Endocrine Disorders and Tumors
Thyroid Gland
Diseases of the thyroid gland are uncommon conditions in children occurring in 37 of 1,000 school-aged children in the USA.1 Most of these lesions are diffuse gland hypertrophy or simple goiters. Thyroiditis is the second most common abnormality, followed by thyroid nodules and functional disorders. The incidence of thyroid cancer is 0.54 per 100,000, though it is more common with multiple endocrine neoplasia (MEN) syndromes and radiation exposure.2 Operative management plays a role in many of these conditions.
Embryology and Physiology
Thyroglobulin is recognized histologically as colloid. Thyroid hormone is synthesized at the interface between the follicular cell and thyroglobulin. The first step in thyroid synthesis is the iodination of tyrosine molecules, which are then coupled, and form the thyroid hormones thyroxine (T4) and triiodothyronine (T3). T4 and T3 are produced at a ratio of ~4 : 1. T3 is more potent than T4 on the target cell. Both T3 and T4 enter the target cell at which point all T4 is converted to T3. T3 then enters the nucleus of the target cell and the T3 molecule interacts with nuclear thyroid receptors. This receptor–T3 complex binds to DNA to regulate genetic transcription.3 Thyroid hormone increases cellular oxygen consumption and basal metabolic rate, stimulates protein synthesis, and influences carbohydrate, lipid, and vitamin metabolism.
Non-neoplastic Thyroid Conditions
Goiter and Thyroiditis
The causes of thyromegaly in one study are listed in Table 76-1.4 Simple adolescent colloid goiters are the most common cause. Physiologically, diffuse thyroid enlargement may be due to a defect in hormone production, related to autoimmune diseases, or a response to an inflammatory condition. Goiters are classified as diffusely enlarged or nodular, and either toxic or euthyroid. Most goiters are euthyroid, and resection is rarely indicated.
TABLE 76-1
Etiology of Thyroid Gland Enlargement (n = 152 Children)
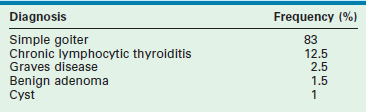
Adapted from Jaksic J, Dumic M, Filipovic B, et al. Thyroid disease in a school population with thyromegaly. Arch Dis Child 1994;70:103–6.
The differential diagnosis for diffuse thyroid enlargement is seen in Box 76-1. Laboratory evaluation should begin with plasma free T4 and TSH levels. With a simple colloid goiter, the patient is euthyroid. Ultrasonography (US) or scintigraphy reveals uniform enlargement, and serum thyroid antibody titers are normal. The etiology of this condition may be an autoimmune process.5 The natural history of colloid goiter is not well known, but one study of adolescents reported that nearly 60% of the glands were normal in size 20 years after diagnosis.1 Exogenous thyroid hormone does not significantly improve resolution of the goiter. In rare cases, resection may be indicated because of size or the suspicion of neoplasia.
Chronic lymphocytic (Hashimoto) thyroiditis is another cause of diffuse thyroid enlargement, occurring most frequently in female adolescents. This condition is part of the spectrum of autoimmune thyroid disorders. It is thought CD4 T-cells are activated against thyroid antigens and recruit cytotoxic CD8 T-cells, which kill thyroid cells, leading to hypothyroidism.6 Children are initially euthyroid and slowly progress to become hypothyroid. However, approximately 10% of children are hyperthyroid, a condition known as ‘hashitoxicosis.’ The thyroid gland is usually pebbly or granular, and may be mildly tender.
Ninety-five per cent of patients with Hashimoto thyroiditis have elevated antithyroid microsomal antibodies or antithyroid peroxidase antibodies. Plasma thyroid hormone levels are normal or low, and TSH levels are elevated in 70% of patients. Thyroid imaging is usually not necessary if clinical and laboratory findings are strongly suggestive of the diagnosis. The radionuclide scan usually shows patchy uptake of the tracer and may mimic the findings in Graves disease or multinodular goiter. The principal ultrasound finding is nonspecific, diffuse thyroid hypoechogenicity. Rarely, autoantibodies cannot be detected and fine needle aspiration is needed to confirm the diagnosis. In as many as one-third of adolescent patients, the thyroiditis resolves spontaneously with the gland becoming normal and the antibodies disappearing. Thus, expectant management should be considered. Exogenous thyroid hormone is administered in the hypothyroid patient. However, in euthyroid children, it is ineffective in reducing the size of the goiter.7
Graves Disease
TSH receptor antibodies are present in more than 95% of patients with active Graves disease, but the inciting event eliciting the antibody response against the TSH receptor is unknown. Infection may produce antibodies that react with the TSH receptor. These antibodies stimulate the thyroid follicles to increase iodide uptake and cyclic adenosine monophosphate production, and induce production and secretion of excess thyroid hormone. Scattered epidemiologic reports of disease clustering supports an infectious etiology for Graves disease.8
Current management includes antithyroid medications, ablation with radioactive 131I, and resection.9 In the USA, most pediatric endocrinologists initiate therapy with methimazole (MTH) or propylthiouracil (PPU) which reduce thyroid hormone production by inhibiting follicle cell organification of iodide and the coupling of iodotyrosines. PPU also inhibits peripheral conversion of T4 to T3, and may be the preferred agent if rapid alleviation of thyrotoxicosis is desired. Both medications possess some immunosuppressive activity as evident by a reduction in antithyroid antibodies. In most cases, MTH is preferred because of its increased potency, longer half-life, and associated improved compliance. The initial adolescent dose is 30 mg once daily, which is reduced if the patient is younger. When the patient becomes euthyroid, as determined by normal T3 and T4 levels, the daily dose of MTH should be reduced to 10 mg. T3 and T4 levels should be monitored. The thyroid gland decreases in size in one-half of patients. Thyroid enlargement with therapy signals either an intensification of the disease or hypothyroidism due to overtreatment.
When treating Graves disease, the goal is to allow natural resolution of the underlying autoimmune process. In general, disease remission rate is approximately 25% after two years of treatment, with a further 25% remission every two years.10 The resolution rate decreases if TSH receptor antibodies persist during and after treatment. The addition of T4 to MTH has had variable results in reducing disease recurrence. However, the use of T4 cannot be recommended in children receiving antithyroid medications.
The thyroid gland must be ablated if resistance or severe reactions to the antithyroid medications develop. Both resection and ablation with radioactive 131I have complications. The advantages of 131I therapy include effectiveness, safety, ease of administration, and relatively low cost.9 Even though the disease recurrence rate is low after 131I treatment, patients risk long-term hypothyroidism. Concerns remain over the possibility of teratogenic or carcinogenic effects of 131I in children and adolescents.11
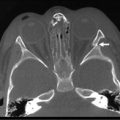
Either a subtotal or total thyroidectomy is indicated for failure or refusal of medical management, or for airway symptoms (Fig. 76-1). Antithyroid medication should be administered to decrease T3 and T4 levels into the normal range before operation. Alternatively, β-blocking agents, such as propranolol, may be used to ameliorate the adrenergic symptoms of hyperthyroidism. In addition, Lugol’s solution, five to ten drops per day, should be administered for 4 to 7 days before thyroidectomy to reduce the vascularity of the gland.
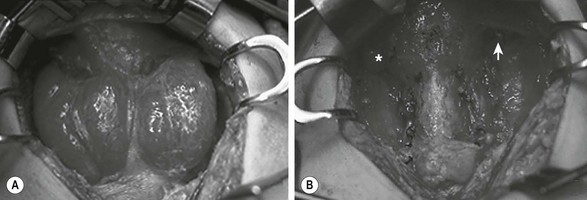
FIGURE 76-1 This teenage girl developed Graves disease and her parents declined radioiodine treatment. (A) The diffusely enlarged, hyperemic thyroid gland is visualized. (B) The thyroid bed is seen after subtotal thyroidectomy. Each upper pole (asterisk, arrow) was left intact.
The incidence of hypothyroidism after subtotal thyroidectomy is variable (10–50%) and the hypothyroidism may be subclinical in up to 45% of children.12 When abnormal TSH levels are considered, the incidence of hyperthyroidism or hypothyroidism is even higher. The rate of recurrent hyperthyroidism is approximately 15%. It is likely the relapse rate increases with time after operation because up to 30% of adult patients exhibit recurrent hyperthyroidism 25 years after their subtotal thyroidectomy.13
Neoplastic Thyroid Conditions
Thyroid Nodules
Thyroid nodules are uncommon in children, but have a 20–25% incidence of malignancy.14,15 Appropriate and prompt evaluation and management are important to decrease disease progression. Various pathology is noted in children who undergo an operation for thyroid nodules (Table 76-2). Other diagnostic possibilities for thyroid nodules include cystic hygroma, thyroglossal duct remnant, and germ cell tumor.
TABLE 76-2
Diagnosis in 251 Pediatric Patients Treated for Thyroid Nodules

Data from Desjardins JG, Khan AH, Montupet P, et al. Management of thyroid nodules in children: A 20-year experience. J Pediatr Surg 1987;22:736–9; Hung W, Anderson KD, Chandra RS, et al. Solitary thyroid nodules in 71 children and adolescents. J Pediatr Surg 1992;27:1407–9; Yip FWK, Reeve TS, Poole AG, et al. Thyroid nodules in childhood and adolescence. Aust N Z J Surg 1994;64:676–8.
As a result of the increased risk of malignancy in younger children, excision of thyroid nodules in prepubescent children is recommended. The natural history of benign lesions in younger children is unknown, and the safety of nonoperative treatment has not been demonstrated. Based on these data, the current recommendation for management of all thyroid nodules in children younger than age 13 is resection. Preoperative ultrasound and thyroid scintigraphy aid in determining the location.14,16
The adolescent spectrum of thyroid disease is similar to that of adults. Thus, fine-needle aspiration is acceptable in evaluating thyroid nodules in this population.17,18 The incidence of malignancy in thyroid nodules in patients age 13 to 18 years is approximately 10%.19 Benign nodules in adolescent patients can be followed with serial examinations and ultrasound. Exogenous thyroid hormone to suppress benign thyroid nodules has not been shown to alter their natural history. Excision should be performed if the nodule is malignant, has indeterminate cytology, increasing size, compressive symptoms, or cosmetic concerns.16 If a cystic thyroid lesion disappears after aspiration, excision can be deferred. If the lesion recurs, it should be removed. Even though cyst fluid can be sent for cytologic analysis, the sensitivity of this test for determining the presence of cancer in children is unknown. Lobectomy can be performed if malignancy is not confirmed preoperatively. If there is a family history, a radiation history, bilateral disease, atypia or suspicious histology, or the lesion is >4 cm, thyroidectomy is recommended.16
Thyroid Carcinoma
Ten per cent of malignant thyroid tumors occur in children, representing about 3% of all pediatric malignancies in the USA. The incidence of childhood thyroid malignancy has decreased in most parts of the world since the mid-1970s due to the reduced use of radiation to treat benign head and neck diseases. However, the incidence may be increasing, perhaps due to improved detection. Overall, mortality remains stable.20
Pediatric thyroid cancer is found more commonly in Caucasians (86%), adolescents (94%), and females (81%). At the time of diagnosis, thyroid cancer is limited to the thyroid gland (42%) or found in the regional lymph nodes (46%). 20 In comparison to adults, children with thyroid carcinoma present with more advanced-stage disease and a higher incidence of lymph node and pulmonary metastases, but have a lower mortality rate.21 Compared to adolescents, prepubertal children have a greater degree of extrathyroid extension, lymph node involvement, and lung metastases at diagnosis.22
Exposure to radiation is a significant risk factor for developing thyroid cancer. A 62-fold increase in thyroid tumors was noted in the Republics of Belarus and Ukraine after the 1986 Chernobyl nuclear power plant catastrophe.23 The tumors were particularly aggressive with increased tumor spread, local invasion, and nodal metastases. The use of radiation for diagnostic purposes has also been linked to increased childhood cancers. It is estimated computed tomography (CT) scans increase risk of malignancy as high as one fatal cancer per 1000 CT scans performed.24,25
Treatment of previous malignancies is another risk factor for thyroid carcinoma. Thyroid cancers constitute about 9% of second malignancies.26 Hodgkin lymphoma is the most common malignancy associated with a subsequent thyroid cancer, followed by non-Hodgkin lymphoma, and leukemia.27 Whereas most thyroid second neoplasms follow previous radiation exposure to the neck, alkylating agents also predispose to thyroid cancer. The mean age at diagnosis of thyroid second neoplasms is 20 years, demonstrating the importance of careful surveillance for second malignancies.
Various molecular biologic events may account for the disparity in behavior of the different histological subtypes of thyroid cancer.28 RAS proto-oncogene mutations are found in about 20% of papillary tumors and 80% of follicular tumors.29 Other studies report RAS is frequently activated in benign follicular adenomas, suggesting this genetic event occurs early in the transformation process.30 An activating mutation of the RET proto-oncogene is found in about 35% of papillary thyroid cancers.31 The RET protein is a receptor tyrosine-kinase molecule, which probably functions within the cell to regulate proliferation or differentiation. This protein may be responsible for the development of medullary thyroid carcinoma (MTC). Specific point mutations are associated with the MEN type 2 (MEN 2A, MEN 2B) syndromes and familial MTC (FMTC). In addition, as many as 40% of patients with sporadic MTCs possess RET mutations.32
Family history is an important factor in pediatric thyroid cancer. Patients with familial nonmedullary thyroid cancer have more aggressive tumors with increased rates of extrathyroid extension, lymph node metastases, and frequently show the phenomenon of ‘anticipation’ (earlier age at disease onset and increased severity in successive generations).33
Thyroid carcinoma usually presents clinically as a thyroid mass, sometimes with enlarged cervical lymph nodes. Regional lymph node metastases are present in three of four children when the disease is first detected (Table 76-3).34,35 Diagnosis of thyroid carcinoma requires appropriate imaging and histological examination. Ultrasound is the diagnostic modality of choice for thyroid lesions with some authors creating Thyroid Imaging Reporting and Data System (TIRADS) to uniformly classify ultrasound characteristics of thyroid nodules.36 In young children, excision is recommended for thyroid nodules after ultrasound evaluation of the thyroid and lymph node basin. In adolescents, ultrasound and ultrasound-guided fine needle aspiration (FNA) is recommended as these patients’ course tends to most resemble adult thyroid disease. US-guided FNA is safe and effective in adolescents when done in experienced centers.16–18,37 As pulmonary metastases are frequent, a preoperative chest radiograph should be obtained in these patients.
TABLE 76-3
Clinical Aspects of Differentiated Thyroid Cancer in Children from Seven Large Pediatric Series133,134
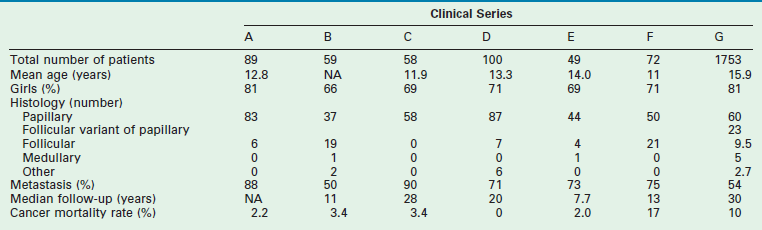
Data from: A, Harness JA, Thompson NW, McLeod MK, et al. Differentiated thyroid carcinoma in children and adolescents. World J Surg 1992;16:547–5542; B, Samuel AM, Sharma SM. Differentiated thyroid carcinomas in children and adolescents. Cancer 1991;67:2186–90; C, Zimmerman D, Hay ID, Gough IR, et al. Papillary thyroid carcinoma in children and adults: Long-term follow-up of 1039 patients conservatively treated at one institution during three decades. Surgery 1988;104:1157–63; D, La Quaglia MP, Corbally MT, Heller G, et al. Recurrence and morbidity in differentiated thyroid carcinoma in children. Surgery 1998;104:1149–56; E, Ceccarelli C, Pacini F, Lippi F, et al. Thyroid cancer in children and adolescents. Surgery 1988;104:1143–8; F, Schlumberger M, De Vathaire F, Travagli JP, et al. Differentiated thyroid carcinoma in childhood: Long-term follow-up in 72 patients. J Clin Endocrinol Metab 1987;65:1088–94; G, Hogan A.R, Zhuge Y, Perez, EA, et al. Pediatric thyroid carcinoma: incidence and outcomes in 1753 patients. J Surg Res 2009;156:167–72.
Treatment for thyroid cancer in children is total or subtotal thyroidectomy. Thyroid cancer is bilateral in up to two-thirds of cases. Approximately 80% of tumors exhibit multifocality. While lobectomy with isthmus resection may be sufficient for isolated lesions, most pediatric surgeons believe total or near-total thyroidectomy should be performed.38,39 No clinical trial has established whether total thyroidectomy, with lymph node dissection if the regional nodes are involved, is better than subtotal thyroidectomy.40 Radioiodine ablative therapy is more effective after removal of the entire gland because functioning thyroid tissue takes up the radionuclide. Surgeons preferring a lesser resection believe differentiated thyroid carcinoma is an indolent disease, and that survival is not related to the extent of gland removal.40,41
If there is regional nodal metastases, a central or modified radial neck dissection is recommended. Prophylactic central node dissection is recommended for T3 or T4 papillary lesions.16 In patients with locally advanced disease, it is imperative to remove as much of the thyroid gland as possible to allow subsequent radioiodine scanning and treatment if the tumor recurs.
Although complications occur less commonly in recent reports, the historical incidence of recurrent laryngeal nerve injury ranges from 0–24%, whereas permanent hypocalcemia occurs in 6–27% of patients undergoing total thyroidectomy.42,43 During dissection, the laryngeal nerve should be identified and protected. When the tumor invades the laryngeal nerve, the nerve can be safely preserved without compromising survival through adjuvant therapy with 131I irradiation to eradicate residual tumor.44 The most reliable way to preserve parathyroid gland function is to identify and preserve the glands at the time of thyroidectomy (Fig. 76-2). If there is apparent devascularization, then one should autotransplant one or two of the glands into the sternocleidomastoid muscle or into the nondominant forearm.45,46
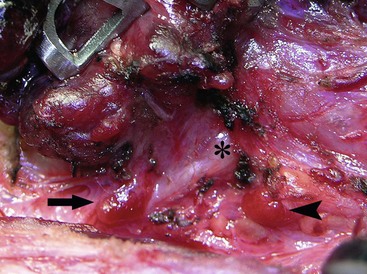
FIGURE 76-2 The relationship of the parathyroid glands to the recurrent laryngeal nerve during prophylactic thyroidectomy for MEN type 2 is seen. The left recurrent laryngeal nerve is marked by an asterisk. The superior parathyroid gland (arrowhead) is typically located posterior to the nerve. The inferior parathyroid gland (arrow) is usually found anterior to the nerve.
The incidence of pulmonary metastases in children at diagnosis is about 6%, but they rarely occur in the absence of significant cervical lymph node metastases.41,47 Pulmonary metastases require treatment with radioiodine. Plain chest films demonstrate the pulmonary disease in only 60% of cases, making scanning with radioiodine necessary. The pulmonary scintiscan can be falsely negative if significant residual thyroid gland remains in the neck.47
In one study, overall survival in non-MTC was 98%.21 A higher rate of recurrence was seen in children who did not receive postoperative 131I than in those who did.21 The time to first recurrence ranged from 8 months to 14.8 years (mean, 5.3 years), thereby emphasizing the importance of long-term follow-up. Prognostic factors associated with recurrence include younger age at diagnosis, histology, capsular or soft tissue invasion, positive margins, and tumor location at diagnosis (thyroid, lymph nodes, lung).21,42 A whole-body 131I scan should be performed approximately six weeks after the initial thyroid resection and followed by therapeutic doses of the radionuclide administered as necessary to ablate residual tissue and treat metastatic disease. Radioactive ablation (RAI) has been shown to decrease the risk of local recurrence, increase the sensitivity of subsequent diagnostic whole-body scans, and improve the utility of serum thyroglobulin as a marker for recurrent or residual disease during long-term follow-up.38 RAI is recommended for all patients with known distant metastases, gross extrathyroidal extension, and primary tumor >4 cm. RAI is recommended for select patients with high risk features such as worrisome histologic subtypes, intrathyroidal vascular invasion, or gross/microscopic multifocal diseases. RAI is not recommended for lesions <1 cm.16 The association of radioactive iodine with second cancers has led to recommendations in the adult literature to avoid radioactive iodine in low risk patients.48
Follow-up for these patients should include physical examination, ultrasound, and thyroglobulin levels. Thyroglobulin can be a useful marker of residual or metastatic thyroid cancer. The plasma level of this protein should be measured yearly, and an elevated value should raise the suspicion of recurrent disease.49 The diagnostic accuracy of this test is decreased in children with residual thyroid tissue or in those who are taking thyroid hormone supplementation. In a long-term study of patients with papillary thyroid cancer, up to 32% of patients had recurrent disease at 40 years follow-up, especially if unilateral lobectomy was performed, emphasizing the need for long-term follow-up.50
MTC accounts for approximately 5% of pediatric thyroid neoplasms. Arising from the parafollicular C cells, MTC may occur either sporadically or in association with MEN 2A, MEN 2B, or the FMTC syndrome. MTC is usually the first tumor to develop in MEN patients and is the most common cause of death in this group. The neoplasm is particularly virulent in patients with MEN 2B and can occur in infancy.51
As with other pediatric thyroid neoplasms, the clinical diagnosis of MTC, not associated with a known familial or MEN syndrome, is usually made only after metastatic spread of the tumor has occurred to the adjacent cervical lymph nodes or to distant sites.52 Resection is the only effective treatment for MTC, underscoring the importance of early diagnosis and therapy before metastases occur.53 For this reason, current management of MTC from MEN 2 and FMTC kindreds relies on the presymptomatic detection of the RET proto-oncogene mutation responsible for the disease. Prophylactic thyroidectomy in young asymptomatic children carrying a mutated allele of the RET proto-oncogene in MEN 2A kindreds has been demonstrated to prevent or cure MTC. In a study of 50 children and teenagers with the RET proto-oncogene mutation who underwent prophylactic thyroidectomy, 66% already had foci of MTC within the thyroid gland.54 However, no child younger than 8 years of age was found to have metastatic disease at surgery. Moreover, when followed for 5 to 8 years, none had evidence of persistent or recurrent disease when screened by physical examination and plasma calcitonin levels obtained after calcium and pentagastrin stimulation. Based on this information, it is recommended that affected children with MEN 2A undergo total thyroidectomy at approximately age 5 years.55–57
Due to the increased virulence of MTC in children with MEN 2B, prophylactic thyroidectomy should be performed at approximately age 1 year. Total thyroidectomy is recommended because of the high incidence of bilateral disease.58 Central lymph node dissection, with removal of lymph nodes medial to the carotid sheaths and between the hyoid bone and the sternum, is likely not necessary except in older children, or when gross lymphadenopathy is discovered at the time of prophylactic thyroidectomy. Early detection by DNA mutation analysis and early operative intervention result in a normal life expectancy.59
Parathyroid Glands
Hyperparathyroidism
The differential diagnosis of hypercalcemia in childhood is shown in Box 76-2. Unlike hypercalcemia in adults, hypercalcemia in children is rarely related to a neoplasm. Rarely, pediatric tumors may secrete a parathyroid-related polypeptide that elevate serum calcium levels. Reported neoplasms producing PTH-like protein include malignant rhabdoid tumor, mesoblastic nephroma, rhabdomyosarcoma, neuroblastoma, and lymphoma. In these patients, the natural PTH level is usually normal or decreased.
Primary Hyperparathyroidism
Primary hyperparathyroidism in childhood typically results from a solitary hyperfunctioning adenoma and more rarely from diffuse hyperplasia of all four glands.60 These lesions are often asymptomatic, but can present with urinary and bone tissue impairment.61 Hyperparathyroidism resulting from hyperplasia in all four glands is a feature of MEN 1. Hyperparathyroidism develops in approximately 30% of MEN 2A patients by their second or third decade of life.62 At the time of prophylactic thyroidectomy for MEN 2, the parathyroid glands can be identified and autotransplanted into the nondominant forearm.45 If hyperparathyroidism develops, a portion of the heterotopic tissue can easily be removed from the forearm.
Primary hyperparathyroidism of infancy is a rare, often fatal, condition usually developing within the first 3 months of life.63,64 Signs include hypotonicity, respiratory distress, failure to thrive, lethargy, and polyuria. The serum PTH level is always elevated, and diffuse parathyroid gland hyperplasia occurs. In about half of the cases, a familial component is found. Early recognition and treatment are essential for normal growth and development.
The management of primary hyperparathyroidism in children is surgical. A solitary hyperfunctioning adenoma accounts for the majority of primary hyperparathyroidism which is often treated safely and effectively with a unilateral neck exploration when preoperative studies demonstrate a single abnormal gland.61,65 Unilateral neck exploration with focused evaluation and excision relies on accurate preoperative localization studies (ultrasound, sestamibi scans, ± CT/MRI) and rapid intraoperative PTH (IO-PTH) assays to confirm that the offending gland or glands are excised. 99mTc-sestamibi is avidly taken up by parathyroid tissue, especially adenomas (Fig. 76-3).66 The sensitivity of detecting abnormal parathyroid glands by sestamibi scan alone is 87%; however, in conjunction with ultrasound, the sensitivity reaches 96%.67,68 After excision of the abnormal parathyroid gland (Fig. 76-4), a baseline serum PTH level is obtained, followed by PTH levels drawn at 5 and 10 minutes. As the intact PTH half-life is only a few minutes, a greater than 50% drop in IO-PTH level within ten minutes signifies successful removal of the hyperfunctioning parathyroid tissue (Fig. 76-5).69 In children, some surgeons feel that due to the rapid IO-PTH decline, the five minute time point is sufficient for determining if the resection is sufficient.70 This approach had the advantage of limiting blood draws and operative time.
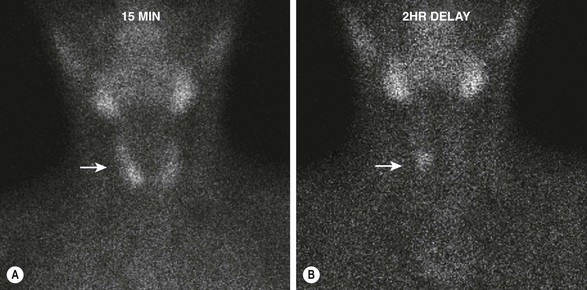
FIGURE 76-3 Early and delayed 99mTc-sestamibi scintigraphy in a 12-year-old boy with primary hyperparathyroidism. (A) The image at 15 minutes demonstrates rapid uptake of the radioisotope by the thyroid gland (arrow). (B) The image at 2 hours reveals that the radioisotope has been washed out of the thyroid, but a focus persists (arrow) that is consistent with a right parathyroid adenoma.
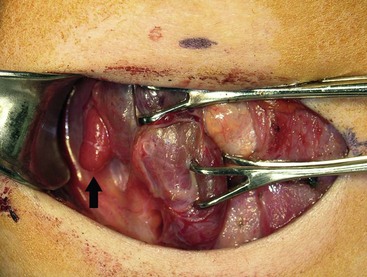
FIGURE 76-4 This intraoperative photograph demonstrates an enlarged right superior parathyroid adenoma (arrow) during unilateral neck exploration that was directed by preoperative localizing studies.
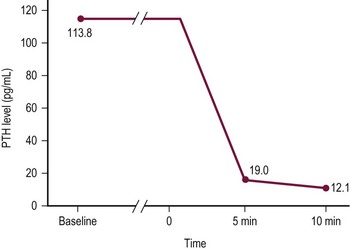
FIGURE 76-5 A rapid intraoperative parathyroid hormone (IO-PTH) assay during unilateral focused parathyroidectomy is depicted. Baseline plasma PTH levels have been drawn. Time 0 represents the moment of resection of the hypersecreting parathyroid gland. PTH levels are drawn at 5 and 10 minutes. A 50% decline in PTH level from baseline at 10 minutes indicates that the offending parathyroid gland or glands have been removed.
Neck exploration with evaluation of all four parathyroid glands is the standard parathyroid operation and should be employed if the patient has an inappropriate response to single gland excision, a single lesion is not identified, parathyroid gland hyperplasia, or suspected MEN. Surgical options for parathyroid gland hyperplasia involving all of the glands include either  gland parathyroidectomy or total parathyroidectomy with heterotopic autotransplantation of some parathyroid tissue back into the nondominant forearm.63 The latter approach is safe in infants and children, and has the advantage of avoiding repeated neck exploration if hyperparathyroidism should recur.45,57 Moreover, total parathyroidectomy with heterotopic autotransplantation results in improved survival in infants with severe hypercalcemia.63 Patients with total parathyroidectomy and autotransplantation require a short period of vitamin D and calcium supplementation until heterotopic tissue begins to function.45
gland parathyroidectomy or total parathyroidectomy with heterotopic autotransplantation of some parathyroid tissue back into the nondominant forearm.63 The latter approach is safe in infants and children, and has the advantage of avoiding repeated neck exploration if hyperparathyroidism should recur.45,57 Moreover, total parathyroidectomy with heterotopic autotransplantation results in improved survival in infants with severe hypercalcemia.63 Patients with total parathyroidectomy and autotransplantation require a short period of vitamin D and calcium supplementation until heterotopic tissue begins to function.45
Familial Hypocalciuric Hypercalcemia
Familial hypocalciuric hypercalcemia is an inherited autosomal dominant disorder caused by a heterozygous mutation in the Ca2+-sensing receptor gene.71 It differs from primary hypoparathyroidism in that the PTH value is normal, but urinary excretion of calcium is low. Patients are usually asymptomatic with an elevated serum calcium level. The serum magnesium level also may be elevated. The parathyroid glands are normal, and usually there is no benefit to parathyroidectomy. If both parents are carriers, severe hypercalcemia may develop in the newborn. These neonates have inherited mutations in both copies of the Ca2+-sensing receptor gene, and often have hyperplasia of all of their parathyroid glands. These infants benefit from parathyroidectomy with transplantation of one gland.
Secondary Hyperparathyroidism
Secondary hyperparathyroidism occurs in children with renal insufficiency or malabsorption. PTH production increases in response to decreased calcium levels. Affected patients typically respond to medical treatment designed to decrease intestinal phosphorus absorption. In rare cases, severe renal osteodystrophy develops, manifested by skeletal fractures and metastatic calcifications. Especially severe cases of secondary hyperparathyroidism may be candidates for total parathyroidectomy with autotransplantation.60
Adrenal Glands
Anatomy and Embryology
The primordial adrenal cortex arises from the coelomic mesoderm and becomes visible between weeks 4 and 6 of development. During embryogenesis, the fetal adrenal gland contains a permanent cortex, fetal cortex, and medulla. The fetal cortex, whose function is unknown, is responsible for the large size of the fetal adrenal gland, with the fetal adrenal gland being four times the size of the fetal kidney at the fourth month of gestation. The fetal cortex decreases in size within hours of birth and disappears by the first year of life. The cells of the permanent cortex are arranged into three separate zones: the zona glomerulosa, zona fasciculata, and zona reticularis. The zona glomerulosa gives rise to the narrowed zona fasciculata and reticularis of the adult cortex. The zona reticularis does not reach adult form until late childhood.72
The adrenal gland is found in several anomalous locations. In adrenal heterotopia, the gland is situated in the normal location, but lies under the capsule of the kidney (adrenal–renal heterotopia) or capsule of the liver (adrenal–hepatic heterotopia). Extra-adrenal tissue (adrenal rest) may be located anywhere in the abdominal cavity, but usually is found along the anatomic derivatives of the urogenital ridge for the adrenal cortex and along the dorsal root ganglia for the medullary tissue. Accessory adrenal glands usually occur without a medullary subcomponent and have been found in 16% of autopsies.73 Another 16% had complete accessory glands.
Adrenal Masses
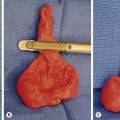
The differential diagnosis of adrenal masses is listed in Box 76-3. Neuroblastoma accounts for more than 90% of adrenal masses. Adrenal masses are detected at a greater rate than previously described because of increased diagnostic testing for other conditions. The significance of such adrenal masses seen on CT is unknown in children. At autopsy, adrenal masses are detected in less than 1% of patients younger than age 30 years.74 This incidence increases to 7% in patients older than 70 years. Follow-up of patients with nonfunctioning adrenal masses demonstrates that 5–25% enlarge by at least 1 cm, and the risk of malignancy is 1 in 1,000. Patients who have an incidentally discovered adrenal mass should undergo hormone evaluation, including a 1 mg dexamethasone suppression test, aldosterone levels, and measurement of plasma free metanephrines.74 Surgical treatment is indicated for all functioning adrenal cortical tumors and pheochromocytoma. In children, most surgeons will resect adrenal tumors regardless of size; however, no clear evidence supports this management over conservative therapy, especially in lesions smaller than 3 cm (Fig. 76-6).75,76
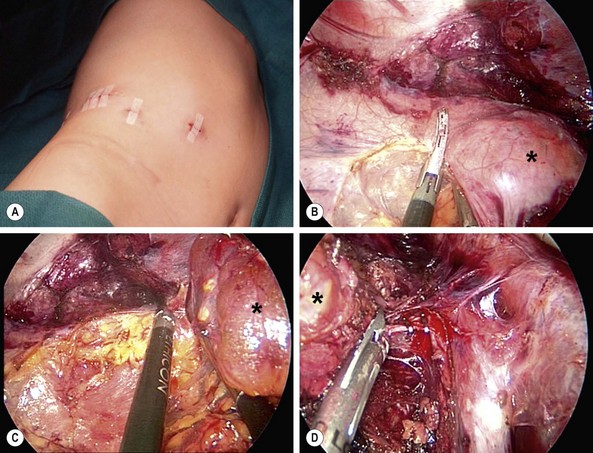
FIGURE 76-6 This teenager was found to have a right adrenal mass measuring 4.5 cm in greatest dimension. Preoperative evaluation did not reveal evidence of a functioning tumor. (A) She was placed in the left lateral decubitus position for the laparoscopic right adrenalectomy. The incisions utilized for the operation are seen. (B) The harmonic scalpel is being used to free the tumor (asterisk) from the lateral surrounding tissue. (C) The tumor (asterisk) has been almost completely mobilized and the cephalad attachments are being lysed with the harmonic scalpel. (D) The tumor (asterisk) is being retracted laterally, and the right adrenal vein is being clipped with endoscopic clips. This tumor was excised uneventfully and was found to be a nonfunctioning adrenal adenoma. The patient was discharged on her first postoperative day.
Adrenal Cortex
Hypercortisolism (Cushing Syndrome)
Hypercortisolism, or Cushing syndrome, describes any form of glucocorticoid excess that can be caused by ACTH secreting pituitary adenomas, adrenal tumors including carcinoma and adenoma, ectopic ACTH syndrome, nodular adrenal hyperplasia, and ACTH-producing tumors (Box 76-4). Additionally, the administration of supraphysiologic quantities of ACTH or glucocorticoids can lead to iatrogenic Cushing syndrome, the most common cause of hypercortisolism in adults and children. Cushing disease is caused by a pituitary microadenoma or, more rarely, by a macroadenoma. It is the second most common cause of Cushing syndrome in children. Ectopic ACTH syndrome is rare in children, but has been reported in infants younger than 1 year. Tumors that can produce ACTH include pulmonary neoplasms, neuroblastomas, pancreatic islet cell carcinomas, thymomas, carcinoids, MTCs, and pheochromocytomas. In children, the most frequent cause of ectopic ACTH is a bronchial carcinoid. ACTH levels are usually ten to 100 times higher than those seen in Cushing disease. These markedly elevated levels of ACTH lead to hypokalemic alkalosis. Additionally, ACTH-independent multinodular adrenal hyperplasia is characterized by hypersecretion of both cortisol and adrenal androgens.
Hypercortisolism is more common in children than previously recognized. In Harvey Cushing’s original description,77 the patient was a 23-year-old woman whose clinical features indicated long-standing disease. In infants and children younger than 7 years, the most common cause of Cushing syndrome is an adrenal tumor (Fig. 76-7). In adults and children older than 7 years, adrenal hyperplasia secondary to hypersecretion of pituitary ACTH predominates.
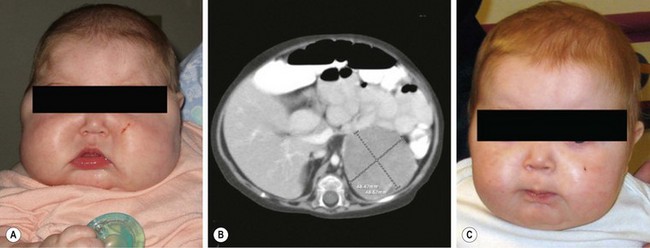
FIGURE 76-7 This 8-month-old child was seen in the emergency department for progressive facial swelling. (A) Examination reveals cushingoid features with moon facies. In addition, she had generalized obesity. Her plasma cortisol level was elevated and her ACTH level was suppressed. (B) CT scan revealed a 4.3 cm × 4.8 × 4.8 cm left adrenal mass that was homogeneous and showed no evidence of invasion of adjacent structures. The infant subsequently underwent laparoscopic left adrenalectomy and was discharged on postoperative day 2. (C) At two-month follow-up, there is marked improvement in the physical manifestations of her Cushing syndrome. (From Kim E, Aguayo P, St. Peter SD, et al. Adrenocortical adenoma expressing glucocorticoid in an 8-month-old female. Eur J Pediatr Surg 2008;18:1–2. Reprinted with permission.)
Clinical features of Cushing syndrome can take five years or longer to develop. Thus, the classic Cushing appearance may not be seen in children. The most frequent and reliable findings with Cushing syndrome are weight gain and growth failure.78 Specifically, any obese child who stops growing should be evaluated for Cushing syndrome.
The initial part of the work-up for Cushing syndrome is to screen for the syndrome followed by localization.79 Screening for Cushing syndrome involves measuring the plasma cortisol at 8:00 am (normal levels, <14 mg/dL) and 6:00 pm (normal levels, <8 mg/dL) to coincide with the diurnal variation. The loss of diurnal rhythm is usually the earliest laboratory evidence of Cushing disease. A single measurement at midnight should be less than 2 mg/dL in normal patients and more than 2 mg/dL in Cushing disease.80 The most sensitive screening test is the 24-hour urinary 17-hydroxycorticosteroid or free cortisol value, which is more than 150 mg/day in patients with Cushing syndrome. The overnight dexamethasone suppression test is performed by administering 1 mg of dexamethasone at 11:00 pm and measuring the plasma cortisol level the following morning at 8:00 am. In normal individuals, ACTH is suppressed and the cortisol level is decreased by 50% or more of baseline (<5 mg/dL). This dose of dexamethasone is insufficient to cause cortisol suppression in patients with Cushing syndrome.
Among children, 80–85% of those with Cushing disease have a surgically identifiable microadenoma81 and transsphenoidal hypophysectomy is indicated. Twenty per cent of patients experience relapse after complete resection and manifest Cushing disease within 5 years. Alternate therapies include pituitary irradiation, adrenalectomy, and drugs inhibiting adrenal function. Of these alternate therapies, adrenalectomy is the preferred treatment when two transsphenoidal procedures fail. Mitotane, an adrenolytic agent, causes a chemical adrenalectomy, but has severe side effects, including nausea, anorexia, and vomiting.
Primary Hyperaldosteronism
Primary hyperaldosteronism is defined as excess production of aldosterone from the adrenal glands with consequent suppression of renin. An adrenal adenoma, or Conn syndrome, is the most common cause of primary hyperaldosteronism in adults, whereas adrenocortical hyperplasia is the most common cause in children.82 Rarely, an adrenal carcinoma can present as primary hyperaldosteronism. Primary hyperaldosteronism is also seen in MEN1-related adrenal tumors.83
After hyperaldosteronism is diagnosed, it is important to distinguish between an aldosterone-secreting adenoma or bilateral adrenal hyperplasia. A solitary adrenal mass greater than 1 cm with a normal-appearing contralateral gland on CT or MRI supports the diagnosis of an adenoma.84 When imaging does not clearly demonstrate a solitary adrenal mass, selective adrenal vein sampling can differentiate unilateral versus bilateral aldosterone hypersecretion. Alternatively, some institutions utilize scintigraphy with 131I-iodomethylnorcholesterol (NP-59), a cholesterol analog taken up as cholesterol in the steroidogenic pathway. Dexamethasone suppression of ACTH-dependent adrenocortical tissue is followed by NP-59 administration. An adenoma is suggested if asymmetric adrenal uptake occurs. Bilateral hyperplasia is suggested if the uptake is symmetric.
The treatment of a functioning adrenal adenoma is resection which can be performed laparoscopically.85,86 The mortality rate following resection is less than 1%, with a cure rate of 75%. Treatment of patients with bilateral adrenal hyperplasia is with spironolactone.
Adrenocortical Carcinoma
Adrenocortical carcinoma is rare. National Cancer Institute’s Surveillance, Epidemiology and End Results data indicate adrenocortical carcinoma represents 1.3% of all carcinomas in children and adolescents younger than 20 years of age in the USA, accounting for only 0.1% of all childhood malignancies.87 There is a female-to-male ratio of 2 : 1 with half of these tumors occurring in patients <5 years of age. The tumors occur equally on the right and left sides, and are hormonally functional in 80–100% of patients. Around the world, there is geographic variability, with southern Brazil having a 15-fold greater incidence compared with other regions. This increased incidence has been attributed to a unique germline missense mutation.88
The etiology of adrenocortical carcinoma is unknown, but its association with several hereditary tumor syndromes gives insight into the molecular pathogenesis of this malignancy. Li–Fraumeni syndrome is an autosomal dominant familial disease characterized by the early onset of tumors, including sarcomas, osteosarcomas, breast and brain cancers, leukemia, and adrenocortical carcinoma. Germ line mutations in the TP53 tumor suppressor gene on chromosome 17p13.1 are found in 70% of affected families.89 These mutations have been found in 20–27% of sporadic cases of adrenocortical carcinomas. Beckwith–Wiedemann syndrome is a congenital overgrowth disorder where patients are predisposed to certain embryonal tumors such as Wilms tumor, hepatoblastoma, neuroblastoma, rhabdomyosarcoma, and adrenocortical carcinoma. Overexpression of insulin-like growth factor (IGF)-2 is believed to contribute to the tumorigenesis in Beckwith–Wiedemann syndrome.90 Several genetic alterations, such as loss of imprinting or loss of heterozygosity of the 11 p15 gene locus causing a strong IGF-2 overexpression, have been demonstrated in the majority of adrenocortical carcinomas.88,91,92 Other hereditary syndromes associated with adrenocortical tumors include Carney complex, MEN 1, and congenital adrenal hyperplasia.83 However, these adrenal tumors are mostly adenomas and rarely carcinoma.
The clinical presentation of adrenocortical carcinoma is usually associated with steroid overproduction. In contrast to adult tumors, most adrenocortical tumors are hormonally active. Virilization is the most frequent presenting feature (66%), whereas the remainder of children will be seen with Cushing syndrome symptoms.93,94 Virilization is secondary to secretion of the adrenal androgens. Features include axillary and pubic hair, deepening of the voice, acne, rapid height growth, hirsutism, enlargement of the penis or clitoromegaly, and development of body odor. Feminization may occur in 2–25% of patients from an overproduction of estrogens, particularly estradiol.95 Only about 5% of pediatric adrenocortical cancers produce no clinical evidence of hormone excess. Accordingly, these patients present with more advanced disease.
As most tumors present with virilization symptoms, evaluation includes measurements of plasma testosterone, and urinary and plasma DHEA and DHEA-S. Urinary 17-ketosteroids are also important because two-thirds of 17-ketosteroids are derived from adrenal androgens. Although the most specific assessment of adrenal androgen production is DHEA-S, 17-ketosteroids are more frequently elevated in malignant disease.96 The clinical presentation of Cushing syndrome is confirmed by hypercortisolism and the loss of diurnal variation. Cortisol excess is determined by elevated plasma cortisol, urinary 17-hydroxycorticosteroids, and urinary free cortisol. Adrenal malignant disease generally causes markedly greater elevations of 17-hydroxycorticosteroids and plasma cortisol than is usually seen with functioning adenomas.
Radiographic studies should proceed concurrently with endocrine evaluation so surgical intervention is expedient. Ultrasound should be used initially for evaluating the adrenal region and for postoperative assessment of recurrence. Smaller lesions are smooth and homogeneous with no pattern of hyperechogenicity or hypoechogenicity. Larger lesions usually demonstrate a ‘scar sign’, radiating linear echoes representing an interphase between separate areas of necrosis, hemorrhage, and neoplasm.97 CT detects tumors as small as 0.5 cm and can identify regional invasion or distant metastases in the liver, lung, or brain. MRI has an accuracy similar to CT with lesions larger than 1–2 cm.98 Finally, adrenal scintigraphy using iodocholesterol-labeled analogs has shown promise in identifying and differentiating functional adrenal lesions.99 Differentiation between hyperplasia, adenoma, and carcinoma is made possible by the inability of carcinoma to concentrate radionuclide. Bilateral symmetric images indicate hyperplasia, unilateral uptake suggests adenoma, and nonvisualization is suggestive of carcinoma.100
Resection offers the only chance for cure. If extensive disease is found at operation, wide en-bloc resection of the tumor, lymph nodes, and involved organs is indicated.101 Higher-volume centers demonstrated improved outcomes with a lower recurrence rate and improved mean time to recurrence.102 For less extensive disease, minimally invasive techniques for adrenalectomy have been advocated, although most authors cite invasive disease as a contraindication to laparoscopic resection.
Adjuvant therapy has marginal results.103 Survival in patients undergoing adjuvant therapy with localized disease has been reported to be 5 years versus 2.3 years for patients who have tumor spread beyond the adrenal gland.101 For patients who underwent further operations for recurrent disease, survival is extended 3.5 years. Pediatric series report the incidence of metastases at diagnosis between 5–64%.89 Mitotane has been the most widely used chemotherapeutic agent. It is used for metastatic disease, for incompletely excised tumors, and for the hormonal effects from the tumors. In adults, tumor response rates to mitotane vary widely (10–60%),89 with a mean response duration of only 10.2 months.104 In the pediatric literature, tumor responses have been reported between 30–40%, though the benefit of adjuvant mitotane is controversial.103,105,106 Additional regimens that have shown some promise include the combination of cisplatin, etoposide, and taxol. The role of radiation therapy has not been well established. In adults, adrenocortical carcinoma is thought to be radioresistant. However, some response has been noted in small series of children with metastatic disease.95 In one report, radiation was used to shrink an unresectable tumor that was subsequently completely excised.89
Patients left untreated with adrenocortical carcinomas have a mean survival of 2.9 months.107 These tumors are highly lethal, with nonfunctional tumors and delay in diagnosis leading to worse prognosis. The range of time from symptoms to diagnosis is 6–36 months.89 The prognosis depends on the child’s age and the resectability of the tumor. In one review of 55 children with adrenal carcinoma, the two-year survival rates were 82% for children <2 years and 29% for children >2 years. Survival rates were more than 67% if the tumors were completely excised, but no survivors were found after partial resection.108
Adrenal Medulla
Pheochromocytoma is a neuroendocrine tumor arising from neural crest–derived chromaffin cells in the adrenal medulla. During embryologic development, chromaffin cells migrate to the adrenal medulla and other areas such as the paraganglia of the carotid arteries, aortic arch, abdominal aorta, and thoracic sympathetic ganglia. Any of these nests of chromaffin cells have the potential to become a tumor, and are the cause of extra-adrenal pheochromocytoma, also termed paraganglioma. The adrenal gland accounts for 85% of all pheochromocytomas in adults. Children have a higher incidence of extra-adrenal pheochromocytoma (30%),109 multifocal, and familial disease,110 but a lower incidence of malignancy compared with adults.111 As in adults, children present with signs and symptoms of catecholamine excess, including hypertension, headaches, sweating, visual complaints, nausea, vomiting, weight loss, polydipsia, and polyuria. Pheochromocytoma should be considered in any child with hypertension.112
The evaluation of pheochromocytoma includes biochemical studies to establish the diagnosis and radiologic imaging to localize the disease.113 Current guidelines, based on the First International Symposium on Pheochromocytoma, recommends initial testing for pheochromocytoma including measurements of fractionated metanephrines (i.e., normetanephrine and metanephrine) in plasma, urine, or both.114 Radiologic imaging options include MRI, CT, and 123I- or 131I-metaiodobenzylguanidine (MIBG) scanning. MRI has the highest sensitivity and is preferred over CT because of decreased radiation exposure.110,115 Although not as sensitive, 131I-MIBG scanning is the most specific imaging technique and is favored when recurrent or metastatic disease is suspected. Most paragangliomas are found in the abdomen and pelvis. When pheochromocytoma is suspected biochemically but not found by imaging, an intrathoracic or intracranial tumor should be suspected.
The perioperative management of patients with pheochromocytoma is critical to a successful outcome. These patients are in a hyperadrenergic state with a low intravascular volume. Catecholamine blockade and intravascular volume expansion are standard preoperative strategies. An α-adrenergic antagonist (e.g., phenoxybenzamine, prazosin, doxazosin, or terazosin) is started a minimum of two weeks before operation. β-adrenergic blockers may be used to treat tachycardia, but should not be used until adequate α blockade has been achieved due to risks of unopposed α-adrenergic stimulation with β-blockade alone. Alternatively, calcium channel blockers have been successfully utilized in the perioperative management of pheochromocytoma and may provide fewer fluctuations in blood pressure as well as cardioprotective effects.111
Historically, pheochromocytoma resection has been performed via an open approach. Even with appropriate preoperative preparation, intraoperative hypertensive and hypotensive episodes are common, and must be anticipated by the anesthesia and surgical team. At operation, it is important to ligate the adrenal vein early and to minimize handling the gland to limit catecholamine surges during the procedure. Laparoscopic transabdominal adrenalectomy has gained significant popularity and been validated for pheochromocytoma resections in adults.116 Although the data in children are currently limited to small series, it appears equally safe and effective in experienced hands (Fig. 76-8).85,86,117,118
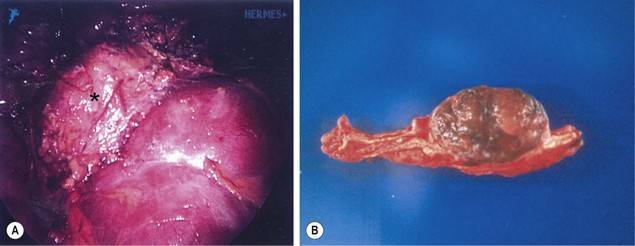
FIGURE 76-8 A teenage boy was found to have marked hypertension and a pheochromocytoma after a metabolic evaluation. He underwent laparoscopic left adrenalectomy and tolerated the procedure nicely. (A) The pheochromocytoma (asterisk) is seen lying on the cephalad portion of the left kidney. The tumor was removed uneventfully. (B) On the cut section of the adrenal gland, the tumor can be seen to be nicely demarcated from the normal adrenal gland.
Up to 25% of pheochromocytomas are familial, and germline mutations in the following genes have been associated with pheochromocytoma: VHL (von Hippel–Lindau syndrome); NF1 (von Recklinghausen neurofibromatosis type 1); RET (MEN 2A or 2B); SDHD and SDHB (familial paragangliomas associated with gene mutations of the mitochondrial succinate dehydrogenase family).110,119 Pheochromocytomas are more frequently associated with MEN 2 syndromes in children than in adults. Also, pheochromocytomas associated with the MEN 2 syndrome are more likely to be bilateral and benign.
In familial pheochromocytoma such as in MEN 2 or VHL syndrome, bilateral tumors inevitably occur. Controversy exists over management of these patients. Bilateral adrenalectomy has been suggested, but predisposes these patients to significant morbidity secondary to corticosteroid replacement as well as complications of medication noncompliance, such as addisonian crises.120 Cortical-sparing adrenalectomies have been proposed for patients with bilateral tumors and those at high risk for developing a metachronous contralateral lesion.121
For patients with metastatic disease, 131I-MIBG scanning and chemotherapy may be effective. Current evidence supports high initial doses of 131I-MIBG for all patients with metastatic lesions who have positive diagnostic 131I-MIBG scans. With tumors responding symptomatically or hormonally to treatment, survival is 4.7 years after treatment.122 Chemotherapy seems to have an additive effect with 131I-MIBG scanning to increase survival in these patients.123 Although serious toxicity may occur, the survival and response rates achieved with high-dose 131I-MIBG suggest its utility in the management of selected patients with metastatic pheochromocytoma and paraganglioma.124
Precocious Puberty
Precocious Puberty in Girls
McCune–Albright syndrome is an interesting disorder causing either true precocious puberty or pseudoprecocious puberty. Patients have the classic triad of precocious puberty, café-au-lait nevi with irregular ‘coast of Maine’ borders, and polyostotic fibrous dysplasia. In these patients, autonomously functioning ovarian follicular cysts can develop, causing precocious puberty. Excess production of LH, FSH, or prolactin by pituitary adenomas also has been described. Other endocrine abnormalities including acromegaly, Cushing syndrome, and hyperthyroidism have been associated with this syndrome.125
Precocious Puberty in Boys
Excess androgen production causing virilization can be caused by congenital adrenal hyperplasia, specifically the 21-hydroxylase or the 11-hydroxylase enzymatic defects. During embryonic development, adrenal rests may be left in the testes. In untreated adrenal hyperplasia, ACTH stimulation can cause enlargement of these adrenal rests and the secretion of androgens. These testes have an irregular appearance. Excess testosterone production also can be caused by interstitial cell tumors of the testes. Finally, exogenous administration of androgens or hCG (for treatment of undescended testes) can cause precocious puberty.
Carcinoid Tumors
Carcinoid tumors comprise less than 0.1% of tumors identified at one large pediatric cancer center,126 though they account for 34% of the primary solid tumors of the colon and rectum.127 These tumors arise from amine uptake and decarboxylation cells, and are usually classified according to their site of origin as foregut, midgut, or hindgut carcinoids.128 Foregut tumors can arise in the bronchus, stomach, or duodenum. Midgut tumors account for 80% to 85% of carcinoid tumors. The majority of carcinoids arise from the appendix (46%), followed by the jejunum and ileum (28%). Hindgut tumors are most commonly located in the rectum (17%). Carcinoid tumors are also found in the biliary ductal system and in ovarian teratomas.
Most patients present with vague symptoms. Only 10% of patients have symptoms of carcinoid syndrome (flushing, diarrhea, abdominal pain, asthma, and right-sided cardiac valvular problems).129 Carcinoids are detected incidentally in up to 60% of patients, usually after appendectomy for acute appendicitis.
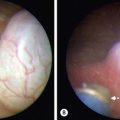
Most children are seen with appendiceal carcinoids smaller than 2 cm (Fig. 76-9). These can be treated with simple appendectomy.127Right hemicolectomy is indicated for tumors larger than 2 cm, close to the cecum, or with mucin production. Metastases are rare, but regular follow-ups with measurement of serotonin and chromogranin A is important.130 Treatment of metastatic disease includes hepatic chemoembolization and resection of isolated hepatic metastases. Long-acting octreotide or 131I-MIBG is used for widely metastatic disease.131
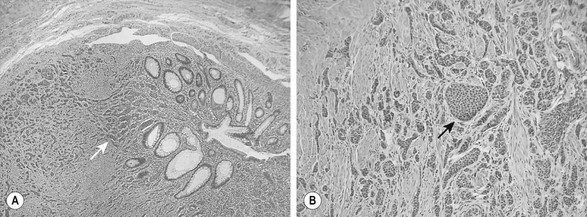
FIGURE 76-9 These two histologic photomicrographs depict a carcinoid tumor of the appendix. (A) Lower-power view shows the nests of neuroendocrine cells (arrow) that are seen invading the muscularis mucosa. (B) Higher-power view shows the solid islands or nests (arrow) of uniform oval to polygonal cells with minimal pleomorphism. There are indistinct cellular borders and round uniform nucleoli with finely granular diffuse chromatin and inconspicuous nucleoli. Mitotic figures are rare.
Carcinoids are fairly indolent tumors. In one retrospective series of 40 children with appendiceal carcinoids, no recurrences or metastases were reported.132 The site of origin has universally been shown to predict survival, with the appendix having the best survival and the midgut or hindgut having the worst.
The most common pulmonary tumor is the bronchial carcinoid. These most frequently appear with recurrent or persistent pneumonia secondary to bronchial obstruction from the tumor. Usually these tumors excrete low levels of serotonin. Children commonly present with wheezing, atelectasis, and weight loss. Also, they can have cough, pneumonitis, and hemoptysis, symptoms that are frequently seen in adults. Bronchial carcinoids can be diagnosed by bronchoscopy. They have a characteristic pink, friable, mulberry appearance. Due to this classic appearance and their propensity to bleed, biopsy is not recommended. If there is no evidence of lymph node involvement, segmental bronchial resection can be performed. However, lobectomy or pneumonectomy is often required for treatment. These tumors are radiosensitive and radiation therapy can be considered for unresectable disease. The prognosis after complete resection is excellent, with a 10-year survival rate of approximately 90%.133,134
References
1. Rallison, ML, Dobyns, BM, Meikle, AW, et al. Natural history of thyroid abnormalities: Prevalence, incidence, and regression of thyroid diseases in adolescents and young adults. Am J Med. 1991; 91:363–370.
2. Davies, L, Welch, HG. Increasing incidence of thyroid cancer in the United States, 1973–2002. JAMA. 2006; 295:2164–2167.
3. Brent, GA. The molecular basis of thyroid hormone action. N Engl J Med. 1994; 331:847–853.
4. Jaksic, J, Dumic, M, Filipovic, B, et al. Thyroid diseases in a school population with thyromegaly. Arch Dis Child. 1994; 70:103–106.
5. Fisher, DA, Pandian, MR, Carlton, E. Autoimmune thyroid disease: An expanding spectrum. Pediatr Clin North Am. 1987; 34:907–918.
6. Dayan, CM, Daniels, GH. Chronic autoimmune thyroiditis. N Engl J Med. 1996; 335:99–107.
7. Rother, KI, Zimmerman, D, Schwenk, WF. Effect of thyroid hormone treatment on thyromegaly in children and adolescents with Hashimoto disease. J Pediatr. 1994; 124:599–601.
8. Phillips, DI, Barker, DJ, Rees Smith, B, et al. The geographical distribution of thyrotoxicosis in England according to the presence or absence of TSH-receptor antibodies. Clin Endocrinol (Oxf). 1985; 23:283–287.
9. Bahn, RS, Burch, HB, Cooper, DS, et al. Hyperthyroidism and other causes of thyrotoxicosis: Management guidelines of the American Thyroid Association and American Association of Clinical Endocrinologists. Endocr Pract. 2011; 17:456–520.
10. Lippe, BM, Landaw, EM, Kaplan, SA. Hyperthyroidism in children treated with long term medical therapy: Twenty-five percent remission every two years. J Clin Endocrinol Metab. 1987; 64:1241–1245.
11. Lang, BH, Wong, IO, Wong, KP, et al. Risk of second primary malignancy in differentiated thyroid carcinoma treated with radioactive iodine therapy. Surgery. 2012; 151:844–850.
12. Waldhausen, JH. Controversies related to the medical and surgical management of hyperthyroidism in children. Semin Pediatr Surg. 1997; 6:121–127.
13. Franklyn, JA. The management of hyperthyroidism. N Engl J Med. 1994; 330:1731–1738.
14. Hung, W, Anderson, KD, Chandra, RS, et al. Solitary thyroid nodules in 71 children and adolescents. J Pediatr Surg. 1992; 27:1407–1409.
15. Desjardins, JG, Khan, AH, Montupet, P, et al. Management of thyroid nodules in children: A 20-year experience. J Pediatr Surg. 1987; 22:736–739.
16. Cooper, DS, Doherty, GM, Haugen, BR, et al. Revised American Thyroid Association management guidelines for patients with thyroid nodules and differentiated thyroid cancer. Thyroid. 2009; 19:1167–1214.
17. Corrias, A, Einaudi, S, Chiorboli, E, et al. Accuracy of fine needle aspiration biopsy of thyroid nodules in detecting malignancy in childhood: Comparison with conventional clinical, laboratory, and imaging approaches. J Clin Endocrinol Metab. 2001; 86:4644–4648.
18. Izquierdo, R, Shankar, R, Kort, K, et al. Ultrasound-guided fine-needle aspiration in the management of thyroid nodules in children and adolescents. Thyroid. 2009; 19:703–705.
19. Yip, FW, Reeve, TS, Poole, AG, et al. Thyroid nodules in childhood and adolescence. Aust N Z J Surg. 1994; 64:676–678.
20. Hogan, AR, Zhuge, Y, Perez, EA, et al. Pediatric thyroid carcinoma: Incidence and outcomes in 1753 patients. J Surg Res. 2009; 156:167–172.
21. Grigsby, PW, Gal-or, A, Michalski, JM, et al. Childhood and adolescent thyroid carcinoma. Cancer. 2002; 95:724–729.
22. Lazar, L, Lebenthal, Y, Steinmetz, A, et al. Differentiated thyroid carcinoma in pediatric patients: Comparison of presentation and course between pre-pubertal children and adolescents. J Pediatr. 2009; 154:708–714.
23. Nikiforov, Y, Gnepp, DR. Pediatric thyroid cancer after the Chernobyl disaster. Pathomorphologic study of 84 cases (1991-1992) from the Republic of Belarus. Cancer. 1994; 74:748–766.
24. Rice, HE, Frush, DP, Farmer, D, et al. Review of radiation risks from computed tomography: Essentials for the pediatric surgeon. J Pediatr Surg. 2007; 42:603–607.
25. Pearce, MS, Salotti, JA, Little, MP, et al. Radiation exposure from CT scans in childhood and subsequent risk of leukaemia and brain tumours: A retrospective cohort study. Lancet. 2012; 380:499–505.
26. Smith, MB, Xue, H, Strong, L, et al. Forty-year experience with second malignancies after treatment of childhood cancer: Analysis of outcome following the development of the second malignancy. J Pediatr Surg. 1993; 1342–1348.
27. Taylor, AJ, Croft, AP, Palace, AM, et al. Risk of thyroid cancer in survivors of childhood cancer: Results from the British Childhood Cancer Survivor Study. Int J Cancer. 2009; 125:2400–2405.
28. Diesen, DL, Skinner, MA. Pediatric thyroid cancer. Semin Pediatr Surg. 2012; 21:44–50.
29. Lemoine, NR, Mayall, ES, Wyllie, FS, et al. Activated ras oncogenes in human thyroid cancers. Cancer Res. 1988; 48:4459–4463.
30. Lemoine, NR, Mayall, ES, Wyllie, FS, et al. High frequency of ras oncogene activation in all stages of human thyroid tumorigenesis. Oncogene. 1989; 4:159–164.
31. Bongarzone, I, Butti, MG, Coronelli, S, et al. Frequent activation of ret protooncogene by fusion with a new activating gene in papillary thyroid carcinomas. Cancer Res. 1994; 54:2979–2985.
32. Eng, C, Smith, DP, Mulligan, LM, et al. Point mutation within the tyrosine kinase domain of the RET proto-oncogene in multiple endocrine neoplasia type 2B and related sporadic tumours. Hum Mol Genet. 1994; 3:237–241.
33. Bonora, E, Tallini, G, Romeo, G. Genetic predisposition to familial nonmedullary thyroid cancer: An update of molecular findings and state-of-the-art studies. J Oncol. 2010; 385–386.
34. Scholz, S, Smith, JR, Chaignaud, B, et al. Thyroid surgery at Children’s Hospital Boston: A 35-year single-institution experience. J Pediatr Surg. 2011; 46:437–442.
35. Roy, R, Kouniavsky, G, Schneider, E, et al. Predictive factors of malignancy in pediatric thyroid nodules. Surgery. 2011; 150:1228–1233.
36. Horvath, E, Majlis, S, Rossi, R, et al. An ultrasonogram reporting system for thyroid nodules stratifying cancer risk for clinical management. J Clin Endocrinol Metab. 2009; 94:1748–1751.
37. Hung, W. Solitary thyroid nodules in 93 children and adolescents. A 35-years experience. Horm Res. 1999; 52:15–18.
38. Hung, W, Sarlis, NJ. Current controversies in the management of pediatric patients with well-differentiated nonmedullary thyroid cancer: A review. Thyroid. 2002; 12:683–702.
39. Haveman, JW, van Tol, KM, Rouwe, CW, et al. Surgical experience in children with differentiated thyroid carcinoma. Ann Surg Oncol. 2003; 10:15–20.
40. Enomoto, Y, Enomoto, K, Uchino, S, et al. Clinical features, treatment, and long-term outcome of papillary thyroid cancer in children and adolescents without radiation exposure. World J Surg. 2012; 36:1241–1246.
41. Zimmerman, D, Hay, ID, Gough, IR, et al. Papillary thyroid carcinoma in children and adults: Long-term follow-up of 1039 patients conservatively treated at one institution during three decades. Surgery. 1988; 104:1157–1166.
42. La Quaglia, MP, Corbally, MT, Heller, G, et al. Recurrence and morbidity in differentiated thyroid carcinoma in children. Surgery. 1988; 104:1149–1156.
43. de Roy van Zuidewijn, DB, Songun, I, Kievit, J, et al. Complications of thyroid surgery. Ann Surg Oncol. 1995; 2:56–60.
44. Nishida, T, Nakao, K, Hamaji, M, et al. Preservation of recurrent laryngeal nerve invaded by differentiated thyroid cancer. Ann Surg. 1997; 226:85–91.
45. Wells, SA, Jr., Farndon, JR, Dale, JK, et al. Long-term evaluation of patients with primary parathyroid hyperplasia managed by total parathyroidectomy and heterotopic autotransplantation. Ann Surg. 1980; 192:451–458.
46. Skinner, MA, Norton, JA, Moley, JF, et al. Heterotopic autotransplantation of parathyroid tissue in children undergoing total thyroidectomy. J Pediatr Surg. 1997; 32:510–513.
47. Vassilopoulou-Sellin, R, Klein, MJ, Smith, TH, et al. Pulmonary metastases in children and young adults with differentiated thyroid cancer. Cancer. 1993; 71:1348–1352.
48. Iyer, NG, Morris, LG, Tuttle, RM, et al. Rising incidence of second cancers in patients with low-risk (T1N0) thyroid cancer who receive radioactive iodine therapy. Cancer. 2011; 117:4439–4446.
49. Kirk, JM, Mort, C, Grant, DB, et al. The usefulness of serum thyroglobulin in the follow-up of differentiated thyroid carcinoma in children. Med Pediatr Oncol. 1992; 20:201–208.
50. Hay, ID, Gonzalez-Losada, T, Reinalda, MS, et al. Long-term outcome in 215 children and adolescents with papillary thyroid cancer treated during 1940 through 2008. World J Surg. 2010; 34:1192–1202.
51. Samaan, NA, Draznin, MB, Halpin, RE, et al. Multiple endocrine syndrome type IIb in early childhood. Cancer. 1991; 68:1832–1834.
52. Gorlin, JB, Sallan, SE. Thyroid cancer in childhood. Endocrinol Metab Clin North Am. 1990; 19:649–662.
53. Kloos, RT, Eng, C, Evans, DB, et al. Medullary thyroid cancer: Management guidelines of the American Thyroid Association. Thyroid. 2009; 19:565–612.
54. Skinner, MA, Moley, JA, Dilley, WG, et al. Prophylactic thyroidectomy in multiple endocrine neoplasia type 2A. N Engl J Med. 2005; 353:1105–1113.
55. Szinnai, G, Meier, C, Komminoth, P, et al. Review of multiple endocrine neoplasia type 2A in children: Therapeutic results of early thyroidectomy and prognostic value of codon analysis. Pediatrics. 2003; 111:E132–E139.
56. Wells, SA, Jr., Chi, DD, Toshima, K, et al. Predictive DNA testing and prophylactic thyroidectomy in patients at risk for multiple endocrine neoplasia type 2A. Ann Surg. 1994; 220:237–247.
57. Skinner, MA, DeBenedetti, MK, Moley, JF, et al. Medullary thyroid carcinoma in children with multiple endocrine neoplasia types 2A and 2B. J Pediatr Surg. 1996; 31:177–181.
58. Telander, RL, Zimmerman, D, van Heerden, JA, et al. Results of early thyroidectomy for medullary thyroid carcinoma in children with multiple endocrine neoplasia type 2. J Pediatr Surg. 1986; 21:1190–1194.
59. Wiersinga, WM. Thyroid cancer in children and adolescents–consequences in later life. J Pediatr Endocrinol Metab. 2001; 5:1289–1296.
60. Ross, AJ, 3rd. Parathyroid surgery in children. Prog Pediatr Surg. 1991; 26:48–59.
61. Li, CC, Yang, C, Wang, S, et al. A 10-year retrospective study of primary hyperparathyroidism in children. Exp Clin Endocrinol Diabetes. 2012; 120:229–233.
62. Howe, JR, Norton, JA, Wells, SA, Jr. Prevalence of pheochromocytoma and hyperparathyroidism in multiple endocrine neoplasia type 2A: Results of long-term follow-up. Surgery. 1993; 114:1070–1077.
63. Ross, AJ, 3rd., Cooper, A, Attie, MF, et al. Primary hyperparathyroidism in infancy. J Pediatr Surg. 1986; 21:493–499.
64. Kulczycha, H, Kaminski, W, Wozniewicz, B, et al. Primary hyperparathyroidism in infancy: Diagnostic and therapeutic difficulties. Klin Padiatr. 1991; 84:116–118.
65. Palazzo, FF, Delbridge, LW. Minimal-access/minimally invasive parathyroidectomy for primary hyperparathyroidism. Surg Clin North Am. 2004; 84:717–734.
66. Coakley, AJ, Kettle, AG, Wells, CP, et al. 99Tcm sestamibi–a new agent for parathyroid imaging. Nucl Med Commun. 1989; 10:791–794.
67. McBiles, M, Lambert, AT, Cote, MG, et al. Sestamibi parathyroid imaging. Semin Nucl Med. 1995; 25:221–234.
68. Arici, C, Cheah, WK, Ituarte, PH, et al. Can localization studies be used to direct focused parathyroid operations? Surgery. 2001; 129:720–729.
69. Garner, SC, Leight, GS, Jr. Initial experience with intraoperative PTH determinations in the surgical management of 130 consecutive cases of primary hyperparathyroidism. Surgery. 1999; 126:1132–1137.
70. Burke JF, Scheider DF, Nichol PF, et al. Analysis of intraoperative parathyroid hormone levels in children: Does standard protocol apply? Presented at the 2012 AAP Meeting.
71. Pollak, MR, Brown, EM, Chou, YH, et al. Mutations in the human Ca(2+)-sensing receptor gene cause familial hypocalciuric hypercalcemia and neonatal severe hyperparathyroidism. Cell. 1993; 75:1297–1303.
72. Sucheston, ME, Cannon, MS. Development of zonular patterns in the human adrenal gland. J Morphol. 1968; 126:477–491.
73. Graham, LS. Celiac accessory adrenal glands. Cancer. 1953; 6:149–152.
74. Grumbach, MM, Biller, BM, Braunstein, GD, et al. Management of the clinically inapparent adrenal mass (‘incidentaloma’). Ann Intern Med. 2003; 138:424–429.
75. Masiakos, PT, Gerstle, JT, Cheang, T, et al. Is surgery necessary for incidentally discovered adrenal masses in children? J Pediatr Surg. 2004; 39:754–758.
76. Stewart, JN, Flageole, H, Kavan, P. A surgical approach to adrenocortical tumors in children: The mainstay of treatment. J Pediatr Surg. 2004; 39:759–763.
77. Cushing, H. The basophil adenomas of the pituitary body and their clinical manifestations. Bull Johns Hopkins Hospital. 1932; 50:137–195.
78. Devoe, DJ, Miller, WL, Conte, FA, et al. Long-term outcome in children and adolescents after transsphenoidal surgery for Cushing’s disease. J Clin Endocrinol Metab. 1997; 82:3196–3202.
79. Nieman, LK, Biller, BM, Findling, JW, et al. The diagnosis of Cushing’s syndrome: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2008; 93:1526–1540.
80. Newell-Price, J, Trainer, P, Besser, M, et al. The diagnosis and differential diagnosis of Cushing’s syndrome and pseudo-Cushing’s states. Endocr Rev. 1998; 19:647–672.
81. Styne, DM, Grumbach, MM, Kaplan, SL, et al. Treatment of Cushing’s disease in childhood and adolescence by transsphenoidal microadenomectomy. N Engl J Med. 1984; 310:889–893.
82. Chudler, RM, Kay, R. Adrenocortical carcinoma in children. Urol Clin North Am. 1989; 16:469–479.
83. Gatta-Cherifi, B, Chabre, O, Murat, A, et al. Adrenal involvement in MEN1. Analysis of 715 cases from the Groupe d’etude des Tumeurs Endocrines database. Eur J Endocrinol. 2012; 166:269–279.
84. Al Fehaily, M, Duh, QY. Clinical manifestation of aldosteronoma. Surg Clin North Am. 2004; 84:887–905.
85. IPEG guidelines for the surgical treatment of adrenal masses in children. J Laparoendosc Adv Surg Tech A. 2010; 20:vii–ix.
86. St Peter, SD, Valusek, PA, Hill, S, et al. Laparoscopic adrenalectomy in children: A multicenter experience. J Laparoendosc Adv Surg Tech A. 2011; 21:647–649.
87. Bernstein, L, Gurney, JG, Carcinomas and other malignant epithelial neoplasms Bethesda, MDRies LAG, Smith MA, Gurney JG, et al, eds. Cancer and survival among children and adolescents: United States SEER program 1975-1995, 1999:139–147.
88. Sutter, JA, Grimberg, A. Adrenocortical tumors and hyperplasias in childhood–etiology, genetics, clinical presentation and therapy. Pediatr Endocrinol Rev. 2006; 4:32–39.
89. Soon, PS, McDonald, KL, Robinson, BG, Sidhu, SB. Molecular markers and the pathogenesis of adrenocortical cancer. Oncologist. 2008; 13:548–561.
90. Liou, LS, Kay, R. Adrenocortical carcinoma in children. Review and recent innovations. Urol Clin North Am. 2000; 27:403–421.
91. Fottner, C, Hoeflich, A, Wolf, E, et al. Role of the insulin-like growth factor system in adrenocortical growth control and carcinogenesis. Horm Metab Res. 2004; 36:397–405.
92. Ilvesmaki, V, Kahri, AI, Miettinen, PJ, et al. Insulin-like growth factors (IGFs) and their receptors in adrenal tumors: High IGF-II expression in functional adrenocortical carcinomas. J Clin Endocrinol Metab. 1993; 77:852–858.
93. Michalkiewicz, E, Sandrini, R, Figueiredo, B, et al. Clinical and outcome characteristics of children with adrenocortical tumors: A report from the International Pediatric Adrenocortical Tumor Registry. J Clin Oncol. 2004; 22:838–845.
94. Hayles, AB, Hahn, HB, Jr., Sprague, RG, et al. Hormone-secreting tumors of the adrenal cortex in children. Pediatrics. 1966; 37:19–25.
95. Stewart, DR, Jones, PH, Jolleys, A. Carcinoma of the adrenal gland in children. J Pediatr Surg. 1974; 9:59–67.
96. Neblett, WW, Frexes-Steed, M, Scott, HW, Jr. Experience with adrenocortical neoplasms in childhood. Am Surg. 1987; 53:117–125.
97. Prando, A, Wallace, S, Marins, JL, et al. Sonographic findings of adrenal cortical carcinomas in children. Pediatr Radiol. 1990; 20:163–165.
98. Glazer, GM. MR imaging of the liver, kidneys, and adrenal glands. Radiology. 1988; 166:303–312.
99. Gross, MD, Shapiro, B, Francis, IR, et al. Scintigraphic evaluation of clinically silent adrenal masses. J Nucl Med. 1994; 35:1145–1152.
100. Thrall, JH, Freitas, JE, Beierwaltes, WH. Adrenal scintigraphy. Semin Nucl Med. 1978; 8:23–41.
101. Cohn, K, Gottesman, L, Brennan, M. Adrenocortical carcinoma. Surgery. 1986; 100:1170–1177.
102. Lombardi, CP, Raffaelli, M, Boniardi, M, et al. Adrenocortical carcinoma: Effect of hospital volume on patient outcome. Langenbecks Arch Surg. 2012; 397:201–207.
103. Grubbs, EG, Callender, GG, Xing, Y, et al. Recurrence of adrenal cortical carcinoma following resection: Surgery alone can achieve results equal to surgery plus mitotane. Ann Surg Oncol. 2010; 17:263–270.
104. Hutter, AM, Jr., Kayhoe, DE. Adrenal cortical carcinoma. Results of treatment with o,p’DDD in 138 patients. Am J Med. 1966; 41:581–592.
105. Mayer, SK, Oligny, LL, Deal, C, et al. Childhood adrenocortical tumors: Case series and reevaluation of prognosis–a 24-year experience. J Pediatr Surg. 1997; 32:911–915.
106. Teinturier, C, Pauchard, MS, Brugieres, L, et al. Clinical and prognostic aspects of adrenocortical neoplasms in childhood. Med Pediatr Oncol. 1999; 32:106–111.
107. Macfarlane, DA. Cancer of the adrenal cortex; the natural history, prognosis and treatment in a study of fifty-five cases. Ann R Coll Surg Engl. 1958; 23:155–186.
108. Sabbaga, CC, Avilla, SG, Schulz, C, et al. Adrenocortical carcinoma in children: Clinical aspects and prognosis. J Pediatr Surg. 1993; 28:841–843.
109. Manger, WM. An overview of pheochromocytoma: History, current concepts, vagaries, and diagnostic challenges. Ann N Y Acad Sci. 2006; 1073:1–20.
110. Lenders, JW, Eisenhofer, G, Mannelli, M, et al. Phaeochromocytoma. Lancet. 2005; 366:665–675.
111. Ross, JH. Pheochromocytoma. Special considerations in children. Urol Clin North Am. 2000; 27:393–402.
112. Januszewicz, P, Wieteska-Klimczak, A, Wyszynska, T. Pheochromocytoma in children: Difficulties in diagnosis and localization. Clin Exp Hypertens A. 1990; 12:571–579.
113. Waguespack, SG, Rich, T, Grubbs, E, et al. A current review of the etiology, diagnosis, and treatment of pediatric pheochromocytoma and paraganglioma. J Clin Endocrinol Metab. 2010; 95:2023–2037.
114. Pacak, K, Eisenhofer, G, Ahlman, H, et al. Pheochromocytoma: Recommendations for clinical practice from the First International Symposium. October 2005. Nat Clin Pract Endocrinol Metab. 2007; 3:92–102.
115. Quint, LE, Glazer, GM, Francis, IR, et al. Pheochromocytoma and paraganglioma: Comparison of MR imaging with CT and I-131 MIBG scintigraphy. Radiology. 1987; 165:89–93.
116. Kercher, KW, Novitsky, YW, Park, A, et al. Laparoscopic curative resection of pheochromocytomas. Ann Surg. 2005; 241:919–926.
117. Miller, KA, Albanese, C, Harrison, M, et al. Experience with laparoscopic adrenalectomy in pediatric patients. J Pediatr Surg. 2002; 37:979–982.
118. Skarsgard, ED, Albanese, CT. The safety and efficacy of laparoscopic adrenalectomy in children. Arch Surg. 2005; 140:905–908.
119. Armstrong, R, Sridhar, M, Greenhalgh, KL, et al. Phaeochromocytoma in children. Arch Dis Child. 2008; 93:899–904.
120. Caty, MG, Coran, AG, Geagen, M. Current diagnosis and treatment of pheochromocytoma in children. Experience with 22 consecutive tumors in 14 patients. Arch Surg. 1990; 125:978–981.
121. Neumann, HP, Bender, BU, Reincke, M, et al. Adrenal-sparing surgery for phaeochromocytoma. Br J Surg. 1999; 86:94–97.
122. Safford, SD, Coleman, RE, Gockerman, JP, et al. Iodine -131 metaiodobenzylguanidine is an effective treatment for malignant pheochromocytoma and paraganglioma. Surgery. 2003; 134:956–962.
123. Sisson, JC, Shapiro, B, Shulkin, BL, et al. Treatment of malignant pheochromocytomas with 131-I metaiodobenzylguanidine and chemotherapy. Am J Clin Oncol. 1999; 22:364–370.
124. Gonias, S, Goldsby, R, Matthay, KK, et al. Phase II study of high-dose [131I]metaiodobenzylguanidine therapy for patients with metastatic pheochromocytoma and paraganglioma. J Clin Oncol. 2009; 27:4162–4168.
125. Rosenfield, RL. Puberty and its disorders in girls. Endocrinol Metab Clin North Am. 1991; 20:15–42.
126. Spunt, SL, Pratt, CB, Rao, BN, et al. Childhood carcinoid tumors: The St Jude Children’s Research Hospital experience. J Pediatr Surg. 2000; 35:1282–1286.
127. Yang, R, Cheung, MC, Zhuge, Y, et al. Primary solid tumors of the colon and rectum in the pediatric patient: A review of 270 cases. J Surg Res. 2010; 161:209–216.
128. Stinner, B, Kisker, O, Zielke, A, et al. Surgical management for carcinoid tumors of small bowel, appendix, colon, and rectum. World J Surg. 1996; 20:183–188.
129. Onaitis, MW, Kirshbom, PM, Hayward, TZ, Quayle, FJ, et al. Gastrointestinal carcinoids: Characterization by site of origin and hormone production. Ann Surg. 2000; 232:549–556.
130. Doede, T, Foss, HD, Waldschmidt, J. Carcinoid tumors of the appendix in children–epidemiology, clinical aspects and procedure. Eur J Pediatr Surg. 2000; 10:372–377.
131. Safford, SD, Coleman, RE, Gockerman, JP, et al. Iodine-131 metaiodobenzylguanidine treatment for metastatic carcinoid. Results in 98 patients. Cancer. 2004; 101:1987–1993.
132. Parkes, SE, Muir, KR, al Sheyyab, M, et al. Carcinoid tumours of the appendix in children 1957-1986: Incidence, treatment and outcome. Br J Surg. 1993; 80:502–504.
133. Wang, LT, Wilkins, EW, Jr., Bode, HH. Bronchial carcinoid tumors in pediatric patients. Chest. 1993; 103:1426–1428.
134. Hancock, BJ, Di Lorenzo, M, Youssef, S, et al. Childhood primary pulmonary neoplasms. J Pediatr Surg. 1993; 28:1133–1136.