CHAPTER 32 Endocrine Tumors of the Pancreas and Gastrointestinal Tract
GENERAL CONSIDERATIONS
HISTORICAL ASPECTS
In 1927,1 five years after the discovery of insulin, the first pancreatic hormone-producing tumor syndrome was described in a patient with a metastatic islet cell tumor and hypoglycemia, and whose tumor extracts had hypoglycemic effects. Numerous other pancreatic endocrine tumors (PETs) have since been described (Table 32-1), with the description of Zollinger-Ellison syndrome in 1955,2 the Verner-Morrison syndrome caused by a diarrheogenic-producing tumor in 1958,3 glucagonoma syndrome by Mallinson in 1974,4 the somatostatinoma syndrome in 1977,5,6 and GRFomas (pancreatic tumors secreting growth hormone-releasing factor) in 1982.7,8 PETs secreting adrenocorticotropic hormone (ACTH; ACTHomas) are also included because 4% to 16% of cases of ectopic Cushing’s syndrome are caused by ACTH-secreting PETs.9–11 PETs causing the carcinoid syndrome (see Chapter 31),12 tumors secreting renin and causing hypertension,13 luteinizing hormone (LH) resulting in masculinization or changes in libido,14 erythropoietin resulting in polycythemia,15 and parathyroid hormone-related protein (PTHrP), resulting in hypercalcemia,16 have also been described. PETs secreting calcitonin17 are proposed to cause a distinct syndrome with diarrhea. However, too few cases have been well described to include this syndrome. Furthermore, other causes of hypercalcitonemia, such as medullary thyroid cancer, are only associated with diarrhea in 25% to 42% of patients.18 Other functional hormonal syndromes have been described, with nonpancreatic, primarily intra-abdominal neuroendocrine and non-neuroendocrine tumors, including secretion of GLP-2 (glucagon-like peptide-2), causing intestinal villous hypertrophy (enteroglucagonomas) and secretion of GLP-1 causing hypoglycemia and delayed GI transit,19 and intestinal and ovarian tumors secreting peptide YY resulting in altered intestinal motility and constipation.20 PETs secreting ghrelin have been described21 but were not associated with acromegaly, increased serum growth hormone levels, or increased insulin growth factor-1 (IGF-1) concentrations.
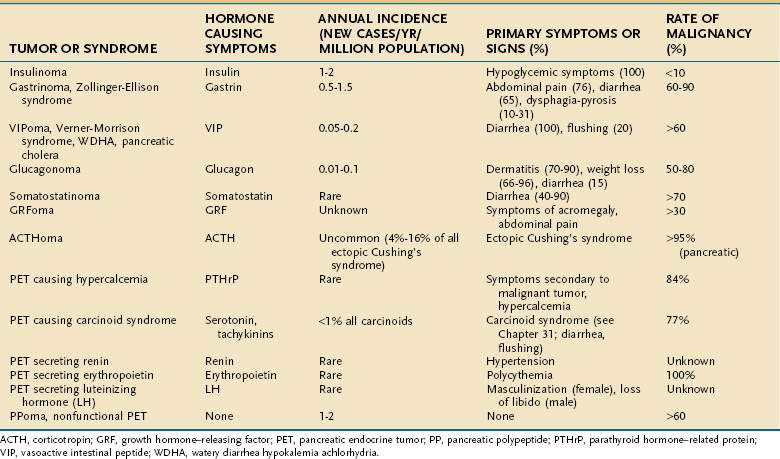
PETs are classified as functional if associated with a clinical syndrome caused by hormone release by the tumor or nonfunctional if not associated with a clinical syndrome caused by hormone release (see Table 32-1). In the nonfunctional category are included nonfunctional PETs (NF-PETS), which have the histologic characteristics of a PET but no associated elevation in plasma hormone levels or clinical syndrome, as well as PETs that release pancreatic polypeptide (PPomas), ghrelin, neurotensin (neurotensinomas), or other peptides that do not cause a distinct clinical syndrome.9,22,23
PREVALENCE AND INCIDENCE
PETs account for 1% to 10% of tumors arising in the pancreas.22,24,25 The overall prevalence of functional PETs is low, reported to be approximately 10/million (1/100,000). In contrast, the prevalence of PETs in autopsy studies is higher, 0.5% to 1.5%.26 The annual incidence of PETs is reported at 1 to 4 cases/million/year.9 Nonfunctional PETs account for 14% to 30% of all PETs in most studies, but they are as high as 60% to 80% in some studies.27 Insulinomas and gastrinomas occur with an equal annual incidence of 0.5 to 3 cases/million.28–30 VIPomas are 12.5% as common and glucagonomas 6% as common as insulinomas and gastrinomas. Somatostatinomas are very rare31,32 and the incidence of GRFomas, PETs secreting renin, erythropoietin, or LH or PETs causing hypercalcemia is unknown (see Table 32-1).
ORIGIN AND HISTOLOGIC FEATURES
PETs are often called islet cell tumors but it is unproven that they originate from the pancreatic islets.9,33,34 These tumors frequently contain ductular structures, produce hormones not normally present in the adult pancreas, such as gastrin and vasoactive intestinal peptide (VIP), and may produce multiple hormones.35 It has been suggested that these tumors represent a dedifferentiation of an immature stem cell. The finding of the ductular structures in many PETs and the budding off of endocrine cells from ductules during ontogenesis of the pancreas has led to the suggestion these tumors are ductular in origin.36
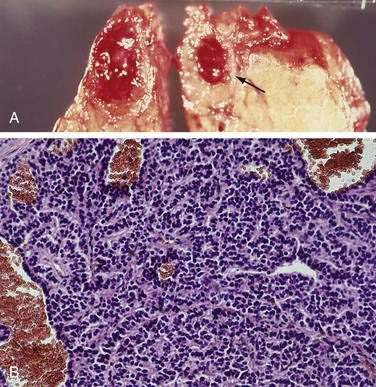
It was originally proposed that PETs might originate from cells that are part of the diffuse neuroendocrine cell system.9,34,37,38 These cells share certain cytochemical properties (amine precursor uptake and decarboxylation) and the tumors have been called APUDomas.33 Ultrastructurally, the cells often have electron-dense granules and produce multiple regulatory hormones and amines, neuron-specific enolase, synaptophysin, and chromogranin A or C. These cells are thought to give rise to carcinoid tumors, medullary carcinomas of the thyroid, melanomas, and pheochromocytomas, and there are marked similarities in the histology of these tumors and PETs. Histologically, PETs consist of a relatively homogeneous sheet of small round cells with uniform nuclei and cytoplasm (Fig. 32-1). Mitotic figures are uncommon.33 Malignancy can be determined only by metastases or invasion and cannot be predicted by light microscopic or ultrastructural studies.20,28 At present, controversy still surrounds the exact cell of origin for PETs, with some recent studies suggesting they originate from islet cells and others supporting a ductular origin.34,36
Most PETs produce multiple gastrointestinal hormones, which can be localized by immunocytochemical methods.9,33 In many studies, most functional and nonfunctional PETs had cells immunoreactive to peptides that were not causing clinical symptoms.35 It is unclear why usually only one or no clinical syndrome is seen, despite the immunochemical occurrence of multiple hormones.28 A functional PET syndrome should be diagnosed only if the appropriate clinical symptoms are present, not only based on immunocytochemistry.
PETs frequently produce chromogranins or the alpha or beta subunit of human chorionic gonadotropin (HCG), which can be localized by immunocytochemistry or by documenting elevated circulating levels.9,28,33 Chromogranins are water-soluble acidic glycoproteins that are present in almost all endocrine or neuronal tissues.26 Plasma chromogranin A levels are elevated in more than 90% of patients with various PETs and carcinoid tumors (see Chapter 31). Although some have suggested that elevations of the alpha or beta subunit of HCG or of chromogranin A may be indicative of malignancy, this is not established.9,28 Furthermore, some studies have reported that serial measurements of chromogranin levels may be useful for monitoring tumor growth39–42 or as a marker of survival.43
In an occasional patient, a second clinical hormone tumor syndrome may be present initially or develop with time.9,28 Whereas one study reported that this occurred in 7% of all patients with PETs during a three-year follow-up,9 another study35 has reported that this is a rare occurrence, occurring at a rate of 2/100 followed over ten years. However, there appears to be a high incidence of the development of Cushing’s syndrome in patients with a functional PET, especially in those with gastrinomas.44,45
CLASSIFICATION
PETs are classified clinically according to the functional syndrome produced (see Table 32-1). Although clinical syndromes have been attributed in some studies to patients with PETs who had elevated plasma levels of neurotensin or pancreatic polypeptide (PP), the existence of these syndromes has not been established with certainty.9,35,46 PETs can be associated with four different inherited disorders: multiple endocrine neoplasia type I (MEN-I), von Hippel-Lindau disease, tuberous sclerosis, and neurofibromatosis-1 (von Recklinghausen’s disease).47–49 These association are important to recognize because family screening may be needed and because these PETs may have a different natural history (see later).28
It has been proposed9,33 that the terms PET and carcinoid tumor be replaced by the term neuroendocrine tumor (NET) and a new classification based on clinical and morphologic categories be used (see Chapter 31). This WHO classification classifies all NETs into well-differentiated endocrine tumors or carcinomas, poorly differentiated endocrine tumors, or mixed exocrine-endocrine tumors, which better allows comparisons of NETs from the pancreas and other gastrointestinal sites. The well-differentiated NETs of the pancreas are specifically divided into well-differentiated tumors and well-differentiated endocrine carcinomas. The well-differentiated endocrine tumors are further divided into those with benign behavior (confined to pancreas, nonaggressive, <2 cm in size, functional or nonfunctional), and uncertain behavior (confined to pancreas, >2 cm in size, or with angioinvasion, functional or nonfunctional). The well-differentiated endocrine carcinomas of the pancreas show low-grade malignancy, with gross invasion and/or metastases and can be functional or nonfunctional. In this chapter, the term PET will be retained because of its widespread use. Recently, for the first time, a TNM classification has been proposed for PETs and for other GI neuroendocrine tumors (see Table 31-1).50 This TNM classification is based on the level of tumor invasion, tumor size, tumor extent, and with grading using the mitotic index or the proliferative index, Ki-67. The importance of these classification systems for predicting prognosis will be discussed in a later section.
PATHOPHYSIOLOGY
In various series, in a small percentage of the patients with a functional syndrome in whom no tumor was found preoperatively and at the time of surgery, hyperplasia of the pancreatic islets was regarded as a possible cause of the disease.9,28 Beta cell hyperplasia or nesidioblastosis, which is a subtype of beta cell hyperplasia consisting of the proliferation of islet cells from pancreatic ducts, is reported to be a cause of hypoglycemia and hyperinsulinemia in a number of infants and newborns. Recently, this condition has been recognized in adolescents and adults and occurs in 5% of patients with hyperinsulinism.51,52 It has been suggested that Zollinger-Ellison syndrome and Verner-Morrison syndrome are caused in up to 10% of cases by hyperplasia of pancreatic ducts, producing gastrin and VIP, respectively. However, this concept has not been substantiated by immunocytochemical studies and thus is not generally accepted.
MOLECULAR PATHOGENESIS
Until recently, the molecular pathogenesis of neuroendocrine tumors (NETs) (carcinoids and PETs) was largely unknown.53,54 Numerous studies have demonstrated that in contrast to most common nonendocrine tumors (e.g., colonic or pancreatic adenocarcinoma), mutations in common oncogenes (e.g., ras, fos, myc, src, jun) and common tumor suppressor genes (e.g., p53, retinoblastoma gene) are uncommon in most NETs (carcinoids, PETs).26,38,55 Recent studies have provide evidence that alterations in the MEN-I gene, p16/MTS1 tumor suppressor gene, DPC4/Smad 4 gene, amplification of the HER-2/neu proto-oncogene, increased expression of growth factors and/or their receptors (endothelial growth factor [EGF], hepatocyte growth factor, platelet-drived growth factor [PDGF]), and deletions of a possible unknown tumor suppressor gene on chromosome 1 or 3p may all be important.26,38,53–57 Alterations in the MEN-I gene occur in up to one third of sporadic (i.e., noninherited) PETs58 and alterations in the p16/MTS1 gene occur in 50% to 92% of PETs and thus may be particularly important. Alterations in the MEN-I gene are discussed in the next section. Genomic-wide allelotyping and comparative genomic hybridization studies have demonstrated that chromosomal losses (especially on 1p, 3p, 3q, 6q, 9q, 12q) and chromosomal gains (especially in 7q, 17q, 17p, 20q) frequently occur in PETs and carcinoids, but their frequency varies markedly in these two gastrointestinal (GI) neuroendocrine tumors, providing evidence that they have a different molecular pathogenesis. Gene expression profiling using microarrays has identified a large number of genes that are altered in PETs in comparison to normal islets or between PETS with different degrees of aggressiveness.26,59–62 At present, it is not clear which of the different genes altered in the different studies will have important prognostic implications or provide important insights into their pathogenesis that will lead to new treatments.
MULTIPLE ENDOCRINE NEOPLASIA
There are three well-established MEN syndromes that can be distinguished by the presence or absence of PETs; medullary thyroid carcinoma, parathyroid disease, pheochromocytoma and a specific phenotype.47,49,63 Each of these syndromes—MEN type I, MEN type IIa, and MEN type IIb—has autosomal dominant inheritance. MEN-I, or Wermer’s syndrome, is considered in detail later; it is characterized by hyperparathyroidism and PETs without the presence of medullary thyroid carcinoma, pheochromocytoma, or unusual phenotype. MEN-IIa, or Sipple’s syndrome, is characterized by bilateral medullary thyroid carcinoma and pheochromocytomas (in 20% to 40%); when they occur, they are bilateral in 70%, with hyperparathyroidism in 17% but without the occurrence of PETs or a specific phenotype. MEN-IIb includes bilateral medullary thyroid carcinoma, which often appears at an early age and appears to be more aggressive than these tumors in patients with MEN-IIa. Pheochromocytomas, when they occur, are bilateral in 70%. Parathyroid disease is seldom present in MEN-IIb, and patients have a characteristic phenotype, with multiple mucosal neuromas, frequently marfanoid habitus, puffy lips, prominent jaw, pes cavus, and medullated corneal nerves, but no PETs. The genetic defect in MEN-I is located on the long arm of chromosome 11 and is caused by mutations in a 10-exon gene encoding for a 610–amino acid protein, MENIN, a nuclear protein that interacts with the AP1 transcription factor, Jun D, nuclear factor κB (NF-κβ), pem, SMAD3, RPA2 (a DNA processing factor), FAN CD2 (a DNA repair factor), nucleoside diphosphate kinase, NM23β, and various cytoskeleton-associated proteins.64
The development of MEN-I endocrine tumors conformed to Knudsen’s9 two-hit model theory of neoplasm, with an inherited mutation in one chromosome unmasked by a somatic deletion or mutation of the other normal chromosome, thereby removing the suppressor effect of the normal gene. In PETs from patients without MEN-I, up to 90% have loss of heterozygosity on chromosome 11 and 27 % to 39% have mutations in the MEN-I gene.26,53,54,58 This suggests that sporadic PETs share a similar tumorigenesis to PETs that occur in patients with MEN-I, which principally involves deletion of a tumor suppressor gene. The MEN-II syndromes are caused by alterations in the pericentromeric region of chromosome 10 in the RET proto-oncogene, which is a 21-exon gene encoding for a tyrosine kinase receptor. Mutations in a cysteine-rich extracellular portion of the receptor primarily cause MEN-IIa, whereas mutations in the gene region encoding the intracellular catalytic core of the tyrosine kinase domain cause MEN-IIb.
In patients with MEN-I, hyperparathyroidism is the most common clinical abnormality, occurring in 78% to 97% (Table 32-2).9,28,47–49 Functional PETs are the second most common clinical abnormality, occurring in 81% to 82% of patients. Gastrinomas occur in 54%, whereas insulinomas, glucagonomas, and VIPomas occur in 18%, 3%, and 3% of patients, respectively.65 Nonfunctional PETs and PPomas may be the most common PET in patients with MEN-I because they are almost always found in histologic studies.66 However, large nonfunctional PETs causing symptoms occur in only 0% to 13% in various series. Many patients without a functional PET do not routinely undergo surgical exploration,56,67,68 and imaging studies routinely miss most small PETs smaller than 1 cm.26,69 Therefore, the true incidence of asymptomatic PETs in these patients is unknown.
Table 32-2 Frequency of Features of Patients with Multiple Endocrine Neoplasia Type I
| FEATURE | AVERAGE FREQUENCY (RANGE), % OF ALL PATIENTS |
|---|---|
| Hyperparathyroidism | 97 (78-100) |
| Pancreatic endocrine tumors | 81-82 |
| Nonfunctional or PPomas | 80-100 (microscopic), 0-13 (symptomatic) |
| Gastrinomas | 54 (20-61) |
| Insulinomas | 18 (7-31) |
| Glucagonomas | 3 (1-6) |
| VIPomas | 1 (1-12) |
| Somatostatinomas | 0-1 |
| GRFoma | <1 |
| Pituitary tumors | 54-65 (15-100) |
| Prolactin-secreting | 15-46 |
| Growth-hormone secreting | 6-20 |
| Cushing’s syndrome | 16 |
| Adrenal tumors | 27-36 (symptomatic, <2%) |
| Cortical adenomas | |
| Hyperplasia, carcinoma (uncommon) | |
| Carcinoid tumors | |
| Gastric (ECLoma) | 7-35 (symptomatic, <5%) |
| Lung | 0-8 |
| Thymic | 0-8 |
| Skin tumors | 40-100 (angiofibromas) |
| Angiofibromas > collagenoma > café-au-lait macules > lipomas | 88, 72, 38, 34 (symptomatic, <1%) |
| CNS tumors—meningiomas, ependymomas, schwannomas | 0-8, 0-1 (symptomatic, <1%) |
| Smooth muscle tumors—leiomyomas, leiomyosarcomas | 1-7 (symptomatic, <1%) |
| Thyroid tumors—adenomas | 0-10 (0-30; symptomatic, <1%) |
CNS, central nervous system; ECL, enterochromaffin-like cell; GRF, growth hormone-releasing factor; PP, pancreatic polypeptide; VIP, vasoactive intestinal peptide.
From references 28, 46–49, and 65.
Pathology studies9,47,48,66 have demonstrated that in almost every patient with MEN-I, the pancreas demonstrates diffuse microadenomatosis, with or without larger tumors. With immunocytochemistry, PP is most frequently seen followed by glucagon and insulin, with gastrin rarely found. These results are consistent with clinical studies that have demonstrated that gastrinomas in more than 80% of patients with MEN-I and Zollinger-Ellison syndrome are located in the duodenum.28,67,68,70,71
MEN-I is present in 20% to 25% of patients with gastrinomas, 4% of patients with insulinomas, 13% to 17% of patients with glucagonomas, 33% of patients with GRFomas, 9% of patients with VIPomas, and 7% with somatostatinomas.9,27,28,47,48,72–78 Characteristically, hyperparathyroidism is the initial manifestation of MEN-I, usually presenting in the third decade of life, followed by the development of a PET in the fourth to fifth decade.49 It is important to recognize whether a patient has MEN-I because patients with and without MEN-I differ in their clinical presentation, in the possibility of surgical cure, and in the clinical and diagnostic approach to the tumor.56,67,68 Patients with MEN-I may develop more than one PET over time so long-term follow-up will differ from that of a patient without MEN-I. Screening of other family members will be indicated in patients with MEN-I, whereas it will not be in patients with sporadic disease. In some PETs, the presence of the hypercalcemia caused by the hyperparathyroidism may affect release of the hormones by the tumor.79
OTHER INHERITED SYNDROMES ASSOCIATED WITH PANCREATIC ENDOCRINE TUMORS
Three inherited phacomatoses have an increased occurrence of PETs—von Hippel-Lindau disease (VHL), von Recklinghausen’s disease (neurofibromatosis-1 [NF-1]), and Bourneville’s disease (tuberous sclerosis).26,46,47 VHL is caused by a defect on chromosome 3p25 encoding for a 232–amino acid protein, pVHL, that forms a complex with a number of proteins, including elongin B and C as well as Cullin 2, which regulate ubiquitin-dependent proteolysis of large cell proteins.80 VHL mutations result in altered transcriptional regulation, resulting in pathologic changes in angiogenic, growth, and mitogenic factors. In 10% to 17% of patients with VHL, a PET is seen which is usually asymptomatic and nonfunctional (>98%), although occasional insulinomas and VIPomas are described. The mean age at diagnosis of a PET in VHL is 29 to 38 years. Most patients have a single PET (67% to 70%). Malignant PETs occurs in 8% to 50% of VHL patients with PETs, with liver metastases in 9% to 37%.
NF-1 is caused by a defect on chromosome 17q11.2 encoding for a 2845–amino acid protein, neurofibromin, which functions as a ras signaling cascade inhibitor.9,81 In various series, from 0% to 10% of NF-1 patients develop a carcinoid tumor, usually in the periampullary region (54%) of the duodenum (see Chapter 31).78,82 Most of these duodenal carcinoids are somatostatinomas by immunocytochemistry, but they rarely produce the somatostatinoma syndrome.47 NF-1 has rarely been associated with Zollinger-Ellison syndrome and insulinomas.46 NF-1 accounts for 48% of all duodenal somatostatinomas and approximately 25% of all ampullary carcinoid tumors. These tumors frequently (63%) show psammoma bodies histologically, and metastases to liver and or lymph nodes occur in 30%.
Tuberous sclerosis is caused by mutations in the 1164–amino acid protein, hamartin (TSC-1), or the 1807–amino acid protein, tuberin (TSC-2).47,83 These two proteins are important in regulating the PI3K signaling cascade (see Chapter 3) and also for regulation of the small GTPase, Rheb, which play important roles in the regulation of protein translation and synthesis, growth, and proliferation, as well as maintenance of cellular energy levels. A few cases of nonfunctional and functional PETs (insulinomas and gastrinomas) have been reported in patients with tuberous sclerosis.26,46
INSULINOMAS
DEFINITION
Insulinomas are insulin-secreting tumors that primarily originate in the pancreas and cause symptoms as a result of hypoglycemia (Table 32-3).
Table 32-3 Frequency of Symptoms and Signs in Patients with Insulinoma
| FINDING | FREQUENCY (%) |
|---|---|
| Any Time During Clinical Course | |
| Neuropsychiatric symptoms (loss of consciousness, confusion, dizziness, diplopia) | 92 |
| Confusion or abnormal behavior | 80 |
| Obesity | 52 |
| Amnesia or coma | 47 |
| Cardiovascular symptoms, palpitations, tachycardia | 17 |
| Convulsions (grand mal) | 12 |
| Gastrointestinal symptoms (hunger, vomiting, pain) | 9 |
| During First Attack | |
| Neuroglycopenic symptoms | |
| Visual disturbances (diplopia, blurred vision) | 59 |
| Confusion | 51 |
| Altered consciousness | 38 |
| Weakness | 32 |
| Transient motor defects, hemiplegia | 29 |
| Dizziness | 28 |
| Fatigue | 27 |
| Inappropriate behavior | 27 |
| Speech difficulty | 24 |
| Headache | 23 |
| Seizure | 23 |
| Syncope | 21 |
| Difficulty concentrating or thinking | 19 |
| Paresthesias | 17 |
| Memory loss | 15 |
| Lethargy | 12 |
| Stupor | 12 |
| Amnesia | 8 |
| Ataxia | 4 |
| Disorientation | 4 |
| Mental change | 4 |
| Adrenergic symptoms | |
| Sweating | 43 |
| Tremulousness | 23 |
| Hunger, nausea | 12 |
| Palpitations | 10 |
PATHOPHYSIOLOGY AND PATHOLOGY
Insulinomas almost always (98.2%) occur in or are attached to the pancreas.9,26,29,30 An occasional insulinoma presenting as a carcinoid tumor has been reported in the duodenum, ileum, and lung, but truly ectopic insulinomas are rare (1% to 3%).84 Insulinomas are evenly distributed in the pancreas, with approximately one third in the pancreatic head, body, and tail.9,26,84–86 Insulinomas are usually small. In one large series, 5% were less than 0.5 cm, 34% were 0.5 to 1 cm, 53% were 1 to 5 cm, and only 8% were more than 5 cm.85
Insulinomas are usually solitary, with multiple tumors occurring in only 2% to 13% of cases.9,26,29,30 If multiple insulinomas are found, MEN-I should be suspected.47 Insulinomas are generally well encapsulated, firmer than normal pancreas, and highly vascular. Only 5% to 16% of insulinomas are malignant.87 Malignant tumors are generally larger, averaging 6 cm in one series, and 5% of patients have metastases at presentation.88 Metastases are usually to the liver (47%), regional lymph nodes (30%), or both.
Among adults with hyperinsulinism and pancreatic islet cell disease, histologic studies have shown a solitary insulinoma in 86% of cases, adenomatosis in 5% to 15%, nesidioblastosis in 4%, and islet hyperplasia in 1%.87 Adenomatosis consists of multiple macroadenomas or microadenomas and occurs especially in patients with MEN-I. A second diffuse lesion is nesioblastosis, a condition in which islet cells bud off from ductular structures and are mixed with lobular elements. This condition previously was reported almost exclusively in infants and children, but has been recognized in 5% of adults and adolescents with hyperinsulinism.9,51,89 Diffuse islet cell hyperplasia, which consists of excessive and diffuse proliferation of beta cells in the islets, has been reported in adults.85 Of 1137 cases of organic hyperinsulinism, only 6% had diffuse islet cell hyperplasia and another 0.6% had both an insulinoma and diffuse islet cell disease. At present, it is unclear whether many of these cases were in fact nesioblastosis because in most cases appropriate immunofluorescence staining methods were not applied.
Insulin is synthesized and stored in beta cells of the pancreatic islets.29,30 Insulin is synthesized in the rough endoplasmic reticulum as preproinsulin, from which proinsulin is liberated and transferred to the Golgi of the cell.90 Proinsulin consists of a 21–amino acid alpha chain and a 30–amino acid beta chain connected by a 33–amino acid connecting peptide (C-peptide). In secretory granules, a protease excises the C-peptide and thus, when secretion occurs, the C-peptide and the double-stranded insulin molecule are released in equimolar amounts. Small amounts of intact proinsulin remain in granules and are also released; this can be detected in the plasma. Proinsulin contains the alpha and beta chains of insulin and, because most insulin antibodies used in radioimmunoassays recognize moieties on these chains, they also recognize proinsulin. Normal subjects have less than 25% of their total serum insulin as proinsulin, whereas over 90% of patients with insulinomas have an elevated proportion of proinsulin relative to total insulin.91
CLINICAL FEATURES
Insulinomas can occur at any age but are rare in adolescents, usually occurring in patients between 20 to 75 years. A large majority occur between the ages of 40 to 45 years and 60% of patients are women.9,26,29,30 Symptoms are caused by hypoglycemia (see Table 32-3) characteristically associated with fasting and thus more frequently occur when a meal is delayed, missed, or before breakfast. Symptoms may also occur during exercise. In one study,87 26% of patients had symptoms during or after an overnight fast, 27% had symptoms prior to lunch or dinner, 8% had symptoms only after a missed meal, 29% had symptoms only before lunch or dinner, and only 9% were uncertain about when their symptoms occurred. The hypoglycemia with fasting or exercise, which is characteristic of insulinomas, differs temporally from hypoglycemia, which occurs after meals (postprandial hypoglycemia). Postprandial hypoglycemia can be caused by a number of other unrelated conditions and is increasingly being reported in patients after various gastric bypass surgery procedures for obesity.92 Most symptoms of insulinomas (82% to 92% of patients)85,93 are caused by neuroglycopenia, because glucose is the main source of energy for the brain. Neuroglycopenic symptoms include somnolence, visual disturbances, irritability, abnormal behavior, confusion, amnesia, paresthesias, stupor, drowsiness, coma, and seizures. Symptoms of hypoglycemia can also be caused by catecholamine release (adrenergic symptoms) and include anxiety, palpitations, weakness, fatigue, headache, tremor, and sweating. Coma occurs in up to 53% of patients and convulsions in 12%. In one study of symptoms of a first attack, 49% of patients initially had both neuroglycopenic and adrenergic symptoms, 38% had neuroglycopenic symptoms only, 12% had adrenergic symptoms only, and 1% had no symptoms. Of the neuroglycopenic symptoms, visual disturbances (57%), confusion (51%), and altered consciousness (38%) are the most common. Of the adrenergic symptoms, sweating (43%) and tremulousness (23%) are the most common. Patients frequently learn to avoid symptoms by eating frequently and obesity may result. In one study,87 40% of patients with organic hypoglycemic were overweight. The average duration of neuroglycopenic symptoms prior to diagnosis is often prolonged, being more than three years in 25% of patients and more than five years in 20%.
DIAGNOSIS AND DIFFERENTIAL DIAGNOSIS
The key to establishing the diagnosis of insulinoma is suspecting by clinical history—that the symptoms could be caused by hypoglycemia—and establishing the relationship of the symptoms to fasting.9,29,30,89 Whipple’s triad, published in 1938, and long used as diagnostic criteria for insulinoma, was based on this association, consisting of characteristic hypoglycemia symptoms, the presence of hypoglycemia (blood sugar < 50 mg/dL), and relief of symptoms following glucose ingestion. Unfortunately, these symptoms are not specific for insulinoma.87
Organic hypoglycemia is generally defined as a fasting blood glucose level lower than 40 mg/dL. In healthy individuals, after an overnight fast, plasma glucose values usually do not decrease below 70 mg/dL.87 After an overnight fast, only 53% of patients with insulinoma have a blood glucose level below 60 mg/dL and only 39% below 50 mg/dL. However, if a blood glucose determination is combined with a concomitant plasma insulin level, this insulin level will be inappropriately elevated in 65% of patients. Hypoglycemia can be classified as a fasting hypoglycemia or postprandial (reactive) hypoglycemia, of which there are a number of different causes (Table 32-4). The distinction can usually be made by a careful clinical history. Because a single overnight fasting blood sugar level, even when combined with a simultaneous plasma insulin level, does not establish the presence of fasting organic hypoglycemia in more than 35% of patients with organic hyperinsulinism, an extended fast is done with blood glucose, plasma insulin, and C-peptide levels measured at three- to six-hour intervals.29,30,89,93 Traditionally, a 72-hour fast is performed, although one study has proposed that a 48-hour fast is sufficient.94 If at any point during the fast the patient becomes symptomatic, plasma insulin and glucose values should be determined before intravenous glucose is given and the test stopped. Within 24 hours of starting the fast, 75% to 80% of patients with an insulinoma will have symptoms and a blood sugar level lower than 40 mg/dL, by 48 hours in 90% to 98%, and by 72 hours in almost 100%.9,86 In nonobese normal subjects, serum insulin concentrations decrease to lower than 6 µU/mL when blood glucose levels decrease to lower than 40 mg/dL, and the ratio of plasma insulin (in µU/mL) to glucose (in mg/dL) remains lower than 0.3. The test is considered positive for insulinoma if the plasma insulin-to-glucose ratio is above 0.3. In some normal obese subjects, because of hyperinsulinemia caused by insulin resistance, the fasting plasma insulin to glucose ratio may be above 0.3 and therefore may mimic the pattern in insulinoma. In these patients, the fasting glucose is normal and above 0.3 and does not decrease to lower than 55 mg/dL with fasting, as occurs in patients with insulinomas.
Table 32-4 Causes of Spontaneous Hypoglycemia
From references 9, 29, 30, 87, and 93.
Two additional important aspects of the diagnosis of insulinoma should be remembered. First, until recently, most the serum insulin levels were determined by radioimmunoassays that in many cases, had some cross-reactivity with proinsulin. Currently, insulin-specific radioimmunoassays (either immunoradiometric assay [IRMA] or immunochemoluminescence), which have no cross-reactivity with proinsulin, are being increasingly used instead of the classic radioimmunoassay.91,93,95 Using these newer assays, insulin levels below 6 µU/mL can be found in more than half of patients with hypoglycemia caused by insulinomas. In a recent study91 of 33 patients with insulinomas and 67 controls, the most sensitive and specific criteria for diagnosis of insulinoma during a 72-hour fast was the combination of a fasting glucose level lower than 45 mg/dL with an elevated proinsulin level. In an occasional patient with classic symptoms of an insulinoma, insulin levels and the insulin-to-glucose ratio remain within the normal range when assessed by the new insulin IRMA assay.96 Such patients can be secreting proinsulin, and serum proinsulin levels should be assessed using a specific proinsulin radioimmunoassay.
A number of conditions (see Table 32-4) in addition to insulinoma can cause fasting hypoglycemia, including organic hyperinsulinism caused by pancreatic islet disease, factitious use of excessive insulin or hypoglycemic agents, and autoantibodies against the insulin receptor or insulin.89,93 To differentiate these conditions, measurements of plasma proinsulin, C-peptide, antibodies to insulin, and plasma sulfonylurea levels are carried out.9,29,30,87 The plasma proinsulin level is elevated in 80% to 90% of patients with insulinoma to more than 22% of the plasma insulin level.87,91,95 In patients with surreptitious use of insulin or oral hypoglycemic agents, the proinsulin level is decreased or normal. C-peptide is released in equimolar quantities to insulin into the plasma; thus, it would be expected to be proportionately elevated with insulin in insulinomas. However, it has a much longer plasma half-life than insulin and thus the plasma level shows less fluctuation than insulin. The measurement of C-peptide has proven useful in differentiating organic hypersecretion of insulin, such as in patients with insulinoma from patients surreptitiously using insulin because commercial insulin preparations contain no C-peptide. In insulinoma, the characteristic findings are an elevated or normal plasma C-peptide concentration, whereas in patients surreptitiously using insulin the plasma insulin level will be high and the C-peptide level low. The C-peptide level does not differentiate patients surreptitiously taking oral hypoglycemic agents from patients with insulinomas, in that both have low blood sugar, elevated insulin, and elevated C-peptide levels.9 Various provocative tests with tolbutamide, leucine, and glucagon and secretin and suppression tests have all been described for the diagnosis of insulinoma; however, each had its limitations and are currently seldom used.
TREATMENT
Treatment of insulinoma consists of two different approaches. Initial treatment is directed at controlling the symptoms of hypoglycemia; then, after tumor localization studies, at a possible surgical cure. Tumor localization of all PETs involves similar approaches (see later). For the 5% to 13% of patients with metastatic insulinoma, chemotherapy or other therapies directed at the tumor itself may need to be considered.88,97 This latter group will be considered in a later section on treatment of metastatic PETs.
Medical Therapy
Hypoglycemia is controlled in most patients by a combination of diet and pharmacologic therapy. The use of appropriately timed feedings with a bedtime snack or midmorning, midafternoon, or 3 am snack may be sufficient to control all symptoms.29,30,87 It is generally advised not to restrict intake to rapidly absorbed carbohydrates because their ingestion may occasionally stimulate insulin secretion from the tumor. More slowly absorbed forms of carbohydrates, such as starches, bread, potatoes and rice, are preferable. During a hypoglycemic episode, however, rapidly absorbable forms of carbohydrate such as fruit juice with glucose or sucrose are preferable. Occasional patients with severe hypoglycemia may require the use of a continuous intravenous infusion of glucose together with an increase in dietary carbohydrates.
A number of drugs have been reported to combat the hyperinsulinemia. Diazoxide, which is a nondiuretic benzothiazide analog, has potent hyperglycemic effects.29,30,98 It directly inhibits insulin release from beta cells through stimulation of α-adrenergic receptors and also has an extrapancreatic hyperglycemic effect that enhances glycogenolysis.87 The major side effects of diazoxide are sodium retention (47%) and even edema, GI symptoms such as nausea, and hirsutism. Addition of a diuretic such as trichlormethiazide, a benzothiadiazide derivative, can correct the edema as well as augment the hyperglycemic effect of diazoxide. The GI side effects can be reduced by taking the diazoxide with a meal. Diazoxide therapy should be initiated with 150 to 200 mg given in two to three divided daily doses and, if not effective, increased to a maximum of 600 to 800 mg/day. Side effects are dose-related and may limit the ability to reach maximal doses. Approximately 60% of patients will respond to diazoxide. Patients have been treated for over 20 years with diazoxide, demonstrating that it can be used long-term.88 Verapamil, propanolol, phenytoin, and glucocorticoids have been reported effective in occasional patients, but they may have only minor hyperglycemic effects and their successes are anecdotal.9
The long-acting somatostatin analog, octreotide, has controlled hypoglycemia in a number of cases of insulinoma.9,99–102 This compound has a half-life of 100 minutes, 33 times longer than that of native somatostatin, and thus can be self-administered two to four times daily subcutaneously. Octreotide controls symptoms and hypoglycemia in 40% to 60% of patients. Somatostatin analogs are thought to act primarily on PETs such as insulinomas by interacting with high-affinity somatostatin receptors on the tumor. Five subtypes of somatostatin receptors exist and octreotide (or lantreotide) has high affinity for subtypes 2 and 5, which have been shown to be present on PETs.103 The response rate of insulinomas to octreotide is likely lower than that of other PETs because they frequently possess low levels of somatostatin receptors, whereas the other PETs possess high densities of receptors, with high affinity for these analogs, in 80% to 90% of cases. Octreotide is generally well tolerated. It is usually given in a starting dose of 50 µg two or three times daily and can be increased to dosages as high as 1500 µg/day. A rather recent significant advance is the availability of long-acting depot forms of somatostatin analogs (octreotide long-acting release [octreotide LAR] and lanreotide slow-release [lanreotide SR] or lanreotide autogel) that last two to four weeks.104 After a 30-mg dose, octreotide LAR maintains a plasma octreotide level of 1 ng/mL or higher for 25 days and therefore can be given monthly. The main side effects of octreotide treatment include gastrointestinal symptoms such as bloating and abdominal cramping. Long-term side effects include malabsorption, cholelithiasis, and, in an occasional patient, worsening of glucose tolerance. In addition to improving symptoms, octreotide decreases plasma insulin levels in 65% of patients. Because somatostatin analogs also decrease glucagon and growth hormone secretion, occasionally their administration may worsen the hypoglycemia.9
Surgical Therapy
Detailed tumor localization studies are important because insulinomas are frequently small and uniformly distributed throughout the pancreas and thus can be difficult to find.29,30,105 In addition, in the uncommon patient with metastatic disease, unnecessary surgery can be avoided. Localization methods will be dealt with in a separate section (see later).
All authorities recommend that if metastatic disease in the liver is not present (>90% of cases), surgical exploration be indicated.29,30,84,106 A careful surgical exploration should be done using the results of the tumor localization studies by a group experienced in treating such patients and with expertise in the use of intraoperative ultrasound.84 In most studies, 70% to 97% of all patients are cured by surgery.9,26 Failure to localize an insulinoma at surgery presents a difficult problem and the role of blind distal pancreatectomy is controversial. Because insulinomas are equally distributed in the pancreas, a pancreatectomy distal to the superior mesenteric vessels gives only a 50% chance of success. To decrease the possibility of a negative laparotomy, the use of intraoperative ultrasound and preoperative venous sampling from portal venous tributaries or hepatic veins for insulin levels after intra-arterial calcium administration should detect tumors in almost every case105,107,108 and therefore obviate the need for a blind resection. Because they are almost invariably intrapancreatic and usually benign, insulinomas are increasingly being resected successfully using a laparoscopic approach if the insulinoma can be localized preoperatively.109
GASTRINOMAS
DEFINITION
Zollinger-Ellison syndrome (ZES) is caused by ectopic secretion of gastrin by a PET (i.e., gastrinoma), which causes excessive gastric acid secretion, characteristically causing peptic disease (often severe) and/or gastroesophageal reflux disease (GERD) (Table 32-5). This disease was first described in 1955 by Zollinger and Ellison, surgeons at Ohio State University, in two patients with extreme acid hypersecretion and intractable peptic ulcer disease caused by a non–beta cell tumor of the pancreas that recurred with any gastric surgical procedure less than a total gastrectomy.2 Most clinicians now generally use gastrinoma and ZES synonymously, although it is important to remember that some pathologists continue to use the term gastrinoma to designate any tumor containing gastrin by immunohistochemistry.110 Gastrin can be detected by immunohistochemical staining in 50% of ovarian cancers, some bronchogenic carcinomas, acoustic neuromas, pheochromocytomas, colorectal cancers, and other PETs; however, except for an occasional ovarian tumor, hypergastrinemia is not seen.111 Therefore, essential to establishing the clinical diagnosis of ZES gastrinoma, it is necessary to demonstrate fasting hypergastrinemia that is inappropriate.28,46,65,71
Table 32-5 Frequency of Clinical and Laboratory Features in Patients with Zollinger-Ellison Syndrome (%)
| FEATURES | Source of Study | |
|---|---|---|
| NIH | LITERATURE | |
| Clinical Features | ||
| Male gender | 56% | 44%-70% |
| Mean age at onset (yr) | 41 | 41-53 |
| Mean duration of symptoms (yr) | 5.2 | 3.2-8.7 |
| Abdominal pain | 75% | 26%-98% |
| Diarrhea | 73% | 17%-73% |
| Heartburn | 44% | 0%-56% |
| Nausea | 30% | 8%-37% |
| Vomiting | 25% | 26%-51% |
| Bleeding | 24% | 8%-75% |
| MEN-I | 22% | 10%-48% |
| History of confirmed peptic ulcer | 71% | 71%-93% |
| Abdominal perforation | 5% | 5%-18% |
| Esophageal stricture | 4% | 4%-6% |
| Laboratory Feature | ||
| Fasting hypergastrinemia | 99% | 96%-100% |
| Positive secretin test (>120-pg/ml increase) | 94% | 94% |
| BAO ≥ 15 mEq/hr (no gastric surgery) | 93% | 44%-100% |
| BAO ≥ 5 mEq/hr (previous gastric surgery) | 93% | 43%-100% |
BAO, basal acid output; MEN-I, multiple endocrine neoplasia type I; NIH, National Institutes of Health.
PATHOPHYSIOLOGY AND PATHOLOGY
Almost all the symptoms listed in Table 32-5 are caused by the gastric acid hypersecretion and only late in the disease course are symptoms such as pain caused by the gastrinoma per se, and then only in a small percentage of patients.28,72 Peptic ulcer disease, GERD, and diarrhea, the most frequent clinical manifestations, all disappear when gastric acid hypersecretion is controlled surgically, medically, or by nasogastric suction.18,26,110,113 The increased parietal cell mass caused by parietal cell hyperplasia driven by hypergastrinemia results in an increased maximal acid output; the hypergastrinemia also results in increased basal acid output. These are characteristic findings in ZES.113 High serum gastrin levels stimulate the growth of the gastric mucosa, resulting in large gastric folds (Fig. 32-2), with not only parietal cell hyperplasia but also proliferation of gastric enterochromaffin-like cells (ECL cells), which secrete histamine.9,112–115 The proliferation of ECL cells results in ECL cell hyperplasia, which can lead to the development of gastric carcinoid tumors (ECLomas), especially in patients with ZES and MEN-I (see Table 31-2).114–118 Recent studies have demonstrated that in sporadic ZES patients and patients with MEN-I–ZES, more than 99% show various degrees of ECL hyperplasia,114,119 with the changes generally being much more advanced in patients with MEN-I–ZES. Furthermore, no gastric carcinoids were seen in sporadic cases, whereas 23% of patients with MEN-I–ZES had type 2 gastric carcinoids. The increased gastric acid secretion results in diarrhea because of direct damaging effects of acid on the small intestinal mucosa; furthermore, the low pH inactivates lipase and can precipitate bile acids. There is no evidence that the chronically high levels of serum gastrin directly contribute to the pathogenesis of the diarrhea by altering intestinal secretion or motility, as was originally proposed.
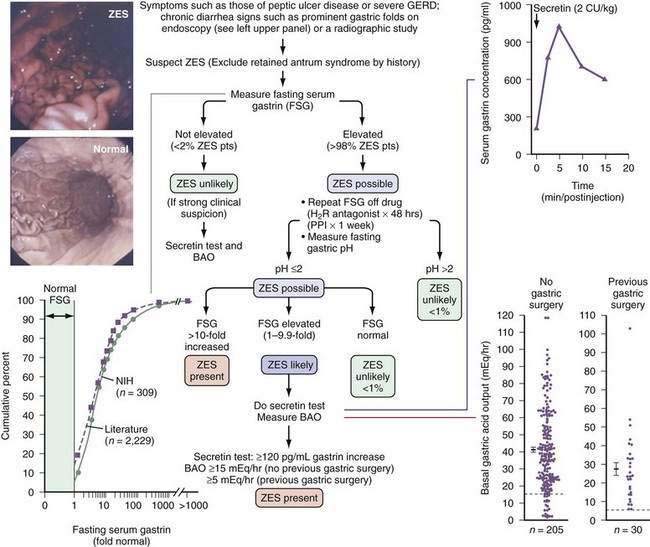
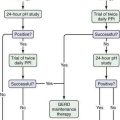
Figure 32-2. Algorithm for the diagnosis of Zollinger-Ellison syndrome (ZES). Right upper panel, Typical ZES patient with a positive secretin test result (i.e., >120-pg/mL increase in fasting gastrin level). Right lower panel, Marked elevation in basal acid output (BAO) seen in ZES patients, with or without previous gastric acid–reducing surgery. Mean + SEM (standard error of the mean) values for patients with ZES, with or without previous gastric acid–reducing surgery, are shown. Dotted horizontal lines show criteria of >15 mEq/hr or >5 mEq/hr proposed to distinguish patients with ZES with or without previous gastric acid–reducing surgery, respectively, from patients without ZES. Left upper panel, Prominent gastric folds found on endoscopy in 92% of ZES patients, as compared with a normal subject. Left lower panel, Fasting serum gastrin levels from 309 National Institutes of Health (NIH) ZES patients and 2229 ZES patients from the literature. Fasting gastrin levels are expressed as a multiple (fold) of the upper limit of normal on the horizontal axis and as the cumulative percentage of patients with the indicated gastrin level on the vertical axis. Only 0.3% of NIH patients and 3.4% of all patients in the literature had normal values; 60% of patients had a <10-fold serum gastrin increase.28,72,112,113,142,144 CU, clinical units; FSG, fasting serum gastrin; GERD, gastroesophageal reflux disease; H2R, histamine H2-receptor; PPI, proton pump inhibitor.
Experimental evidence demonstrates that gastrin-like peptides can stimulate growth and development of colorectal cancers.116,118 Whether gastrin-like peptides are important to growth or development of colorectal tumors in humans is controversial. Increased proliferative rates of colonocytes and rectal mucosal cells have been reported in patients with ZES.9,116 However, epidemiologic studies in hypergastrinemic patients with pernicious anemia or ZES have not shown an increased risk of colorectal cancer.111,120
Gastrinomas were originally reported to be non–beta islet cell tumors or neuroendocrine tumors of the pancreas.2,28,121 However, in most current series, more than 50% of gastrinomas are located in the duodenum and duodenal gastrinomas outnumber pancreatic gastrinomas by two- to five-fold in different series.46,56,65,68,71,110,122–124 Within the duodenum, gastrinomas are more frequent proximally (D1, 56%; D2, 32%; D3, 6%; and D4, 6%). Within the pancreas, gastrinomas show a pancreatic head-to-body-to-tail ratio of approximately 1 : 1 : 2. The increased identification of duodenal gastrinomas accounts for the fact that 60% to 90% of gastrinomas are now found in the “gastrinoma triangle.”124–127 This is an area formed by the junction of the cystic and common bile ducts posteriorly, the junction of the second and third parts of the duodenum inferiorly, and the junction of the pancreatic neck and body medially. Gastrinomas originate in a nonduodenal-nonpancreatic abdominal location in 2% to 24% of patients in different series, including in the ovary, liver and biliary tract, jejunum, mesentery, renal capsule, omentum, and pylorus.128
Lymph node primary gastrinomas are reported in up to 11% of sporadic cases of ZES (i.e., non–MEN-I).110,129,130 The issue of lymph node primary gastrinomas remains controversial, even though patients with sporadic ZES have remained cured, with follow-up to 20 years postresection of only lymph node(s) containing gastrinoma. Pathology studies reported endocrine cell nests in lymph nodes of patients without gastrinomas that could possibly give rise to gastrinomas. Two extra-abdominal locations have been reported to be the primary site of the gastrinoma in 0.5% of patients—namely, the cardiac intraventricular septum and non–small cell lung cancer.131,132
At presentation, 70% of 221 National Institutes of health (NIH) patients with ZES had localized disease, with a primary tumor only in 36% (literature range, 23% to 51%) or with a primary tumor and lymph node metastases in 28% (literature range, 8% to 61%).110 In these 221 patients, no primary tumor was located in 13% (literature range, 7% to 48%). This failure to locate a primary tumor is unlikely to be the result of some other entity such as nesioblastosis causing hypergastrinemia and mimicking ZES, because in our surgical series at the NIH, gastrinoma was found in the last 81 patients, However, 16% of these patients only had positive lymph nodes without a primary tumor found and were not cured by resection, suggesting that a small primary tumor was missed.129 Of the NIH patients, 17% had metastatic disease to the liver (literature range, 13% to 53%), with 3% (literature range, 4% to 14%) having liver metastases only found, and 14% (literature range, 4% to 23%) having lymph node metastases only.
In older studies, gastrinomas were malignant in 60% to 90% of patients28,133 based on the occurrence of lymph node or hepatic metastases. These data suggest that all gastrinomas should be considered potentially malignant.110 As with other PETs, only the presence of metastases or gross invasion of normal tissues remains the generally accepted criterion for malignancy.45 At present, it is not known how frequently, if at all, hepatic metastases develop without lymph node involvement. Studies have demonstrated that bone metastases also occur more frequently than previously reported in patients with advanced tumors; in one study,134 115 patients with ZES were examined prospectively with serial bone scans, octreotide scans, and magnetic resonance imaging (MRI) of the spine and bone. No bone metastases were seen in patients without liver metastases. Of all patients, 7% had bone metastases and 31% had liver metastases. The most frequent areas of bone involvement were the pelvis (75%), scapula, and ribs.
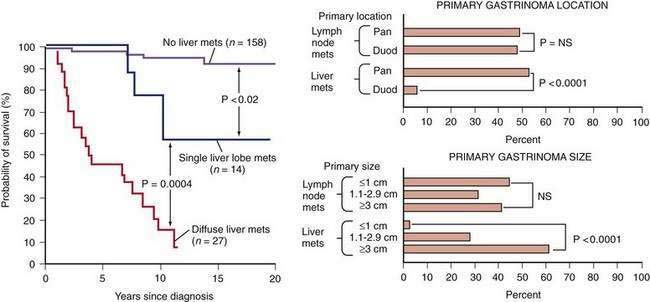
Two large NIH studies68,135 have supported the proposal of older studies121 that gastrinomas demonstrate two general growth patterns, an aggressive growth pattern in 25% and a nonaggressive growth pattern in 75%. The 10-year survival in patients with tumors showing aggressive growth was only 30%, whereas it was 96% in those with tumors without aggressive growth. Aggressive disease was defined by the initial presence (19% of patients) or the development (5% of patients) of liver metastases. The presence and extent of liver metastases was the most important prognostic factor (Fig. 32-3).25 Particularly important predictors of the presence of liver metastases, but not lymph node metastases, were the presence of a pancreatic as opposed to a nonpancreatic gastrinoma and size larger than 3 cm. Most duodenal gastrinomas are small (92% < 1 cm) compared with only 8% of pancreatic gastrinomas. The number of large duodenal gastrinomas or small pancreatic gastrinomas was not sufficient to determine whether gastrinoma size and location are independent predictors of the development of liver metastases. The percentage of pancreatic and duodenal gastrinomas with lymph node metastases was similar. Therefore, the development of lymph node metastases was independent of primary gastrinoma location, as it was of primary gastrinoma size. The difference in biological behavior of duodenal and pancreatic gastrinomas has been used to support the hypothesis that they have different origins.136
Another important finding from the natural history studies of gastrinomas that has important clinical applications is that even in patients with gastrinomas that have metastasized to the liver, the tumor growth rate is highly variable. In one study of 19 patients with liver metastases with ZES receiving no treatment, the metastatic tumor showed no growth in 26% of patients over a 29-month period, showed slow growth in 32%, and showed rapid growth in 42%.137 In patients with rapid tumor growth, 62% died during follow-up, whereas none died in the no-growth or slow-growth group. Therefore, the rate of growth of the metastases is an important prognostic factor and not all patients with liver metastases require equally aggressive antitumor treatment.
The development of bone metastases or ectopic Cushing’s syndrome caused by release of ACTH by the gastrinoma is associated with a poor prognosis.25,68,110,135 Survival in patients developing bone metastases or ectopic Cushing’s syndrome was 1.9 ± 0.4 years and 1.7 ± 0.4 years, respectively, after their diagnosis.
CLINICAL FEATURES
The principal clinical features of ZES are summarized in Table 32-5.72 There is a slight male predominance, with a mean age of onset of 41 years and a mean delay in diagnosis of five to six years. Abdominal pain primarily caused by peptic ulcer disease remains the most frequent early symptom, although increasingly diarrhea or GERD symptoms are reported early in the course of the disease. The abdominal pain that patients with ZES initially develop is clinically indistinguishable from that seen in patients with other forms of peptic ulcer disease.28,110 Later, the symptoms may become persistent, refractory to antisecretory medications, or associated with complications, which may suggest the diagnosis (Table 32-6). Diarrhea initially may be the only symptom in 9% to 10% of patients and occurs with abdominal pain in 28% to 56%.46,65,72 Symptoms of GERD are being increasingly recognized, with up to 31% presenting with GERD as the initial manifestation and 49% to 61% of patients having symptoms of GERD or esophageal lesions at initial evaluation.138 Most patients today with ZES have a typical duodenal ulcer at diagnosis, and 18% to 29% have no ulcer at diagnosis. This is an important difference from older studies, in which more than 90% of patients with ZES presented with peptic ulcers, and multiple ulcers or ulcers in atypical locations were frequent.112,133 In contrast to older studies, in which up to 100% of patients presented with or developed a complication of advanced acid peptic disease (bleeding, penetration, esophageal stricture, perforation, obstruction), at present less than 30% of patients develop these complications, even though 71% have a confirmed history of peptic ulcer disease. Patients presenting with duodenal gastrinomas do not different clinically from patients with pancreatic tumors.124
Table 32-6 Historical Features, Symptoms, and Signs That Suggest Zollinger-Ellison Syndrome
| Historical Feature |
| In Patients with PUD or GERD |
GERD, gastroesophageal reflux disease; PUD, peptic ulcer disease; UGI, upper gastrointestinal; ZES, Zollinger-Ellison syndrome.
From reference 72.
In the 25% of patients with ZES who have MEN-I syndrome, the clinical presentation is similar to that of those with sporadic disease (see Table 32-5).47,48,67,72 Some important clues that should suggest MEN-I are the following: (1) a history of nephrolithiases and/or renal colic is much more frequent in patients with ZES with MEN-I than with sporadic ZES (47% vs. 4%); (2) patients with MEN-I present with ZES at a younger age (34 vs. 43 years); and (3) 72% have a family history of endocrinopathies. In a review of 107 patients with ZES with MEN-I prospectively studied at the NIH and 1007 cases from the literature,48 88% to 94% developed hyperparathyroidism, 31% to 60% pituitary disease, 6% to 30% various carcinoids (gastric, bronchial, thymic), and 6% to 16% other functional PETs, each of which may mask the diagnosis of ZES and be the presenting feature of the syndrome.
DIAGNOSIS AND DIFFERENTIAL DIAGNOSIS
Despite the widespread availability of gastrin radioimmunoassays and the publication of more than 3300 articles on ZES, as well as most physicians’ general awareness of the features of ZES, the diagnosis of ZES continues to be delayed four to six years after onset of symptoms (see Table 32-5).46,48,72 This likely occurs for a number of reasons. ZES is an uncommon cause of peptic disease (1 to 3 new cases/million/year). Initially, it is frequently clinically indistinguishable from patients with peptic ulcer disease (2300 cases/million/year), and GERD, which can affect 3% to 4% of the population.28,67,112 In contrast to older studies, the initial symptoms of abdominal pain or GERD symptoms that most ZES patients currently exhibit are generally indistinguishable from those seen in peptic disease. Also, the characteristics of the ulcer itself (single, duodenal in location) do not distinguish it from those typically seen in patients with duodenal ulcer disease. Furthermore, the widespread use of proton pump inhibitors (PPIs) is complicating and delays the diagnosis of ZES. One study139 has analyzed the number of patients referred and diagnosed with ZES in two well-established referral centers (Università La Sapienza, Rome, and the NIH) before and after the introduction of PPIs. The widespread use of PPIs decreased referrals of patients with possible ZES by 62%, decreased by 40% the number of cases of ZES diagnosed, and was associated with a two- to six-fold increase in patients with a false diagnosis of ZES referred to the U.S. center. This false diagnosis occurred because chronic treatment with PPIs causes hypergastrinemia in 80% to 100% of all patients with peptic ulcer disease or GERD and it frequently reaches five times normal levels, a range seen in 60% of patients with ZES. The use of histamine H2 receptor antagonists often suggests the diagnosis of ZES because they often do not control acid hypersecretion in ZES with the conventional doses used to treat routine peptic ulcer disease or GERD. Thus, treatment failure with these drugs leads to a suspicion of the diagnosis of ZES.9 In contrast, PPIs mask the diagnosis of ZES because they control symptoms in most patients with ZES with conventional doses used in the treatment of peptic ulcer disease or GERD, and therefore a treatment failure rarely occurs with PPIs.71,140
There are a number of distinctive clinical and laboratory features that should suggest the diagnosis of ZES in a patient with acid peptic disease or diarrhea (see Table 32-6). A few important points will be briefly discussed. Diarrhea is now infrequent in patients with peptic disease or GERD because higher doses of antacids are rarely used, so its presence should lead to a suspicion of ZES because it occurs in 73% of ZES patients (see Tables 32-5 and 32-6). Diarrhea alone can be the presenting symptom in up to 27% of patients with ZES.28,72,112 MEN-I is a cause of ZES in 20% to 25% of patients and is characterized by endocrine abnormalities in multiple endocrine glands (parathyroid > pancreas > pituitary). Therefore, anyone with a personal or family history of endocrinopathies, or laboratory evidence of endocrinopathies, should be suspect of having ZES.46,48 Helicobacter pylori is an important cause of peptic ulcer disease (present in 80% to 100%); however, it is present in only 10% to 50% of patients with ZES.141 Prominent gastric folds on upper gastrointestinal (UGI) endoscopy should suggest the diagnosis of ZES (see Fig. 32-2). Whereas enlarged gastric folds were reported in only 13% to 30% in older studies of patients with ZES, an NIH prospective study72 involving 261 patients, in which all patients underwent UGI endoscopy, reported this finding in 94% of cases. This endoscopic finding particularly contrasts with the loss of gastric folds reported in many patients with acid hyposecretory disorders, resulting in fasting hypergastrinemia (e.g., atropic gastritis, pernicious anemia; see Chapter 51), and therefore should be an important clue to the possible presence of ZES.
The diagnosis of ZES requires the demonstration of acid hypersecretion in the presence of hypergastrinemia (see Fig. 32-2).28,46,65,112,113,124 Therefore, to diagnose ZES, assessments of fasting serum gastrin levels and acid secretion are required. At the NIH, over 99% of 309 ZES patients studied had fasting hypergastrinemia and, in 2229 cases from the literature, 97% had fasting hypergastrinemia.142 Hence, if the fasting serum gastrin level is normal, especially in repeated determinations, the diagnosis of ZES is very unlikely. There are two specific exceptions to this general conclusion. First, in MEN-I patients after parathyroidectomy, correction of hyperparathyroidism may result in normalization of fasting gastrin levels in patients with ZES.67,79,143 Second, after gastrinoma resection, the fasting serum gastrin level may be normalized, even though the patient is not cured.144 In these latter two special cases and the rare case in which ZES is suspected clinically even though the fasting gastrin level is normal, a secretin provocative test should be performed.
Because PPIs can elevate fasting serum gastrin levels up to three- to five-fold, a range that overlaps with gastrin levels in 60% of patients with ZES, it is difficult to diagnose ZES when the patient is taking PPIs.9,46,71,139 Therefore, if the fasting gastrin level is elevated while taking a PPI, the fasting gastrin level should be performed after stopping the PPI for at least one week (see Fig. 32-2). However, abruptly stopping PPIs can lead to the rapid development of peptic complications in a small percentage of ZES patients.
In one prospective study of acid secretion in 235 ZES patients and review of 984 cases from the literature,113 99% of patients had a fasting gastric pH lower than 2; therefore, gastric pH should be determined when the fasting gastrin is repeated (see Fig. 32-2) to exclude physiologic hypergastrinemia (i.e., caused by hypochlorhydria or achlorhydria). The most common causes of physiological hypergastrinemia include atrophic gastritis, H. pylori infection, pernicious anemia, the use of potent acid suppressant drugs (particularly PPIs), chronic renal failure, and following gastric acid-reducing surgery.28,46,65,71 In patients with fasting serum gastrin levels less than 10-fold increased (i.e., usually <1000 pg/mL) and gastric pH lower than 2, which includes 60% of those with ZES, other conditions such as gastric outlet obstruction, H. pylori infection, rarely renal failure or short bowel syndrome, antral G cell hyperfunction or hyperplasia, and retained gastric antrum syndrome can mimic ZES and need to be considered. In such patients, secretin test and basal acid output (BAO) tests should be performed.142,144,145
Historically three different gastrin provocative tests have been used for the diagnosis of ZES—secretin, calcium, and standard meal gastrin provocation tests.28,112,144,145 Both the secretin and calcium provocation studies are based on the finding that gastrinomas release an exaggerated amount of gastrin in response to these agents, which is likely caused by the presence of specific receptors for these agents on tumor cells.146,147 The secretin test is the only one widely used at present (see Fig. 32-2). Until recently, the most widely used criterion was an increase of 200 pg/mL or more following secretin.144,145 However, a recent study144 of 293 NIH ZES patients and 537 ZES patients from the literature, demonstrates that this criterion (200 pg/mL or more) does not have the highest sensitivity and specificity and therefore should be replaced. A gastrin increase of 120 pg/mL or more after secretin has a significantly higher sensitivity than the 200-pg/mL or more criterion (94% vs. 83%), with no loss of specificity (100%); thus, it is the criterion that is currently recommended.144
In a hypergastrinemic patient with gastric pH lower than 2, no false-positive secretin tests have been reported and, as noted, 94% of patients with ZES will have a 120-pg/mL or more increase in fasting gastrin with secretin provocation.28,46,65,144,145 The BAO is elevated in more than 90% of patients with ZES; it is more than 15 mEq/hr in patients without previous gastric surgery and more than 5 mEq/hr in patients with previous acid-reducing surgery (see Fig. 32-2 and Table 32-5).113
Gastric acid secretion is now uncommonly measured. However, a recent study has reported that it can be measured during UGI endoscopic evaluation, which may facilitate its more general use.148 The results of a prospective study of gastric acid secretion in 235 patients with ZES and a comparison with 984 ZES cases from the literature provided some important insights into when ZES should be suspected.113 The BAO did not differ between patients with duodenal or pancreatic gastrinoma.124 BAO was slightly higher in patients with sporadic ZES than in those with ZES–MEN-I in one study, but not in another.67 The mean BAO for patients without previous gastric surgery was 42.3 mEq/hr (range, 1.6 to 118 mEq/hr), with 94%, 90%, and 86% having a BAO more than 10, 15, or 18 mEq/hr, respectively, criteria proposed for diagnosis of ZES in different studies (see Fig. 32-2). In patients with previous gastric acid-reducing surgery, the mean BAO was 27.6 mEq/hr (range, 5.9 to 103 mEq/hr); 100% and 73% exceeded 5 mEq/hr and 14.4 mEq/hr, criteria proposed for diagnosis in different studies (see Fig. 32-2). These results, as well as those from 984 cases from the literature,113 confirm the reports of older, smaller studies28 that most ZES patients have marked acid hypersecretion. Therefore, prompt and effective treatment of the gastric acid hypersecretion is essential for the management of these patients46,65,124 (see later).
TREATMENT
ZES requires immediate and long-term control of the gastric hypersecretion and then treatment directed against the gastrinoma.28,46,56,65,71,122,124 Treatment for the gastric hypersecretion is required because almost every patient with ZES has acid hypersecretion and in more than 50% of cases it is more than five times normal.113 Older studies have demonstrated that if the acid hypersecretion is not controlled, complications of peptic disease almost invariably occur and can frequently be life-threatening.112,133 Furthermore, immediate treatment is needed for the hypersecretion because life-threatening complications can occur rapidly. Currently, even though most patients receive some form of gastric antisecretory treatment prior to the diagnosis of ZES, 24% develop bleeding, bowel perforations develop in 6% to 7%, and 8% to 10% develop GERD severe enough to result in strictures.9,72 Treatment also needs to be directed against the gastrinoma because its removal may result in cure,68 and 60% to 90% of gastrinomas were malignant in older studies.25,133 With increased ability to control the gastric hypersecretion medically, the natural history of the gastrinoma is becoming one of the major determinants of long-term survival.45,135
Pharmacologic Treatment of Gastric Acid Hypersecretion
Acid hypersecretion in patients with ZES can now be controlled with oral antisecretory drugs both acutely and long term in all patients, with the exception of the rare patient who will not or cannot take oral medications regularly.28,46,65,71,110,124,149 PPIs, because of their long duration of action (i.e., >48 hrs) and potency, allow once or twice daily dosing in most patients (>95%) and are now the drugs of choice.140,150 Histamine H2 receptor antagonists are also effective; however, higher than conventional doses are required (mean dosages—cimetidine, 3.6 g/day; ranitidine, 1.2 g/day; famotidine, 0.25 g/day).110
All PPIs have been shown to be effective in ZES (omeprazole, lansoprazole, pantoprazole, esomeprazole, rabeprazole).149–152 PPIs are usually started with a dosage equivalent to 60 mg/day of omeprazole, except in patients with complicated ZES–MEN-I, severe GERD, or previous Billroth II resection, for whom an initial dose equivalent to 60 mg twice daily of omeprazole is recommended.138 Sufficient antisecretory drug needs to be given to reduce acid hypersecretion to lower than 10 mEq/hr in patients without previous gastric acid-reducing surgery (<5 mEq/hr in patients with previous gastric acid-reducing surgery) for the hour prior to the next dose of drug. This degree of suppression allows peptic disease and GERD lesions to heal and also prevents their recurrence. Although initial lower doses of PPIs are frequently used, because of the desirability of rapidly controlling the acid hypersecretion in ZES patients, these higher doses are recommended. This recommendation was supported by a study153 that demonstrated that a low initial dose of omeprazole (i.e., 20 mg/day) controlled acid hypersecretion within 24 hours in only 68% of ZES patients. Patients with previous Billroth II resection or moderate to severe GERD with ZES require greater acid suppression, and antisecretory drugs should be increased to control symptoms and heal all mucosal lesions, which frequently require suppression to 1 to 2 mEq/hr or even lower.154 Patients with ZES and MEN-I with hyperparathyroidism also require higher doses of antisecretory drugs. Studies have demonstrated that parathyroidectomy for hyperparathyroidism in MEN-I–ZES patients reduces the fasting gastrin level, reduces the BAO, and increases the sensitivity to a given dose of antisecretory drug, which facilitates medical management.67,79,143 Acid secretory control in especially important for MEN-I–ZES patients, because a recent study has reported155 that these patients, compared with those with sporadic ZES, have a three-fold higher rate of esophageal strictures, five-fold higher rate of Barrett’s esophagus, and eight-fold higher incidence of dysplasia, which was thought in part caused by inadequate control of the acid hypersecretion early in these patients’ course.
Occasional patients with ZES require parenteral control of the gastric acid hypersecretion caused by surgery, vomiting, chemotherapy, or gastric outlet obstruction. Parenteral histamine H2 receptor antagonists (cimetidine, ranitidine)9,46 and PPIs (omeprazole, pantoprazole)156–158 have been reported to be effective. Several IV PPI preparation are approved in the United States and are effective at controlling acid hypersecretion in patients with ZES—for example, the usual dose of pantoprazole IV is 80 mg given every 8 hours by 15-minute IV infusion. Parenteral histamine H2 receptor antagonists are effective; however, continuous infusions of relatively high doses are required to control hypersecretion (mean dose, 1 mg/kg/hr of ranitidine).
Long-term oral antisecretory treatment with high doses of histamine H2 receptor antagonists or PPIs has remained effective for more than 10 years without the development of tachyphylaxis that limited effectiveness in most patients.28,46,65,110,140,149 Because antisecretory drug requirements can change with time, it is recommended that patients with ZES have their acid secretory control checked after six months initially, after starting histamine H2 receptor antagonists, or yearly after starting PPIs. A recent study has reported that this can be reliably performed during UGI endoscopic studies.148 High doses of cimetidine can cause antiandrogen side effects in males, and high doses of some histamine H2 receptor antagonists can interfere with cytochrome P450 metabolizing enzymes or result in central nervous system (CNS) symptoms in patients with hepatic or renal failure. Long-term treatment with PPIs rarely has dose-related side effects, although potential side effects have been raised because of their effects on nutrient absorption.159,160 In studies of long-term PPI treatment in ZES patients, some patients developed decreased serum vitamin B12 levels, but not decreased body iron stores.161 This likely occurs because the absorption of protein-bound vitamin B12 in foods require the presence of acid and pepsin and therefore potent acid suppressants such as PPIs can interfere with vitamin B12 absorption. At present, it is unclear if long-term PPI treatment in patients with ZES will result in clinically significant vitamin B12 deficiency.
In addition, long-term PPI treatment may interfere with calcium absorption. A recent epidemiologic study has reported that long-term high-dose PPI usage is associated with an increased incidence of hip fractures.159 There is no evidence that long-term treatment with PPIs increases the rate of development of gastric carcinoids in patients with or without ZES or other tumors.116,118,162 These observations are important because the risk of developing gastric carcinoids on PPIs in animal studies is related to the severity of the hypergastrinemia. Hence, the chronic use of PPIs, by causing physiologic hypergastrinemia, could contribute to the extent of hypergastrinemia in ZES patients.114–118,163 In a detailed study of 106 patients with sporadic ZES, many of whom had been treated long-term with PPIs, no gastric carcinoids were found, even though over 99% had ECL hyperplasia and 7% had dysplasia.114 In contrast, in a similar study in MEN-I–ZES patients, 53% had advanced ECL changes and 23% had gastric carcinoids, demonstrating that gastric carcinoids are more than 70 times more common in MEN-I–ZES.119 However, in this latter study, there was no evidence that the use of PPIs increased the rate of development of gastric carcinoids, but was instead best correlated with the extent of hypergastrinemia and presence of the MEN-I. Furthermore, animal and cell biology studies have provide evidenced for a possible role of gastrin-related peptides in growth and development of other tumors, including colorectal cancer.164 Although studies in ZES patients9 have shown increased proliferative rates of colonic mucosal cells, epidemiologic studies have not shown an increased association of colorectal cancer and hypergastrinemia.120,162 Therefore, in ZES patients there is no evidence that chronic hypergastrinemia, whether caused only by the gastrinoma or possibly contributed to by chronic use of PPIs, increases the development and/or growth of colorectal cancer.
Surgical Treatment of Gastric Hypersecretion
Total gastrectomy, the only effective means to treat the gastric acid hypersecretion seen in ZES for many years, is now rarely used.2,28,56,65,71,133,165 At present, total gastrectomy is relatively safe, with an overall mortality of 5.8% in 248 cases since 1980 and 2.4% for elective cases.47 However, the long-term morbidity of total gastrectomy in ZES remains unclear.110
Vagotomy decreases gastric acid hypersecretion in patients with ZES.9,28,110,166 Parietal cell vagotomy decreased BAO by 41% and decreased the dose of oral histamine H2 receptor antagonist by 40%. Long-term, 36% of patients were able to discontinue antisecretory drugs and 80% continued to have a decrease in BAO. It has been proposed that at the time of laparotomy for possible cure, a parietal cell vagotomy should be performed to decrease antisecretory drug requirements in the 70% of patients who are not cured long term.68
Parathyroidectomy in patients with ZES–MEN-I with hyperparathyroidism decreases fasting gastrin levels, decreases gastrin increases after secretin injection, decreases BAO, and increases sensitivity to antisecretory drugs.28,67,79,110,143 This latter observation is particularly important because patients with ZES–MEN-I with hyperparathyroidism can be relatively more resistant to PPIs, and higher and more frequent doses are required in these patients.140,150 This may lead to a significantly increased expense for antisecretory drugs in these patients, which can affect compliance.
Surgical Treatment of Gastrinoma
Because gastrinomas are malignant in 60% to 90% of cases, detailed tumor imaging is essential to determine tumor extent and location of the primary tumor.26,28,68,69,110,122 Localization methods and results are discussed in a later section.
Most but not all authorities recommend that if diffuse metastatic disease to the liver is not present and the patient does not have MEN-I, surgical exploration is indicated.9,28,56,68,71,110,122,167 In a large prospective study from the NIH68 involving 151 patients with ZES, patients with sporadic ZES (i.e., no MEN-I), gastrinomas were found in 92%; 51% were disease-free immediately after tumor resection and 34% were disease-free at 10 years. This cure rate is higher than the 0% to 20% rate reported in many earlier studies and is, in large part, the result of finding increased numbers of small duodenal gastrinomas.125,126 It is essential to perform a routine duodenotomy at surgery, which identifies 20% to 25% more duodenal tumors than other commonly used methods.110,122, A prospective NIH study123 has demonstrated that the routine use of duodenotomy increases the short- and long-term cure rates (Fig. 32-4). Two NIH studies have demonstrated that routine surgical resection, primarily in sporadic ZES patients, not only decreases the rate of development of liver metastases, but also increases survival.168,169
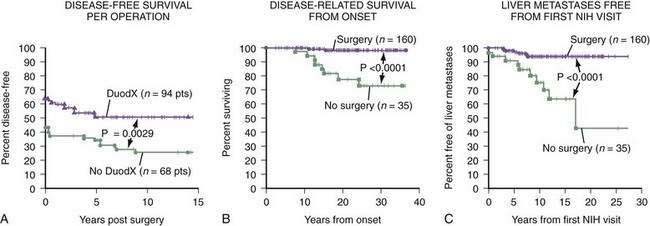
In patients with MEN-I and ZES, the role of surgery for cure of the gastrinoma is controversial.47,56,67,68,71,170 This controversy has occurred because the disease-free rate for ZES in patients with MEN-I is very low (0% to 5%) because most these patients have multiple duodenal gastrinomas and frequently have lymph node metastases at surgery.171 Studies have shown that gastrinomas and other pancreatic endocrine tumors can show aggressive growth in 15% of the patients, with hepatic metastases in 23%, and the rate of progression of these tumors is becoming an important determinant of the long-term survival of these patients.67,170,172 The role of surgery is controversial in part because it is unclear whether early surgical resection will alter survival in MEN-I–ZES; without aggressive resections, it does not cure these patients. Some recommend routine exploration in all patients with MEN-I–ZES, others recommend no exploration and still others recommend exploration only if lesions larger than 2 to 3 cm are seen.28,173 Although Whipple resection can cure a high proportion of MEN-I–ZES patients, it is not generally recommended because of the excellent long-term prognosis of unoperated patients with small PETs (<2 cm) and because of the side effects of Whipple resections. Studies have shown that patients with PETs smaller than 2 cm with MEN-I–ZES had a 100% survival rate at 15 years,170 which is similar to results in MEN-I patients with nonfunctional PETs smaller than 2 cm who had no increase mortality compared with MEN-I patients without PETs.174 At present, we recommend surgical exploration only if lesions larger than 2 cm are imaged in MEN-I–ZES patients. At surgery, we do not recommend routine distal pancreatectomy; this will rarely result in cure but because larger lesions are associated with more frequent liver metastases, it may decrease the liver metastases rate.9,175
A small percentage of patients (10% to 15%) with ZES develop limited metastatic disease in the liver and a number of studies recommend surgical resection in these patients.28,122,128,176,177 Although this will result in cure in only a small percentage of patients, it has been proposed that it may extend survival, though there are no prospective studies that establish this.
GLUCAGONOMAS
DEFINITION
Glucagonomas are PETs that secrete excessive amounts of glucagon and cause a distinct syndrome characterized by a specific dermatitis (migratory necrolytic erythema), weight loss, glucose intolerance, and anemia (Table 32-7). Although Mallinson and colleagues4 specifically established the association of the rash with glucagon-producing tumors of the pancreas when they reported nine cases in 1974, the disease had been described earlier by others, although the association with glucagon release was not appreciated. In 1942, Becker and associates described the association of a PET with a skin rash.178 In 1966, McGarvan and coworkers reported a patient with an elevated fasting glucagon level, dermatitis, diabetes, and a PET.179 In 1973, Wilkinson and colleagues80 described the rash as necrolytic migratory erythema.
Table 32-7 Frequency of Clinical Features and Laboratory Abnormalities in Patients with Glucagonoma
| PARAMETER | FREQUENCY (%) |
|---|---|
| Clinical Features | |
| Dermatitis | 54-90 |
| Diabetes mellitus, glucose intolerance | 22-90 |
| Weight loss | 56-96 |
| Glossitis, stomatitis, cheilitis | 29-40 |
| Diarrhea | 14-15 |
| Abdominal pain | 12 |
| Thromboembolic disease | 12-35 |
| Venous thrombosis | 4-24 |
| Pulmonary emboli | 11 |
| Psychiatric disturbance | Low |
| Laboratory Abnormalities | |
| Anemia | 33-85 |
| Hypoaminoacidemia | 26-100 |
| Hypocholesterolemia | 80 |
| Renal glycosuria | Unknown |
PATHOPHYSIOLOGY AND PATHOLOGY
In contrast to insulinomas, most glucagonomas are large at the time of diagnosis, with the average size between 5 and 10 cm (range, 0.4 to 35 cm).73,74,76,181 Similar to other PETs except insulinoma, 50% to 80%9,182,183 of glucagonomas had evidence of metastatic spread or invasion. The most common site of metastatic spread was to the liver (43% to 82%), with lymph nodes (38%), bone, and mesentery less commonly.9 Most glucagonomas occur in the pancreas (>97%); however, a glucagonoma associated with the typical clinical syndrome was found in the proximal duodenum. Glucagonomas usually occur as a single tumor, although 10% to 12% of patients in one series had multiple tumors or diffuse involvement by a single mass.181
The pathophysiology of the glucagonoma syndrome is related to the known actions of glucagon. Glucagon stimulates glycogenolysis, gluconeogenesis, ketogenesis, lipolysis, and insulin secretion, as well as having inhibitory effects on pancreatic and gastric secretion and on gut motility.184 Hyperglycemia results from the increased hepatic glycogenolysis and gluconeogenesis. Because glucagon also increases secretion of insulin, which prevents lipolysis and maintains normal free fatty acid concentrations, ketonemia usually does not develop.9 The weight loss has been attributed to the known catabolic effects of glucagon.181 However, severe anorexia and adipsia are seen in rats with transplanted glucagonomas and studies have suggested that the tumor is producing a novel anorectic substance. It is not clearly established that the skin rash is caused by the hyperglucagonemia per se because numerous patients have been given large doses of glucagon over extended periods and the skin rash did not develop. However, in two studies, prolonged hyperglucagonemia caused by glucagon administration did cause a typical skin rash.185,186 It is possible that hypoaminoacidemia, which develops in 80% to 90% of glucagonoma patients,182 or essential fatty acid deficiency may be involved in the genesis of the rash because, if these metabolic derangements are corrected, the dermatitis may improve without changing plasma glucagon levels. The similarity of the skin lesions to those seen in patients with zinc deficiencies has resulted in trials of zinc, with some responses. However, in some patients, the rash has resolved with rehydration and glucose solution; therefore, there may be differing contributing factors in different patients. The severe hypoaminoacidemia is believed secondary to the hyperglucagonemia, and glucagon infusions can alter amino acid metabolism. This conclusion is further supported by findings that total pancreatectomy or somatostatin administration, which decreases plasma glucagon levels, increases plasma amino acid levels, whereas glucagon administration decreases plasma amino acid levels. The role of glucagon per se in causing the thromboembolic phenomenon is not clear. Glucagon is known to affect coagulation parameters; however, the relationship of this to the thromboembolic events is not known. The anemia may be caused by the glucagon excess because prolonged treatment with a long-acting glucagon preparation decreases erythropoiesis in animals.
Immunocytochemical and histologic studies of glucagonomas show results typical of PETs. Glucagon is one of the most commonly seen peptides in immunocytochemical studies of PETs, but in many cases it is not associated with any syndrome. In one series of 1366 autopsy cases, a frequency of 0.8% adenomas was reported and all contained glucagon-producing cells.9 In glucagonomas, as with other PETs, multiple GI hormones are frequently seen on immunocytochemical studies.73 The morphology of most glucagon-producing tumors demonstrates no histologic features that distinguish them from other PETs. Even though the tumors are usually malignant, mitotic figures and nuclear atypia are uncommon. With electron microscopy, readily identifiable A granules, typical of those seen in normal alpha cells, are recognizable in PETs; they stain positive for glucagon and yet do not cause the glucagonoma syndrome, whereas in patients with a glucagonoma syndrome, atypical granules are usually seen.181
CLINICAL FEATURES
Glucagonomas usually occur in middle-aged or older adults.9,73,76,181,182 No cases have been reported in individuals younger than 19 years; only 16% of all cases occur in those younger than 40 years, with most cases occurring in individuals 50 to 70 years old. Glucagonomas occur slightly more commonly in women. Cutaneous lesions are one of the most common manifestations of the disease, occurring in 53% to 90% (Fig. 32-5 and see Table 32-7).74–76 Cutaneous lesions often precede the diagnosis of the syndrome for long periods, with a mean of six to eight years in one study and a maximum of 18 years.187 In 70% of patients, the skin lesion is the presenting sign of the disease and has been reported up to three years before the PET is found.188 Skin lesions may wax and wane and may be misdiagnosed as pemphigus foliaceous, pemphigoid, vasculitis, acrodermatitis enteropathica, psoriasis, herpes, seborrheic or contact dermatitis, eczema, pellagra, or even a chemical burn.189 Excellent descriptions of the typical rash, necrolytic migratory erythema associated with glucagonoma, have been published.180,181 Characteristically, the skin lesion starts as an erythematous area typically at periorofacial or intertriginous areas, such as the groin, buttocks, thighs or perineum, and then spreads laterally. The lesions subsequently become raised, with superficial central blistering. The top of the bullae frequently detach or rupture, leaving eroded areas that crust. The lesions tend to heal in the center while the edges continue to spread, with a crusting well-defined edge. Healing is associated with the development of hyperpigmentation. This entire sequence characteristically takes one to two weeks and while some new lesions are developing, others are healing. Therefore, a mixed pattern of erythema, bullous formation with epidermal separation, crusting, and hyperpigmentation, together with normal skin, can occur. The histopathology can be as varied as the clinical presentation.9 In its classic form, early lesions demonstrate a superficial spongiosis and necrosis, with subcorneal and midepidermal bullae. Fusiform keratinacytes with pyknotic nuclei are often seen, as are mononuclear inflammatory infiltrates.4,180 This characteristic histologic pattern is best seen in an early lesion. Glossitis or angular stomatitis is reported to occur in 34% to 68% of patients.73,182 In addition, some patients develop a nail dystrophy, with brittleness and crumbling of the nails. In a review187 of 13 cases of necrolytic migratory erythema, it was reported that patients frequently present with noncharacteristic clinical and histologic features and the diagnosis of glucagonoma was delayed.
Glucose intolerance with or without frank diabetes mellitus occurs in 22% to 90% of cases (see Table 32-7).9,73–76,181,182 In one series, 42% of patients required oral hypoglycemic agents and 24% required insulin. The onset of the diabetes mellitus preceded the diagnosis of the glucagonoma by up to 10 years in one study, with an average time of five years. A number of patients with hyperglucagonemia do not have diabetes mellitus. Furthermore, although correlations between plasma glucagon levels and changes in plasma glucose have been described in a few cases, this correlation was not easily demonstrated when a number of proven cases were analyzed. Tumor resection and normalization of blood glucagon may not result in normalization of the glucose intolerance.82 In some patients, removal of the glucagonoma improved glucose tolerance.4 In various patients with glucagonomas, plasma insulin concentrations are normal or elevated, but circulating plasma insulin and glucagon levels are not correlated. The relationship of the diabetes mellitus to the hyperglucagonemia remains unclear.
Hypoaminoacidemia occurs in 26% to 100% of patients with a clinical glucagonoma4,9,73 and essential fatty acid deficiencies are also reported. Plasma concentrations of amino acids are frequently less than 25% of normal, with glycogenic amino acids most affected, whereas branched chain amino acids are reported to be less affected.181 The degree of hypoaminoacidemia may vary with the intensity of the disease. The hypoaminoacidemia is reported to be associated with the pathogenesis of the skin rash (see earlier).
Weight loss is a prominent feature of the glucagonoma syndrome, occurring in 56% to 96% of patients (see Table 32-7).9,73–76,181 A number of observations suggest that the weight loss is a unique aspect of the syndrome. Weight loss is seen even in patients with small tumors without metastatic spread.4,9,182 The weight loss is often associated with anorexia and may be profound, with a mean weight loss of 20 kg in 44 cases, with a maximal loss of 30 kg.
Venous thromboembolism is common in patients with glucagonoma, occurring in 4% to 35% of patients in various series.9,74–76,182 Venous thrombosis occurred in 24% of patients and pulmonary emboli in 12%. This complication is thought to be related to the glucagonoma syndrome because it is not seen as frequently in other PETs. Anemia occurs in 33% to 85% of cases. The anemia is usually normocytic and normochromic (in 73% of cases in one study) and may be severe, but usually is not. When serum iron, folate, and vitamin B12 levels have been measured, they are usually normal. The anemia is reported to respond to successful tumor therapy. Psychiatric symptoms, although mentioned in several reports,4 appear to be relatively infrequent. Depression is the most commonly reported abnormality. Whether psychiatric disturbances occur with increased frequency compared with other comparable debilitating illnesses is unclear. Abdominal pain without specific identifying characteristics is reported in approximately 12% of patients. Diarrhea, weight loss, and hepatomegaly are more frequent in patients with metastatic disease.73 Diarrhea is reported in 14% to 15% of patients181 with severe steatorrhea. Diarrhea is reported far more commonly than constipation (15% vs. 4% in one study)9 and the cause of the diarrhea remains unclear. Jejunal biopsies have been reported to be normal or to show hypertrophic folds. It remains possible that other hormones may also be secreted by the tumor that could contribute to the diarrhea, but this possibility has not been extensively studied. Other laboratory findings reported to be abnormal in patients with glucagonoma include the presence of renal glycosuria and hypocholesterolemia. Renal glycosuria may occur early, and may represent a direct renal effect of glucagon. Hypocholesteremia was reported in 80% of cases.
DIAGNOSIS AND DIFFERENTIAL DIAGNOSIS
Glucagonomas are usually suspected because of the skin rash, although occasionally the diagnosis is suspected in a patient with a pancreatic mass with weight loss or diabetes.73,74,76,181,187 The skin lesion is most frequently confused with pemphigus foliaceous, although a number of dermatologic lesions have also been misdiagnosed. A number of cases of patients with a typical necrolytic migratory erythema have been described who do not have glucagonoma.9,182 Necrolytic migratory erythema is therefore not pathognomonic for glucagonoma. The rash can be seen in myelodysplastic disorders,190 short bowel syndrome,191 hepatitis B infections, malnutrition,192 cirrhosis, celiac disease, other malignancies, malabsorptive syndromes, nutritional deficiencies, and inflammatory bowel diseases.193 In some studies, up to 20% of patients with glucagonomas have Zollinger-Ellison syndrome; 13% to 17% have MEN-I whereas 0% to 3% of MEN-I patients develop glucagonomas.47,194
Once the diagnosis is suspected, it can be confirmed by demonstrating an increase in plasma glucagon concentration. In most laboratories, the upper limit of normal for fasting glucagon concentration using antibody 30 K of Unger is 150 to 200 pg/mL. In one large review, only two patients had a plasma glucagon level of 200 to 500 pg/mL, four cases had 500 to 1000 pg/mL, and 52 cases more than 1000 pg/mL.181 These results are in close agreement with another study9 in which the mean plasma glucagon concentration in 73 cases of glucagon was 2110 pg/mL, with a range of 550 to 6600 pg/mL, with 30% between 500 and 1000 pg/mL and the remaining 70% more than 1000 pg/mL. However, in some studies,74–76,195 patients with the glucagonoma syndrome with only mildly elevated plasma glucagon levels have been described; in one recent study,76 25% of patients had plasma glucagon levels lower than 500 pg/mL. Hyperglucagonemia is reported to occur in cirrhosis, chronic renal insufficiency, diabetic ketoacidosis, prolonged starvation, acute pancreatitis, acromegaly, hypercorticism, septicemia, severe burns, severe stress (trauma, exercise), celiac disease, and familial hyperglucagonemia and with danazol therapy and hepatic disease.9,179,180,182 It has been reported that the plasma glucagon level in these conditions does not exceed 500 pg/mL. It therefore has been recommended that a plasma glucagon concentration more than 1000 pg/mL is diagnostic of glucagonoma. The one reported exception to this is patients with cirrhosis, in whom the plasma level of glucagon can be more than 1000 pg/mL. Because a necrolytic migratory erythematous-like rash has been reported in patients with hepatic disease, diagnostic confusion could occasionally result. However, in some studies, no overlap in plasma glucagon levels was found between patients with cirrhosis (or any other of the conditions listed) and the values seen in patients with glucagonomas.
Various provocative tests have been described, such as the use of secretin to cause a paradoxical increase in glucagon release or a mixed or carbohydrate-rich meal.9 However, at present, none of these tests are sufficiently reliable to differentiate glucagonoma with normal plasma glucagon elevations from these other conditions. Patients with familial hyperglucagonemia have been described. These patients are asymptomatic and can be distinguished from patients who have the glucagonoma syndrome. In addition, fractionation of the plasma glucagon immunoreactivity gives a different pattern than that seen in normals or patients with glucagonoma; specifically, an increased percentage of the high molecular weight big plasma glucagon peak is described in familial cases.181
TREATMENT
Medical Treatment
Preoperative medical control of symptoms is important in these patients, because they are generally poor operative risks. The catabolic effects of glucagon, combined with glucose intolerance and diabetes mellitus (see Table 32-7), can markedly affect the nutritional status of these patients. They have an increased incidence of venous thromboembolism, increasing the postoperative risk. To improve the metabolic status of these patients prior to surgery, blood transfusions in those with severe anemia and a period of extended hyperalimentation are recommended. Parenteral nutrition with restoration of plasma amino acid levels and/or essential fatty acid levels to normal is also reported to have an excellent effect in healing the dermatitis.9
The long-acting somatostatin analog, octreotide, has been useful in controlling symptoms in patients with glucagonoma.74–76,100–102,181 The rash improved with octreotide treatment in 54% to 90% of patients, with complete disappearance in up to 30%. Octreotide generally improved the symptoms of weight loss, abdominal pain, and diarrhea as well. Diarrhea sometimes even resolves. Diabetes mellitus was not improved with octreotide treatment. Plasma glucagon levels decreased in 80% to 90% of patients but only decreased into the normal range in 10% to 20% of patients with octreotide treatment. In most studies, 100 to 400 µg/day of octreotide was used.9,183 However, now that long-acting formulations of octreotide (octreotide LAR) and lanreotide (Autogel or lanreotide SR) are available, it is likely these will be predominantly used in the future. It has been reported that octreotide LAR and lanreotide SR are effective at reducing symptoms and serum glucagon levels in a few patients with glucagonoma.196 In some patients, with continued treatment, the dosage had to be increased to continue to control symptoms.
Surgical Treatment
Approximately 50% to 90% of patients with glucagonomas have metastases at the time of diagnosis.9,73,74,74–76,197 Surgical resection has been successful in a number of cases.180–182 The exact percentage of cases that can be cured is unknown; however, with the syndrome currently recognized relatively late in most patients, it is likely that fewer than 20% of patients can be cured. A number of operated patients developed recurrence with elevated plasma glucagon elevations after what was thought to be complete tumor removal.82 However, even if a patient eventually develops a postoperative recurrence or if surgical debulking is performed, an extended disease-free interval may be attained. A number of studies have reported a benefit to patients even if surgical debulking only can be done. In patients with widely metastatic disease in whom surgical debulking is not possible, various chemotherapeutic agents are frequently used and will be dealt with in a latter section on treatment of advanced disease.