients with sporadic gastrinomas.60 We therefore recommend surgical exploration only for those patients with ZES/MEN1 and an imageable tumour > 2 cm. The indication in this case is to prevent metastatic spread of tumour to the liver, not to cure ZES.
Outcome

In patients with sporadic ZES, an immediate postoperative cure rate of 60% can be obtained if all identifiable tumour is resected, and approximately 40% of these patients remain free of disease at 5-year follow-up.60 With regard to tumour-related mortality, surgery to remove gastrinoma has been shown to improve survival in patients with sporadic ZES and patients with MEN1/ZES who have tumour > 2.5 cm. Specifically, surgery for gastrinoma has been demonstrated to result in a lower rate of liver metastasis (5% vs. 29%, P = 0.0002) and lower rate of disease-related death (1% vs. 23%, P < 0.00001), translating into a statistically significant 15-year survival difference of 93% vs. 73% (P = 0.0002).77 These data were collected prospectively from groups with equivalent patient and tumour characteristics, who differed only in whether or not they underwent surgery. Patients who have liver metastases at time of presentation have an overall survival of only 20–38%.69 Therefore, surgical exploration can be recommended for all patients with sporadic ZES, and all patients with MEN1/ZES who have a tumour larger than 2.5 cm.
Non-functional pNETs
pNETs that do not cause a syndrome of hormone excess were previously estimated to occur with an incidence of 1–2 cases per million population per year.78 However, this incidence has grown in recent years, as tumours of this type are discovered with increasing frequency on cross-sectional imaging obtained for another purpose. Although commonly classified as non-functional pNETs (NFpNETs), these tumours may in fact produce multiple hormones and peptides, including neurotensin, pancreatic polypeptide, chromogranin A and neuron-specific enolase.78 On immunohistochemistry they may stain for insulin, gastrin or somatostatin, although serum levels of the hormones are not elevated and associated clinical syndromes are absent. Plasma levels of chromogranin A are elevated in 60–100% of patients with NFpNETs and may be useful as a tumour marker to follow disease progression, relapse and response to therapy.79,80
Symptomatic NFpNETs are typically larger at presentation than functional pNETs, as they present with symptoms of mass effect, including abdominal pain, jaundice and obstruction. Greater than 60% of patients will have liver metastases at the time of diagnosis. Because aggressive surgical management including resection of localised liver metastases is indicated for NFpNETs, it is important to distinguish these tumours from pancreatic adenocarcinoma. SRS and biopsy are useful in this regard.81
The management of smaller, incidentally discovered NFpNETs is the subject of controversy. Some authors have advocated observation of small lesions. However, there is increasing evidence that even small NFpNETs may have aggressive behaviour and may give rise to lymph node and liver metastases.82 Furthermore, histological appearance of the tumour (well differentiated vs. poorly differentiated) appears to correlate poorly with prognosis. For this reason we advocate at least enucleation of these lesions and potentially formal resection to facilitate evaluation of lymph nodes.
Other rare endocrine tumours of the pancreas
Other pNETs include VIPoma, glucagonoma, somatostatinoma, growth hormone-releasing factor (GRF)-oma, adrenocorticotropic hormone (ACTH)-oma, parathyroid hormone (PTH)-like-oma and neurotensinoma (Table 5.1). These neoplasms occur in less than 0.2 persons per million per year. In general, these tumours resemble gastrinoma in that all are associated with a high incidence of malignancy. Each can also arise in association with MEN1. The hormones, symptoms and signs, diagnostic tests, sites of occurrence, proportions malignant and frequency of associated MEN1 for each tumour are given in Table 5.1.
Malignant pNETs
No diagnostic histological criteria from examination of tumour biopsy samples or resected primary tumours exist to define malignancy for pNETs. Malignancy is definitively established with surgical exploration and histological evidence of tumour remote from the primary lesion, usually in peripancreatic lymph nodes or the liver. Recurrence of tumour at a location distant from a resected primary tumour site also definitively indicates malignancy. Gross invasion of blood vessels, surrounding tissues or adjacent organs usually suggests a malignant tumour. IOUS showing a pancreatic tumour with indistinct margins may imply local invasion and malignancy. Very large tumours (> 5 cm) have an increased risk of being malignant.44 Tumour DNA ploidy and tumoral growth fraction determined by flow cytometry may provide an indication of the biological behaviour of some of these tumours. Because islet cell tumours generally grow slowly, metastases may not become evident until years after the initial primary tumour resection.
Evaluation of metastatic disease
Evaluation of a patient with a malignant neuroendocrine tumour begins by assessing the extent of disease using radiological imaging studies. SRS seems to be the single best imaging study to select patients for aggressive surgery to remove metastatic disease.1 If the tumour binds this isotope, then disease anywhere in the body can be identified. Miliary or extensive bilobar hepatic disease and distant metastases are considered inoperable and, if identified preoperatively, can prevent unnecessary surgery. CT or MRI may identify disease in the chest and abdomen. Specific complaints of bone pain are elicited and, if present, evaluated with bone scan and radiography.
Malignant primary insulinomas are relatively large (≈ 6 cm) and can usually be readily detected by non-invasive imaging studies.23 Gastrinomas may metastasise to regional lymph nodes when only millimetres in size. In one study, duodenal primary gastrinomas have been found to have a higher incidence of lymph node metastases (55%) than pancreatic gastrinomas (22%).1 Some suggest that rare gastrinomas to the left of the superior mesenteric artery in the pancreatic tail are always malignant and more commonly produce liver metastases. Metastatic tumour must be distinguished from multiple tumours, which occur simultaneously. If multiple insulinomas or gastrinomas are found in a patient, then MEN1 should be suspected.
Surgical management
Pancreatic neuroendocrine carcinomas have a better prognosis than adenocarcinoma of the exocrine pancreas and are often managed with aggressive surgical resection.71 Surgery is undertaken to decrease tumour bulk so that hormonal syndromes are more effectively controlled by medical management, to relieve symptoms of mass effect, and/or to eliminate malignant tissue and improve disease-free or overall survival. Preoperative staging studies are important to exclude patients from surgery who would not benefit from resection.
Limited metastases as well as the primary tumour should be resected to adequately debulk tumour and to eliminate the hormonal syndrome. Some patients with MEN1 and ZES have had more aggressive tumours, as evidenced by larger size, liver metastases and higher serum levels of gastrin. Resection of advanced disease, including vascular reconstruction, has been performed safely and is suggested to improve survival.83 Incomplete tumour resection may improve the ability to control the hormonal syndrome medically. For medically fit patients with metastatic insulinoma in whom hypoglycaemia is poorly controlled by medical management, tumour debulking may control symptoms for prolonged time periods, even in the setting of distant metastases. Approximately 50% of patients with metastatic insulinoma undergoing resection have complete biochemical remission.84 Surgical resection of the primary tumour and aggressive resection of liver metastases are both associated with prolonged survival.
Although treatment is generally palliative and not curative for patients with locally advanced tumours and limited metastatic disease, surgery may be the only therapy that effectively ameliorates life-threatening symptoms. It may also increase survival because these tumours are generally indolent, slow-growing neoplasms. Limited regional metastatic disease can often be successfully resected and may be curative if no liver metastases are present. Complete resection of localised or regional nodal metastases with negative margins at the initial surgery provides the highest probability of cure.1 Although disease-free survival is prolonged in most patients, most eventually develop recurrent tumour.
Approximately 30% of patients with metastatic insulinoma can undergo complete resection of tumour.85 Median survival is increased from 11 months in patients with metastatic insulinoma who cannot undergo resection to 4 years in those in whom tumour debulking is possible. Palliative re-resection of recurrent tumour extends median survival from 11–19 months to 4 years.86 Surgery may also be the most effective treatment for patients with metastatic gastrinoma if most or all of the tumour can be resected.1 Aggressive resection of liver metastases of gastrinoma, considered resectable by preoperative radiological imaging studies, improves 5-year survival from 28% in patients with inoperable metastases to 79–85%.87,88 Surgical resection of other liver metastatic neuroendocrine tumours besides gastrinoma is also associated with a 5-year survival of 73%.89
Non-surgical management
Symptoms from extensive metastases may respond to chemotherapy or octreotide, but these treatments are not curative.1 Treatment with octreotide results in unpredictable responses, causing decreased tumour growth in some patients and having no effect in others.90 The addition of interferon-α to octreotide therapy may benefit a subgroup of patients with advanced metastatic disease that is unresponsive to octreotide monotherapy.91 Octreotide may ameliorate symptoms, especially in patients with malignant VIPoma (Fig. 5.8), and when symptoms are adequately controlled, patients can live comfortably and productively for many years despite metastatic disease.
Key points
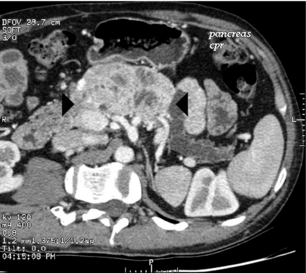
Figure 5.8 Pancreatic protocol CT scan of a glucagonoma within the body of the pancreas (arrows). The tumour was abutting the superior mesenteric vein and causing obstruction of the pancreatic duct with atrophy of the tail of the pancreas.
• With the exception of insulinomas, most pNETs are malignant.
• The diagnosis of insulinoma is established through a supervised 72-hour fast.
• Virtually all insulinomas may be localised by the experienced surgeon with the aid of preoperative and intraoperative modalities. Blind distal pancreatectomy is never indicated.
• Medical control of acid hypersecretion in ZES should be achieved with a PPI to maintain BAO < 15 mEq/h.
• Somatostatin receptor scintigraphy is the first-line preoperative localisation study for gastrinoma.
• Duodenotomy has been shown to improve cure rates in ZES and should be performed during every exploration for gastrinoma.
• Surgical exploration for cure is recommended for all patients with sporadic ZES. In addition, surgery has been demonstrated to improve long-term survival in patients with sporadic ZES and patients with MEN1/ZES and tumour > 2.5 cm.
• Patients with pNETs that are locally advanced or have limited metastatic spread appear to benefit from aggressive surgical resection.
References
1. Norton, J.A., Neuroendocrine tumors of the pancreas and duodenum. Curr Probl Surg. 1994;31(2):77–156. 7904550
2. Doherty, G.M., Doppman, J.L., Shawker, T.H., et al, Results of a prospective strategy to diagnose, localize, and resect insulinomas. Surgery. 1991;110(6):989–997. 1745987 Excellent paper describing the results of diagnostic testing in 25 patients with insulinoma.
3. Dizon, A.M., Kowalyk, S., Hoogwerf, B.J., Neuroglycopenic and other symptoms in patients with insulinomas. Am J Med. 1999;106(3):307–310. 10190379
4. Gorden, P., Skarulis, M.C., Roach, P., et al, Plasma proinsulin-like component in insulinoma: a 25-year experience. J Clin Endocrinol Metab. 1995;80(10):2884–2887. 7559869
5. Grunberger, G., Weiner, J.L., Silverman, R., et al, Factitious hypoglycemia due to surreptitious administration of insulin. Diagnosis, treatment, and long-term follow-up. Ann Intern Med. 1988;108(2):252–257. 3277509
6. Glaser, B., Hirsch, H.J., Landau, H., Persistent hyperinsulinemic hypoglycemia of infancy: long-term octreotide treatment without pancreatectomy. J Pediatr. 1993;123(4):644–650. 8410523
7. Thornton, P.S., Alter, C.A., Katz, L.E., et al, Short-and long-term use of octreotide in the treatment of congenital hyperinsulinism. J Pediatr. 1993;123(4):637–643. 8410522
8. Service, G.J., Thompson, G.B., Service, F.J., et al, Hyperinsulinemic hypoglycemia with nesidioblastosis after gastric-bypass surgery. N Engl J Med. 2005;353(3):249–254. 16034010
9. Grant, C.S., Insulinoma. Surg Oncol Clin N Am. 1998;7(4):819–844. 9735136
10. Arnold, R., Frank, M., Kajdan, U., Management of gastroenteroPETs: the place of somatostatin analogues. Digestion. 1994;55(Suppl. 3):107–113. 7698532
11. Hiramoto, J.S., Feldstein, V.A., LaBerge, J.M., et al, Intraoperative ultrasound and preoperative localization detects all occult insulinomas. Arch Surg. 2001;136(9):1020–1026. 11529824
12. Grant, C.S., van Heerden, J., Charboneau, J.W., et al, Insulinoma. The value of intraoperative ultrasonography. Arch Surg. 1988;123(7):843–848. 2838008
13. Rodallec, M., Vilgrain, V., Zins, M., et al, Helical CT of PETs. J Comput Assist Tomogr. 2002;26(5):728–733. 12439306
14. Boukhman, M.P., Karam, J.M., Shaver, J., et al, Localization of insulinomas. Arch Surg. 1999;134(8):818–823. 10443803
15. Owens, L.V., Huth, J.F., Cance, W.G., Insulinoma: pitfalls in preoperative localization. Eur J Surg Oncol. 1995;21(3):326–328. 7781809
16. Gauger, P.G., Scheiman, J.M., Wamsteker, E.J., et al, Role of endoscopic ultrasonography in screening and treatment of PETs in asymptomatic patients with multiple endocrine neoplasia type 1. Br J Surg. 2003;90(6):748–754. 12808627
17. Kann, P.H., Rothmund, M., Zielke, A., Endoscopic ultrasound imaging of insulinomas: limitations and clinical relevance. Exp Clin Endocrinol Diabetes. 2005;113(8):471–474. 16151982
18. Glover, J.R., Shorvon, P.J., Lees, W.R., Endoscopic ultrasound for localisation of islet cell tumours. Gut. 1992;33(1):108–110. 1310948
19. Rosch, T., Lightdale, C.J., Botet, J.F., et al, Localization of PETs by endoscopic ultrasonography. N Engl J Med. 1992;326(26):1721–1726. 1317506
20. McLean, A.M., Fairclough, P.D., Endoscopic ultrasound in the localisation of pancreatic islet cell tumours. Best Pract Res Clin Endocrinol Metab. 2005;19(2):177–193. 15763694
21. Richards, M.L., Gauger, P.G., Thompson, N.W., et al, Pitfalls in the surgical treatment of insulinoma. Surgery. 2002;132(6):1040–1049. 12490853
22. Cohen, M.S., Picus, D., Lairmore, T.C., et al, Prospective study of provocative angiograms to localize functional islet cell tumors of the pancreas. Surgery. 1997;122(6):1091–1100. 9426424
23. Norton, J.A., Cromack, D.T., Shawker, T.H., et al, Intraoperative ultrasonographic localization of islet cell tumors. A prospective comparison to palpation. Ann Surg. 1988;207(2):160–168. 2829761
24. Gianello, P., Gigot, J.F., Berthet, F., et al, Pre- and intraoperative localization of insulinomas: report of 22 observations. World J Surg. 1988;12(3):389–397. 2840779
25. Norton, J.A., Sigel, B., Baker, A.R., et al, Localization of an occult insulinoma by intraoperative ultrasonography. Surgery. 1985;97(3):381–384. 2983451
26. Doppman, J.L., Chang, R., Fraker, D.L., et al, Localization of insulinomas to regions of the pancreas by intra-arterial stimulation with calcium. Ann Intern Med. 1995;123(4):269–273. 7611592
27. Veldhuis, J.D., Norton, J.A., Wells, S.A., Jr., et al, Surgical versus medical management of multiple endocrine neoplasia (MEN) type I. J Clin Endocrinol Metab. 1997;82(2):357–364. 9024218
28. Dexter, S.P., Martin, I.G., Leindler, L., et al, Laparoscopic enucleation of a solitary pancreatic insulinoma. Surg Endosc. 1999;13(4):406–408. 10094758
29. Gagner, M., Pomp, A., Herrera, M.F., Early experience with laparoscopic resections of islet cell tumors. Surgery. 1996;120(6):1051–1054. 8957494
30. Pierce, R.A., Spitler, J.A., Hawkins, W.G., et al, Outcomes analysis of laparoscopic resection of pancreatic neoplasms. Surg Endosc. 2007;21(4):579–586. 17180287
31. Toniato, A., Meduri, F., Foletto, M., et al, Laparoscopic treatment of benign insulinomas localized in the body and tail of the pancreas: a single-center experience. World J Surg. 2006;30(10):1916–1921. 16855802
32. Sheppard, B.C., Norton, J.A., Doppman, J.L., et al, Management of islet cell tumors in patients with multiple endocrine neoplasia: a prospective study. Surgery. 1989;106(6):1108–1118. 2573957
33. Eriksson, B., Oberg, K., Skogseid, B., Neuroendocrine pancreatic tumors. Clinical findings in a prospective study of 84 patients. Acta Oncol. 1989;28(3):373–377. 2472825
34. Zollinger, R.M., Ellison, E.H., Primary peptic ulcerations of the jejunum associated with islet cell tumors of the pancreas. Ann Surg. 1955;142(4):709–728. 13259432
35. Cisco, R.M., Norton, J.A., Surgery for gastrinoma. Adv Surg 2007; 41:165–176. 17972563
36. Wolfe, M.M., Jensen, R.T., Zollinger–Ellison syndrome. Current concepts in diagnosis and management. N Engl J Med. 1987;317(19):1200–1209. 3309661
37. Fox, P.S., Hofmann, J.W., Decosse, J.J., et al, The influence of total gastrectomy on survival in malignant Zollinger–Ellison tumors. Ann Surg. 1974;180(4):558–566. 4416322
38. Zollinger, R.M., Ellison, E.C., O’Dorisio, T.M., et al, Thirty years’ experience with gastrinoma. World J Surg. 1984;8(4):427–435. 6148806
39. Fraker, D.L., Norton, J.A., Alexander, H.R., et al, Surgery in Zollinger–Ellison syndrome alters the natural history of gastrinoma. Ann Surg. 1994;220(3):320–330. 7916560
40. Norton, J.A., Melcher, M.L., Gibril, F., et al, Gastric carcinoid tumors in multiple endocrine neoplasia-1 patients with Zollinger–Ellison syndrome can be symptomatic, demonstrate aggressive growth, and require surgical treatment. Surgery. 2004;136(6):1267–1274. 15657586
41. Yu, F., Venzon, D.J., Serrano, J., et al, Prospective study of the clinical course, prognostic factors, causes of death, and survival in patients with long-standing Zollinger–Ellison syndrome. J Clin Oncol. 1999;17(2):615–630. 10080607
42. Alexander, H.R., Fraker, D.L., Norton, J.A., et al, Prospective study of somatostatin receptor scintigraphy and its effect on operative outcome in patients with Zollinger–Ellison syndrome. Ann Surg. 1998;228(2):228–238. 9712569
43. Lamberts, S.W., Bakker, W.H., Reubi, J.C., et al, Somatostatin-receptor imaging in the localization of endocrine tumors. N Engl J Med. 1990;323(18):1246–1249. 2170840 Original description of somatostatin receptor scintigraphy.
44. Peplinski, G.R., Norton, J.A., Gastrointestinal endocrine cancers and nodal metastasis: biologic significance and therapeutic implications. Surg Oncol Clin N Am. 1996;5(1):159–171. 8789500
45. Thompson, N.W., Pasieka, J., Fukuuchi, A., Duodenal gastrinomas, duodenotomy, and duodenal exploration in the surgical management of Zollinger–Ellison syndrome. World J Surg. 1993;17(4):455–462. 8103249
46. Sutliff, V.E., Doppman, J.L., Gibril, F., et al, Growth of newly diagnosed, untreated metastatic gastrinomas and predictors of growth patterns. J Clin Oncol. 1997;15(6):2420–2431. 9196158
47. Wank, S.A., Doppman, J.L., Miller, D.L., et al, Prospective study of the ability of computed axial tomography to localize gastrinomas in patients with Zollinger–Ellison syndrome. Gastroenterology. 1987;92(4):905–912. 3556996
48. Gibril, F., Reynolds, J.C., Doppman, J.L., et al, Somatostatin receptor scintigraphy: its sensitivity compared with that of other imaging methods in detecting primary and metastatic gastrinomas. A prospective study. Ann Intern Med. 1996;125(1):26–34. 8644985
49. Meko, J.B., Doherty, G.M., Siegel, B.A., et al, Evaluation of somatostatin-receptor scintigraphy for detecting neuroendocrine tumors. Surgery. 1996;120(6):975–984. 8957483
50. Gibril, F., Reynolds, J.C., Chen, C.C., et al, Specificity of somatostatin receptor scintigraphy: a prospective study and effects of false-positive localizations on management in patients with gastrinomas. J Nucl Med. 1999;40(4):539–553. 10210211
51. McLean, A.M., Fairclough, P.D., Endoscopic ultrasound in the localisation of pancreatic islet cell tumours. Best Pract Res Clin Endocrinol Metab. 2005;19(2):177–193. 15763694
52. Norton, J.A., Alexander, H.R., Fraker, D.L., et al, Does the use of routine duodenotomy (DUODX) affect rate of cure, development of liver metastases, or survival in patients with Zollinger–Ellison syndrome? Ann Surg. 2004;239(5):617–626. 15082965 Demonstrates that duodenotomy not only localises more gastrinomas, but also improves cure rates.
53. Norton, J.A., Alexander, H.R., Fraker, D.L., et al, Possible primary lymph node gastrinoma: occurrence, natural history, and predictive factors: a prospective study. Ann Surg. 2003;237(5):650–659. 12724631
54. Perrier, N.D., Batts, K.P., Thompson, G.B., et al, An immunohistochemical survey for neuroendocrine cells in regional pancreatic lymph nodes: a plausible explanation for primary nodal gastrinomas? Mayo Clinic Pancreatic Surgery Group. Surgery. 1995;118(6):957–966. 7491540
55. Thompson, N.W., Vinik, A.I., Eckhauser, F.E., Microgastrinomas of the duodenum. A cause of failed operations for the Zollinger–Ellison syndrome. Ann Surg. 1989;209(4):396–404. 2930285
56. Pipeleers-Marichal, M., Donow, C., Heitz, P.U., et al, Pathologic aspects of gastrinomas in patients with Zollinger–Ellison syndrome with and without multiple endocrine neoplasia type I. World J Surg. 1993;17(4):481–488. 8103250
57. Stabile, B.E., Morrow, D.J., Passaro, E., Jr., The gastrinoma triangle: operative implications. Am J Surg. 1984;147(1):25–31. 6691547
58. Pipeleers-Marichal, M., Somers, G., Willems, G., et al, Gastrinomas in the duodenums of patients with multiple endocrine neoplasia type 1 and the Zollinger–Ellison syndrome. N Engl J Med. 1990;322(11):723–727. 1968616
59. Frucht, H., Norton, J.A., London, J.F., et al, Detection of duodenal gastrinomas by operative endoscopic transillumination. A prospective study. Gastroenterology. 1990;99(6):1622–1627. 2227278
60. Norton, J.A., Fraker, D.L., Alexander, H.R., et al, Surgery to cure the Zollinger–Ellison syndrome. N Engl J Med. 1999;341(9):635–644. 10460814 Results of a prospective surgical trial to cure ZES.
61. Bornman, P.C., Marks, I.N., Mee, A.S., et al, Favourable response to conservative surgery for extra-pancreatic gastrinoma with lymph node metastases. Br J Surg. 1987;74(3):198–201. 3567510
62. Norton, J.A., Doppman, J.L., Collen, M.J., et al, Prospective study of gastrinoma localization and resection in patients with Zollinger–Ellison syndrome. Ann Surg. 1986;204(4):468–479. 3532971
63. Wolfe, M.M., Alexander, R.W., McGuigan, J.E., Extrapancreatic, extraintestinal gastrinoma: effective treatment by surgery. N Engl J Med. 1982;306(25):1533–1536. 6281644
64. Arnold, W.S., Fraker, D.L., Alexander, H.R., et al, Apparent lymph node primary gastrinoma. Surgery. 1994;116(6):1123–1130. 7985097
65. Herrmann, M.E., Ciesla, M.C., Chejfec, G., et al, Primary nodal gastrinomas. Arch Pathol Lab Med. 2000;124(6):832–835. 10835515
66. Wu, P.C., Alexander, H.R., Bartlett, D.L., et al, A prospective analysis of the frequency, location, and curability of ectopic (nonpancreaticoduodenal, nonnodal) gastrinoma. Surgery. 1997;122(6):1176–1182. 9426435
67. Noda, S., Norton, J.A., Jensen, R.T., et al, Surgical resection of intracardiac gastrinoma. Ann Thorac Surg. 1999;67(2):532–533. 10197684
68. Ahn, Y.J., Kim, S.W., Park, Y.C., et al, Duodenal-preserving resection of the head of the pancreas and pancreatic head resection with second-portion duodenectomy for benign lesions, low-grade malignancies, and early carcinoma involving the periampullary region. Arch Surg. 2003;138(2):162–168. 12578411
69. Ellison, E.C., Forty-year appraisal of gastrinoma. Back to the future. Ann Surg. 1995;222(4):511–524. 7574931
70. Kisker, O., Bastian, D., Bartsch, D., et al, Localization, malignant potential, and surgical management of gastrinomas. World J Surg. 1998;22(7):651–658. 9606277
71. Norton, J.A., Sugarbaker, P.H., Doppman, J.L., et al, Aggressive resection of metastatic disease in selected patients with malignant gastrinoma. Ann Surg. 1986;203(4):352–359. 2938550
72. Jaskowiak, N.T., Fraker, D.L., Alexander, H.R., et al, Is reoperation for gastrinoma excision indicated in Zollinger–Ellison syndrome? Surgery. 1996;120(6):1055–1063. 8957495
73. Norton, J.A., Cornelius, M.J., Doppman, J.L., et al, Effect of parathyroidectomy in patients with hyperparathyroidism, Zollinger–Ellison syndrome, and multiple endocrine neoplasia type I: a prospective study. Surgery. 1987;102(6):958–966. 2891201
74. MacFarlane, M.P., Fraker, D.L., Alexander, H.R., et al, Prospective study of surgical resection of duodenal and pancreatic gastrinomas in multiple endocrine neoplasia type 1. Surgery. 1995;118(6):973–980. 7491542
75. Thompson, N.W., Current concepts in the surgical management of multiple endocrine neoplasia type 1 pancreatic–duodenal disease. Results in the treatment of 40 patients with Zollinger–Ellison syndrome, hypoglycaemia or both. J Intern Med. 1998;243(6):495–500. 9681848
76. Thompson, N.W., Management of PETs in patients with multiple endocrine neoplasia type 1. Surg Oncol Clin N Am. 1998;7(4):881–891. 9735139
77. Norton, J.A., Fraker, D.L., Alexander, H.R., et al, Surgery increases survival in patients with gastrinoma. Ann Surg. 2006;244(3):410–419. 16926567 Study demonstrating improved survival in patients who undergo surgical resection for gastrinoma.
78. Jensen, R.T., PETs: recent advances. Ann Oncol. 1999;10(Suppl. 4):170–176. 10436815
79. Baudin, E., Gigliotti, A., Ducreux, M., et al, Neuron-specific enolase and chromogranin A as markers of neuroendocrine tumours. Br J Cancer. 1998;78(8):1102–1107. 9792158
80. Nobels, F.R., Kwekkeboom, D.J., Bouillon, R., et al, Chromogranin A: its clinical value as marker of neuroendocrine tumours. Eur J Clin Invest. 1998;28(6):431–440. 9693933
81. van Eijck, C.H., Lamberts, S.W., Lemaire, L.C., et al, The use of somatostatin receptor scintigraphy in the differential diagnosis of pancreatic duct cancers and islet cell tumors. Ann Surg. 1996;224(2):119–124. 8757373
82. Haynes, A.B., Deshpande, V., Ingkakul, T., et al, Implications of incidentally discovered, nonfunctioning pancreatic endocrine tumors: short-term and long-term patient outcomes. Arch Surg. 2011;146(5):534–538. 21576607
83. Gibril, F., Venzon, D.J., Ojeaburu, J.V., et al, Prospective study of the natural history of gastrinoma in patients with MEN1: definition of an aggressive and a nonaggressive form. J Clin Endocrinol Metab. 2001;86(11):5282–5293. 11701693
84. Rothmund, M., Stinner, B., Arnold, R., Endocrine pancreatic carcinoma. Eur J Surg Oncol. 1991;17(2):191–199. 1849844
85. Modlin, I.M., Lewis, J.J., Ahlman, H., et al, Management of unresectable malignant endocrine tumors of the pancreas. Surg Gynecol Obstet. 1993;176(5):507–518. 8386860
86. Zogakis, T.G., Norton, J.A., Palliative operations for patients with unresectable endocrine neoplasia. Surg Clin North Am. 1995;75(3):525–538. 7538230
87. Danforth, D.N., Jr., Gorden, P., Brennan, M.F., Metastatic insulin-secreting carcinoma of the pancreas: clinical course and the role of surgery. Surgery. 1984;96(6):1027–1037. 6095477
88. Norton, J.A., Doherty, G.M., Fraker, D.L., et al, Surgical treatment of localized gastrinoma within the liver: a prospective study. Surgery. 1998;124(6):1145–1152. 9854596
89. Norton, J.A., Warren, R.S., Kelly, M.G., et al, Aggressive surgery for metastatic liver neuroendocrine tumors. Surgery. 2003;134(6):1057–1065. 14668741
90. Mozell, E., Woltering, E.A., O’Dorisio, T.M., et al, Effect of somatostatin analog on peptide release and tumor growth in the Zollinger–Ellison syndrome. Surg Gynecol Obstet. 1990;170(6):476–484. 2188384
91. Frank, M., Klose, K.J., Wied, M., et al, Combination therapy with octreotide and alpha-interferon: effect on tumor growth in metastatic endocrine gastroenteropancreatic tumors. Am J Gastroenterol. 1999;94(5):1381–1387. 10235222
[/level-membership-for-endocrinology-diabetes-and-metabolism-category][not-level-membership-for-endocrinology-diabetes-and-metabolism-category]5
Endocrine tumours of the pancreas
Introduction
Pancreatic neuroendocrine tumours (pNETs) are the most common endocrine tumours found in the abdomen. They may be broadly classified into two groups: functional tumours, which cause clinical syndromes due to hormone secretion; and non-functional tumours, which may cause symptoms through mass effect or metastatic spread, or may be discovered incidentally. The most common functional pNETs are gastrinomas and insulinomas, but a variety of other rare hormone secreting tumours also occur (Table 5.1). Identification of clinical syndromes and hormone production allows the classification of functional pNETs into specific types. Potentially life-threatening situations caused by hormone overproduction are a major reason to identify and resect these neoplasms. Additionally, pNETs have malignant potential. However, they are less malignant than pancreatic adenocarcinoma and surgical extirpation is beneficial in most instances.
Insulinoma
Endogenous hyperinsulinism was first described in 1927, and was the first syndrome of excessive pancreatic hormone production to be recognised.1 Hyperinsulinaemia and consequent hypoglycaemia is the major cause of morbidity and potential mortality associated with insulinoma. Insulinoma occurs in approximately one person per million population per year (Table 5.1).1 Hyperinsulinaemic hypoglycaemia associated with insulinoma is not well controlled by medical therapy, and surgery has remained the cornerstone of treatment over the past 80 years. Insulinomas are unique among pNETs in that 90% of insulinomas are benign, solitary growths that occur uniformly throughout and almost exclusively within the pancreas, without evidence of local invasion or locoregional lymph node metastases. Tumours may be as small as 6 mm in diameter and are usually less than 2 cm in size, making localisation challenging in many cases.2
Presentation
Excessive and physiologically uncontrolled secretion of insulin by the tumour causes episodes of symptomatic hypoglycaemia and leads patients to seek medical evaluation. Acute neuroglycopenia induces anxiety, dizziness, obtundation, confusion, unconsciousness, personality changes and seizures.2 Symptoms commonly occur during early morning hours, when glucose reserves are low after a period of overnight fasting during which insulin overproduction has continued. Most patients (80%) experience major weight gain as they attempt to treat these symptoms with increased caloric intake. They may first present with symptoms of hypoglycaemia when food intake is then decreased in an attempt to lose weight. A majority (60–75%) of patients are women, and many have undergone extensive psychiatric evaluation before the correct diagnosis is reached. Other patients will have been misdiagnosed with neurological conditions such as seizure disorders, cerebrovascular accidents or transient ischaemic attacks. Potentially life-threatening symptoms may be present for several years prior to diagnosis.2 In a review of 59 patients with insulinoma, the interval from onset of symptoms to time of diagnosis ranged from 1 month to 30 years, with the median time to diagnosis being 2 years.3 Because insulinoma is rare and neuroglycopenic symptoms are relatively non-specific, a high index of suspicion for insulinoma is necessary when other explanations for these symptoms are not evident. The identification of symptomatic patients and the liberal use of simple and precise biochemical tests result in accurate diagnosis of insulinoma prior to life-threatening sequelae.
Diagnosis
The diagnosis of insulinoma is established through a 72-hour supervised fast. The most definitive biochemical result for insulinoma is a plasma insulin concentration above 5 μU/mL in the presence of symptomatic hypoglycaemia.2,4 Factitious hypoglycaemia must be excluded.5
The classic diagnostic triad, proposed by Whipple in 1935 based on his observations in 32 patients, consists of symptoms of hypoglycaemia during a fast, a concomitant blood glucose concentration less than 3 mmol/L and relief of the hypoglycaemic symptoms after glucose administration.1 Factitious hypoglycaemia due to clandestine administration of exogenous insulin or oral hypoglycaemic drugs may also result in this same presentation and can lead to a misdiagnosis of insulinoma.5 Factitious hypoglycaemia classically occurs in patients associated with the medical profession or patients who have relatives with diabetes. The diagnosis of insulinoma must be reached in each patient by performing a 72-hour supervised fast with appropriate biochemical measurements. Urinary sulphonylurea concentrations (to exclude oral hypoglycaemic drugs) should be measured by gas chromatography–mass spectroscopy.
Supervised standard fasting test
The standard fasting test is carried out in a hospital setting and begins with a baseline examination in which memory, calculations and coordination are documented (Fig. 5.1). An intravenous catheter with a heparin lock is then placed, and the patient is allowed to drink only non-caloric beverages. Close observation is necessary. Blood is collected every 6 hours for measurement of serum glucose and immunoreactive insulin concentrations. As the blood glucose level falls below 3 mmol/L, blood samples are collected more frequently (every hour or less) and the patient is observed more closely. When neuroglycopenic symptoms appear, blood is collected immediately for determination of serum insulin, glucose, C-peptide and proinsulin concentrations (Fig. 5.1). Glucose is then administered and the fast is terminated. If a patient remains symptom-free for the entire 72 hours, the test is terminated and the above blood concentrations are measured.
Figure 5.1 Results of supervised fast in a patient with insulinoma. The patient, a 48-year-old woman, experienced acute onset of confusion and blurred vision 16 hours into a supervised fast in the ICU (arrow). Plasma glucose was 40 mg/dL at this time and plasma insulin was 12 μU/mL. Proinsulin measured simultaneously was 87 pmol/L (normal: 3–20 pmol/L). Symptoms rapidly resolved following intravenous administration of dextrose. The most diagnostic biochemical result in a patient with insulinoma is an inappropriately increased plasma insulin concentration above 5 μU/mL at the time of documented hypoglycaemia and symptoms.
Neuroglycopenic symptoms manifest in approximately 60% of patients with insulinomas within 24 hours after fasting begins.4 Approximately 16% of patients with insulinoma develop symptoms when the blood glucose concentration is greater than 2.5 mmol/L.2 The blood glucose concentration eventually decreases below 2.5 mmol/L in approximately 85% of patients with insulinomas during the 72-hour fast (Table 5.2). The most definitive diagnostic biochemical test for insulinoma is an inappropriately increased plasma immunoreactive insulin concentration above 5 μU/mL at the time of documented hypoglycaemia and symptoms.4 The plasma insulin concentration is usually greater than 10 μU/mL.2 Although prolonged maximal stimulation of insulin secretion in normal subjects does not cause the release of the insulin precursor molecule proinsulin, some insulinomas secrete large amounts of uncleaved proinsulin. Patients with high proinsulin-producing tumours may remain euglycaemic and asymptomatic for longer periods during the fast because proinsulin is not biologically active. The proinsulin-like component (PLC) is measured at the time of symptomatic hypoglycaemia and termination of the fast. A value greater than 25% or an increased PLC: total immunoreactive insulin ratio is abnormal and consistent with the diagnosis of insulinoma.2,4 Hypersecretion of endogenous insulin also results in increases of the circulating concentration of C-peptide, a biologically inactive by-product of enzymatic insulin cleavage from the precursor proinsulin molecule. Most patients with insulinomas have C-peptide concentrations greater than 1.7 ng/mL.2
Table 5.2
Standard fasting test results and the differentiation of insulinoma from factitious hypoglycaemia
Increased serum concentrations of proinsulin or C-peptide during hypoglycaemia effectively exclude the diagnosis of factitious hypoglycaemia because exogenously administered insulin does not contain these proteins and actually suppresses their production. However, approximately 13–22% of patients with insulinoma do not have increased serum proinsulin or C-peptide concentrations, and a supervised fast prohibiting exogenous insulin administration remains the best test to diagnose insulinoma and conclusively exclude factitious hypoglycaemia. The biochemical parameters measured during the standard fasting test cannot discriminate between patients with MEN1 and patients with sporadic insulinoma.4
Nesidioblastosis
Insulinoma must be distinguished from nesidioblastosis, a condition of islet cell hyperfunction and malregulation that occurs primarily in infants and causes hyperinsulinaemic hypoglycaemia. Age at the time of presentation is the most important distinguishing factor, as nesidioblastosis occurs most commonly in children under the age of 18 months. Approximately half of infants with nesidioblastosis require a spleen-preserving near-total pancreatectomy, in which 95% of the pancreas is removed, because this disorder affects the entire pancreas diffusely.6,7
Adult nesidioblastosis has been reported and was recently described in a small series of adult patients following gastric bypass surgery for morbid obesity.8 However, nesidioblastosis in adults is exceedingly rare. In a review of over 300 patients with hyperinsulinaemic hypoglycaemia evaluated at the Mayo Clinic since 1927, only five adult patients had a reasonably confirmed diagnosis of nesidioblastosis.9 Furthermore, biochemical tests (blood glucose, insulin and C-peptide) do not reliably distinguish hyperinsulinaemic hypoglycaemia caused by an insulinoma from that attributed to nesidioblastosis, and an insulinoma may be present in a patient who has islet cell hyperplasia. Therefore, the diagnosis of nesidioblastosis in the adult must be critically suspect and should not preclude attempts to localise an insulinoma.
Management
Medical management of hypoglycaemia
Medical management aims to prevent hypoglycaemia caused by hyperinsulinism so that symptoms and life-threatening sequelae are avoided. In patients with acute hypoglycaemia, blood glucose concentrations are normalised initially with an intravenous dextrose infusion. To prevent hypoglycaemic episodes during diagnosis, tumour localisation and the preoperative period, euglycaemia is maintained by giving frequent feeds of a high-carbohydrate diet, including a night feed. For patients who continue to become hypoglycaemic between feedings, diazoxide may be added to the treatment regimen at a dose of 400–600 mg orally each day. Diazoxide inhibits insulin release in approximately 50% of patients with insulinoma; however, side-effects of oedema, weight gain and hirsutism occur in 50% of patients, and nausea occurs in over 10%.9 Diazoxide should be discontinued 1 week prior to surgery as it may contribute to intraoperative hypotension. Calcium-channel blockers or phenytoin may also suppress insulin production in some patients. Long-term control of hypoglycaemic symptoms with medical management has generally been ineffective for patients with insulinoma. The surgeon must know the patient’s response to medical management in order to gauge the urgency of surgical intervention.
Octreotide, a synthetic, long-acting analogue of the naturally occurring hormone somatostatin, may be useful for treating symptoms caused by vasoactive intestinal peptide (VIP)-omas and carcinoid tumours but is not generally recommended for insulinomas because its efficacy in inhibiting insulin release is unpredictable.10 The usefulness of radiolabelled octreotide in imaging insulinomas has been equally disappointing. Therefore, long-term medical management of hypoglycaemia in patients with insulinomas generally is reserved for the few patients (< 5%) with unlocalised, unresected tumours after thorough preoperative testing and exploratory laparotomy, and for patients with metastatic, unresectable malignant insulinoma. Patients with malignant insulinomas and refractory hypoglycaemia may even require the placement of implantable glucose pumps for continuous glucose infusion.9
Preoperative tumour localisation
Virtually all insulinomas may be localised by the experienced surgeon through the combination of preoperative modalities and intraoperative ultrasound.11 Blind pancreatic resection is not indicated.
Non-invasive imaging studies: An initial attempt should be made to localise the tumour and identify metastatic disease by using non-invasive tests. Computed tomography (CT) and magnetic resonance imaging (MRI) are both capable of identifying pancreatic tumours as small as 1 cm in diameter (Table 5.3). If CT is elected, a pancreatic protocol study with biphasic contrast injection and fine cuts through the pancreas should be used. Tumours will appear hypervascular on arterial phase images (Fig. 5.2a). The sensitivity of CT for insulinoma is 40–80% in recent series.12,13 MRI may image an islet cell tumour based on increased signal intensity (brightness) on T2-weighted images (Fig. 5.2b). The sensitivity of MRI is similar to that of CT and, as expected, the accuracy of both CT and MRI increases with larger tumour size.14
Figure 5.2 A 1.3 × 1.3 cm insulinoma at the junction of the body and tail of pancreas as visualised on preoperative and intraoperative imaging (tumour is identified by arrows). (a) Pancreatic protocol CT demonstrates the hypervascular tumour following administration of intravenous contrast. (b) On T2-weighted MRI images the tumour has increased signal intensity (brightness). (c) Intraoperative ultrasound demonstrates the insulinoma, which is sonolucent compared with the more echodense pancreas.
Invasive localising procedures: Up to 50% of insulinomas are too small to be detected by non-invasive imaging. A variety of more sensitive invasive tests are used to localise these tumours preoperatively. Endoscopic ultrasound (EUS) is safe and highly effective in experienced hands and is the modality of choice for patients whose insulinoma is not localised by CT.15 It has been demonstrated to detect tumours as small as 2–3 mm, well below the limits of detection of CT or MRI,16,17 and it is the most sensitive modality for detection of intrapancreatic tumours.
To perform EUS of the pancreas, the endoscope is passed into the duodenum and a saline-filled balloon is inflated against the intestinal wall. A 5- to 10-MHz transducer is used to generate an image of the pancreas through the intestinal and stomach walls. Tumours as small as 2–3 mm in diameter can be identified at the pancreatic head by moving the transducer through the duodenum at the junction with the pancreas. The endoscope must be passed well into the third portion of the duodenum to adequately visualise the uncinate process. Insulinomas in the pancreatic body and tail are imaged by positioning the transducer in the stomach and scanning through its posterior wall. Insulinomas will appear homogeneously hypoechoic, well circumscribed and round, and are typically easily distinguishable from the surrounding pancreatic parenchyma. Sensitivity of EUS for insulinoma ranges from 70% to 94%.18,19 Rates of detection are highest in the head of the pancreas (83–100%) because the head can be viewed from three angles (from the third portion of the duodenum, through the bulb of the duodenum and through the stomach). They are lower (37–60%) in the body and tail, which can only be viewed through the stomach.20
Despite the tremendous potential and proven benefit of EUS for insulinoma, there are some limitations. First, there may be false positives, which include accessory spleens and intrapancreatic lymph nodes. Further, EUS is limited in assessment of malignancy, identification of pedunculated tumours, and differentiation of large tumours from the pancreatic parenchyma.21
In patients with negative results after non-invasive imaging studies and EUS, calcium arteriography may be useful (Fig. 5.3). This study relies on the functional activity of the insulinoma (i.e. excessive insulin production) and not on the ability to image the tumour (i.e. tumour size). Arteries that perfuse the pancreatic head (gastroduodenal artery and superior mesenteric artery) and the body/tail (splenic artery) are selectively catheterised, and a small amount of calcium gluconate (0.025 mEq Ca2 +/kg body weight) is injected into each artery during sequential runs. A catheter positioned in the right hepatic vein is then used to collect blood for measurement of insulin concentrations. Calcium stimulates a marked increase in insulin secretion from the insulinoma. A greater than twofold increase in the hepatic vein insulin concentration indicates localisation of the tumour to the area of the pancreas being perfused by the injected artery (Fig. 5.3). In this way, calcium provocation may identify the region of the pancreas containing the tumour (head, body or tail). Additionally, injection of contrast may reveal a tumour blush, confirming the location of the insulinoma by imaging the tumour. These combined features are particularly useful in identifying the insulinoma in patients with MEN1 who may have multiple imaged pNETs. The reported sensitivity of calcium stimulation is between 88% and 94% and few false-positive results occur (Table 5.3).22
Figure 5.3 Calcium angiogram in a patient with an insulinoma localised to the pancreatic tail. Intra-arterial calcium was selectively injected into splenic, superior mesenteric, hepatic and gastroduodenal arteries in four different runs. Blood samples were taken serially from hepatic vein and insulin concentrations measured at 0, 1 and 2 minutes before and after calcium injection. After injection into the splenic artery there was a rapid marked increase in hepatic vein insulin concentrations at 1 and 2 minutes. This finding localised the insulinoma to the pancreatic tail.
A small proportion of insulinomas will remain unlocalised even after all preoperative studies are obtained. When the diagnosis is certain on the basis of the results of the supervised fast, surgical exploration with careful inspection, palpation and intraoperative ultrasonography (IOUS) of the pancreas is still indicated. Most of these patients (> 90%) will still have an insulinoma identified and removed by the experienced surgeon.1,23 Retrospective reviews have shown that the combination of careful surgical exploration with IOUS will identify almost all insulinomas.20,24
Operative management
Open exploration: In contradistinction to gastrinomas, virtually all insulinomas are located within the pancreas and are uniformly distributed throughout the entire gland.2 Therefore, in the patient with unlocalised insulinoma, the head, body and tail of the pancreas should be sufficiently mobilised to permit evaluation of the entire organ. This requires an extended Kocher manoeuvre, to adequately lift the head of the pancreas out of the retroperitoneum, and division of attachments at the inferior and posterior border of the pancreas, to permit evaluation of the posterior body and tail (Fig. 5.4). The entire pancreatic surface should then be inspected, as an insulinoma may appear as a brownish-red purple mass. Because the head of the pancreas is thick, small tumours that are centrally located may not be easily palpated. The entire pancreatic head must be sufficiently mobilised so that the posterior surface can be adequately examined visually and palpated between the thumb and forefinger. The splenic ligaments may be divided to completely mobilise the spleen out of the retroperitoneum for full examination and palpation of the pancreatic tail.
Figure 5.4 Intraoperative manoeuvres to identify insulinoma. (a) Kocher manoeuvre with careful palpation of head of pancreas. (b) Opening gastrocolic ligament, superior retraction of stomach, inferior retraction of transverse colon, and careful palpation of body and tail of pancreas after incision along inferior border.
IOUS is the best intraoperative method to idenitify insulinomas. It is performed by placing the transducer on the surface of the pancreas, which is covered in a pool of saline to maximise image quality. A 10- or 7.5-MHz real-time probe is used, which has a short focal length and high resolution. An insulinoma appears as a sonolucent mass with margins distinct from the uniform, more echodense pancreatic parenchyma (Fig. 5.2c). IOUS can localise an occult insulinoma that has not been identified preoperatively and can identify tumours that are not visible or palpable.25 It is particularly helpful in evaluation of the pancreatic head.2,23 The sensitivity for detecting insulinomas using IOUS is greater than 75%12,24 and approaches 100%2,26 (Table 5.3). A study of 37 consecutive patients showed that IOUS correctly identified 35 (95%). The two tumours that were missed were in the pancreatic tail.11
Resection of insulinoma: Enucleation of the tumour with removal of minmal adjacent pancreatic tissue is the operation of choice for benign insulinomas. However, tumour size, location and surrounding anatomy may dictate that formal pancreatic resection is necessary. It is important to consider the relationship of the tumour to the pancreatic duct by imaging both structures with IOUS prior to tumour excision. Small tumours that are separated from the pancreatic duct and major vessels by normal pancreas can be safely enucleated. If a clear margin of normal pancreatic tissue does not exist between the insulinoma and other structures, then a spleen-preserving distal or subtotal pancreatectomy is advised.11,25 Evidence of malignancy, such as involvement of peripancreatic lymph nodes or tumour invasion, also mandates pancreatic resection. Rarely, pancreatico-duodenectomy is indicated if enucleation cannot be performed safely. Frozen-section confirmation of neuroendocrine tumour is traditionally used as the end-point of surgery.
Insulinoma and MEN1
Approximately 10% of insulinomas occur in the setting of MEN1, and 20% of patients with MEN1 develop insulinomas. Insulinomas in MEN1 may be multiple and may occur simultaneously and diffusely throughout the pancreas. In addition, patients with MEN1 may have multiple non-functioning pNETs. The goal of treatment in this scenario is to ameliorate the hypoglycaemia by eliminating the source of insulin hypersecretion. Difficulty may arise in identifying which tumour produces the excessive insulin. Calcium angiogram is useful to determine if the dominant imaged tumour is responsible for the excessive secretion of insulin. If the insulinoma(s) arises within the body or tail of the pancreas, then a subtotal or distal pancreatectomy is indicated because multiple other islet cell tumours are virtually always present. A tumour that arises in the head of the pancreas is enucleated, if possible, or alternatively resected by pancreatico-duodenectomy. As in the case of sporadic insulinoma, medical management for this condition is reserved for those occasional patients who have failed surgical therapy, those who are poor surgical candidates, or those in whom a single source of hyperinsulinism cannot be found.27
Laparoscopic surgery
Laparoscopic surgery offers many advantages to patients, including shorter hospital stay, reduced pain, smaller incisions and faster recovery. Because insulinomas are typically small, benign and limited to the pancreas, laparoscopic surgery can offer an ideal way to treat these patients. Both enucleation and subtotal pancreatectomy, with or without splenectomy, may be performed laparoscopically.28,29 Good candidates for a minimally invasive approach have benign-appearing tumours that are well localised on preoperative studies. Some recommend that candidates have tumour confined to the body and tail of the pancreas; however, it is also possible to laparoscopically enucleate an insulinoma in the pancreatic head. Even after successful preoperative localisation, tumour location should be confirmed intraoperatively through the use of laparoscopic IOUS. Because palpation is not possible during laparoscopic exploration, the availability of laparoscopic IOUS and an experienced operator is essential. Several published series indicate that laparoscopic pancreatic resection is safe in experienced hands, although operation times are typically longer than for an open procedure.30,31 Complications include pancreatic fistula, and some procedures will require conversion to open surgery.
Outcome
Most patients with insulinoma are cured of hypoglycaemia and return to a fully functional lifestyle with normal long-term survival (Table 5.4). Appropriate localisation of sporadic insulinoma and complete surgical resection results in a cure rate of greater than 95%.2,11 Symptoms resolve postoperatively and the fasting serum concentration of glucose normalises. Although successful resection of the tumour(s) responsible for hyperinsulinism renders most MEN1 patients asymptomatic postoperatively,32 persistent or recurrent hypoglycaemia due to a missed insulinoma or metastatic disease from the original tumour may develop.
Gastrinoma
Each year, approximately 0.1–3 people per million population develop gastrinoma, the second most common functional pNET (Table 5.1).33 The clinical features of this tumour were first described by Zollinger and Ellison in 1955.34 Because of an increased awareness of Zollinger–Ellison syndrome (ZES) and the widespread availability of accurate immunoassays to measure serum concentrations of gastrin, gastrinoma is increasingly diagnosed and treated at an early stage of disease. However, the mean time from symptoms to diagnosis is still as long as 8 years, so improvements in detection are needed.
Patient presentation
Gastrinomas secrete excessive amounts of the hormone gastrin, causing acid hypersecretion, which results in epigastric pain, diarrhoea and oesophagitis. The most common presenting symptoms are those of peptic ulcer disease. Diarrhoea, caused by gastrin-induced hypersecretion and increased bowel motility, is the second most common symptom and may be the only manifestation of ZES in 20% of patients. Oesophagitis with or without stricture occurs with more severe forms of the syndrome. Approximately 20% of patients with ZES will have it as part of MEN1,1
[/not-level-membership-for-endocrinology-diabetes-and-metabolism-category]
