Endocrine Disorders
Several endocrine glands have significant effects on skeletal growth, maturation, and modeling, and understanding these effects facilitates correct radiographic interpretation, allowing for optimal therapy. The parathyroid glands are also intimately involved in mineral homeostasis and have major effects on bone mineralization and resorption.1,2 Because of the importance of hyperparathyroidism (HPTH) in its pathophysiology, renal osteodystrophy is also included in this chapter as are other related conditions such as those causing neonatal hypercalcemia and disorders of bone resorption.
Hyperparathyroidism and Renal Osteodystrophy
Etiology: The parathyroid glands act to maintain a normal circulating calcium concentration. Synthesis and secretion of parathyroid hormone (PTH) are regulated by the parathyroid calcium sensing receptor, which increases PTH in response to hypocalcemia. PTH then has many effects that function to restore the serum calcium concentration. PTH promotes bone resorption to mobilize calcium. In the kidney, PTH upregulates renal 1-α-hydroxylase to produce calcitriol (1,25[OH]2-vitamin D) which then acts on the gut to absorb calcium. PTH also decreases renal calcium excretion and increases phosphate excretion.
Renal osteodystrophy (ROD)3–5 comprises a variety of skeletal manifestations of chronic renal failure, the most important of which is secondary HPTH. In chronic renal failure, as the glomerular filtration rate falls, less phosphate is filtered. Phosphate retention and the slight decrease in the ionized calcium concentration caused by it, each stimulates PTH which then acts to promote phosphaturia and increase calcium. Although this initially normalizes calcium and phosphate concentrations, it does so at the expense of elevating PTH and mobilizing calcium and phosphate from bone. With advancing renal failure, decreased renal mass also reduces calcitriol synthesis, leading to rickets and osteomalacia. However, this effect on the skeleton is less pronounced than is HPTH, and many of the manifestations of “renal rickets” are actually those of HPTH.
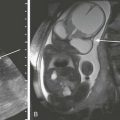
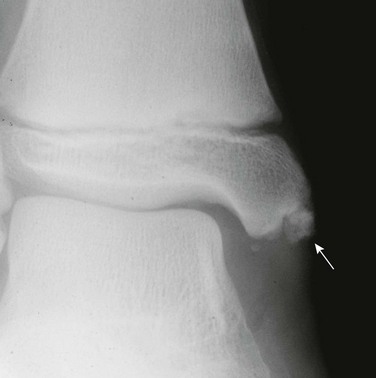
Imaging: PTH stimulation of osteoclasts leads to bone resorption at multiple sites.3–6 The most specific radiographic manifestation of HPTH is subperiosteal resorption, initially seen along the radial aspects of the index and the middle finger middle phalanges (Fig. 141-1). The distal phalangeal terminal tufts are also involved relatively early. With progression, subperiosteal resorption is noted along the ulnar aspects of the phalanges and other bones including the medial aspects of the humeral and femoral necks and the medial aspects of the proximal tibial metaphyses. Although less specific than subperiosteal resorption, HPTH may lead to intracortical resorption, or “tunneling,” causing a striated appearance of the cortex, as well as endosteal, subchondral, and subligamentous resorption (Fig. 141-2). Subphyseal resorption (a form of subchondral resorption) leads to resorption of the zone of provisional calcification and metaphyseal bone beneath the physis, simulating the appearance of rickets. Additional findings of HPTH include resorption of the lamina dura, which forms the thin opaque line surrounding tooth roots (Figs. 141-3 and 141-4), cystic-appearing “brown tumors” (osteoclastomas), and bone sclerosis. Although brown tumors are more characteristic of primary HPTH, overall, they are seen more often in secondary HPTH because it is much more common than primary HPTH in children. Osteosclerosis, most often seen with renal osteodystrophy, may be generalized or most pronounced subjacent to the vertebral end plates, resulting in a “rugger jersey” appearance (Fig. 141-5).
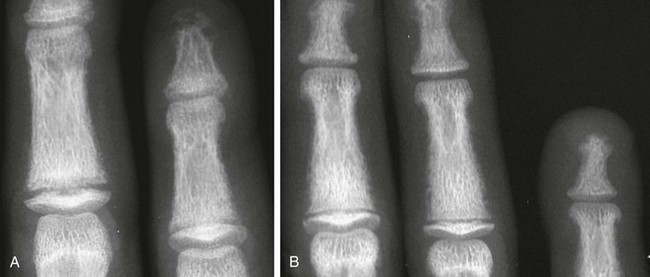
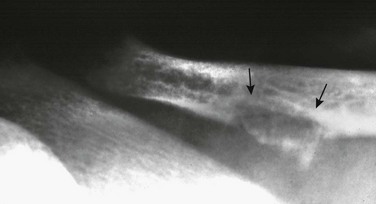
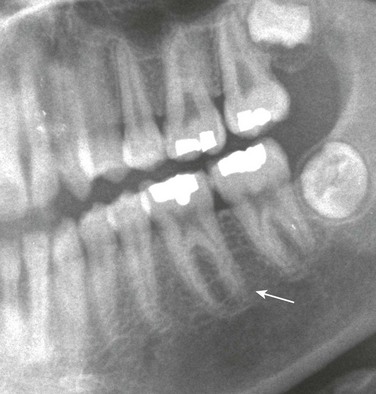
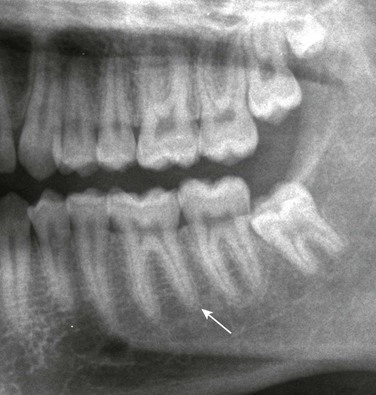
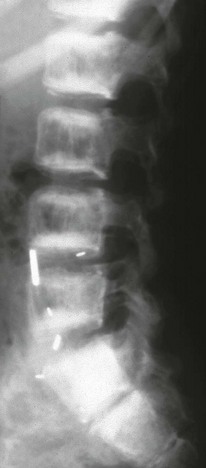
Figure 141-5 Rugger jersey spine.
The lateral view of the lumbar spine shows vertebral end plate sclerosis producing rugger jersey appearance.
Slipped epiphyses (e-Fig. 141-6) are an important complication of renal osteodystrophy in children, accounting for more cases than other predisposing conditions such as hypothyroidism, hypogonadism, and growth hormone deficiency. Although subphyseal resorption can mimic the radiographic appearance of rickets, the radiolucent material beneath the growth plate is fibrous tissue of osteitis fibrosa cystica, which is weaker than the nonmineralized cartilage and osteoid in true rickets, accounting for slipped epiphyses in renal osteodystrophy but not true rickets. Radiographic findings indicating a particularly high risk of slippage include coxa vara with reorientation of the physis from horizontal toward vertical, increased width of the growth plate, and subperiosteal resorption of the adjacent metaphysis. The risk of epiphyseal slippage is particularly high when growth hormone is used to treat short stature, a major clinical problem in children with chronic renal failure. Although slipped epiphyses are seen most frequently in the proximal femur, many other sites may also be involved.
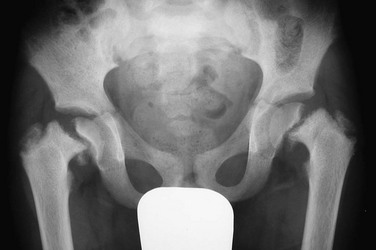
e-Figure 141-6 Bilateral slipped capital femoral epiphyses in 11-year-old with renal osteodystrophy.
This finding was made at the time of presentation with previously undiagnosed end-stage renal failure.
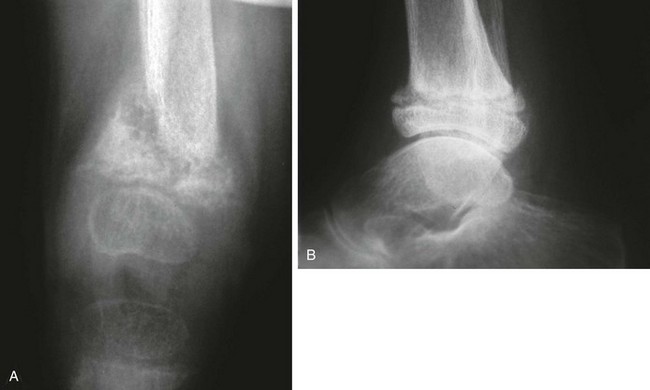
True rickets and osteomalacia may also be seen from decreased production of calcitriol. It is often not possible to distinguish true rickets from the rachitic appearance of osteitis fibrosa cystica. Looser zones, if present, indicate osteomalacia. Renal osteodystrophy usually manifests a “high turnover” state from HPTH. Less often, an adynamic “low turnover” state may also be seen. Previously, adynamic bone disease was often due to aluminum toxicity, which occurred as a complication of dialysis or aluminum-based phosphate binders. Presently, causes of adynamic bone disease include calcitriol therapy (which suppresses PTH), malnutrition, immobilization, corticosteroid therapy, and prior parathyroidectomy. Fractures may occur in renal osteodystrophy with HPTH predisposing to metaphyseal fractures (e-Fig. 141-7).
Treatment and Follow-up: Treatment of secondary HPTH in children with chronic renal disease is directed toward normalization of mineral metabolism to improve growth, decrease skeletal deformity and fragility, and prevent extraskeletal calcification, most importantly vascular calcification.4,7 Treatment is often initiated at stage 3 chronic renal disease (GFR <60 mL/min/1.73 m2) when positive phosphate balance and calcitriol deficiency appear. Dietary phosphate restriction is often difficult to achieve while maintaining adequate protein intake. Hence, intestinal phosphate binders are used. Although calcium-containing phosphate binders such as calcium carbonate have been used extensively, they pose a risk of hypercalcemia and vascular calcification, particular when used along with calcitriol. Aluminum-containing phosphate binders may cause aluminum bone disease and should be avoided. Sevelamer is a newer phosphate binder that does not contain calcium and has been shown to lower phosphate and control the skeletal lesions of HPTH without the adverse effects on vascular calcification that are seen with calcium-containing binders.
Other Disorders of Hyperparathyroidism and Causes of Bone Resorption
Primary HPTH in neonates is quite rare and is usually caused by hyperplasia rather than parathyroid adenoma.8 Some cases, neonatal HPTH may result from homozygosity of the gene for familial hypocalciuric hypercalcemia, which usually causes asymptomatic hypercalcemia in adults.9 Additional causes of HPTH in neonates include Jansen metaphyseal chondrodysplasia and I-cell disease. Although Williams syndrome may also cause neonatal hypercalcemia, PTH is not elevated.
Jansen Metaphyseal Chondrodysplasia
Jansen metaphyseal chondrodysplasia is an autosomal dominant condition with hypercalcemia and other findings suggestive of severe HPTH during the neonatal period and short-limbed dwarfism later in life. It is caused by a mutation of the receptor for PTH and PTH-related peptide (PTHrP) that causes the receptor to be constantly activated.10,11 Hence, even though no PTH or PTHrP are detectable, PTH signaling is increased, producing effective HPTH with hypercalcemia and bone resorption, which are most prominent early in life (Fig. 141-8). Similarly, constitutive activation of the PTH/PTHrP receptor also leads to excessive PTHrP signaling, which inhibits endochondral ossification by preventing proliferating chondrocytes from entering hypertrophic differentiation.12 This causes an ossification defect leading to buildup of a large amount nonossified cartilage and creating a lucent gap between the epiphysis and the ossified portion of the shaft (see Fig. 141-8). Subsequently, bizarre chondroid calcifications develop within these regions, which become widened and dysplastic. Eventually, the skeleton becomes fully ossified with residual shortening and deformity. The opposite effects are seen in Blomstrand lethal chondrodysplasia with an inactivating mutation of the PTH/PTHrP receptor.13 The lack of PTHrP signaling leads to accelerated endochondral ossification, causing severe growth failure from loss of proliferating chondrocytes and markedly premature physeal closure.
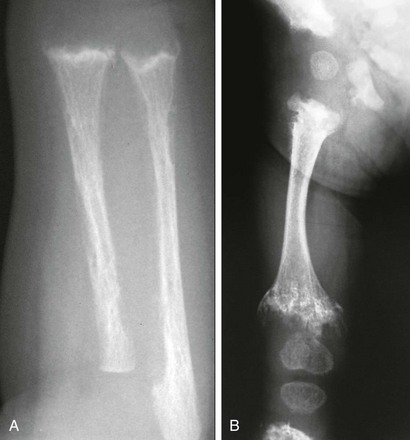
Figure 141-8 Jansen metaphyseal chondrodysplasia, with manifestations of parathyroid hormone/parathyroid hormone–related peptide (PTH/PTHrP) receptor activation.
At 5 days of age, the forearm shows severe demineralization and subperiosteal resorption indicative of a hyperparathyroid state from increased PTH signaling. Also prominent are rachitic findings in the metaphyses. Impaired endochondral ossification from excessive PTHrP signaling is demonstrated in the right femur at 15 months with a large lucent zone between the shaft and epiphyses, comprising nonossified cartilage. These lucent zones are beginning to fill in with disorganized chondroid calcifications, which are most apparent distally. This disorganized pattern of ossification leads to the marked metaphyseal dysplasia of this condition.
Mucolipidosis II
Mucolipidosis II (I-cell disease) is an autosomal recessive storage disorder with clinical and radiographic features similar to those of the mucopolysaccharidosis Hurler syndrome. In addition, some infants with mucolipidosis II have skeletal features of HPTH, including generalized demineralization and subperiosteal bone resorption.14 Although PTH is elevated, calcium is not, indicating that this is a form of second-degree HPTH. It is speculated that placental involvement by this storage disorder impairs calcium transport leading to fetal and neonatal calcium deficiency causing second-degree HPTH.
Other Disorders of Bone Resorption
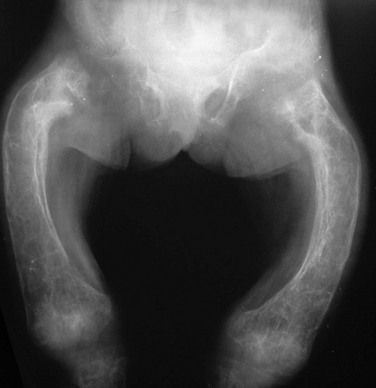
Abnormal bone resorption may also be seen in conditions other than HTPH. Decreased resorption, usually caused by a defect in osteoclastic acidification, leads to osteopetrosis. Increased resorption is seen in hyperphosphatasia and some endocrine disorders such as glucocorticoid excess. Hyperphosphatasia, also known as juvenile Paget disease, is a rare autosomal recessive condition with increased bone turnover caused by deficiency of osteoprotegerin.15,16 Osteoprotegerin normally suppresses bone turnover by acting as a “decoy receptor” for the osteoclast differentiation factor RANKL. In the absence of this decoy receptor, excessive RANKL signaling stimulates osteoclast differentiation and activity, causing accelerated bone resorption and hence bone turnover. Hyperphosphatasia often presents at 2 to 3 years of age with short stature, bone fragility, and deformity, including long bone bowing, kyphoscoliosis, and pectus carinatum. Unlike adult Paget disease, which involves limited portions of the skeleton, hyperphosphatasia is more generalized. The radiographic features may be striking, with overall demineralization, coarse trabecular pattern, cortical thickening, widened cylindrical long bones with decreased tubulation, and markedly variable bone texture with osteosclerosis intermixed with regions of cystic-appearing lucency (Fig. 141-9). Bowing and pathologic fractures are common. The skull shows thickening and regions of lucency combined with “cottonwool” sclerosis, similar to adult Paget disease. Familial expansile osteolysis is a related condition with deafness, loss of teeth, expansion of bones, painful phalanges, and episodic hypercalcemia caused by duplication of the gene for the osteoclast membrane-bound receptor that is activated by RANKL.17
Hypoparathyroidism
Etiology: Idiopathic hypoparathyroidism in children is most commonly part of polyglandular autoimmune disease type I.18 This autosomal recessive condition targets multiple endocrine glands, most frequently the parathyroids and adrenals. Other causes of hypoparathyroidism in children include congenital absence of the parathyroids, which may accompany thymic absence in DiGeorge syndrome, surgical removal during thyroidectomy, and transient suppression of fetal parathyroids by maternal HPTH.19
Imaging: Skeletal manifestations of hypoparathyroidism include osteosclerosis, dense metaphyseal bands, cranial vault thickening, intracranial calcifications such as basal ganglia and choroid plexus, and dental abnormalities. Vertebral findings include marginal sclerosis, “bone within bone” appearance (Fig. 141-10), and paravertebral ossification.
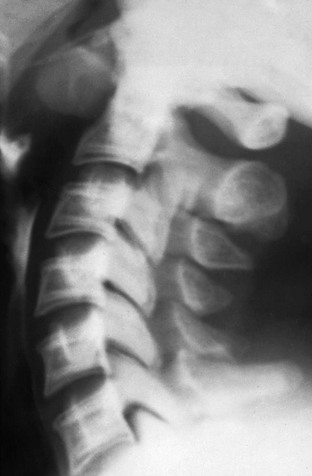
Pseudohypoparathyroidism
Etiology: Pseudohypoparathyroidism is characterized by variable resistance to PTH and, less often, other hormones, as well as typical somatic features known as Albright hereditary osteodystrophy (AHO).18,20 The hormone resistance results from an inactivating mutation of the GNAS1 gene, which is involved in G-protein biosignaling. In pseudopseudohypoparathyroidism, the somatic features of AHO are seen without hormone resistance.
Imaging: The somatic features of AHO include short stature, obesity, a round face, variable degrees of subnormal intelligence, subcutaneous calcification and ossification, small exostoses, and brachydactyly of hands and feet (Fig. 141-11). The brachydactyly is often associated with premature growth plate closure that results from impaired PTHrP signaling leading to accelerated endochondral ossification. Brachydactyly E, Turner syndrome, and acrodysostosis are differential considerations. Resistance to PTH leads to manifestations of hypoparathyroidism including hypocalcemia and soft tissue and basal ganglia calcifications despite elevated PTH levels. Although hormone resistance often involves both kidney and bone, skeletal resistance is variable. With skeletal responsiveness to PTH, manifestations of HPTH such as subperiosteal bone resorption and brown tumors (Fig. 141-12) may be seen.21,22
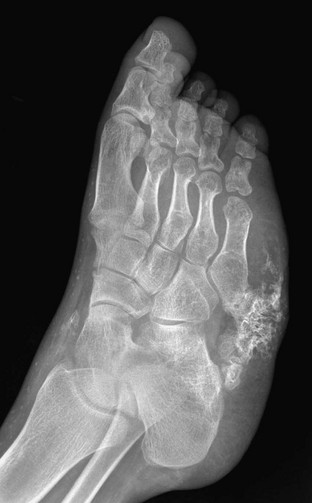

Nonparathyroid Endocrine Disorders
Growth Hormone Disorders
Etiology: Growth hormone (GH) is an important positive regulator of postnatal longitudinal growth, acting directly as well as indirectly through insulin-like growth factor (IGF), to stimulate proliferation of physeal chondrocytes.23–25 Prenatal growth depends mostly on IGF, accounting for the relative preservation of prenatal growth in GH deficiency. In GH deficiency, skeletal growth and maturation are retarded to a similar degree, resulting in a similar height age and bone age. Maturation of the carpal bones is often more delayed compared with the phalanges. The retarded growth and maturation of GH deficiency may be mimicked by deprivational dwarfism (“psychosocial dwarfism”) although the presence of multiple growth restart lines favors deprivational dwarfism (e-Fig. 141-13).26 Slipped capital femoral epiphysis (SCFE) may be seen with growth hormone deficiency either prior to or following growth hormone therapy.
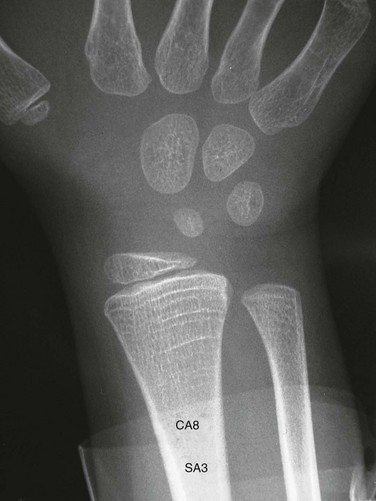
e-Figure 141-13 Deprivational dwarfism.
Delayed skeletal maturation with a skeletal age of 3 years (SA3) and chronologic age of 8 years (CA8). There are multiple growth restart lines in the distal radial metaphysis. Similar findings were also present in the distal femur (not shown). (Courtesy Andrew K. Poznanski, MD, Chicago, IL.)
Imaging: The effects of growth hormone excess are different in children than in adults. Prior to skeletal maturity, GH excess causes gigantism with increased linear growth, a relatively rare condition and usually caused by an isolated pituitary adenoma. Approximately 20% of cases are associated with McCune-Albright syndrome with growth hormone excess caused by either adenoma or hyperplasia. In most cases, skeletal maturation is normal. With increased linear growth and advanced skeletal maturation, other endocrinopathies such as excessive gonadal or adrenal androgens should be considered. After skeletal maturity has been attained, increased GH leads to increased periosteal bone formation, reactivation of endochondral bone formation at chondro-osseous junctions, and cartilage and soft tissue hypertrophy. These processes lead to acromegaly, which is predominantly an adult disorder.
Hypothyroidism
Etiology: Thyroid hormone (TH) is essential for normal brain development during the first 3 years of life, as well for normal skeletal growth and maturation.23,24,27–29 Although TH does not directly affect chondrocyte proliferation, the positive effects of GH and IGF on proliferation require the presence of TH. Additionally, TH has a major effect on skeletal maturation by inhibiting PTHrP signaling which ordinarily retards endochondral ossification. Hence increased PTHrP signaling in hypothyroidism causes impaired endochondral ossification.
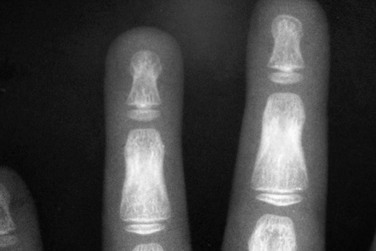
Imaging: Skeletal maturation is delayed to an even greater degree than linear growth, whereas in GH deficiency, growth and maturation are proportionately diminished. Impaired ossification not only delays the appearance of epiphyses but also leads to an abnormal pattern with multiple fragmented ossification centers, which is known as epiphyseal dysgenesis. Although these eventually coalesce, the epiphysis often has uneven opacity and irregular margins (Fig. 141-14). Although the term stippled is often used for these epiphyses, the appearance is distinct from chondrodysplasia punctata and associated disorders. Epiphyseal dysgenesis is particularly common in the hips. With treatment, the findings of epiphyseal dysgenesis resolve. SCFE may be a presenting manifestation of hypothyroidism, often bilateral and occurring at a younger age than idiopathic SCFE.30,31 In the skull, hypothyroidism may cause multiple wormian bones, enlargement of the sella from pituitary hyperplasia, and brachycephaly from decreased growth at the spheno-occipital synchondrosis. Radiographs of hands may show small projections of bone extending from the distal phalangeal metaphyses into the growth plates (Fig. 141-15).32
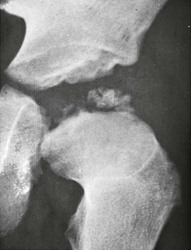
Figure 141-14 Epiphyseal dysgenesis in a 9-year-old hypothyroid girl who had started treatment 1 year earlier.
The femoral head is small, flattened, and contains multiple small fragmented ossification centers. At the beginning of treatment 1 year earlier, no ossification of the femoral heads was seen. With follow-up, these ossification centers were observed to be coalesced, but the femoral heads remained flattened. Similar finding were also present in several major epiphyses.
Hyperthyroidism
Etiology: As discussed for hypothyroidism, TH inhibits PTHrP and hence promotes hypertrophic differentiation of proliferating chondrocytes leading to accelerated skeletal maturation. Hyperthyroidism in the pediatric age range is most frequent in adolescent girls. Hyperthyroidism may also occur as a manifestation of McCune-Albright syndrome, although precocious puberty is most common.
Imaging: Skeletal maturation in hyperthyroidism may be normal or mildly advanced. Accelerated maturation may be seen in infants born to mothers with uncontrolled hyperthyroidism during the last trimester of pregnancy. Although hyperthyroidism is uncommon in infancy, acceleration of skeletal maturation is greatest in this group and may include premature craniosynostosis.33 Other skeletal manifestations of hyperthyroidism include calcification of costal cartilage and tracheal rings, diffuse osteopenia, and cortical striations, which are indicative of increased bone turnover. Hyperthyroidism may mimic osteomalacia, including the presence of Looser transformation zones, although excessive nonmineralized osteoid is the result of overproduction of osteoid rather than impairment of its mineralization. Although it is predominantly an adult disorder, thyroid acropachy may be seen in adolescents with spiculated or feathery appearing periosteal new bone involving the metacarpals, metatarsals, and phalanges.
Glucocorticoid Excess
Etiology: Cushing syndrome refers to the systemic effects of glucocorticoid excess. Most cases of Cushing syndrome in children are iatrogenic and result from pharmacologic dosages of glucocorticoids used for treatment of chronic inflammatory or autoimmune disorders. Endogenous Cushing syndrome in children is uncommon and usually caused by primary adrenal lesions in those below 5 years of age and pituitary lesions in those above 5 years. Glucocorticoids decrease linear growth and maturation, acting at multiple levels to negatively regulate physeal chondrogenesis by decreasing GH secretion and downregulating GH and IGF receptors in growth plate chondrocytes.24
Glucocorticoids also impair skeletal mineralization.34,35 Systemically, they negate the positive influence of the sex steroids on mineralization and decrease calcium absorption in the gut. Glucocorticoids also act directly on bone to decrease bone formation and increase osteoclastic resorption by increasing RANKL and decreasing osteoprotegerin expression. Osteonecrosis is an additional complication of endogenous or exogenous glucocorticoid excess. Its pathogenesis is not well understood; potential mechanisms include compromise of intraosseous perfusion caused by increased intraosseous pressure from fat deposition in marrow and fat embolization resulting from fatty liver.
Imaging: The skeletal findings of endogenous or exogenous corticosteroid excess are mostly nonspecific generalized osteopenia with cortical thinning, trabecular rarefaction, and vertebral compression fracture. In addition, more specific findings include vertebral end plate sclerosis, abundant callus formation at fracture sites, and osteonecrosis. Excessive callus formation associated with compression fractures of vertebral bodies probably accounts for end plate sclerosis, which may be a clue in the diagnosis of Cushing syndrome in a patient with osteopenia. Glucocorticoid induced osteonecrosis most frequently involves the femoral head, with the humeral head and femoral condyles being next in frequency. Radiographic findings include subcortical fracture, bone collapse, fragmentation, and patchy osteosclerosis.
Adrenal Androgen Excess
Etiology: Adrenogenital syndrome, the most frequent cause of excessive adrenal androgens excess, results from a biosynthetic defect in the production of cortisol with compensatorily increased adrenocorticotropic hormone driving excessive production of adrenal androgens.35 Excessive amounts of adrenal androgens may also be secreted by adrenal cortical neoplasms. Although they are weaker than testosterone, adrenal androgens promote virilization and accelerate skeletal maturation.
Imaging: Although growth is initially accelerated, this effect is less pronounced than is advancement of maturation. Hence, physeal closure occurs prematurely, and final height is decreased rather than increased. Advancement of skeletal maturation caused by excessive androgens is more pronounced than that caused by hyperthyroidism.
Gonadal Disorders
Etiology: Although previously thought that normal skeletal maturation was mediated by estrogens in females and androgens in males, the effects of sex steroids on physeal fusion and bone mineral accretion are currently believed to be caused by estrogen, with these processes mediated in males by estrogens formed from peripheral conversion of androgens.36–38
Bastepe, M. The GNAS locus and pseudohypoparathyroidism. Adv Exp Med Biol. 2008;626:27–40.
Dabbagh, S. Renal osteodystrophy. Curr Opin Pediatr. 1998;10:190–196.
Loder, RT, Wittenberg, B, DeSilva, G. Slipped capital femoral epiphysis associated with endocrine disorders. J Pediatr Orthop. 1995;15:349–356.
Sanchez, CP. Mineral metabolism and bone abnormalities in children with chronic renal failure. Rev Endocr Metab Disord. 2008;9:131–137.
Weinstein, LS, Liu, J, Sakamoto, A, et al. Mini-review: GNAS: normal and abnormal functions. Endocrinology. 2004;145:5459–5464.
References
1. Nissenson, RA, Jüppner, H. Parathyroid hormone. In: Rosen CJ, ed. Primer on the Metabolic bone diseases and disorders of mineral metabolism. 7th ed. Washington, DC: American Society for Bone and Mineral Research; 2008:123–127.
2. Ross, FP. Osteoclast biology and bone resorption. In: Rosen CJ, ed. Primer on the metabolic bone diseases and disorders of mineral metabolism. 7th ed. Washington, DC: American Society for Bone and Mineral Research; 2008:16–22.
3. Goodman, WG, Coburn, JW, Slatopolsky, E, et al. Renal osteodystrophy in children and adults. In: Favis MJ, ed. Primer on the metabolic bone diseases and disorders of mineral metabolism. 5th ed. Washington, DC: American Society for Bone and Mineral Research; 2003:430–447.
4. Hruska, KA, Mathew, S. Chronic kidney disease mineral bone disorder (CKD-MBD). In: Rosen CJ, ed. Primer on the metabolic bone diseases and disorders of mineral metabolism. 7th ed. Washington, DC: American Society for Bone and Mineral Research; 2008:343–348.
5. Resnick, D. Parathyroid disorders and renal osteodystrophy. In: Resnick D, ed. Diagnosis of bone and joint disorders. 4th ed. Philadelphia, PA: WB Saunders; 2002:2043–2111.
6. Pugh, DG. Subperiosteal resorption of bone: roentgenologic manifestation of primary hyperparathyroidism and renal osteodystrophy. Am J Roentgenol Radium Ther Nucl Med. 1951;66:577–586.
7. Wesseling, K, Bakkaloglu, S, Salusky, I. Chronic kidney disease mineral and bone disorder in children. Pediatr Nephrol. 2008;23:195–207.
8. Langman, CB. Hypercalcemic syndromes in infants and children. In: Favis MJ, ed. Primer on the metabolic bone diseases and disorders of mineral metabolism. 5th ed. Washington, DC: American Society for Bone and Mineral Research; 2003:267–270.
9. Arnold, A, Marx, SJ. Familial hyperparathyroidism (including MEN, FHH, and HPT-JT). In: Rosen CJ, ed. Primer on the metabolic bone diseases and disorders of mineral metabolism. 7th ed. Washington, DC: American Society for Bone and Mineral Research; 2008:361–366.
10. Schipani, E, Langman, CB, Parfitt, AM, et al. Constitutively activated receptors for parathyroid hormone and parathyroid hormone-related peptide in Jansen’s metaphyseal chondrodysplasia. N Engl J Med. 1996;335:708–714.
11. Schipani, E, Provot, S. PTHrP, PTH, and the PTH/PTHrP receptor in endochondral bone development. Birth Defects Res C Embryo Today. 2003;69:352–362.
12. Kronenberg, HM. PTHrP and skeletal development. Ann N Y Acad Sci. 2006;1068:1–13.
13. Zhang, P, Jobert, AS, Couvineau, A, et al. A homozygous inactivating mutation in the parathyroid hormone/parathyroid hormone-related peptide receptor causing Blomstrand chondrodysplasia. J Clin Endocrinol Metab. 1998;83:3365–3368.
14. Unger, S, Paul, DA, Nino, MC, et al. Mucolipidosis II presenting as severe neonatal hyperparathyroidism. Eur J Pediatr. 2005;164:236–243.
15. Eroglu, M, Taneli, NN. Congenital hyperphosphatasia (juvenile Paget’s disease): eleven years follow-up of three sisters. Ann Radiol (Paris). 1977;20:145–150.
16. Whyte, MP, Obrecht, SE, Finnegan, PM, et al. Osteoprotegerin deficiency and juvenile Paget’s disease. N Engl J Med. 2002;347:175–184.
17. Whyte, MP, Mills, BG, Reinus, WR, et al. Expansile skeletal hyperphosphatasia: a new familial metabolic bone disease. J Bone Miner Res. 2000;15:2330–2344.
18. Rubin, MR, Levine, MA. Hypoparathyroidism and pseudohypoparathyroidism. In: Rosen CJ, ed. Primer on the metabolic bone diseases and disorders of mineral metabolism. 7th ed. Washington, DC: American Society for Bone and Mineral Research; 2008:354–361.
19. DiGeorge, AM. Congenital absence of the thymus and its immunologic consequences: concurrence with congenital hypoparathyroidism. Birth Defects. 1968;4:116–123.
20. Levine, MA. Parathyroid hormone resistance syndromes. In: Favis MJ, ed. Primer on the metabolic bone diseases and disorders of mineral metabolism. 5th ed. Washington, DC: American Society for Bone and Mineral Research; 2003:278–285.
21. Frame, B, Hanson, CA, Frost, HM, et al. Renal resistance to parathyroid hormone with osteitis fibrosa: pseudohypohyperparathyroidism. Am J Med. 1972;52:311–321.
22. Burnstein, MI, Kottamasu, SR, Peffifor, JM, et al. Metabolic bone disease in pseudohypoparathyroidism: radiologic features. Radiology. 1985;155:351–356.
23. Siebler, T, Robson, H, Shalet, SM, et al. Glucocorticoids, thyroid hormone and growth hormone interactions: implications for the growth plate. Horm Res. 2001;56(suppl 1):7–12.
24. Nilsson, O, Marino, R, De Luca, F, et al. Endocrine regulation of the growth plate. Horm Res. 2005;64:157–165.
25. Resnick, D. Pituitary disorders. In: Resnick D, ed. Diagnosis of bone and joint disorders. 4th ed. Philadelphia, PA: WB Saunders; 2002:2003–2025.
26. Hernandez, RJ, Poznanski, AK, Hopwood, NJ, et al. Incidence of growth lines in psychosocial dwarfs and idiopathic hypopituitarism. AJR Am J Roentgenol. 1978;131:477–479.
27. Robson, H, Siebler, T, Stevens, DA, et al. Thyroid hormone acts directly on growth plate chondrocytes to promote hypertrophic differentiation and inhibit clonal expansion and cell proliferation. Endocrinology. 2000;141:3887–3897.
28. Stevens, DA, Hasserjian, RP, Robson, H, et al. Thyroid hormones regulate hypertrophic chondrocyte differentiation and expression of parathyroid hormone-related peptide and its receptor during endochondral bone formation. J Bone Miner Res. 2000;15:2431–2442.
29. Resnick, D. Thyroid disorders. In: Resnick D, ed. Diagnosis of bone and joint disorders. 4th ed. Philadelphia, PA: WB Saunders; 2002:2026–2042.
30. Heyerman, W, Weiner, D. Slipped epiphysis associated with hypothyroidism. J Pediatr Orthop. 1984;4:569–573.
31. Loder, RT, Wittenberg, B, DeSilva, G. Slipped capital femoral epiphysis associated with endocrine disorders. J Pediatr Orthop. 1995;15:349–356.
32. Hernandez, RJ, Poznanski, AK. Distinctive appearance of the distal phalanges in children with primary hypothyroidism. Radiology. 1979;132:83–84.
33. Riggs, W, Jr., Wilroy, RS, Jr., Etteldorf, JN. Neonatal hyperthyroidism with accelerated skeletal maturation, craniostenosis and brachydactyly. Radiology. 1972;105:621–625.
34. Weinstein, RS. Glucocorticoid induced osteoporosis. In: Rosen CJ, ed. Primer on the metabolic bone diseases and disorders of mineral metabolism. 7th ed. Washington, DC: American Society for Bone and Mineral Research; 2008:267–272.
35. Resnick, D. Disorders of other endocrine glands and of pregnancy. In: Resnick D, ed. Diagnosis of bone and joint disorders. 4th ed. Philadelphia, PA: WB Saunders; 2002:2112–2143.
36. Morishima, A, Grumbach, MM, Simpson, ER, et al. Aromatase deficiency in male and female siblings caused by a novel mutation and the physiological role of estrogens. J Clin Endocrinol Metab. 1995;80:3689–3698.
37. Smith, EP, Boyd, J, Frank, GR, et al. Estrogen resistance caused by a mutation in the estrogen-receptor gene in a man. N Engl J Med. 1994;331:1056–1061.
38. Weise, M, De-Levi, S, Barnes, KM, et al. Effects of estrogen on growth plate senescence and epiphyseal fusion. Proc Natl Acad Sci U S A. 2001;98:6871–6876.