[level-membership-for-internal-medicine-category]
Chapter 19 Endocrine disease
Introduction
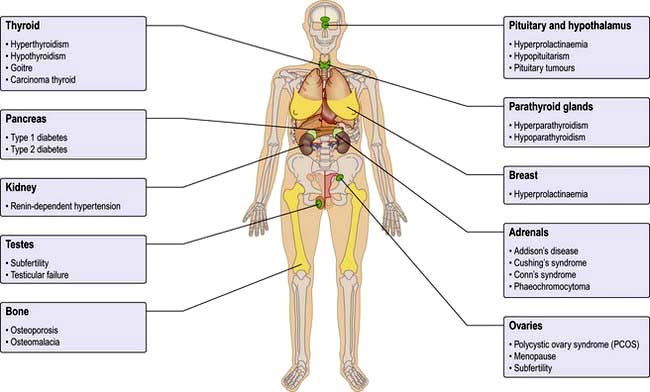
The endocrine system consists of glands that exert their actions at distant parts of the body via the production of biologically active hormones secreted into the bloodstream. Unlike the neurological system, which produces an immediate response, the endocrine system typically has a slower and longer lasting effect on the body. The main endocrine glands are the pituitary, thyroid, adrenals, gonads, parathyroids and pancreas and the common endocrine problems seen in clinical practice are shown in Figure 19.1. The pituitary gland, a pea-sized structure situated at the base of the brain, plays a key role in the control and feedback mechanisms of the endocrine system and has been termed the ‘conductor of the endocrine orchestra’.
Clinical presentation of endocrine disease
History



 |
Common endocrine conditions
The most common endocrine disorders, excluding obesity (Ch. 5) and diabetes mellitus (Ch. 20), are:






Aetiology of endocrine disease
Aetiological mechanisms common to many endocrine disorders include:
Autoimmune disease
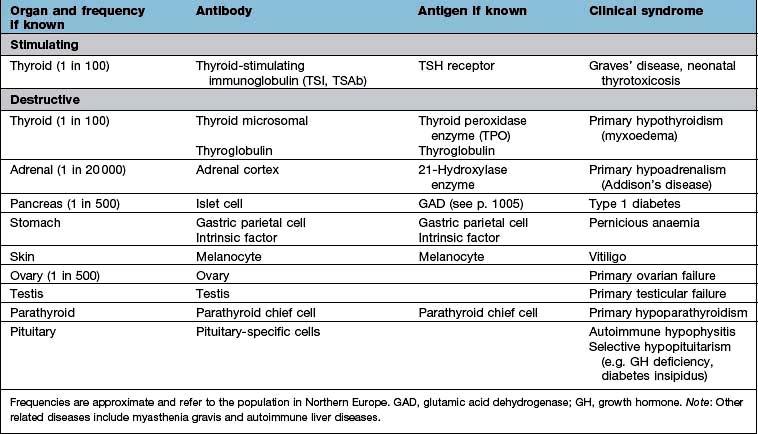
Organ-specific autoimmune diseases can affect every major endocrine organ (Table 19.2). They are characterized by the presence of specific antibodies in the serum, often present years before clinical symptoms are evident, are usually more common in women and have a strong genetic component, often with an identical-twin concordance rate of 50% and with HLA associations (see individual diseases). Several of the autoantigens have been identified.
Endocrine tumours
Most endocrine tumours are benign, although a cytological or histological diagnosis may be needed if there is clinical or radiological suspicion of malignancy. Clinical presentation depends on whether the tumour is functional or non-functional, the latter presenting only as a mass clinically or on imaging. Palpable thyroid nodules are common, and mass effects are a frequent presentation of pituitary adenomas, but the increased use of high-resolution ultrasound and detailed cross-sectional imaging has revealed a very high prevalence of asymptomatic, incidentally-discovered thyroid, adrenal and pituitary lesions, commonly termed ‘incidentalomas’ (see p. 989).
Endocrine adenomas typically present in a single gland, although rarer multiple endocrine neoplasia (MEN) syndromes exist due to very specific mutations of a single gene, such as the mutations of the RET proto-oncogene in MEN 2 or the MEN1 gene mutation in MEN 1 (see p. 997).
Enzyme defects
The biosynthesis of most hormones involves many stages. Deficient or abnormal enzymes can lead to absent or reduced production of the secreted hormone. In general, severe deficiencies present early in life with obvious signs; partial deficiencies usually present later with mild signs or are only evident under stress. An example of an enzyme deficiency is congenital adrenal hyperplasia (CAH), where the molecular basis has also been identified as mutations or deletions of the gene encoding the relevant enzymes (see p. 987).
Receptor abnormalities
There are rare conditions in which hormone secretion and control are normal but the receptors are defective; thus, if androgen receptors are defective, normal levels of androgen will not produce masculinization (e.g. testicular feminization). There are also a number of rare syndromes of diabetes and insulin resistance from receptor abnormalities (see p. 1006); other examples include nephrogenic diabetes insipidus, pseudohypoparathyroidism and thyroid hormone resistance which can cause an unusual pattern of thyroid blood results.
Hormonal activity
Hormone action and receptors
Hormone receptors are broadly divided into:
 Cell surface or membrane receptors: typically transmembrane receptors which contain hydrophobic sections spanning the lipid-rich plasma membrane and which trigger internal cellular messengers (see also p. 18)
Cell surface or membrane receptors: typically transmembrane receptors which contain hydrophobic sections spanning the lipid-rich plasma membrane and which trigger internal cellular messengers (see also p. 18)
 Nuclear receptors which typically bind hormones and translocate them to the nucleus where they bind hormone response elements of nuclear DNA via characteristic amino-acid sequences (e.g. so-called ‘zinc fingers’, see p. 27).
Nuclear receptors which typically bind hormones and translocate them to the nucleus where they bind hormone response elements of nuclear DNA via characteristic amino-acid sequences (e.g. so-called ‘zinc fingers’, see p. 27).
Abnormal receptors are an occasional cause of endocrine disease (see p. 43).
Mechanisms of hormone-receptor action
Common structural mechanisms of hormone-receptor action are illustrated in Figure 2.9 (p. 25) and include:
 stimulate cyclic AMP (cAMP) generation by adenylate cyclase – activating further intracellular kinases and leading to phosphorylation
stimulate cyclic AMP (cAMP) generation by adenylate cyclase – activating further intracellular kinases and leading to phosphorylation

 lead to diacylglycerol (DAG) activation of C-kinase and subsequent protein phosphorylation.
lead to diacylglycerol (DAG) activation of C-kinase and subsequent protein phosphorylation.




Control and feedback
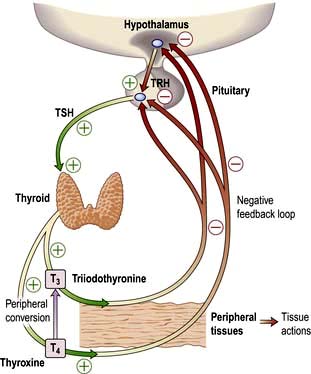
Most hormone systems are under tight regulatory control (typically by the hypothalamo-pituitary (HP) axis) by a system known as negative feedback. An example of the negative feedback system in the hypothalamo-pituitary-thyroid axis is demonstrated in Figure 19.2 and described here:








Primary and secondary gland failure
 ‘Primary’ hormone deficiency due to a disease process in the endocrine end-organ (thyroid, adrenal or gonad) will lead to a loss of negative feedback and subsequent elevation in the corresponding anterior pituitary hormone. Conversely, an abnormal hormone excess due to a disease process in the primary endocrine gland, or excess amount of exogenous hormone, will lead to increased negative feedback and suppression of the corresponding pituitary hormones.
‘Primary’ hormone deficiency due to a disease process in the endocrine end-organ (thyroid, adrenal or gonad) will lead to a loss of negative feedback and subsequent elevation in the corresponding anterior pituitary hormone. Conversely, an abnormal hormone excess due to a disease process in the primary endocrine gland, or excess amount of exogenous hormone, will lead to increased negative feedback and suppression of the corresponding pituitary hormones.

Measurement of hormones
Hormones are measured in routine clinical practice by biochemical assays in the laboratory. It is possible to measure pituitary trophic hormones and the hormones produced by the end-organ glands, but hypothalamic hormones are not routinely measured in practice because of their low concentration and local action within the hypothalamo-pituitary axis. Circulating levels of most hormones are very low (10−9– 10−12 mol/L) and cannot be measured by simple chemical techniques. Hormones are therefore usually measured by immunoassays, which rely on highly specific polyclonal or monoclonal antibodies, which bind to the hormone being measured during the assay incubation. This hormone-antibody interaction is measured by use of labelled hormone after separation of bound and free fractions (Fig. 19.3).
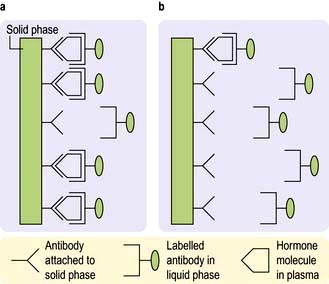
Hormone binding proteins
Many hormones are transported in the bloodstream from the primary gland to their distant target organ attached to a specific binding protein (p. 940). It is more helpful to measure the free hormone rather than total bound hormone level, as this is the part that is biologically active. Some modern assays attempt to measure the free hormone level directly (e.g. free T4) and are therefore a more accurate reflection of biological activity, although there are often technical problems with this approach and many assays still measure total hormone level.
Patterns of hormonal secretion
Hormone secretion can be continuous or intermittent, for example:
 Continuous secretion is shown by the thyroid hormones, with a half-life of 7–10 days for T4 and 6–10 hours for T3, and with little variation in levels over the day, month and year
Continuous secretion is shown by the thyroid hormones, with a half-life of 7–10 days for T4 and 6–10 hours for T3, and with little variation in levels over the day, month and year

Biological rhythms
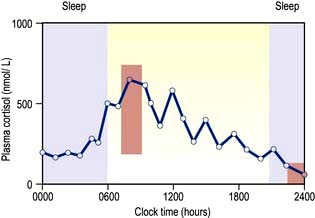
Circadian means changes over the 24 hours of the day–night cycle and is best shown for the pituitary–adrenal axis. Figure 19.4 shows plasma cortisol levels measured over 24 hours – levels are highest in the early morning and lowest overnight. Additionally, cortisol release is pulsatile, following the pulsatility of pituitary ACTH. Thus ‘normal’ cortisol levels vary during the day and great variations can be seen in samples taken only 30 minutes apart.
The menstrual cycle is an example of a longer and more complex (28-day) biological rhythm (see p. 972).



Testing endocrine function
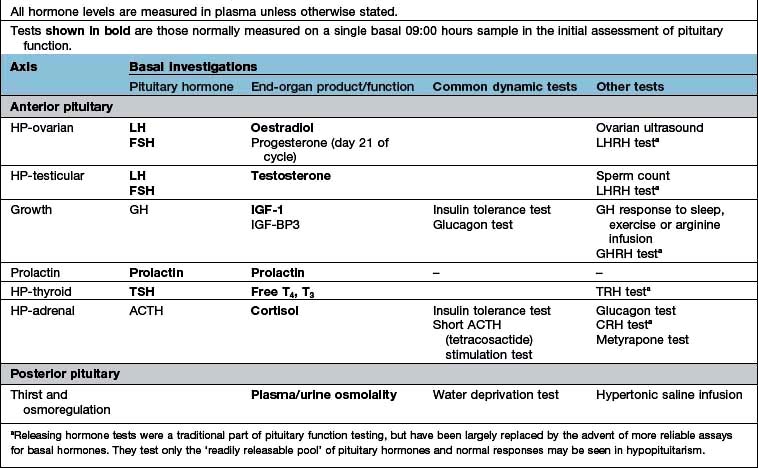
Basal blood levels
Assays for all clinically relevant pituitary and end-organ hormones are available.
Stimulation and suppression tests
For example, where the secretory capacity of a gland is damaged, maximal stimulation by the trophic hormone will give a diminished output. Thus, in the short ACTH stimulation test for adrenal reserve (Box 19.1, Fig. 19.5a), the healthy subject shows a normal response while the subject with primary hypoadrenalism (Addison’s disease) demonstrates an impaired cortisol response to tetracosactide (an ACTH analogue).
 Box 19.1
Box 19.1






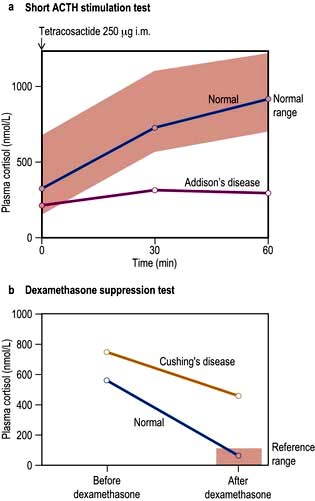
A patient with a hormone-producing tumour usually fails to show normal negative feedback. A patient with Cushing’s disease (excess pituitary ACTH) will thus fail to suppress ACTH and cortisol production when given a dose of synthetic steroid, in contrast to normal subjects. Figure 19.5b shows the response of a normal subject given dexamethasone 1 mg at midnight; cortisol is suppressed the following morning. The subject with Cushing’s disease shows inadequate suppression.
The pituitary gland and hypothalamus
Anatomy
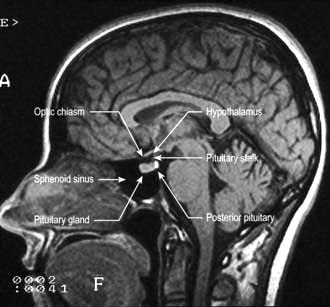
The anatomical relations of the hypothalamus and pituitary (Fig. 19.6) include the optic chiasm just above the pituitary fossa; any expanding lesion from the pituitary or hypothalamus can thus produce visual field defects by pressure on the chiasm. Such upward expansion of the gland through the diaphragma sellae is termed ‘suprasellar extension’. Lateral extension of pituitary lesions may involve the vascular and nervous structures in the cavernous sinus and may rarely reach the temporal lobe of the brain. The pituitary is itself encased in a bony box, therefore any lateral, anterior or posterior expansion must cause bony erosion.
Physiology
Hypothalamus
Hypothalamic neurones secrete pituitary hormone-releasing and -inhibiting factors and hormones (Table 19.3) into the portal system which run down the stalk to the pituitary. As well as the classical hormones illustrated in Figure 19.7, the hypothalamus also contains large amounts of other neuropeptides and neurotransmitters such as neuropeptide Y, vasoactive intestinal peptide (VIP) and nitric oxide that can also alter pituitary hormone secretion.
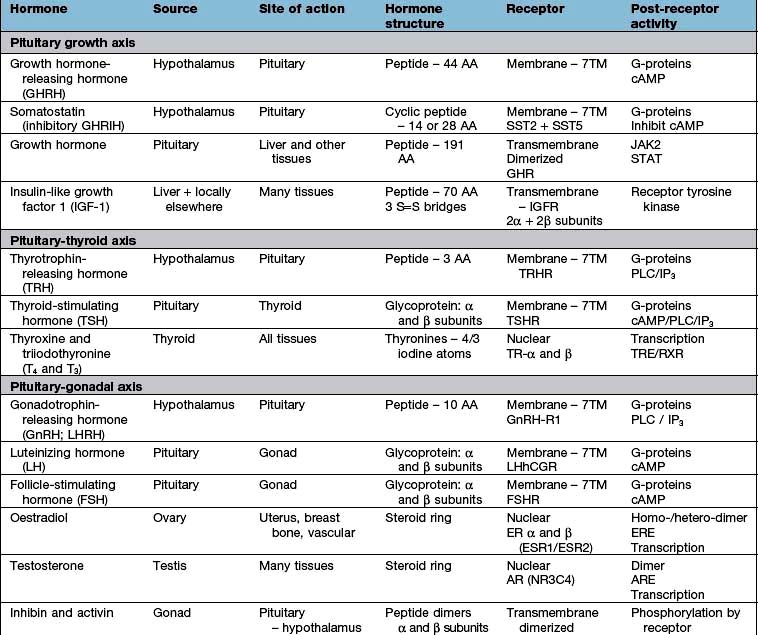
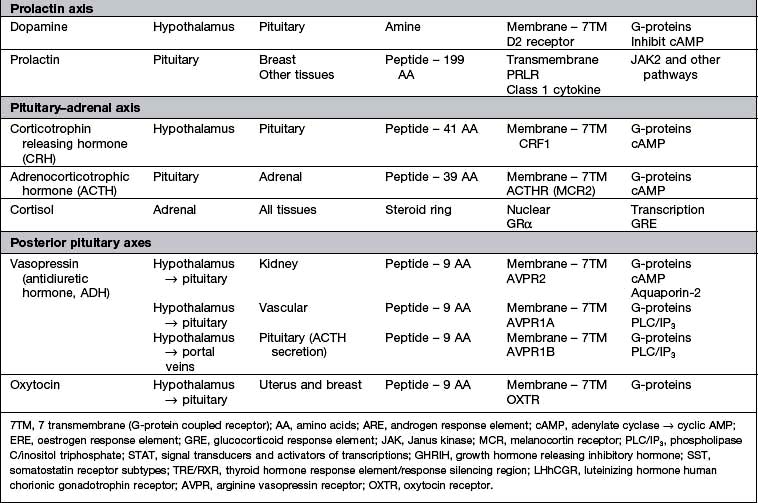

Posterior pituitary
The posterior pituitary is neuro-anatomically connected to specific hypothalamic nuclei, and acts merely as a storage organ. Antidiuretic hormone (ADH, also called vasopressin) and oxytocin, both nonapeptides, are synthesized in the supraoptic and paraventricular nuclei in the anterior hypothalamus. They are then transported along the axon and stored in the posterior pituitary (Fig. 19.7). This means that damage to the stalk or pituitary alone does not prevent synthesis and release of ADH and oxytocin. ADH is discussed on page 991; oxytocin produces milk ejection and uterine myometrial contraction.
Presentations of pituitary and hypothalamic disease
Pituitary space-occupying lesions and tumours
Pituitary tumours (Table 19.4) are the most common cause of pituitary disease, and the great majority of these are benign pituitary adenomas, usually monoclonal in origin. Problems are caused by:



Table 19.4 Characteristics of common pituitary and related tumours
| Tumour or condition | Usual size | Most common clinical presentation |
|---|---|---|
|
Prolactinoma |
Most <10 mm (microprolactinoma) |
Galactorrhoea, amenorrhoea, hypogonadism, erectile dysfunction |
|
|
Some >10 mm (macroprolactinoma) |
As above plus headaches, visual field defects and hypopituitarism |
|
Acromegaly |
Few mm to several cm |
Change in appearance, visual field defects and hypopituitarism |
|
Cushing’s disease |
Most small: few mm (some cases are hyperplasia) |
Central obesity, cushingoid appearance (local symptoms rare) |
|
Nelson’s syndrome |
Often large: >10 mm |
Post-adrenalectomy, pigmentation, sometimes local symptoms |
|
Non-functioning tumours |
Usually large: >10 mm |
Visual field defects; hypopituitarism (microadenomas may be incidental finding) |
|
Craniopharyngioma |
Often very large and cystic (skull X-ray abnormal in >50%; calcification common) |
Headaches, visual field defects, growth failure (50% occur below age 20; about 15% arise from within sella) |
Investigations (of a possible or proven mass)
Is there a tumour?
 the visual pathways, with field defects and visual loss (most common)
the visual pathways, with field defects and visual loss (most common)
 the cavernous sinus, with III, IV and VI cranial nerve lesions
the cavernous sinus, with III, IV and VI cranial nerve lesions
 bony structures and the meninges surrounding the fossa, causing headache
bony structures and the meninges surrounding the fossa, causing headache

 the ventricles, causing interruption of cerebrospinal fluid (CSF) flow leading to hydrocephalus
the ventricles, causing interruption of cerebrospinal fluid (CSF) flow leading to hydrocephalus

Investigations
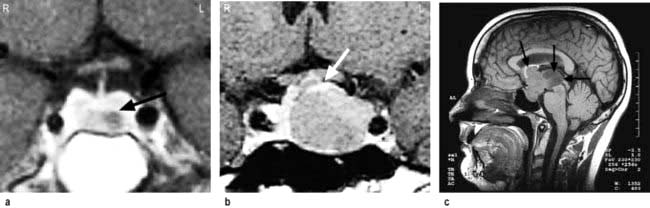
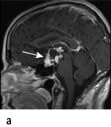
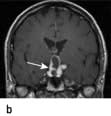
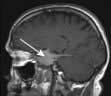
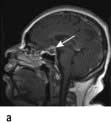
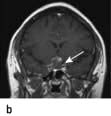
 MRI of the pituitary. MRI is superior to CT scanning (Fig. 19.8) and will readily show any significant pituitary mass. Small lesions within the pituitary fossa on MRI consistent with small pituitary microadenomas are very common (10% of normal individuals in some studies). Such small lesions are sometimes detected during MRI scanning of the head for other reasons – so-called ‘pituitary incidentalomas’.
MRI of the pituitary. MRI is superior to CT scanning (Fig. 19.8) and will readily show any significant pituitary mass. Small lesions within the pituitary fossa on MRI consistent with small pituitary microadenomas are very common (10% of normal individuals in some studies). Such small lesions are sometimes detected during MRI scanning of the head for other reasons – so-called ‘pituitary incidentalomas’.
 Visual fields. These should be plotted formally by automated computer perimetry or Goldmann perimetry, but clinical assessment by confrontation using a small red pin as target is also sensitive and valuable. Common defects are upper temporal quadrantanopia and bitemporal hemianopia (see p. 1073).
Visual fields. These should be plotted formally by automated computer perimetry or Goldmann perimetry, but clinical assessment by confrontation using a small red pin as target is also sensitive and valuable. Common defects are upper temporal quadrantanopia and bitemporal hemianopia (see p. 1073).
Is there a hormonal excess?
 Prolactin excess (prolactinoma or hyperprolactinaemia): histologically, prolactinomas are ‘chromophobe’ adenomas (a description of their appearance on classical histological staining)
Prolactin excess (prolactinoma or hyperprolactinaemia): histologically, prolactinomas are ‘chromophobe’ adenomas (a description of their appearance on classical histological staining)
 GH excess (acromegaly or gigantism): somatotroph adenomas, usually ‘acidophil’, and sometimes due to specific G-protein mutations (see p. 945)
GH excess (acromegaly or gigantism): somatotroph adenomas, usually ‘acidophil’, and sometimes due to specific G-protein mutations (see p. 945)

The clinical features of acromegaly, Cushing’s disease or hyperprolactinaemia are usually (but not always) obvious, and are discussed on pages 953, and 957. Hyperprolactinaemia may be clinically ‘silent’. Tumours producing LH, FSH or TSH are well described but very rare.
Is there a deficiency of any hormone?
Clinical examination may give clues; thus, short stature in a child with a pituitary tumour is likely to be due to GH deficiency. A slow, lethargic adult with pale skin is likely to be deficient in TSH and/or ACTH. Milder deficiencies may not be obvious, and require specific testing (see Table 19.7).
Treatment
Treatment depends on the type and size of tumour (Table 19.5). In general, therapy has three aims:
Table 19.5 Comparisons of primary treatments for pituitary tumours
| Treatment method | Advantages | Disadvantages |
|---|---|---|
|
Surgical |
|
|
|
Trans-sphenoidal adenomectomy or hypophysectomy |
Relatively minor procedure Potentially curative for microadenomas and smaller macroadenomas |
Some extrasellar extensions may not be accessible |
|
Transcranial (usually transfrontal) |
Good access to suprasellar region |
Major procedure; danger of frontal lobe damage |
|
Radiotherapy |
|
|
|
External (40–50 Gy) |
Non–invasive |
Slow action, often over many years |
|
Stereotactic |
Precise administration of high dose to lesion |
Long-term follow-up data limited |
|
Medical |
|
|
|
Dopamine agonist therapy (e.g. bromocriptine, cabergoline) |
Non-invasive; reversible |
Usually not curative; significant side-effects in minority |
|
Somatostatin analogue therapy (octreotide, lanreotide) |
Non-invasive; reversible |
Usually not curative; gallstones; expensive |
|
Growth hormone receptor antagonist (pegvisomant) |
Highly selective |
Usually not curative; very expensive |
Removal/control of tumour
 Surgery via the trans-sphenoidal route is usually the treatment of choice. Very large tumours are occasionally removed via the open transcranial (usually transfrontal) route.
Surgery via the trans-sphenoidal route is usually the treatment of choice. Very large tumours are occasionally removed via the open transcranial (usually transfrontal) route.

 Medical therapy with somatostatin analogues and/or dopamine agonists sometimes causes shrinkage of specific types of tumour (see p. 954) and if successful can be used as primary therapy.
Medical therapy with somatostatin analogues and/or dopamine agonists sometimes causes shrinkage of specific types of tumour (see p. 954) and if successful can be used as primary therapy.
Reduction of excess hormone secretion
Reduction is usually obtained by surgical removal but sometimes by medical treatment. Useful control can be achieved with dopamine agonists for prolactinomas or somatostatin analogues for acromegaly, but ACTH secretion usually cannot be controlled by medical means. Growth hormone antagonists are also available for acromegaly (p. 955).
Replacement of hormone deficiencies
Replacement of hormone deficiencies, i.e. hypopituitarism, is discussed below (see Table 19.8).
| Axis | Usual replacement therapies |
|---|---|
|
Adrenal |
Hydrocortisone 15–40 mg daily (starting dose 10 mg on rising/5 mg lunchtime/5 mg evening) |
|
(Normally no need for mineralocorticoid replacement) |
|
|
Thyroid |
Levothyroxine 100–150 µg daily |
|
Gonadal |
|
|
Male |
Testosterone intramuscularly, orally, transdermally or implant |
|
Female |
Cyclical oestrogen/progestogen orally or as patch |
|
Fertility |
HCG plus FSH (purified or recombinant) or pulsatile GnRH to produce testicular development, spermatogenesis or ovulation |
|
Growth |
Recombinant human GH used routinely to achieve normal growth in children |
|
Also advocated for replacement therapy in adults where GH has effects on muscle mass and wellbeing |
|
|
Thirst |
Desmopressin 10–20 µg one to three times daily by nasal spray or orally 100–200 µg three times daily |
|
Carbamazepine, thiazides and chlorpropamide are very occasionally used in mild diabetes insipidus |
|
|
Breast (prolactin inhibition) |
Dopamine agonist (e.g. cabergoline, 500 µg weekly) |
Differential diagnosis of pituitary or hypothalamic masses
Other tumours
 Craniopharyngioma (1–2%), a usually cystic hypothalamic tumour, often calcified, arising from Rathke’s pouch, often mimics an intrinsic pituitary lesion. It is the most common pituitary tumour in children but may present at any age.
Craniopharyngioma (1–2%), a usually cystic hypothalamic tumour, often calcified, arising from Rathke’s pouch, often mimics an intrinsic pituitary lesion. It is the most common pituitary tumour in children but may present at any age.

Hypopituitarism
Causes
Disorders causing hypopituitarism are listed in Table 19.6. Pituitary and hypothalamic tumours, and surgical or radiotherapy treatment, are the most common.
|
Congenital |
Traumatic |
|
Isolated deficiency of pituitary hormones (e.g. Kallmann’s syndrome) |
Skull fracture through base |
|
Infiltrations |
|
|
Infective |
Sarcoidosis |
|
Basal meningitis (e.g. tuberculosis) |
|
|
Vascular |
|
|
Pituitary apoplexy |
|
|
Others |
|
|
Radiation damage |
|
|
Immunological |
‘Functional’ |
|
Autoimmune (lymphocytic) hypophysitis |
Anorexia nervosa |
|
Neoplastic |
|
|
Pituitary or hypothalamic tumours |
|
|
Craniopharyngioma |
|
|
Meningiomas |
|
|
Gliomas |
|
|
Pinealoma |
|
|
Secondary deposits, especially breast |
|
|
Lymphoma |
Clinical features
 Secondary hypothyroidism and adrenal failure both lead to tiredness and general malaise.
Secondary hypothyroidism and adrenal failure both lead to tiredness and general malaise.
 Hypothyroidism causes weight gain, slowness of thought and action, dry skin and cold intolerance.
Hypothyroidism causes weight gain, slowness of thought and action, dry skin and cold intolerance.


 Hyperprolactinaemia may cause galactorrhoea and hypogonadism.
Hyperprolactinaemia may cause galactorrhoea and hypogonadism.
 GH deficiency causes growth failure in children and impaired wellbeing in some adults.
GH deficiency causes growth failure in children and impaired wellbeing in some adults.


Kallmann’s syndrome. This syndrome is isolated gonadotrophin (GnRH) deficiency (p. 976).This syndrome arises due to mutations in the KAL1 gene which is located on the short (p) arm of the X chromosome. Kallmann’s is classically characterized by anosmia because the KAL1 gene provides instructions to make anosmin, which has a role in development of both the olfactory system as well as migration of GnRH secreting neurones.
Investigations
Tests range from the simple basal levels (e.g. free T4 for the thyroid axis), to stimulatory tests for the pituitary, and tests of feedback for the hypothalamus (Table 19.7). Assessment of the hypothalamic-pituitary-adrenal axis is complex: basal 09:00 hours cortisol levels above 400 nmol/L usually indicate an adequate reserve, while levels below 100 nmol/L predict an inadequate stress response. In many cases basal levels are equivocal and a dynamic test is essential: the insulin tolerance test (Box 19.2) is widely regarded as the ‘gold standard’ but the short ACTH stimulation test (Box 19.1), though an indirect measure, is used by many as a routine test of hypothalamic-pituitary-adrenal status. Occasionally, the difference between ACTH deficiency and normal HPA axis can be subtle, and the assessment of adrenal reserve is best left to an experienced endocrinologist.
 Box 19.2
Box 19.2
Insulin tolerance test

Procedure
 Test explained to patient and consent obtained
Test explained to patient and consent obtained
 Should only be performed in experienced, specialist units
Should only be performed in experienced, specialist units

 Intravenous hydrocortisone and glucose available for emergency
Intravenous hydrocortisone and glucose available for emergency
 Overnight fast, begin at 08:00–09:00 hours
Overnight fast, begin at 08:00–09:00 hours
 Soluble insulin, 0.15 U/kg, i.v. at time 0
Soluble insulin, 0.15 U/kg, i.v. at time 0
 Glucose, cortisol and GH levels at 0, 30, 45, 60, 90, 120 min
Glucose, cortisol and GH levels at 0, 30, 45, 60, 90, 120 min



Treatment
 Steroid and thyroid hormones are essential for life. Both are given as oral replacement drugs, as in primary thyroid and adrenal deficiency, aiming to restore the patient to clinical and biochemical normality (Table 19.8) and levels are monitored by routine hormone assays. Note: Thyroid replacement should not commence until normal glucocorticoid function has been demonstrated or replacement steroid therapy initiated, as an adrenal ‘crisis’ may otherwise be precipitated.
Steroid and thyroid hormones are essential for life. Both are given as oral replacement drugs, as in primary thyroid and adrenal deficiency, aiming to restore the patient to clinical and biochemical normality (Table 19.8) and levels are monitored by routine hormone assays. Note: Thyroid replacement should not commence until normal glucocorticoid function has been demonstrated or replacement steroid therapy initiated, as an adrenal ‘crisis’ may otherwise be precipitated.




FURTHER READING
Dattani MT. Growth hormone deficiency and combined pituitary deficiency: does the genotype matter? Clin Endocrinol 2005; 63:121–130.
National Institute for Health and Clinical Excellence. Human growth hormone (somatotropin) in adults with growth hormone deficiency; 2003: Technology appraisal 64; http://guidance.nice.org.uk/TA64/Guidance/pdf/English
Schneider HJ, Aimaretti G, Kreitschmann-Andermahr I et al. Hypopituitarism. Lancet 2007; 369:1151–1470.
Growth and abnormal stature
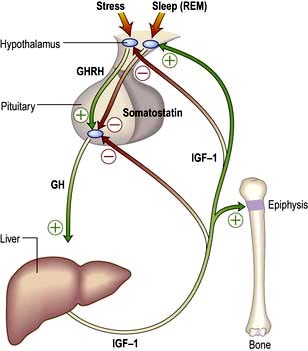
Physiology and control of growth hormone (GH) (Fig. 19.9)
GH is the pituitary factor responsible for stimulation of body growth in humans. Its secretion is stimulated by GHRH, released into the portal system from the hypothalamus; it is also under inhibitory control by somatostatin. A separate GH stimulating system involves a distinct receptor (GH secretogogue receptor), which interacts with ghrelin (see p. 259). It is not known how these two systems interact but because ghrelin is synthesized in the stomach, it suggests a nutritional role for GH.
 GH acts by binding to a specific (single transmembrane) receptor located mainly in the liver (Table 19.3). This induces an intracellular phosphorylation cascade involving the JAK/STAT (Janus kinase/signal transducing activators of transcription) pathway (p. 32). STAT proteins are translocated from the cytoplasm into the cell nucleus and cause GH-specific effects by binding to nuclear DNA.
GH acts by binding to a specific (single transmembrane) receptor located mainly in the liver (Table 19.3). This induces an intracellular phosphorylation cascade involving the JAK/STAT (Janus kinase/signal transducing activators of transcription) pathway (p. 32). STAT proteins are translocated from the cytoplasm into the cell nucleus and cause GH-specific effects by binding to nuclear DNA.




Normal growth
There are factors other than GH involved in linear growth in the human.
 Genetic factors. Children of two short parents will probably be short and vice-versa.
Genetic factors. Children of two short parents will probably be short and vice-versa.


 Intrauterine growth retardation. These infants often grow poorly in the long term, while infants with simple prematurity usually catch up. There is some evidence that low birthweight may predispose to hypertension, diabetes and other health problems in later adult life (p. 195).
Intrauterine growth retardation. These infants often grow poorly in the long term, while infants with simple prematurity usually catch up. There is some evidence that low birthweight may predispose to hypertension, diabetes and other health problems in later adult life (p. 195).

The relevant aspects of history and examination in the assessment of problems are shown in Box 19.3.
 Box 19.3
Box 19.3











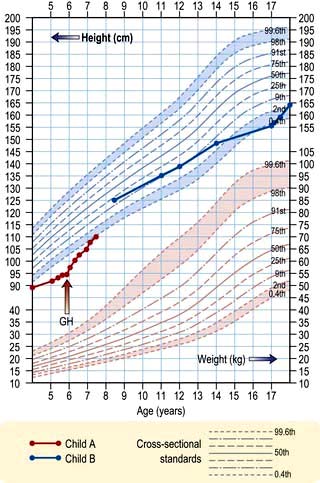
Assessment of growth
Charts showing normal centiles of height and weight are essential to monitor growth; they are available for normal British children (Fig. 19.10) and many other national and ethnic groups. Height must be measured, ideally at the same time of day on the same instrument by the same observer.
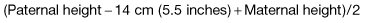
Growth failure: short stature
When children or their parents complain of short stature, particular attention should focus on:
 Intrauterine growth retardation, weight and gestation at birth
Intrauterine growth retardation, weight and gestation at birth
 Possible systemic disorders – any system, but especially small bowel disease
Possible systemic disorders – any system, but especially small bowel disease
 Evidence of skeletal, chromosomal or other congenital abnormalities
Evidence of skeletal, chromosomal or other congenital abnormalities
 Endocrine status – particularly thyroid
Endocrine status – particularly thyroid
 Dietary intake and use of drugs, especially steroids for asthma
Dietary intake and use of drugs, especially steroids for asthma

Consistently slow-growing children require full endocrine assessment. Features of the more common causes of growth failure are given in Table 19.9.
Investigations
Systemic disease having been excluded, perform:
 Thyroid function tests: serum TSH and free T4 to exclude hypothyroidism
Thyroid function tests: serum TSH and free T4 to exclude hypothyroidism
 GH status. Basal levels are of little value. Dynamic tests include the GH response to insulin (the ‘gold standard’; Box 19.2), glucagon, arginine, exercise and clonidine. Tests should only be performed in centres experienced in their use and interpretation. Normal responses depend on test and GH assay used
GH status. Basal levels are of little value. Dynamic tests include the GH response to insulin (the ‘gold standard’; Box 19.2), glucagon, arginine, exercise and clonidine. Tests should only be performed in centres experienced in their use and interpretation. Normal responses depend on test and GH assay used


 Karyotyping in females. Turner’s syndrome (p. 978) is associated with short stature. It is thought that this is due to a defect in the short stature homeobox (SHOX) gene which has a role in non-GH mediated growth.
Karyotyping in females. Turner’s syndrome (p. 978) is associated with short stature. It is thought that this is due to a defect in the short stature homeobox (SHOX) gene which has a role in non-GH mediated growth.
Treatment
Systemic illness should be treated and primary hypothyroidism treated with levothyroxine.
GH treatment in so-called ‘short normal’ children has not been shown to produce any worthwhile increase in final height. In Turner’s syndrome (see p. 983) large doses of GH are effective in increasing final height, especially in combination with appropriate very-low-dose oestrogen replacement. Familial cases of resistance to GH owing to an abnormal GH receptor (Laron-type dwarfism) are well described. They are very rare but may respond to therapy with synthetic IGF-1 (mecasermin).
Pituitary hypersecretion syndromes
Acromegaly and gigantism
Clinical features
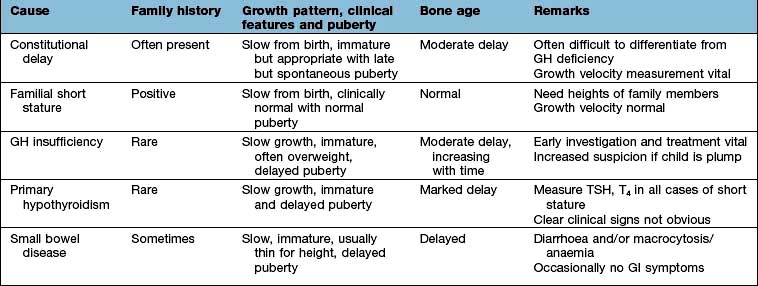
Symptoms and signs of acromegaly are shown in Figure 19.11. One-third of patients present with changes in appearance, one-quarter with visual field defects or headaches; in the remainder the diagnosis is made by an alert observer in another clinic, e.g. GP, diabetic, hypertension, dental, dermatology. Sleep apnoea is common and requires investigation and treatment if there are suggestive symptoms (see p. 818). Sweating, headaches and soft tissue swelling are particularly useful symptoms of persistent growth hormone secretion. Headache is very common in acromegaly and may be severe even with small tumours; it is often improved after surgical cure or with somatostatin analogues.
Investigations
 GH levels may exclude acromegaly if undetectable but a detectable value is non-diagnostic taken alone. Normal adult levels are <0.5 µg/L for most of the day except during stress or a ‘GH pulse’.
GH levels may exclude acromegaly if undetectable but a detectable value is non-diagnostic taken alone. Normal adult levels are <0.5 µg/L for most of the day except during stress or a ‘GH pulse’.


 Visual field examination: defects are common, e.g. bitemporal hemianopia.
Visual field examination: defects are common, e.g. bitemporal hemianopia.
 MRI scan of pituitary if above tests abnormal. This will almost always reveal the pituitary adenoma.
MRI scan of pituitary if above tests abnormal. This will almost always reveal the pituitary adenoma.
 Pituitary function: partial or complete anterior hypopituitarism is common.
Pituitary function: partial or complete anterior hypopituitarism is common.
 Prolactin: mild to moderate hyperprolactinaemia occurs in 30% of patients (see Fig. 19.13). In some, the adenoma secretes both GH and prolactin.
Prolactin: mild to moderate hyperprolactinaemia occurs in 30% of patients (see Fig. 19.13). In some, the adenoma secretes both GH and prolactin.
Management and treatment
When present, hypopituitarism should be corrected (see p. 950) and concurrent diabetes and/or hypertension should be treated conventionally; both usually improve with treatment of the acromegaly.
The general advantages and disadvantages of surgery, radiotherapy and medical treatment are discussed on page 947. Progress can be assessed by monitoring GH and IGF-1 levels.
 Surgery. Trans-sphenoidal surgery is the appropriate first-line therapy. It will result in clinical remission in a majority of cases (60–90%) with pituitary microadenoma, but in only 50% of those with macroadenoma. Very high preoperative GH and IGF-1 levels are also poor prognostic markers of surgical cure. Surgical success rates are variable and highly dependent upon experience, and a specialist pituitary surgeon is essential. Transfrontal surgery is rarely required except for massive macroadenomas. There is approximately a 10% recurrence rate.
Surgery. Trans-sphenoidal surgery is the appropriate first-line therapy. It will result in clinical remission in a majority of cases (60–90%) with pituitary microadenoma, but in only 50% of those with macroadenoma. Very high preoperative GH and IGF-1 levels are also poor prognostic markers of surgical cure. Surgical success rates are variable and highly dependent upon experience, and a specialist pituitary surgeon is essential. Transfrontal surgery is rarely required except for massive macroadenomas. There is approximately a 10% recurrence rate.


Hyperprolactinaemia
The hypothalamic-pituitary control of prolactin secretion is illustrated in Figure 19.12. Prolactin is a large peptide secreted in the pituitary and acts via a transmembrane receptor stimulating JAK2 and other pathways (Table 19.3).
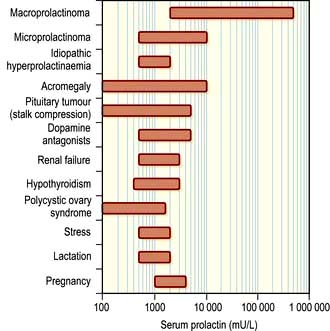
Physiological hyperprolactinaemia occurs in pregnancy, lactation and severe stress, as well as during sleep and coitus. The range of serum prolactin seen in common causes of hyperprolactinaemia is illustrated in Figure 19.13. Mildly increased prolactin levels (400–600 mU/L) may be physiological and asymptomatic but higher levels require a diagnosis. Levels above 5000 mU/L always imply a prolactin-secreting pituitary tumour.
Clinical features








Investigations
Further tests are appropriate after physiological and drug causes have been excluded:
 Visual fields should be checked.
Visual fields should be checked.
 Primary hypothyroidism must be excluded since this is a cause of hyperprolactinaemia.
Primary hypothyroidism must be excluded since this is a cause of hyperprolactinaemia.
 Anterior pituitary function should be assessed if there is any clinical evidence of hypopituitarism or radiological evidence of a pituitary tumour (Table 19.7; Box 19.1, Box 19.2).
Anterior pituitary function should be assessed if there is any clinical evidence of hypopituitarism or radiological evidence of a pituitary tumour (Table 19.7; Box 19.1, Box 19.2).

In the presence of a pituitary mass on MRI, the level of prolactin helps determine whether the mass is a prolactinoma or a non-functioning pituitary tumour causing stalk-disconnection hyperprolactinaemia: levels of above 5000 mU/L in the presence of a macroadenoma, or above 2000 mU/L in the presence of a microadenoma (or with no radiological abnormality), strongly suggest a prolactinoma (see p. 946). Macroprolactinoma refers to tumours above 10 mm diameter, microprolactinoma to smaller ones.
Treatment
Medical treatment. Hyperprolactinaemia is controlled with a dopamine agonist.
 Cabergoline (500 µg once or twice a week judged on clinical response and prolactin levels) is the best tolerated and longest-acting drug and is the first drug of choice.
Cabergoline (500 µg once or twice a week judged on clinical response and prolactin levels) is the best tolerated and longest-acting drug and is the first drug of choice.


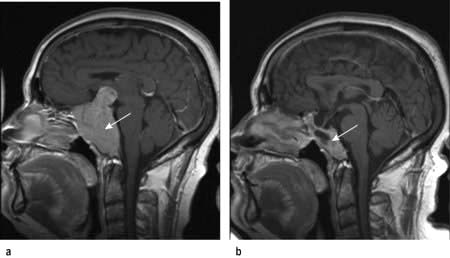
In most cases a dopamine agonist will be the first and only therapy and can be used in the long term. Prolactinomas usually shrink in size on a dopamine agonist, and in macroadenomas any pituitary mass effects commonly resolve (Fig. 19.14). Microprolactinomas may not recur after several years of dopamine agonist therapy in a minority of cases, but in the majority hyperprolactinaemia will recur if treatment is stopped.
Cushing’s syndrome
Pathophysiology and causes
Spontaneous Cushing’s syndrome is rare, with an incidence of <5/million per year.
Causes of Cushing’s syndrome are usually subdivided into two groups (Table 19.10):
1. Increased circulating ACTH from the pituitary (65% of cases), known as Cushing’s disease, or from an ‘ectopic’, non-pituitary, ACTH-producing tumour elsewhere in the body (10%) with consequent glucocorticoid excess (‘ACTH dependent’ Cushing’s)
2. A primary excess of endogenous cortisol secretion (25%) by an adrenal tumour or nodular hyperplasia, with subsequent (physiological) suppression of ACTH (‘non-ACTH-dependent’ Cushing’s). Rare cases are due to aberrant expression of receptors for other hormones (e.g. glucose-dependent insulinotrophic peptide (GIP), LH or catecholamines) in adrenal cortical cells.
Clinical features
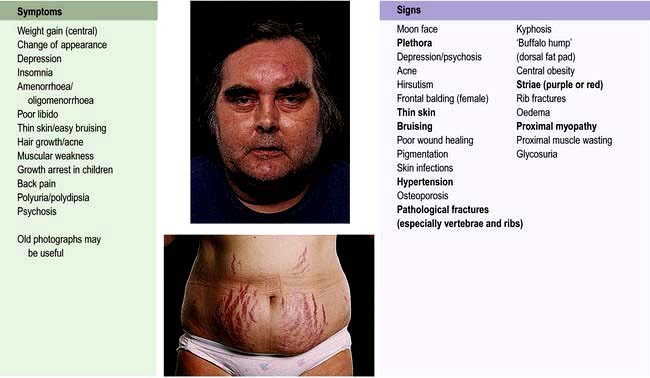
The clinical features of Cushing’s syndrome are those of glucocorticoid excess and are illustrated in Figure 19.15.
 Pigmentation occurs only with ACTH-dependent causes.
Pigmentation occurs only with ACTH-dependent causes.

 Impaired glucose tolerance or frank diabetes is common, especially in the ectopic ACTH syndrome.
Impaired glucose tolerance or frank diabetes is common, especially in the ectopic ACTH syndrome.


Diagnosis
There are two phases to the investigation.
1. Confirmation
Investigations to confirm the diagnosis include:
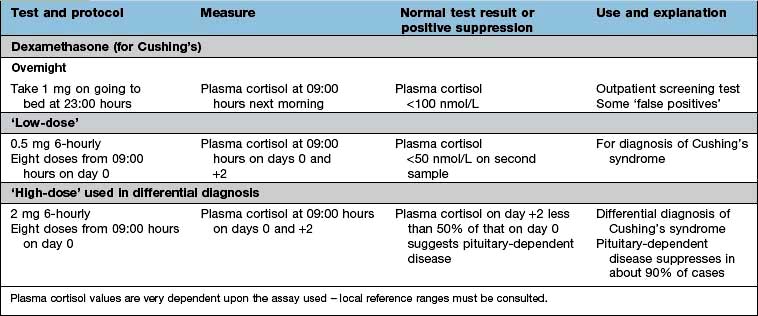
 48-hour low-dose dexamethasone test (see Table 19.11). Normal individuals suppress plasma cortisol to <50 nmol/L. People with Cushing’s syndrome fail to show complete suppression of plasma cortisol levels (although levels may fall substantially in a few cases). This test is highly sensitive (>97%). The overnight dexamethasone test is slightly simpler, but has a higher false-positive rate.
48-hour low-dose dexamethasone test (see Table 19.11). Normal individuals suppress plasma cortisol to <50 nmol/L. People with Cushing’s syndrome fail to show complete suppression of plasma cortisol levels (although levels may fall substantially in a few cases). This test is highly sensitive (>97%). The overnight dexamethasone test is slightly simpler, but has a higher false-positive rate.

 Circadian rhythm. After 48 hours in hospital, cortisol samples are taken at 09:00 hours and 24:00 hours (without warning the patient). Normal subjects show a pronounced circadian variation (see Fig. 19.4, p. 942); those with Cushing’s syndrome have high midnight cortisol levels (>100 nmol/L), though the 09:00 hours value may be normal. Midnight salivary cortisol collected at home gives the same information more simply where the assay is available.
Circadian rhythm. After 48 hours in hospital, cortisol samples are taken at 09:00 hours and 24:00 hours (without warning the patient). Normal subjects show a pronounced circadian variation (see Fig. 19.4, p. 942); those with Cushing’s syndrome have high midnight cortisol levels (>100 nmol/L), though the 09:00 hours value may be normal. Midnight salivary cortisol collected at home gives the same information more simply where the assay is available.
 Other tests. There are frequent exceptions to the classic responses to diagnostic tests in Cushing’s syndrome. If any clinical suspicion of Cushing’s remains after preliminary tests then specialist investigations are still indicated. These may include insulin stress test, desmopressin stimulation test (p. 993) and CRH tests.
Other tests. There are frequent exceptions to the classic responses to diagnostic tests in Cushing’s syndrome. If any clinical suspicion of Cushing’s remains after preliminary tests then specialist investigations are still indicated. These may include insulin stress test, desmopressin stimulation test (p. 993) and CRH tests.
2. Differential diagnosis of the cause
Biochemical and radiological procedures for diagnosis include:
 Plasma ACTH levels. Low or undetectable ACTH levels (<10 ng/L) on two or more occasions are a reliable indicator of non-ACTH-dependent disease.
Plasma ACTH levels. Low or undetectable ACTH levels (<10 ng/L) on two or more occasions are a reliable indicator of non-ACTH-dependent disease.



 High-dose dexamethasone test (Table 19.11). Failure of significant plasma cortisol suppression suggests an ectopic source of ACTH or an adrenal tumour.
High-dose dexamethasone test (Table 19.11). Failure of significant plasma cortisol suppression suggests an ectopic source of ACTH or an adrenal tumour.


Treatment
Choice of further treatment depends upon the cause.
Cushing’s disease (pituitary-dependent hyperadrenalism)
 Trans-sphenoidal removal of the tumour is the treatment of choice. Selective adenomectomy nearly always leaves the patient ACTH deficient immediately postoperatively, and this is a good prognostic sign. Overall, pituitary surgery results in remission in 75–80% of cases, but results vary considerably and an experienced surgeon is essential.
Trans-sphenoidal removal of the tumour is the treatment of choice. Selective adenomectomy nearly always leaves the patient ACTH deficient immediately postoperatively, and this is a good prognostic sign. Overall, pituitary surgery results in remission in 75–80% of cases, but results vary considerably and an experienced surgeon is essential.



The thyroid axis
Physiology
The thyroid synthesizes two hormones:


Inorganic iodide is trapped by the gland by an enzyme dependent system, oxidized and incorporated into the glycoprotein thyroglobulin to form mono- and diiodotyrosine and then T4 and T3 (Fig. 19.16).
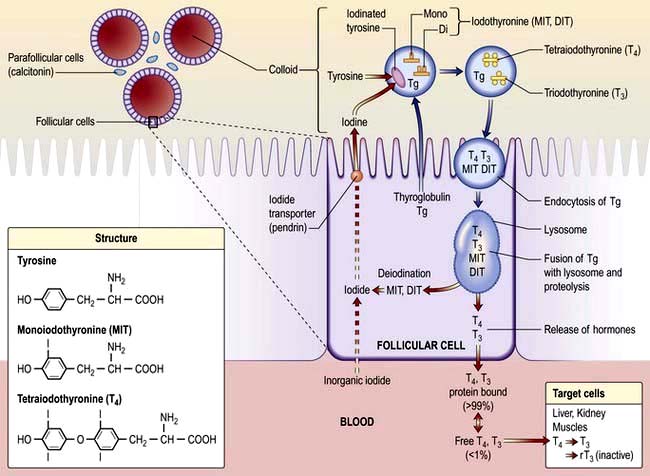
Control of the hypothalamic-pituitary-thyroid axis. Thyrotrophin-releasing hormone (TRH), a peptide produced in the hypothalamus, stimulates the pituitary to secrete thyroid-stimulating hormone (TSH) (see Fig. 19.2). TSH in turn stimulates growth and activity of the thyroid follicular cells via the G-protein coupled TSH membrane receptor (see Table 19.3). The T3 and T4 subsequently secreted into the circulation by follicular cells exert negative feedback on the hypothalamus, as described on page 941.
Circulating T4 is peripherally deiodinated to T3 which binds to the thyroid hormone nuclear receptor (TR) on target organ cells to cause modified gene transcription. There are two TR receptors (TR-α and TR-β) and the tissue-specific effects of T3 are dependent upon the local expression of these TR receptors. TR-α knockout mice show poor growth, bradycardia and hypothermia, whilst TR-β knockout mice show thyroid hyperplasia and high T4 levels in the presence of inappropriately normal circulating TSH, suggesting a role for the latter receptors in thyroid hormone resistance (see p. 967).
Physiological effects of thyroid hormones. The physiological effects of thyroid hormones are summarized in Table 19.12.
| Target | Effect |
|---|---|
|
Cardiovascular system |
Increases heart rate and cardiac output |
|
Bone |
Increases bone turnover and resorption |
|
Respiratory system |
Maintains normal hypoxic and hypercapnic drive in respiratory centre |
|
Gastrointestinal system |
Increases gut motility |
|
Blood |
Increases red blood cell 2,3-BPGa facilitating oxygen release to tissues |
|
Neuromuscular function |
Increases speed of muscle contraction and relaxation and muscle protein turnover |
|
Carbohydrate metabolism |
Increases hepatic gluconeogenesis/glycolysis and intestinal glucose absorption |
|
Lipid metabolism |
Increases lipolysis and cholesterol synthesis and degradation |
|
Sympathetic nervous system |
Increases catecholamine sensitivity and β-adrenergic receptor numbers in heart, skeletal muscle, adipose cells and lymphocytes |
Thyroid function tests
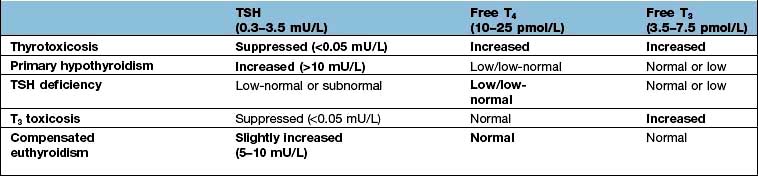
Immunoassays for free T4, free T3 and TSH are widely available. There are only minor circadian rhythms, and measurements may be made at any time. Particular uses of the tests are summarized in Table 19.13, with typical findings in common disorders.
Table 19.13 Characteristics of thyroid function tests in common thyroid disorders (the clinically most informative tests in each situation are shown in bold)
Problems in interpretation of thyroid function tests
There are three major areas of difficulty.
1.Serious acute or chronic illness





Hypothyroidism
Pathophysiology
Underactivity of the thyroid is usually primary, from disease of the thyroid, but may be secondary to hypothalamic-pituitary disease (reduced TSH drive) (Table 19.14). Primary hypothyroidism is one of the most common endocrine conditions with an overall UK prevalence of over 2% in women, but under 0.1% in men; lifetime prevalence for an individual is higher – perhaps as high as 9% for women and 1% for men with mean age at diagnosis around 60 years. The worldwide prevalence of subclinical hypothyroidism varies from 1% to 10%.
|
Primary disease of thyroid |
InfectivePost-subacute thyroiditis |
|
Congenital |
|
|
Agenesis |
Post-surgery |
|
Post-irradiation |
|
|
Defects of hormone synthesis |
|
|
Iodine deficiency |
|
|
Infiltration |
|
|
Tumour |
|
|
Secondary (to hypothalamic-pituitary disease) |
|
|
Autoimmune |
|
|
Hypopituitarism |
|
|
Isolated TSH deficiency |
|
|
Peripheral resistance to thyroid hormone |
Causes of primary hypothyroidism (Table 19.14)
Autoimmune
Atrophic (autoimmune) hypothyroidism. This is the most common cause of hypothyroidism and is associated with antithyroid autoantibodies leading to lymphoid infiltration of the gland and eventual atrophy and fibrosis. It is six times more common in females and the incidence increases with age. The condition is associated with other autoimmune disease such as pernicious anaemia, vitiligo and other endocrine deficiencies (p. 939). Occasionally intermittent hypothyroidism occurs with subsequent recovery; antibodies which block the TSH receptor may sometimes be involved in the aetiology.
Defects of hormone synthesis
Iodine deficiency. Dietary iodine deficiency still exists (p. 213) in some areas as ‘endemic goitre’ where goitre, occasionally massive, is common. The patients may be euthyroid or hypothyroid depending on the severity of iodine deficiency. The mechanism is thought to be borderline hypothyroidism leading to TSH stimulation and thyroid enlargement in the face of continuing iodine deficiency. Iodine deficiency is still a problem in the Netherlands, Western Pacific, India, South East Asia, Russia and parts of Africa. Efforts to prevent deficiency by providing iodine in salt continue worldwide but often with incomplete success. Even in the late 20th century of the 500 million with iodine deficiency in India, about 2 million had cretinism (see below).
Dyshormonogenesis. This rare condition is due to genetic defects in the synthesis of thyroid hormones; patients develop hypothyroidism with a goitre. One particular familial form is associated with sensorineural deafness due to a deletion mutation in chromosome 7, causing a defect of the transporter pendrin (Pendred’s syndrome) (see Fig. 19.16).
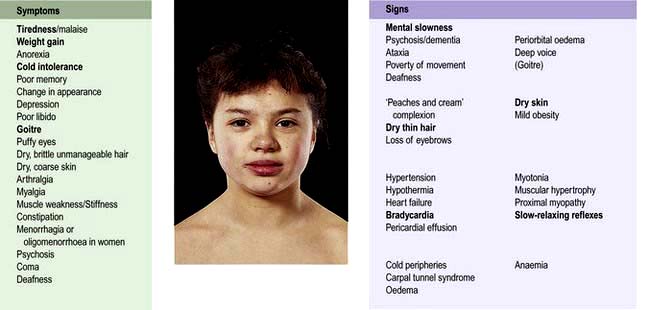
Clinical features (Fig. 19.17)
Special difficulties in diagnosis may arise in certain circumstances:



Investigation of primary hypothyroidism
 Anaemia, which is usually normochromic and normocytic in type but may be macrocytic (sometimes this is due to associated pernicious anaemia) or microcytic (in women, due to menorrhagia)
Anaemia, which is usually normochromic and normocytic in type but may be macrocytic (sometimes this is due to associated pernicious anaemia) or microcytic (in women, due to menorrhagia)
 Increased serum aspartate transferase levels, from muscle and/or liver
Increased serum aspartate transferase levels, from muscle and/or liver
 Increased serum creatine kinase levels, with associated myopathy
Increased serum creatine kinase levels, with associated myopathy
 Hypercholesterolaemia and hypertriglyceridaemia
Hypercholesterolaemia and hypertriglyceridaemia
 Hyponatraemia due to an increase in ADH and impaired free water clearance.
Hyponatraemia due to an increase in ADH and impaired free water clearance.
Myxoedema coma





Screening for hypothyroidism
 The incidence of congenital hypothyroidism is approximately 1 in 3500 births. Untreated, severe hypothyroidism produces permanent neurological and intellectual damage (‘cretinism’). Routine screening of the newborn using a blood spot, as in the Guthrie test, to detect a high TSH level as an indicator of primary hypothyroidism is efficient and cost-effective; cretinism is prevented if T4 is started within the first few months of life.
The incidence of congenital hypothyroidism is approximately 1 in 3500 births. Untreated, severe hypothyroidism produces permanent neurological and intellectual damage (‘cretinism’). Routine screening of the newborn using a blood spot, as in the Guthrie test, to detect a high TSH level as an indicator of primary hypothyroidism is efficient and cost-effective; cretinism is prevented if T4 is started within the first few months of life.

Hyperthyroidism
Hyperthyroidism (thyroid overactivity, thyrotoxicosis) is common, affecting perhaps 2–5% of all females at some time and with a sex ratio of 5 : 1, most often between the ages of 20 and 40 years. Nearly all cases (>99%) are caused by intrinsic thyroid disease; a pituitary cause is extremely rare (Table 19.15).
|
|
Graves’ disease
FURTHER READING
Bartalena L, Baldeschi L, Dickinson A et al. Consensus statement of the European Group on Graves’ orbitopathy (EUGOGO) on the management of GO. Eur J Endocrinol 2008; 158:273–285.
Brent GA. Graves’ disease. N Engl J Med 2008; 358:2594–2605.
Brent GA. Thyroid function and screening in pregnancy. N Engl J Med 2012; 366:562–568.
Other causes of hyperthyroidism/thyrotoxicosis
Clinical features of hyperthyroidism
The symptoms and signs of hyperthyroidism affect many systems (Fig. 19.18).

Figure 19.18 Hyperthyroidism: symptoms and signs. Bold type indicates symptoms or signs of greater discriminant value.
Symptomatology and signs vary with age and with the underlying aetiology.





Investigations
 Serum TSH is suppressed in hyperthyroidism (<0.05 mU/L), except for the very rare instances of TSH hypersecretion.
Serum TSH is suppressed in hyperthyroidism (<0.05 mU/L), except for the very rare instances of TSH hypersecretion.

 Thyroid peroxidase (TPO) and thyroglobulin antibodies are present in most cases of Graves’ disease.
Thyroid peroxidase (TPO) and thyroglobulin antibodies are present in most cases of Graves’ disease.
TSHR-Ab are not measured routinely, but are commonly present: thyroid-stimulating immunoglobulin (TSI) 80% positive, TSH-binding inhibitory immunoglobulin (TBII) 60–90% in Graves’ disease (see p. 961).
Treatment
Antithyroid drugs
\xEF”\xBF\xEF”\xBFCarbimazole\xEF”\xBF\xEF”\xBF is most often used in the UK, and propylthiouracil (PTU) is also used. Thiamazole (methimazole), the active metabolite of carbimazole, is used in the USA. These drugs inhibit the formation of thyroid hormones and also have other minor actions; carbimazole/thiamazole is also an immunosuppressive agent. Initial doses and side-effects are detailed in Table 19.16.
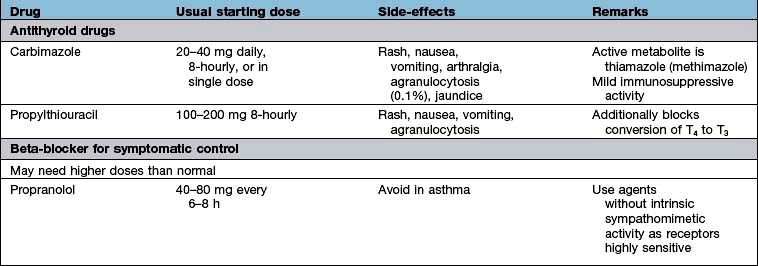
Although thyroid hormone synthesis is reduced very quickly, the long half-life of T4 (7 days) means that clinical benefit is not apparent for 10–20 days. As many of the manifestations of hyperthyroidism are mediated via the sympathetic system, beta-blockers are used to provide rapid partial symptomatic control; they also decrease peripheral conversion of T4 to T3. Drugs preferred are those without intrinsic sympathomimetic activity, e.g. propranolol (Table 19.16). They should not be used alone for hyperthyroidism except when the condition is self-limiting, as in subacute thyroiditis.
Dosage regimen
1. Start carbimazole 20–40 mg daily.
2. Review after 4–6 weeks and reduce the dose of carbimazole, depending on clinical state and fT4/fT3 levels. TSH levels may remain suppressed for several months and are unhelpful at this stage.
3. When clinically and biochemically euthyroid, stop beta-blockers.
4. Review thyroid function regularly during the planned course of treatment (typically 18 months – but some use courses between 6 and 24 months).
5. Reduce carbimazole if fT4 falls below or TSH rises above normal – and when approaching the end of the planned course.
6. Increase carbimazole if fT4 or fT3 are above normal (and consider if TSH remains suppressed after several months with a normal fT4).
7. Stop treatment at end of course if the patient is euthyroid on 5 mg daily carbimazole.
Radioactive iodine
Early discomfort in the neck and immediate worsening of hyperthyroidism are sometimes seen; if worsening occurs, the patient should receive propranolol (Table 19.16); if necessary carbimazole can be restarted. Euthyroidism normally returns in 2–3 months. People with dysthyroid eye disease are more likely to show worsening of eye problems after radioiodine than after antithyroid drugs; this represents a partial contraindication to RAI, although worsening can usually be prevented by steroid administration.
Surgery
 Early postoperative bleeding causing tracheal compression and asphyxia is a rare emergency requiring immediate removal of all clips/sutures to allow escape of the blood/haematoma.
Early postoperative bleeding causing tracheal compression and asphyxia is a rare emergency requiring immediate removal of all clips/sutures to allow escape of the blood/haematoma.

 Transient hypocalcaemia occurs in up to 10% but with permanent hypoparathyroidism in fewer than 1%.
Transient hypocalcaemia occurs in up to 10% but with permanent hypoparathyroidism in fewer than 1%.

Indications for either surgery or radioiodine are given in Box 19.4.
 Box 19.4
Box 19.4




Special situations in hyperthyroidism
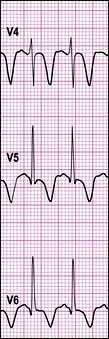
The fetus and maternal Graves’ disease
Untreated neonatal hyperthyroidism is probably associated with hyperactivity in later childhood.
Thyroid hormone resistance
Thyroid hormone resistance is an inherited condition caused by an abnormality of the thyroid hormone receptor. Mutations to the receptor (TR β) result in the need for higher levels of thyroid hormones to achieve the same intracellular effect. As a result, the normal feedback control mechanisms (see Fig. 19.2, p. 941) result in high blood levels of T4 with a normal TSH in order to maintain a euthyroid state. This has two consequences:
1. Thyroid function tests appear abnormal even when the patient is euthyroid and requires no treatment. Specialist review is required to differentiate from hyperthyroidism due to inappropriate TSH secretion.
2. Different tissues contain different thyroid hormone receptors and, in some families, receptors in certain tissues may have normal activity. In this case the level of thyroid hormones to maintain euthyroidism at pituitary and hypothalamic levels (which controls secretion of TSH) may be higher than that required in other tissues such as heart and bone, so that these tissues may exhibit ‘thyrotoxic’ effects in spite of a normal serum TSH. This ‘partial thyroid hormone resistance’ can be very difficult to manage effectively.
Thyroid eye disease
This is also known as dysthyroid eye disease or ophthalmic Graves’ disease.
Pathophysiology
The ophthalmopathy of Graves’ disease is due to a specific immune response that causes retro-orbital inflammation (Fig. 19.19). Swelling and oedema of the extraocular muscles lead to limitation of movement and to proptosis which is usually bilateral but can sometimes be unilateral. Ultimately increased pressure on the optic nerve may cause optic atrophy. Histology of the extraocular muscles shows focal oedema and glycosaminoglycan deposition followed by fibrosis. The precise autoantigen which leads to the immune response remains to be identified, but it appears to be an antigen in retro-orbital tissue with similar immunoreactivity to the TSH receptor.
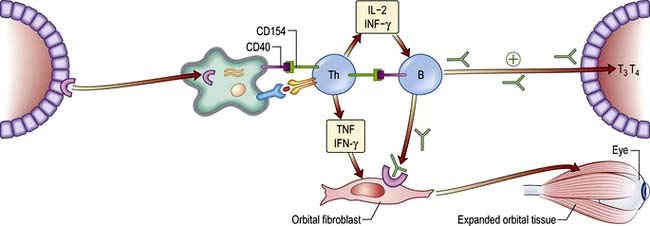
Eye disease is a manifestation of Graves’ disease and can occur in patients who may be hyperthyroid, euthyroid or hypothyroid. Thyroid dysfunction and ophthalmopathy usually occur within two years of each other although sometimes a gap of many years is seen. TSH receptor antibodies are almost invariably found in the serum but their role in the pathogenesis is becoming clearer (Fig. 19.19). Ophthalmopathy is more common and more severe in smokers.
Clinical features
The clinical appearances are characteristic (Fig. 19.18) but thyroid eye disease demonstrates a wide range of severity. A high proportion of people with Graves’ disease notice some soreness, painful watering or prominence of the eyes, and the ‘stare’ of lid retraction is relatively common. More severe proptosis occurs in a minority of cases, and limitation and discomfort of eye movement and visual impairment due to optic nerve compression are relatively uncommon. Proptosis and lid retraction may limit the ability to close the eyes completely so that corneal damage may occur. There is periorbital oedema and conjunctival oedema and inflammation.









Goitre (thyroid enlargement)
Goitre is more common in women than in men and may be either physiological or pathological.
Clinical features
Particular points of note are:
 Puberty and pregnancy may produce a diffuse increase in size of the thyroid.
Puberty and pregnancy may produce a diffuse increase in size of the thyroid.
 Pain in a goitre may be caused by thyroiditis, bleeding into a cyst or (rarely) a thyroid tumour.
Pain in a goitre may be caused by thyroiditis, bleeding into a cyst or (rarely) a thyroid tumour.
 Excessive doses of carbimazole or PTU will induce goitre.
Excessive doses of carbimazole or PTU will induce goitre.
 Iodine deficiency and dyshormonogenesis (see above) can also cause goitre.
Iodine deficiency and dyshormonogenesis (see above) can also cause goitre.
Assessment
There are two major aspects of any goitre: its pathological nature and the patient’s thyroid status.
The nature can often be judged clinically. Goitres (Table 19.17) are usually separable into diffuse and nodular types, the causes of which differ.
|
Diffuse |
Nodular |
|
Simple |
Multinodular goitre |
|
Physiological (puberty, pregnancy) |
Solitary nodular |
|
Tumours |
|
|
Adenomas |
|
|
Miscellaneous |
|
|
Sarcoidosis |
Investigations
Clinical findings will dictate appropriate initial tests:
 Thyroid function tests: TSH plus free T4 or T3 (see Table 19.7).
Thyroid function tests: TSH plus free T4 or T3 (see Table 19.7).



 Fine-needle aspiration (FNA). In people with a solitary nodule or a dominant nodule in a multinodular goitre, there is a 5% chance of malignancy; in view of this, FNA should be performed in the outpatient clinic or during ultrasound. Cytology in expert hands can usually differentiate the suspicious or definitely malignant nodule.
Fine-needle aspiration (FNA). In people with a solitary nodule or a dominant nodule in a multinodular goitre, there is a 5% chance of malignancy; in view of this, FNA should be performed in the outpatient clinic or during ultrasound. Cytology in expert hands can usually differentiate the suspicious or definitely malignant nodule.
 Thyroid scan (99mTc, 125I or 131I) can be useful to distinguish between functioning (hot) or non-functioning (cold) nodules. A hot nodule is only rarely malignant; however, a cold nodule is malignant in only 10% of cases and FNA has largely replaced isotope scans in the diagnosis of thyroid nodules.
Thyroid scan (99mTc, 125I or 131I) can be useful to distinguish between functioning (hot) or non-functioning (cold) nodules. A hot nodule is only rarely malignant; however, a cold nodule is malignant in only 10% of cases and FNA has largely replaced isotope scans in the diagnosis of thyroid nodules.



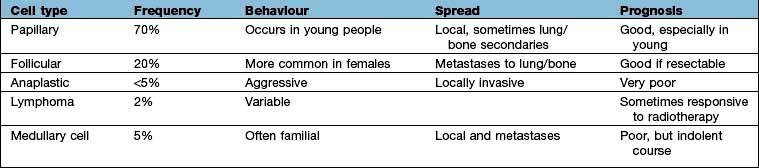
Thyroid carcinoma
Types of thyroid carcinoma, their characteristics and treatment are listed in Table 19.18. While not common, these tumours are responsible for 400 deaths annually in the UK and an annual incidence of 30 000 cases in the USA. Over 75% occur in women. In 90% of cases they present as thyroid nodules (see above), but occasionally with cervical lymphadenopathy (about 5%), or with lung, cerebral, hepatic or bone metastases.
Anaplastic carcinomas and lymphoma
These do not respond to radioactive iodine, and external radiotherapy produces only a brief respite.
Medullary carcinoma
Medullary carcinoma (MTC) is a neuroendocrine tumour of the calcitonin-producing C cells of the thyroid. This condition is often associated with multiple endocrine neoplasia type 2 (MEN 2, see p. 998) – approximately 25% of patients diagnosed with MTC have a mutation of the RET proto-oncogene, although the other manifestations of MEN 2 may be absent, hence the importance of genetic counselling and family screening. People with MEN2 mutations are advised to have a prophylactic thyroidectomy as early as 5 years of age to prevent the development of MTC.
FURTHER READING
British Thyroid Association and Royal College of Physicians Guidelines 2007. The management of thyroid cancer 2007. London: BTA
Wartofsky, L. Highlights of the American Thyroid Association Guidelines for patients with thyroid nodules or differentiated thyroid carcinoma: the 2009 revision. Thyroid 2009; 19:1139–43.
Reproduction and sex
Terminology in reproductive medicine is shown in Box 19.5.
 Box 19.5
Box 19.5
Definitions in reproductive medicine
|
Erectile dysfunction |
Inability of the male to achieve or sustain an erection adequate for satisfactory intercourse |
|
Azoospermia |
Absence of sperm in the ejaculate |
|
Oligospermia |
Reduced numbers of sperm in the ejaculate |
|
Libido |
Sexual interest or desire; often difficult to assess and is greatly affected by stress, tiredness and psychological factors |
|
Menarche |
Age at first period |
|
Primary amenorrhoea |
Failure to begin spontaneous menstruation by age 16 |
|
Secondary amenorrhoea |
Absence of menstruation for 3 months in a woman who has previously had cycles |
|
Oligomenorrhoea |
Irregular long cycles; often used for any length of cycle above 32 days |
|
Dyspareunia |
Pain or discomfort in the female during intercourse |
|
Menstruation |
Onset of spontaneous (usually regular) uterine bleeding in the female |
|
Virilization |
Occurrence of male secondary sexual characteristics in the female |
Embryology


Physiology
The male
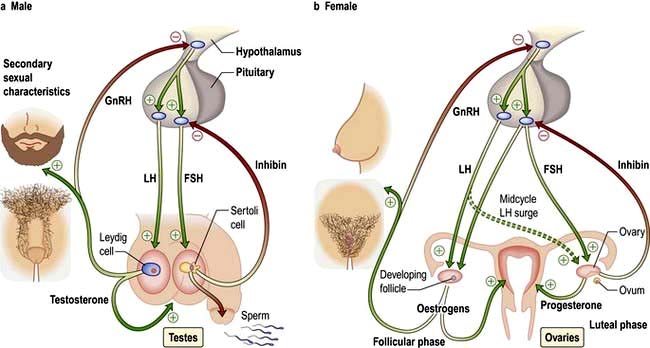
An outline of the hypothalamic-pituitary-gonadal axis is shown in Figure 19.20.
1. Pulses of gonadotrophin-releasing hormone (GnRH) are released from the hypothalamus and stimulate LH and FSH release from the pituitary. LH and FSH are composed of two glycoprotein chains (α and β subunits). The α subunits are identical and are shared with TSH, whilst the β subunit confers specific biological activity.
2. LH stimulates testosterone production from Leydig cells of the testis.
3. Testosterone acts via nuclear androgen receptors which interact with coregulatory proteins to produce the appropriate tissue responses: male secondary sexual characteristics, anabolism and the maintenance of libido. It also acts locally within the testis to aid spermatogenesis. Testosterone circulates largely bound to sex hormone-binding globulin (SHBG) (see p. 942). Testosterone feeds back on the hypothalamus/pituitary to inhibit GnRH secretion.
4. FSH stimulates the Sertoli cells in the seminiferous tubules to produce mature sperm and the inhibins A and B.
5. Inhibin feeds back to the pituitary to decrease FSH secretion. Activin, a related peptide, counteracts inhibin.
The female
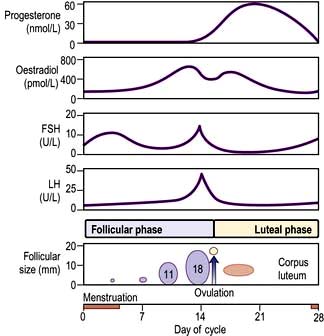
Female physiology is more complex (Figs 19.20, 19.21).
1. In the adult female, higher brain centres impose a menstrual cycle of 28 days upon the activity of hypothalamic GnRH.
2. Pulses of GnRH, at about 2-hour intervals, stimulate release of pituitary LH and FSH.
3. LH stimulates ovarian androgen production by the ovarian theca cells.
4. FSH stimulates follicular development and aromatase activity (an enzyme required to convert ovarian androgens to oestrogens) in the ovarian granulosa cells. FSH also stimulates release of inhibin from ovarian stromal cells, which inhibits FSH release. Activin counteracts inhibin (Fig. 19.20).
5. Although many follicles are ‘recruited’ for development in early folliculogenesis, by day 8–10 a ‘leading’ (or ‘dominant’) follicle is selected for development into a mature Graafian follicle.
6. Oestrogens have a double feedback action on the pituitary (Fig. 19.20). Initially they inhibit gonadotrophin secretion (negative feedback), but later high-level exposure results in increased GnRH secretion and increased LH sensitivity to GnRH (positive feedback), which leads to the mid-cycle LH surge inducing ovulation from the leading follicle (Fig. 19.21).
7. The follicle then differentiates into a corpus luteum, which secretes both progesterone and oestradiol during the second half of the cycle (luteal phase).
8. Oestrogen initially and then progesterone cause uterine endometrial proliferation in preparation for possible implantation; if implantation does not occur, the corpus luteum regresses and progesterone secretion and inhibin levels fall so that the endometrium is shed (menstruation) allowing increased GnRH and FSH secretion.
9. If implantation and pregnancy follow, human chorionic gonadotrophin (HCG) production from the trophoblast maintains corpus luteum function until 10–12 weeks of gestation, by which time the placenta will be making sufficient oestrogen and progesterone to support itself.
Puberty
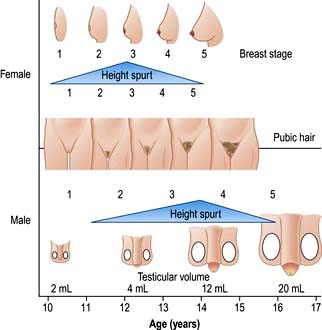
LH and FSH are both low in the prepubertal child. In early puberty, FSH begins to rise first, initially in nocturnal pulses; this is followed by a rise in LH with a subsequent increase in testosterone/oestrogen levels. The milestones of puberty in the two sexes are shown in Figure 19.22.
Precocious puberty
Other forms of precocity include:
 Cerebral precocity. Many causes of hypothalamic disease, especially tumours, present in this way. In boys this must be rigorously excluded. MRI scan is almost always indicated to exclude this diagnosis.
Cerebral precocity. Many causes of hypothalamic disease, especially tumours, present in this way. In boys this must be rigorously excluded. MRI scan is almost always indicated to exclude this diagnosis.
 McCune–Albright syndrome. This usually occurs in girls, with precocity, polyostotic fibrous dysplasia and skin pigmentation (café-au-lait). See also page 998.
McCune–Albright syndrome. This usually occurs in girls, with precocity, polyostotic fibrous dysplasia and skin pigmentation (café-au-lait). See also page 998.


The menopause
Clinical features and treatment
Women show a rapid loss of bone density in the 10 years following the menopause (osteoporosis, see p. 552) and the premenopausal protection from ischaemic heart disease disappears.
Hormone replacement therapy (HRT). Symptomatic patients should usually be treated but the previous widespread use of HRT has been thrown into doubt by a number of large prospective studies which have reported in recent years. Although scientific debate continues, the overall benefits and risks are summarized as in Box 19.6.
 Box 19.6
Box 19.6
Risks and benefits of hormone replacement therapya
Potential benefits
 Symptomatic improvement in most menopausal symptoms for the majority of women. Oestrogen-deficient symptoms respond well to oestrogen replacement, the vaguer general symptoms may or may not improve. Vaginal symptoms also respond to local oestrogen preparations.
Symptomatic improvement in most menopausal symptoms for the majority of women. Oestrogen-deficient symptoms respond well to oestrogen replacement, the vaguer general symptoms may or may not improve. Vaginal symptoms also respond to local oestrogen preparations.
 Protection against fractures of wrist, spine and hip, secondary to osteoporosis (~24–33%) (see p. 552), owing to protection of predominantly trabecular bone (p. 549). However, this is not a recommended indication for therapy.
Protection against fractures of wrist, spine and hip, secondary to osteoporosis (~24–33%) (see p. 552), owing to protection of predominantly trabecular bone (p. 549). However, this is not a recommended indication for therapy.
 Significant reduction in the risk of large bowel cancer (~33%).
Significant reduction in the risk of large bowel cancer (~33%).

Potential risks
 Significant increase in the risk of breast cancer (+26%), but no change in breast cancer mortality. This is primarily a risk of combined oestrogen-progesterone HRT. Some studies suggest that breast cancers diagnosed on HRT are easier to treat effectively.
Significant increase in the risk of breast cancer (+26%), but no change in breast cancer mortality. This is primarily a risk of combined oestrogen-progesterone HRT. Some studies suggest that breast cancers diagnosed on HRT are easier to treat effectively.

 Significant increase in the risk of ischaemic heart disease (+29%) and stroke (+41%).
Significant increase in the risk of ischaemic heart disease (+29%) and stroke (+41%).




FURTHER READING
Beral V; Million Women Study Collaborators. Breast cancer and hormone-replacement therapy in the Million Women Study. Lancet 2003; 362:419–427.
Chlebowski RT, Schwartz AG, Wakelee H et al. Oestrogen plus progestin and lung cancer in postmenopausal women (Women’s Health Initiative trial): a post-hoc analysis of a randomised controlled trial. Lancet 2009; 374:1243–1251.
Palmert MR, Dunkel L. Delayed puberty. N Engl J Med 2012; 336:443–453.
Clinical features of disorders of sex and reproduction
A detailed history and examination of all systems is required (Box 19.7). A man having regular satisfactory intercourse or a woman with regular ovulatory periods is most unlikely to have significant endocrine disease, assuming the history is accurate.
 Box 19.7
Box 19.7













Tests of gonadal function
 Low testosterone or oestradiol with high gonadotrophins indicates primary gonadal disease.
Low testosterone or oestradiol with high gonadotrophins indicates primary gonadal disease.


 Pregnancy provides complete demonstration of normal male and female function.
Pregnancy provides complete demonstration of normal male and female function.

| Test | Uses/comments |
|---|---|
|
Male |
|
|
Basal testosterone |
Normal levels exclude hypogonadism |
|
Sperm count |
Normal count excludes deficiencyMotility and abnormal sperm forms should be noted |
|
Female |
|
|
Basal oestradiol |
Normal levels exclude hypogonadism |
|
Luteal phase progesterone (days 18–24 of cycle) |
If >30 nmol/L, suggests ovulation |
|
Ultrasound of ovaries |
To confirm ovulation |
|
Both sexes |
|
|
Basal LH/FSH |
Demonstrates state of feedback system for hormone production (LH) and germ cell production |
|
(FSH) |
|
|
HCG test (testosterone or oestradiol measured) |
Response shows potential of ovary or testis; failure demonstrates primary gonadal problem |
|
Clomifene test (LH and FSH measured) |
Tests hypothalamic negative feedback system; clomifene is oestrogen antagonist and causes LH/FSH to rise |
|
LHRH test (rarely used) |
Shows adequacy (or otherwise) of LH and FSH stores in pituitary |
[/level-membership-for-internal-medicine-category][not-level-membership-for-internal-medicine-category]
Chapter 19 Endocrine disease
Introduction
The endocrine system consists of glands that exert their actions at distant parts of the body via the production of biologically active hormones secreted into the bloodstream. Unlike the neurological system, which produces an immediate response, the endocrine system typically has a slower and longer lasting effect on the body. The main endocrine glands are the pituitary, thyroid, adrenals, gonads, parathyroids and pancreas and the common endocrine problems seen in clinical practice are shown in Figure 19.1. The pituitary gland, a pea-sized structure situated at the base of the brain, plays a key role in the control and feedback mechanisms of the endocrine system and has been termed the ‘conductor of the endocrine orchestra’.
Clinical presentation of endocrine disease
History
|
|
Common endocrine conditions
The most common endocrine disorders, excluding obesity (Ch. 5) and diabetes mellitus (Ch. 20), are:
Aetiology of endocrine disease
Aetiological mechanisms common to many endocrine disorders include:
Autoimmune disease
Organ-specific autoimmune diseases can affect every major endocrine organ (Table 19.2). They are characterized by the presence of specific antibodies in the serum, often present years before clinical symptoms are evident, are usually more common in women and have a strong genetic component, often with an identical-twin concordance rate of 50% and with HLA associations (see individual diseases). Several of the autoantigens have been identified.
Endocrine tumours
Most endocrine tumours are benign, although a cytological or histological diagnosis may be needed if there is clinical or radiological suspicion of malignancy. Clinical presentation depends on whether the tumour is functional or non-functional, the latter presenting only as a mass clinically or on imaging. Palpable thyroid nodules are common, and mass effects are a frequent presentation of pituitary adenomas, but the increased use of high-resolution ultrasound and detailed cross-sectional imaging has revealed a very high prevalence of asymptomatic, incidentally-discovered thyroid, adrenal and pituitary lesions, commonly termed ‘incidentalomas’ (see p. 989).
Endocrine adenomas typically present in a single gland, although rarer multiple endocrine neoplasia (MEN) syndromes exist due to very specific mutations of a single gene, such as the mutations of the RET proto-oncogene in MEN 2 or the MEN1 gene mutation in MEN 1 (see p. 997).
Enzyme defects
The biosynthesis of most hormones involves many stages. Deficient or abnormal enzymes can lead to absent or reduced production of the secreted hormone. In general, severe deficiencies present early in life with obvious signs; partial deficiencies usually present later with mild signs or are only evident under stress. An example of an enzyme deficiency is congenital adrenal hyperplasia (CAH), where the molecular basis has also been identified as mutations or deletions of the gene encoding the relevant enzymes (see p. 987).
Receptor abnormalities
There are rare conditions in which hormone secretion and control are normal but the receptors are defective; thus, if androgen receptors are defective, normal levels of androgen will not produce masculinization (e.g. testicular feminization). There are also a number of rare syndromes of diabetes and insulin resistance from receptor abnormalities (see p. 1006); other examples include nephrogenic diabetes insipidus, pseudohypoparathyroidism and thyroid hormone resistance which can cause an unusual pattern of thyroid blood results.
Hormonal activity
Hormone action and receptors
Hormone receptors are broadly divided into:
Cell surface or membrane receptors: typically transmembrane receptors which contain hydrophobic sections spanning the lipid-rich plasma membrane and which trigger internal cellular messengers (see also p. 18)
Nuclear receptors which typically bind hormones and translocate them to the nucleus where they bind hormone response elements of nuclear DNA via characteristic amino-acid sequences (e.g. so-called ‘zinc fingers’, see p. 27).
Abnormal receptors are an occasional cause of endocrine disease (see p. 43).
Mechanisms of hormone-receptor action
Common structural mechanisms of hormone-receptor action are illustrated in Figure 2.9 (p. 25) and include:
stimulate cyclic AMP (cAMP) generation by adenylate cyclase – activating further intracellular kinases and leading to phosphorylation
lead to diacylglycerol (DAG) activation of C-kinase and subsequent protein phosphorylation.
Control and feedback
Most hormone systems are under tight regulatory control (typically by the hypothalamo-pituitary (HP) axis) by a system known as negative feedback. An example of the negative feedback system in the hypothalamo-pituitary-thyroid axis is demonstrated in Figure 19.2 and described here:
Primary and secondary gland failure
‘Primary’ hormone deficiency due to a disease process in the endocrine end-organ (thyroid, adrenal or gonad) will lead to a loss of negative feedback and subsequent elevation in the corresponding anterior pituitary hormone. Conversely, an abnormal hormone excess due to a disease process in the primary endocrine gland, or excess amount of exogenous hormone, will lead to increased negative feedback and suppression of the corresponding pituitary hormones.
Measurement of hormones
Hormones are measured in routine clinical practice by biochemical assays in the laboratory. It is possible to measure pituitary trophic hormones and the hormones produced by the end-organ glands, but hypothalamic hormones are not routinely measured in practice because of their low concentration and local action within the hypothalamo-pituitary axis. Circulating levels of most hormones are very low (10−9– 10−12 mol/L) and cannot be measured by simple chemical techniques. Hormones are therefore usually measured by immunoassays, which rely on highly specific polyclonal or monoclonal antibodies, which bind to the hormone being measured during the assay incubation. This hormone-antibody interaction is measured by use of labelled hormone after separation of bound and free fractions (Fig. 19.3).
Hormone binding proteins
Many hormones are transported in the bloodstream from the primary gland to their distant target organ attached to a specific binding protein (p. 940). It is more helpful to measure the free hormone rather than total bound hormone level, as this is the part that is biologically active. Some modern assays attempt to measure the free hormone level directly (e.g. free T4) and are therefore a more accurate reflection of biological activity, although there are often technical problems with this approach and many assays still measure total hormone level.
Patterns of hormonal secretion
Hormone secretion can be continuous or intermittent, for example:
Continuous secretion is shown by the thyroid hormones, with a half-life of 7–10 days for T4 and 6–10 hours for T3, and with little variation in levels over the day, month and year
Biological rhythms
Circadian means changes over the 24 hours of the day–night cycle and is best shown for the pituitary–adrenal axis. Figure 19.4 shows plasma cortisol levels measured over 24 hours – levels are highest in the early morning and lowest overnight. Additionally, cortisol release is pulsatile, following the pulsatility of pituitary ACTH. Thus ‘normal’ cortisol levels vary during the day and great variations can be seen in samples taken only 30 minutes apart.
The menstrual cycle is an example of a longer and more complex (28-day) biological rhythm (see p. 972).
Testing endocrine function
Basal blood levels
Assays for all clinically relevant pituitary and end-organ hormones are available.
Stimulation and suppression tests
For example, where the secretory capacity of a gland is damaged, maximal stimulation by the trophic hormone will give a diminished output. Thus, in the short ACTH stimulation test for adrenal reserve (Box 19.1, Fig. 19.5a), the healthy subject shows a normal response while the subject with primary hypoadrenalism (Addison’s disease) demonstrates an impaired cortisol response to tetracosactide (an ACTH analogue).
A patient with a hormone-producing tumour usually fails to show normal negative feedback. A patient with Cushing’s disease (excess pituitary ACTH) will thus fail to suppress ACTH and cortisol production when given a dose of synthetic steroid, in contrast to normal subjects. Figure 19.5b shows the response of a normal subject given dexamethasone 1 mg at midnight; cortisol is suppressed the following morning. The subject with Cushing’s disease shows inadequate suppression.
The pituitary gland and hypothalamus
Anatomy
The anatomical relations of the hypothalamus and pituitary (Fig. 19.6) include the optic chiasm just above the pituitary fossa; any expanding lesion from the pituitary or hypothalamus can thus produce visual field defects by pressure on the chiasm. Such upward expansion of the gland through the diaphragma sellae is termed ‘suprasellar extension’. Lateral extension of pituitary lesions may involve the vascular and nervous structures in the cavernous sinus and may rarely reach the temporal lobe of the brain. The pituitary is itself encased in a bony box, therefore any lateral, anterior or posterior expansion must cause bony erosion.
Physiology
Hypothalamus
Hypothalamic neurones secrete pituitary hormone-releasing and -inhibiting factors and hormones (Table 19.3) into the portal system which run down the stalk to the pituitary. As well as the classical hormones illustrated in Figure 19.7, the hypothalamus also contains large amounts of other neuropeptides and neurotransmitters such as neuropeptide Y, vasoactive intestinal peptide (VIP) and nitric oxide that can also alter pituitary hormone secretion.
Posterior pituitary
The posterior pituitary is neuro-anatomically connected to specific hypothalamic nuclei, and acts merely as a storage organ. Antidiuretic hormone (ADH, also called vasopressin) and oxytocin, both nonapeptides, are synthesized in the supraoptic and paraventricular nuclei in the anterior hypothalamus. They are then transported along the axon and stored in the posterior pituitary (Fig. 19.7). This means that damage to the stalk or pituitary alone does not prevent synthesis and release of ADH and oxytocin. ADH is discussed on page 991; oxytocin produces milk ejection and uterine myometrial contraction.
Presentations of pituitary and hypothalamic disease
Pituitary space-occupying lesions and tumours
Pituitary tumours (Table 19.4) are the most common cause of pituitary disease, and the great majority of these are benign pituitary adenomas, usually monoclonal in origin. Problems are caused by:
Table 19.4 Characteristics of common pituitary and related tumours
| Tumour or condition | Usual size | Most common clinical presentation |
|---|---|---|
|
Prolactinoma |
Most <10 mm (microprolactinoma) |
Galactorrhoea, amenorrhoea, hypogonadism, erectile dysfunction |
|
|
Some >10 mm (macroprolactinoma) |
As above plus headaches, visual field defects and hypopituitarism |
|
Acromegaly |
Few mm to several cm |
Change in appearance, visual field defects and hypopituitarism |
|
Cushing’s disease |
Most small: few mm (some cases are hyperplasia) |
Central obesity, cushingoid appearance (local symptoms rare) |
|
Nelson’s syndrome |
Often large: >10 mm |
Post-adrenalectomy, pigmentation, sometimes local symptoms |
|
Non-functioning tumours |
Usually large: >10 mm |
Visual field defects; hypopituitarism (microadenomas may be incidental finding) |
|
Craniopharyngioma |
Often very large and cystic (skull X-ray abnormal in >50%; calcification common) |
Headaches, visual field defects, growth failure (50% occur below age 20; about 15% arise from within sella) |
Investigations (of a possible or proven mass)
Is there a tumour?
the visual pathways, with field defects and visual loss (most common)
the cavernous sinus, with III, IV and VI cranial nerve lesions
bony structures and the meninges surrounding the fossa, causing headache
the ventricles, causing interruption of cerebrospinal fluid (CSF) flow leading to hydrocephalus
Investigations
MRI of the pituitary. MRI is superior to CT scanning (Fig. 19.8) and will readily show any significant pituitary mass. Small lesions within the pituitary fossa on MRI consistent with small pituitary microadenomas are very common (10% of normal individuals in some studies). Such small lesions are sometimes detected during MRI scanning of the head for other reasons – so-called ‘pituitary incidentalomas’.
Visual fields. These should be plotted formally by automated computer perimetry or Goldmann perimetry, but clinical assessment by confrontation using a small red pin as target is also sensitive and valuable. Common defects are upper temporal quadrantanopia and bitemporal hemianopia (see p. 1073).
Is there a hormonal excess?
Prolactin excess (prolactinoma or hyperprolactinaemia): histologically, prolactinomas are ‘chromophobe’ adenomas (a description of their appearance on classical histological staining)
GH excess (acromegaly or gigantism): somatotroph adenomas, usually ‘acidophil’, and sometimes due to specific G-protein mutations (see p. 945)
The clinical features of acromegaly, Cushing’s disease or hyperprolactinaemia are usually (but not always) obvious, and are discussed on pages 953, and 957. Hyperprolactinaemia may be clinically ‘silent’. Tumours producing LH, FSH or TSH are well described but very rare.
Is there a deficiency of any hormone?
Clinical examination may give clues; thus, short stature in a child with a pituitary tumour is likely to be due to GH deficiency. A slow, lethargic adult with pale skin is likely to be deficient in TSH and/or ACTH. Milder deficiencies may not be obvious, and require specific testing (see Table 19.7).
Treatment
Treatment depends on the type and size of tumour (Table 19.5). In general, therapy has three aims:
Table 19.5 Comparisons of primary treatments for pituitary tumours
| Treatment method | Advantages | Disadvantages |
|---|---|---|
|
Surgical |
|
|
|
Trans-sphenoidal adenomectomy or hypophysectomy |
Relatively minor procedure Potentially curative for microadenomas and smaller macroadenomas |
Some extrasellar extensions may not be accessible |
|
Transcranial (usually transfrontal) |
Good access to suprasellar region |
Major procedure; danger of frontal lobe damage |
|
Radiotherapy |
|
|
|
External (40–50 Gy) |
Non–invasive |
Slow action, often over many years |
|
Stereotactic |
Precise administration of high dose to lesion |
Long-term follow-up data limited |
|
Medical |
|
|
|
Dopamine agonist therapy (e.g. bromocriptine, cabergoline) |
Non-invasive; reversible |
Usually not curative; significant side-effects in minority |
|
Somatostatin analogue therapy (octreotide, lanreotide) |
Non-invasive; reversible |
Usually not curative; gallstones; expensive |
|
Growth hormone receptor antagonist (pegvisomant) |
Highly selective |
Usually not curative; very expensive |
Removal/control of tumour
Surgery via the trans-sphenoidal route is usually the treatment of choice. Very large tumours are occasionally removed via the open transcranial (usually transfrontal) route.
Medical therapy with somatostatin analogues and/or dopamine agonists sometimes causes shrinkage of specific types of tumour (see p. 954) and if successful can be used as primary therapy.
Reduction of excess hormone secretion
Reduction is usually obtained by surgical removal but sometimes by medical treatment. Useful control can be achieved with dopamine agonists for prolactinomas or somatostatin analogues for acromegaly, but ACTH secretion usually cannot be controlled by medical means. Growth hormone antagonists are also available for acromegaly (p. 955).
Replacement of hormone deficiencies
Replacement of hormone deficiencies, i.e. hypopituitarism, is discussed below (see Table 19.8).
| Axis | Usual replacement therapies |
|---|---|
|
Adrenal |
Hydrocortisone 15–40 mg daily (starting dose 10 mg on rising/5 mg lunchtime/5 mg evening) |
|
(Normally no need for mineralocorticoid replacement) |
|
|
Thyroid |
Levothyroxine 100–150 µg daily |
|
Gonadal |
|
|
Male |
Testosterone intramuscularly, orally, transdermally or implant |
|
Female |
Cyclical oestrogen/progestogen orally or as patch |
|
Fertility |
HCG plus FSH (purified or recombinant) or pulsatile GnRH to produce testicular development, spermatogenesis or ovulation |
|
Growth |
Recombinant human GH used routinely to achieve normal growth in children |
|
Also advocated for replacement therapy in adults where GH has effects on muscle mass and wellbeing |
|
|
Thirst |
Desmopressin 10–20 µg one to three times daily by nasal spray or orally 100–200 µg three times daily |
|
Carbamazepine, thiazides and chlorpropamide are very occasionally used in mild diabetes insipidus |
|
|
Breast (prolactin inhibition) |
Dopamine agonist (e.g. cabergoline, 500 µg weekly) |
Differential diagnosis of pituitary or hypothalamic masses
Other tumours
Craniopharyngioma (1–2%), a usually cystic hypothalamic tumour, often calcified, arising from Rathke’s pouch, often mimics an intrinsic pituitary lesion. It is the most common pituitary tumour in children but may present at any age.
Hypopituitarism
Causes
Disorders causing hypopituitarism are listed in Table 19.6. Pituitary and hypothalamic tumours, and surgical or radiotherapy treatment, are the most common.
|
Congenital |
Traumatic |
|
Isolated deficiency of pituitary hormones (e.g. Kallmann’s syndrome) |
Skull fracture through base |
|
Infiltrations |
|
|
Infective |
Sarcoidosis |
|
Basal meningitis (e.g. tuberculosis) |
|
|
Vascular |
|
|
Pituitary apoplexy |
|
|
Others |
|
|
Radiation damage |
|
|
Immunological |
‘Functional’ |
|
Autoimmune (lymphocytic) hypophysitis |
Anorexia nervosa |
|
Neoplastic |
|
|
Pituitary or hypothalamic tumours |
|
|
Craniopharyngioma |
|
|
Meningiomas |
|
|
Gliomas |
|
|
Pinealoma |
|
|
Secondary deposits, especially breast |
|
|
Lymphoma |
Clinical features
Secondary hypothyroidism and adrenal failure both lead to tiredness and general malaise.
Hypothyroidism causes weight gain, slowness of thought and action, dry skin and cold intolerance.
Hyperprolactinaemia may cause galactorrhoea and hypogonadism.
GH deficiency causes growth failure in children and impaired wellbeing in some adults.
Kallmann’s syndrome. This syndrome is isolated gonadotrophin (GnRH) deficiency (p. 976).This syndrome arises due to mutations in the KAL1 gene which is located on the short (p) arm of the X chromosome. Kallmann’s is classically characterized by anosmia because the KAL1 gene provides instructions to make anosmin, which has a role in development of both the olfactory system as well as migration of GnRH secreting neurones.
Investigations
Tests range from the simple basal levels (e.g. free T4 for the thyroid axis), to stimulatory tests for the pituitary, and tests of feedback for the hypothalamus (Table 19.7). Assessment of the hypothalamic-pituitary-adrenal axis is complex: basal 09:00 hours cortisol levels above 400 nmol/L usually indicate an adequate reserve, while levels below 100 nmol/L predict an inadequate stress response. In many cases basal levels are equivocal and a dynamic test is essential: the insulin tolerance test (Box 19.2) is widely regarded as the ‘gold standard’ but the short ACTH stimulation test (Box 19.1), though an indirect measure, is used by many as a routine test of hypothalamic-pituitary-adrenal status. Occasionally, the difference between ACTH deficiency and normal HPA axis can be subtle, and the assessment of adrenal reserve is best left to an experienced endocrinologist.
Box 19.2
Insulin tolerance test
Procedure
Test explained to patient and consent obtained
Should only be performed in experienced, specialist units
Intravenous hydrocortisone and glucose available for emergency
Overnight fast, begin at 08:00–09:00 hours
Soluble insulin, 0.15 U/kg, i.v. at time 0
Glucose, cortisol and GH levels at 0, 30, 45, 60, 90, 120 min
Treatment
Steroid and thyroid hormones are essential for life. Both are given as oral replacement drugs, as in primary thyroid and adrenal deficiency, aiming to restore the patient to clinical and biochemical normality (Table 19.8) and levels are monitored by routine hormone assays. Note: Thyroid replacement should not commence until normal glucocorticoid function has been demonstrated or replacement steroid therapy initiated, as an adrenal ‘crisis’ may otherwise be precipitated.
[/not-level-membership-for-internal-medicine-category]
